

Summary
Lysosome function is essential for cellular homeostasis but quality control mechanisms that maintain healthy lysosomes remain poorly characterized. Here we developed a method to measure lysosome turnover and use this to identify a selective mechanism of membrane degradation that involves lipidation of the autophagy protein LC3 onto lysosomal membranes and the formation of intraluminal vesicles through microautophagy. This mechanism is induced in response to metabolic stress resulting from glucose starvation, or by treatment with pharmacological agents that induce osmotic stress on lysosomes. Cells lacking ATG5, an essential component of the LC3 lipidation machinery, show reduced ability to regulate lysosome size and degradative capacity in response to activation of this mechanism. These findings identify a selective mechanism of lysosome membrane turnover that is induced by stress and uncover a function for LC3 lipidation in regulating lysosome size and activity through microautophagy.
Keywords: autophagy, lysosome, LC3, LAP, microautophagy
Introduction
Quality control mechanisms contribute to maintaining cellular function by regulating the turnover of selected intracellular substrates, from individual proteins and protein complexes, to protein aggregates and whole organelles. Two major degradative pathways contribute to quality control: the ubiquitin proteasome pathway that regulates the turnover of short-lived proteins, and the autophagy-lysosome pathway that targets long-lived proteins and larger substrates such as protein aggregates and organelles (Dikic, 2017; Wong and Cuervo, 2010). Through autophagy, substrates are degraded in the lysosome following sequestration into double-membrane vesicles called autophagosomes.
While lysosome-mediated degradation is one major mechanism that contributes to the turnover of selected intracellular substrates, little is known about how the lysosome itself is turned-over. Lysosomes are critical for maintaining endocytic and autophagy flux, and they provide a platform for metabolic signaling pathways (Lawrence and Zoncu, 2019). Lysosome dysfunction contributes to cellular aging, and leads to the development of lysosomal storage diseases that are linked to inefficient substrate degradation (Lawrence and Zoncu, 2019; Sun, 2018). The regulation of lysosome quality is therefore important for maintaining healthy cells and tissues. While recent findings have uncovered transcriptional programs that allow cells to respond to stress on the lysosome network by generating more lysosomes, controlled by transcription factor EB and related factors (Raben and Puertollano, 2016), conversely how lysosomes are eliminated from cells and turned-over in support of optimal network function is less clear. Here we sought to identify mechanisms that control lysosome turnover by (1) developing a method to monitor lysosome network degradation and (2) using this system to interrogate mechanisms of lysosome turnover in response to stress.
Results
Glucose starvation induces selective lysosome turnover.
To study mechanisms controlling lysosome turnover, we first established reporters of lysosomal membrane degradation. The turnover of damaged lysosomes has been measured previously by expression of a cytosolic reporter, GFP-Galectin3 (GFP-Gal3), that forms puncta in response to the rupture of lysosomes by binding to β-galactosides on the luminal face of the membrane (Maejima et al., 2013). We sought to establish a method that could measure the turnover of lysosomes even in the absence of membrane rupture. To measure lysosome turnover, GFP-tagged lysosomal transmembrane proteins were expressed in cells and their degradation was quantified by measuring the accumulation of untagged GFP protein by western blotting, a strategy that takes advantage of the resistance of GFP to cathepsin-mediated digestion, which has been used to report the lysosomal turnover of other organelles such as the endoplasmic reticulum (Cheong et al., 2011; Mochida et al., 2015). Five different lysosomal transmembrane proteins, including the structural protein lysosomal-associated membrane protein 1 (LAMP1), amino acid transporters PQ-loop repeat-containing protein 2 (PQLC2 or SLC66A1), putative sodium-coupled neutral amino acid transporter 7 (SNAT7 or SLC38A7) and cystinosin (CTNS or SLC66A4), and the ion channel transient receptor potential cation channel, mucolipin subfamily, member 1 (TRPML1), were expressed as GFP fusions and their degradation was examined in cells treated with a known lysosome damaging agent, L-Leucyl-L-Leucine methyl ester (LLOMe), which ruptures lysosomal membranes and induces a form of autophagic lysosome turnover called lysophagy (Maejima et al., 2013). As shown in Figure 1A and B, LLOMe treatment led to the appearance of free GFP from all five reporter proteins, consistent with turnover of whole lysosomes in response to LLOMe-mediated lysosome rupture.
Figure 1. Whole organelle and selective lysosome turnover measured by the degradation of lysosomal transmembrane proteins.
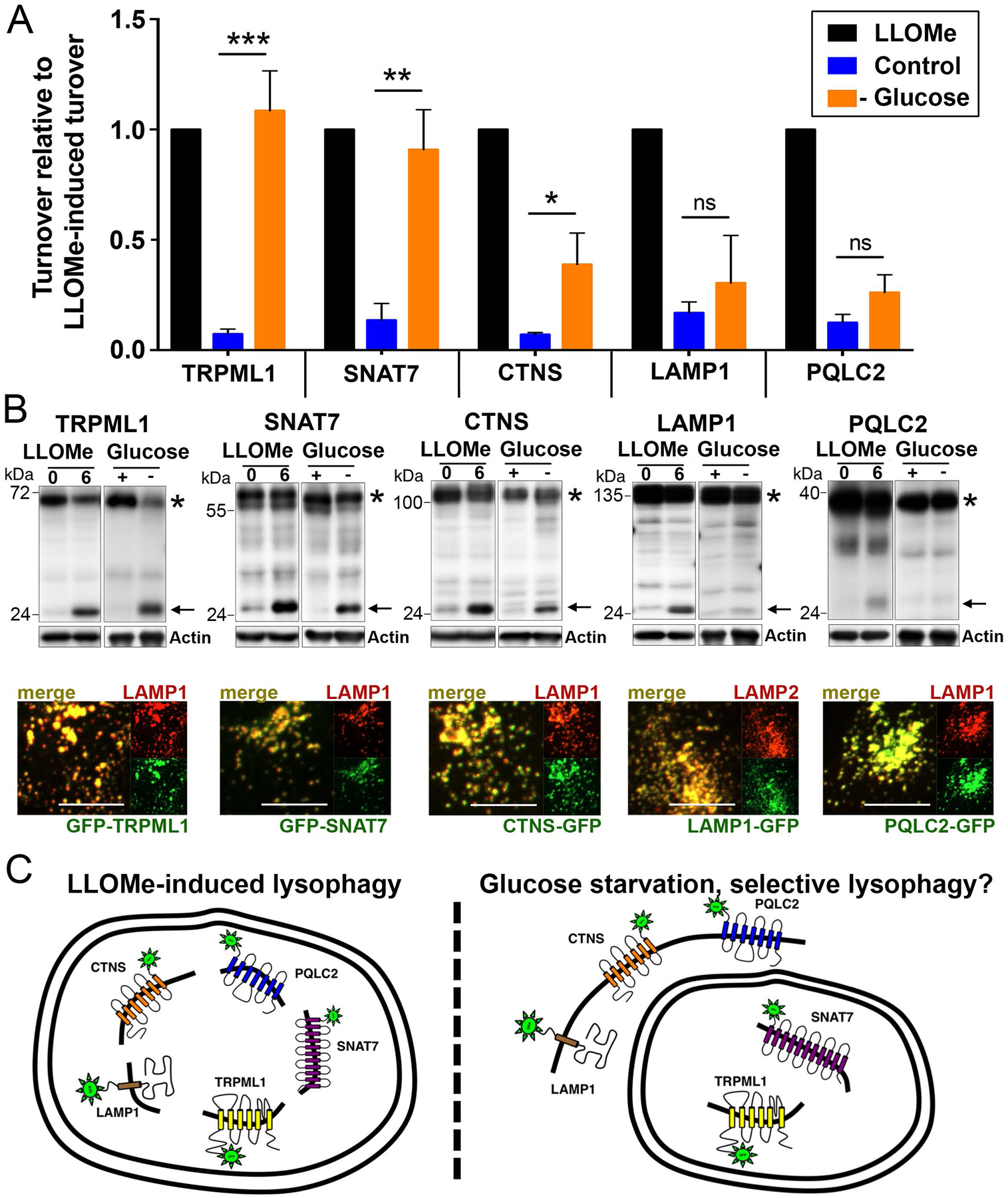
(A) Graph shows relative turnover of the indicated GFP-tagged lysosomal transmembrane proteins normalized to turnover in response to LLOMe. Turnover was quantified as the percentage of free GFP over the total full-length protein plus free GFP, in control, LLOMe-treated, or glucose-starved conditions. Cells were starved for glucose for 24 hours or treated with LLOMe for one hour followed by a six-hour chase. Graph shows data from three or more independent experiments for each reporter; error bars show SEM. *p<.05; **p<.01; ***p<.001. (B) Representative western blots from the data graphed in part A. For each reporter, free GFP is indicated with an arrow, and full-length protein is indicated with an asterisk. For GFP-TRPML1, full length protein indicated by asterisk is cleavage product, as reported (Kiselyov et al., 2005). Actin loading controls are also shown. Images below the blots show colocalization of GFP-tagged reporter proteins with endogenous LAMP1 or LAMP2 lysosomal proteins detected by immunostaining. Scale bars = 10μm. (C) Working models for whole lysosome turnover through lysophagy in response to lysosome rupture with LLOMe (left), or selective turnover observed in response to starvation for glucose (right). Double-membrane structure on left depicts autophagosome surrounding whole damaged lysosome. Double membrane structure on right is drawn to surround only those reporter proteins that undergo selective turnover in glucose-starved cells.
We next used these reporters to examine if lysosomes might undergo turnover in response to stress resulting from nutrient starvation. While the starvation of cells for amino acids is known to induce autophagy and lysosome biogenesis, due to a loss of activity of the mechanistic target of rapamycin complex 1 (mTORC1) nutrient-sensing kinase, amino acid starvation did not lead to detectable lysosome turnover (Supplemental Fig.1). By contrast, starvation for glucose led to significant turnover of the GFP-TRPML1 and SNAT7-GFP lysosomal reporters, which were degraded to a similar extent as in LLOMe-treated cells (Fig. 1A, B). Intriguingly, glucose starvation did not induce measureable turnover of LAMP1-GFP and PQLC2-GFP, and the CTNS-GFP reporter was also degraded significantly less in glucose-starved cells than in cells treated with LLOMe (Fig. 1A, B). These data demonstrate that while amino acid starvation does not induce lysosome turnover, glucose-starved cells exhibit significant but selective turnover of lysosomal membrane proteins (Fig. 1C).
Selective turnover occurs through a mechanism regulated by ATG5.
To elucidate how glucose starvation leads to selective lysosome turnover, we first examined if lysosomes become damaged when cells are cultured under glucose-free conditions. Treatment with LLOMe is known to induce lysosome membrane rupture that is marked by the appearance of GFP-Gal3 puncta (Fig. 2A). Unlike LLOMe, glucose starvation did not induce the appearance of GFP-Gal3 puncta in cells, suggesting that lysosomes do not undergo rupture under this condition (Fig. 2A). Although lysosomes did not show evidence of rupture, which is a known trigger of lysophagy, we further examined if the autophagy pathway might still play a role in selective turnover. Intriguingly, while loss of the essential autophagy gene ATG5 significantly reduced GFP-TRPML1 turnover in response to glucose starvation, we found that loss of another canonical autophagy gene that functions in the pre-initiation complex, ATG13, had no effect (Fig. 2B, C). Similarly, treatment with an inhibitor of the ULK1 kinase (SBI-0206965) (Egan et al., 2015), which functions in the same complex as ATG13 (Jung et al., 2009), failed to inhibit GFP-TRPML1 turnover in response to glucose starvation, despite inhibiting the induction of starvation-induced autophagy (Fig. 2D–F). As the ULK1 kinase complex plays a role in the initiation of canonical autophagy (Florey and Overholtzer, 2012), these data demonstrate that a non-canonical mechanism, related to autophagy and involving ATG5, but not requiring the ULK1 pre-initiation complex, controls selective lysosome turnover in response to glucose starvation.
Figure 2. Selective turnover occurs through an ATG5-regulated mechanism.
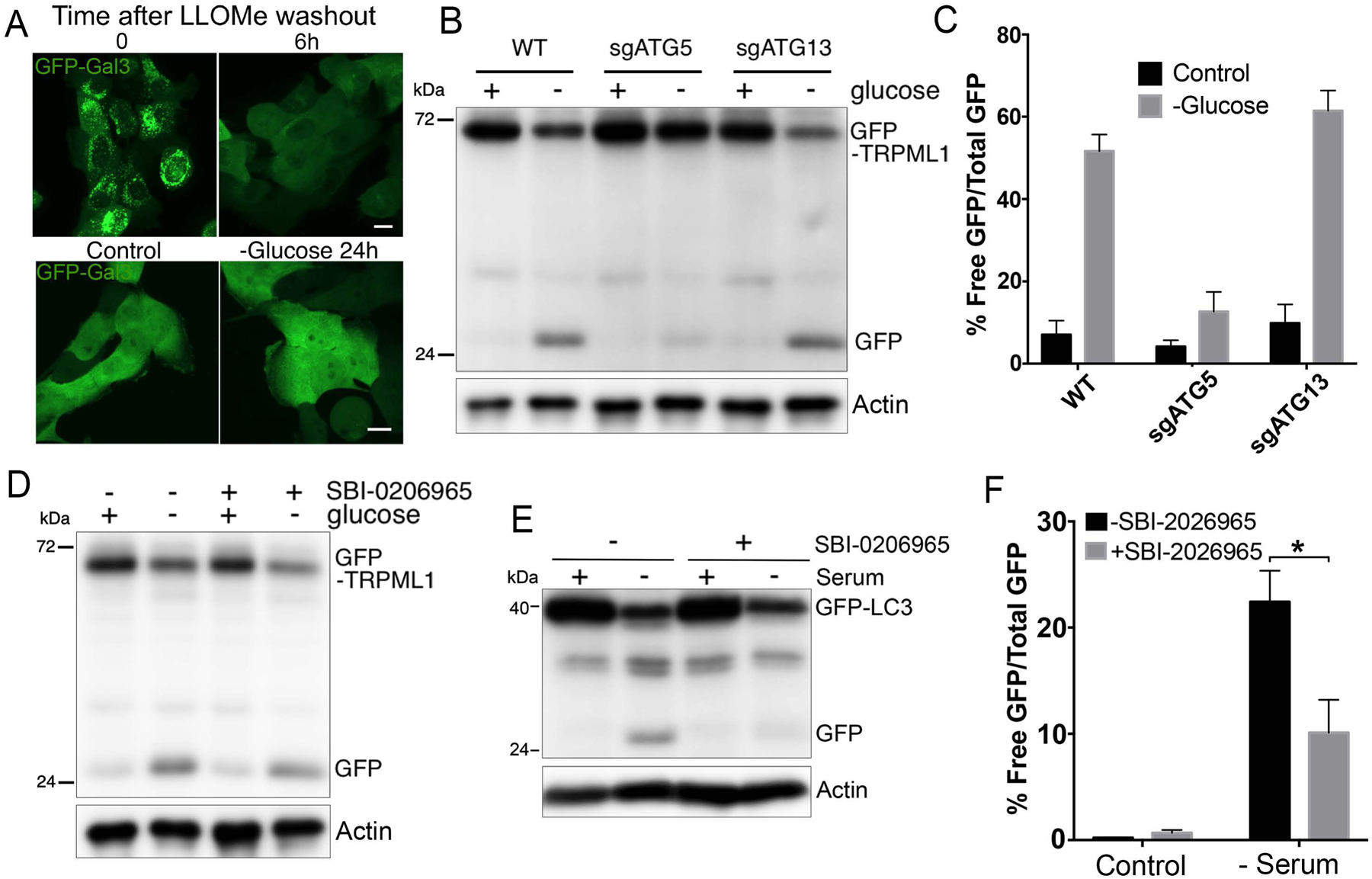
(A) Lysosomes undergo rupture in response to LLOMe but not glucose starvation. Images show GFP-Gal3 fluorescence in LLOMe-treated cells at time zero after treatment (top left), or six hours after LLOMe washout (top right), and in control or cells starved of glucose for 24 hours (bottom images). (B) GFP-TRPML1 turnover in response to glucose starvation is regulated by ATG5 but not ATG13. Western blot shows GFP-TRPML1 cleavage after 24 hours of glucose starvation. GFP-TRPML1 and free GFP are indicated. Actin loading controls are also shown. (C) Quantification of data shown in part B from three independent experiments; error bars show SEM. (D) GFP-TRPML1 turnover in glucose-starved cells (24 hours) is not inhibited with treatment with the ULK1 kinase inhibitor SBI-0206965. Actin loading controls are also shown. (E) Autophagy flux in response to serum starvation for 10 hours, measured by GFP-LC3 cleavage leading to the generation of free GFP, is inhibited by treatment with SBI-0206965. Actin loading controls are also shown. (F) Quantification of data shown in part E from three independent experiments; error bars show SEM. *p<.05.
Selective turnover involves endolysosomal LC3 lipidation and glutamine catabolism.
One autophagy-related mechanism that is regulated by ATG5 but occurs independently of the ULK1 kinase complex is the lipidation of the autophagy protein microtubule-associated protein A light chain 3 (LC3) onto endosomal membranes, including phagosomes (Florey et al., 2011; Martinez et al., 2011; Sanjuan et al., 2007), macropinosomes (Florey et al., 2011), and also endosomes and lysosomes (Florey et al., 2015; Heckmann et al., 2019), instead of autophagosomes (Florey and Overholtzer, 2012). This mechanism is sometimes referred to as LC3-Associated Phagocytosis, or LAP, signifying its original discovery on phagosome membranes (Sanjuan et al., 2007). Studies identifying stimulators of lysosomal LC3 lipidation have identified several ionophores and lysosomotropic compounds that can induce this process (Jacquin et al., 2017). To examine if inducers of lysosomal LC3 lipidation can also stimulate lysosome turnover, we treated cells with the ionophores nigericin and monensin, and also the weak base ammonium chloride, which are reported to induce this form of LC3 lipidation (Jacquin et al., 2017). Treatment with nigericin, monensin or ammonium chloride led to LC3 lipidation on lysosomal membranes in ATG13 knockout cells (Fig. 3A), and like starvation for glucose, induced the turnover of GFP-TRPML1 in a manner that was significantly reduced by the loss of ATG5, but occurred independently of ATG13 (Fig. 3B and Supplemental Fig. 2A, B). We further examined additional components of the LC3 lipidation machinery, and found that the ATG7 and ATG3 genes, which encode the E1- and E2-like enzymes that regulate LC3 lipidation upstream of ATG5, also regulate GFP-TRPML1 turnover, which was reduced significantly upon their siRNA-mediated depletion (Supplemental Fig. 2C, D).
Figure 3. Endolysomal LC3 lipidation induces selective lysosome turnover.
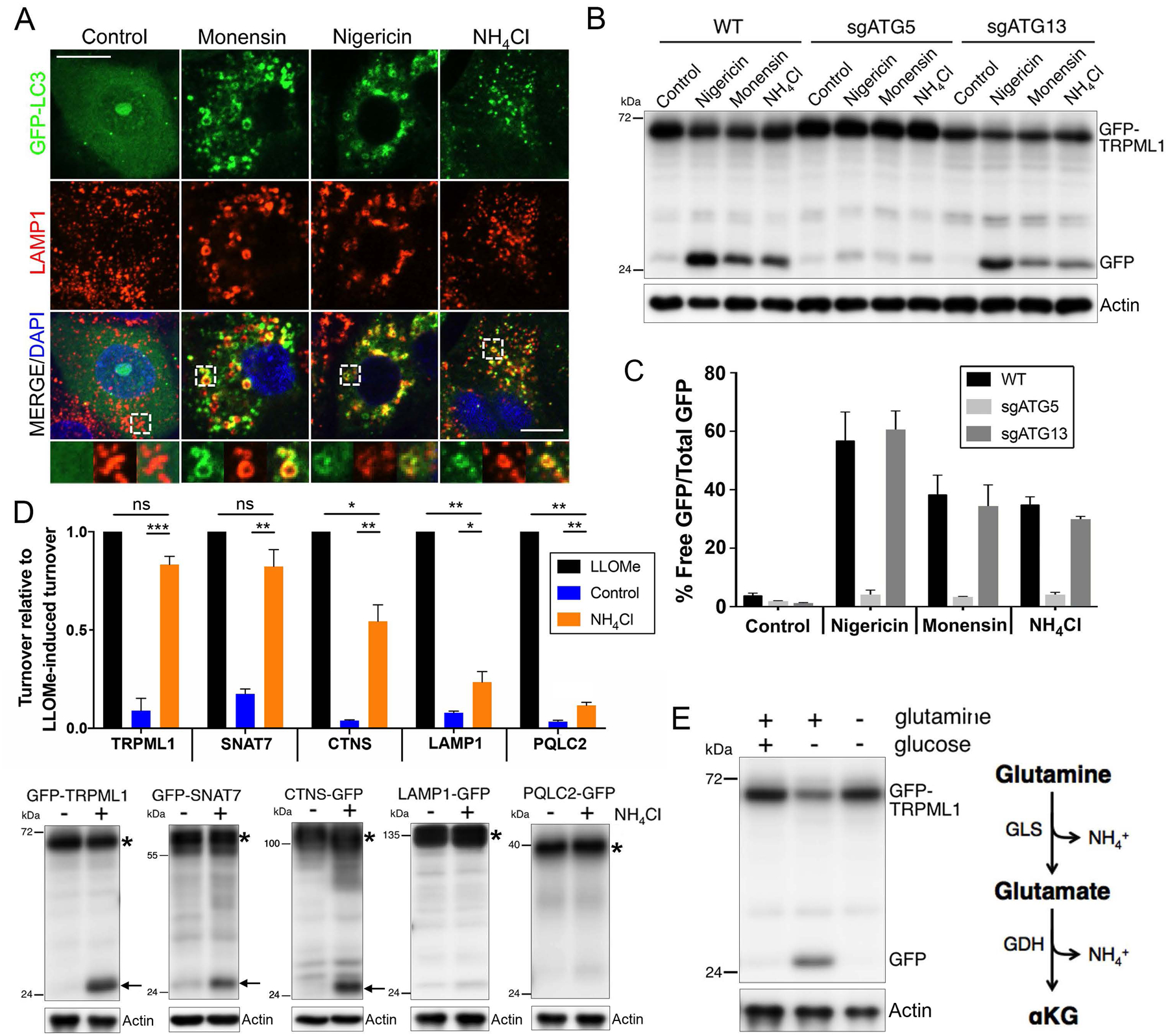
(A) Treatment with monensin, nigericin, or ammonium chloride for two hours induces colocalization of GFP-LC3 (green) with LAMP1 (red) on enlarged endolysosomes. Insets at bottom are indicated by white hatched boxes. Scale bar = 10μm. (B) Treatment with monensin, nigericin, or ammonium chloride for eight hours induces turnover of GFP-TRPML1 in an ATG5-regulated, ATG13-independent manner. Actin loading controls are also shown. (C) Graph shows quantification of data from part B from three independent experiments; error bars show SEM. (D) Treatment with ammonium chloride for 24 hours induces selective turnover of GFP-TRPML1 and GFP-SNAT7. Graph shows relative turnover of the indicated GFP-tagged lysosomal transmembrane proteins in control or ammonium-treated cells, normalized to turnover in response to LLOMe. Data are from three independent experiments for each reporter; error bars show SEM. *p<.05; **p<.01; ***p<.001. Note that CTNS-GFP, LAMP1-GFP, and PQLC2-GFP turnover to a significantly lesser extent in response to ammonium as compared to LLOMe. Representative western blots are shown below the graph. For each reporter, free GFP is indicated with an arrow and full-length protein is indicated with an asterisk. (E) GFP-TRPML1 turnover in cells starved for glucose for 24 hours requires the presence glutamine. Right schematic: glutamine catabolism generates ammonium.
To further examine how glucose starvation might be linked to selective turnover involving endolysosomal LC3 lipidation, we considered a potential role for the catabolism of glutamine, which is known to support metabolism under glucose-deprived conditions and to generate ammonium as a byproduct (Cheong et al., 2011; Le et al., 2012). Treatment of cells with ammonium in ranges shown previously to accumulate in glucose-starved cells (Cheong et al., 2011) indeed induced GFP-TRPML1 turnover in a dose-dependent manner (Supplemental Fig. 3). Ammonium also induced selective turnover, as GFP-TRPML1 and GFP-SNAT7 were turned-over to a similar manner as in response to LLOMe, while CTNS-GFP showed an intermediate level of turnover, and the LAMP1-GFP and PQLC2-GFP reporters showed minimal evidence of turnover (Fig. 3D). Moreover, while amino acid starvation did not cause lysosome turnover (Supplemental Fig. 1), starvation for the amino acid glutamine actually blocked turnover that was induced by glucose withdrawal (Fig. 3E), demonstrating a requirement for glutamine to support selective lysosome turnover. Taken together, these data support a model that ammonium generated by the catabolism of glutamine induces selective lysosome turnover in glucose-starved cells that results from ATG5-regulated endolysosomal LC3 lipidation.
Selective lysosome turnover involves microautophagy.
The discovery that inducers of LC3 lipidation on the lysosome membrane lead to selective transmembrane protein turnover prompted us to consider whether modification of the membrane in this manner could be associated with microautophagy, a process whereby luminal vesicles are formed by invagination of the limiting membrane. To examine this hypothesis, we imaged endolysosomal ultrastructure by electron microscopy to determine if treatment with ammonium would result in the appearance of multivesicular structures. As shown in Figure 4A and B, ammonium treatment significantly increased the number of multivesicular endosomes, in an ATG5-regulated manner, consistent with the induction of microautophagy activity. As microautophagy involves invagination of the limiting lysosomal membrane, we next examined if selective turnover requires intact microtubules, as this activity is predicted to occur independently of vesicle trafficking (Sahu et al., 2011). Treatment with the microtubule depolymerizing agent nocodazole inhibited autophagy flux but had no effect on ammonium-induced selective lysosome turnover, demonstrating that while microtubules are required for autophagy, they are not required for ATG5-regulated selective lysosome turnover (Fig. 4C–E).
Figure 4. Selective turnover occurs through microautophagy.
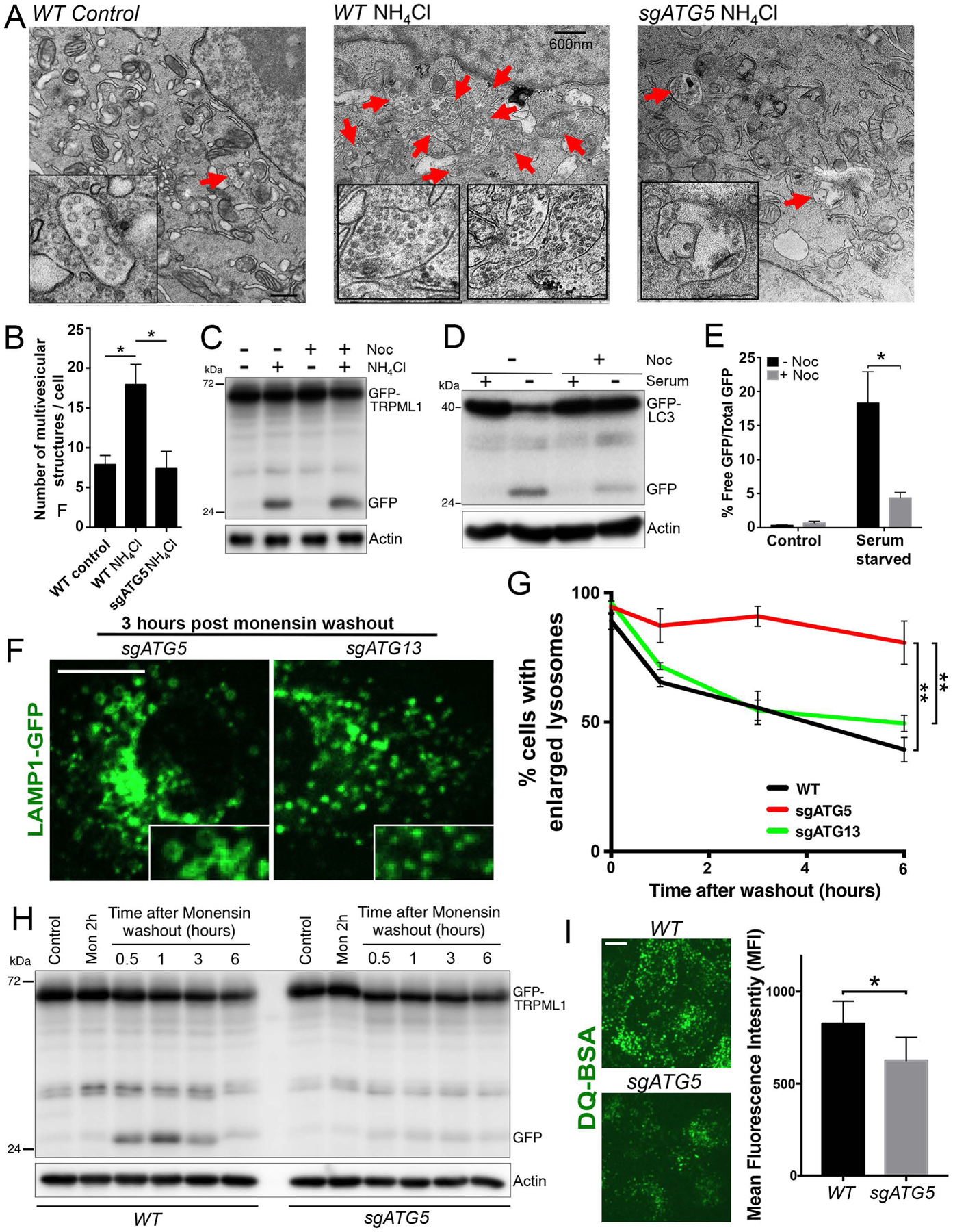
(A) Treatment with ammonium induces the appearance of multivesicular endosomes (indicated with red arrows) in an ATG5-regulated manner. Electron micrographs show control untreated WT cells (left), or WT cells (middle) or sgATG5 cells (right) treated with ammonium for 24 hours. Scale bar = 600nm. (B) Quantification of data from part A from >10 independent cells per condition; error bars show SEM. *p<.05. (C) GFP-TRPML1 turnover induced by ammonium treatment for 24 hours is unaffected by treatment with nocodazole (Noc). Actin loading controls are also shown. (D) Autophagy flux induced by serum starvation for 10 hours and measured by GFP-LC3 cleavage is inhibited by treatment with nocodazole. Actin loading controls are also shown. (E) Quantification of data from part D from three independent experiments; error bars show SEM. *p<.05. (F) Lysosomes undergo ATG5-regulated shrinkage after treatment with monensin. Images show lysosomes marked by LAMP1-GFP expression (green), three hours post monensin washout after a two-hour treatment. Note lysosomes in sgATG5 cells appear larger than in sgATG13 cells (inset). Scale bar = 10μm. (G) Quantification of lysosome sizes over time in WT, sgATG5 and sgATG13 cells after washout of monensin following a two-hour treatment. Data from three independent experiments are quantified and graphed as percent cells with five or more lysosomes of 2μm diameter or larger. In total >150 cells were analyzed for each condition across three independent experiments. **p<.01. (H) GFP-TRPML1 undergoes turnover following monensin washout after a two-hour treatment. GFP-TRPML1 and GFP are indicated. Actin loading controls are also shown. (I) sgATG5 cells exhibit reduced DQ-BSA cleavage compared to WT cells four hours after monensin washout. Images show DQ-BSA fluorescence (green). Scale bar = 10μm. Graph shows DQ-BSA fluorescence intensities quantified by flow cytometry from three independent experiments. *p<.05.
Microautophagy regulates lysosome size and function.
Because numerous inducers of lysosomal LC3 lipidation are known to lead to increased lysosome size, resulting from swelling induced by osmotic stress and lysosome fusion (Florey et al., 2015), we examined if the LC3-associated microautophagy mechanism we identified might play a role in regulating lysosome size in this context. Cells expressing LAMP1-GFP to label lysosomes were treated with the ionophore monensin for two hours to induce lysosome swelling, followed by monensin washout and quantification of lysosome sizes as they reduced over time. As shown in Figure 4F and G, lysosome sizes decreased after monensin washout, and this effect was significantly inhibited by loss of ATG5, but not ATG13, consistent with a role for endolysosomal LC3 lipidation in controlling lysosome size. The ATG5-regulated turnover of the GFP-TRPML1 lysosomal reporter occurred within 30 minutes to one hour following monensin washout, in a manner correlating with a reduction in lysosome size (Fig. 4H), suggesting that microautophagy-mediated membrane turnover contributes significantly to lysosome shrinkage. To further examine the effect of this activity on lysosome function, cells were treated with the lysosomal substrate DQ-BSA that fluoresces in response to cathepsin-mediated cleavage. As shown in Figure 4I, following a two-hour monensin treatment and four-hour washout, to induce LC3 lipidation and subsequent ATG5-regulated lysosome shrinkage, ATG5-knockout cells were inhibited for lysosome function compared to wild-type cells, as measured by reduced DQ-BSA fluorescence. By contrast, ATG5-knockout cells cultured under normal conditions showed no evidence of lysosome dysfunction (Supplemental Fig. 4). To examine ATG5-regulated formation of multivesicular endosomes under these conditions, cells expressing GFP-TRPML1 were treated with monensin, and then lysosomes were further enlarged by co-treatment with an inhibitor of the PIKfyve kinase, apilimod, as reported (Cai et al., 2013; Choy et al., 2018), to enable the visualization of intraluminal vesicles by light microscopy. As shown in Figure 5A and Supplemental Movie 1, monensin treatment led to the appearance of GFP-TRPML1 intraluminal vesicles inside of enlarged lysosomes in an ATG5-regulated manner. Together these data demonstrate that ATG5-regulated microautophagy occurs in response to monensin treatment and contributes to regulating lysosome size and function in response to osmotic stress on the lysosome network.
Figure 5. ATG5-regulated microautophagy.
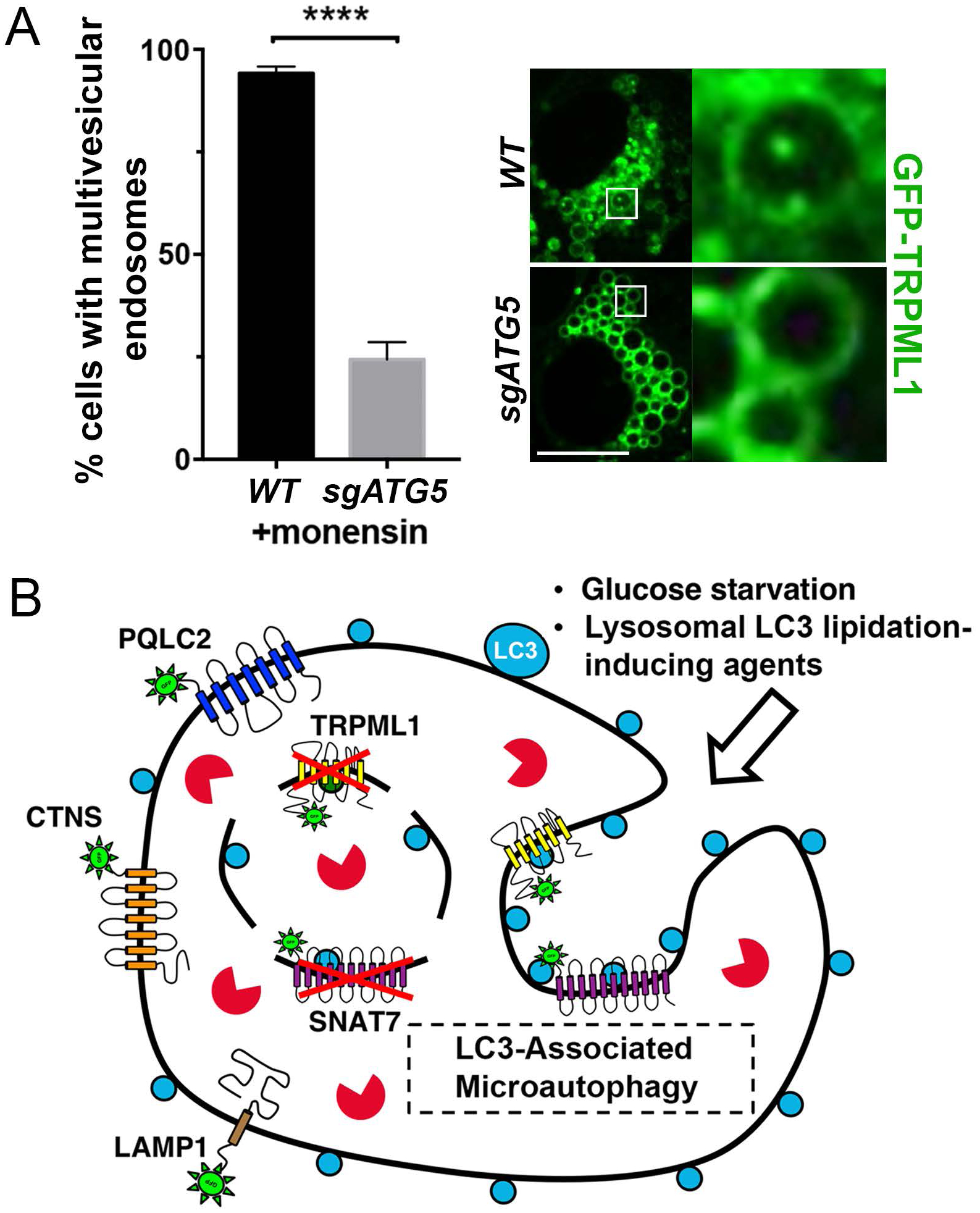
(A) ATG5-regulated formation of intraluminal vesicles is induced by monensin treatment. Graph shows quantification of intraluminal vesicles in monensin-treated WT and sgATG5 cells. In total >180 cells were analyzed for each condition across four independent experiments. Error bars show SEM. ****p<.0001. Images show enlarged lysosomes from WT and sgATG5 GFP-TRPML1 expressing cells treated with monensin for one hour, followed by apilimod for two hours. See Supplemental Movie 1. Scale bar = 10μm. (B) Glucose-starved cells and cells treated with pharmacological agents that induce endolysosmal LC3 lipidation exhibit ATG5-regulated microautophagy, involving the formation of intraluminal vesicles (arrow) and selective turnover of lysosomal membrane proteins including TRPML1 and SNAT7.
Discussion
Here we developed a reporter system to measure lysosome turnover that involves the cleavage of GFP from tagged lysosomal transmembrane proteins and utilized this system to discover a mechanism that stimulates turnover of the lysosome membrane. Using this approach, we show that lysophagy occurring in response to membrane damage leads to the turnover of whole organelles, while starvation of cells for nutrients induces either no measurable turnover, in the case of amino acid withdrawal, or induces selective turnover in response to starvation for glucose. In glucose-starved cells, we identify a mechanism that controls lysosome membrane turnover that involves the core machinery of autophagy acting through a non-canonical mechanism to direct lipidation of the autophagy protein LC3 onto the lysosomal membrane. Lysosomal LC3 lipidation is associated with the formation of intraluminal vesicles, and ultimately, the selective turnover of lysosomal membrane proteins (Fig. 5B).
The lipidation of LC3 onto endosomal membranes was first discovered through studies of phagocytosis (Sanjuan et al., 2007), and was subsequently shown to occur on macropinosome-derived vesicles (Florey et al., 2011), entotic vacuoles (Florey et al., 2011), endosomes (Heckmann et al., 2019), and lysosomes (Florey et al., 2015). Our finding that selective lysosomal membrane turnover involves LC3 lipidation and requires core autophagy genes, including those that encode components of the E1 (ATG7), E2 (ATG3) and E3-like (ATG5) enzymes, but does not require the ULK pre-initiation complex, is consistent with LAP-like LC3 lipidation leading to selective membrane turnover (Florey et al., 2011; Martinez et al., 2011). These findings uncover a potential direct consequence of LC3 lipidation on endosomal membranes. Future studies may examine which of the numerous ATG8 orthologs or LC3-family proteins in mammalian cells may participate in this mechanism (Shpilka et al., 2011).
Interestingly this mechanism of selective turnover may have similarities with a recently described function for LC3 in controlling the formation and loading of exosome vesicles derived from late endosomes (Leidal et al., 2020). In future studies it may be of interest to examine whether selectivity defined by our measurements of membrane protein turnover may be related to selectivity occurring during the release of exosomes through lysosomal exocytosis.
Microautophagy is a poorly understood process in mammalian cells. Studies in yeast have shown that infolding of the vacuolar membrane can control the selective turnover of captured cytoplasmic substrates, including regions of the ER (Schuck et al., 2014), or the turnover of a vacuolar transmembrane protein, the amino acid transporter YPQ1, when yeast are starved for its substrate amino acid (Li et al., 2015; Zhu et al., 2017). A recent report also identified amino acid signaling through TORC1 as contributing to the turnover of additional vacuolar membrane proteins in yeast (Yang et al., 2020). In mammalian cells, microautophagy has been shown to occur on late endosomes where it can regulate the turnover of cytosolic substrates that are taken into intraluminal vesicles, including numerous metabolic enzymes (Sahu et al., 2011), autophagy receptor proteins (Mejlvang et al., 2018), and also synaptic proteins in neurons (Uytterhoeven et al., 2015). The microautophagy activity that we find occurring on lysosomes may differ significantly from these processes as it is regulated by autophagy proteins and involves the direct lipidation of LC3 onto the lysosomal membrane, whereas endosomal microautophagy has been shown to occur in an autophagy protein-independent manner and is not known to involve membrane selectivity (Sahu et al., 2011). Our study also suggests that unlike vacuolar protein turnover in yeast, lysosome membrane protein turnover in mammalian cells is not induced by a loss of amino acid signaling but is more specifically linked to metabolic adaptations that occur in response to starvation for glucose. Future studies may uncover how lysosomal remodeling through selective microautophagy contributes to supporting lysosome function during stress.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Michael Overholtzer. (overhom1@mskcc.org)
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact with completed Materials Transfer Agreement.
Data and Code Availability
This study did not generate any unique datasets or code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines
MCF10A cells stably expressing GFP-tagged proteins were generated by retroviral transduction using the pBabe-puro expression plasmid, as previously described (Florey et al., 2011). GFP-TRPML1 (gift from Dr. Shmuel Muallem), PQLC2-GFP, LAMP1-GFP (Addgene), and GFP-Galectin3 (Addgene) inserts were PCR amplified, purified, and cloned into pBabe-puro using Gibson Assembly (New England Biolabs). SNAT7 ORF (NM_018231.3) was obtained through Standard Gene Synthesis from Genewiz. SNAT7 and GFP inserts were amplified by PCR, purified, and cloned into pBabe-puro using Gibson Assembly, with the GFP at the N-terminus. CTNS insert was amplified by PCR from MCF10A cDNA library using the following primers:
Forward: 5’-ctccatagaagattctagagATGATAAGGAATTGGCTGAC-3’
Reverse: 5’-ccgcggtaccGTTCAGCTGGTCATACCC-3’
Insert was cloned into pCDH-puro expression plasmid using Gibson Assembly, with the GFP insert at the C-terminus. GFP-positive cells were selected by 2μg/ml puromycin and/or fluorescence-activated cell sorting (Flow Cytometry Core, MSKCC). GFP-LC3 expressing MCF10A cells were previously generated (Krishna et al., 2016). ATG5 and ATG13 knockout MCF10A cells were generated using CRISPR/Cas9 (Hawk et al., 2018). CRISPR guide sequence, 5’-GTGATTGTCCAGGCTCGGCT-3’, was used to target ATG13. MCF10A HA-ATG5 re-expressing ATG5 knockout cells were previously generated (Hawk et al., 2018). MCF10A cells generated for this study were cultured at 37°C, 5% CO2, in DMEM/F12 supplemented with 5% horse serum, 20ng/ml epidermal growth factor, 10μg/ml insulin, 0.5μg/ml hydrocortisone, and 100ng/ml cholera toxin.
METHOD DETAILS
Nutrient starvation and drug treatments
For nutrient starvation experiments, cells were washed twice with PBS before incubation in starvation media. Glucose/pyruvate-free DMEM/F12 media was used for glucose-starvation experiments. Base DMEM/F12 media was used for serum-free conditions. Glucose/pyruvate-free, amino acid-free, glucose/glutamine-free DME/F12 media (Media Preparation Core, MSKCC) were supplemented with 5% dialyzed horse serum, 20ng/ml epidermal growth factor, 10μg/ml insulin, 0.5μg/ml hydrocortisone, and 100ng/ml cholera toxin. For lysophagy experiments, cells were treated with 0.5mM L-Leucyl-L-Leucine methyl esther (LLOMe) (Cayman chemicals) for one hour to induce lysosome damage and then washed twice with media and then cultured in growth media. For experiments with nocodazole and SBI-0206965, cells were pre-treated for one hour with either 1μg/ml nocodazole or 10μM SBI-0206965, and these agents were also added for the duration of the experiment. For endolysosomal LC3 lipidation imaging, 50μM Monensin, 25μM nigericin, and 10mM NH4Cl were used. For lysosome turnover, 10μM monensin, 5μM nigericin, and 5mM NH4Cl were used.
siRNA
ON-TARGETplus SMARTpool siRNAs against human ATG3 and ATG7 were obtained from Horizon Discovery. Cells were seeded in a 6-well plate at 2 × 105 cells per well and transfected with 100nM siRNA using Lipofectamine 3000 (Thermo Fisher Scientific), following manufacturer’s protocol. Cells were assayed 48 hours post-transfection.
Western Blotting
Cells were harvested in lysis buffer (50mM Tris-Cl pH 6.8, 10% glycerol, 2% SDS), boiled at 100°C for 5-min, and vigorously pipetted to remove viscosity. Protein concentrations were measured by BCA assay (Thermo Fisher Scientific) and all samples were prepared to equal protein amount. Western blot was performed following standard protocol and analyzed using the following primary antibodies: GFP (1:500; Cell Signaling Technology (CST)), GFP (1:1000; Roche, used to detect GFP-SNAT7 and CTNS-GFP), ATG3 (1:1000; CST), ATG7 (1:500; CST), ATG5 (1:1000; CST), ATG13 (1:1000; CST), β-Actin (1:2000; Sigma). Chemiluminescence was visualized using the Amersham Imager 600 (GE Healthcare) and densitometry analysis was performed on unsaturated images using ImageStudio (LICOR).
Live Cell Confocal Microscopy
Live cell confocal microscopy was carried out using the Ultraview Vox spinning-disc confocal system (PerkinElmer) equipped with a Yokogawa CSU-X1 spinning-disc head and electron-multiplying charge-coupled device camera (Hamamatsu C9100–13) coupled to a Nikon Ti-E microscope. Cells were cultured on 35mm glass-bottomed dishes (MatTek) and imaged in live-cell incubation chambers maintained at 37°C and 5% CO2. For GFP-Galectin3 imaging experiments, GFP-Galectin3 expressing MCF10A cells were treated with 0.5mM LLOMe for one hour to induce lysosome damage and then washed twice with media and cultured in growth media. For lysosome size control experiments, LAMP1-GFP expressing MCF10A cells were treated with 50μM monensin for two hours to enlarge lysosomes and were then washed twice with growth media and cultured in growth media. Dishes were immediately taken to the microscope at the indicated times for image acquisition. Image acquisition and lysosome size analyses were carried out using Volocity software (PerkinElmer).
DQ-BSA
Cells were treated with 50μM monensin for two hours and then washed twice with growth media before incubation with 20μg/ml DQ-BSA Green (Thermo Fisher Scientific) for four hours. For imaging, DQ-BSA was washed out with growth media and immediately imaged by confocal microscopy. DQ-BSA fluorescence intensities were measured by flow cytometry on LSRII (BD Biosciences; Flow Cytometry Core, MSKCC), with 100,000 cells analyzed for each condition per experiment. Data were analyzed on FCS Express 7 (De Novo Software).
Immunofluorescence
Cells seeded on 35mm glass-bottomed dishes (MatTek) were washed once with PBS and then fixed in 1:1 methanol/acetone for 5-min at −20°C, followed by three 5-min PBS washes, one hour in blocking solution (5% BSA, 100mM glycine in PBS), and then overnight incubation with primary antibodies at 4°C. Samples were then washed three times with PBS for 5-min each and then incubated with secondary antibodies for one hour at room temperature, followed by three 5-min PBS washes. Antibodies used were anti-LAMP1 (1:200; BD Biosciences), anti-LAMP2 (1:200; Santa Cruz), and Alexa Fluor 568 goat anti-mouse secondary (1:500; Life Technologies). Images were acquired using confocal microscopy.
Intraluminal vesicle imaging
One day prior to treatment, 200,000 cells were plated on 35mm glass-bottomed dishes. Cells were treated with 50μM monensin for one hour, followed by the addition of 200nM Apilimod (Axon Medchem) for an additional two hours to enlarge lysosomes and visualize intraluminal vesicles. Cells were then immediately imaged by confocal microscopy. Quantification was performed by counting the percentage of cells that had 50 percent or more of their enlarged GFP-TRPML1-labeled endosomes containing intraluminal vesicles, indicated by luminal motile GFP puncta.
Transmission Electron Microscopy
Cells were fixed in 2.5% Glutaraldehyde in cacodylate buffer, pH 7.4 and post-fixed using 1% osmium tetroxide containing 1.5% potassium ferrocyanide. The samples were washed with water and stained with 1% aqueous uranyl acetate. Samples were then dehydrated in a graded series of ethanol and infiltrated using Eponate 12 (Electron Microscopy Sciences) using a microwave (Pelco Biowave, Ted Pella). Sections were cut at 70nm and imaged at 80kV on a JEOL 100CX transmission electron microscope with an XR41-C AMT digital imaging system (Advantage Microscopy Technology Corp, Woburn, MA).
QUANTIFICATION AND STATISTICAL ANALYSIS
Graphs were assembled and statistics were performed using Prism 7 (GraphPad). Error bars are shown as mean ± SEM from n=3 independent experiments, unless otherwise indicated. P values were calculated using Student’s t-test (***, p<0.001; **, p<0.01; *, p<0.05; ns, not significant), unless otherwise indicated.
Supplementary Material
Supplemental Movie 1. ATG5-regulated formation of intraluminal vesicles is induced by monensin treatment, Related to Figure 5. Time-lapse confocal images show enlarged lysosomes from GFP-TRPML1 expressing WT (left) and sgATG5 (right) cells treated with monensin for one hour, followed by apilimod for two hours. Arrows indicate examples of multivesicular structures in the WT cell; no multivesicular structures are observed in the sgATG5 cell. Images were acquired every 100ms.
Acknowledgements
This research was supported by grants from the National Cancer Institute (CA154649 to M.O.) and the SKI Basic Research Innovation Award Initiative (BRIA) and Dalio Explore Fund (to M.O.). We would like to thank past and present members of the Overholtzer laboratory for helpful discussions.
Footnotes
Declaration of Interests
The authors declare no competing interests.
References
- Cai X, Xu Y, Cheung AK, Tomlinson RC, Alcazar-Roman A, Murphy L, Billich A, Zhang B, Feng Y, Klumpp M, et al. (2013). PIKfyve, a class III PI kinase, is the target of the small molecular IL-12/IL-23 inhibitor apilimod and a player in Toll-like receptor signaling. Chem Biol 20, 912–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong H, Lindsten T, Wu J, Lu C, and Thompson CB (2011). Ammonia-induced autophagy is independent of ULK1/ULK2 kinases. Proc Natl Acad Sci U S A 108, 11121–11126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choy CH, Saffi G, Gray MA, Wallace C, Dayam RM, Ou ZA, Lenk G, Puertollano R, Watkins SC, and Botelho RJ (2018). Lysosome enlargement during inhibition of the lipid kinase PIKfyve proceeds through lysosome coalescence. J Cell Sci 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikic I (2017). Proteasomal and Autophagic Degradation Systems. Annu Rev Biochem 86, 193–224. [DOI] [PubMed] [Google Scholar]
- Egan DF, Chun MG, Vamos M, Zou H, Rong J, Miller CJ, Lou HJ, Raveendra-Panickar D, Yang CC, Sheffler DJ, et al. (2015). Small Molecule Inhibition of the Autophagy Kinase ULK1 and Identification of ULK1 Substrates. Mol Cell 59, 285–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florey O, Gammoh N, Kim SE, Jiang X, and Overholtzer M (2015). V-ATPase and osmotic imbalances activate endolysosomal LC3 lipidation. Autophagy 11, 88–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florey O, Kim SE, Sandoval CP, Haynes CM, and Overholtzer M (2011). Autophagy machinery mediates macroendocytic processing and entotic cell death by targeting single membranes. Nat Cell Biol 13, 1335–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florey O, and Overholtzer M (2012). Autophagy proteins in macroendocytic engulfment. Trends Cell Biol 22, 374–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckmann BL, Teubner BJW, Tummers B, Boada-Romero E, Harris L, Yang M, Guy CS, Zakharenko SS, and Green DR (2019). LC3-Associated Endocytosis Facilitates beta-Amyloid Clearance and Mitigates Neurodegeneration in Murine Alzheimer’s Disease. Cell 178, 536–551 e514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquin E, Leclerc-Mercier S, Judon C, Blanchard E, Fraitag S, and Florey O (2017). Pharmacological modulators of autophagy activate a parallel noncanonical pathway driving unconventional LC3 lipidation. Autophagy 13, 854–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, Kundu M, and Kim DH (2009). ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell 20, 1992–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiselyov K, Chen J, Rbaibi Y, Oberdick D, Tjon-Kon-Sang S, Shcheynikov N, Muallem S, and Soyombo A (2005). TRP-ML1 is a lysosomal monovalent cation channel that undergoes proteolytic cleavage. J Biol Chem 280, 43218–43223. [DOI] [PubMed] [Google Scholar]
- Lawrence RE, and Zoncu R (2019). The lysosome as a cellular centre for signalling, metabolism and quality control. Nat Cell Biol 21, 133–142. [DOI] [PubMed] [Google Scholar]
- Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J, Tsukamoto T, Rojas CJ, Slusher BS, Zhang H, et al. (2012). Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab 15, 110–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leidal AM, Huang HH, Marsh T, Solvik T, Zhang D, Ye J, Kai F, Goldsmith J, Liu JY, Huang YH, et al. (2020). The LC3-conjugation machinery specifies the loading of RNA-binding proteins into extracellular vesicles. Nat Cell Biol 22, 187–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Rong Y, Chuang YS, Peng D, and Emr SD (2015). Ubiquitin-dependent lysosomal membrane protein sorting and degradation. Mol Cell 57, 467–478. [DOI] [PubMed] [Google Scholar]
- Maejima I, Takahashi A, Omori H, Kimura T, Takabatake Y, Saitoh T, Yamamoto A, Hamasaki M, Noda T, Isaka Y, et al. (2013). Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. EMBO J 32, 2336–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez J, Almendinger J, Oberst A, Ness R, Dillon CP, Fitzgerald P, Hengartner MO, and Green DR (2011). Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc Natl Acad Sci U S A 108, 17396–17401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mejlvang J, Olsvik H, Svenning S, Bruun JA, Abudu YP, Larsen KB, Brech A, Hansen TE, Brenne H, Hansen T, et al. (2018). Starvation induces rapid degradation of selective autophagy receptors by endosomal microautophagy. J Cell Biol 217, 3640–3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochida K, Oikawa Y, Kimura Y, Kirisako H, Hirano H, Ohsumi Y, and Nakatogawa H (2015). Receptor-mediated selective autophagy degrades the endoplasmic reticulum and the nucleus. Nature 522, 359–362. [DOI] [PubMed] [Google Scholar]
- Raben N, and Puertollano R (2016). TFEB and TFE3: Linking Lysosomes to Cellular Adaptation to Stress. Annu Rev Cell Dev Biol 32, 255–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahu R, Kaushik S, Clement CC, Cannizzo ES, Scharf B, Follenzi A, Potolicchio I, Nieves E, Cuervo AM, and Santambrogio L (2011). Microautophagy of cytosolic proteins by late endosomes. Dev Cell 20, 131–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjuan MA, Dillon CP, Tait SW, Moshiach S, Dorsey F, Connell S, Komatsu M, Tanaka K, Cleveland JL, Withoff S, et al. (2007). Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 450, 1253–1257. [DOI] [PubMed] [Google Scholar]
- Schuck S, Gallagher CM, and Walter P (2014). ER-phagy mediates selective degradation of endoplasmic reticulum independently of the core autophagy machinery. J Cell Sci 127, 4078–4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shpilka T, Weidberg H, Pietrokovski S, and Elazar Z (2011). Atg8: an autophagy-related ubiquitin-like protein family. Genome Biol 12, 226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun A (2018). Lysosomal storage disease overview. Ann Transl Med 6, 476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uytterhoeven V, Lauwers E, Maes I, Miskiewicz K, Melo MN, Swerts J, Kuenen S, Wittocx R, Corthout N, Marrink SJ, et al. (2015). Hsc70–4 Deforms Membranes to Promote Synaptic Protein Turnover by Endosomal Microautophagy. Neuron 88, 735–748. [DOI] [PubMed] [Google Scholar]
- Wong E, and Cuervo AM (2010). Integration of clearance mechanisms: the proteasome and autophagy. Cold Spring Harb Perspect Biol 2, a006734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Zhang W, Wen X, Bulinski PJ, Chomchai DA, Arines FM, Liu YY, Sprenger S, Teis D, Klionsky DJ, et al. (2020). TORC1 regulates vacuole membrane composition through ubiquitin- and ESCRT-dependent microautophagy. J Cell Biol 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L, Jorgensen JR, Li M, Chuang YS, and Emr SD (2017). ESCRTs function directly on the lysosome membrane to downregulate ubiquitinated lysosomal membrane proteins. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Movie 1. ATG5-regulated formation of intraluminal vesicles is induced by monensin treatment, Related to Figure 5. Time-lapse confocal images show enlarged lysosomes from GFP-TRPML1 expressing WT (left) and sgATG5 (right) cells treated with monensin for one hour, followed by apilimod for two hours. Arrows indicate examples of multivesicular structures in the WT cell; no multivesicular structures are observed in the sgATG5 cell. Images were acquired every 100ms.
Data Availability Statement
This study did not generate any unique datasets or code.