

Abstract
Aims
Lung adenocarcinoma (LUAD) cells could escape from the monitoring of immune cells and metastasize rapidly through immune escape. Therefore, we aimed to develop a method to predict the prognosis of LUAD patients based on immune checkpoints and their associated genes, thus providing guidance for LUAD treatment.
Methods
Gene sequencing data were downloaded from the Cancer Genome Atlas (TCGA) and analyzed by R software and R Bioconductor software package. Based on immune checkpoint genes, kmdist clustering in ConsensusClusterPlus R software package was utilized to classify LUAD. CIBERSORT was used to quantify the abundance of immune cells in LUAD samples. LM22 signature was performed to distinguish 22 phenotypes of human infiltrating immune cells. Gene set variation analysis (GSVA) was performed on immune checkpoint cluster and immune checkpoint score using GSVA R software package. The risk score was calculated by LASSO regression coefficient. Gene Ontology (GO), Hallmark, and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis were performed. PROC was performed to generate the ROC curve and calculate the area under the curve (AUC).
Results
According to the immune checkpoint, LUAD was classified into clusters 1 and 2. Survival rate, immune infiltration patterns, TMB, and immune score were significantly different between the two clusters. Functional prediction showed that the functions of cluster 1 focused on apoptosis, JAK/STAT signaling pathway, TNF-α/NFκB signaling pathway, and STAT5 signaling pathway. The risk score model was constructed based on nine genes associated with immune checkpoints. Survival analysis and ROC analysis showed that patients with high-risk score had poor prognosis. The risk score was significantly correlated with cancer status (with tumor), male proportion, status, tobacco intake, and cancer stage. With the increase of the risk score, the enrichment of 22 biological functions increased, such as p53 signaling pathway. The signature was verified in IMvigor immunotherapy dataset with excellent diagnostic accuracy.
Conclusion
We established a nine-gene signature based on immune checkpoints, which may contribute to the diagnosis, prognosis, and clinical treatment of LUAD.
1. Background
Lung adenocarcinoma (LUAD) accounts for 40% of lung cancer patients and is one of the main subtypes of lung cancer [1]. LUAD is characterized by diffuse metastasis and poor prognosis. The survival rate of patients is less than five years [2]. Because of the high resistance of LUAD to radiotherapy and chemotherapy [3], it is urgent to look for treatment from other directions.
Recently, immunotherapy has become an emerging strategy for cancer treatment [4]. Immune system is involved in the growth and metastasis of cancer cells. For example, cancer cell antigens are recognized by dendritic cells and other antigen presenting cells, which promote the maturation of the corresponding cytotoxic CD8+ T lymphocytes [5, 6]. However, any mistakes in this process might lead to the failure of antigen presenting cells to present antigen normally. It is well known that the complex tumor microenvironment is an important reason for the failure of immunotherapy. As T cells failure to reach the tumor area, the immune system was unable to recognize and remove cancer cells [7]. Wu et al. developed a scoring system based on the expression of genes related to tumor metabolism in LUAD that is suitable not only for predicting prognosis in patients with LUAD but also for predicting LUAD response to checkpoint immunotherapy [8]. Currently, based on the blockade of immune checkpoint, tumor immunotherapy has made a breakthrough progress.
Immune checkpoints were defined as ligand receptor pairs that inhibit or stimulate immune responses [9]. Tumor cells evaded the attack of immune cells by activating immune checkpoints [10]. Currently, some drugs have been developed to block the immune checkpoint. The combination of ipilimumab and placebo can prevent CTLA-4 from binding to its ligand, thus blocking the immune checkpoint [11]. High expression of PD-1 could inhibit the function of T cells and promote the immune escape of tumor cells [12, 13]. Nivolumab is an antibody against PD-1, which could play an antitumor role in clinical trials. However, patients have side effects of loss of appetite and diarrhea [14]. Therefore, to develop better targeted drugs, we need to find more representative immune checkpoints.
TCGA database is often utilized to predict reliable biomarkers [15]. Based on the analysis of TCGA dataset, we classified LUAD according to immune checkpoint genes and detected the clinical characteristics of patients after classification. Simultaneously, based on immune checkpoint-related genes, we established a risk score model and analyzed its correlation with the clinical characteristics and signaling pathways of LUAD samples.
2. Materials and Methods
2.1. LUAD Dataset and Preprocessing
The Cancer Genome Atlas (TCGA) dataset was downloaded from UCSC Xena (https://xenabrowser.net/). RNA sequencing (RNA-seq) data was downloaded from TCGA data portal. Then, the fragments per kilobase million (FPKM) were converted to transcripts per million (TPM). The microarray dataset GSE68465 was downloaded from Gene-Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) as an external validation set. The raw data from the microarray dataset was generated by Affymetrix. Then, the RMA algorithm in Affy package was applied to process the original data from Affymetrix for quantile normalization and background correction. All data were analyzed by R software (version 3.6.1) and R Bioconductor software package.
2.2. Identification of Related Classification of LUAD Immune Checkpoint
Sixty-five immune checkpoint genes were obtained for subsequent clustering. The filtering procedure was performed. The kmdist clustering method in ConsensuClusterPlus R software package was utilized to classify LUAD by immune checkpoint genes [16], so as to determine the patterns related to the immune checkpoint, and the patients were grouped for further analysis.
2.3. Estimation of Immune Infiltration
CIBERSORT algorithm was used to quantify the abundance of immune cells in LUAD samples using Leukocyte signature matrix (LM22) as an eigenmatrix [17], since LM22 gene signature could sensitively and specifically distinguish 22 phenotypes of human infiltrated immune cells. The algorithm was run with an LM22 signature and 1000 permutations. Gene expression profiles have been uploaded to CIBERSORT web portal (http://cibersort.stanford.edu/).
2.4. Pathway Analysis
All gene sets were downloaded from MSigDB database [18]. Gene Set Variation Analysis (GSVA) R software package was utilized to analyze the GSVA of immune checkpoint cluster and immune checkpoint score [19], including Gene Ontology Biological Process (GO BP), Kyoto Encyclopedia of Genes and Genomes (KEGG), and Hallmark gene sets. The selection criteria of immune checkpoint cluster related pathway was based on the corrected p < 0.05. The selection criteria of immune checkpoint score related pathway was based on the correlation analysis p < 0.05.
2.5. Establishment of Immune Checkpoint Score
Limma package in R software was performed to identify differential genes (DEG) associated with two immune checkpoint-related patterns [20]. The corrected p value <0.05 and | logFC | < 1 were set as the significance criteria to determine DEG in immune checkpoint subtypes. Univariate Cox regression analysis was utilized to certificate the representative DEG, and then, random forest in machine learning method of caret package was used to reduce gene dimension. The TCGA dataset was randomly divided into two parts, each of them containing 250 samples. One part of the data was performed to train the model, and the other part and the total data were used to verify the model. In the training set, the dimensionality reduced genes were utilized for single-factor Cox analysis to screen meaningful genes, and then, the highest lambda value (“min” lambda) was selected through 1000 cross-validation in LASSO method. We obtained a group of prognostic genes and their LASSO regression coefficients. Risk score was the sum of the expression value of genes screened by LASSO regression coefficient.
Risk score = (−0.0044)∗
CCR7 (gene expression level) + 0.0323∗
CPS1 + (−0.1006)∗
LILRB1 + 0.1460∗
GOS2 + (−0.2027)∗
HLA-DOB + (−0.2116)∗
CCR2 + (−0.1181)∗
CLEC7A + 0.3002∗
BIRC3 + (−0.0286)∗
CD1E.
2.6. Survival Curve and ROC Analysis
The pROC package was used to generate receiver operating characteristic (ROC) curve and calculate area under curve (AUC) [21]. Kaplan–Meier was utilized to generate and visualize survival curves of subgroups. The statistical significance of the differences in each dataset was determined by log rank test. All survival curves were generated by R package survminer. All heatmaps were generated based on pheatmap. All statistical analysis were analyzed in R (https://www.r-project.org/, version 3.6.1).
2.7. Statistical Analysis
The Shapiro–Wilk test was utilized to assess the normality of variables. For normally distributed variables, unpaired Student's t-test was used to compare the differences between the two groups. Wilcoxon test was utilized to compare variables with nonnormal distribution. For multiple groups, ANOVA was performed as the parametric method to compare the mean value, while Kruskal–Wallis test was used as the nonparametric method. Pearson correlation and distance correlation analysis were used to calculate the correlation coefficient. According to the risk score of dichotomy, patients were divided into high- or low-risk score of each dataset. Ggplot2 was used of visualization in the R program. In the analysis of differentially expressed genes, we utilized Benjamini-Hochberg, which transformed p value into FDR to identify important genes. All the tests were two-sided, and p values <0.05 were considered statistically significant.
3. Results
3.1. Distribution of Immune Checkpoints after Gene Clustering
We clustered LUAD by immune checkpoint. The results showed that the boundaries of LUAD clustering into two categories were clear (Figure S1A), while the boundaries of lung adenocarcinoma clustering into three or four categories were fuzzy (Figures S1B and S1C). Consistency cluster analysis (consensus CDF) was used for further analysis, and the best classification was defined as the one with the smallest slope of curve decline in the abscissa range of 0.1–0.9. The results showed that when LUAD was divided into two categories, clusters 1 and 2, according to immune checkpoints, the descending slope of curve was the smallest. The correlation between immune checkpoint-related gene expression and clinical characteristics in the two clusters was shown in Figure 1(a). Most genes were highly expressed in cluster 1 and low expressed in cluster 2. We also found that the expression of molecules related to antigen presentation (HLA-C, HLA-B), ligands (CXCL5, CXCL10), receptors (ADORA2A, BTLA), inhibitors (btn3a1, btn3a2), activators (CD28, CD80), and cell adhesion (ICAM1, ITGB2) in cluster 1 was significantly higher than that in cluster 2. Other proteins involved in immune regulation, such as IDO1, GZMA, and PRF1, were also highly expressed in cluster 1 compared with cluster 2 (Figure S2). We used the survival curve to analyze the survival difference between clusters 1 and 2. It was found that the survival rate of patients in cluster 2 decreased faster than that in cluster 1 over time. This indicated that the prognosis of patients in cluster 2 was worse than that in cluster 1. PCA image showed that clusters 1 and 2 can be separated (Figure 1(c)), which proved that the classification of LUAD was meaningful. Different types of immune cells were distributed differently in the two clusters (Figure 1(d)). For example, the distribution of B cells memory, dendritic cells resting, macrophages M1 and M2, T cells CD4 memory resting, and T cells CD8 in cluster 1 were more abundant than those in cluster 2. While the distribution of plasma cells, dendritic cells activated, macrophages M0, mast cells activated, and NK cells resting in cluster 2 were contrary.
Figure 1.
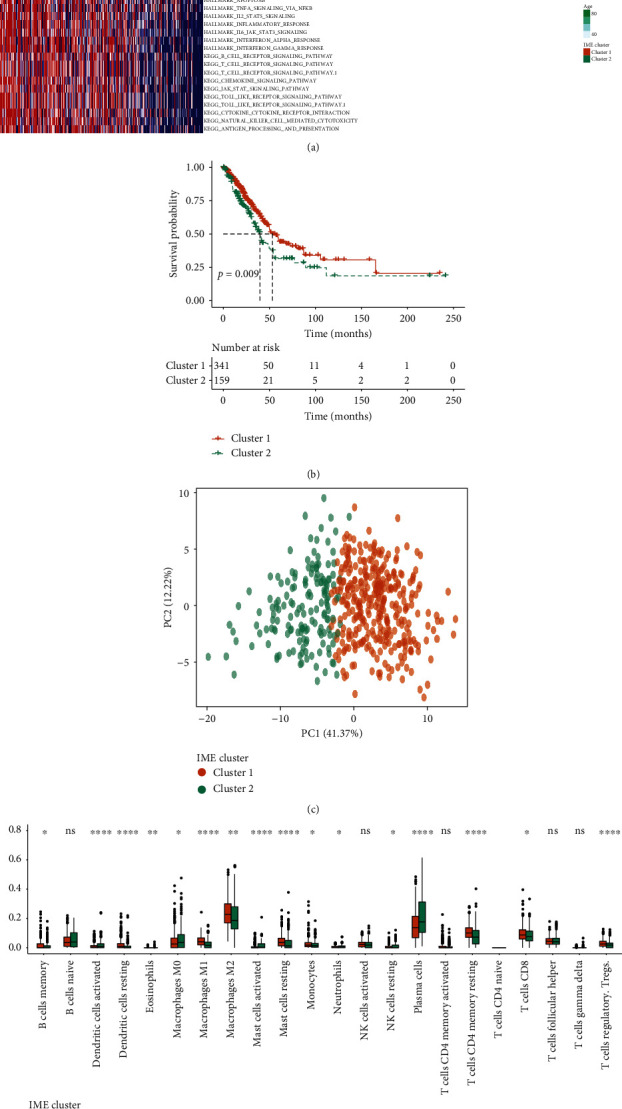
The characteristics of clusters 1 and 2. (a) Correlation between immune checkpoint-related gene expression and clinical characteristics in two clusters. (b) Survival analysis of clusters 1 and 2. (c) PCA analysis of clusters 1 and 2. (d) Comparison of the distribution of immune cells in clusters 1 and 2.
3.2. Functional Analysis of Checkpoints in Clusters 1 and 2
We performed GO analysis, Hallmark, and KEGG pathway to analyze the function of immune checkpoint-related genes in clusters 1 and 2 (Figure 2(a)). GO analysis results showed that mitochondrial RNA catabolic process was significantly enriched in cluster 2. The related genes of cluster 1 were mainly involved in macrophage proliferation, positive regulation of inflammatory response to antigenic stimulus, CD8 positive alpha beta T cell activation, and microglial cell activation. Hallmark results showed that cluster 2 genes were involved in Myc targets V2, G2/M checkpoint, and DNA repair. The genes in cluster 1 were mainly involved in apoptosis, TNFa signaling via NFkB, and IL2 STAT5 signaling. In addition, KEGG pathway showed that, compared with cluster 2, genes in cluster 1 were mainly enriched in T and B cell receptor signaling pathway, chemokine signaling pathway, JAK/STAT signaling pathway, and toll-like receptor signaling pathway. These results indicated that immune-related pathway scores were significantly higher in cluster 1 than in cluster 2. Then, we examined the tumor mutation burden (TMB) of clusters 1 and 2, and the results showed that cluster 1 was higher than cluster 2 (Figure 2(b)). Finally, we found that the ESTIMTE score, immune score, and stromal score of cluster 1 were significantly higher than those of cluster 2 (p < 2.22e − 16) (Figure 2(c)). These results suggested that the survival and prognosis of cluster 1 patients were better than those of cluster 2 patients.
Figure 2.
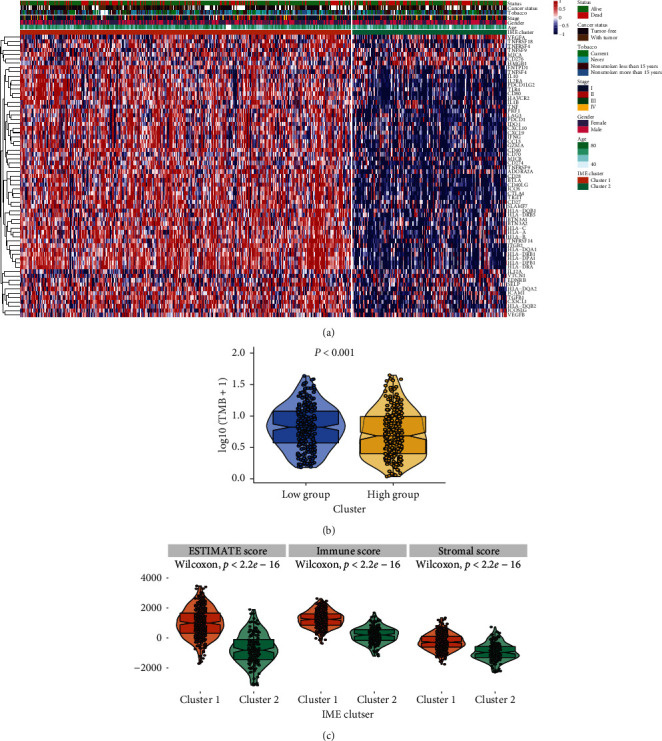
Functional analysis and immune score of clusters 1 and 2. (a) Functional analysis of the two clusters was based on GO enrichment, Hallmark, and KEGG pathway. (b) Tumor mutation burden of clusters 1 and 2. (c) Immune infiltration assessment in clusters 1 and 2.
3.3. Screening of Genes in Clusters 1 and 2 and Establishment of Prognosis Model
The total number of genes differing between the two clusters was 552 (Figure 3(a)). Later, we screened 164 genes by univariate analysis in TCGA dataset and further obtained 132 genes by random forest dimensionality reduction (Figure 3(b)). Through single factor analysis of 132 genes in the modeling set, 77 genes were obtained (Figure 3(c)), Subsequently, nine genes, including BIRC3, G0S2, CCR7, CPS1, CLEC7A, LILRB1, CCR2, HLA-DOB, and CD1E, were screened by LASSO method (Figure 3(d)). These nine genes were divided into high-risk group and low-risk group, to establish the risk score formula: risk score = (−0.0044)∗
Figure 3.
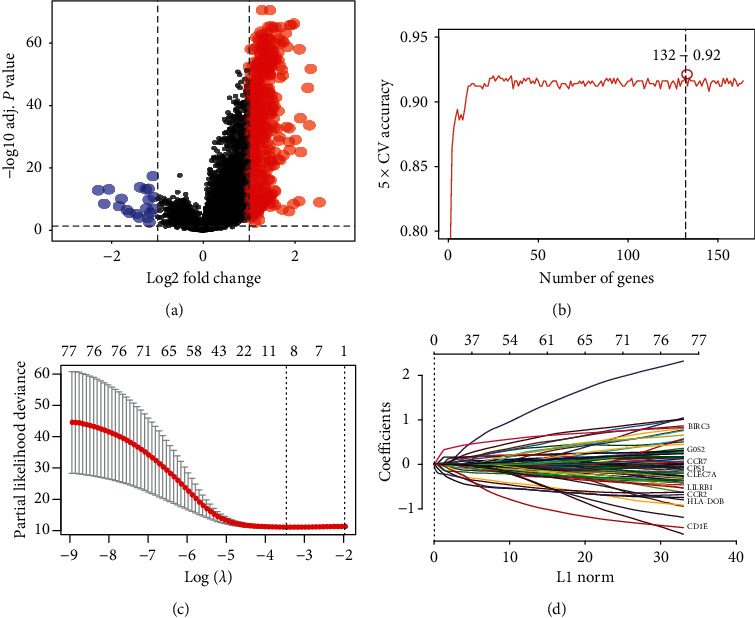
Screening of immune checkpoint-related genes. (a) Differential expressed genes between the two clusters. (b) The number of genes obtained by random forest dimensionality reduction. (c) Partial likelihood deviance coefficient profiles of 77 immune checkpoint genes via 10-fold cross-validation. (d) Least absolute shrinkage and selection operator (LASSO) profiles of the selected genes.
CCR7 (gene expression level) + 0.0323∗
CPS1 + (−0.1006)∗
LILRB1 + 0.1460∗
GOS2 + (−0.2027)∗
HLA-DOB + (−0.2116)∗
CCR2 + (−0.1181)∗
CLEC7A + 0.3002∗
BIRC3 + (−0.0286)∗
CD1E. Then, we used the risk score model to analyze the survival rate of patients with these nine genes in the training set (Figure 4(a)), testing set (Figure 4(b)), overall TCGA set (Figure 4(c)), and external validation set (Figure 4(d)). Survival analysis showed that patients with high-risk score had a poor prognosis (p < 0.05). Time-dependent ROC analysis showed that the one-year AUC reached 0.755 in all datasets. It suggested that the efficacy of the model was satisfactory. To ensure the robustness of the model, we verified the LUAD model with IMvigor immunotherapy dataset, as shown in Figure S3. The survival rate of the high-risk group was lower than that of the low-risk group. It demonstrated the accuracy of our model in predicting prognosis.
Figure 4.
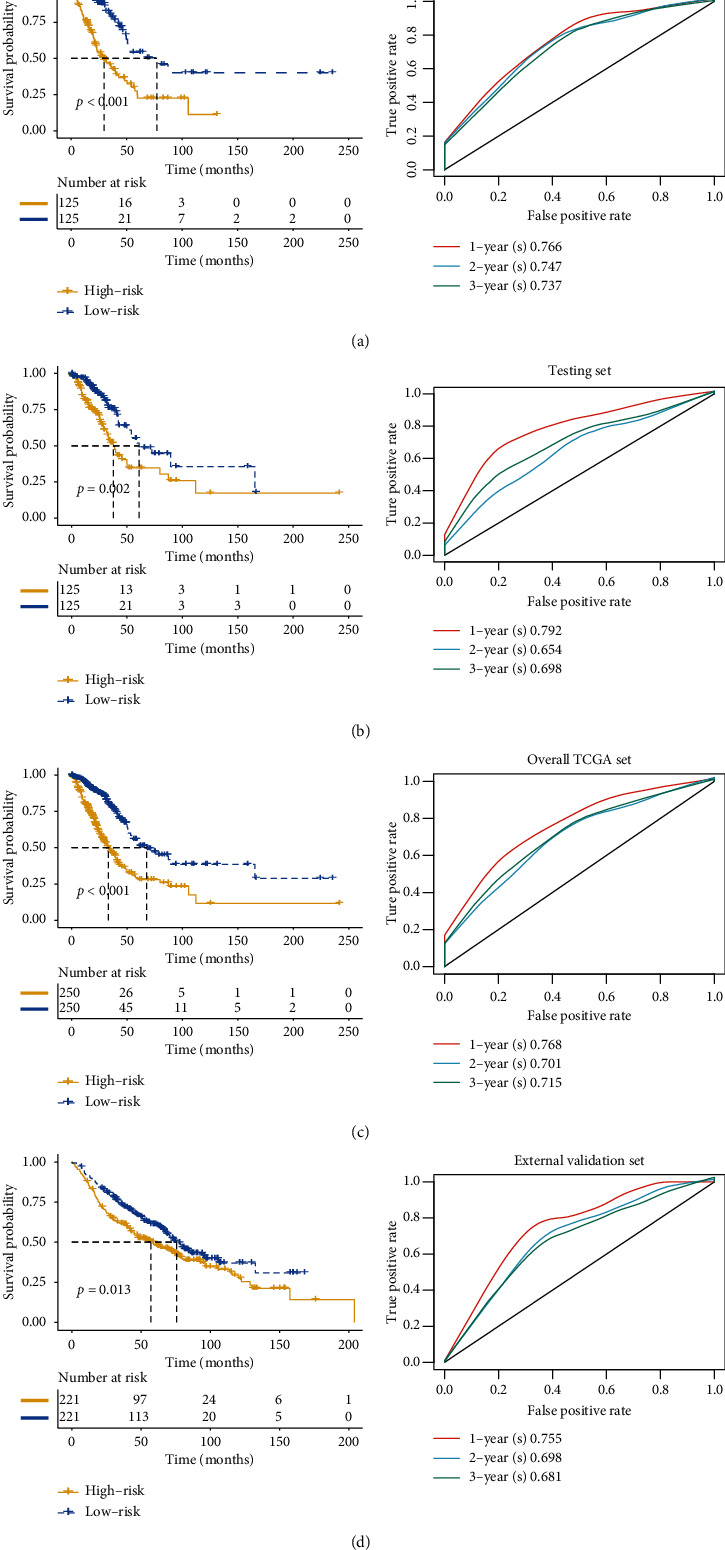
Survival curve analysis and ROC analysis of the nine-gene signature. (a) Survival analysis of two risk groups and ROC analysis of the nine-gene signature in training set. (b) Survival curve of two risk groups and ROC curve of the nine-gene signature in testing set. (c) Survival status of two risk groups and ROC analysis of the nine-gene signature in overall TCGA set. (d) Survival analysis and ROC curve of the nine-gene signature in the external validation set.
3.4. Analysis of the Relationship between Risk Score, Clinical Characteristics, and Signaling Pathways
According to the risk score of these nine genes, we detected the changes of different clinical characteristics of the patients. The results showed that the TMB value of the low-risk score group was higher than that of the high-risk score group (Figure 5(a)). Then, we evaluated the relationship between risk score and clinical characteristics. There was no significant difference between high-risk score and low-risk score in age (p=0.11). The risk score exhibited statistical differences in cancer status (tumor-free, with tumor), gender (female, male), status (alive, dead), tobacco (current smoker, never smoke, nonsmoker: less than 15 years, and nonsmoker: more than 15 years, and stage (I, II, III, IV) (p < 0.05) (Figure 5(b)). We also evaluated the relationship between the risk score and biological function (Figure 5(c)). GO analysis showed that the expression of cell cycle related pathways and DNA repair increased as the risk score increased gradually, while the expression of MHC type II protein decreased. Hallmark results showed that the expression of E2F target, G2/M checkpoint, and Myc target increased, and the expression of KRAS signaling pathway decreased, when the risk score gradually increased. KEGG pathway showed that seven pathways, including base excision repair, cell cycle, and DNA replication, were positively correlated with the risk score. B cell receptor signaling pathway, chemokine signaling pathway, and intestinal immune network for IgA production were negatively correlated with the risk score.
Figure 5.
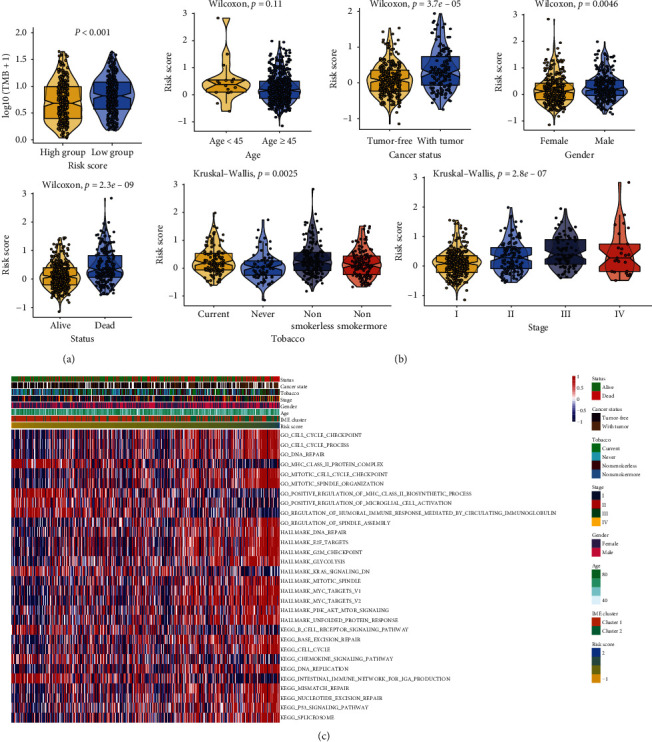
Analysis of the relationship between risk score, clinical characteristics, and signaling pathways. (a) Comparison of TMB between the two risk groups. (b) The relationship between risk score model and clinical characteristics, including age, cancer status, gender, status, tobacco, and stage. (c) Function prediction of GO, Hallmark, and KEGG pathways based on risk score model.
4. Discussion
LUAD has the characteristics of poor prognosis and rapid metastasis, which leads to high mortality. Many studies have shown that tumor development is controlled by the patient's immune system, which has important implications for patient prognosis and response to drug therapy [22]. Therefore, we classified LUAD according to immune checkpoint. After classification, we found that the expression of molecules related to antigen presentation, ligands, receptors, inhibitors, activators, and cell adhesion in cluster 1 was significantly higher than that in cluster 2. Simultaneously, the survival rate of patients in cluster 1 was higher than that in cluster 2. The results of immunocyte distribution showed that there were more immunocytes in cluster 1, but the dendritic cells were dormant. In cluster 2, the dendritic cells were activated, but the cells with lethality were less. Thus, we suspect that there is something wrong with the antigen presentation or immune checkpoint.
In our study, the results of pathway prediction showed that the expression of immune cell activation related pathways in cluster 1 was significantly higher than that in cluster 2. Therefore, we speculated that this may lead to more immune cells in cluster 1 than in cluster 2. In addition, the function of checkpoint in cluster 1 was also enriched in apoptosis, JAK/STAT signaling pathway, TNF-α/NF κ B signaling pathway, and STAT5 signaling pathway. The continuous activation of JAK/STAT signaling pathway was closely related to many immune and inflammatory diseases, which was closely related to the recognition of tumor cells by immune cells and the immune escape process of tumor [23]. Taking NK cells as an example, the early maturation, development, and functional activation of NK cells were strictly regulated by cytokines of JAK/STAT pathway [24]. IL-2 and IL-15 promoted NK cell homeostasis, proliferation, and function through STAT5 transduction [25] NFκB signaling pathway was also involved in the regulation of JAK/STAT pathway on immune cells [18].
We screened the immune checkpoint-related genes, BIRC3, G0S2, CCR7, CPS1, CLEC7A, LILRB1, CCR2, HLA-DOB, and CD1E, and detected the effect of these genes on the survival rate of patients. When the risk index of these genes increased, the survival rate of patients decreased. After literature search, it was found that these genes can be used as therapeutic targets for cancer. For example, birc3 was associated with chemoimmunotherapy resistance, and its inactivation also affects tumor cells that depend on NFκB pathway to survive [26]. Wang et al. found that hypoxia could induce birc3 expression through HIF1 alpha signal transduction mechanism in glioblastoma cells [27]. The sensitivity of G0/G1 transition gene 2 (G0S2) breast cancer cells to tamoxifen was relatively increased, which makes G0S2 an antitumor breast cancer target and biomarker of recurrence and therapeutic response [28, 29]. CCR7 and CD163 could be used as markers of macrophage polarization in lung cancer microenvironment [30]. We speculated that these genes may regulate the activity of immune cells in tumor microenvironment. After that, we will further explore the specific regulatory role of these genes on immune cells. We verified the clinical changes and related pathway changes of LUAD patients after the rise of the risk score of nine genes. We found that the high-risk group showed lower TMB. Ouyang et.al. showed that TMB was related to the immunotherapy response of colon adenocarcinoma. In the group with high TMB level, higher infiltration of CD8+ T cells, CD4+ T cells, and other immune cells could be observed in the cancer tissue [31]. This suggests that when the risk score is increased, the distribution of immune cells in the tumor may decrease correspondingly, leading to immune escape of tumor cells. In our results, the risk score was positively correlated with KARs pathway activation. KARS gene was a proto oncogene. Smoking could cause KARS mutation and carcinogenesis. Studies have shown that curcumin can be used as a sensitizer to overcome the resistance of NSCLC patients with wild-type EGFR and/or KRAS mutations [32]. However, it is worth noting that p53 is a tumor suppressor protein, and gene therapy has been found to increase the expression of p53 [33]. The increased expression of p53 pathway in our experiment still needs further study.
Current radiotherapy and chemotherapy are difficult to meet the needs of the treatment of LUAD. Currently, bioinformatics technology has been used to screen biomarkers of LUAD, which was more targeted for the treatment of the disease. Song et al. established a gene model consisting of 30 immune-related genes to predict the prognosis of LUAD [34]. In this study, we established a nine-gene prognostic model for LUAD based on immune checkpoints. The IMvigor dataset verified its prognostic value. From the perspective of clinical application, nine key genes associated with immune checkpoints identified in this study may be easier to be detected in clinical practice.
There are still some limitations in this study. First, our data comes from a public database, and potential selection bias cannot be ruled out. Second, the robustness of our results needs to be further validated in prospective studies. In addition, as there are not enough clinical samples, we have not tested this model in clinical trials. Therefore, we will clarify the biological function of the predicted genes on the basis of the present work in combination with experimental verification.
5. Conclusion
In conclusion, we identified a nine-gene signature based on immune checkpoints that may be used to aid prognostic analysis in patients with LUAD. Our study may provide several valuable molecular targets for immunotherapy of LUAD.
Acknowledgments
This study was funded by National Natural Science Foundation of China (nos. U20A20408 and 82074450), Natural Science Foundation of Hunan Province (no. 2020JJ4066), Hunan Province Research and Innovation Projects for Postgraduates (no. CX20190541), Hunan University Undergraduate Research Learning and Innovative Experiment Project (no. 201609030114), and Hospital Level Project of Hunan Brain Hospital (nos. 2018B01 and 2018B14).
Contributor Information
Xuefei Tian, Email: txf1718@163.com.
Liang Li, Email: superliliang@126.com.
Data Availability
The data of this study were taken from the TCGA and GEO databases.
Conflicts of Interest
The authors declare that there are no conflicts of interest regarding the publication of this paper.
Supplementary Materials
Figure S1: cluster analysis of lung adenocarcinoma. (A) Consensus matrix, K = 2. (B) Consensus matrix, K = 3. (C) Consensus matrix, K = 4. (D) Empirical CDFs corresponding to the entries of consensus matrix for K = 2, 3,… and 10. Figure S2: the expression of immune process related proteins in clusters 1 and 2. (A) Antigen present. (B) Ligand. (C) Receptor. (D) Coinhibitor. (E) Costimulator. (F) Cell adhesion. (G) Others. Figure S3: Kaplan–Meier survival curves showed the effectiveness of the risk prediction model in IMvigor immunotherapy dataset.
References
- 1.Santarpia M., Aguilar A., Chaib I., et al. Non-small-cell lung cancer signaling pathways, metabolism, and PD-1/PD-L1 antibodies. Cancers. 2020;12(6) doi: 10.3390/cancers12061475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang H., Guo L., Chen J. Rationale for lung adenocarcinoma prevention and drug development based on molecular biology during carcinogenesis. OncoTargets and Therapy. 2020;13:3085–3091. doi: 10.2147/ott.s248436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Spella M., Stathopoulos G. T. Immune resistance in lung adenocarcinoma. Cancers. 2021;13(3) doi: 10.3390/cancers13030384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kennedy L. B., Salama A. K. S. A review of cancer immunotherapy toxicity. CA: A Cancer Journal for Clinicians. 2020;70(2):86–104. doi: 10.3322/caac.21596. [DOI] [PubMed] [Google Scholar]
- 5.van der Leun A. M., Thommen D. S., Schumacher T. N. CD8+ T cell states in human cancer: insights from single-cell analysis. Nature Reviews Cancer. 2020;20(4):218–232. doi: 10.1038/s41568-019-0235-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Met Ö., Jensen K. M., Chamberlain C. A., Donia M., Svane I. M. Principles of adoptive T cell therapy in cancer. Seminars in Immunopathology. 2019;41(1):49–58. doi: 10.1007/s00281-018-0703-z. [DOI] [PubMed] [Google Scholar]
- 7.Xia A., Zhang Y., Xu J., Yin T., Lu X.-J. T cell dysfunction in cancer immunity and immunotherapy. Frontiers in Immunology. 2019;10:p. 1719. doi: 10.3389/fimmu.2019.01719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu X. N., Su D., Mei Y.-D., et al. Identified lung adenocarcinoma metabolic phenotypes and their association with tumor immune microenvironment. Cancer Immunology, Immunotherapy. 2021 doi: 10.1007/s00262-021-02896-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang Y., Zheng J. Functions of immune checkpoint molecules beyond immune evasion. Advances in Experimental Medicine and Biology. 2020;1248:201–226. doi: 10.1007/978-981-15-3266-5_9. [DOI] [PubMed] [Google Scholar]
- 10.Darvin P., Toor S. M., Sasidharan Nair V., Elkord E. Immune checkpoint inhibitors: recent progress and potential biomarkers. Experimental & Molecular Medicine. 2018;50(12):1–11. doi: 10.1038/s12276-018-0191-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hellmann M. D., Paz-Ares L., Bernabe Caro R., et al. Nivolumab plus ipilimumab in advanced non-small-cell lung cancer. The New England Journal of Medicine. 2019;381(21):2020–2031. doi: 10.1056/NEJMoa1910231. [DOI] [PubMed] [Google Scholar]
- 12.Wartewig T., Kurgyis Z., Keppler S., et al. PD-1 is a haploinsufficient suppressor of T cell lymphomagenesis. Nature. 2017;552(7683):121–125. doi: 10.1038/nature24649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang H., Dai Z., Wu W., et al. Regulatory mechanisms of immune checkpoints PD-L1 and CTLA-4 in cancer. Journal of Experimental & Clinical Cancer Research. 2021;40(1):p. 184. doi: 10.1186/s13046-021-01987-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smith K. M., Desai J. Nivolumab for the treatment of colorectal cancer. Expert Review of Anticancer Therapy. 2018;18(7):611–618. doi: 10.1080/14737140.2018.1480942. [DOI] [PubMed] [Google Scholar]
- 15.Blum A., Wang P., Zenklusen J. C. SnapShot: TCGA-analyzed tumors. Cell. 2018;173(2):p. 530. doi: 10.1016/j.cell.2018.03.059. [DOI] [PubMed] [Google Scholar]
- 16.Monti S., Tamayo P., Mesirov J. P., Golub T. R. Consensus clustering: a resampling-based method for class discovery and visualization of gene expression microarray data. Machine Learning. 2003;52(1):91–118. doi: 10.1023/a:1023949509487. [DOI] [Google Scholar]
- 17.Newman A. M., Liu C. L., Green M. R., et al. Robust enumeration of cell subsets from tissue expression profiles. Nature Methods. 2015;12(5):453–457. doi: 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fisher D. A. C., Miner C. A., Engle E. K., et al. Cytokine production in myelofibrosis exhibits differential responsiveness to JAK-STAT, MAP kinase, and NFκB signaling. Leukemia. 2019;33(8):1978–1995. doi: 10.1038/s41375-019-0379-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Subramanian A., Tamayo P., Mootha V. K., et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ritchie M. E., Phipson B., Wu D., et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Research. 2015;43(7):p. e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Robin X., Turck N., Hainard A., et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics. 2011;12(1):p. 77. doi: 10.1186/1471-2105-12-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blanco B., Domínguez-Alonso C., Álvarez-Vallina L. Bispecific immunomodulatory antibodies for cancer immunotherapy. Clinical Cancer Research. 2021 doi: 10.1158/1078-0432.ccr-20-3770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Owen K. L., Brockwell N. K., Parker B. S. JAK-STAT signaling: a double-edged sword of immune regulation and cancer progression. Cancers. 2019;11(12) doi: 10.3390/cancers11122002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gotthardt D., Trifinopoulos J., Sexl V., Putz E. M. JAK/STAT cytokine signaling at the crossroad of NK cell development and maturation. Frontiers in Immunology. 2019;10:p. 2590. doi: 10.3389/fimmu.2019.02590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gotthardt D., Putz E. M., Grundschober E., et al. STAT5 is a key regulator in NK cells and acts as a molecular switch from tumor surveillance to tumor promotion. Cancer Discovery. 2016;6(4):414–429. doi: 10.1158/2159-8290.cd-15-0732. [DOI] [PubMed] [Google Scholar]
- 26.Frazzi R. BIRC3 and BIRC5: multi‐faceted inhibitors in cancer. Cell & Bioscience. 2021;11(1):p. 8. doi: 10.1186/s13578-020-00521-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang D., Berglund A. E., Kenchappa R. S., MacAulay R. J., Mulé J. J., Etame A. B. BIRC3 is a biomarker of mesenchymal habitat of glioblastoma, and a mediator of survival adaptation in hypoxia-driven glioblastoma habitats. Scientific Reports. 2017;7(1):p. 9350. doi: 10.1038/s41598-017-09503-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yim C. Y., Bikorimana E., Khan E., et al. G0S2 represses PI3K/mTOR signaling and increases sensitivity to PI3K/mTOR pathway inhibitors in breast cancer. Cell Cycle. 2017;16(21):2146–2155. doi: 10.1080/15384101.2017.1371884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Páez-Pérez E. D., Llamas-García M. L., Benítez-Cardoza C. G., Montero-Morán G. M., Lara-González S. Bioinformatic analysis and biophysical characterization reveal structural disorder in G0S2 protein. ACS Omega. 2020;5(40):25841–25847. doi: 10.1021/acsomega.0c03171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kwiecień I., Polubiec-Kownacka M., Dziedzic D., Wołosz D., Rzepecki P., Domagała-Kulawik J. CD163 and CCR7 as markers for macrophage polarization in lung cancer microenvironment. Central-European Journal of Immunology. 2019;44(4):395–402. doi: 10.5114/ceji.2019.92795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ouyang R., Li Z., Peng P., et al. Exploration of the relationship between tumor mutation burden and immune infiltrates in colon adenocarcinoma. International Journal of Medical Sciences. 2021;18(3):685–694. doi: 10.7150/ijms.51918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen P., Huang H.-P., Wang Y., et al. Curcumin overcome primary gefitinib resistance in non-small-cell lung cancer cells through inducing autophagy-related cell death. Journal of Experimental & Clinical Cancer Research. 2019;38(1):p. 254. doi: 10.1186/s13046-019-1234-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kanapathipillai M. Treating p53 mutant aggregation-associated cancer. Cancers. 2018;10(6) doi: 10.3390/cancers10060154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Song Q., Shang J., Yang Z., et al. Identification of an immune signature predicting prognosis risk of patients in lung adenocarcinoma. Journal of Translational Medicine. 2019;17(1):p. 70. doi: 10.1186/s12967-019-1824-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: cluster analysis of lung adenocarcinoma. (A) Consensus matrix, K = 2. (B) Consensus matrix, K = 3. (C) Consensus matrix, K = 4. (D) Empirical CDFs corresponding to the entries of consensus matrix for K = 2, 3,… and 10. Figure S2: the expression of immune process related proteins in clusters 1 and 2. (A) Antigen present. (B) Ligand. (C) Receptor. (D) Coinhibitor. (E) Costimulator. (F) Cell adhesion. (G) Others. Figure S3: Kaplan–Meier survival curves showed the effectiveness of the risk prediction model in IMvigor immunotherapy dataset.
Data Availability Statement
The data of this study were taken from the TCGA and GEO databases.