

A locomotor locus shows high divergence and signatures of introgression among Heliconius butterflies.
Abstract
Heliconius butterflies have undergone adaptive radiation and therefore serve as an excellent system for exploring the continuum of speciation and adaptive evolution. However, there is a long-lasting paradox between their convergent mimetic wing patterns and rapid divergence in speciation. Here, we characterize a locus that consistently displays high divergence among Heliconius butterflies and acts as an introgression hotspot. We further show that this locus contains multiple genes related to locomotion and conserved in Lepidoptera. In light of these findings, we consider that locomotion traits may be under selection, and if these are heritable traits that are selected for, then they might act as species barriers.
INTRODUCTION
Speciation involves the complex interplay of multiple factors. During this process, selection acts as a main driving force to shape incipient species, resulting in the aggregation of differentiated morphological and physiological traits (1). A few traits are directly under directional selection and play a major role in speciation, such as beak size in Darwin’s finches and skeletal shape in threespine sticklebacks (2, 3). Some of these speciation traits are ecological traits that trigger reproductive isolation, whereas others are subject to directional selection and associated with a mating cue or directly contribute to assortative mating, known as classic or automatic “magic traits” (4).
The underlying mechanisms of speciation traits have also been illustrated at the molecular level, with an increasing number of speciation genes being identified and characterized. Many of these studies have focused on a single gene or tightly linked genes such as the hybrid sterility gene Overdrive and the hybrid male sterility 1 (HMS1) region (encoding DNA binding proteins) in Drosophila (5, 6). Limited case studies have documented the interactions of genetic loci associated with different traits, such as the genetic coupling of vision and pigmentation genes and the linkage of mate preference and wing pattern loci in Heliconius butterflies (7, 8). Disentangling the relationships of multiple ecological and mating traits triggering reproductive isolation or involved in early speciation might provide a more comprehensive understanding of speciation.
Neotropical butterflies in the Heliconius genus exhibit Müllerian wing patterns for predator warning and have provided an ideal system for exploring the continuum of speciation and adaptive evolution (9, 10). Their wing patterns have also contributed to reproductive isolation as a critical mating cue (8, 11). Therefore, the genetic basis of their characteristics has drawn considerable attention since the 1950s (12) and more than 30 wing pattern loci have been documented such as the B/D locus for red pigmentation, Yb for yellow hindwing bars, and K for forewing band color (8, 13–15). Notably, there remains a long-lasting paradox between the convergence of mimetic wing patterns and the rapid divergence and speciation in this genus (16). Besides wing patterns, other behavior and ecological factors may contribute to build and establish the species barriers, such as mating preference, chemical sensing, and habitat selection. For example, from a holistic perspective, butterfly wings also exhibit behavioral and dynamic traits, and the literature documents the existence of synergy between behavior and morphology, such as the behavioral similarity of mimetic butterflies (10). A series of behavioral and genomics studies indicate that wing locomotion might be involved in Heliconius adaptive radiation (17–19). Recently, studies based on multiomics data also provide insights into assortative mating (20, 21) and chemosensory communication (22), but the genetic mechanisms of other traits remain unclear.
Here, with the aim of addressing speciation and adaptation of Heliconius butterflies, we examine the genome-wide patterns of differentiation in multiple Heliconius races/species and identify a locus, L, that displays strong linkage disequilibrium (LD) and high divergence. We elucidate the complex evolutionary history of L and reveal that it is a hotspot region for introgression. We also show that the L locus contains a group of locomotion-related genes clustered only in Lepidoptera. Our work has systematically characterized the function and evolution of a locomotor locus and provided a fresh perspective regarding the role of biomechanical traits in butterfly adaptation and speciation.
RESULTS
Taxon sampling and whole-genome phylogenetic analysis
The highly divergent and rapidly evolving Heliconius genus is a model system for understanding the speciation process (Fig. 1A). With the aim of characterizing genetic regions involved in different stages of divergence and isolation, we first obtained multiple whole-genome resequencing datasets of representative taxa in Heliconius genus from previous studies and generated a whole-genome phylogeny using 105.2 Mb of genome-wide single-nucleotide polymorphism (SNP) data with sequences aligned to the Heliconius melpomene reference genome (Fig. 1B) (23). See Materials and Methods for detailed information of data sources. This genome-wide phylogeny yielded the same tree topology for the silvaniform-cydno-melpomene clade as reported in the previous study (18) but yielded a different topology for the Heliconius erato subspecies (24). To avoid additional missing data and biased sampling, we aligned the H. erato samples to the H. erato reference genome (24) for downstream analyses. We also obtained additional H. e. emma samples from different populations from a previous study (24) and added them to examine this discordant tree topology (fig. S1). The frequent incongruence in H. erato subspecies, likely owing to the complex history of divergent regions across the genome, is consistent with the view resulting from a recent genomics study indicating that the phylogeny of Heliconius is not a simple bifurcating tree (25). See Supplementary Results for a complete description of the phylogenetic incongruence in H. erato subspecies.
Fig. 1. The geographical distribution and genome-wide phylogeny of Heliconius butterflies.
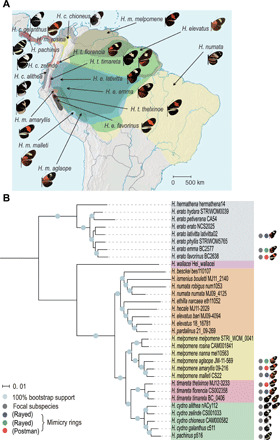
(A) The distributions of sampled subspecies/species are shown in different colors, along with wing pattern images. (B) The maximum likelihood phylogenetic tree was constructed on the basis of 102.5 Mb of genome-wide SNP data. The color ranges represent major clades. The scale bar represents the percentage of substitutions per site. The round labels indicate the selected subspecies used in downstream analyses, among which the three H. melpomene subspecies form co-mimicry pairs with selected H. erato subspecies and H. timareta subspecies.
According to the geographical distribution and phylogenetic results, we focused on four taxa of Heliconius genus—Heliconius timareta, Heliconius cydno and Heliconius pachinus, H. melpomene, and H. erato (Fig. 1B)—and evaluated the genomic differentiations between the geographical races, incipient species, and sympatric sister species across the speciation continuum. Each group contained subspecies displaying similar or different wing patterns and formed comimetic pairs with species from other groups (Fig. 1A). Among these species, H. cydno and H. melpomene are sister species belonging to the melpomene-cydno-timareta clade. They coexist in Central America and the western slopes of the Andes and have incomplete reproductive isolation with occasional hybridization (26). Relative to H. melpomene, H. timareta is genetically closer to H. cydno and is distributed across the eastern Andes (27). However, sympatric races of H. timareta and H. melpomene show remarkable signatures of introgression (27). These three distinct but closely related species split into a series of subspecies across their geographic ranges. H. erato is distantly related to the melpomene-cydno-timareta clade, but its races have undergone parallel radiation with H. melpomene races throughout the Neotropics (28). Thus, pairwise comparisons of three timareta races, four cydno races, five melpomene races, and three erato races were made to locate the genomic islands with increased divergence. We also compared the melpomene samples at the scale of allopatric races from the eastern and western slopes of the Andes, compared more established species from the cydno and melpomene clades, and divided samples of H. e. emma into two populations according to the phylogeny (fig. S1). The small genome-wide FST values for comparisons in each group suggested that these samples were all in the early stage but at different levels of speciation (table S1).
Genetic divergence across the L locus and wing pattern loci
We calculated FST for every 50-kb window across the genome between races/species in each group. The low FST throughout the genome indicated reduced divergence between incipient species, whereas a few outlier peaks displayed substantially higher values, including two peaks corresponding to two known color pattern and mimicry loci, Yb and B/D, respectively (Fig. 2). However, regardless of the wing patterns, we observed a consistent peak that stood out on chromosome one in both incipient and more established species and showed FST values greater than the FST at B/D and Yb, ranked in the top 0.05 to 0.1% in H. timareta, the top 0.05 to 0.5% in H. cydno and H. pachinus except between H. cydno galanthus and H. cydno chioneus, and the top 1% in the comparison of H. c. galanthus and H. melpomene rosina (Fig. 2, fig. S2, and tables S1 and S2). Notably, this region was the only outlier region shared by H. timareta and H. cydno comparisons (fig. S3). For melpomene races from Peru and Colombia, the differentiation patterns were mainly explained by B/D and Yb locus, but we captured a pattern that stood out in this region that had a greater FST value than the genome-wide mean value (Mann-Whitney U test, P < 0.001; Fig. 2 and table S1) in the comparison of allopatric melpomene races from the western and eastern slopes of the Andes (Fig. 2, fig. S4, and table S2). In the erato group, the corresponding peak regions were relatively smaller but still noticeable, so we calculated FST values for every 20-kb window instead and observed the corresponding regions for which the FST values were significantly higher among all the erato comparisons (Mann-Whitney U test, P < 0.001; fig. S4 and table S1). In particular, the corresponding regions were ranked in the top 1% in the comparison of the two H. e. emma populations (table S2). In addition, this outlier region also exhibited elevated LD (r2) relative to neighboring regions in the timareta and cydno groups (Fig. 3A and fig. S5). Owing to the relatively fewer SNPs in this region, the LD pattern was not discernible in the melpomene and erato groups (fig. S5). Given that these FST outlier regions on chromosome one show elevated LD in multiple Heliconius races/species, we propose that this region likely acts as a single locus and named it the L locus. The divergence across L has been repeatedly observed among incipient species in both the melpomene-cydno-timareta and erato clades and between more established species H. cydno and H. melpomene, suggesting a consistent role of L that has contributed to large genomic differences between multiple Heliconius races/species. Notably, some of these incipient species with divergent L display similar wing patterns (e.g., between H. cydno subspecies, between H. t. florencia and H. t. timareta, and between H. e. emma populations) (Fig. 1). These findings indicate a quite different evolutionary history of L from that of wing pattern loci.
Fig. 2. Genomic divergence during early speciation.
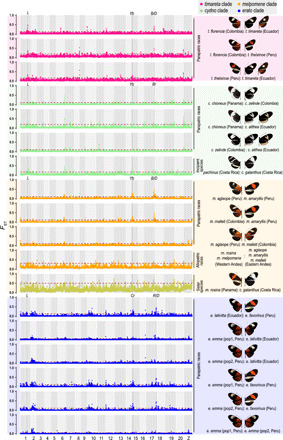
Pairwise FST values are calculated across the genome in 50-kb sliding windows with a step size of 20 kb, between subspecies of H. timareta, H. cydno and H. pachinus, and H. melpomene; between the eastern and western populations of H. melpomene; and between H. m. rosina and H. c. galanthus. Twenty-kilobase sliding windows are used for the calculation of FST values between subspecies of H. erato. Results of comparisons within each clade are grouped in the same color. The red dashed lines represent the top 1% threshold across the genome. The L locus and other wing patterning loci are labeled with gray bars. In H. erato, the orthologous loci of Yb and B/D are known as Cr and R/D, respectively.
Fig. 3. Local patterns of LD, genetic diversity, and phylogenetic trees along the L locus.
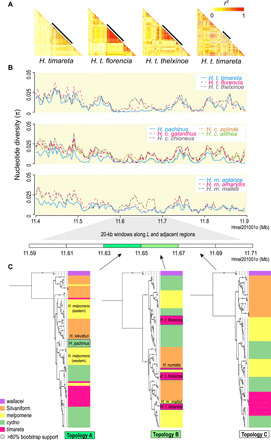
(A) Pairwise LD, measured as r2, is estimated among biallelic SNPs along the L locus and its adjacent regions for all the H. timareta samples, with the L locus indicated by bold black bars. (B) Values of nucleotide diversity (π) are calculated along the L locus for the subspecies of H. timareta, H. cydno and H. pachinus, and H. melpomene. (C) Maximum likelihood phylogenetic trees are constructed for every 20-kb window along the L locus and adjacent regions. The H. timareta subspecies are grouped polyphyletically in the window of topology B, with signatures of introgression between H. t. thelxinoe and H. numata and between H. t. timareta and H. m. malleti. The eastern and western melpomene populations join the silvaniform and cydno-timareta clades, respectively, in the window of topology A, with signatures of introgression between eastern melpomene and H. elevatus and between western melpomene and H. pachinus, respectively.
Evidence of selection and introgression at the L locus
Focusing on the L locus, we calculated the nucleotide diversity (π) for each comparison of the four groups, and we observed significantly reduced diversity at L, comparable to the reduced diversity at B/D and Yb (Mann-Whitney U test, P < 0.001; Fig. 3B and table S4). These results suggest that the L locus was subject to strong selection. We also annotated a list of protein-coding genes located within the L locus (fig. S5 and table S3). Notably, the L locus contains two collagen type IV genes previously reported to be involved in monarch butterfly migration (29) and introgression among distantly related Heliconius butterflies (table S3) (18). In addition, given that the widespread reticulate evolution is observed across the Heliconius genus (25), we constructed sliding-window phylogenetic trees along the L locus and observed different topologies from the species tree and the tree of the color pattern locus B/D (Fig. 3C and fig. S6). In particular, the three timareta subspecies became polyphyletic within a 20-kb interval ranging from 11.65 to 11.67 Mb on the scaffold Hmel201001o on chromosome one, where they were grouped separately within the cydno, melpomene, and silvaniform clades (Fig. 3C). Likewise, the western and eastern melpomene populations joined the cydno-timareta and silvaniform clades, respectively, within a 20-kb interval ranging from 11.63 to 11.65 Mb on Hmel201001o, and the western melpomene was grouped with H. pachinus in this region (Fig. 3C). Another instance of topological discordance was observed in the erato group, in which H. e. lativitta was found to group with H. e. favorinus within a 20-kb interval ranging from 15.26 to 15.28 Mb on the scaffold Herato0101 on chromosome one, whereas H. e. favorinus was closer to H. e. emma in the species tree (Fig. 1B and figs. S7 and S8). Given that both introgression and incomplete lineage sorting can result in discordant topologies, we tested the patterns of introgression at L by integrating the D-statistic and fd-statistic (30) and then distinguished between introgression and ancestral variation by comparing the absolute divergence (dxy) (table S5) (31). We combined these results and interpreted the typical patterns of introgression along the L locus between H. t. timareta and H. m. malleti and between H. t. thelxinoe and Heliconius numata, which explained the polyphyletic topology of the three H. timareta subspecies within L (fig. S9 and table S5). We also found evidence of possible introgression between eastern H. melpomene and Heliconius elevatus, between western H. melpomene and H. pachinus, and between H. e. lativitta and H. e. favorinus, indicating an even more complex evolutionary history of L (fig. S9 and table S5). In summary, we observed multiple signatures of gene flow at L between H. t. timareta and H. m. malleti, between H. t. thelxinoe and H. numata, between eastern H. melpomene and silvaniform species, and between H. pachinus and H. melpomene. All these cases suggest that additional divergence could be introduced via hybridization.
Given that we did not detect introgression involved in the differentiation of L among H. cydno subspecies, we used them to tease apart the role of L in ecological speciation in the absence of gene flow. Genome-wide pairwise FST between H. cydno subspecies showed signatures of isolation by distance, which were linearly and positively correlated with geographic distance. FST of Yb and Br loci also yielded weak albeit constant signatures, whereas FST of L increased with geographic distance and was approximately eightfold faster than the genome-wide FST (Fig. 4). These results suggested that L was likely resistant to gene flow between parapatric races and/or subject to strong directional selection in different populations, revealing its possible role in H. cydno local adaptation and speciation.
Fig. 4. Correlations between genetic differentiation and geographical distance in H. cydno subspecies.
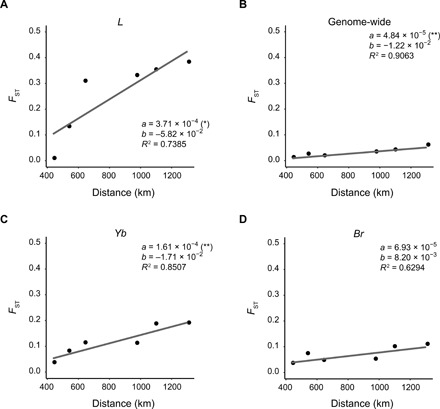
Point plots show the correlation of pairwise genetic differentiation and geographical distance in H. cydno subspecies. The distribution center of each H. cydno subspecies was measured as the geographic midpoint of sampled individuals, and pairwise geodesic distances between H. cydno subspecies were calculated. FST for specific loci, L (A) and two wing patterning loci Yb (C) and Br (D), and genome-wide FST (B) are plotted against geographical distances. FST of loci L and Yb are significantly and positively correlated with distances, as well as the genome-wide mean FST. The differentiation of L shows the strongest evidence for differentiation by distance with a slope eightfold greater than the genome-wide mean FST and twofold greater than FST of Yb in the linear fitting. **P < 0.01; *P < 0.05.
Functional implications of the L locus in Heliconius butterflies
Given the strong LD around the L locus, our a priori hypothesis was that these tightly linked genes might coordinately drive differentiation and speciation in Heliconius butterflies. According to orthologous gene annotation in Drosophila melanogaster computed at FlyBase (http://flybase.org/), at least four genes were considered to be related to locomotion including Col4α1 and Vkg, related to extracellular matrix organization (32, 33) and skeletal muscle tissue development (34); Oseg4, related to indirect flight muscle and wing structure (34, 35); and na, related to adult locomotor behavior (36, 37), although direct evidence is still lacking (table S3). To test this hypothesis, we analyzed wing motion in Heliconius species and observed significantly distinct wing beat frequencies (WBFs) between H. cydno and H. melpomene and between H. m. malleti (eastern melpomene) and H. m. rosina (western melpomene) (Fig. 5A), in line with their remarkable genetic differentiation at L (Fig. 2). We also examined the effect of races and sexes on WBF using a subset of data including individuals with known gender, but the significant difference was only found between races rather than between sexes (fig. S10). Moreover, we analyzed the expression patterns of focal genes in wing development of melpomene races. Consistent with the sequence divergence and WBF difference between eastern and western melpomene, they showed significantly different expression pattern of Col4α1 and Vkg in pupal wing discs, which were similar within either eastern or western melpomene likely because of local adaptation (Fig. 5B and fig. S11). Likewise, we found that the similarity of the expression levels of Col4α1 and Vkg in pupal wing discs of Heliconius himera and H. e. lativitta was greater than the similarity of either of these patterns to that of H. e. petiverana, consistent with their phylogenetic relationship at L (fig. S12). See Supplementary Results for a complete description of genetic differentiation and gene expression at the L locus in H. himera and other H. erato subspecies. We further investigated the expression levels in different tissues of one H. melpomene race and revealed that the expression profiles of Col4α1 and Vkg were similar, likely owing to the two chains forming a heterotrimer molecule. Oseg4 and na also showed similar patterns with wing disc and antenna-biased expression, indicating a possible role of sensory function (Fig. 5C). Together, these results suggest a correlation between levels of genetic differentiation and gene expression of L during development of Heliconius wings.
Fig. 5. Functional characterization of the L locus in Heliconius butterflies.
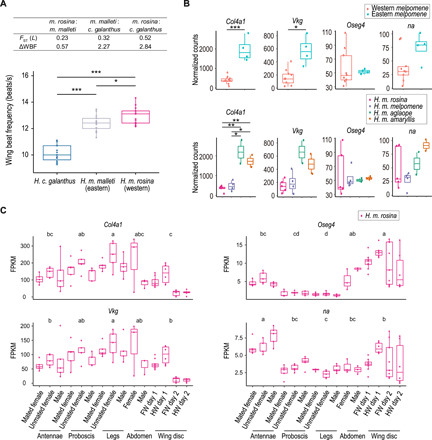
(A) Boxplots show the WBFs of H. c. galanthus, H. m. malleti, and H. m. rosina. Center line indicates median; box extends to upper and lower quartile, and whisker extends to the minimum and the maximum. ***P < 0.001; *P < 0.05 (Tukey’s test). H. cydno beat wings significantly slower than H. melpomene in free-moving flight [one-way analysis of variance (ANOVA), F2,26 = 3.37, P < 0.001]. The table summarizes the differences of WBF and the genetic differentiations at the L locus between pairs of races. (B) Expression levels of Col4α1, Vkg, Oseg4, and na in western and eastern melpomene races. Col4α1 and Vkg have significantly lower expression in western melpomene in comparison with that of eastern melpomene. The boxplots summarize the normalized counts (DESeq2’s median of ratios). ***P < 0.001, **P < 0.01, and *P < 0.05. (C) Tissue-specific expression levels of Col4α1, Vkg, Oseg4, and na in H. m. rosina. Boxplots show the ranges of fragments per kilobase of transcript per million mapped reads (FPKM). Different letters (a, b, c, and d) indicate statistically significant differences across groups (P < 0.05; Scheffe’s test). FW, forewing; HW, hindwing.
To further dissect the function of L, we focused on the orthologs of Col4α1, Vkg, Oseg4, and na in D. melanogaster and performed functional validation via the tissue-specific knockdown (KD) of the genes in flight-associated organs. To avoid off-target effects of RNA interference (RNAi), at least two RNAi lines targeting distinct regions in the individual gene locus were used (fig. S13). Flight in Drosophila is powered by the synchronized movement of the indirect flight muscles of the thorax, and a recent study suggested that flight behaviors additionally require scutellum-mediated wing-wing coordination (38). The KD of all four genes resulted in similar notum phenotypes, such as an altered structure of scutellum accompanied by a reduced number of mechanosensory bristles (Fig. 6, A to O′, and fig. S13). Notably, the resulting KD flies displayed a stretched out or curly wing morphology (fig. S13). In addition, ectopic veins and hairs as well as misarranged sensory bristles were easily observable. Consistent with the aforementioned adult wing phenotypes, the expression pattern of Senseless, a transcriptional factor essential for sensory organ development, was altered in the third-instar wing imaginal discs of KD flies. As scutellum-mediated wing-wing coordination is essential for flight control and stereotypically organized mechano- and chemosensory bristles are optimally designed for efficient mechanical force and chemical detection, KD flies were accordingly found to lose their flight ability (Fig. 6, P and Q). Specifically, Fisher’s exact test revealed a significant difference between control and KD flies in terms of flight ability (Fig. 6, P and Q, and table S6). See Supplementary Results for a complete description of significantly different flight abilities between control and KD flies. In summary, our studies provide strong evidence that the Drosophila orthologous genes within the L locus are functionally related to locomotion.
Fig. 6. Functional implication of the orthologous genes of the L locus in D. melanogaster.
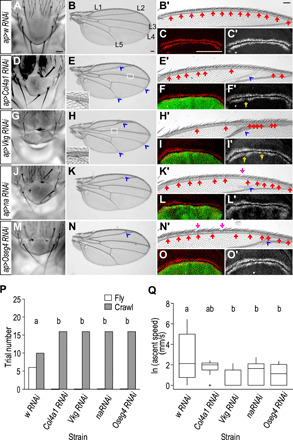
(A to C′) The adult Drosophila scutellum, showing two pairs of mechanosensory bristles, is required for wing-wing coordination during flight (A). The adult wing blade contains five longitudinal veins (L1 to L5) (B) and one dorsal anterior marginal vein [(B) and enlarged in (B′)], embedded with tracheal tubes for air exchange. Notably, wing mechano- and gustatory (chemosensory) sensilla, specified by Senseless at the third-instar imaginal discs (C and C′), are closely associated with the marginal vein (B′). The expression of w RNAi by ap-Gal4 does not produce any obvious defect (A to C′). (D to O′) Knocking down either Col4α1, Vkg, na, or Oseg4 produces similar defects associated with locomotion. The scutellum structure is altered, and the number of mechanosensory bristles is reduced (D, G, J, and M). Ectopic veins, hairs (E, H, K, and N) (blue arrowheads), and sensory bristles [insets in (E) and (H)] are produced on wing blade. The pattern of gustatory bristles is misarranged (E′, H′, K′, and N′) (red arrows), mechanosensory bristles are occasionally lost (K′ and N′) (purple arrows), and Senseless is altered consistently (F′, I′, L′, and O′) (yellow arrows). The effects of knocking down the four candidate genes on flight ability (N = 80) are shown in (P) and (Q). The KD flies fail to fly in response to the dropping stimulus (P). The ascent speed of KD flies is slower than that of non-KD individuals (Q). Different letters (a and b) indicate statistically significant differences across groups (P < 0.05) (P and Q). Scale bars, 100 μm.
Although the gene sequences at L are conserved, we observed a pattern of gene rearrangement by drawing a synteny map for L between multiple species in different orders (Fig. 7 and table S3). The abovementioned four genes were clustered at L in all lepidopteran species but scattered on different chromosomes in other orders (Fig. 7 and table S3), suggesting the cis-regulation and tight linkage of L specifically involved in lepidopteran evolution. Besides, there were two small genes, PITHD1 and SetT, adjacent to Vkg, likely hitchhiked with Vkg during the lepidopteran rearrangements, as they are located on the same chromosome with Vkg in the two outgroup species D. melanogaster and Tribolium castaneum (table S3). Furthermore, we disrupted the ortholog of Oseg4 in another nymphalid butterfly species, Kallima inachus, and observed slower but not significantly different WBFs in somatic mosaic knockouts (mKOs), likely owing to limited mutants with weak mosaic phenotypes but revealing a possible functional constraint of L in Lepidoptera (fig. S14). In addition, we observed that one Oseg4 mKO had abnormally shorter antennae, indicating its role in sensory function (fig. S14). Together, our results elucidate the potential function of the L locus by showing that the four main genes at this locus might all contribute to locomotion but in different ways. We therefore consider L to very likely be an important locus for controlling lepidopteran locomotion.
Fig. 7. Multiple alignments of the L locus.
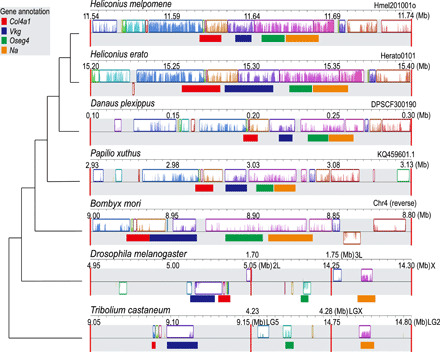
The multiple alignment of the L locus between several insect genomes suggests that it is a conserved genomic region only in Lepidoptera. Locally collinear blocks are shown as rectangular blocks, with four orthologous genes labeled by horizontal bars in different colors. The interchromosomal boundaries are indicated by red vertical bars.
DISCUSSION
In this study, we characterized a genetic locus that displays high divergence among Heliconius butterflies and acted as an introgression hotspot. We further showed that it is likely a lepidopteran gene cluster containing multiple genes related to locomotion. These findings involve a handful of fundamental issues in evolution, including speciation, adaptation, and hybridization. Understanding the role of locomotion appears to be the key to disentangling this complexity. Butterfly wings exhibit a variety of functions and have been favored by natural selection and sexual selection, and wing patterns have been recognized as an important morphological trait involved in speciation and adaptation. Although locomotion is a less intuitive biomechanical feature, our results decode the role of wing locomotion that cannot be overlooked: it has been favored by selection in multiple Heliconius species and likely plays an important role in ecological speciation and local adaptation.
We found high divergence of the L locus between incipient Heliconius species in the present study, which indicates that locomotion might be involved in multiple stages and lineages during Heliconius speciation. Directional selection serves as an important driving force for ecological speciation. This process could be promoted by either a strong single trait or multiple traits (39). If the latter, multiple traits could be involved either simultaneously or in a particular order, depending on their roles and other stochastic factors. In Heliconius butterflies, wing patterns have been favored by natural selection. Ample evidence supports the functional role of wing patterns (9, 13), suggesting that they have served to promote biodiversity and speciation. In addition to wing patterns, our results suggest that other traits, such as locomotion, might have been co-opted along with wing patterns or could be independently involved in Heliconius speciation as an ecological trait during multiple stages of speciation. Different locomotion abilities might facilitate Heliconius butterflies to explore and become established in different niches, resulting in assortative mating as a by-product of allopatric speciation. Beyond the Heliconius genus, we showed that the origin of L was likely due to gene rearrangement in Lepidoptera, indicating an even more prevalent role of wing locomotion in butterflies and moths.
Besides, our results suggest an additional scenario of speciation with gene flow: Islands of divergence could originate via hybridization with a third-party species. The role of hybridization in speciation has been debated for decades, leading to two widely accepted hypotheses. Hybridization may either facilitate speciation by weeding out uncompetitive hybrids (known as reinforcement) or hinder speciation by breaking down species barriers (40). In the first scenario, islands of divergence increase by increased genetic hitchhiking, eventually blocking hybridization. However, how these islands of divergence originate remains an unresolved question. As a logical principle, islands of divergence will include the first regions to avoid hybridization and will therefore serve as islands of speciation. Our results show that hybridization can fuel divergence, and some islands of divergence might have a greater chance than expected to participate in hybridization as introgression hotspots in the early stage of speciation. Therefore, in this scenario, introgression serves as a direct source of divergence instead of weeding out hybrids. Recent studies on Heliconius butterflies have also shown the wide impact of introgression in adaptive radiation (25). Our observation of both high divergence and typical signatures of introgression across L between multiple Heliconius races provides detailed evidence illustrating that divergence introduced via introgression might particularly facilitate speciation among incipient species under rapid radiation.
On a broader scale, the locomotor organs of a butterfly can be considered a combination of the wings, thorax, and brain; therefore, genes involved in locomotion are likely associated with multiple functional categories. The functional validation of orthologous genes in the model organism D. melanogaster also showed the complex locomotor function of L (Fig. 6). The genes located at L are tightly packed with reduced recombination but still have the chance to undergo recombination (e.g., recombination via introgression). The complex function and the chance of recombination enable L to be favored by selection and to play multiple roles in different evolutionary processes. Given that the L locus is conserved across Lepidoptera, further evidence from other lepidopteran species is still required to fully elucidate the role of wing locomotion and its relationship with other traits.
MATERIALS AND METHODS
Data collection and genotype calling
We downloaded 123 individual genome resequencing datasets from National Center for Biotechnology Information (NCBI) PRJEB12740 (41), PRJNA73595 (27), PRJEB1749 (42), PRJEB8011 (43), PRJNA324415 (24), PRJNA308754 (18), PRJEB21091 (44), PRJNA471310 (45), and PRJEB11772 (23). We performed quality control by removing raw reads with an average quality below 10 using Trimmomatic v0.38 (46). We aligned qualified reads to the reference genomes of H. melpomene v2.5 (23) and H. e. demophoon v1 (24) using Bowtie2 v2.3.4 (47) with the parameter -very-sensitive-local and reordered alignments and removed polymerase chain reaction (PCR) duplicates using Picard v1.96 (http://broadinstitute.github.io/picard/). We realigned indels using RealignerTargetCreator and IndelRealigner in GATK v3.7 (48) and called genotypes using UnifiedGenotyper in GATK v3.7 with the following parameters: heterozygosity, 0.05; stand_call_conf, 50.0; and dcov, 250. We extracted genotype calls with a good quality (Qual > 50) for the subsequent analyses (table S7).
Phylogenetic and population genetic analyses
We aligned and concatenated SNPs with good quality (Qual > 50, approximately 105.2 Mb) from individual samples with similar sequencing coverage and constructed maximum likelihood phylogenetic trees with the GTRGAMMA model and 100 bootstrap replicates using Randomized Axelerated Maximum Likelihood (RAxML) (49). The tree files were visualized using Interactive Tree Of Life (iTOL) (50).
The FST values and nucleotide diversity (π) were calculated using VCFtools (51). For the genome-wide estimates, we used a block size of 50 kb by filtering out windows with SNP counts below 100 and then calculated the SE using a jackknife approach. For the local genomic regions, we used a block size of 500 base pairs and calculated the SE using a moving block bootstrap approach (18).
The square of the correlation coefficient (r2) was estimated to measure pairwise LD using Haploview v4.2 (52) with the following parameters: -maxdistance, 200, and -dprime and -minMAF, 0.10. The heatmaps of the LD statistics were generated using LDheatmap in R (53).
We used both Patterson’s D-statistic and a modified f-statistic (fd) to test for introgression. The D-statistic was used to identify an excess of shared derived alleles supporting either the discordant ABBA or BABA pattern (54, 55), whereas fd was used to estimate the fraction of admixture in an unbiased manner (30). The D-statistic and fd were calculated using ABBABABAwindows.py (30), and two-tailed z tests were performed to determine whether the SE for each test was significantly different from zero, thus suggesting potential introgression. We also calculated pairwise sequence divergence (dxy) to distinguish incomplete lineage sorting from putative introgression using a customized script adjusted on the basis of popgenWindows.py (https://github.com/simonhmartin/genomics_general).
Wing motion analyses
We filmed the flight of free-moving butterfly individuals in H. c. galanthus (n = 15), H. m. malleti (n = 16), and H. m. rosina (n = 15) using a digital camera (30 or 60 frames per second). For each individual, the butterfly was allowed to fly freely in the greenhouse or the insectary, and the uninterrupted flights were captured. The flight sequences were analyzed using the DLTdv5 digitizing package in MATLAB (version R2019b) (56). The flight trajectories were digitized frame by frame, during which the WBF was recorded. We performed a one-way analysis of variance (ANOVA) to examine the WBF difference among groups, with the WBF as the dependent variable and species as the independent variable. Shapiro-Wilk normality test and Levene’s test were carried out to confirm that the data meet the assumptions of normality and homogeneity. Tukey’s paired comparisons were conducted to determine differences between different taxa using the function “glht” in the R package multcomp (57).
Differential gene expression analyses
RNA sequencing (RNA-seq) datasets for H. melpomene and H. erato were downloaded from PRJNA577441 (22), PRJEB2745 (27), PRJNA552081 (58), and PRJNA435610 (59). RNA-seq data for adult antennae, proboscis, legs and abdomen tissues of H. m. rosina, and mid-pupal forewings and hindwings of H. m. rosina, H. m. melpomene, H. m. aglaope, H. m. amaryllis, H. himera, H. e. lativitta, and H. e. petiverana were obtained and analyzed (table S7). Raw reads were trimmed and aligned to the reference genome of H. melpomene v2.5 (23) and H. e. demophoon v1 (24) using STAR (60) with gene models obtained from Lepbase (http://lepbase.org/). The abundances of each gene were quantified by RSEM (RNA-Seq by Expectation Maximization) (61). Count normalization and differential expression analyses were carried out using DESeq2 (62). The cutoffs of differentially expressed genes were set to an adjusted P value less than 0.05 and an absolute value of log2 fold change greater than one. The SNP sites of H. erato transcriptomes were called using GATK v3.7 (48). We extracted coding DNA sequences of each gene at the L locus and constructed gene trees using RAxML (49).
Multiple alignment and synteny mapping for the L locus
Using BlastP (63), we identified the orthology of genes at the L locus, including Col4α1, Vkg, Oseg4, and na, in multiple insect reference genomes, including H. e. demophoon v1 (24), D. plexippus v3 (64), P. xuthus v1.0 (65), B. mori (66), D. melanogaster v6.22 (67), T. castaneum v5.2 (68), A. mellifera HAv3.1 (69), A. lucorum (70), and L. migratoria (71). We extracted 200-kb regions containing these four orthologous genes and performed multiple alignments to the reference genomes of H. melpomene v2.5 (23) to determine locally collinear blocks using Mauve (72) with default parameters. We modified annotation tracts from Lepbase v4 (73) and Ensembl Metazoa release 45 (74) and inferred a phylogenetic diagram according to the phylogenetic relationships among major insect lineages (75).
Fly genetics
All fly crosses were maintained at 29°C unless otherwise noted. The phenotypes of the adult scutellum, wing, and larval wing discs are all fully penetrant (n > 20). The original fly strains used in this study are as follows: Drosophila, ap-Gal4 (BDSC3041, Bloomington), Drosophila, UAS-mcd8-gfp (BDSC5137, Bloomington), Drosophila, UAS-w RNAiSH00017.N (THU0558, Tsinghua Fly Center), Drosophila, UAS-Col4α1 RNAiGD12784 (VDRC28369, Vienna Drosophila RNAi Center), Drosophila, UAS-Col4α1 RNAiSH04419.N (TH01836.N, Tsinghua Fly Center), Drosophila, UAS-Vkg RNAiKK111668 (VDRC106812, Vienna Drosophila RNAi Center), Drosophila, UAS-Vkg RNAiSH04246.N (TH01665.N, Tsinghua Fly Center), Drosophila, UAS-na RNAiGD1172 (VDRC3306, Vienna Drosophila RNAi Center), Drosophila, UAS-na RNAiTR01764P.1 (THU3978, Tsinghua Fly Center), Drosophila, UAS-Oseg4 RNAi2069R-1 (NIG2069R-1, National Institute of Genetics, Kyoto), and Drosophila, UAS-Oseg4 RNAiTR01764P.1 (THU3978, Tsinghua Fly Center).
Intermediate strains were constructed on the basis of these strains. Detailed genotypes of animals in all experiments are as follows: For Fig. 6 (A to C′) and fig. S13B, ap-Gal4 x UAS-w RNAiSH00017.N; for Fig. 6 (D, E, and E′) and fig. S13D, ap-Gal4, UAS-mcd8-gfp x UAS-Col4α1 RNAiSH04419.N; for Fig. 6 (F and F′) and fig. S13C, ap-Gal4, UAS-mcd8-gfp x UAS-Col4α1 RNAiGD12784; for Fig. 6 (G to I′) and fig. S13F, ap-Gal4, UAS-mcd8-gfp x UAS-Vkg RNAiSH04246.N; for fig. S13E, ap-Gal4, UAS-mcd8-gfp x UAS-Vkg RNAiKK111668; for Fig. 6 (J, K, and K′) and fig. S13G, ap-Gal4, UAS-mcd8-gfp x UAS-na RNAiTR01764P.1; for Fig. 6 (L and L′) and fig. S13H, ap-Gal4, UAS-mcd8-gfp x UAS-na RNAiGD1172; for Fig. 6 (M to O′) and fig. S13J, ap-Gal4, UAS-mcd8-gfp x UAS-Oseg4 RNAiTR01764P.1; and for fig. S13I, ap-Gal4, UAS-mcd8-gfp x UAS-Oseg4 RNAi2069R-1.
Immunohistochemistry
For Senseless immunofluorescence staining, fly wing discs dissected from third-instar fly larvae were fixed in 4% paraformaldehyde, blocked in 0.2% bovine serum albumin, and incubated overnight at 4°C with guinea pig anti-Sens (1:500). The wing discs were incubated with Alexa Fluor–conjugated secondary antibodies (1:400; Invitrogen) for 1 hour at room temperature before mounting. Fluorescence images were acquired with a Leica SP8 confocal microscope. The figures were assembled in Adobe Photoshop CS5. Minor image adjustments (brightness and/or contrast) were performed in Photoshop.
Flight ability tests and statistical analyses
We conducted flight ability tests on 20 individuals from each of the four treated fly strains—Col4α1 RNAi, Vkg RNAi, na RNAi, and Oseg4 RNAi—and a control strain, w RNAi. For each trial, one individual was introduced into a 55 mm–by–55 mm transparent acrylic box, in which the fly was allowed to move freely. After 3 min of acclimatization, the acrylic box containing the individual was lifted to a height of approximately 30 mm and dropped on the ground. The trial started when the individual dropped to the bottom surface of the acrylic box and ended after 30 s of free movement. The container was cleaned with 75% ethanol to remove any chemical pheromones between trials. All trials were video recorded using a Nikon D850 camera (1920 × 1080 pixel resolution; 120 frames per second). Video analyses were conducted using Tracker v5.1.3 (76). Specifically, for each trial, we recorded (i) whether the individual exhibited flight behavior during the test period; (ii) how fast the individual recovered movement (i.e., movement latency), defined as the time period from the start of the trial to the time point at which the individual started to move; and (iii) ascent speed, defined as how quickly the individual reached the highest point during the test period.
We analyzed the behavior data in the program R v3.6.2 (www.r-project.org). The data were log-transformed to meet the assumptions of parametric tests when required. We first conducted Fisher’s exact test to examine whether knocking down the candidate genes resulted in a deficiency of flight ability, followed by a post hoc analysis using the function “pairwiseNominalIndependence” in the rcompanion package (https://cran.r-project.org/package=rcompanion) with a corrected P for multiple pairwise comparisons between strains. We then tested for ascent speed using one-way ANOVA, with ascent speed as the dependent variable and strain as the independent variable. The ascent speed data were log-transformed. Tukey’s paired comparisons were used to determine differences between strains using the function glht in the multcomp package (57). We performed a nonparametric Kruskal-Wallis test to determine the effect of knocking down candidate genes on movement latency because the movement latency data were not normally distributed. Post hoc analyses were conducted using Dunn’s test for multiple comparisons.
CRISPR-Cas9 genome editing
For CRISPR-Cas9 experiments, single-guide RNA (sgRNA) of Oseg4 was designed using sgRNAcas9 V3.0 (77) against the reference genome of K. inachus (78) to avoid off-target effect. Following the protocol for CRISPR-Cas9 genome editing in Lepidoptera (79), the mix of sgRNA and Cas9 protein was injected into developing embryos 1 to 2 hours after oviposition. Larvae of K. inachus were reared to adults at 26° to 28°C with 70 relative humidity. Genotyping primers were designed outside the target region, and CRISPR mutants were identified by bidirectional Sanger sequencing of PCR amplicons. PCR amplicons were then cloned into pEASY-T1 vector (TransGen Biotech) and sequenced. The primers used for gene editing and genotyping are as follows: guide RNA, GACCATAAGTACGCCTTCTA; genotyping primer forward, ATGACCAATCCACAACTACGTACA; and genotyping primer reverse, GTTCTGGATGCATGTTGAACAGTTA.
Free-moving flight of mosaic mutants (n = 6) and wild types (n = 6) were filmed using a Nikon D850 camera (1920 × 1080 pixel resolution; 120 frames per second). The wing motion was recorded on the 2 and 6 days after emergence with three reduplicates for each individual. The flight sequences were analyzed using Tracker v5.1.3 (76), and WBFs were calculated. A linear mixed-effects model was performed to examine the relationship between WBF and genetic background considering intraindividual variations. The model was conducted using the R package “lmerTest” (80) with WBF as the dependent variable, genetic background as the fixed effect, and individual as the random effect. The validity of the mixed-effect model was assessed by comparing the model with the fixed effect to the null model with only the random effect using the function “anova.”
Acknowledgments
We thank M. Kronforst, N. Wu, and F. Li for helpful discussions and for providing a number of valuable suggestions. Funding: This project was supported by grants from the Beijing Natural Science Foundation (JQ19021 to W.Z.), the National Natural Science Foundation of China (31871271 to W.Z. and 31725019 to A.J.Z.), the Peking-Tsinghua Center for Life Science to W.Z. and A.J.Z., the State Key Laboratory of Protein and Plant Gene Research and Qidong-SLS Innovation Fund to W.Z., and the Ministry of Education Key Laboratory of Cell Proliferation and Differentiation to A.J.Z. Stocks obtained from the Bloomington Drosophila Stock Center (National Institutes of Health grant P40OD018537), National Institute of Genetics, Tsinghua Fly Center, and Vienna Drosophila RNAi Center were used in this study. Author contributions: W.Z. conceived the study. W.Z. and A.J.Z. designed the study. Y.Z., D.T., W.L., M.L., H.Z., E.W., S.P., L.S., and L.C. analyzed the data. W.Z., Y.Z., and M.L. wrote the manuscript with input from A.J.Z., D.T., W.L., H.Z., E.W., S.P., L.S., and L.C. All the authors proofread and approved the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: Sequence data are available from NCBI SRA: https://trace.ncbi.nlm.nih.gov/Traces/sra/ (NCBI PRJEB12740, PRJNA73595, PRJEB1749, PRJEB8011, PRJNA324415, PRJNA308754, PRJEB21091, PRJNA471310, PRJEB11772, PRJNA577441, PRJEB2745, PRJNA552081, and PRJNA435610). All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/32/eabh2340/DC1
REFERENCES AND NOTES
- 1.Higham T. E., Rogers S. M., Langerhans R. B., Jamniczky H. A., Lauder G. V., Stewart W. J., Martin C. H., Reznick D. N., Speciation through the lens of biomechanics: Locomotion, prey capture and reproductive isolation. Proc. Biol. Sci. 283, 20161294 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lamichhaney S., Berglund J., Almén M. S., Maqbool K., Grabherr M., Martinez-Barrio A., Promerová M., Rubin C. J., Wang C., Zamani N., Grant B. R., Grant P. R., Webster M. T., Andersson L., Evolution of Darwin’s finches and their beaks revealed by genome sequencing. Nature 518, 371–375 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Peichel C. L., Nereng K. S., Ohgi K. A., Cole B. L. E., Colosimo P. F., Buerkle C. A., Schluter D., Kingsley D. M., The genetic architecture of divergence between threespine stickleback species. Nature 414, 901–905 (2001). [DOI] [PubMed] [Google Scholar]
- 4.Servedio M. R., Van Doorn G. S., Kopp M., Frame A. M., Nosil P., Magic traits in speciation: ‘Magic’ but not rare? Trends Ecol. Evol. 26, 389–397 (2011). [DOI] [PubMed] [Google Scholar]
- 5.Phadnis N., Orr H. A., A single gene causes both male sterility and segregation distortion in Drosophila hybrids. Science 323, 376–379 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lienard M. A., Araripe L. O., Hartl D. L., Neighboring genes for DNA-binding proteins rescue male sterility in Drosophila hybrids. Proc. Natl. Acad. Sci. U.S.A. 113, E4200–E4207 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hench K., Vargas M., Hoppner M. P., McMillan W. O., Puebla O., Inter-chromosomal coupling between vision and pigmentation genes during genomic divergence. Nat. Ecol. Evol. 3, 657–667 (2019). [DOI] [PubMed] [Google Scholar]
- 8.Kronforst M. R., Young L. G., Kapan D. D., McNeely C., O’Neill R. J., Gilbert L. E., Linkage of butterfly mate preference and wing color preference cue at the genomic location of wingless. Proc. Natl. Acad. Sci. U.S.A. 103, 6575–6580 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Merrill R. M., Dasmahapatra K. K., Davey J. W., Dell’Aglio D. D., Hanly J. J., Huber B., Jiggins C. D., Joron M., Kozak K. M., Llaurens V., Martin S. H., Montgomery S. H., Morris J., Nadeau N. J., Pinharanda A. L., Rosser N., Thompson M. J., Vanjari S., Wallbank R. W. R., Yu Q., The diversification of Heliconius butterflies: What have we learned in 150 years? J. Evol. Biol. 28, 1417–1438 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Bates H. W., XXXII. Contributions to an insect fauna of the Amazon Valley. Lepidoptera: Heliconidæ. Trans. Linn. Soc. Lond. 23, 495–566 (1862). [Google Scholar]
- 11.Jiggins C. D., Naisbit R. E., Coe R. L., Mallet J., Reproductive isolation caused by colour pattern mimicry. Nature 411, 302–305 (2001). [DOI] [PubMed] [Google Scholar]
- 12.Beebe W., Polymorphism in reared broods of Heliconius butterflies from Surinam and Trinidad. Fortschr. Zool. 40, 139–143 (1955). [Google Scholar]
- 13.Kronforst M. R., Papa R., The functional basis of wing patterning in Heliconius butterflies: The molecules behind mimicry. Genetics 200, 1–19 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Joron M., Papa R., Beltrán M., Chamberlain N., Mavárez J., Baxter S., Abanto M., Bermingham E., Humphray S. J., Rogers J., Beasley H., Barlow K., H. ffrench-Constant R., Mallet J., McMillan W. O., Jiggins C. D., A conserved supergene locus controls colour pattern diversity in Heliconius butterflies. PLOS Biol. 4, e303 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reed R. D., Papa R., Martin A., Hines H. M., Counterman B. A., Pardo-Diaz C., Jiggins C. D., Chamberlain N. L., Kronforst M. R., Chen R., Halder G., Nijhout H. F., McMillan W. O., optix drives the repeated convergent evolution of butterfly wing pattern mimicry. Science 333, 1137–1141 (2011). [DOI] [PubMed] [Google Scholar]
- 16.Joron M., Mallet J. L., Diversity in mimicry: Paradox or paradigm? Trends Ecol. Evol. 13, 461–466 (1998). [DOI] [PubMed] [Google Scholar]
- 17.Srygley R. B., Evolution of the wave: Aerodynamic and aposematic functions of butterfly wing motion. Proc. Biol. Sci. 274, 913–917 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang W., Dasmahapatra K. K., Mallet J., Moreira G. R., Kronforst M. R., Genome-wide introgression among distantly related Heliconius butterfly species. Genome Biol. 17, 25 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Srygley R. B., Locomotor mimicry in Heliconius butterflies: Contrast analyses of flight morphology and kinematics. Phil. Trans. R. Soc. B Biol. Sci. 354, 203–214 (1999). [Google Scholar]
- 20.Merrill R. M., Rastas P., Martin S. H., Melo M. C., Barker S., Davey J., McMillan W. O., Jiggins C. D., Genetic dissection of assortative mating behavior. PLOS Biol. 17, e2005902 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rossi M., Hausmann A. E., Thurman T. J., Montgomery S. H., Papa R., Jiggins C. D., McMillan W. O., Merrill R. M., Visual mate preference evolution during butterfly speciation is linked to neural processing genes. Nat. Commun. 11, 4763 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Schooten B., Meléndez-Rosa J., van Belleghem S. M., Jiggins C. D., Tan J. D., McMillan W. O., Papa R., Divergence of chemosensing during the early stages of speciation. Proc. Natl. Acad. Sci. U.S.A. 117, 16438–16447 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Davey J. W., Chouteau M., Barker S. L., Maroja L., Baxter S. W., Simpson F., Merrill R. M., Joron M., Mallet J., Dasmahapatra K. K., Jiggins C. D., Major improvements to the Heliconius melpomene genome assembly used to confirm 10 chromosome fusion events in 6 million years of butterfly evolution. G3 (Bethesda) 6, 695–708 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Van Belleghem S. M., Rastas P., Papanicolaou A., Martin S. H., Arias C. F., Supple M. A., Hanly J. J., Mallet J., Lewis J. J., Hines H. M., Ruiz M., Salazar C., Linares M., Moreira G. R. P., Jiggins C. D., Counterman B. A., Millan W. O. M., Papa R., Complex modular architecture around a simple toolkit of wing pattern genes. Nat. Ecol. Evol. 1, 52 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Edelman N. B., Frandsen P. B., Miyagi M., Clavijo B., Davey J., Dikow R. B., García-Accinelli G., van Belleghem S. M., Patterson N., Neafsey D. E., Challis R., Kumar S., Moreira G. R. P., Salazar C., Chouteau M., Counterman B. A., Papa R., Blaxter M., Reed R. D., Dasmahapatra K. K., Kronforst M., Joron M., Jiggins C. D., McMillan W. O., di Palma F., Blumberg A. J., Wakeley J., Jaffe D., Mallet J., Genomic architecture and introgression shape a butterfly radiation. Science 366, 594–599 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mallet J., Beltrán M., Neukirchen W., Linares M., Natural hybridization in heliconiine butterflies: The species boundary as a continuum. BMC Evol. Biol. 7, 28 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heliconius Genome Consortium , Butterfly genome reveals promiscuous exchange of mimicry adaptations among species. Nature 487, 94–98 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hines H. M., Counterman B. A., Papa R., Albuquerque de Moura P., Cardoso M. Z., Linares M., Mallet J., Reed R. D., Jiggins C. D., Kronforst M. R., McMillan W. O., Wing patterning gene redefines the mimetic history of Heliconius butterflies. Proc. Natl. Acad. Sci. U.S.A. 108, 19666–19671 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhan S., Zhang W., Niitepõld K., Hsu J., Haeger J. F., Zalucki M. P., Altizer S., de Roode J. C., Reppert S. M., Kronforst M. R., The genetics of monarch butterfly migration and warning colouration. Nature 514, 317–321 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martin S. H., Davey J. W., Jiggins C. D., Evaluating the use of ABBA-BABA statistics to locate introgressed loci. Mol. Biol. Evol. 32, 244–257 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith J., Kronforst M. R., Do Heliconius butterfly species exchange mimicry alleles? Biol. Lett. 9, 20130503 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fessler J. H., Fessler L. I., Drosophila extracellular matrix. Annu. Rev. Cell Biol. 5, 309–339 (1989). [DOI] [PubMed] [Google Scholar]
- 33.Natzle J. E., Monson J. M., McCarthy B. J., Cytogenetic location and expression of collagen-like genes in Drosophila. Nature 296, 368–371 (1982). [DOI] [PubMed] [Google Scholar]
- 34.Schnorrer F., Schönbauer C., Langer C. C. H., Dietzl G., Novatchkova M., Schernhuber K., Fellner M., Azaryan A., Radolf M., Stark A., Keleman K., Dickson B. J., Systematic genetic analysis of muscle morphogenesis and function in Drosophila. Nature 464, 287–291 (2010). [DOI] [PubMed] [Google Scholar]
- 35.Balmer S., Dussert A., Collu G. M., Benitez E., Iomini C., Mlodzik M., Components of intraflagellar transport complex A function independently of the cilium to regulate canonical Wnt signaling in Drosophila. Dev. Cell 34, 705–718 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moose D. L., Haase S. J., Aldrich B. T., Lear B. C., The narrow abdomen ion channel complex is highly stable and persists from development into adult stages to promote behavioral rhythmicity. Front. Cell. Neurosci. 11, 159 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nash H. A., Scott R. L., Lear B. C., Allada R., An unusual cation channel mediates photic control of locomotion in Drosophila. Curr. Biol. 12, 2152–2158 (2002). [DOI] [PubMed] [Google Scholar]
- 38.Deora T., Singh A. K., Sane S. P., Biomechanical basis of wing and haltere coordination in flies. Proc. Natl. Acad. Sci. U.S.A. 112, 1481–1486 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nosil P., Harmon L. J., Seehausen O., Ecological explanations for (incomplete) speciation. Trends Ecol. Evol. 24, 145–156 (2009). [DOI] [PubMed] [Google Scholar]
- 40.Mallet J., Hybridization as an invasion of the genome. Trends Ecol. Evol. 20, 229–237 (2005). [DOI] [PubMed] [Google Scholar]
- 41.Nadeau N. J., Pardo-Diaz C., Whibley A., Supple M. A., Saenko S. V., Wallbank R. W. R., Wu G. C., Maroja L., Ferguson L., Hanly J. J., Hines H., Salazar C., Merrill R. M., Dowling A. J., ffrench-Constant R. H., Llaurens V., Joron M., McMillan W. O., Jiggins C. D., The gene cortex controls mimicry and crypsis in butterflies and moths. Nature 534, 106–110 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martin S. H., Dasmahapatra K. K., Nadeau N. J., Salazar C., Walters J. R., Simpson F., Blaxter M., Manica A., Mallet J., Jiggins C. D., Genome-wide evidence for speciation with gene flow in Heliconius butterflies. Genome Res. 23, 1817–1828 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wallbank R. W. R., Baxter S. W., Pardo-Diaz C., Hanly J. J., Martin S. H., Mallet J., Dasmahapatra K. K., Salazar C., Joron M., Nadeau N., McMillan W. O., Jiggins C. D., Evolutionary novelty in a butterfly wing pattern through enhancer shuffling. PLOS Biol. 14, e1002353 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Enciso-Romero J., Pardo-Díaz C., Martin S. H., Arias C. F., Linares M., McMillan W. O., Jiggins C. D., Salazar C., Evolution of novel mimicry rings facilitated by adaptive introgression in tropical butterflies. Mol. Ecol. 26, 5160–5172 (2017). [DOI] [PubMed] [Google Scholar]
- 45.Jay P., Whibley A., Frézal L., de Cara M. Á. R., Nowell R. W., Mallet J., Dasmahapatra K. K., Joron M., Supergene evolution triggered by the introgression of a chromosomal inversion. Curr. Biol. 28, 1839–1845.e3 (2018). [DOI] [PubMed] [Google Scholar]
- 46.Bolger A. M., Lohse M., Usadel B., Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Langmead B., Salzberg S. L., Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M. A., The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stamatakis A., RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Letunic I., Bork P., Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 47, W256–W259 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Danecek P., Auton A., Abecasis G., Albers C. A., Banks E., DePristo M. A., Handsaker R. E., Lunter G., Marth G. T., Sherry S. T., McVean G., Durbin R.; 1000 Genomes Project Analysis Group , The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Barrett J. C., Fry B., Maller J., Daly M. J., Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics 21, 263–265 (2005). [DOI] [PubMed] [Google Scholar]
- 53.Shin J.-H., Blay S., McNeney B., Graham J., LDheatmap: An R function for graphical display of pairwise linkage disequilibria between single nucleotide polymorphisms. J. Stat. Softw. 16, 1–10 (2006). [Google Scholar]
- 54.Durand E. Y., Patterson N., Reich D., Slatkin M., Testing for ancient admixture between closely related populations. Mol. Biol. Evol. 28, 2239–2252 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Green R. E., Krause J., Briggs A. W., Maricic T., Stenzel U., Kircher M., Patterson N., Li H., Zhai W., Fritz M. H. Y., Hansen N. F., Durand E. Y., Malaspinas A. S., Jensen J. D., Marques-Bonet T., Alkan C., Prufer K., Meyer M., Burbano H. A., Good J. M., Schultz R., Aximu-Petri A., Butthof A., Hober B., Hoffner B., Siegemund M., Weihmann A., Nusbaum C., Lander E. S., Russ C., Novod N., Affourtit J., Egholm M., Verna C., Rudan P., Brajkovic D., Kucan Z., Gusic I., Doronichev V. B., Golovanova L. V., Lalueza-Fox C., de la Rasilla M., Fortea J., Rosas A., Schmitz R. W., Johnson P. L. F., Eichler E. E., Falush D., Birney E., Mullikin J. C., Slatkin M., Nielsen R., Kelso J., Lachmann M., Reich D., Paabo S., A draft sequence of the Neandertal genome. Science 328, 710–722 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hedrick T. L., Software techniques for two- and three-dimensional kinematic measurements of biological and biomimetic systems. Bioinspir. Biomim. 3, 034001 (2008). [DOI] [PubMed] [Google Scholar]
- 57.Hothorn T., Bretz F., Westfall P., Simultaneous inference in general parametric models. Biom. J. 50, 346–363 (2008). [DOI] [PubMed] [Google Scholar]
- 58.Hanly J. J., Wallbank R. W. R., McMillan W. O., Jiggins C. D., Conservation and flexibility in the gene regulatory landscape of heliconiine butterfly wings. EvoDevo 10, 15 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lewis J. J., Reed R. D., Genome-wide regulatory adaptation shapes population-level genomic landscapes in Heliconius. Mol. Biol. Evol. 36, 159–173 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dobin A., Davis C. A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T. R., STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li B., Dewey C. N., RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Altschul S. F., Madden T. L., Schäffer A. A., Zhang J., Zhang Z., Miller W., Lipman D. J., Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhan S., Reppert S. M., MonarchBase: The monarch butterfly genome database. Nucleic Acids Res. 41, D758–D763 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li X., Fan D., Zhang W., Liu G., Zhang L., Zhao L., Fang X., Chen L., Dong Y., Chen Y., Ding Y., Zhao R., Feng M., Zhu Y., Feng Y., Jiang X., Zhu D., Xiang H., Feng X., Li S., Wang J., Zhang G., Kronforst M. R., Wang W., Outbred genome sequencing and CRISPR/Cas9 gene editing in butterflies. Nat. Commun. 6, 8212 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kawamoto M., Jouraku A., Toyoda A., Yokoi K., Minakuchi Y., Katsuma S., Fujiyama A., Kiuchi T., Yamamoto K., Shimada T., High-quality genome assembly of the silkworm, Bombyx mori, Bombyx mori. Insect Biochem. Mol. Biol. 107, 53–62 (2019). [DOI] [PubMed] [Google Scholar]
- 67.Hoskins R. A., Carlson J. W., Wan K. H., Park S., Mendez I., Galle S. E., Booth B. W., Pfeiffer B. D., George R. A., Svirskas R., Krzywinski M., Schein J., Accardo M. C., Damia E., Messina G., Méndez-Lago M., de Pablos B., Demakova O. V., Andreyeva E. N., Boldyreva L. V., Marra M., Carvalho A. B., Dimitri P., Villasante A., Zhimulev I. F., Rubin G. M., Karpen G. H., Celniker S. E., The Release 6 reference sequence of the Drosophila melanogaster genome. Genome Res. 25, 445–458 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shelton J. M., Coleman M. C., Herndon N., Lu N., Lam E. T., Anantharaman T., Sheth P., Brown S. J., Tools and pipelines for BioNano data: Molecule assembly pipeline and FASTA super scaffolding tool. BMC Genomics 16, 734 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wallberg A., Bunikis I., Pettersson O. V., Mosbech M. B., Childers A. K., Evans J. D., Mikheyev A. S., Robertson H. M., Robinson G. E., Webster M. T., A hybrid de novo genome assembly of the honeybee, Apis mellifera, with chromosome-length scaffolds. BMC Genomics 20, 275 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liu Y., Liu H., Wang H., Huang T., Liu B., Yang B., Yin L., Li B., Zhang Y., Zhang S., Jiang F., Zhang X., Ren Y., Wang B., Wang S., Lu Y., Wu K., Fan W., Wang G., Apolygus lucorum genome provides insights into omnivorousness and mesophyll feeding. Mol. Ecol. Resour. 21, 287–300 (2021). [DOI] [PubMed] [Google Scholar]
- 71.Wang X., Fang X., Yang P., Jiang X., Jiang F., Zhao D., Li B., Cui F., Wei J., Ma C., Wang Y., He J., Luo Y., Wang Z., Guo X., Guo W., Wang X., Zhang Y., Yang M., Hao S., Chen B., Ma Z., Yu D., Xiong Z., Zhu Y., Fan D., Han L., Wang B., Chen Y., Wang J., Yang L., Zhao W., Feng Y., Chen G., Lian J., Li Q., Huang Z., Yao X., Lv N., Zhang G., Li Y., Wang J., Wang J., Zhu B., Kang L., The locust genome provides insight into swarm formation and long-distance flight. Nat. Commun. 5, 2957 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Darling A. C., Mau B., Blattner F. R., Perna N. T., Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Challis R. J., Kumar S., Dasmahapatra K. K. K., Jiggins C. D., Blaxter M., Lepbase: The Lepidopteran genome database. BioRxiv, 056994 (2016). [Google Scholar]
- 74.Kersey P. J., Allen J. E., Allot A., Barba M., Boddu S., Bolt B. J., Carvalho-Silva D., Christensen M., Davis P., Grabmueller C., Kumar N., Liu Z., Maurel T., Moore B., McDowall M. D., Maheswari U., Naamati G., Newman V., Ong C. K., Paulini M., Pedro H., Perry E., Russell M., Sparrow H., Tapanari E., Taylor K., Vullo A., Williams G., Zadissia A., Olson A., Stein J., Wei S., Tello-Ruiz M., Ware D., Luciani A., Potter S., Finn R. D., Urban M., Hammond-Kosack K. E., Bolser D. M., de Silva N., Howe K. L., Langridge N., Maslen G., Staines D. M., Yates A., Ensembl Genomes 2018: An integrated omics infrastructure for non-vertebrate species. Nucleic Acids Res. 46, D802–D808 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Misof B., Liu S., Meusemann K., Peters R. S., Donath A., Mayer C., Frandsen P. B., Ware J., Flouri T., Beutel R. G., Niehuis O., Petersen M., Izquierdo-Carrasco F., Wappler T., Rust J., Aberer A. J., Aspock U., Aspock H., Bartel D., Blanke A., Berger S., Bohm A., Buckley T. R., Calcott B., Chen J., Friedrich F., Fukui M., Fujita M., Greve C., Grobe P., Gu S., Huang Y., Jermiin L. S., Kawahara A. Y., Krogmann L., Kubiak M., Lanfear R., Letsch H., Li Y., Li Z., Li J., Lu H., Machida R., Mashimo Y., Kapli P., McKenna D. D., Meng G., Nakagaki Y., Navarrete-Heredia J. L., Ott M., Ou Y., Pass G., Podsiadlowski L., Pohl H., von Reumont B. M., Schutte K., Sekiya K., Shimizu S., Slipinski A., Stamatakis A., Song W., Su X., Szucsich N. U., Tan M., Tan X., Tang M., Tang J., Timelthaler G., Tomizuka S., Trautwein M., Tong X., Uchifune T., Walzl M. G., Wiegmann B. M., Wilbrandt J., Wipfler B., Wong T. K. F., Wu Q., Wu G., Xie Y., Yang S., Yang Q., Yeates D. K., Yoshizawa K., Zhang Q., Zhang R., Zhang W., Zhang Y., Zhao J., Zhou C., Zhou L., Ziesmann T., Zou S., Li Y., Xu X., Zhang Y., Yang H., Wang J., Wang J., Kjer K. M., Zhou X., Phylogenomics resolves the timing and pattern of insect evolution. Science 346, 763–767 (2014). [DOI] [PubMed] [Google Scholar]
- 76.Brown D., Cox A. J., Innovative uses of video analysis. Phys. Teach. 47, 145–150 (2009). [Google Scholar]
- 77.Xie S., Shen B., Zhang C., Huang X., Zhang Y., sgRNAcas9: A software package for designing CRISPR sgRNA and evaluating potential off-target cleavage sites. PLOS ONE 9, e100448 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yang J., Wan W., Xie M., Mao J., Dong Z., Lu S., He J., Xie F., Liu G., Dai X., Chang Z., Zhao R., Zhang R., Wang S., Zhang Y., Zhang W., Wang W., Li X., Chromosome-level reference genome assembly and gene editing of the dead-leaf butterfly Kallima inachus. Mol. Ecol. Resour. 20, 1080–1092 (2020). [DOI] [PubMed] [Google Scholar]
- 79.T. Sekimura, H. Frederik Nijhout, Diversity and Evolution of Butterfly Wing Patterns: An Integrative Approach (Springer Nature, 2017). [Google Scholar]
- 80.Kuznetsova A., Brockhoff P. B., Christensen R. H. B., lmerTest Package: Tests in linear mixed effects models. J. Stat. Softw. 82, 1–26 (2017). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/32/eabh2340/DC1