

The exon junction complex helicase eIF4A3 orchestrates Pol I and Pol II posttranscriptional events.
Abstract
Eukaryotic initiation factor 4A-III (eIF4A3), a core helicase component of the exon junction complex, is essential for splicing, mRNA trafficking, and nonsense-mediated decay processes emerging as targets in cancer therapy. Here, we unravel eIF4A3’s tumor-promoting function by demonstrating its role in ribosome biogenesis (RiBi) and p53 (de)regulation. Mechanistically, eIF4A3 resides in nucleoli within the small subunit processome and regulates rRNA processing via R-loop clearance. EIF4A3 depletion induces cell cycle arrest through impaired RiBi checkpoint–mediated p53 induction and reprogrammed translation of cell cycle regulators. Multilevel omics analysis following eIF4A3 depletion pinpoints pathways of cell death regulation and translation of alternative mouse double minute homolog 2 (MDM2) transcript isoforms that control p53. EIF4A3 expression and subnuclear localization among clinical cancer specimens correlate with the RiBi status rendering eIF4A3 an exploitable vulnerability in high-RiBi tumors. We propose a concept of eIF4A3’s unexpected role in RiBi, with implications for cancer pathogenesis and treatment.
INTRODUCTION
Ribosome biogenesis (RiBi) refers to a multistep process involving a range of factors to perform and coordinate ribosomal DNA (rDNA) transcription, ribosomal RNA (rRNA) processing, and assembly with ribosomal proteins (RPs) into ribonucleoprotein (RNP) complexes that eventually form the ribosome (1). Aberrations in any of these steps may lead to a condition known as RiBi stress often manifested by alterations in nucleolar morphology, deregulated RNA polymerase I (Pol I) transcription, tempered translation, and activation of the impaired RiBi checkpoint (IRBC), whereby 5S rRNA together with RPs (mainly L5 and L11 also known as uL18 and uL5), which are released from the nucleolus, binds to and thereby sequesters MDM2 (mouse double minute 2 homolog), the negative regulator of p53. Consequently, p53 becomes stabilized and transcriptionally active, triggering major, context-dependent, and cell fate decisions (2). Perturbations in RiBi due to RP haploinsufficiency or oncogene activation can lead to cancer via elevated global protein synthesis and proteotoxic stress, metabolic rewiring, and enhanced genomic instability (3). Moreover, abnormalities in RiBi are directly connected to a group of diseases collectively known as ribosomopathies with developmental defects in various tissues and enhanced susceptibility to specific cancer types (4). Elevated Pol I activity due to activation of oncogenes or ablation of tumor suppressors has emerged as a previously unidentified cancer vulnerability, a finding that led to the discovery of Pol I chemical inhibitors that are being assessed as candidate drugs in hemato-oncology clinical trials (5).
Eukaryotic initiation factor 4A-III (eIF4A3) (also known as DDX48) is an adenosine 5′-triphosphate (ATP)–dependent RNA helicase, member of the eIF4A family. Being part of the exon junction complex (EJC), it participates in various posttranscriptional regulatory processes, such as splicing and nonsense-mediated mRNA decay (NMD) (6), the latter representing an essential quality control mechanism for the newly translated mRNAs and a promising target in cancer (7). Reduced expression or mutations of eIF4A3 are connected to abnormal neural crest development (8) and cause the Richieri-Costa-Pereira syndrome (RCPS), a pathological entity characterized by craniofacial and limb developmental defects (9). On the contrary, eIF4A3 mRNA levels have been found elevated in cancer (10), where the helicase regulates the expression and tumor-promoting activity of various noncoding RNAs including circular, micro-, or long noncoding RNAs (11). At the molecular level, eIF4A3 deficiency hinders cell migration and impairs cell viability (12), two aspects that inspired the development of eIF4A3 chemical inhibitors for cancer treatment (13). Notably, p53 ablation could only partially rescue the microcephaly of eIF4A3 haploinsufficient mice (8), leaving elusive the exact mechanisms of excessive cell death following reduced eIF4A3 expression, an intriguing issue addressed in our present study. Furthermore, the similarities of RCPS with some of the ribosomopathies [e.g., Treacher Collins syndrome (14)] and the fact that eIF4A3 has previously been implicated in rRNA processing (15), albeit through a poorly understood mechanism, inspired us to examine the role of eIF4A3 in Pol I biology and RiBi. Our results from these analyses and considerations of how these new findings advance the current understanding of eIF4A3 function and potential pathophysiological impact of its malfunction are presented in the following sections of this study.
RESULTS
Elevated eIF4A3 expression correlates with high RiBi and poor prognosis in cancer
The emerging role of NMD in cancer biology (7) led to its exploitation in the production of new candidate drugs for cancer treatment. Being part of the EJC complex and therefore important for NMD (6), eIF4A3 has already been chemically targeted with promising results for cancer therapy (13). A mutational analysis of eIF4A3 in various cancer types shows great heterogeneity, with amino acid residues known to affect NMD being less prone to become mutational hotspots than those residues critical for eIF4A3’s helicase activity or interaction with other members of the EJC complex (fig. S1A) pinpointing a potential non-NMD therapeutic target window. Analysis of publicly available cancer patient data shows that in most cases studied (16), eIF4A3 expression is higher in cancer compared to the normal tissue counterparts (Fig. 1A), a finding we could further support in a panel of cancer versus normal/nontransformed (diploid) human cell types, both at the RNA (fig. S1, B and C) and the protein level (fig. S1D). In addition, lower eIF4A3 mRNA levels correlate with better prognosis in most of The Cancer Genome Atlas (TCGA)–registered cancer types represented in LinkedOmics (Fig. 1B, fig. S1E, and table S1A) (17). These findings suggest that elevated eIF4A3 levels may be selected for during tumor development and benefit cancer cell survival and growth. To find previously unknown eIF4A3-related cancer vulnerabilities, we performed a pathway enrichment analysis among the top 100 genes whose dependency scores were positively correlated with that of eIF4A3 among 793 different cancer cell lines (DepMap, CRISPR, and Avana 20Q3) (Fig. 1C and table S1, B and C) (18). Apart from mRNA splicing, NMD, and p53 signaling, we noticed a correlation with genes implicated in rRNA processing [e.g., uL5 (RPL11), RLPL17, and SNU13]. Using a curated RiBi gene signature (table S1D) and RNA sequencing (RNA-seq) data from the DepMap consortium, we then categorized all cell lines into low and high RiBi (fig. S1F) and found that eIF4A3 expression is higher in cell lines with high RiBi (Fig. 1D), further supporting a role of the helicase in this process. Moreover, the effect of eIF4A3 knockdown on cell survival (DepMap and DEMETER2) was inversely correlated with the cytotoxicity of RiBi stress–related chemical compounds [e.g., oxaliplatin (19) or the MDM2-p53 inhibitors CGM097 and idasanutlin], strongly implicating eIF4A3 in their mechanism of action (Fig. 1E and table S1E). In conclusion, eIF4A3 expression might serve as a predictive marker in high-RiBi cancers and its underlying role in RiBi points toward a previously unidentified cancer vulnerability with a therapeutic potential.
Fig. 1. Elevated eIF4A3 expression in cancer is correlated with high RiBi rate and poor prognosis.
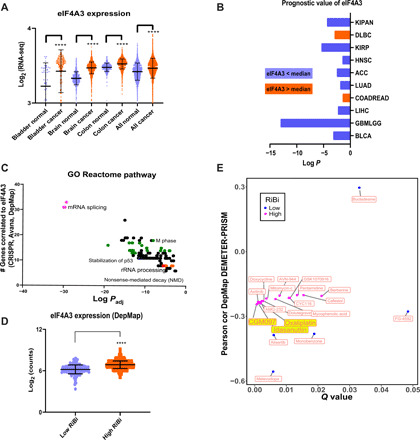
(A) Representative GENT2 (Gene Expression database of Normal and Tumor tissues 2)-derived comparison of eIF4A3 mRNA levels in various cancer types and their normal counterpart tissues (****P < 0.001). (B) Correlation of eIF4A3 mRNA expression levels to survival rates in all TCGA cancer types analyzed in LinkedOmics (abbreviations can be found in table S1A). Only cases with P < 0.05 (Cox regression analysis) are shown in color (blue, better prognosis correlates with lower than the median expression values of eIF4A3; orange, opposite kinetics). (C) Reactome pathway enrichment analysis of the 100 highest correlated genes to eIF4A3 in dependency score. Data were collected from 789 different cancer cell lines (DepMap and Avana 20Q3) and processed with ClueGO in Cytoscape. (D) eIF4A3 mRNA expression in low versus high RiBi addicted cell lines (DepMap and CCLE 2019). (E) Correlation of eIF4A3 dependency score (DepMap and DEMETER2) to the cytotoxicity of chemical compounds from the PRISM database (DepMap; cutoff, Q < 0.05; Pearson r < 0.2).
EIF4A3 partially resides in nucleoli and its depletion alters nucleolar structure
EIF4A3 has been shown to reside, as part of the EJC, in perispeckles, nuclear subdomains formed around splicing centers (20). Using SC35 (serine and arginine rich splicing factor 2) as a surrogate marker for nuclear speckles, we confirmed the presence of eIF4A3 in these domains by immunofluorescence (IF), using a U2OS cell line ectopically expressing green fluorescent protein (GFP)–tagged eIF4A3 (fig. S2A). The perispeckle staining pattern did not change upon ribosomal stress induced by low concentration of actinomycin D (5 nM; henceforth ActDL) (21). Besides perispeckles, however, we also noticed variable nucleolar localization of eIF4A3 (Fig. 2A), consistent with an in-depth analysis of the nucleolar proteome, which indicated that the helicase resides also in nucleoli (22). In our cell culture experiments, treatment with ActDL or BMH-21 (two inhibitors of Pol I) (23) reduced the amount of eIF4A3 colocalizing with the nucleolar marker fibrillarin (FBL) or UBF (upstream binding transcription factor), supporting the notion that active Pol I transcription is required for nucleolar localization of eIF4A3 (Fig. 2, A and B, and fig. S2B). Staining of U2OS cells for endogenous Y14 showed that other components of the EJC complex were also detected in the nucleolus (fig. S2C) in agreement with a previous report (24). Upon ActDL treatment, however, Y14 did not show the same spatial pattern and relocalization kinetics as eIF4A3, an observation that suggested potentially separate, nucleolar roles for these proteins.
Fig. 2. EIF4A3 localizes in transcriptionally active nucleoli and safeguards the nucleolar structure.

(A) Representative IF images of eIF4A3 localization in U2OS cells left either untreated or stimulated with the ribosomal stress facilitators ActDL and BMH-21. Insets depict magnifications of the regions designated in squares. Scale bar, 5 μm. DMSO, dimethyl sulfoxide. (B) Violin plot of high-content microscopy–based eIF4A3 nucleolar signal quantification under the experimental conditions described in (A). Nucleolar signal was calculated as the mean integrated intensity of endogenous eIF4A3 colocalizing with the nucleolar marker FBL. A total of 1000 to 2000 cells were analyzed per experiment (data are shown as means ± SD; n = 3 biological replicates; ****P < 0.001). a.u., arbitrary units. (C) Representative IF images of nucleolar structure in U2OS left untreated or treated with sieIF4A3, ActDL, or their combination. FBL and UBF were used as fibrillar center and dense fibrillar component markers, respectively. Insets depict magnifications of the regions designated in squares. Scale bar, 5 μm. (D) Representative IF images of the nucleolus under the same experimental conditions (A) using nucleophosmin 1 (NPM1) as a granular component (GC) marker. Scale bar, 5 μm. (E) Electron microscopy (EM) images showing the impact of sieIF4A3 ± ActDL on nucleolar morphology of U2OS cells. Dashed borders designate nucleoli. Scale bar, 2 μm. (F) Detection of eIF4A3 protein levels with immunocytochemistry in samples from patients with normal brain or glioblastoma. Regions in yellow squares are presented magnified in the bottom panel. Scale bar, 100 μm. (G) Same methodology in normal cervix or cervical squamous cell carcinoma specimens.
Aberrant rRNA processing is commonly associated with altered nucleolar structure (25). We therefore assessed the nucleolar integrity upon eIF4A3 knockdown by IF, using FBL and UBF proteins as nucleolar markers (fig. S2D) and treatment with ActDL as a positive control for nucleolar stress. In U2OS (Fig. 2C) and A549 cells (fig. S2E), eIF4A3 knockdown resulted in reshaping of the nucleolus into a necklace-like structure indicative of aberrantly altered rRNA processing (26), whereas ActDL administration caused formation of nucleolar caps due to Pol I transcriptional inhibition (27). We further analyzed the structure of the outer, granular component (GC), region of the nucleolus where rRNA processing takes place (fig. S2D) (28). Staining for the GC-resident protein nucleophosmin 1 (NPM1) (Fig. 2D) showed a malformed nucleolar exterior in small interfering RNA (siRNA) targeting eIF4A3 (sieIF4A3)–treated U2OS cells, again in contrast to the diffused NPM1 pattern following treatment with ActDL. Electron microscopy (EM) analysis of cells under the same experimental conditions showed enlarged and deformed nucleoli both in sieIF4A3- and eIF4A3 inhibitor (eiF4A3i)–treated samples (Fig. 2E and fig. S2F, respectively) (29), whereas ActDL induced highly dense and compacted nucleoli reflecting Pol I inhibition (30). Overall, these findings support a potential protective role of eIF4A3 in the maintenance of nucleolar function and point toward nucleolar stress as a contributing plausible cause of sieIF4A3-mediated induction of p53.
Nucleolar localization of EIF4A3 in cells on human clinical specimens from diverse cancer types
To assess relevance of the above model experiments and potential nucleolar role of eIF4A3 for clinical settings, we next performed a large-scale immunohistochemical analysis of the eIF4A3 protein abundance and subcellular localization, with special emphasis on the nucleolus. Our panel of human tissue sections from archival paraffin blocks included normal tissues and cohorts of tumor specimens from the brain (glioblastoma), colon (adenomas and carcinomas), urinary bladder cancer (early preinvasive and invasive stages), and uterine cervix (dysplasias and squamous cell carcinomas). Representative examples of the eIF4A3 staining patterns are shown in Fig. 2 (F and G) and fig. S2G, and the overall graphical summary from all cancer types is presented in fig. S2H. Notably, consistent with our experimental results so far, eIF4A3 was detectable in nucleoli of both normal cells and tumor cells, in the vast majority of cases with a similar staining intensity in both the nucleoplasm and the nucleolus. This subcellular localization of eIF4A3 was seen also in neurons of normal brain tissue (Fig. 2F), a finding that could indicate a default nucleolar role of eIF4A3 even in nonproliferating yet metabolically highly active normal cells. The staining intensity and patterns of subnuclear localization of eIF4A3 varied widely among individual cells or cell clusters in a given tissue section (Fig. 2, F and G, and fig. S2G) including subsets of cells either with nucleoli being either selectively negative or in contrast nucleoli showing a much stronger signal than the nucleoplasm. We detected the latter pattern of preferential nucleolar localization of eIF4A3 with a higher frequency especially among the high-grade cervical cancer specimens compared to their low-grade counterparts or normal tissue (fig. S2H).
Together, these results were consistent with a potential function of eIF4A3 in the nucleolus and an apparently dynamic localization pattern with even selectively nucleolar localization in subsets of tumor cells. These findings validated the potential pathophysiological relevance of nucleolar eIF4A3 in human cells and tissues, prompting us to deepen our mechanistic insights into such role using model systems and a variety of experimental approaches, as described below.
EIF4A3 depletion alters expression of genes involved in rRNA processing and triggers a p53 response
Ablation of p53 was previously shown to rescue the neurological defects caused by eIF4A3 haploinsufficiency in mice (8). However, the functional relationship of eIF4A3 and p53 remained unexplored. To address this issue, we engineered human U2OS cells to express a double doxycycline (DOX)–inducible system carrying short hairpin–mediated RNAs (shRNAs) against endogenous eIF4A3 (DOX-sheIF4A3-U2OS) together with a FLAG-tagged eIF4A3 cassette for ectopic reexpression of the helicase (DOX-FLAG.WT.eIF4A3-sheIF4A3-U2OS, Fig. 3A). Using two different shRNAs, targeting either eIF4A3 3′ untranslated region (3′UTR) (#1) or its cds (coding sequence) (#2) (Fig. 3A and table S4B), we knocked down eIF4A3 and measured p53 protein levels by immunoblotting. A 80 to 90% reduction in eIF4A3 levels was followed by induction of p53 (Fig. 3B, lanes 1, 3, and 5), whereas ectopic reexpression of eIF4A3 almost completely reversed the induction of p53, indicating a causal link between eIF4A3 ablation and p53 accumulation (lanes 2, 4, and 6). p53 accumulation was not a result of increased TP53 mRNA translation but rather a consequence of enhanced protein stability (fig. S3, A to C), followed by p53 activation as evidenced by the rise of total protein levels of its downstream target, p21 (fig. S3D).
Fig. 3. eIF4A3 knockdown induces p53 and alters the expression of genes regulating rRNA processing.

(A) Illustration depicting U2OS carrying the double DOX-inducible system (table S4B). Administration of DOX suppresses the endogenous eIF4A3, while inducing the ectopic expression of WT FLAG-eIF4A3. ev, empty vector. (B) p53 and eIF4A3 protein levels measured by immunoblotting in engineered U2OS following DOX-induced eIF4A3 knockdown using two different shRNAs and concomitant ectopic expression of FLAG-tagged eIF4A3. Arrows depict endogenous and ectopically expressed FLAG-eIF4A3. L, low exposure; H, high exposure. (C) Reactome pathway enrichment analysis of DE genes between siCtr- versus sieIF4A3-treated cells (DESeq2). Z score shows the overall expression trend (up- or down-regulation) of the genes included in a specific gene ontology (GO) term. Magenta points refer to cell cycle terms and green points to splicing. Data were produced with ClueGO in Cytoscape using the default parameters. (D) GO biological process (BP) analysis of genes referring to the term RNA metabolism (C) using ClueGO and Cytoscape. (E) Log2 fold change (log2FC) in mRNA levels of genes referring to term rRNA processing (D). (F) Starburst plot comparing expression of DE genes following sieIF4A3 or ActDL treatment of U2OS cells (R = 0.57, P < 0.001). (G) Reactome pathway enrichment analysis in common up-regulated genes among sieIF4A3 or ActDL (F, points shown in quadrant I). The inset explains the scaling used. TNF, tumor necrosis factor.
To uncover abnormalities that may explain the p53 induction, we explored the effect of eIF4A3 knockdown (sieIF4A3) on the transcriptome of U2OS cells (fig. S3E). Differential expression (DE) analysis revealed 4493 transcripts (table S2A, Padj ≤ 0.05, log2 fold change ≥ |1|), and pathway enrichment analysis indicated an overall suppression of genes regulating the cell cycle, extracellular matrix organization, splicing, and DNA damage response, while “translation,” “RNA metabolism,” and “p53 signaling” terms were enriched among the up-regulated genes (Fig. 3C and table S2B). While these gene expression results are largely consistent with previous reports (8), little is known about any potential role of eIF4A3 in RiBi. A closer inspection of the RNA metabolism gene signature uncovered a positive trend for genes associated with rRNA processing [e.g., small subunit (SSU) components DHX37, UTP3, EXOSC4, and RRP9], while genes regulating RNA nuclear transport were found mostly down-regulated (Fig. 3, D and E, and table S2C). EIF4A3’s involvement in RiBi was further supported by the fact that Reactome pathway enrichment analysis among the 100 most variable genes in human HCT116 colon carcinoma cells treated or not with an inhibitor of eIF4A3 (31) further attested to a plausible role for eIF4A3 in RiBi (fig. S3F and table S2D).
To identify unique molecular signatures related to RiBi, we compared our RNA-seq results obtained from eIF4A3-depleted cells with those produced upon ActDL known to block Pol I activity. Figure 3F shows that the two experimental conditions share 440 up-regulated genes (quadrant I) whose pathway enrichment analysis points toward p53 signaling (Fig. 3G and table S2, E and F). This and the fact that eIF4A3 knockdown and ActDL both induce p53 and p21 in a similar fashion (fig. S3G) further implicated eIF4A3 in RiBi. Although eIF4A3 knockdown affects splicing (32) and leads to production of aberrant RNA species such as NMD candidates (fig. S3, H and I, and table S7B), chemical inhibition of neither splicing [by pladienolide B (pladB)] nor nuclear export [by leptomycin B (LMB)], two manipulations known to activate p53 (33, 34), altered the observed sieIF4A3-mediated p53 induction, suggesting that these inhibited processes are unlikely the triggers of p53 activation in cells depleted of eIF4A3 (fig. S3J). Collectively, our data point toward perturbations in RiBi as a plausible mechanism for p53 induction.
EIF4A3 associates with the SSU processome and regulates rRNA processing via clearance of excessive R loops
Most genes affected by small interfering RNA (siRNA)–mediated eIF4A3 depletion suggest an impact of eIF4A3 on the early steps of rRNA processing (Fig. 3). This and the fact that eIF4A3 localizes in the nucleolus (Fig. 2) prompted us next to investigate the potential nucleolar function of the helicase. To validate biochemically our RNA-seq findings, we used primers covering precursor and mature rRNA regions (30) and detected a decrease in the levels of early rRNA species both in sieIF4A3-treated and in eIF4A3i-treated samples (Fig. 4A and fig. S4A, respectively). To exclude that changes in the levels of various rRNA species follow deregulated/aberrantly enhanced Pol I transcription, we used a luciferase-based assay using a Pol I–targeted promoter (35) and found that eIF4A3 knockdown did not significantly change expression levels from this rDNA reporter promoter, in contrast to treatment with ActDL (Fig. 4B). This result was further validated by high-content imaging following a short pulse of EU (5-ethynyl uridine) (Fig. 4C) or an antibody against 5.8S rRNA (Fig. 4D). rRNA expression changes in the helicase-depleted cells could not be reversed by the addition of ActDL, suggesting an altered posttranscriptional “flow” of rRNA processing rather than inhibition of Pol I transcription per se.
Fig. 4. EIF4A3 binds the SSU processome and clears excessive R loops to secure unperturbed rRNA processing.

(A) Expression levels of different rRNA species in U2OS treated with siCtr or sieIF4A3 using multiple primer sets. The inset below the graph depicts primer positioning relative to 47S rRNA. RQ, relative quantification. (B) rDNA transcription rate in U2OS cells treated with siRNA against eIF4A3 ± ActDL measured with a luciferase reporter attached to rDNA promoter. (C) EU levels calculated following IF and high-content imaging of U2OS undergoing same treatments as in (B). A total of 1000 to 2000 cells were analyzed per experiment. (D) 5.8S rRNA levels calculated following IF and high-content imaging of U2OS undergoing same treatments as in (B). A total of 1000 to 2000 cells were analyzed per experiment. (E) U3 snoRNA expression levels following immunoprecipitation of eIF4A3 (RIP). (F) 18S rRNA expression levels following immunoprecipitation of eIF4A3 (RIP). (G) Scatterplot showing DE genes following cross-linking immunoprecipitation sequencing (CLIP-seq) data comparison between eIF4A3 and CASC3. The red dot designates U3 snoRNA (SNORD3A) enriched only in eIF4A3 CLIP. (H) Correlation plot between eIF4A3 and early rRNA processing factor BMS1 (DepMap and CCLE 2019). (I) Nucleolar R loop quantification following sieIF4A3 treatment ± RNAse H1 ectopic expression via DOX administration. R loops were detected using S9.6 IF and high-content imaging. All data in (B), (F), and (I) are shown as means ± SD, n = 3 biological replicates, *P < 0.05, **P < 0.01, ***P = 0.01, and ****P < 0.001; nonsignificant values are not shown.
As the early rRNA processing events are executed by the SSU processome (36), we then asked whether eIF4A3 could be part of this complex. To address this notion, we performed RNA immunoprecipitation (RIP) between eIF4A3 and the main SSU small nucleolar RNA (snoRNA), U3. Figure 4E shows that eIF4A3 interacts specifically with U3 snoRNA in fixed U2OS cells in agreement with earlier findings in HeLa cells (37), and their interaction is extended onto the rRNA as seen from RIP assays between eIF4A3 and 18S rRNA (Fig. 4F). DE analysis following cross-linking immunoprecipitation sequencing (CLIP-seq) among eIF4A3 and CASC3 (CASC3 Exon Junction Complex Subunit) RNA interactors (Fig. 4G and table S3A) revealed also U3 snoRNA (SNORD3A) as a specific eIF4A3 interactor, supporting our findings and pinpointing that this interaction is irrelevant to eIF4A3’s role in NMD (where CASC3 participates). Moreover, we found that BMS1 (BMS1 Ribosome Biogenesis Factor), which regulates early rRNA processing via SSU (36), is the most positively correlated RiBi gene to eIF4A3 (Fig. 4H and table S3B), further supporting a role for eIF4A3 in rRNA metabolism.
Given the role of some DEAD helicases in R loop clearance (38) and the fact that nucleoli are susceptible to R loop formation (39), we then reasoned that eIF4A3’s role in SSU may relate to the clearance of excessive R loops that could potentially block rRNA processing and lead to genotoxic transcription-replication collision events. To address this, we used an engineered U2OS cell line expressing a DOX-inducible ribonuclease H1 (RNase H1) cassette (40) and measured R loop abundance in nucleoli with IF, using the widely used antibody S9.6, followed by high-content microscopy. We found that whereas sieIF4A3 enhanced the accumulation of R loops, ectopic expression of RNase H1 reversed this effect, thereby directly implicating eIF4A3 in nucleolar R loop clearance (Fig. 4I). Our findings were further validated using a cell line constitutively expressing catalytically inactive GFP-tagged RNase H1 (fig. S4B) (40), to overcome any technical and/or interpretation difficulties linked to the use of the S9.6 antibody (41). sieIF4A3-triggered R loop accumulation in the nucleolus was related to active Pol I, since treatment of U2OS with ActDL known to selectively block Pol I activity reduced the overall S9.6 signal (fig. S4C). Excessive R loops can cause DNA damage (42), and since our RNA-seq analysis has uncovered relevant gene signatures, we decided to study this notion further. eIF4A3 knockdown led to induced levels of the DNA damage marker γH2AX, shown both by immunoblotting (fig. S4D) and high-content microscopy (fig. S4E). Focusing on the nucleoli, we found that ActDL treatment partially prevented the sieIF4A3-evoked H2AX phosphorylation, a finding consistent with eIF4A3’s involvement in preventing nucleolar R loop accumulation that can lead to DNA damage (fig. S4F). In conclusion, we show here that eIF4A3 is part of the SSU and clears excessive R loops to ensure proper rRNA processing following Pol I transcription (fig. S4G).
EIF4A3’s EJC and RNA binding functions guard against activation of the IRBC
Perturbations in RiBi cause p53 activation via the RPL5/RPL11–5S rRNA–MDM2 complex (IRBC) (2). To assess potential activation of this checkpoint, we measured p53 and p21 levels in U2OS cells depleted of eIF4A3, with or without concomitant depletion of components of the IRBC, and treated or not by ActDL. Figure 5A shows that sieIF4A3 evoked p53 induction that was partially prevented by concomitant depletion of RPL5 (uL18), RPL11 (uL5), or 5S rRNA (lanes 2 and 6 to 8), supporting the notion that depletion of eIF4A3 caused ribosomal biogenesis stress followed by p53 activation. Similar preventive effect was also observed at the additively enhanced level of p53 in cells depleted of eIF4A3 and treated with ActDL (lanes 10 and 14 to 16), indicating that the helicase may be involved in mitigating nucleolar stress caused by exogenous factors such as drugs. Analogous data were reproduced in human A549 lung carcinoma cells, documenting that the observed effects were not cell type–restricted (fig. S5A).
Fig. 5. EIF4A3’s EJC and RNA binding domains are essential for IRBC-mediated p53 induction.
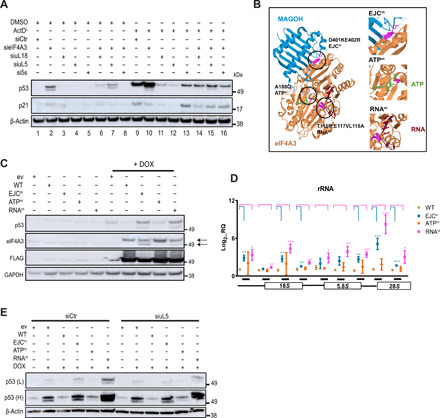
(A) p53 and p21 levels measured by immunoblotting in U2OS treated with sieIF4A3 ± siRNAs against all components of the 5S RNP complex (uL18, uL5, and 5S rRNA) in the presence of ActDL or DMSO vehicle. (B) Left: Graphic illustration of the structural interaction between eIF4A3 (gold) and MAGOH (blue). Encircled residues in magenta refer to eIF4A3 amino acid positions subjected to mutagenesis. Right: Amplification of the mutated regions (green, ATP; red, RNA) (eIF4A3EJCm, D401KE402R; eIF4A3ATPm, A188Q; eIF4A3RNAm, T115R;E117VL118A; table S4A). (C) Immunoblotting showing p53, eIF4A3, and FLAG protein levels in engineered U2OS cells carrying WT or mutant eIF4A3 before and after DOX induction. Arrows depict endogenous and ectopically expressed FLAG-eIF4A3. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. (D) Quantification of different rRNA species in U2OS ectopically expressing WT or mutant eIF4A3 using multiple primer sets. The inset below the graph depicts primer positioning relative to 47S rRNA. Data are shown as means ± SD; n = 3 biological replicates; *P < 0.05, **P < 0.01, and ***P = 0.01; nonsignificant values are not shown. (E) p53 protein levels in engineered U2OS cells following DOX-mediated induction of WT or mutant eIF4A3 ± siRNAs against uL5. (L), low exposure, (H), high exposure.
To clarify which traits of eIF4A3 mediate the suppressive effect on p53 induction, we expressed eIF4A3 mutated in diverse domains, with selectively disabled functional aspects of the protein, in the DOX-sheIF4A3-U2OS cells. DOX treatment of these cells induced expression of the ectopic FLAG-tagged eIF4A3 versions carrying individual mutations in three sites essential for its major activities of RNA binding, interaction with the EJC complex and ATP binding, respectively (DOX-FLAG.mut.eIF4A3-sheIF4A3-U2OS, Fig. 5B, fig. S5B, and table S4A) (43). As can be seen in Fig. 5C, ectopic expression of either the EJC-binding incompetent (D401KE402R, henceforth referred to as eIF4A3EJCm) or the RNA binding incompetent (T115R;E117VL118A, henceforth referred to as eIF4A3RNAm) mutants failed to rescue the effect of knocking down the endogenous wild-type (WT) eIF4A3 on p53 induction, whereas the ATP-binding incompetent mutant (A188Q, henceforth referred to as eIF4A3ATPm) efficiently rescued the impact of lacking the endogenous eIF4A3. The mutant eIF4A3 proteins defective in EJC binding and RNA binding, respectively, were also impaired in terms of their nucleolar residence (fig. S5, C and D), and they caused perturbed rRNA processing (Fig. 5D). The capacity of these two mutants to cause p53 induction was partially reversed in cells treated with siRNA against RPL11 (Fig. 5E), implicating eIF4A3’s EJC and RNA binding capacity in eIF4A3’s nucleolar activity and prevention of the IRBC-mediated p53 up-regulation. In conclusion, eIF4A3 knockdown causes activation of p53 via IRBC, and the ability to reverse this impact by reexpression of eIF4A3 requires the functional EJC and RNA binding domains of the protein.
Residual translation in eIF4A3-depleted cells alters production of stress-related proteins
To determine whether sieIF4A3 affects translation downstream of perturbed RiBi, we used initially a puromycin pulse-chase assay in U2OS cells treated with ActDL or siRNA against eIF4A3. Figure S6A shows that whereas exposure to ActDL substantially reduced global translation, eIF4A3 knockdown showed a less pronounced effect. Quantification of the translation rate using O-propargyl-puromycin (OPP; Fig. 6A) and HPG (‘L-homopropargylglycine) (fig. S6B) click chemistry followed by high-content microscopy indicated a statistically significant but less severe effect of sieIF4A3 on translation compared to that of the protein synthesis inhibitor cycloheximide (CHX) (20 to 25% versus 50%). Furthermore, polysome profiling of U2OS and HeLa cells revealed a higher 80S monosome peak in cells depleted of eIF4A3 (Fig. 6B and fig. S6C, respectively) reminiscent of translation arrest (44).
Fig. 6. eIF4A3 knockdown promotes 80S monosome–mediated translation of stress-related genes and induces p53-mediated cell cycle arrest.
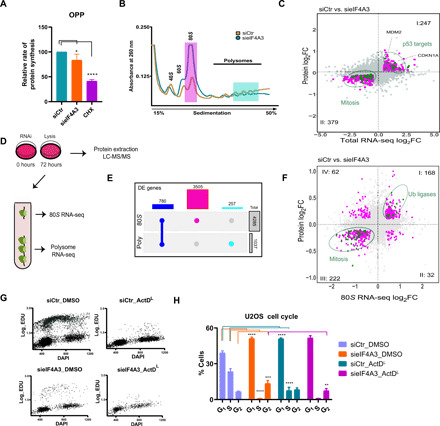
(A) OPP incorporation followed by high-content microscopy for the quantification of translation rates in U2OS treated with sieIF4A3 or CHX as control. A total of 1000 to 2000 cells were analyzed per experiment (data are shown as means ± SD; n = 3 biological replicates; *P < 0.05, ****P < 0.001). (B) Polysome profiling of U2OS cells following eIF4A3 knockdown and separation of the cell lysates in sucrose gradients. High-lightened regions depict fractions used in downstream analyses (magenta, 80S monosome; cyan, polysomes consisted of more than three ribosomes). (C) Starburst plot comparing DE genes in RNA and protein level following sieIF4A3 treatment of U2OS cells (R = 0.56, P < 0.001). (D) Graphic illustration of the experimental design for polysome profile RNA-seq and proteomic analysis. (E) UpSet plot among the DE genes affected by sieIF4A3 in the 80S monosome or polysome translation level. Blue, the common targets; magenta, the genes associated with 80S monosomes; cyan, the ones bound to the polysomes. (F) Starburst plot comparing DE genes in the translation (80S monosome) and protein level following sieIF4A3 treatment of U2OS cells (R = 0.45, P < 0.001, axis of quadrants I to III). (G) Representative 5-ethynyl-2′-deoxyuridine (EdU)–4′,6-diamidino-2-phenylindole (DAPI) IF intensity scatter plots following Click-IT EdU immunostaining of U2OS cells treated with sieIF4A3 ± ActDL (>1000 cells were analyzed per condition). Green points represent cells in S phase. (H) Cell cycle staging in U2OS following treatment with sieIF4A3 ± ActDL. The analysis was based on EdU incorporation and cyclin A1 immunostaining. A total of 1000 to 2000 cells were analyzed per experiment (data are shown as means ± SD; n = 3 biological replicates; **P < 0.01, ***P = 0.01, and ****P < 0.001; nonsignificant values are not shown).
To see the extent of any residual ongoing translation in sieIF4A3-treated cells, we performed liquid chromatography–mass spectrometry (LC-MS) in U2OS treated with sieIF4A3 or control siRNA and run a proteomics DE analysis. We found around 1700 differentially expressed proteins [false discovery rate (FDR) < 0.05] with gene ontology (GO) analysis pointing to cell cycle modulators (table S5, A and B). To exclude the effect of posttranslational events on protein expression, we then integrated these results with our transcriptomic analysis (Fig. 3) and run a pathway enrichment analysis on genes showing monotonic deregulation (Fig. 6C). Among the up-regulated genes (e.g., MDM2 and CDKN1A), p53 signaling was the most prominent GO term, while, in the down-regulated ones, “cell cycle” and “DNA damage” were among the most enriched terms (table S5C). No “rRNA processing”–related terms were detected, indicating translational buffering events of relevant DE genes at the RNA level (Fig. 3) (45) and further supporting our findings regarding a direct role of eIF4A3 in rRNA metabolism via RNA-protein interactions.
Next, we performed RNA-seq analysis on the polysome profile fractions to understand the net effect of translation on the proteome landscape of sieIF4A3-treated cells (Fig. 6D). Given that 80S monosomes may be translationally active (46), we assessed both the translatome of monosomes (Fig. 6B, magenta box) and polysomes (>3 ribosomes; Fig. 6B, cyan box). Following DE analysis, we separated the transcripts into monosome- and polysome-associated, respectively, and found that the majority are bound to 80S monosomes (circa 10 times more compared to polysomes and 5 times more compared to common regulated transcripts; Fig. 6E). To see whether the 80S-bound transcripts correlate with their protein products, we compared the 80S translatome to our proteomics data (Fig. 6F and table S5D). Unexpectedly, most of the correlated transcripts showed monotonic up- or down-regulation (Fig. 6F, quadrants I and III) supporting active translation in monosomes. Among the up-regulated genes, we detected diverse ubiquitin ligases with seminal roles in cell physiology such as MDM2 and RNF135 (ring finger protein 135), while down-regulated genes were enriched in cell cycle modulators and particularly mitotic factors (Fig. 6F and table S5, E and F). Together, we conclude that eIF4A3 depletion hinders partially protein synthesis while allowing some residual translation to take place via both polysomes and monosomes, with cell cycle modulators representing the functional category most prominently affected by this altered translation.
EIF4A3 guards against p53-mediated cell cycle arrest
Given the profound effect of sieIF4A3 on genes regulating cell cycle, we next investigated this aspect in greater detail. Using 5-ethynyl-2′-deoxyuridine (EdU) incorporation followed by high-content microscopy, we first examined the cell cycle progression of nonsynchronized U2OS cells depleted of eIF4A3. Figure 6 (G and H) shows that eIF4A3 knockdown reduced the proportion of S phase cells, similarly to treatment with ActDL, leading to accumulation of U2OS cells in G1 and G2 phases. The concomitant G1 and G2 arrest suggests activation of multiple cell cycle checkpoints, an assumption that was validated by the inability of the eIF4A3i-treated cells, synchronized either in G1 (serum starvation) or in G2 (nocodazole block), to progress into the subsequent cell cycle phases upon release of the inhibitory treatment (fig. S6, D and E). Using FUCCI (fluorescent ubiquitination-based cell cycle indicator) reporter expressing U2OS cells, we showed that the effect of sieIF4A3 on the G1-S transition is the dominant one (fig. S6F). G1 arrest can follow aberrant mitosis (47), and mitotic dysfunction in the absence of eIF4A3 was evident from our omics results. Accordingly, the inability of sieIF4A3-treated cells to undergo proper mitosis was validated using an engineered U2OS cell line stably expressing GFP-tagged histone H2B (fig. S6G) and was further substantiated by the elevated number of micronuclei in cells depleted of eIF4A3 (fig. S6H). The observed G1 arrest of eIF4A3-depleted cells could reflect p53-mediated G1 checkpoint activation including p21 up-regulation (Fig. 3) and the ensuing inhibition of the G1-S transition promoting cyclin E/cdk2 kinase complex, analogous to the effect of DNA damage on cells (48). In support of this notion, we could rescue the cell cycle inhibitory effect of sieIF4A3 (fig. S6I), using a U2OS cell line carrying a dominant negative C-terminal p53 fragment capable of binding to and inhibiting the endogenous WT p53 (ddp53-U2OS). G2 arrest was also rescued, suggesting that p53 is instrumental in both checkpoints. In summary, cells depleted of eIF4A3 show a p53-mediated cell cycle arrest.
EIF4A3 knockdown induces cell death via multiple mechanisms
Our omics analyses revealed an apoptotic gene signature upon sieIF4A3 treatment with genes including FAS, BBC3, and BAX found up-regulated both in monosome and polysome fractions (Fig. 7A and table S6A). This observation prompted us to further study the role of eIF4A3 in cell survival downstream of perturbed cell cycle. EIF4A3 depletion reduced cell survival in a time-dependent fashion (Fig. 7B) while promoting apoptosis evidenced by up-regulation of apoptotic genes, caspase activation, poly(ADP-ribose) polymerase (PARP) cleavage, and diminished colony formation capacity (Fig. 7, C to E, and fig. S7, A to D). Using TP53 knockout HCT116 colon carcinoma cells and the ddp53-U2OS cells, it was possible to partially reverse the effect of sieIF4A3 on cell survival by absence of or interference with p53 (Fig. 7, D and E), indicating that the proapoptotic pathways triggered by eIF4A3 depletion encompass both p53-dependent and p53-independent mechanisms. This concept of dual proapoptotic pathways triggered in the absence of eIF4A3 was further corroborated by the fact that using separately either catalytically inactive p53 (Fig. 7F, lanes 2 and 8) or knockdown of the apoptotic genes BAX and BBC3 (PUMA) (lanes 2, 5, and 6) partially rescued the effect of sieIF4A3 on PARP cleavage, while a combination of these manipulations showed no additive effect (lanes 5, 6, 11, and 12). Moreover, sieIF4A3-mediated FAS up-regulation was apparent also in ddp53-U2OS cells, further supporting the notion that some p53-independent proapoptotic program also becomes active in cells depleted of eIF4A3 (Fig. 7G). Using our total and polysome profiling RNA-seq data, we then performed the deltaTE analysis (45) that identifies transcripts regulated at different levels: transcriptional (buffered and offset), translational (exclusive), or both (forward and intensified) (fig. S7E and table S6B). When we plotted the results of this analysis against our proteomics data, we found that apoptotic genes (e.g., BBC3, FAS, and BAX) were monotonically up-regulated both in monosomes and polysomes (Fig. 7, H and I), a finding that could be validated biochemically (fig. S7, F and G) and indicated bimodal translation of these genes. We further noticed that the regulation mode (intensified versus forward) was different among the monosome and the polysome fractions (Fig. 7, H and I). For example, BAX shows intensified expression (regulated both at the transcriptional level and the translational level) only in the monosome fraction, whereas BBC3 follows the same regulation mode both in monosomes and polysomes (Fig. 7, H and I). This concept of intensified expression was then biochemically validated in the case of BBC3. Whereas p53 is essential for sieIF4A3-mediated BBC3 induction, it is redundant for PUMA protein accumulation under the same experimental conditions (fig. S7, H and I). This suggests that PUMA expression is regulated both at the transcriptional and the translational level, overcoming the “p53 bottleneck.” These findings support a multimodal regulation of cell death genes, consistent with concomitant p53-dependent and p53-independent aspects of cell death induction upon depletion of eIF4A3.
Fig. 7. Loss of eIF4A3 triggers apoptosis in a dual p53/non-p53–dependent manner.

(A) Starburst plot comparing expression of DE genes regulated translationally by the 80S monosome or the polysomes following sieIF4A3 treatment of U2OS cells. Common targets with monotonic deregulation are shown in orange (R = 0.79, P < 0.01). (B) Resazurin time course survival assay in U2OS deprived of eIF4A3 using two different shRNAs. (C) Percentage of caspase 3/7–positive apoptotic cells following eIF4A3 knockdown in U2OS cells. (D) Resazurin assay of WT or TP53 knockout HCT116 cells following eIF4A3 knockdown. (E) Resazurin assay in U2OS or ddp53-U2OS cells deprived of eIF4A3 with siRNA. (F) Immunoblotting of cleaved PARP in U2OS or ddp53-U2OS cells following knockdown of eIF4A3 ± siBAX [siRNA targeting BAX (BCL2 associated X, apoptosis regulator)] or/and siPUMA. (G) Comparative analysis of FAS protein levels between U2OS or ddp53-U2OS cells measured by Western blot following sieIF4A3 or siCtr treatment. Neocarzinostatin was used a positive control for induction of apoptosis. The numbers show quantitation of FAS/β-actin signal. (H) Starburst plot comparing DE genes between 80S monosome–based translational and the protein level. Green dots indicate genes affected from the transcriptional throughout the translational and protein level (forward). Magenta dots show genes that are regulated transcriptionally but are subjected to extratranslational regulation control (intensified) (R = 0.83, P < 0.001). (I) Starburst plot comparing DE genes between polysome-based translational and the protein level. The color coding is the same as in (H) (R = 0.76, P < 0.001). All data in (B) to (E) are shown as means ± SD; n = 3 biological replicates; **P < 0.01 and ****P < 0.001).
EIF4A3 loss alters MDM2 transcript and protein repertoire following p53 activation
On the basis of our findings, we hypothesized that the net transcriptional outcome of p53 activation affecting cell fate could be modulated at the protein translation level (Figs. 6 and 7). Given the effect of sieIF4A3 on posttranscriptional mRNA regulation (6), we then asked whether sieIF4A3 could alter the repertoire of the translated p53 targets. For this purpose, we examined MDM2, a p53 target that was present throughout our omics analyses and was found to be associated with 80S monosomes (Fig. 8A). Under unperturbed growth conditions, MDM2 controls p53 levels via ubiquitination-mediated degradation (49). Degradation or competitive binding of MDM2 to other proteins such as the 5S RNP complex upon RiBi stress (2) causes p53 stabilization. We initially noticed that eIF4A3 ablation (or chemical inhibition) caused production of unique MDM2 protein isoforms not evident following administration of other p53 inducers (Fig. 8, B and C). These isoforms were not byproducts of degradation known to induce p53 (fig. S8, A and B) (50) and could result from translation of alternative MDM2 mRNA isoforms. Using DEXSeq, a differential exon usage–based algorithm, to analyze our RNA-seq data (51), we observed these alternative isoforms that could be also validated by reverse transcription polymerase chain reaction (RT-PCR) (Fig. 8D, fig. S8C, and table S7A). These results are reminiscent of published data regarding the effect of Y14 (another EJC component) on MDM2 splicing (32), likely indicating a universal role for EJC in MDM2 regulation. Integrative analysis of our proteomics data and our 80S monosome RNA-seq data following a transcript-based DE analysis (Salmon; table S7B) revealed that (i) different MDM2 isoforms show different transcription-translation kinetics and (ii) there is at least one NMD MDM2 isoform that is loaded onto the 80S monosomes and could potentially give a protein product (Fig. 8E).
Fig. 8. Depletion of eIF4A3 causes p53-mediated production of aberrant MDM2 transcript isoforms.

(A) Venn diagram depicting p53 targets deregulated by sieIF4A3 treatment of U2OS in the translational (80S monosome and polysomes) and the protein level. MDM2 is altered both in the translatome (80S monosomes) and the protein level. (B) MDM2 protein levels in U2OS treated with sieIF4A3, camptothecin (CPT), 5- fluorouracil (5-FU), or ActDL. The arrows depict the sieIF4A3-specific MDM2 protein products, and the asterisk indicates the main MDM2 protein product. n.s., nonspecific band. (C) MDM2 protein levels following eIF4A3 chemical inhibition in U2OS. The arrows depict the sieIF4A3-specific MDM2 protein products, and the asterisk indicates the main MDM2 protein product. (D) MDM2 mRNA levels measured with RT-PCR following sieIF4A3 ± ActDL treatment of U2OS cells. Arrows indicate MDM2 transcript isoforms present only in samples treated with siRNA against eIF4A3. MW, molecular weight. (E) Starburst plot comparing sieIF4A3-induced DE genes in the RNA (Salmon) and the translational level of 80S monosomes. Orange dots designate genes of all biotypes commonly affected in both levels and cyan the common genes that are also NMD targets (R = 0.24, P < 0.001).
Given that MDM2 mRNA up-regulation following eIF4A3 ablation is p53-dependent (fig. S8D), we hypothesized that production of aberrant mRNA/protein isoforms follows the initial p53 activation that could reflect RiBi stress. In support of this hypothesis, concomitant knockdown of uL5 (RPL11), a component of the 5S RNP complex responsible for the IRBC-mediated p53 up-regulation, prevented the sieIF4A3-mediated induction of all MDM2 isoforms (fig. S8E). In a similar fashion, ectopic expression of the eIF4A3 mutants incompetent in binding either EJC or RNA (eIF4A3EJCm and eIF4A3RNAm, respectively) did not reverse the phenotype of aberrant MDM2 isoform pattern seen upon depletion of endogenous WT eIF4A3. In contrast, replacing endogenous eIF4A3 by the mutant that does not bind ATP (eIF4A3ATPm) could rescue the MDM2 isoform pattern (fig. S8F). As with eIF4A3 knockdown, the production of these alternative MDM2 species was partially reversed by concomitant knockdown of uL5. These isoforms could affect MDM2’s functional interaction with p53, a possibility consistent with the finding that sieIF4A3 treatment markedly enhanced p53 stability (fig. S3A) and augmented the effect of MDM2 chemical inhibition on p53 total levels (fig. S8G). In conclusion, eIF4A3 knockdown leads to both the production of aberrant MDM2 mRNA species (probably via alternative splicing) and their differential translation. It is likely that at least some of these MDM2 species are incompetent to bind p53 (or antagonize normal MDM2 transcripts), rendering the MDM2-p53 feedback loop dysfunctional (52) while, at the same time, sensitizing the cells to stressful stimuli that can induce p53, such as ActDL. Targeting eIF4A3 in cancer can thus be beneficial in two ways: (i) through activation of p53 via IRBC and (ii) by sustaining p53 in an active state via attenuation of the MDM2-p53 feedback loop.
DISCUSSION
In this study, we identified and validated some previously unidentified and unexpected functional features of the human eIF4A3 helicase known for its roles in RNA metabolism, including mRNA splicing via EJC, mRNA nuclear export, and RNA surveillance through nonsense- mediated RNA decay (6). The major advances contributed by our present work include characterization of the so-far elusive and multifaceted role of eIF4A3 in RiBi, including molecular and phenotypic consequences of eIF4A3 depletion in several models, with relevance to human cancer and potential innovative treatments in oncology. Thus, we have unraveled the full spectrum of genes affected by eIF4A3 knockdown, with impact seen at both the RNA and protein levels and uncovered a so-far unanticipated functional requirement for eIF4A3 in rRNA processing through R loop clearance. Our immunohistochemical findings on a range of normal human tissues and tumors further support this nucleolar function of eIF4A3, with even preferential nucleolar localization in subsets of more aggressive lesions. Furthermore, our study explains in detail how p53 is stabilized following eIF4A3 loss through a combination of induced ribosomal stress checkpoint and deregulated MDM2 translation.
Conceptually and mechanistically, our findings expand the previously suggested but unclear role of eIF4A3 in Pol I posttranscriptional events (15) by providing robust functional insight and firmly connecting it to the p53 surveillance pathway. Notably, RiBi perturbations in eIF4A3-depleted cells were followed by translational repression with ensuing bimodal residual protein synthesis taking place both in monosomes and polysomes. Most of the transcripts were found to be 80S-bound, enriched in cell cycle regulators, and monotonically up- or down-regulated throughout the transcriptome-translatome-proteome axis, consistent with the observed consequences for the p53-mediated G1 and G2 arrest.
However, another contribution of our present work is the dissection of the cell death phenotype observed in subsets of eIF4A3-deficient cells. Whereas p53 was essential for the cell cycle delay/arrest in sieIF4A3-treated cells, it was partly redundant for induction of cell death. Integrating polysome profile RNA-seq with proteomics, we found that the sieIF4A3-mediated cell death reflects a dual p53-dependent and p53-independent regulation with some p53 targets (e.g., FAS and BBC3) being regulated both at the transcriptional and the translational level. p53 redundancy may reflect its deregulated control and function, a hypothesis that was tested by looking into its master regulator, MDM2. Depleting cells of eIF4A3 produced various MDM2 translatable transcripts and impeded the MDM2-p53 feedback loop, a fact that could affect the overall impact of p53 on cell fate (52). Particularly relevant to the human RCPS, where an eIF4A3 induced p53 ribosomal stress response, could be rendered more complex by an additional layer of MDM2 alterations.
On the basis of our data presented here, we propose the following model that highlights eIF4A3 as a bridging node between Pol I and Pol II posttranscriptional events (Fig. 9, left): Under normal conditions, nucleolar eIF4A3 supports unperturbed rRNA processing, alongside the canonical EJC-assisted nucleoplasmic splicing. Mature rRNAs bind RPs to form the ribosomal subunits that relocate to the cytoplasm, where they clamp mRNA molecules to translate them. When eIF4A3 levels drop [e.g., RNA interference (RNAi)] (Fig. 9, right), splicing (32) becomes deregulated, allowing the production of aberrant mRNA species. Concomitantly, rRNA processing is interrupted via accumulation of R loops, thereby leading to (i) activation of the IRBC followed by p53 induction and (ii) enhancement of 80S monosome–based translation that allows production of stress-related proteins (e.g., cell cycle and apoptosis) and alternative mRNA isoforms (e.g., alt-MDM2). p53 activation, even when insufficient to induce cell death, is still pivotal in cell cycle delay or arrest, providing another checkpoint and thus opportunity for restoration of cell homeostasis. Accordingly, cancer cells may become addicted to eIF4A3 to evade checkpoints triggered by aberrant Pol I regulation that follows excessive RiBi rates due to their augmented metabolic/energetic demands.
Fig. 9. Illustrated model summary of the role of eIF4A3 in splicing, RiBi, and MDM2-p53 control.

Normal condition (left): eIF4A3 resides both at perispeckles where it assists Pol II–transcribed mRNA splicing and at the nucleoli where it mediates rRNA processing by clearing excessive R loops. Processed rRNA binds RPs to produce ribosomes that subsequently exit the nucleus to translate mRNAs in the cytoplasm. This previously unidentified role of eIF4A3 in RiBi is hijacked in a wide spectrum of human tumors, by mutations, overexpression, or selective nucleolar localization (right): In cases of reduced eIF4A3 levels [e.g., RNAi and RCPS (Richieri Costa-Pereira syndrome)], rRNA processing is compromised and nucleolar morphology changes. This causes ribosomal stress followed by p53-mediated cell cycle arrest and translational reprogramming that favors the production of stress-related proteins (e.g., cell cycle and apoptosis), allowing the cell to adequately react to stress. At the same time, the absence of eIF4A3 affects splicing downstream of Pol II transcription and allows production of alterative transcript isoforms, which, in the case of MDM2 (alt-MDM2), affect the negative p53-MDM2 feedback loop keeping p53 levels high (circled arrows). Nu, nucleolus; Np, nucleoplasm; Cp, cytoplasm; FC, fibrillar center; DFC, dense fibrillar component; GC, granular component.
From a broader, translational perspective, our study opens up previously unidentified avenues for cancer treatment by targeting eIF4A3. While the exon-junction complex and its component eIF4A3 are known to secure the transcriptome, preserving the expression of correctly processed full-length mRNAs (53), we define additional critical roles for eIF4A3 in protecting rRNA processing during RiBi and in maintaining a well-functioning MDM2-p53 axis. Notably, the robustly up-regulated RiBi rates in certain cancers, coupled to the dependence of cancer cells on eIF4A3 for growth and survival as reported herein, highlight a previously unknown mechanistic understanding of an emerging cancer vulnerability with the potential to inspire future therapeutic strategies in oncology.
MATERIALS AND METHODS
Cell culture, cloning, and generation of engineered cell lines
U2OS was purchased from the American Type Culture Collection. A549, SaOS-2, HeLa, MCF7-1, RPE1, human embryonic kidney (HEK) 293T, and GFP.RNase H1.U2OS were provided by T. Helleday [Karolinska Institutet (KI)]. IMR90 and CCD841 were provided by M. Bienko (KI); Cl2.6 by M. Lindström (KI); and BJ, HT29, ddp53-U2OS, HCT116, and HCT116 TP53−/− by J. Bartek (Danish Cancer Society). All cell lines were maintained in Dulbecco’s modified Eagle’s medium GlutaMAX (Gibco) supplemented with 10% fetal bovine serum (Sigma-Aldrich). Cell lines were tested for mycoplasma on a monthly basis (Lonza). shRNAs against eIF4A3 (table S4B, uppercase is the backbone and lowercase is the target sequence) were cloned into a U6-Tet-shRNA:Ef1a-TetR-Puro-iRFP670 vector using Age I/Eco RI, as previously described in (54). Viral particles were produced by transfecting the lentiviral and corresponding packaging plasmids (encoding polymerase and envelope proteins) into HEK293T cells. Supernatants were collected two times with 24-hour intervals, filtered, and used to infect U2OS. The cells (DOX-sheIF4A3-U2OS) were selected in the presence of puromycin (1 μg/ml) for 72 hours or until all nontransduced control cells were killed. For the production of DOX-FLAG-eIF4A3(WT/mutant)–sheIF4A3-U2OS cells, EIF4A3 was PCR-amplified from pCMV6-eIF4A3 (Origene, vector: PS100001) using the primer set provided in table S4B and cloned into a N-FLAG–tagged pENTR4 plasmid, which was produced by inserting an oligo-annealed V5-sequence into a pENTR4 vector (55) using Sal I/Not I. EIF4A3 was then subcloned into an DOX-inducible destination vector with blasticidin resistance. The destination vector was produced by inserting an Ef1a-promoter-tta3G-P2A-blasticidin cassette (GenScript) into a Tet-on-inducible plasmid (Addgene #27567) using Pml I/Kpn I. For the production of eIF4A3 mutants, point mutations were inserted into pENTR-eIF4A3 using the GeneArt site-directed mutagenesis kit (Thermo Fisher Scientific, A13282), and the primers are shown in table S4A. Viral particle production and cell transduction were conducted in the same fashion as with shRNA carrying plasmids, and stably transduced cells were selected in the presence of blasticidin (20 μg/ml) for 72 hours or until all nontransduced control cells were killed. GFP-eIF4A3-U2OS cells were produced by transduction of U2OS with pLenti-C-GFP-eIF4A3 (Origene, vector: PS100065).
Cell treatments
Cells were treated with 5 nM ActDL (Sigma-Aldrich, A1410) or 2 μM BMH-21 (Sigma-Aldrich, SML1183) for 24 hours. CHX (Sigma-Aldrich, C4859) was used at a working concentration of 100 μg/ml for 30 min or the indicated time points. For DOX induction (Sigma-Aldrich, D9891), cells were treated with DOX (1 μg/ml; Sigma-Aldrich, D9881) for 48 to 72 hours, unless otherwise indicated. Pan-caspase inhibitor Z-VAD-FMK (R&D Systems, FMK001) and caspase 2 inhibitor Z-VDVAD-FMK (R&D Systems, FMK003) were used at a final concentration of 20 μM for 1 hour or the indicated time points. MG132 (Sigma-Aldrich, M7449), Nutlin-3 (Selleckchem, S1061), and MLN4924 (Selleckchem, S7109) were added to cells in a final concentration of 10, 5, and 1 μM, respectively, for 24 hours. Camptothecin (Sigma-Aldrich, C9911) was used at a working concentration of 5 μM and 5-fluorouracil (Sigma-Aldrich, F6627) at 10 μg/ml. Neocarzinostatin was used at 0.5 μg/ml for 24 hours, LMB was used at 20 ng/ml for 6 hours, and pladB was used at 1 μM for 6 hours. For chemical inhibition of eIF4A3, 52a, or 52b, 53a from (31) was used at 5 μM for 24 hours. For cell synchronization at G2, cells were treated with nocodazole (Sigma-Aldrich, M1404) at 400 nM for 16 hours.
Antibodies
The antibodies used are as follows: mouse monoclonal SC35 (Abcam, ab11826), mouse monoclonal eIF4A3 (Santa Cruz Biotechnology, sc-365549), rabbit monoclonal α-tubulin (Abcam, ab176560), mouse monoclonal α-phospho-H2A.X (Ser139) (Sigma-Aldrich, 05-636), mouse monoclonal UBF (Santa Cruz Biotechnology, sc-13125), rabbit polyclonal FBL (Abcam, ab5821), rabbit polyclonal RBM8A (Y14) (Thermo Fisher Scientific, PA5-53790), mouse monoclonal nucleophosmin (Abcam, ab10530), rabbit polyclonal nucleolin (Abcam, ab22758), mouse monoclonal puromycin (Kerafast, EQ0001), rabbit polyclonal RPL5 (uL18) (Abcam, ab86863), mouse monoclonal RPL11 (uL5) (1:200; Life Technologies, 373000), mouse monoclonal p53 (1:1000; Abcam, ab1101), mouse monoclonal FLAG (1:1000; Sigma-Aldrich, F3165), rabbit monoclonal p21 (Cell Signaling Technology, 2947), rabbit polyclonal cyclin A1 (Abcam, ab53699), mouse monoclonal phospho–serine 10 H3 (Abcam, ab14955), rabbit polyclonal FAS (Abcam, ab82419), rabbit monoclonal PARP (Abcam, ab32138), mouse monoclonal MDM2 (Santa Cruz Biotechnology, sc965), mouse monoclonal MDM2m (mixture of 2A9, 4B2, and 4B11 clones, provided by M. Oren), mouse monoclonal MDM2 (SMP14, sc-965), and mouse S9.6 (provided by O. F. Capetillo). Unless otherwise stated, all antibodies were used at a working dilution of 1:500 for Western blotting and 1:400 for IF. Secondary antibodies used are as follows: mouse Alexa Fluor 647 (Thermo Fisher Scientific, A-21235), rabbit Alexa Fluor 647 (Thermo Fisher Scientific, A-21244), rabbit Alexa Fluor 488 (Thermo Fisher Scientific, A-11008), mouse Alexa Fluor 488 (Thermo Fisher Scientific, A-11029), mouse horseradish peroxidase (HRP; Sigma-Aldrich, A9044), and rabbit HRP (Sigma-Aldrich, A6154). Secondary antibodies for IF were used in dilution 1:500 and for immunoblotting in dilution 1:10,000.
Gene silencing
Commercially available SMARTpool ON-TARGET oligonucleotides targeting human eIF4A3 (L-020762), uL5 (RPL11) (L-013703), CASP8 (L-003466), CASP9 (L-003309), TP53 (L-003329), BAX (L-003308), BBC3 (L-004380), and nontargeting siRNA (D-001810) were purchased by Horizon Discovery (Dharmacon). siRNAs against 5S rRNA and RPL5 (uL18) were custom made Horizon Discovery (Dharmacon) according to (56) and (30), respectively. Cells were transfected with 20 nM siRNA using Lipofectamine RNAiMAX reagent (Thermo Fisher Scientific, 13778150) according to the manufacturer’s instructions. After 48 hours or the times indicated, cells were harvested and lysed for RNA or protein extraction.
Cell survival assays
Cell proliferation was assessed using the resazurin assay. Cells were seeded at a density of 2000 cells per well in 96-well plates; the next day, they were transfected with siRNAs for 48 to 72 hours, while chemical addition took place within the last 24 hours before measuring optical density (OD). Resazurin working solution (Sigma-Aldrich, R7017) was added to supernatants to a final concentration of 50 μg/ml. The plates were further incubated for 4 hours at 37°C, and the emitted fluorescence was measured with a microplate reader using the 560-nm excitation/590-nm emission filter set (Tecan Infinite M1000 Pro). All growth assays were performed a minimum of three times using independent platings of cells to ensure reproducibility. Colony formation assays were performed according to Franken et al. (57).
Preparation of cell extracts and Western blotting
Following chemical administration or transfection, subconfluent cells were lysed in radioimmunoprecipitation assay (RIPA) buffer supplemented with a cocktail of protease and phosphatase inhibitors (Thermo Fisher Scientific, 78444) and sonicated for five cycles of 30-s on and 15-s off, in a Bioruptor (Diagenode). Following lysate clearance with centrifugation for 10 min at 13,000 rpm and 4°C, protein quantification was performed with the DC Protein Assay Kit II (Bio-Rad, 5000112). Cell lysate (20 μg) was boiled in Laemmli sample buffer for 5 min at 95°C, loaded onto SDS–polyacrylamide gel electrophoresis gels and transferred onto nitrocellulose or polyvinylidene difluoride membranes. Chemiluminescence signal was detected using SuperSignal West Dura (Thermo Fisher Scientific, 34076). Images were acquired with an Amersham Imager 600 scanner. Cell fractionation was performed as previously described (58). Data regarding eIF4A3 subcellular localization were acquired from PepTracker (59). Trichloroacetic acid precipitation was used for protein extraction following polysome profiling.
Quantitative real-time PCR
Total RNA extraction was performed with the PureLink RNA Mini Kit (Thermo Fisher Scientific, 12183025), and RNA extraction from polysome profiling fractions was achieved with TRIzol, followed by clarification with the Norgen Biotek Corp RNA Clean Up/Concentration kit (Norgen Biotek, 298-23600). Quantitative RT-PCR was conducted with the TaqMan RNA-to-CT 1-Step Kit (Thermo Fisher Scientific, 4392938) in a StepOnePlus Real-Time PCR System (Applied Biosystems). TaqMan probes used in this study are listed below. For identification of MDM2 mRNA species, RNA was reverse-transcribed using SuperScript IV Reverse Transcriptase (Thermo Fisher Scientific, 18090050) using random hexamer primers to generate cDNA. The cDNA was treated with 5 U of RNase H (New England Biolabs, M027) for 30 min at 37°C and then analyzed by PCR using the external primer set from Sigalas et al. (60) and a Veriti 96-Well Thermal Cycler (Thermo Fisher Scientific) [30 cycles; Tm (melting temperature): 55°C]. Primers and PCR conditions for rRNA species and apoptotic genes were previously described (30). TaqMan probes (Thermo Fisher Scientific): EIF4A3: 4331182 (Hs01556773_m1); and MDM2: 4400291 (Hs02970282_cn), FAS (Hs00236330_m1), BBC3 (Hs00248075_m1), and TP53 (Hs01034249_m1).
Transcriptomics
RNA-seq was performed by National Genomics Infrastructure (NGI) at Science for Life Laboratory Stockholm, Sweden. Briefly, following RNA quality assessment by TapeStation electrophoresis (Agilent), libraries were prepared with Illumina RiboZero TruSeq Stranded mRNA, and samples were sequenced in a HiSeq 2500 system (Illumina). Data preprocessing was performed in NGI through nf-core/rnaseq (61). DE analysis was performed using DESeq2 (v1.24.0) (62). Transcript level quantification was performed with Salmon (63), followed by DESeq2-mediated DE. Exon-based differential analysis was achieved with DEXSeq (51) and splicing analysis with IsoformSwitchAnalyzeR (64). For downstream GO analysis, DESeq2-produced data were filtered using a log2 fold change > |1| and P < 0.05 threshold and processed via ClueGO (v2.5.4, ontologies update 09 April 2018) (65) in Cytoscape (66) or g:Profiler (67). Z score in GO analysis gives the likelihood that a specific GO term is increased or decreased and is described in (68). Transcription factor enrichment analysis was executed with iRegulon (v1.3) (69) in Cytoscape. CLIP-seq data from (70) were analyzed with DESeq2. RNA-seq data following eIF4A3 chemical inhibition were incorporated from (31). All analyses were performed in RStudio (v1.1.453).
Proteomics analysis
Samples were lysed by 4% SDS lysis buffer and prepared for MS analysis using a modified version of the SP3 protein clean-up and digestion protocol (71). Peptides were labeled with TMT10plex reagent according to the manufacturer’s protocol (Thermo Fisher Scientific) and separated by immobilized pH gradient–isoelectric focusing (IPG-IEF) on 3 to 10 strips as described previously (72). Extracted peptide fractions from the IPG-IEF were separated using an online 3000 RSLCnano system coupled to a Thermo Scientific Q Exactive-HF. MSGF+ and Percolator in the Galaxy platform was used to match MS spectra to the Ensembl_92 Homo sapiens protein database (73). DE analysis was performed with DEP (74) in RStudio (v1.1.453). DE proteins passing the threshold of α = 0.85 and lfc (log fold change) = 0.1 were used subsequently for STRING (v. 11) (75) and GO analyses.
RIP assay
RIP was performed according to previously published data (76). Briefly, cells were transfected with 20 nM control siRNA or siRNA against eIF4A3 for 48 hours. The cells were then fixed in 1% formaldehyde and lysed. Following sonication and DNA digestion, equal amounts of lysate were immunoprecipitated with either 3 μg of mouse α-eIF4A3 (Santa Cruz Biotechnology, sc-365549) or mouse immunoglobulin G (IgG) using G-agarose magnetic beads. The next day, the beads were washed thoroughly, and following elution of RNA-protein complexes, RNA was isolated with phenol/chloroform and used as template in qPCR for U3 snoRNA (table S4C) or 18S rRNA (30).
Microscopy
Cells were seeded on 6-well plates containing coverslips (Thermo Scientific Nunc Thermanox Coverslips, 12-565-88) or 96-well plates 1 day before treatment. Following treatment, cells were washed three times with phosphate-buffered saline (PBS), fixed in 4% formaldehyde (Sigma-Aldrich, F8775) for 10 min, and permeabilized for 10 min using PBS with 0.5% Triton X-100 (Sigma-Aldrich, X100). They were subsequently washed three times with PBS and incubated for 1 hour at room temperature in blocking solution [3% bovine serum albumin (BSA) in PBS] (Sigma-Aldrich, A7906). Fixed cells were incubated at room temperature with primary antibodies for 1 hour at dilution 1:400 (unless otherwise stated), washed three times in PBS, and incubated with secondary antibodies for 1 hour at room temperature (dilution, 1:500). Last, cells were stained with 10 μM Hoechst (Thermo Fisher Scientific, 62249) for 30 min at room temperature, and the cover slips were mounted onto slides. FBL was used as a marker for nucleolar segregation. Images were acquired using a Nikon Eclipse Ti2 inverted epifluorescence microscope or an inverted Zeiss LSM 780 confocal microscope. EdU/EU staining was performed with the Click-iT EdU Cell Proliferation Kit for Imaging and Alexa Fluor 647 dye (Thermo fisher Scientific, C10340) or Click-iT RNA Alexa Fluor 488 Imaging Kit (Thermo fisher Scientific, C10329), respectively, OPP staining with the Click-iT Plus OPP Alexa Fluor 488 Protein Biogenesis Assay Kit (Thermo Fisher Scientific, C10456) and HPG staining with the Click-iT HPG Alexa Fluor 488 Protein Synthesis Assay Kit (Thermo Fisher Scientific, C10428) according to the manufacturer’s instructions. Images were acquired using an IN Cell Analyzer 2200 (GE Healthcare) and analyzed using Cell Profiler, Fiji (Image J), and RStudio (v1.1.453).
Translation monitoring
For puromycin pulse-chase assays, cells were incubated with 1 μM puromycin (Sigma-Aldrich, P8833) for 60 min and lysed in RIPA containing a cocktail of protease and phosphatase inhibitors (Thermo Fisher Scientific, 78444). Puromycin incorporation was visualized by Western blotting with an antibody against puromycin. For polysome profiling, cells in 70 to 80% confluent 15-cm dishes were washed twice in PBS containing CHX (100 μg/ml) and lysed after treatment in hypotonic buffer supplemented with CHX (Sigma-Aldrich, C7968) (44). Lysates were cleared by centrifugation, and equal amounts (adjusted by 260-nm OD measurement) were loaded onto 5 to 50% sucrose gradients produced in sucrose gradient buffer with a gradient forming unit (BioComp). Samples were centrifuged at 36,000 rpm for 3 hours using a SW41Ti rotor in an Optima XE-90 ultracentrifuge (Beckman Coulter). The samples were then analyzed in a piston gradient fractionator (BioComp) using the company’s software. Twenty-eight fractions were collected at a pace of 2.86 mm per fraction (absorbance at 540 nm) and were further used for protein extraction (see section ‘Preparation of cell extracts and Western blotting’).
Polysome profiling RNA-seq
RNA was isolated from 80S monosomes and polysomes (>3 ribosomes) fractions by using phenol/chloroform extraction method and purified further with Agencourt AMPure RNAClean XP beads (Beckman Coulter, A62987). RNA-seq libraries were prepared using a TruSeq Stranded Total RNA library preparation kit (Illumina, 20020596) and TruSeq RNA Single Indexes (Illumina, 20020492) according to the manufacturer’s protocol, starting with 100 ng of RNA. SuperScript IV Reverse Transcriptase (Thermo Fisher Scientific, 1809001) was used for cDNA synthesis, and DNA was purified with Agencourt AMPure XP beads (Beckman Coulter, A63881). Quality assessment and concentration estimation of the purified RNA and DNA were performed using the Qubit 3 (Life Technologies) and BioAnalyzer 2100 (Agilent). Each library was diluted to 4 nM and combined equimolar into a single pool before sequencing on the Illumina NextSeq500 platform (Illumina) using the NextSeq 500/550 High Output Kit v2.5 (150 cycles) (Illumina, 20024907). RNA-seq data were processed using nf-core/rnaseq (61) pipeline using GRCh37 genome build. Following initial multidimensional scaling analysis, one sample (from polysome fraction) was omitted in downstream DE analyses due to poor alignment efficiency. DE analysis was performed using DESeq2 (v1.24.0) (62) and deltaTE (45).
Flow cytometry
Following treatment, live FUCCI-U2OS cells were washed twice and resuspended in PBS containing 10% BSA. Samples of 200 to 250 μl (5000 events) in 96-well round-bottom plates were analyzed with the Guava easyCyte 5HT HPL Benchtop Flow Cytometer (Millipore). Cell cycle stages were determined using the InCyte 3.3 software with the gain controls set to 7.66 (forward scatter), 1.0 (side scatter), 3.83 (red/green fluorescence), 3.36 (yellow fluorescence), and 7.8% yellow-green compensation.
Apoptosis assay
Cells were seeded in 96-well plates at low density (2000 cells per well), treated as indicated, and stained for caspase 3/7 with CellEvent Caspase-3/7 Green Detection Reagent (Thermo Fisher Scientific, C10423) according to the manufacturer’s guides. Cells were counterstained with 10 μM Hoechst (Thermo Fisher scientific, 62249) for 30 min, and images were acquired with IN Cell Analyzer 2200 (GE Healthcare) and analyzed using Cell Profiler.
Electron microscopy
Cells were fixed, after treatment, in 2.5% glutaraldehyde/0.1 M phosphate buffer (pH 7.4) at room temperature for 30 min and were then collected and in Eppendorf tubes and incubated overnight at 4°C. Cell pellets were subsequently rinsed in PBS, fixed in 2% osmium tetroxide (TAAB, Berks, England)/0.1 M phosphate buffer (pH 7.4) at 4°C for 2 hours, dehydrated in ethanol/acetone, and embedded in LX-112 (Ladd, Burlington, Vermont, USA). Ultrathin sections (~50 to 60 nm) were cut with Leica EM UC 6 (Leica Wien, Austria), incubated in uranyl acetate and lead citrate, and examined with a Hitachi HT 7700 (Tokyo, Japan) at 80 kV. Digital images were acquired with a Veleta camera (Olympus Soft Imaging Solutions, GmbH, Münster, Germany). Transmission EM was performed in EMil Core Facility at KI.
Data analysis and illustration
RNA-seq and CLIP-seq DE analysis was performed using the R packages DESeq2 (v1.25.7) (62) and DEXSEq (v1.31.0) (51) and the bash-scripting software for transcript quantification Salmon (v0.14.1) (63). Polysome profile RNA-seq DE analysis was performed using the R packages deltaTE (45). Proteomics analysis was achieved via the R package DEqMS (77). Graphs were produced in ggplot2 (v3.2.0) (78) or GraphPad Prism (v8.0.1) and graphical illustrations in Adobe Illustrator.
Chemical biogenesis
Synthesis of eIF4A3 chemical inhibitors (4-(4-bromobenzoyl)-3-(4-chlorophenyl)piperazin-1-yl)(6-bromopyrazolo[1,5-a]pyridin-3-yl)methanone and 3-(4-(4-(4-bromobenzoyl)-3-(4-chlorophenyl)piperazine-1-carbonyl)-5-methyl-1H-pyrazol-1-yl)benzonitrile (compounds 52a and 52b, respectively) were prepared as racemic compounds according to (79). Both compounds were separated into enantiomers using preparative supercritical fluid chromatography (SFC). Both enantiomers were collected for each compound and were analyzed by LC-MS and nuclear magnetic resonance. All fractions were analyzed by chiral SFC and 52b and 52a by optical rotation. Analytical data were in accordance with (79). Compound 52a was separated into enantiomers using a CHIRALPAK ID column (10 mm by 250 mm) eluting with 55% CO2 and 45% MeOH, a flow of 15 ml/min, and a temperature of 45°C. The first eluting fraction was assigned as 52a, (R)-(4-(4-bromobenzoyl)-3-(4-chlorophenyl)piperazin-1-yl)(6-bromopyrazolo[1,5-a]pyridin-3-yl)methanone, and the second fraction was assigned as 52b (R)-(4-(4-bromobenzoyl)-3-(4-chlorophenyl)piperazin-1-yl)(6-bromopyrazolo[1,5-a]pyridin-3-yl)methanone. Compound 53 was separated into enantiomers using an YMC Chiral Cellulose SB-column (10 mm by 250 mm) eluting with 70% CO2 and 30% MeOH, a flow of 15 ml/min, and a temperature of 45°C. The first eluting fraction was assigned as 53a (S)-3-(4-(4-(4-bromobenzoyl)-3-(4-chlorophenyl)piperazine-1-carbonyl)-5-methyl-1H-pyrazol-1-yl)benzonitrile, and the second fraction was assigned as 53b (R)-3-(4-(4-(4-bromobenzoyl)-3-(4-chlorophenyl)piperazine-1-carbonyl)-5-methyl-1H-pyrazol-1-yl)benzonitrile. Compound specificity was described in (79).
Tumor tissues and immunohistochemistry
All tissues used for immunohistochemical analysis were archival specimens routinely fixed in buffered formalin and embedded in paraffin. The information about cohorts of human tissue and tumor specimens used in the present study can be found in our published studies on DNA damage response markers on these tissues, as follows: urinary bladder (normal tissue, n = 12; early Ta-T1 lesions, n = 222; and invasive T2-T4 tumors, n = 230) (80), colon (normal mucosa, n = 10; adenomas, n = 19; and colon carcinomas, n = 15) (81), uterine cervix (normal tissue, n = 3; dysplasia, n = 4; squamous carcinoma in situ, n = 5; and invasive squamous carcinoma, n = 15) (82), and brain tissue and glioblastomas (normal brain, n = 6; and glioblastoma, n = 43) (83). The tumors were classified and staged following the official international guidelines for grading from the World Health Organization. The studies were approved by the relevant ethical committees in Denmark. To detect the eIF4A3 protein and its staining patterns in the above listed human tissue and tumor specimens, we used our well-established sensitive immunohistochemical staining protocol (80–82, 84). Standard deparaffinization of the archival formalin-fixed, paraffin-embedded tissue sections was followed by antigen unmasking in the citrate buffer (pH 6, for 15 min in a microwave). After overnight incubation with the primary antibody, samples were processed for the indirect streptavidin-biotin-peroxidase method using the VECTASTAIN Elite Kit (Vector Laboratories) and nickel sulfate–based chromogen enhancement detection as previously described (83), without nuclear counterstaining (80–82, 84). The primary antibody against eIF4A3 used here was a rabbit polyclonal antibody used at 1:250 dilution (Santa Cruz Biotechnology, sc67369). For negative controls, sections were incubated with nonimmune rabbit serum. For positive controls, a rabbit antibody against human phospho-histone H2A.X (Ser139) was used. Results were evaluated by a senior oncopathologist, and data were expressed in three scoring categories based on the percentage of positive tumor cells with defined subnuclear expression patterns of the eIF4A3 protein as follows: category (I) pan-nuclear, with eIF4A3 detected with similar intensity in both nucleolus and nucleoplasm in 95% or more cells; category (II) eIF4A3 preferentially accumulated in nucleoli over nucleoplasm in at least 6% cells (6 to 100% of cells per lesion); or category (III) eIF4A3 being selectively excluded from nucleoli in at least 6% cells (6 to 100% of cells per lesion), respectively.
Mining public databases
To determine the mutational status of eIF4A3 in cancer, we combined TCGA data describing eIF4A3 mutations in multiple cancer types with point mutations presented by Bono et al. (43). Correlation of eIF4A3 mRNA levels and TCGA cancer patient survival rates was accomplished with LinkedOmics (17). Comparison of eIF4A3 mRNA expression between cancer and normal tissue was performed with GENT2 (Gene Expression database of Normal and Tumor tissues 2) (16). For the division of cancer cell lines in low- and high-RiBi groups, we used RNA-seq expression data from DepMap (v.20Q3) (18). For each gene of a curated (RiBi-related) gene list (based on Reactome pathways: RHSA73864 and RHSA72312 excluding the histones; table S1D), the median was calculated, and cell lines were characterized as high RiBi when the number of the genes scoring higher than each median was bigger than the number of the genes scoring lower than each median and vice versa. Compound cytotoxicity data were extracted from PRISM primary repurposing drug screen (DepMap, 19Q4).
Statistics and reproducibility
RNA-seq and proteomics experiments were performed in biological triplicates, and statistical analysis followed the tests provided in the relevant DE analysis packages in R. quantitative RT-PCR–, image-, survival-, and apoptosis-associated experiments were performed a minimum of three times with independent biological samples. Parametric (N > 100) or nonparametric t test (N < 100) or analysis of variance (ANOVA) was used for statistical testing. Sample size (N) and P (or FDR) values are provided in the corresponding figures or figure legends.
Acknowledgments
We would like to thank C. Navarro for the support in bioinformatics, M. Oren (Weizmann Institute of Science) for providing MDM2m antibody, and O. F. Capetillo for providing the S9.6 antibody. Moreover, we thank T. Helleday for providing A549, SaOS-2, HeLa, MCF7, RPE1, HEK293T, and GFP.RNAseH1.U2OS and M. Bienko for sharing IMR90 and CCD841 cell lines. MS analysis was performed by the Clinical Proteomics Mass Spectrometry facility, KI/Karolinska University Hospital/Science for Life Laboratory. We would like to acknowledge also the support from the NGI and Uppmax (Science for Life Laboratory) for providing assistance in massive parallel sequencing and computational infrastructure. Last, we would like to thank A. S. Nilsson and J. F. Martinez for technical assistance with cell cultures and P. Moberg and the Drug Discovery and Development Platform at Science for Life Laboratory, Sweden, for assisting with development of the eIF4A3is. Funding: This work was funded by the following grants: the Swedish Cancer Society (grant number 170176) and the Swedish Research Council (VR-MH 2014-46602-117891-30) (both granted to J. Bartek.). S.J.E. acknowledges funding by Vetenskapsrådet (2015-04815), H2020 ERC Starting Grant (715024 RAPID), and Cancerfonden (2015/430). A.-M.K. acknowledges funding from Barncancerfonden (TJ2016-0056). Author contributions: Initial conceptualization: D.C.K. and J. Bartek. Evolution of conceptualization: D.C.K., J.A.E., M.S.L., and J. Bartek. Methodology: D.C.K., J.A.E., A.-M.K., E.S., J. Bartkova, and M.A. Formal analysis: D.C.K., E.S., and S.J.E. Investigation: D.C.K., J.A.E., A.Z., E.S., J. Boström, A.-M.K, J.Bartkova, M.A., S.J.E., and M.S.L., and J. Bartek. Resources: L.D., H.B., M.S.L., and J. Bartek. Data curation: D.C.K., J.A.E., and E.S. Writing—review and editing: all authors. Visualization: D.C.K., J.A.E., and E.S. Supervision: M.S.L. and J. Bartek. Project administration: J. Bartek. Funding acquisition: J. Bartek. Competing interests: The authors declare that they have no competing interests. Data and materials availability: The RNA-seq FASTQ files generated specifically for this study can be accessed from the NCBI’s BioProject repository using the IDs PRJNA579088 and PRJNA645345 and the proteomics data from ProteomeXchange under the accession number PXD016005. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIAL
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/32/eabf7561/DC1
REFERENCES AND NOTES
- 1.Peña C., Hurt E., Panse V. G., Eukaryotic ribosome assembly, transport and quality control. Nat. Struct. Mol. Biol. 24, 689–699 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Turi Z., Lacey M., Mistrik M., Moudry P., Impaired ribosome biogenesis: Mechanisms and relevance to cancer and aging. Aging 11, 2512–2540 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bursać S., Prodan Y., Pullen N., Bartek J., Volarević S., Dysregulated ribosome biogenesis reveals therapeutic liabilities in cancer. Trends Cancer 7, 57–76 (2021). [DOI] [PubMed] [Google Scholar]
- 4.Danilova N., Gazda H. T., Ribosomopathies: How a common root can cause a tree of pathologies. Dis. Model. Mech. 8, 1013–1026 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khot A., Brajanovski N., Cameron D. P., Hein N., Maclachlan K. H., Sanij E., Lim J., Soong J., Link E., Blombery P., Thompson E. R., Fellowes A., Sheppard K. E., McArthur G. A., Pearson R. B., Hannan R. D., Poortinga G., Harrison S. J., First-in-human RNA polymerase I transcription inhibitor CX-5461 in patients with advanced hematologic cancers: Results of a phase I dose-escalation study. Cancer Discov. 9, 1036–1049 (2019). [DOI] [PubMed] [Google Scholar]
- 6.Le Hir H., Saulière J., Wang Z., The exon junction complex as a node of post-transcriptional networks. Nat. Rev. Mol. Cell Biol. 17, 41–54 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Popp M. W., Maquat L. E., Nonsense-mediated mRNA decay and cancer. Curr. Opin. Genet. Dev. 48, 44–50 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mao H., McMahon J. J., Tsai Y.-H., Wang Z., Silver D. L., Haploinsufficiency for core exon junction complex components disrupts embryonic neurogenesis and causes p53-mediated microcephaly. PLOS Genet. 12, e1006282 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Favaro F. P., Alvizi L., Zechi-Ceide R. M., Bertola D., Felix T. M., de Souza J., Raskin S., Twigg S. R. F., Weiner A. M. J., Armas P., Margarit E., Calcaterra N. B., Andersen G. R., McGowan S. J., Wilkie A. O. M., Richieri-Costa A., de Almeida M. L. G., Passos-Bueno M. R., A noncoding expansion in EIF4A3 causes Richieri-Costa-Pereira syndrome, a craniofacial disorder associated with limb defects. Am. J. Hum. Genet. 94, 120–128 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin Y., Zhang J., Cai J., Liang R., Chen G., Qin G., Han X., Yuan C., Liu Z., Li Y., Zou D., Mao Y., Systematic analysis of gene expression alteration and co-expression network of eukaryotic initiation factor 4A-3 in cancer. J. Cancer 9, 4568–4577 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang R., Zhang S., Chen X., Li N., Li J., Jia R., Pan Y., Liang H., EIF4A3-induced circular RNA MMP9 (circMMP9) acts as a sponge of miR-124 and promotes glioblastoma multiforme cell tumorigenesis. Mol. Cancer 17, 166 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miller E. E., Kobayashi G. S., Musso C. M., Allen M., Ishiy F. A. A., de Caires L. C., Goulart E., Griesi-Oliveira K., Zechi-Ceide R. M., Richieri-Costa A., Bertola D. R., Passos-Bueno M. R., Silver D. L., EIF4A3 deficient human iPSCs and mouse models demonstrate neural crest defects that underlie Richieri-Costa-Pereira syndrome. Hum. Mol. Genet. 26, 2177–2191 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mizojiri R., Nakata D., Satoh Y., Morishita D., Shibata S., Iwatani-Yoshihara M., Kosugi Y., Kosaka M., Takeda J., Sasaki S., Takami K., Fukuda K., Kamaura M., Sasaki S., Arai R., Cary D. R., Imaeda Y., Discovery of novel 5-(piperazine-1-carbonyl)pyridin-2(1H)-one derivatives as orally eIF4A3-selective inhibitors. ACS Med. Chem. Lett. 8, 1077–1082 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Trainor P. A., Dixon J., Dixon M. J., Treacher Collins syndrome: Etiology, pathogenesis and prevention. Eur. J. Hum. Genet. 17, 275–283 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alexandrov A., Colognori D., Steitz J. A., Human eIF4AIII interacts with an eIF4G-like partner, NOM1, revealing an evolutionarily conserved function outside the exon junction complex. Genes Dev. 25, 1078–1090 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Park S.-J., Yoon B.-H., Kim S.-K., Kim S.-Y., GENT2: An updated gene expression database for normal and tumor tissues. BMC Med. Genomics 12, 101 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vasaikar S. V., Straub P., Wang J., Zhang B., LinkedOmics: Analyzing multi-omics data within and across 32 cancer types. Nucleic Acids Res. 46, D956–D963 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ghandi M., Huang F. W., Jané-Valbuena J., Kryukov G. V., Lo C. C., McDonald E. R., Barretina J., Gelfand E. T., Bielski C. M., Li H., Hu K., Andreev-Drakhlin A. Y., Kim J., Hess J. M., Haas B. J., Aguet F., Weir B. A., Rothberg M. V., Paolella B. R., Lawrence M. S., Akbani R., Lu Y., Tiv H. L., Gokhale P. C., de Weck A., Mansour A. A., Oh C., Shih J., Hadi K., Rosen Y., Bistline J., Venkatesan K., Reddy A., Sonkin D., Liu M., Lehar J., Korn J. M., Porter D. A., Jones M. D., Golji J., Caponigro G., Taylor J. E., Dunning C. M., Creech A. L., Warren A. C., McFarland J. M., Zamanighomi M., Kauffmann A., Stransky N., Imielinski M., Maruvka Y. E., Cherniack A. D., Tsherniak A., Vazquez F., Jaffe J. D., Lane A. A., Weinstock D. M., Johannessen C. M., Morrissey M. P., Stegmeier F., Schlegel R., Hahn W. C., Getz G., Mills G. B., Boehm J. S., Golub T. R., Garraway L. A., Sellers W. R., Next-generation characterization of the cancer cell line encyclopedia. Nature 569, 503–508 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bruno P. M., Liu Y., Park G. Y., Murai J., Koch C. E., Eisen T. J., Pritchard J. R., Pommier Y., Lippard S. J., Hemann M. T., A subset of platinum-containing chemotherapeutic agents kills cells by inducing ribosome biogenesis stress. Nat. Med. 23, 461–471 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Daguenet E., Baguet A., Degot S., Schmidt U., Alpy F., Wendling C., Spiegelhalter C., Kessler P., Rio M.-C., Le Hir H., Bertrand E., Tomasetto C., Perispeckles are major assembly sites for the exon junction core complex. Mol. Biol. Cell 23, 1765–1782 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bensaude O., Inhibiting eukaryotic transcription: Which compound to choose? How to evaluate its activity? Transcription 2, 103–108 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Andersen J. S., Lam Y. W., Leung A. K. L., Ong S.-E., Lyon C. E., Lamond A. I., Mann M., Nucleolar proteome dynamics. Nature 433, 77–83 (2005). [DOI] [PubMed] [Google Scholar]
- 23.Golomb L., Volarevic S., Oren M., p53 and ribosome biogenesis stress: The essentials. FEBS Lett. 588, 2571–2579 (2014). [DOI] [PubMed] [Google Scholar]
- 24.Tatsuno T., Ishigaki Y., C-terminal short arginine/serine repeat sequence-dependent regulation of Y14 (RBM8A) localization. Sci. Rep. 8, 612 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nicolas E., Parisot P., Pinto-Monteiro C., de Walque R., De Vleeschouwer C., Lafontaine D. L. J., Involvement of human ribosomal proteins in nucleolar structure and p53-dependent nucleolar stress. Nat. Commun. 7, 11390 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Latonen L., Phase-to-phase with nucleoli — Stress responses, protein aggregation and novel roles of RNA. Front. Cell. Neurosci. 13, 151 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shav-Tal Y., Blechman J., Darzacq X., Montagna C., Dye B. T., Patton J. G., Singer R. H., Zipori D., Dynamic sorting of nuclear components into distinct nucleolar caps during transcriptional inhibition. Mol. Biol. Cell 16, 2395–2413 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krüger T., Zentgraf H., Scheer U., Intranucleolar sites of ribosome biogenesis defined by the localization of early binding ribosomal proteins. J. Cell Biol. 177, 573–578 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen Y.-J. C., Wang H.-J., Jauh G.-Y., Dual role of a SAS10/C1D family protein in ribosomal RNA gene expression and processing is essential for reproduction in Arabidopsis thaliana. PLOS Genet. 12, e1006408 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Espinoza J. A., Zisi A., Kanellis D. C., Carreras-Puigvert J., Henriksson M., Hühn D., Watanabe K., Helleday T., Lindström M. S., Bartek J., The antimalarial drug amodiaquine stabilizes p53 through ribosome biogenesis stress, independently of its autophagy-inhibitory activity. Cell Death Differ. 27, 773–789 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mazloomian A., Araki S., Ohori M., El-Naggar A. M., Yap D., Bashashati A., Nakao S., Sorensen P. H., Nakanishi A., Shah S., Aparicio S., Pharmacological systems analysis defines EIF4A3 functions in cell-cycle and RNA stress granule formation. Commun. Biol. 2, 165 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fukumura K., Wakabayashi S., Kataoka N., Sakamoto H., Suzuki Y., Nakai K., Mayeda A., Inoue K., The exon junction complex controls the efficient and faithful splicing of a subset of transcripts involved in mitotic cell-cycle progression. Int. J. Mol. Sci. 17, 1153 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Allende-Vega N., Dayal S., Agarwala U., Sparks A., Bourdon J.-C., Saville M. K., p53 is activated in response to disruption of the pre-mRNA splicing machinery. Oncogene 32, 1–14 (2013). [DOI] [PubMed] [Google Scholar]
- 34.Lecane P. S., Kiviharju T. M., Sellers R. G., Peehl D. M., Leptomycin B stabilizes and activates p53 in primary prostatic epithelial cells and induces apoptosis in the LNCaP cell line. Prostate 54, 258–267 (2003). [DOI] [PubMed] [Google Scholar]
- 35.Ghoshal K., Majumder S., Datta J., Motiwala T., Bai S., Sharma S. M., Frankel W., Jacob S. T., Role of human ribosomal RNA (rRNA) promoter methylation and of methyl-CpG-binding protein MBD2 in the suppression of rRNA gene expression. J. Biol. Chem. 279, 6783–6793 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barandun J., Hunziker M., Klinge S., Assembly and structure of the SSU processome—A nucleolar precursor of the small ribosomal subunit. Curr. Opin. Struct. Biol. 49, 85–93 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shao Z., Flynn R. A., Crowe J. L., Zhu Y., Liang J., Jiang W., Aryan F., Aoude P., Bertozzi C. R., Estes V. M., Lee B. J., Bhagat G., Zha S., Calo E., DNA-PKcs has KU-dependent function in rRNA processing and haematopoiesis. Nature 579, 291–296 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Song C., Hotz-Wagenblatt A., Voit R., Grummt I., SIRT7 and the DEAD-box helicase DDX21 cooperate to resolve genomic R loops and safeguard genome stability. Genes Dev. 31, 1370–1381 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lindström M. S., Jurada D., Bursac S., Orsolic I., Bartek J., Volarevic S., Nucleolus as an emerging hub in maintenance of genome stability and cancer pathogenesis. Oncogene 37, 2351–2366 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Björkman A., Johansen S. L., Lin L., Schertzer M., Kanellis D. C., Katsori A. M., Christensen S. T., Luo Y., Andersen J. S., Elsässer S. J., Londono-Vallejo A., Bartek J., Schou K. B., Human RTEL1 associates with Poldip3 to facilitate responses to replication stress and R-loop resolution. Genes Dev. 34, 1065–1074 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smolka J. A., Sanz L. A., Hartono S. R., Chédin F., Recognition of RNA by the S9.6 antibody creates pervasive artifacts when imaging RNA:DNA hybrids. J. Cell Biol. 220, e202004079 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Skourti-Stathaki K., Proudfoot N. J., A double-edged sword: R loops as threats to genome integrity and powerful regulators of gene expression. Genes Dev. 28, 1384–1396 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bono F., Ebert J., Lorentzen E., Conti E., The crystal structure of the exon junction complex reveals how it maintains a stable grip on mRNA. Cell 126, 713–725 (2006). [DOI] [PubMed] [Google Scholar]
- 44.Gandin V., Sikström K., Alain T., Morita M., McLaughlan S., Larsson O., Topisirovic I., Polysome fractionation and analysis of mammalian translatomes on a genome-wide scale. J. Vis. Exp. 2014, 51455 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chothani S., Adami E., Ouyang J. F., Viswanathan S., Hubner N., Cook S. A., Schafer S., Rackham O. J. L., deltaTE: Detection of translationally regulated genes by integrative analysis of Ribo-seq and RNA-seq data. Curr. Protoc. Mol. Biol. 129, e108 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Heyer E. E., Moore M. J., Redefining the translational status of 80S monosomes. Cell 164, 757–769 (2016). [DOI] [PubMed] [Google Scholar]
- 47.Blagosklonny M. V., Mitotic arrest and cell fate: Why and how mitotic inhibition of transcription drives mutually exclusive events. Cell Cycle 6, 70–74 (2007). [DOI] [PubMed] [Google Scholar]
- 48.Bartek J., Lukas J., Pathways governing G1/S transition and their response to DNA damage. FEBS Lett. 490, 117–122 (2001). [DOI] [PubMed] [Google Scholar]
- 49.Moll U. M., Petrenko O., The MDM2-p53 interaction. Mol. Cancer Res. 1, 1001–1008 (2003). [PubMed] [Google Scholar]
- 50.Oliver T. G., Meylan E., Chang G. P., Xue W., Burke J. R., Humpton T. J., Hubbard D., Bhutkar A., Jacks T., Caspase-2-mediated cleavage of Mdm2 creates a p53-induced positive feedback loop. Mol. Cell 43, 57–71 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Anders S., Reyes A., Huber W., Detecting differential usage of exons from RNA-seq data. Genome Res. 22, 2008–2017 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cheng T.-H., Cohen S. N., Human MDM2 isoforms translated differentially on constitutive versus p53-regulated transcripts have distinct functions in the p53/MDM2 and TSG101/MDM2 feedback control loops. Mol. Cell. Biol. 27, 111–119 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Boehm V., Britto-Borges T., Steckelberg A.-L., Singh K. K., Gerbracht J. V., Gueney E., Blazquez L., Altmüller J., Dieterich C., Gehring N. H., Exon junction complexes suppress spurious splice sites to safeguard transcriptome integrity. Mol. Cell 72, 482–495.e7 (2018). [DOI] [PubMed] [Google Scholar]
- 54.Eshtad S., Mavajian Z., Rudd S. G., Visnes T., Boström J., Altun M., Helleday T., HMYH and hMTH1 cooperate for survival in mismatch repair defective T-cell acute lymphoblastic leukemia. Oncogenesis 5, e275 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Campeau E., Ruhl V. E., Rodier F., Smith C. L., Rahmberg B. L., Fuss J. O., Campisi J., Yaswen P., Cooper P. K., Kaufman P. D., A versatile viral system for expression and depletion of proteins in mammalian cells. PLOS ONE 4, e6529 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li M., Gu W., A critical role for noncoding 5S rRNA in regulating Mdmx stability. Mol. Cell 43, 1023–1032 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Franken N. A. P., Rodermond H. M., Stap J., Haveman J., van Bree C., Clonogenic assay of cells in vitro. Nat. Protoc. 1, 2315–2319 (2006). [DOI] [PubMed] [Google Scholar]
- 58.Boisvert F.-M., Lam Y. W., Lamont D., Lamond A. I., A quantitative proteomics analysis of subcellular proteome localization and changes induced by DNA damage. Mol. Cell. Proteomics 9, 457–470 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brenes A., Afzal V., Kent R., Lamond A. I., The encyclopedia of proteome dynamics: A big data ecosystem for (prote)omics. Nucleic Acids Res. 46, D1202–D1209 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sigalas I., Calvert A. H., Anderson J. J., Neal D. E., Lunec J., Alternatively spliced mdm2 transcripts with loss of p53 binding domain sequences: Transforming ability and frequent detection in human cancer. Nat. Med. 2, 912–917 (1996). [DOI] [PubMed] [Google Scholar]
- 61.Ewels P. A., Peltzer A., Fillinger S., Alneberg J., Patel H., Wilm A., Garcia M. U., Di Tommaso P., Nahnsen S., nf-core: Community curated bioinformatics pipelines. bioRxiv , 610741 (2019). [DOI] [PubMed] [Google Scholar]
- 62.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Patro R., Duggal G., Love M. I., Irizarry R. A., Kingsford C., Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vitting-Seerup K., Sandelin A., IsoformSwitchAnalyzeR: Analysis of changes in genome-wide patterns of alternative splicing and its functional consequences. Bioinformatics 35, 4469–4471 (2019). [DOI] [PubMed] [Google Scholar]
- 65.Bindea G., Mlecnik B., Hackl H., Charoentong P., Tosolini M., Kirilovsky A., Fridman W.-H., Pagès F., Trajanoski Z., Galon J., ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 25, 1091–1093 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shannon P., Lejay L. V., Dean A., Chiaromonte F., Shasha D. E., Coruzzi G. M., Amin N., Schwikowski B., Ideker T., Stitt M., Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Raudvere U., Kolberg L., Kuzmin I., Arak T., Adler P., Peterson H., Vilo J., G:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 47, W191–W198 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Walter W., Sánchez-Cabo F., Ricote M., GOplot: An R package for visually combining expression data with functional analysis. Bioinformatics 31, 2912–2914 (2015). [DOI] [PubMed] [Google Scholar]
- 69.Janky R., Verfaillie A., Imrichová H., Van de Sande B., Standaert L., Christiaens V., Hulselmans G., Herten K., Naval Sanchez M., Potier D., Svetlichnyy D., Kalender Atak Z., Fiers M., Marine J.-C., Aerts S., iRegulon: From a gene list to a gene regulatory network using large motif and track collections. PLOS Comput. Biol. 10, e1003731 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hauer C., Sieber J., Schwarzl T., Hollerer I., Curk T., Alleaume A. M., Hentze M. W., Kulozik A. E., Exon junction complexes show a distributional bias toward alternatively spliced mRNAs and against mRNAs coding for ribosomal proteins. Cell Rep. 16, 1588–1603 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Moggridge S., Sorensen P. H., Morin G. B., Hughes C. S., Extending the compatibility of the SP3 paramagnetic bead processing approach for proteomics. J. Proteome Res. 17, 1730–1740 (2018). [DOI] [PubMed] [Google Scholar]
- 72.Branca R. M. M., Orre L. M., Johansson H. J., Granholm V., Huss M., Pérez-Bercoff A., Forshed J., Käll L., Lehtiö J., HiRIEF LC-MS enables deep proteome coverage and unbiased proteogenomics. Nat. Methods 11, 59–62 (2014). [DOI] [PubMed] [Google Scholar]
- 73.Boekel J., Chilton J. M., Cooke I. R., Horvatovich P. L., Jagtap P. D., Käll L., Lehtiö J., Lukasse P., Moerland P. D., Griffin T. J., Multi-omic data analysis using Galaxy. Nat. Biotechnol. 33, 137–139 (2015). [DOI] [PubMed] [Google Scholar]
- 74.Zhang X., Smits A. H., van Tilburg G. B., Ovaa H., Huber W., Vermeulen M., Proteome-wide identification of ubiquitin interactions using UbIA-MS. Nat. Protoc. 13, 530–550 (2018). [DOI] [PubMed] [Google Scholar]
- 75.Szklarczyk D., Gable A. L., Lyon D., Junge A., Wyder S., Huerta-Cepas J., Simonovic M., Doncheva N. T., Morris J. H., Bork P., Jensen L. J., von Mering C., STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–D613 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gagliardi M., Matarazzo M. R., RIP: RNA immunoprecipitation. Methods Mol. Biol. 1480, 73–86 (2016). [DOI] [PubMed] [Google Scholar]
- 77.Zhu Y., Orre L. M., Zhou Tran Y., Mermelekas G., Johansson H. J., Malyutina A., Anders S., Lehtiö J., DEqMS: A method for accurate variance estimation in differential protein expression analysis. Mol. Cell. Proteomics 19, 1047–1057 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.W. Hadley, ggplot2: Elegant Graphics for Data Analysis (Springer-Verlag, 2016). [Google Scholar]
- 79.Ito M., Tanaka T., Cary D. R., Iwatani-Yoshihara M., Kamada Y., Kawamoto T., Aparicio S., Nakanishi A., Imaeda Y., Discovery of novel 1,4-diacylpiperazines as selective and cell-active eIF4A3 inhibitors. J. Med. Chem. 60, 3335–3351 (2017). [DOI] [PubMed] [Google Scholar]
- 80.Bartkova J., Horejsí Z., Koed K., Krämer A., Tort F., Zieger K., Guldberg P., Sehested M., Nesland J. M., Lukas C., Ørntoft T., Lukas J., Bartek J., DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 434, 864–870 (2005). [DOI] [PubMed] [Google Scholar]
- 81.Tort F., Bartkova J., Sehested M., Orntoft T., Lukas J., Bartek J., Retinoblastoma pathway defects show differential ability to activate the constitutive DNA damage response in human tumorigenesis. Cancer Res. 66, 10258–10263 (2006). [DOI] [PubMed] [Google Scholar]
- 82.Gudjonsson T., Altmeyer M., Savic V., Toledo L., Dinant C., Grøfte M., Bartkova J., Poulsen M., Oka Y., Bekker-Jensen S., Mailand N., Neumann B., Heriche J. K., Shearer R., Saunders D., Bartek J., Lukas J., Lukas C., TRIP12 and UBR5 suppress spreading of chromatin ubiquitylation at damaged chromosomes. Cell 150, 697–709 (2012). [DOI] [PubMed] [Google Scholar]
- 83.Bartkova J., Hamerlik P., Stockhausen M.-T., Ehrmann J., Hlobilkova A., Laursen H., Kalita O., Kolar Z., Poulsen H. S., Broholm H., Lukas J., Bartek J., Replication stress and oxidative damage contribute to aberrant constitutive activation of DNA damage signalling in human gliomas. Oncogene 29, 5095–5102 (2010). [DOI] [PubMed] [Google Scholar]
- 84.Vanzo R., Bartkova J., Merchut-Maya J. M., Hall A., Bouchal J., Dyrskjøt L., Frankel L. B., Gorgoulis V., Maya-Mendoza A., Jäättelä M., Bartek J., Autophagy role(s) in response to oncogenes and DNA replication stress. Cell Death Differ. 27, 1134–1153 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/32/eabf7561/DC1