

Abstract
A number of cancer drugs activate innate immune pathways in tumor cells but unfortunately also compromise anti-tumor immune function. We discovered that inhibition of Carm1, an epigenetic enzyme and co-transcriptional activator, elicited beneficial anti-tumor activity in both cytotoxic T cells and tumor cells. In T cells, Carm1 inactivation substantially enhanced their anti-tumor function and preserved memory-like populations required for sustained anti-tumor immunity. In tumor cells, Carm1 inactivation induced a potent type 1 interferon response that sensitized resistant tumors to cytotoxic T cells. Substantially increased numbers of dendritic cells, CD8 T cells and NK cells were present in Carm1-deficient tumors, and infiltrating CD8 T cells expressed low levels of exhaustion markers. Targeting of Carm1 with a small molecule elicited potent anti-tumor immunity and sensitized resistant tumors to checkpoint blockade. Targeting of this co-transcriptional regulator thus offers an opportunity to enhance immune function while simultaneously sensitizing resistant tumor cells to immune attack.
Keywords: Cancer immunology, in vivo CRISPR screen, resistance, T cell exhaustion, type I interferon response, CARM1
Introduction
Many cancer drugs have been developed that induce apoptosis of tumor cells, for example by inducing DNA damage or inhibiting key signaling pathways required for cell proliferation (1). While such drugs can induce substantial tumor shrinkage, recurrence is a major challenge due to outgrowth of drug-resistant tumor cells. The immune system could potentially target residual disease, but many of these tumor cell-targeted drugs also compromise immune cell survival/function or the production of immune cells by the hematopoietic system. For example, chemotherapy drugs not only kill dividing tumor cells, but also rapidly dividing hematopoietic precursors and immune cells (2,3).
Chemotherapy-induced DNA damage can induce activation of innate immune pathways in tumor cells, including the cGAS – STING pathway (4). The cGAS enzyme is activated by cytosolic double-stranded DNA, resulting in the synthesis of the cyclic dinucleotide cGAMP which activates the STING receptor and thereby induces a powerful type 1 interferon response through the IRF3 transcription factor. Importantly, type 1 interferons also induce maturation of dendritic cells, a key step for T cell-mediated immunity (5). Some chemotherapy drugs are being used in combination with immunotherapy agents. For example, the combination of nab-paclitaxel and a PD-L1 blocking mAb was recently approved by the FDA for the treatment of metastatic triple-negative breast cancer (TNBC) but only a small fraction of treated patients benefitted from this combination regimen compared to monotherapy with nab-paclitaxel (6). It is important to develop tumor cell-targeted drugs which enhance rather than compromise immune function.
The present study focuses on CARM1, an arginine methyltransferase that introduces asymmetric methylation of arginine residues in histone H3 and other chromatin-associated proteins. Asymmetric methylation refers to a highly specific modification in which two methyl groups are attached to one of the two nitrogen atoms of the arginine side chain (7). CARM1 acts as a transcriptional co-activator for nuclear hormone receptors and other transcription factors. It is recruited to chromatin by a member of the p160 family of proteins which also recruits the p300/CBP histone acetyltransferases. Following recruitment, CARM1 enhances the activity of this co-activation complex by methylation of arginine residues in p160, p300/CBP and histone H3 (on residues H3R17 and H3R26) (8–10). Overexpression of CARM1 mRNA has been reported for many human cancer types, and in breast and prostate cancers it serves as a co-activator of transcription for estrogen alpha and androgen receptors (11,12).
Using an in vivo CRISPR/Cas9 screen, we discovered that inactivation of the Carm1 gene in T cells enhanced their anti-tumor function and increased the pool of tumor-infiltrating memory-like T cells which are known to be required for sustained immunity. Recent work demonstrated that effector T cell populations are only maintained in tumors when sufficient numbers of tumor-specific memory-like cells are present in the microenvironment (13,14). Inactivation of Carm1 in tumor cells elicited a potent T cell-dependent immune attack associated with greatly increased infiltration of tumors by CD8 T cells and dendritic cells. These data demonstrate that targeting of Carm1 induces potent tumor immunity by sensitizing resistant tumors to immune attack and enhancing anti-tumor T cell function.
Results
Discovery of Carm1 as a negative regulator of tumor-infiltrating T cells
We performed an in vivo CRISPR/Cas9 screen in tumor-specific T cells to discover negative regulators of anti-tumor immunity. A gRNA library targeting epigenetic regulators was delivered into CD8 T cells using a lentiviral vector. These T cells originated from mice transgenic for Cas9 and the OT-I T cell receptor (TCR), thus yielding a gene-edited library of T cells with defined antigen specificity (Fig. 1A). A total of 426 genes representing annotated epigenetic regulators were evaluated using three gRNA pools (5 gRNAs/gene plus 100 negative control gRNAs). Edited T cells were transferred to immunocompetent mice bearing subcutaneous B16F10 melanomas which expressed the Ova antigen recognized by the OT-I TCR. Targeting of key negative regulators enhanced T cell proliferation/survival within tumors and thus resulted in enrichment of the corresponding gRNAs. Representation of gRNAs was quantified after 10 days by deep sequencing of the gRNA cassette in T cells isolated from tumors or a control organ (spleen). gRNAs targeting the positive control genes Pdcd1 and Cblb (encoding PD-1 and Cbl-b proteins, respectively) were among the top enriched gRNAs in each of the three pools, demonstrating that key negative regulators could be reproducibly identified (Fig. 1B–C, Table S1). The top hit from the primary screen was the Carm1 gene, which encodes an arginine methyltransferase that introduces asymmetric dimethylation of histone H3 (H3R17 and H3R26 residues) and other nuclear proteins. This effect was specific for Carm1 and not seen for genes encoding other arginine methyltransferases: while gRNAs targeting Carm1 were enriched tumor-infiltrating T cells relative to spleen, gRNAs targeting Prmt1, Prmt2, Prmt5 and Prmt6 were depleted; also, no enrichment was observed for gRNAs targeting Prmt3, Prmt7 and Prmt8. We validated these results using a targeted gRNA library representing 31 top candidate genes, two positive control genes (Pdcd1 and Cblb) and a set of control gRNAs. Carm1 was again identified as the top hit in this validation screen (Supplementary Fig. S1A, Table S1).
Figure 1. Carm1 is an epigenetic inhibitor in tumor-specific T cells.
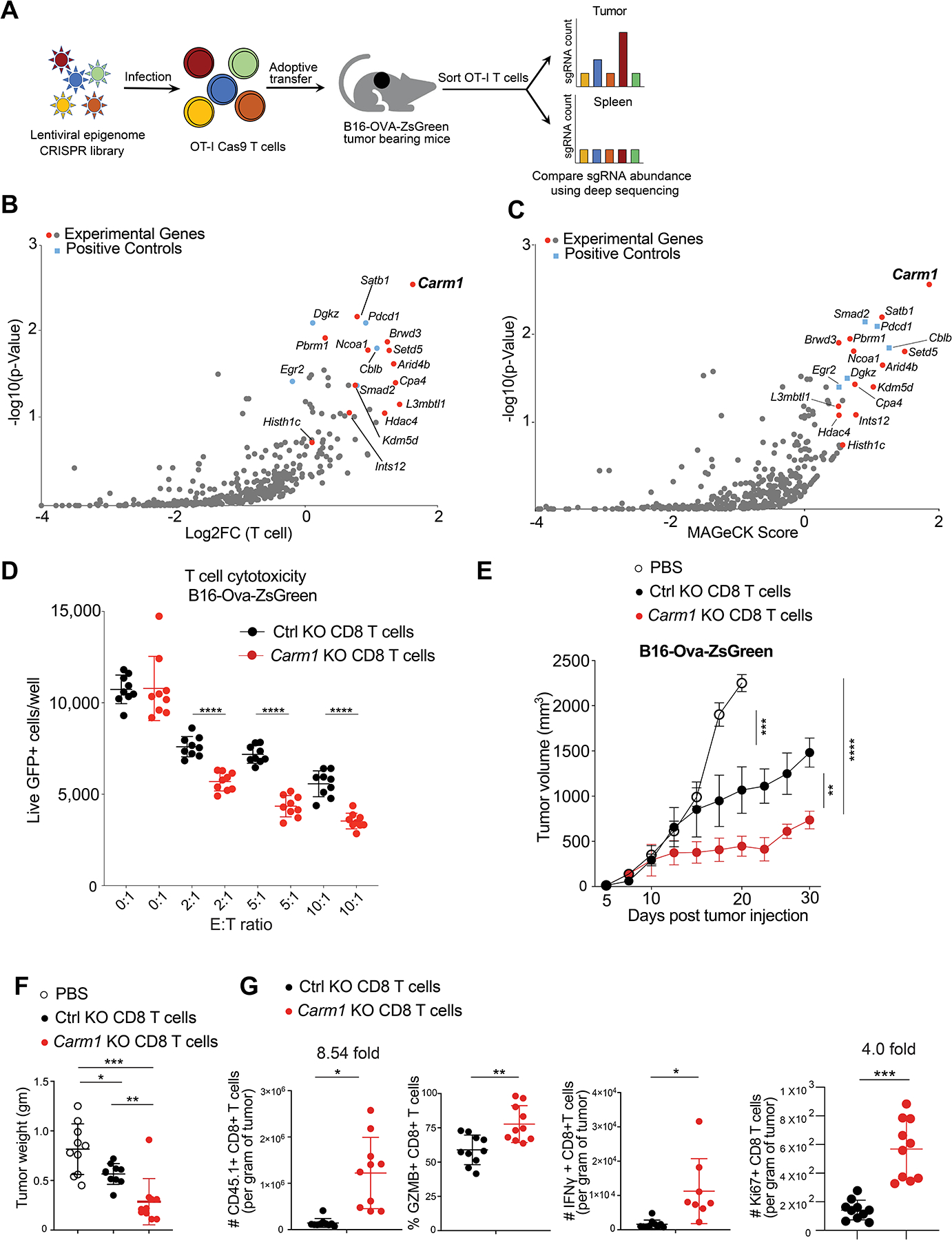
A. Experimental design for in vivo discovery of epigenetic regulators that inhibit CD8 T cell accumulation in tumors.
B. In vivo CRISPR screen with epigenetic gRNA library in tumor specific CD8 T cells. gRNA quantification in CD8 T cells was compared in tumors (experimental site) and spleens (control organ) (log2 fold change). Major experimental genes and positive control genes were highlighted in red and blue, respectively.
C. MAGeCK (Model-based Analysis of Genome-Wide CRIPSR-Cas9 knockout) analysis of in vivo CRISPR screen data; MAGeCK score provides integrated readout for strength of gene effects.
D. T cell cytotoxicity assay with Carm1-KO and control-KO OT-I CD8 T cells. CD8 T cells were edited by electroporation with Cas9 protein and bound gRNA, and cells were grown in IL-15 + IL-7 containing T cell media for 5 days. T cells were co-cultured with B16F10-OVA-ZsGreen tumor cells at indicated effector to target (E: T) ratios (n=8–10/replicates per condition); 24 hours later live GFP-positive tumor cells were counted using a Celigo image cytometer. Data are representative of three experiments and shown as mean ± SEM. ****p < 0.0001, by unpaired two-sided Mann-Whitney test.
E. Anti-tumor activity of adoptively transferred Carm1-KO or control-KO OT-I CD45.1 CD8 T cells. B16-OVA-ZsGreen tumor cells (0.1×106) were implanted subcutaneously. On day 7 post tumor cell inoculation, edited CD8 T cells (1×106) were transferred via tail vein injection. Tumor size was recorded; n=8–10 mice per group.
F. Tumor weights 7 days following adoptive T cell transfer for experiment shown in E.
G. Flow cytometry analysis of tumor-infiltrating Carm1-KO or control-KO CD8 T cells following adoptive transfer of edited OT-I CD45.1 CD8 T cells (n=10 mice/group) with gating on CD45.1 and CD8 T cell markers. Quantification of CD8 T cell infiltration and expression of effector (granzyme B, IFNγ) as well as proliferation (Ki-67) markers
Carm1-KO T cells were generated for functional experiments by electroporation of OT-I T cells with ribonucleoprotein complexes (RNPs) composed of Cas9 protein and bound gRNAs. This transient editing procedure was effective, as shown by sequencing of genomic DNA and loss of Carm1 protein following editing with two different gRNAs (Supplementary Fig. S1B–C). A cytotoxicity assay demonstrated that Carm1-KO T cells were more effective in killing B16F10-Ova melanoma cells compared to control edited T cells (Fig. 1D and Supplementary Fig. S1D–F). Following co-culture with B16F10-Ova tumor cells, Carm1-KO compared to control-KO T cells expressed higher levels of the CD69 activation marker, the granzyme B cytotoxicity protein and the cytokines IL-2, IFNγ and TNFα (Supplementary Fig. S1G–H) and showed enhanced antigen-induced proliferation (Supplementary Fig. S1I). These data demonstrated that Carm1 was a negative regulator of tumor-specific T cells.
Carm1 inhibition in CD8 T cells enhances their anti-tumor function
Carm1-KO T cells were found to confer more effective anti-tumor immunity than control-KO CD8 T cells against B16F10-OVA tumors (Fig. 1E–F and Supplementary Fig. S2A). Flow cytometry analysis revealed greatly enhanced tumor infiltration by Carm1-KO compared to control-KO CD45.1+ CD8+ T cells, including increased accumulation of T cells that expressed the effector molecules granzyme B and IFNγ as well as the proliferation marker Ki67 (Fig. 1G and Supplementary Fig. S2B). We confirmed enhanced anti-tumor immunity by Carm1-KO T cells using a second gRNA (Supplementary Fig. S2C–D).
Seven days following editing, OT-I CD8 T cells were co-cultured for 24 hours with B16F10-Ova tumor cells. RNA-seq analysis demonstrated striking changes in gene expression, including upregulation of 1,143 genes and downregulation of 1,199 genes in Carm1-KO compared to control T cells (Fig. 2A–B, Table S2). Upregulated genes encoded chemokine receptors that mediate T cell recruitment into tumors (Cxcr3) and key genes required for maintenance of memory T cell populations (transcription factors Tcf7, Myb, Bcl6; surface receptors Itgae and Cd27). Downregulated genes included those associated with terminal differentiation (Klrg1), inhibition of cytokine signaling (Socs1, Socs3) and T cell dysfunction within tumors (Egr2). To validate the RNA-seq results, we performed qPCR analysis using two different gRNAs and found that Tcf7, Myb, Bcl6 and Itgae (associated with T cell memory) were upregulated in Carm1-KO compared to control-KO CD8 T cells while Klrg1 (associated with terminal differentiation) and Havcr2 (associated with dysfunction) were downregulated in Carm1-KO T cells (Fig. 2C).
Figure 2. Carm1 inhibition in CD8 T cells enhances their anti-tumor function.
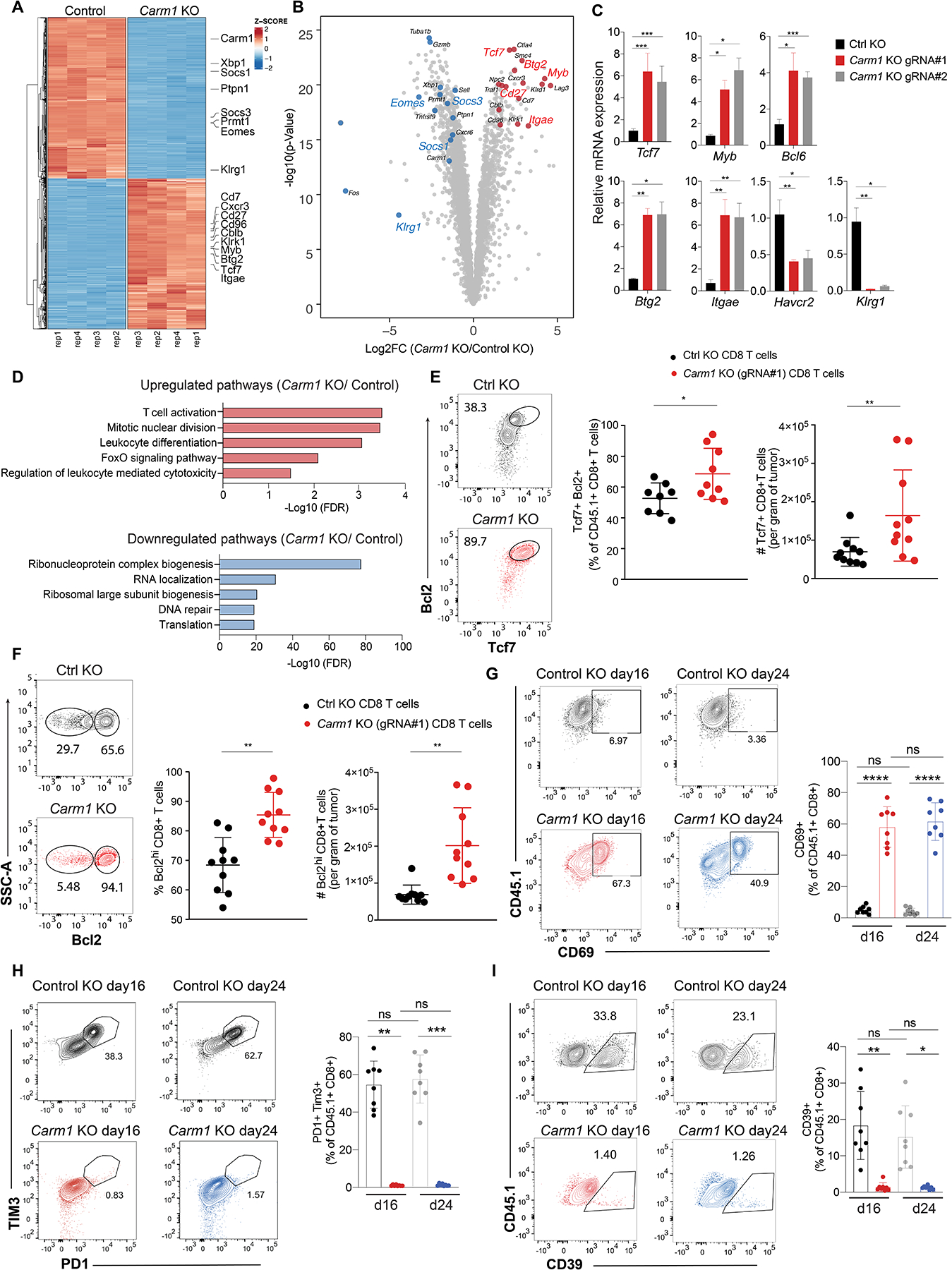
A. NA-seq analysis of differentially expressed genes in Carm1-KO or control-KO OT-I CD8 T cells co-cultured for 24 hours with B16F10-Ova tumor cells (four biological replicates per condition). Color code represents Z-scores for differential gene expression.
B. Volcano plot of all differentially expressed genes between Carm1-KO and control-KO OT-I CD8 T cells. Statistical significance (log10 adjusted P value) was plotted against log2 fold change of gene expression levels (Carm1-KO/control-KO cells).
C. RT-qPCR analysis of Tcf7, Myb, Bcl6, Btg2, Itgae, Havcr2 and Klrg1 mRNA levels in Carm1-KO and control-KO CD8 T cells (targeting of Carm1 with two different gRNAs, triplicate measurements).
D. Gene Ontology (GO) analysis of significantly upregulated/downregulated pathways in Carm1-KO versus control-KO T cells.
E. Tumor-infiltrating Carm1-KO or control-KO CD8 T cells following adoptive transfer of edited OT-I CD45.1 CD8 T cells (n=10 mice/group) with gating on CD45.1 and CD8 T cell markers. Quantification of Tcf7+ T cells with high Bcl2 protein levels and Tcf7+ CD8 T cells.
F. Quantification of Bcl2 high tumor-infiltrating Carm1-KO or control-KO CD8 T cells.
G-I. Tumor-infiltrating Carm1-KO or control-KO CD8 T cells were analyzed 16 or 24 days following adoptive transfer of edited OT-I CD45.1 CD8 T cells (n=8 mice/group) with gating on CD45.1 and CD8 T cell markers. Quantification of CD8 T activation marker (CD69) (G) and markers of T cell exhaustion (H-I).
Data shown are representative of two experiments. Two-way ANOVA was used to determine statistical significance for time points when all mice were viable for tumor measurement. Graphs shown represent data summarized as mean ± S.D. and were analyzed by unpaired two-sided Mann-Whitney test, ****P < 0.0001; ***P < 0.001; **P < 0.01; *P < 0.05.
Gene set enrichment analysis (GSEA) highlighted ‘T cell activation’, ‘mitotic nuclear division’, ‘Foxo signaling’ pathway and ‘regulation of leukocyte mediated cytotoxicity’ among the top pathways for genes overexpressed in Carm1-KO compared to control T cells. Downregulated pathways related to RNA biology, protein translation and DNA repair (Fig. 2D). Genes in the Foxo signaling pathway identified by GSEA included Bcl6 and Il7r, both of which play important roles in T cell memory. A large fraction of tumor-infiltrating Carm1-KO CD8 T cells were positive for Tcf7 protein and also had high levels of Bcl-2, consistent with an increased pool of memory-like cells for Carm1-KO compared to control-KO T cells (Fig. 2E). Also, the number of tumor-infiltrating Bcl-2hi cells was substantially higher for Carm1-KO compared to control-KO CD8 T cells (Fig. 2F). We investigated T cell persistence by studying tumor-infiltrating Carm1-KO and control-KO CD8 T cells at early (d16) and late (d24) time points following T cell transfer. A significantly larger population of Carm1-KO CD8 T cells expressed the CD69 activation marker at both time points (Fig. 2G). Conversely, only a small percentage of Carm1-KO T cells co-expressed the PD-1 and Tim-3 inhibitory receptors or CD39, which are markers of exhausted T cells (Fig. 2H–I). These data indicated that targeting of Carm1 in CD8 T cells enhanced their anti-tumor function and maintained a substantial population of tumor-infiltrating T cells that expressed memory markers.
Inactivation of Carm1 gene in tumor cells elicits tumor immunity
Analysis of RNA-seq data from 1,208 human cancer cell lines (Cancer Cell Line Encyclopedia, CCLE) revealed high-level CARM1 expression across a diverse range of human cancer cell lines (Fig. 3A). We therefore interrogated the role of Carm1 in tumor cells by inactivating the Carm1 gene in murine cancer cell lines resistant to checkpoint blockade, including B16F10 melanoma and 4T1 breast cancer models (Fig. 3B–C). In vitro proliferation and survival of B16F10 and 4T1 cells was not affected by inactivation of the Carm1 gene, but in vivo growth of Carm1-KO tumor cells was greatly diminished compared to control-KO tumor cells (Supplementary Fig. S2E, Fig. 3D and Supplementary Fig. S3A). Importantly, depletion of CD8 T cells restored the in vivo growth of Carm1-KO B16F10 cells, indicating that Carm1 inactivation in tumor cells elicited potent T cell-mediated tumor immunity (Fig. 3D and Supplementary Fig. S3A). This conclusion was validated by comparing B16F10 tumor growth in immunocompetent wild-type and T cell-deficient T cell receptor α (Tcrα) KO mice (Fig. 3E). Inactivation of the Carm1 gene in the 4T1 model of triple-negative breast cancer (TNBC) and the MC38 colon cancer model also substantially slowed tumor growth and conferred a survival benefit (Fig. 3F–H). Importantly, the number of spontaneous pulmonary metastasis was substantially reduced following orthotopic implantation of Carm1-KO compared to control-KO 4T1 tumor cells (Fig. 3G).
Figure 3. Inactivation of Carm1 gene in tumor cells elicits tumor immunity.
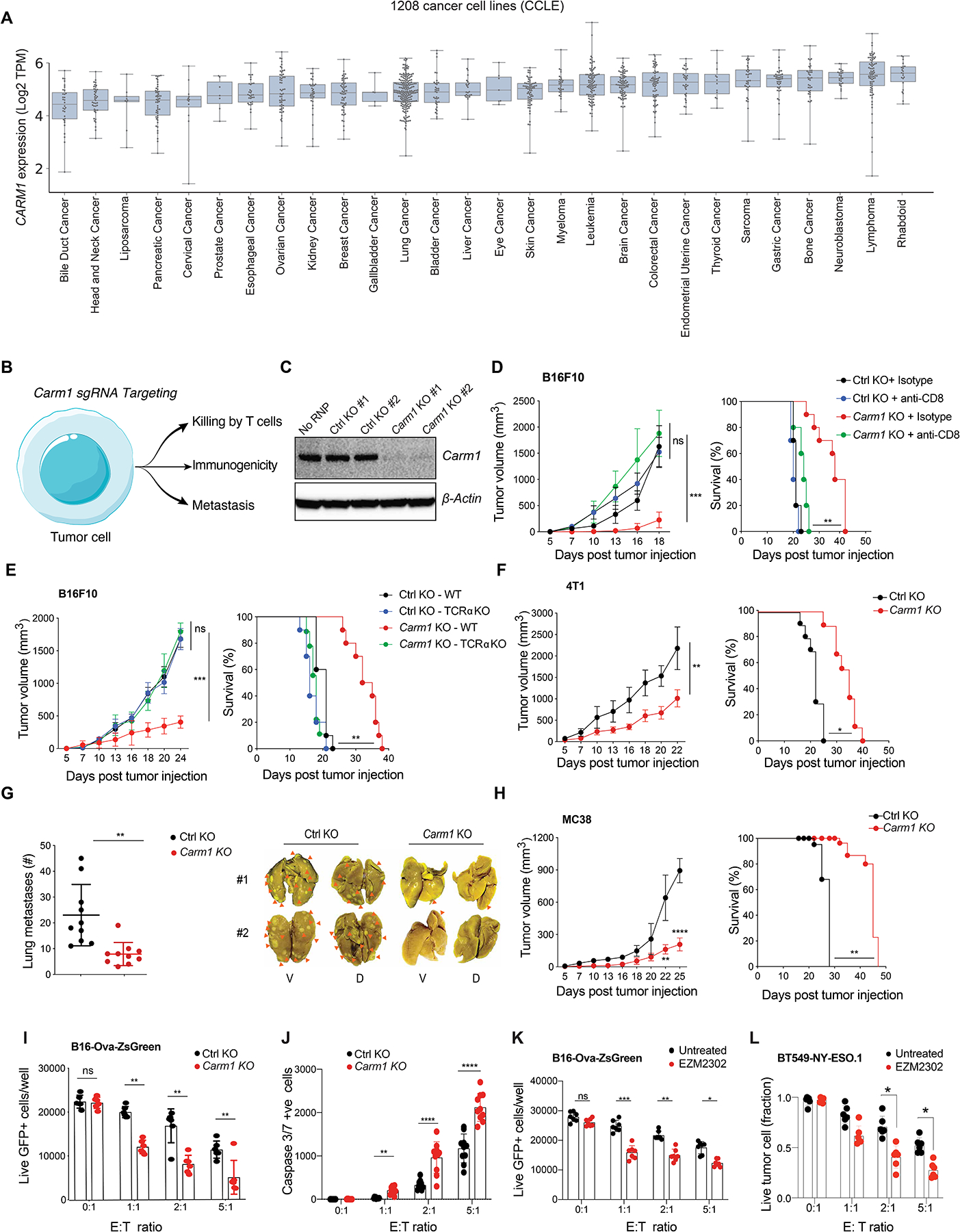
A. CARM1 mRNA levels in a diverse panel of 1,208 cancer cell lines from the Cancer Cell Line Encyclopedia (CCLE). Tumor cell lines were grouped based on cancer type.
B. Strategy for targeting Carm1 in tumor cells to study impact on T cell-mediated tumor immunity.
C. Western blot analysis of Carm1 protein in B16F10 melanoma cells following electroporation with RNPs composed of Cas9 protein and bound gRNAs (control, Carm1); two different control and Carm1 gRNAs were evaluated.
D. Growth of Carm1-KO and control-KO B16F10 tumors (left) and survival of tumor bearing mice (right). Mice (n=8–10/group) were treated with CD8 depleting or isotype control antibodies. This in vivo phenotype was confirmed with a second Carm1 gRNA (Fig. S3A).
E. Growth of Carm1-KO tumors in T cell-deficient mice. Carm1-KO and control-KO B16F10 tumor cells (0.2×106) were implanted into immunocompetent or immunodeficient (Tcra KO) mice (n=8–10 mice/group); tumor growth (left) and survival (right) were recorded.
F. Growth of Carm1-KO or control-KO 4T1 tumor cells following implantation into the mammary fat pad (n=8–10 mice/group); tumor growth (left) and survival (right) were recorded
G. Quantification of spontaneous lung metastases formed by Carm1-KO or control-KO 4T1 tumors in immunocompetent mice. Representative images of lung metastases (V, ventral; D, dorsal) (right).
H. Tumor growth (left) and survival (right) following implantation of Carm1-KO and control-KO MC38 tumor cells (n=8–10 mice/group).
I. T cell cytotoxicity assay with Carm1-KO or control-KO B16F10-OVA-ZsGreen tumor cells. Tumor cells were co-cultured for 24 hours with OT-I CD8 T cells at indicated effector to target (E:T) ratios (n=8–10 replicates per condition).
J. Induction of tumor cell apoptosis (Carm1-KO or control-KO B16F10-OVA-ZsGreen cells) by CD8 T cells (as described in I), measured with a Caspase-3/7 dye at different E:T ratios (n = 8–10 replicates /group).
K. Sensitization of tumor cells to T cells with a CARM1 inhibitor. B16F10-OVA-ZsGreen tumor cells were pretreated with CARM1 inhibitor (EZM2302, 0.1μM) for 24 hours. Vehicle or inhibitor treated tumor cells were co-cultured with OT-I CD8 T cells at indicated E:T ratios (n=7–8 replicates/condition).
L. T cell cytotoxicity assay with human BT549 TNBC and human CD8 T cells that expressed a NY-ESO-1 TCR. Tumor cells were pretreated with CARM1 inhibitor (EZM2302, 0.1μM) for 24 hours (n=7–10 replicates/group); numbers of surviving tumor cells were quantified after 24 hours of co-culture.
Two-way ANOVA was used to determine statistical significance for tumor measurements at time points when all mice were alive. Statistical significance for survival of mice in each treatment group were calculated by Log-rank (Mantel-Cox) test. Bar graphs represent data summarized as mean ± S.E.M. and were analyzed by unpaired two-sided Mann-Whitney test. Data shown are representative of three experiments. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns (non-significant).
Next, we used an imaging-based T cell cytotoxicity assay to examine whether inactivation of the Carm1 gene sensitized tumor cells to killing by CD8 T cells. Indeed, Carm1-KO B16F10-Ova tumor cells were significantly more sensitive to T cell-mediated cytotoxicity than control-KO tumor cells, as shown by quantification of surviving live ZsGreen+ tumor cells or dying tumor cells labelled with a caspase 3/7 cell death reporter (Fig. 3I–J). A high-affinity small molecule inhibitor of CARM1 was reported (EZM2302) (15). Pre-treatment of B16F10-Ova tumor cells with EZM2302 also sensitized them to CD8 T cells (Fig. 3K). This CARM1 inhibitor also sensitized a human TNBC cell line to cytotoxic T cells. BT549 TNBC cells were co-cultured with human CD8 T cells that expressed a TCR specific for a NY-ESO-1 peptide presented by HLA-A2:01. Pre-treatment of these tumor cells with the CARM1 inhibitor (EZM2302) enhanced CD8 T cell-mediated cytotoxicity (Fig. 3L). These data demonstrated that inactivation of the Carm1 gene in tumor cells induced potent T cell-mediated immunity and that Carm1 deficient tumor cells were more sensitive to T cell-mediated cytotoxicity.
Innate immune activation in Carm1 deficient tumor cells
RNA-seq analysis demonstrated striking changes in the transcriptome of Carm1-KO versus control-KO B16F10 tumor cells. In particular, we observed increased expression of many interferon response genes in Carm1-KO compared to control-KO tumor cells, even though these tumor cells had not been exposed to exogenous type 1 interferon or IFNγ (Fig. 4A, Supplementary Fig. S3B, Table S3). Gene set enrichment analysis (GSEA) also highlighted transcriptional activation of the IFN α/γ and p53 pathways in Carm1-KO tumor cells (Fig. 4B, Supplementary Fig. S3C). In human melanoma, a higher type 1 interferon gene expression signature was found to correlate with increased CD8 T cell infiltration (16). Importantly, there was little overlap in the genes that were differentially expressed as a consequence of Carm1 gene inactivation in tumor cells compared to T cells (Fig. 4C). In particular, the p53 pathway and interferon α/γ response pathways were only transcriptionally activated in Carm1-KO tumor cells but not Carm1-KO T cells. Validation by RT-qPCR using two Carm1 targeting gRNAs demonstrated that multiple interferon stimulated genes (ISGs) including Irf7, CCl5, Cxcl10, Ifit1, Oasl1, and Tap1 were expressed at two to seven-fold higher levels in Carm1-KO compared to control-KO tumor cells (Fig. 4D). Importantly, pre-treatment of B16F10 cells with the CARM1 inhibitor EZM2302 also significantly increased the mRNA levels of these ISGs (Fig. 4E, Supplementary Fig. S3D). EZM2302 also induced the expression of a similar set of ISGs in human breast and melanoma cancer cell lines (Supplementary Fig. S3E–F).
Figure 4. Innate immune activation in Carm1 deficient tumor cells.
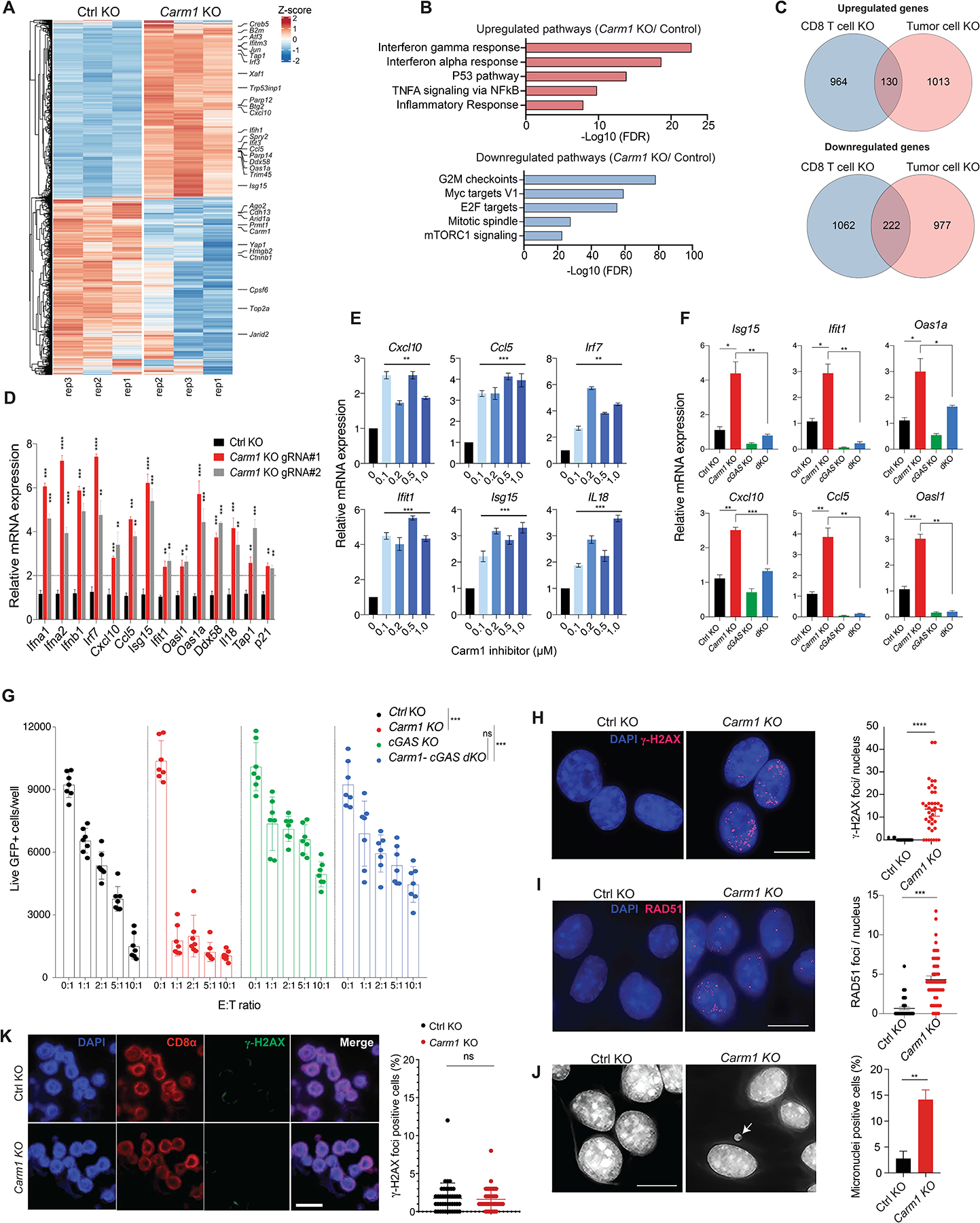
A. RNA-seq analysis of differentially expressed genes in Carm1-KO and control-KO B16F10 tumor cells (n=3/group). Data are representative of two independent experiments.
B. Gene Ontology (GO) analysis of significantly upregulated/downregulated genes in Carm1-KO compared to control-KO B16F10 tumor cells.
C. Venn diagram representing number of overlapping differentially expressed genes in Carm1-KO tumor and CD8 T cells.
D. Validation of interferon inducible genes (ISGs) by RT-qPCR in Carm1-KO compared to control-KO B16F10 cells (n=3/group).
E. RT-qPCR analysis of ISG mRNA levels following treatment of B16F10 cells with CARM1 inhibitor EZM2302 (0.1 – 1μM) or solvent control for 7 days (n=3/group).
F. Expression of selected ISGs in control-KO, Carm1-KO, cGAS-KO and Carm1/cGAS double-KO (dKO) B16F10 cells analyzed by RT-qPCR (n=3/group).
G. T cell cytotoxicity assay with control-KO, Carm1-KO, cGAS-KO and Carm1/cGAS dKO B16F10 cells. Tumor cells were co-cultured with OT-I CD8 T cells at indicated E:T ratios for 24 hours (n=7–10 replicates/condition); live GFP positive tumor cells were counted using a Celigo image cytometer. Data are shown as mean ± SEM. ***p < 0.001, by unpaired two-sided Mann-Whitney test.
H-I. dsDNA damage in Carm1-KO versus control-KO B16F10 tumor cells based on labeling with γH2AX (H) and RAD51 (I) Abs. Representative immunofluorescence images (left) of γH2AX or RAD51 antibody labeling (red); nuclei labeled with DAPI. Quantification of number of γH2AX or RAD51 foci/nucleus (right). Data are shown as mean ± SEM, ***p < 0.001, by unpaired two-sided Mann-Whitney test. Scale bar – 10μM.
J. Detection of micronuclei in Carm1-KO and control-KO B16F10 tumor cells. DNA was labeled with DAPI; representative images (left) and quantification of cells positive for micronuclei (right). Data are shown as mean ± SEM, **p < 0.01, by unpaired two-sided Mann-Whitney test. Scale bar – 10μM.
K. Analysis of Carm1-KO versus control-KO CD8+ T cells for dsDNA damage. OT-I CD8 T cells were plated 7 days posted editing using CRIPSR-Cas9 as previously described and stained for CD8α, γH2AX and DAPI. Representative immunofluorescence images of CD8α antibody labeling (red), γH2AX antibody labeling (green); nuclei stained with DAPI (blue). Scale bar – 20μM.
Data shown in D-J are representative of three independent experiments. Bar graphs represent data summarized as mean ± S.E.M. and were analyzed by unpaired two-sided Mann-Whitney test. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns (non-significant). Error bars for all qPCR data represent SD with three replicates per group.
The gene sets induced by type 1 interferons and IFNγ overlap substantially, and IFNγ secreted by activated T cells is an essential cytokine for protective tumor immunity (17). We therefore investigated whether inactivation of the Carm1 gene in tumor cells enhanced their transcriptional response to IFNγ. Indeed, Carm1-KO B16F10 and 4T1 cells showed a heightened transcriptional response to IFNγ for all tested ISGs compared to control-KO tumor cells (Supplementary Fig. S3G–H). IFNγ stimulation also induced higher levels of STAT1 phosphorylation in Carm1-KO compared to control-KO B16F10 cells, even though total STAT1 and STAT2 protein levels were similar between the cell lines (Supplementary Fig. S4A). IFNγ inhibited the proliferation of Carm1-KO tumor cells more significantly than control-KO cells (Supplementary Fig. S4B). Carm1-KO tumor cells expressed moderately higher levels of MHC class I protein (H2-Kb) both in the absence and following stimulation with IFNγ; PD-L1 levels were slightly higher for Carm1-KO compared to control-KO tumor cells following IFNγ stimulation (Supplementary Fig. S4C–D). The enhanced transcriptional response of Carm1-KO tumor cells to IFNγ was attenuated when the Ifnar1 gene was inactivated in Carm1-KO tumor cells implicating enhanced type 1 interferon signaling in this process (Supplementary Fig. S4E). These data indicated that Carm1-KO tumor cells showed an increased responsiveness to IFNγ, an important cytokine secreted by activated T cells.
We next investigated which innate immune sensor was required for the interferon gene expression signature identified in Carm1-KO tumor cells. Inactivation of the Mavs gene, which encodes an essential adaptor protein for the double-stranded RNA sensors Rig-I and Mda-5, had no impact on expression of ISGs (Supplementary Fig. S5A–B). In striking contrast, inactivation of the Cgas gene greatly diminished mRNA levels of ISGs, indicating that the cGAS enzyme represented a key element in the innate immune pathway activated in Carm1-KO tumor cells (Fig. 4F and Supplementary Fig. S5C–D). We also tested whether activation of cGAS could explain the enhanced sensitivity of Carm1-KO tumor cells to cytotoxic T cells. Indeed, Carm1-KO tumor cells were highly sensitive to CD8 T cells, while inactivation of Cgas in Carm1-KO tumor cells rendered them substantially more resistant to cytotoxic T cells (Fig. 4G). Inactivation of Cgas (without inactivation of Carm1) also rendered B16F10-Ova tumor cells more resistant to T cells compared to wild-type B16F10-Ova cells. These data indicated that the enhanced sensitivity of Carm1-KO compared to control-KO tumor cells to cytotoxic T cells required a functional cGAS-STING pathway, and that a lower level of activation of the cGAS-STING pathway in control-KO tumor cells was relevant for T cell-mediated killing.
These results strongly suggested that inactivation of Carm1 induced a DNA damage response in tumor cells. DNA damage induces rapid phosphorylation of histone H2AX (γH2AX) which provides a sensitive readout for double-stranded DNA (dsDNA) breaks (18). Immunofluorescence analysis showed that a substantial fraction of Carm1-KO B16F10 cells had multiple nuclear foci labeled with a γH2AX antibody while such foci were detected in only a small percentage of control-KO tumor cells (Fig. 4H). This conclusion was confirmed by labeling with an antibody specific for RAD51, another marker for dsDNA breaks (Fig. 4I). dsDNA breaks can induce chromosome mis-segregation during mitosis and formation of cytosolic micronuclei (19). Such micronuclei tend to have fragile nuclear envelopes, resulting in exposure of their dsDNA to cGAS (20). DAPI+ micronuclei were detected in a significantly larger percentage of Carm1-KO compared to control-KO tumor cells (Fig. 4J). A subset of these cytosolic DAPI+ micronuclei were positive for cGAS when an epitope-tagged version of cGAS was expressed in tumor cells (Supplementary Fig. S5E). CARM1 inhibitor treatment also induced γH2AX foci accumulation in B16F10 melanoma cells (Supplementary Fig. S5F). Using a gRNA targeting an intergenic region in the mouse genome, we confirmed that CRISPR/Cas9 based gene editing (>1 week prior to analysis) did not result in substantial dsDNA damage or ISG expression (Supplementary Fig. S5G–H). These results demonstrated that inactivation of the Carm1 gene induced innate immune activation within tumor cells due to activation of the cGAS-STING pathway. CARM1 and p300 were previously shown to cooperate with BRCA1 and p53 to induce expression of the cell cycle inhibitor p21CIP1 (CDKN1A) (21). Failure of cell cycle inhibition following DNA damage can result in chromosome segregation during mitosis, formation of micronuclei and cGAS activation (22).
Interestingly, unlike tumor cells, Carm1 ablation did not induce dsDNA damage in CD8 T cells (Fig. 4K). Also, Carm1 inactivation resulted in distinct gene expression changes in T cells versus tumor cells (Fig. 4C, Fig. 2A–C), suggesting that Carm1 induced cell type specific consequences in T cells (enhanced function, preserved pool of memory-like cells) compared to tumor cells (DNA damage and induction of cGAS-STING signaling).
Carm1 inhibition overcomes resistance to checkpoint blockade
Many human tumor types resistant to checkpoint blockade with CTLA-4 or PD-1 mAbs are poorly infiltrated by CD8 T cells (‘cold tumors’). Poor infiltration by CD8 T cells is associated with an absence of a type 1 interferon gene expression signature (23,24). We hypothesized that treatment with a CARM1 inhibitor could be effective in checkpoint blockade-resistant tumors by enhancing the function of tumor-specific T cells and also increasing the sensitivity of tumor cells to cytotoxic T cells. B16F10 melanomas are resistant to monotherapy with CTLA-4 or PD-1 mAbs and even combination therapy with both checkpoint antibodies (25). Treatment of Carm1-KO tumors with CTLA-4 or PD1 blockade substantially reduced tumor growth and conferred a significant survival benefit (Fig. 5A and Fig. S6A). Importantly, small molecule-mediated Carm1 inhibition (EZM2302) also sensitized B16F10 melanomas to checkpoint blockade with a CTLA-4 mAb and resulted in a significant survival benefit (Fig. 5B). The inhibitor was administered at a dose of 150 mg/kg but optimization of drug dose for this application was not feasible due to limited availability of this compound. This inhibitor was previously only evaluated in an immunodeficient mouse model and shown to moderately slow the in vivo growth of a human multiple myeloma cell line (RPMI-8226) (15). The highly metastatic 4T1 model of TNBC is also resistant to CTLA-4 blockade. Inactivation of the Carm1 gene sensitized 4T1 tumors to CTLA-4 blockade and conferred a marked survival benefit when compared to the three other experimental groups (Fig. 5C). CTLA-4 mAb treated mice with Carm1-KO 4T1 tumors also showed a substantial reduction in the number of spontaneous lung metastases, again in comparison to all other treatment groups (Fig. 5D).
Figure 5. Carm1 inhibition overcomes resistance to checkpoint blockade.
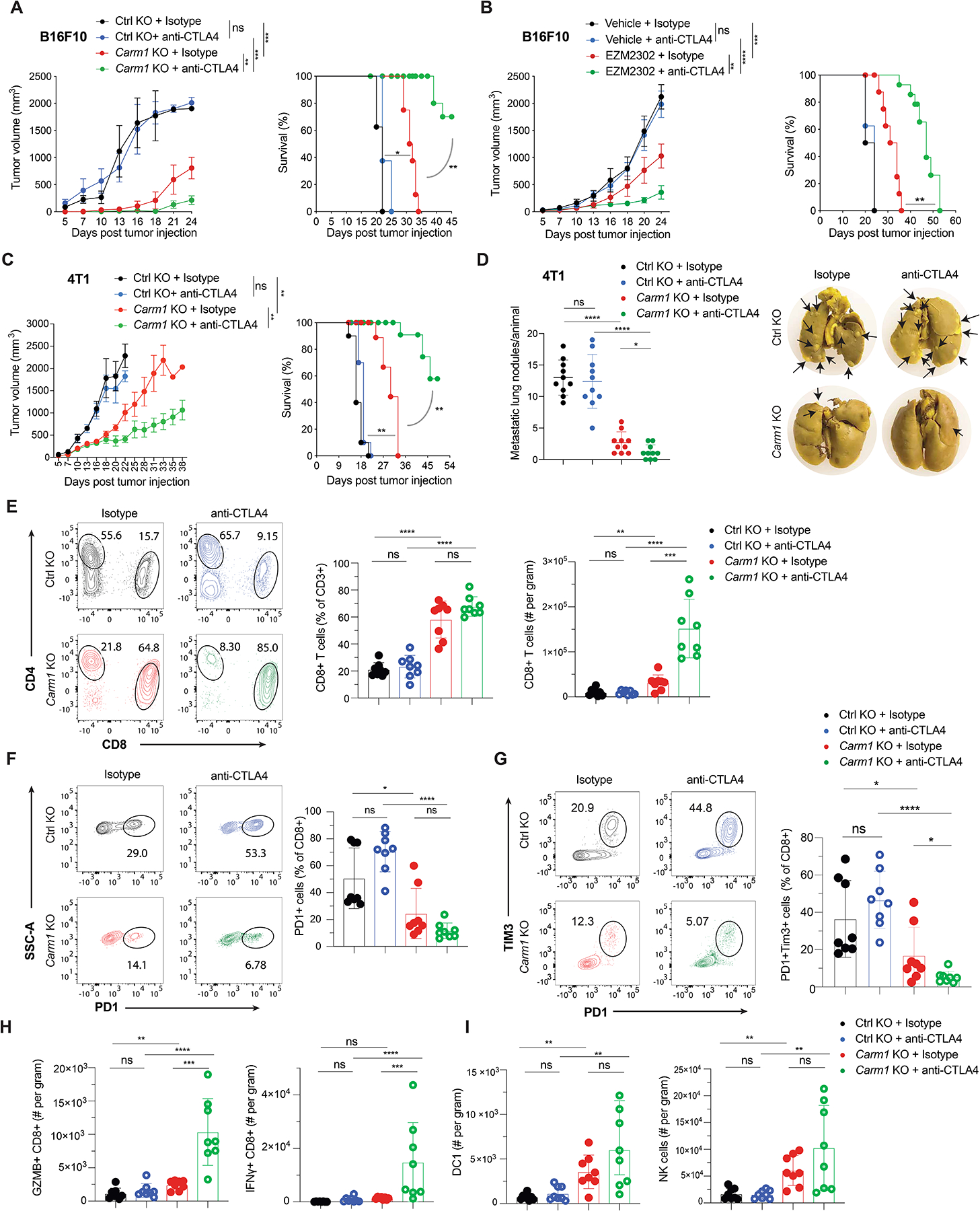
A. reatment of Carm1-KO or control-KO B16F10 tumors with CTLA-4 or isotype control antibodies (n=8 mice/group). Tumor growth (left) and survival of tumor bearing mice (right) are shown. Mice bearing comparable tumor volume (~50mm3) were randomized into different treatment groups.
B. Treatment of B16F10 tumors with CARM1 inhibitor (EZM2302) or vehicle control in combination with CTLA-4 or isotype control antibodies (n=8 mice/group). EZM2302 (150mg/kg b.i.d.) was orally administered for 2 weeks (days 7–21).
C. Treatment of Carm1-KO or control-KO 4T1 tumors with anti-CTLA-4 or isotype control antibodies (n=8 mice/group).
D. hange in number of spontaneous lung metastases (left) and representative images of lung metastases formed by 4T1 tumors treated as described in C (right) (n=8 mice/group).
E. Quantification of tumor-infiltrating CD8 T cells in Carm1-KO and control-KO B16F10 tumors (n=8 mice/group) following treatment with CTLA-4 or isotype control antibodies (day 18 post tumor cell implantation). Representative flow plots (left) and quantification of CD8 T cells as percentage of CD3+ cells and per gram of tumor (middle and right, respectively).
F-G. Quantification of PD-1 positive and PD-1/Tim-3 double positive tumor-infiltrating CD8+ T cells for experiment described in E.
H-I. Quantification of CD8 T cells expressing effector markers (GZMB and IFNγ), migratory cross-presenting DCs (CD45+/CD3-/F4/80-/CD11c+/MHC-IIhigh/CD103+/CD11b-) and NK cells (CD45+/CD3-/CD49b+) per gram of tumor for experiment described in E.
Data shown are representative of two experiments. Two-way ANOVA was used to determine statistical significance for time points when all mice were viable for tumor measurement. Log-rank (Mantel-Cox) test was used to determine statistical significance for survival of mice. Bar graphs represent data summarized as mean ± S.E.M. and were analyzed by unpaired two-sided Mann-Whitney test, ****P < 0.0001; ***P < 0.001; **P < 0.01; *P < 0.05; n.s., non-significant.
Analysis of Carm1-KO B16F10 tumors showed a striking increase in the number of infiltrating CD8 T cells and a substantial reduction in the percentage of CD8 T cells that expressed the inhibitory PD-1 and Tim-3 inhibitory receptors (Fig. 5E–G). In contrast, the number of tumor-infiltrating CD4 T cells (calculated per gram of tumor) was similar between all treatment groups, although the percentage of CD4 T cells in the total T cell pool was lower in Carm1-KO compared to control-KO tumors due to the striking increase in CD8 T cell accumulation (Supplementary Fig. S6B, Fig. 5E). CTLA-4 mAb treatment further enhanced CD8 T cell infiltration into Carm1-KO B16F10 tumors compared to the three other experimental groups and reduced the percentage of CD8 T cells double positive for the PD-1 and Tim-3 inhibitory receptors (Fig. 5E–G). Also, a substantially larger percentage of CD8 T cells from Carm1-KO compared to control-KO CTLA-4 treated tumors were positive for key functional markers including granzyme B and IFNγ (Fig. 5H and Supplementary Fig. S6C). Carm1-KO tumors (treated with control or CTLA-4 mAbs) were also infiltrated by a larger number of dendritic cells, including cross-presenting cDC1 cells, as well as NK cells, when compared to control-KO tumors treated with either mAb (Fig. 5I, Supplementary Fig. S6D). Similar changes were observed in Carm1-KO 4T1 tumors in particular following treatment with a CTLA-4 mAb (Supplementary Fig. S6E–I). These data demonstrated that tumor cell-intrinsic inactivation of the Carm1 gene induced multiple significant changes in the tumor microenvironment, including enhanced infiltration by CD8 T cells, NK cells and dendritic cells. Also, CD8 T cells expressed substantially lower levels of the PD-1 and Tim-3 inhibitory receptors consistent with a reduced level of T cell exhaustion.
We also investigated the impact of CARM1 inhibitor treatment on the tumor immune microenvironment, both as monotherapy as well as in combination with PD-1 or CTLA-4 mAbs (Supplementary Fig. S7, S8 and S9). In toxicity studies with this CARM1 inhibitor, we did not observe any histological changes in a comprehensive analysis of major organs or weight loss between the CARM1 inhibitor versus solvent control treatment groups (Supplementary Fig. S7A–B). CD8 T cell infiltration was substantially increased following monotherapy with the CARM1 inhibitor and was even higher when the inhibitor was combined with PD-1 or CTLA-4 mAbs (Supplementary Fig. S8A). In contrast, CD8 T cell infiltration was not increased following PD-1 or CTLA-4 monotherapy compared to the isotype control antibody group. We also observed a striking increase in the number of CD8 T cells (per gram of tumor) that expressed granzyme B, IL-2, IFNγ, and perforin in all three CARM1 inhibitor treatment groups (Supplementary Fig. S8B–E). In contrast, PD-1 expression by CD8 T cells was reduced in CARM1 inhibitor treatment groups compared the vehicle control group (Supplementary Fig. S8F). CARM1 inhibitor treatment did not change the number of tumor-infiltrating CD4 T cells or FoxP3+ Tregs. However, the CD8/FoxP3 Treg ratio was substantially increased in all three Carm1 treatment groups due to the increase in CD8 T cell infiltration (Supplementary Fig. S9A–D). Interestingly, tumor-infiltrating NK cells were also increased in all three CARM1 inhibitor treatment groups and were highest when the CARM1 inhibitor was combined with PD-1 or CTLA-4 mAbs. cDC1 cells were increased when the CARM1 inhibitor was combined with PD-1 or CTLA-4 mAbs, but no changes in macrophage numbers were detected across any of the treatment groups (Supplementary Fig. S9E). These data demonstrated that small molecule-mediated inhibition of Carm1 induced major favorable changes in the tumor immune microenvironment, in particular for CD8 T cells, NK cells and cDC1. These favorable changes were further enhanced when the CARM1 inhibitor was combined with PD-1 or CTLA-4 mAbs.
To examine whether re-expression of Carm1 in tumor cells reversed the phenotype induced by Carm1 inactivation, we introduced a Carm1 cDNA under the control of a doxycycline (Dox)-inducible promoter into Carm1-KO B16F10 tumor cells (Supplementary Fig. S10A–B). Dox treatment suppressed mRNA levels of IFNγ inducible genes in Carm1-KO cells, consistent with our finding that control-KO tumor showed lower responsiveness to IFNγ than Carm1-KO tumor cells (Supplementary Fig. S10C and S3G–H). Control-KO B16F10 tumor cells transduced with the empty vector showed rapid growth, regardless of whether mice were treated with vehicle or Dox (Supplementary Fig. S10D). As expected, Carm1-KO tumor cells transduced with the Carm1 vector but treated with vehicle grew slowly, with similar kinetics as Carm1-KO tumor cells transduced with the empty vector. In contrast, Dox treatment substantially accelerated growth of Carm1-KO tumor cells transduced with the Carm1 vector. Importantly, key aspects of the tumor microenvironment were also reversed by Dox-induced re-expression of Carm1 in Carm1-KO tumor cells, including the striking degree of CD8 T cell infiltration, lower levels PD-1 expression by CD8 T cells and the increase in cDC1 infiltration (Supplementary Fig. S11A–D).
Tdrd3 and Med12 are effector molecules of the Carm1 pathway
Carm1 deposits H3R17me2a and H3R26me2a methylarginine marks on histone tails that are recognized by the Tudor domain-containing protein Tdrd3 (26). Carm1 also methylates a number of other nuclear proteins, including Med12, a component of the regulatory arm of the Mediator complex (27). We found that inactivation of either Tdrd3 or Med12 genes in B16F10 cells increased mRNA levels of multiple ISGs, similar to inactivation of Carm1 (Fig. 6A–B, Supplementary Fig. S12A–D). Furthermore, this ISG gene expression signature was lost when the Cgas gene was also inactivated in Tdrd3-KO or Med12-KO B16F10 cells (Fig. 6A–B). Similar to Carm1 ablation, Tdrd3 or Med12 inactivation resulted in higher sensitivity to IFNγ and led to enhanced IFNγ-induced expression of ISGs (Supplementary Fig. S12E–F). Inactivation of genes encoding other Tdrd3 or Med12 associated proteins (Top3b, Top1 and Med13) did not result in a substantial increase in ISG mRNA levels except for some upregulation of ISGs in Med13-KO tumor cells (Supplementary Fig. S12G–I). Inactivation of Tdrd3 or Med12 genes in B16F10 tumor cells also resulted in accumulation of γH2AX positive nuclear foci and cytosolic micronuclei, as described above for Carm1-KO B16F10 cells (Fig. 6C–D). Finally, we found that Tdrd3-KO tumors showed substantially attenuated growth, and that this phenotype was again CD8 T cell dependent (Fig. 6E–F). CTLA-4 mAb treatment further inhibited the growth of Tdrd3-KO tumor cells and resulted in survival of a large fraction of the mice (Fig. 6G).
Figure 6. Tdrd3 and Med12 are downstream effectors of Carm1.
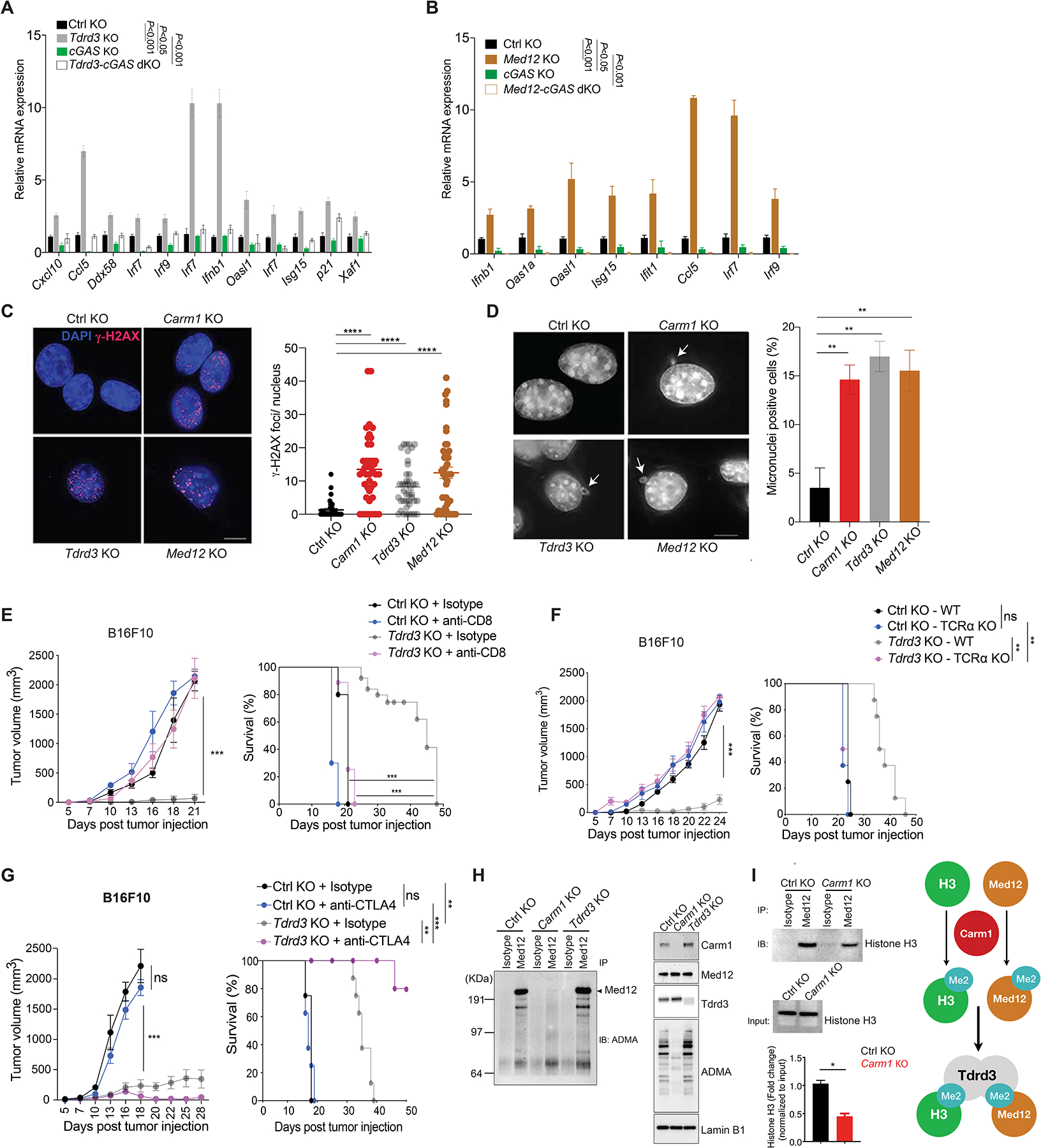
A. Comparison of ISG expression in control-KO, Tdrd3-KO, cGAS-KO and Tdrd3/cGAS dKO B16F10 cells by RT-qPCR (n=3/group).
B. Comparison of ISG expression in control-KO, Med12-KO, cGAS-KO and Med12/cGAS dKO B16F10 cells by RT-qPCR (n=3/group).
C. Immunofluorescence analysis of dsDNA damage by γH2AX antibody staining (red foci; nuclei labeled with DAPI) in control-KO, Carm1-KO, Tdrd3-KO and Med12-KO B16F10 tumor cells (left). Images for control-KO and Carm1-KO reshown from Fig. 4 to illustrate comparison to other KO tumor cell lines. Quantification of number of γH2AX foci/nucleus for each cell line (right). Scale bar – 10μM.
D. Analysis of micronuclei in control-KO, Carm1-KO, Tdrd3-KO and Med12-KO B16F10 tumor cells using DAPI as a DNA stain. Representative images (left) and quantification of percentage of cells with micronuclei (right). Scale bar – 10μM.
E. Tumor growth and survival of mice bearing control-KO and Tdrd3-KO B16F10 tumors. Mice were treated with CD8 T cell depleting or isotype control antibodies (n=8–10 mice/group).
F. Tumor growth and survival of control-KO and Tdrd3-KO B16F10 tumors in immunocompetent and T cell deficient (Tcra KO) mice (n=8–10 mice/group).
G. Tumor growth and survival of anti-CTLA-4 or isotype control antibody treated of mice bearing Tdrd3-KO or control-KO B16F10 tumors (n=8 mice/group).
H. Arginine methylation of Med12 by Carm1. Immunoprecipitation of Med12 protein from nuclear extracts of control-KO, Carm1-KO or Tdrd3-KO B16F10 tumor cells, followed by Western blot detection with an antibody specific for asymmetric dimethylation of arginine residues (ADMA, left). Western blot analysis of nuclear extracts from the same cell lines with antibodies for the indicated proteins (right).
I. Effect of Carm1 on interaction of Med12 with histone H3. Immunoprecipitation of Med12 protein from control-KO or Carm1-KO B16F10 tumor cells, followed by Western blot detection with histone H3 antibody. Input levels of histone H3 in immunoprecipitated samples is shown (middle); quantification of histone H3 bound to Med12 normalized to total histone H3 (bottom). Graph shows proposed biochemical interactions (right).
Two-way ANOVA was used to determine statistical significance for time points when all mice were viable for tumor measurement. Statistical significance for survival of mice in each treatment group was calculated by Log-rank (Mantel-Cox) test. Bar graphs represent data summarized as mean ± S.E.M. and were analyzed by unpaired two-sided Mann-Whitney test. Data are representative of three (A-H) and two (I) experiments, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns (non-significant).
Biochemical studies showed that Med12 was indeed a direct target of Carm1. When Med12 was immunoprecipitated from control-KO B16F10 cells, asymmetric methylation of arginine residues of Med12 could be readily detected by Western blot analysis (Fig. 6H), consistent with a previous report that identified Med12 as a target of Carm1 (27). This asymmetric methylation of arginine residues of Med12 was absent in Carm1-KO B16F10 tumor cells (but not in Tdrd3-KO tumor cells, as expected). Furthermore, immunoprecipitation of Med12 from nuclear lysates showed that less histone H3 was bound to Med12 in Carm1-KO compared to control-KO cells, indicating that Carm1 facilitated recruitment of Med12 to histone H3 (Fig. 6I). These results demonstrated that inactivation of the Tdrd3 and Med12 genes resulted in a similar immune-mediated phenotype as inactivation of the Carm1 gene.
We further investigated whether RNA Pol II mediated transcription was altered in Carm1-KO compared to control-KO tumor cells. Western blot analysis of the C-terminal domain (CTD) of RNA Pol II showed increased phosphorylation of Ser2 (p-Ser2, a mark of transcriptional elongation) (28) and Ser5 in nuclear lysates from Carm1-KO compared with control-KO cells (Supplementary Fig. S13A). We systematically investigated Pol II phosphorylation using mammalian native elongating transcript sequencing (mNET-Seq) (29). Inactivation of the Carm1 gene increased the normalized read density of p-Ser2 CTD Pol II tags relative total Pol II tags around immediate promoter regions of genes (Supplementary Fig. S13B–C). Also, ~25% of the genes upregulated in Carm1-KO compared to control-KO cells were associated with higher normalized RNA Pol II pSer2 status (Supplementary Fig. S13D). GSEA analysis of these overlap genes showed an enrichment in the p53 pathway, consistent with the data presented in Fig. 4 (Supplementary Fig. S13E). These data provided evidence for altered transcriptional regulation in Carm1 deficient tumor cells, consistent with the identification of Med12 as a Carm1 target. Furthermore, increased abundance of alternatively spliced genes (exon gains in 638 genes and exon losses in 708 genes) were found in Carm1-KO tumor cells. Pathways analysis using these alternatively spliced genes identified DNA repair, regulation of transcription and chromatin organization to be among top enriched pathways in Carm1-KO tumor cells (Supplementary Fig. S13F). Co-transcriptional R-loop structures have been linked to genome instability and are thought to be resolved through TDRD3 and TOP3B (30). We used S9.6-based DNA:RNA Immunoprecipitation approaches (DRIP-seq) (31) to determine whether R-loops were increased as a result of Carm1-KO. No significant trend towards R-loop gains was observed (Supplementary Fig. S13G), arguing that co-transcriptional R-loops over genic regions were not a source of the genomic instability observed in Carm1-KO cells.
Relevance of CARM1 in human cancers
We next investigated the relevance of CARM1 in human cancers, including a potential role of CARM1 in tumor cells. We found that gene signatures for a number of important pathways were downregulated in human cancer cell lines with high versus low CARM1 mRNA levels, including the p53, MHC class I antigen presentation and interferon-related pathways (Fig. 7A, Table S4). These results suggested that CARM1 also served an important role in human tumor cells by inhibiting important immune pathways.
Figure 7. Relevance of CARM1 in human cancers.
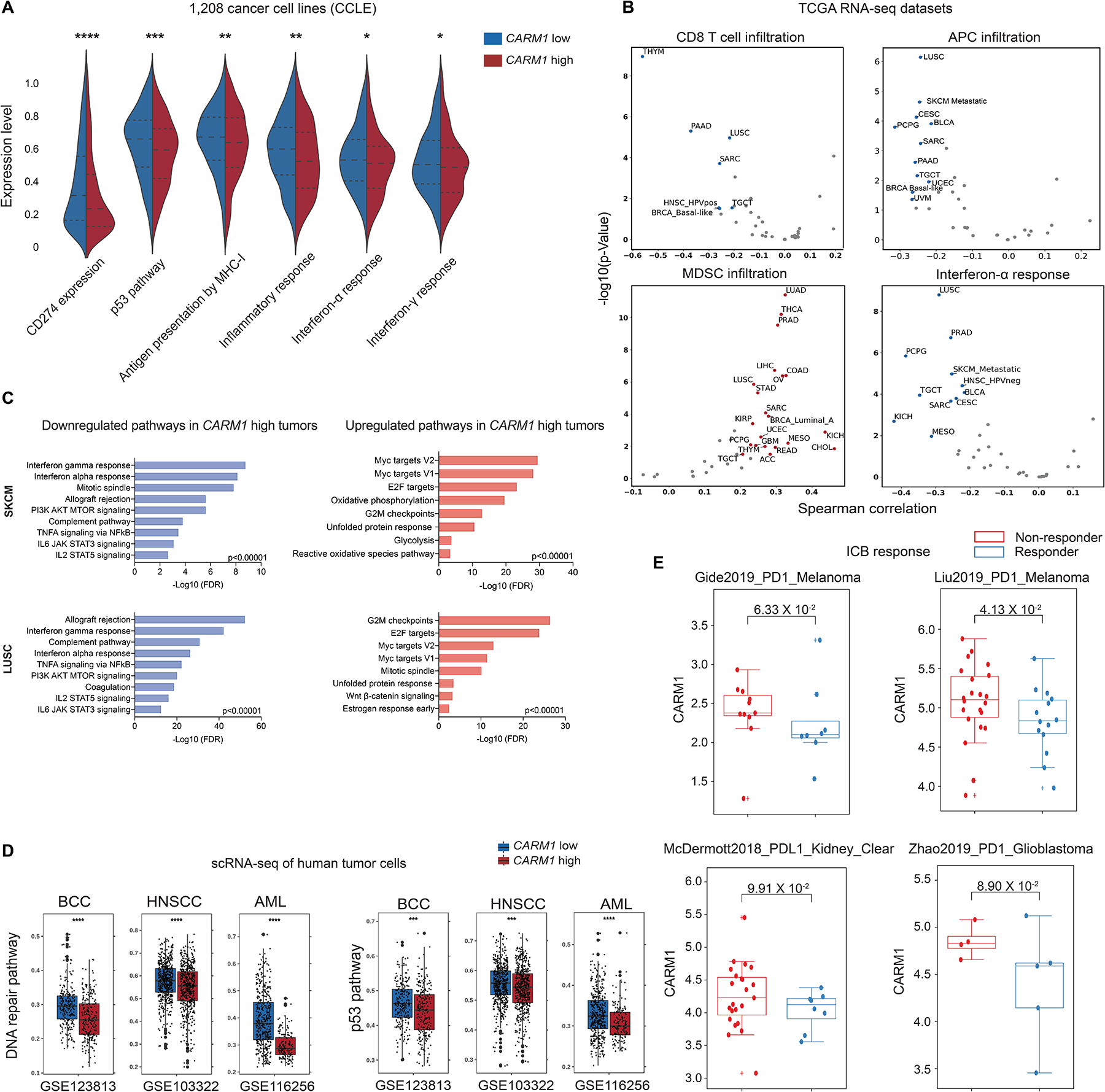
A. Analysis of indicated pathways across a diverse panel of 1,208 human cancer cell lines (Cancer Cell Line Encyclopedia, CCLE). Symmetric violin plots illustrate stratifications for CARM1 high and low cell lines using median expression levels.
B. Analysis of TCGA RNA-seq data across human cancer types. Correlation of CARM1 mRNA levels with indicated pathways. Plots show Spearman’s correlation and estimated statistical significance for indicated pathways in different cancer types adjusted for tumor purity. Each dot represents a cancer type in TCGA.
C. Gene Ontology (GO) analysis of significantly upregulated/downregulated genes in CARM1 high tumor cells in skin cutaneous melanoma (SKCM, N=442 patients) and lung squamous cell carcinoma (LUSC, N=363 patients) datasets (TCGA PanCancer Atlas).
D. Analysis of scRNA-seq data of malignant cells from three human cancer cohorts (GSE123813: basal cell carcinoma; GSE103322: head and neck cancer; GSE116256: AML). Scores for DNA repair, and p53 pathways are shown. Data were stratified by CARM1 high and low groups using median expression levels. Statistical comparisons were made using two-sided unpaired Mann-Whitney tests.
E. Correlation of CARM1 mRNA levels with response to ICB (immune checkpoint blockade) in cancer patients treated with PD-1 or PD-L1 mAbs. The analysis is shown for tumors with low (<median) MED12 mRNA levels. CARM1 mRNA levels did not correlate with response to ICB in tumors with high (>median) MED12 mRNA levels. The p-values were inferred by Mann-Whitney U test.
Analysis of The Cancer Genome Atlas (TCGA) RNA-seq datasets indicated that CARM1 may regulate immune responses in human cancers. In a large number of human cancer types, CARM1 mRNA levels were negatively correlated with gene signatures of tumor infiltration by T cells and APCs as well as response to IFNγ and IFNα (Fig. 7B, Supplementary Fig. S14A). Furthermore, CARM1 mRNA levels positively correlated with a gene signature for tumor infiltration by immunosuppressive myeloid derived suppressor cells (MDSC). To better understand the effect of CARM1 expression in human tumors, we performed an analysis of differentially regulated pathways for two of the cancer types investigated above [TCGA skin cutaneous melanoma (SKCM) and lung squamous cell carcinoma (LUSC)]. Tumors with high versus low CARM1 mRNA levels showed downregulation of interferon gamma and alpha response pathways, while pathways related to Myc targets V1/V2 and G2M checkpoints were upregulated (Fig. 7C).
RNA-seq data from TCGA enable analysis of a large number of tumors but lack single cell resolution. We therefore also investigated the malignant cell populations in independent human tumor scRNA-seq datasets from multiple cancer types. Again, we found that high CARM1 mRNA levels negatively correlated with p53 and DNA repair pathways as well as key immune pathways (APC infiltration and response to IFNα/IFNγ); high CARM1 mRNA levels were also associated with reduced survival in a number of human cancer types (Fig. 7D, Supplementary Fig. S14B–C, and S15A–B).
We also examined CARM1 gene expression in clinical immune checkpoint blockade (ICB) datasets, analyzing 16 clinical trial datasets. Low expression of CARM1 per se was not associated with ICB response. Given that MED12 was a downstream target of CARM1, we investigated the role of MED12 and CARM1 mRNA levels on response to PD-1 pathway inhibition: we first separated the cohorts based on MED12 mRNA levels, and then examined the correlation of CARM1 mRNA levels in MED12 low (<median) and MED12 high (>median) cohorts. In four clinical cohorts, we found that ICB responders had significantly lower expression levels of both MED12 and CARM1 compared to non-responders (two trials of PD-1 blockade in melanoma, one trial of PD-L1 blockade in kidney cancer, one trial of PD-1 blockade in glioblastoma) (Fig. 7E). In addition, higher levels of a Carm1-KO gene expression signature were associated with response to ICB in four clinical cohorts; this signature was obtained from RNA-seq analysis of Carm1-KO versus control-KO B16F10 tumor cells (Supplementary Fig. S16A, Table S5). In TCGA RNA-seq datasets, this Carm1-KO signature positively correlated with gene signatures of CTL infiltration, antigen processing and presentation as well as interferon γ response (Supplementary Fig. S16B). These data provided evidence that CARM1 expression was associated with major immune pathways in human cancers.
Discussion
In this study, we report a novel immunotherapy approach for tumors resistant to checkpoint blockade. Many human cancers fail to respond to PD-1 and/or CTLA-4 antibodies, and these resistant tumors frequently lack significant infiltration by CD8 T cells (‘cold’ tumors) (32). In essence, such tumors are not sufficiently immunogenic to elicit a spontaneous T cell response that can be enhanced by checkpoint blockade. Priming of a tumor-specific cytotoxic T cell response requires recruitment of cross-presenting dendritic cells (cDC1) into tumors, followed by activation and migration of dendritic cells into tumor-draining lymph nodes where they prime naïve CD8 T cells (33). These cellular events require activation of innate immune signals that induce production of key chemokines and cytokines, including type 1 interferons that activate dendritic cells. We show here that this barrier to effective cancer immunotherapy can be overcome by inhibition of Carm1, an epigenetic regulator. Interestingly, Carm1 inhibition in either tumor cells or T cells had major beneficial effects on T cell-mediated tumor immunity. Inactivation of Carm1 in tumor cells induced a type 1 interferon response and resulted in substantially increased numbers of tumor infiltrating CD8 T cells, NK cells and dendritic cells. Also, these tumor-infiltrating CD8 T cells showed higher functionality and lower expression of exhaustion markers. Inactivation of Carm1 in T cells preserved a substantial pool of tumor-infiltrating memory-like CD8 T cells with enhanced anti-tumor function. These findings are significant because T cell exhaustion and loss of tumor-infiltrating memory populations are considered to represent major barriers to protective tumor immunity. We provide substantial evidence that this pathway is relevant in human cancers. Analysis of TCGA data demonstrated high CARM1 mRNA levels across a wide range of human cancer types, including human cancers that have thus far been largely resistant to checkpoint blockade. CARM1 mRNA levels were negatively correlated with gene expression signatures of key immune pathways, including the MHC class I antigen presentation, type 1 interferon and IFNγ pathways. These human data are consistent with previous publications which demonstrated that a type 1 gene expression signature is associated with T cell inflamed (‘hot’) as opposed to non-T cell inflamed (‘cold’) tumors (32).
Why does inhibition of Carm1 elicit such distinct responses in T cells versus tumor cells? Inactivation of Carm1 in T cells greatly increased the CD8 T cell accumulation in tumors. RNA-seq analysis of Carm1-KO compared to control-KO T cells indicated that Carm1 inactivation reduced terminal effector differentiation (reduced expression of Klrg1) which is known to impair T cell-mediated tumor immunity. Rather, Carm1-KO T cells expressed higher levels of transcription factors critical for differentiation, self-renewal and persistence of memory T cells, including Tcf7 and Myb (34). A recent single-cell RNA-seq analysis of CD8 T cells in human melanomas demonstrated that higher expression of TCF7 by CD8 T cells predicted a positive outcome in patients treated with checkpoint blockade (35). Carm1-KO T cells expressed higher levels of Myb, which encodes a transcription factor that promotes memory T cell formation by transcriptional activation of Tcf7 and repression of Zeb2. Myb overexpression was previously shown to enhance CD8 T cell memory formation, poly-functionality and promote protective anti-tumor immunity (36). These data are consistent with the hypothesis that Carm1 acts as a co-transcriptional activator that promotes terminal differentiation of tumor-infiltrating T cells.
In contrast, inactivation of Carm1, Tdrd3 and Med12 in tumor cells resulted in induction of a type 1 interferon response that sensitized tumors to T cell-mediated attack. This type 1 interferon response was accompanied by DNA damage, formation of micronuclei and cGAS-STING activation. These findings are consistent with the previous finding that CARM1 cooperates with BRCA1 and p53 to induce expression of the cell cycle inhibitor p21CIP1 (CDKN1A) (21). Cell cycle progression despite the presence of double-stranded DNA breaks can result in chromosome mis-segregation during mitosis and the formation of micronuclei that activate the cGAS-STING pathway (37). Interestingly, no DNA damage was observed in T cells, even though these cells can undergo rapid proliferation following triggering of the T cell receptor. We considered the possibility that the DNA damage phenotype in tumor cells was caused by accumulation of genic R-loops but DRIP-seq did not demonstrate increased formation of R-loops in Carm1-KO compared to control-KO tumor cells. Many genes involved in the DNA damage response represent tumor suppressors and are inactivated in tumor cells due to mutations or epigenetic mechanisms. We hypothesize that Carm1 inactivation in tumor cells amplifies pre-existing DNA damage by interfering with p53-induced inhibition of cell cycle progression. This hypothesis could explain why Carm1 inhibition induces cGAS-STING activation in tumor cells but not T cells.
Some chemotherapy drugs can induce activation of the cGAS-STING pathway in tumor cells but targeting of the cell cycle is detrimental to hematopoietic precursors and proliferating tumor-specific T cells. Also, a number of small molecule STING agonists have been developed which are delivered by intra-tumoral injection (38). The approach presented here may be particularly relevant in the setting of metastatic disease resistant to checkpoint blockade by sensitizing tumor cells to T cells and improving persistence of cytotoxic T cells. Inactivation of the Carm1 gene in tumor cells combined with CTLA-4 blockade induced a substantial survival benefit in B16F10 melanoma and 4T1 TNBC models. Importantly, a small molecule inhibitor of Carm1 also showed synergy with a CTLA-4 blocking mAb in the B16F10 melanoma model that is resistant even to the combination of PD-1 and CTLA-4 mAbs. This inhibitor greatly increased tumor infiltration by CD8 T cells, NK cells and cross-presenting dendritic cells. These data provide the rationale for targeting of CARM1 in human cancers resistant to current immunotherapies. This approach may not only be useful for checkpoint blockade therapy, as illustrated here, but also other immunotherapy approaches in which T cells serve as key effector cells, including neoantigen-based cancer vaccines and CAR T cell therapies for solid tumor indications. In adoptive T cell therapies, CARM1 inhibition may not only sensitize resistant solid tumors to cytotoxic T cells but also enhance T cell memory and persistence, which are critical for sustained clinical benefit with such cellular therapies (39).
Methods
Cell lines
B16F10, 4T1, MC38 parental cell lines were purchased from American Type Culture Collection (ATCC). B16-OVA-ZsGreen cells were generated by lentiviral transduction of the parental line with a pHAGE expression vector driving expression of an N-terminally truncated variant of chicken ovalbumin (subcloned from pcDNA3-deltaOVA, Addgene plasmid #6459525). zsGreen+ cells were sorted to purity to establish the cell line. B16-OVA-zsGreen cells were validated for expression of zsGreen and cell surface presentation of the OVA peptide SIINFEKL in complex with H2-Kb. Furthermore, the cells were tested for their ability to activate OT-I CD8 T cells. Murine Carm1 cDNA was synthesized and cloned into pINDUCER21-ORF-EG (Addgene Plasmid # 46948) using gBlocks™ from IDT to generate the Dox-Carm1 construct. Empty vector or Dox- Carm1 transduced B16F10 control-KO or Carm1-KO cells were sorted for purity based on GFP expression. Dox-inducible Carm1 protein expression was confirmed by Western blotting. B16F10 and 4T1 cells were grown in DMEM and RPMI media, respectively, supplemented with 10% FBS and 1% Penicillin/Streptomycin. MC38 cells were grown in DMEM media supplemented with 10% fetal bovine serum (FBS), 100 IU/ml Penicillin/Streptomycin, 5 mM non-essential amino acids, 5 mM sodium pyruvate and 5 mM HEPES at 37°C with 5% CO2. Cell lines were validated for Mycoplasma contamination using the ATCC Universal Mycoplasma Detection Kit.
Mice
6–8-week-old male mice were used for all experiments. WT C57BL/6 mice (JAX stock #000664), Balb/c (JAX stock #000651), and Tcrα-KO mice (JAX stock #002116) were purchased from the Jackson Laboratory. OT-I (JAX stock #003831) were crossed with the CD45.1 congenic strain (JAX #002014). OT-I Cas9 double transgenic mice were generated by crossing OT-I mice (JAX stock #003831) with mice carrying a Rosa26-targeted knock-in of Streptococcus pyogenes Cas9 (JAX stock #024858) with constitutive Cas9 expression. The presence of the Cas9 transgene was verified according to genotyping protocols published by Jackson Labs. All purchased mice were acclimated for one week to housing conditions at the Dana-Farber Cancer Institute Animal Resource Facility prior to all experiments. Colonies for each strain of mice were maintained in the same animal facility. Mice were housed in pathogen-free conditions and in accordance with the animal care guidelines from the Dana-Farber Cancer Institute standing committee on Animals and the National Institutes of Health. Animal protocols were approved by the DFCI IACUC.
In vivo CRISPR/Cas9 screen in tumor infiltrating CD8 T cells
A. Cloning of epigenetic gRNA library and virus production
For the primary screen, we constructed three lentiviral plasmid libraries of gRNA sequences targeting a total of 426 genes that encoded epigenetic regulators. Each library contained five unique gRNA sequences that targeted 142 candidate genes. In addition, gRNAs targeting six genes previously shown to inhibit CD8 T cell accumulation in tumors were included as positive controls (Pdcd1, Ctla4, Cblb, Egr2, Smad2 and Ppp2r2d). We also included 100 gRNA sequences as negative controls. These gRNA libraries were cloned into the lentiviral plasmid vector pLKO-gRNA-Thy1.1 that drove expression of the Thy1.1 surface marker. For this purpose, the pLKO.3G vector (Addgene plasmid #14748) was modified by replacing eGFP with the Thy1.1 coding sequence. gRNA libraries were then cloned into the resulting lentiviral expression vector.
For the validation screen, a new gRNA library was constructed by targeting 31 genes selected from the top hits of the primary screens as well as Pdcd1 and Cblb as positive control genes (six gRNA/gene) (Genetic Perturbation Platform, Broad Institute). As negative controls, 186 gRNAs were added (93 non-targeting plus 93 intergenic gRNAs).
To generate lentivirus for transduction with pooled gRNA libraries, the following transfection mix was generated for each 162 cm2 tissue culture flask of HEK293T cells: 7 μg of pLKO-gRNA-Thy1.1 plasmid prep containing lentiviral gRNA sequence libraries, 7 μg of pCMV-DR.9.1 and 0.7 μg of pCMV-VSV-G in 700 μl of OPTI-MEM serum-free media (Gibco) plus 42 μl of TransiT-293 transfection reagent (Mirus). This transfection mix was added to low-passage HEK293T cells (80–90% confluence) in 162 cm2 tissue culture flasks followed by overnight incubation. The next day, the media was removed and replaced with 20 ml of RPMI supplemented with 20% FCS. Viral supernatants were collected at 48- and 72-hours post transduction (2 × 20 ml supernatant total per 162 cm2 flask), passed through a 0.45 μM filtration unit (ThermoFisher) and concentrated by ultracentrifugation at 112,000 x g. Viral titers were determined by transducing HEK293T cells with serial dilutions of a small aliquot of the concentrated prep and measuring the percentage of Thy1.1 positive HEK cells by flow cytometry.
B. Transduction of OT-I Cas9 CD8 T cells
Spleens and peripheral lymph nodes (inguinal, axillary and cervical nodes) from OT-I Cas9 mice were mechanically dissociated using 70 μm cell strainers in complete RPMI medium [containing 10% FBS + 1x GlutaMax™ (Gibco), 100 U/ml penicillin-streptomycin, 1 mM sodium pyruvate, 20 mM HEPES, and 50 μM 2-mercaptoethanol]. OT-I Cas9 CD8 T cells (≥ 97% purity) were isolated from single-cell suspensions using an EasySep™ Mouse CD8+ T Cell Isolation Kit (Stemcell Technologies) according to the manufacturer’s instructions. T cells were cultured in complete RPMI media supplemented with 100 ng/ml IL-15 (Biolegend) and 5 ng/ml IL-7 (Biolegend) for 48 hours. T cells were then transduced with lentiviral gRNA libraries by spin-infection with concentrated lentivirus preps (MOI = 15) using retronectin-coated (Takara Bio) 24-well non-tissue culture treated plates (2 × 106 cells/well). Spin-infection was done at 2,000 rpm for 1.5–2 hours at 32°C in a total volume of 1 ml of virus prep plus complete RPMI media supplemented with 5 μg/ml protamine sulphate (Sigma-Aldrich). Cells were cultured for 72 hours post-transduction in complete RPMI media supplemented with 50 ng/ml IL-15 and 2.5 ng/ml IL-7 (Biolegend) before magnetic enrichment of Thy1.1+ cells using an EasySep™ Mouse CD90.1 Positive Selection Kit (StemCell Technologies); this approach resulted in purity of Thy1.1+ cells of ≥ 93%.
C. In vivo screen with tumor infiltrating CD8 T cells
gRNA transduced Thy1.1+ OT-I Cas9 CD8 T cells (5×106) were injected intravenously into each of 10–12 C57BL/6 mice with B16-OVA-ZsGreen tumors (≥25 mm2 tumor area). On day 10 following T cell transfer, tumors and spleens were isolated for recovery of transferred Thy1.1+ CD8 T cells. Tumors were dissociated using GentleMACS C tubes (Miltenyi Biotec) on a GentleMACS dissociator (Miltenyi Biotec) with the ‘37C_m_TDK_1’ program and an enzyme mix containing 1 mg/ml collagenase D (Sigma-Aldrich), 20 U/ml DNase I (Sigma Aldrich) and 100 μg/ml hyaluronidase type V (Sigma Aldrich) in RPMI media (without additional supplements). Total tumor cell suspensions were then centrifuged at 50 x g for 5 minutes, and supernatants were collected. Spleens were mechanically dissociated using 70 μm cell strainers, and total CD8 T cells were then isolated using EasySep™ Mouse CD8+ T Cell Isolation Kits (Stemcell Technologies) according to the manufacturer’s instructions. Live singlet Thy1.1+ TCRβ+ CD8+ CD4− cells were sorted from tumor and spleen suspensions using a FACSAria IIIu cell sorter (BD) equipped with a 70 μm nozzle and the ‘Yield’ purity mask to ensure complete collection of events. Cell pellets were washed once with cold PBS, and genomic DNA was extracted with a Zymogen Quick-gDNA Microprep Kit following the manufacturer’s protocol for suspension cells.
D. Sequencing of gRNA libraries and quantification of gRNA representation
Genomic DNA isolated from tumor-infiltrating OT-I Thy1.1+ CD8 T cells was subjected to PCR amplification of the gRNA cassette for Illumina sequencing of gRNA representation by the Genetic Perturbation Platform of the Broad Institute of MIT and Harvard (Cambridge, MA). Protocols for PCR amplification and Illumina sequencing are described in detailed at https://portals.broadinstitute.org/gpp/public/resources/protocols.
Data analysis was performed using MaGeCK (Model-based Analysis of Genome-wide CRISPR-Cas9 Knockout) (40). For candidate gene discovery, the normalized gRNA count table was loaded into MaGeCK by comparing tumor (experimental) and spleen (control) conditions described above. Top genes were determined based on mean log2 fold change (LFC) for all gRNAs and false discovery rate (FDR).
Assays with edited T cells and tumor cells
Editing of genes in tumor cells
Cells were edited by electroporation with ribonucleoprotein complexes (RNP) composed of Cas9 protein with bound gRNAs. 20μM of gRNA (designed using Genomic Perturbation Platform of Broad Institute) was mixed at equimolar ratio with Cas9 protein (obtained from UC Berkeley). Editing of genes in tumor cells was performed by nucleofection using the SF cell line 96-well nucleofector kit on 105 tumor cells per nucleofection reaction. Gene editing efficiency was determined by DNA sequencing and subsequent TIDE analysis as well as Western blot analysis.
Adoptive transfer of edited T cells into tumor-bearing mice
Editing of OT-I CD45.1 CD8 T cells was performed by electroporation with RNP composed of Cas9 protein (20μM) and bound gRNA (20μM) using the P3 primary cell 96-well nucleofector kit (Lonza) (2×106 cells per electroporation condition). Freshly isolated naive OT-I CD45.1+ CD8 T cells were edited and then cultured with CD3+CD28+Dynabeads (Invitrogen) for 24h in complete RPMI media [RPMI 1640 medium (Life Technologies 11875119) containing 1% penicillin/streptomycin (Life Technologies 15140122), 50 μM β-mercaptoethanol (Life Technologies 21985023), and 1% L-glutamine (Life Technologies 25030081) supplemented with 10% FBS (Life Technologies 10437028) and 2 ng/ml IL-2 + 2.5 ng/mL IL-7 + 50 ng/mL IL-15]. Dynabeads were then removed and cells were cultured for five additional days in fresh media containing 2.5 ng/mL IL-7 + 50 ng/mL IL-15. Editing efficiency with Carm1 gRNA was confirmed by Western blot analysis. Carm1 or control edited T cells (1×106 cells in 100μL of PBS) were then adoptively transferred to C57BL/6 mice bearing B16-OVA-ZsGreen tumors. An additional group of mice was injected with 100 μL of PBS (no T cell control). Tumor size was measured every third day using a digital caliper.
Generation of primary human CD8 T cells expressing NY-ESO-1 specific TCR
Peripheral blood mononuclear cells (PBMCs) were isolated by Ficoll density gradient centrifugation from leukapheresis collars of healthy donors (Brigham and Women’s Hospital Blood Bank). CD8 T cells were purified from PBMCs using CD8 Dynabeads (StemCell #19053) following the manufacturer’s instructions. Isolated CD8 cells were activated for 48 hours with αCD3/αCD28 beads (Life Technologies #11132D, 1:2 ratio of beads to T cells) and grown in the presence of 30U/mL of human IL-2 for one week. Expanded CD8 T cells were transduced with the lentivirus by spin infection to introduce the NY-ESO-1 TCR. A non-tissue culture treated 24 well plate was coated with 0.8 ml of 15 μg/ml Retronectin (Takara; Kyoto, Japan) overnight at 4°C. Wells were blocked with sterile 2% BSA for 15 minutes at room temperature and gently washed once with PBS. Next, lentivirus was added to wells of the retronectin-coated plate at a multiplicity of infection (MOI) of 15, and plates were spun for 2.5 hours at 2,000 x g, 32°C. The supernatant in the wells was then carefully decanted, and wells were gently washed with 0.5 ml of PBS. 0.5 × 106 T cells were transferred to wells containing 10 μg/ml protamine sulfate (Sigma-Aldrich) in RPMI-1640 media containing 30 U/ml IL-2 and cultured for three days. NY-ESO-1 TCR+ T cells were isolated to >90% purity by FACS and expanded with Dynabeads and IL-2 (30 U/ml).
T cell cytotoxicity assays
A Celigo image cytometer (Nexcelom) was used to study the killing of fluorescent tumor cells by CD8 T cells. Carm1-KO or control-KO B16-OVA-ZsGreen cells were washed with PBS, and 5,000 tumor cells were added per well in flat-bottom 96-well plates (8–10 replicates/group). OT-I CD8 T cells were added at different effector to target ratios. Following 24 or 48 hours of co-culture, the media was removed, and wells were washed with PBS to remove dead tumor cells and CD8 T cells. Live, adherent tumor cells were then counted using the Celigo image cytometer. As an alternative approach, apoptotic tumor cells were counted based on Caspase 3/7 activation. A Caspase-3/7 reagent (Essen Bioscience) was added directly to the culture media (0.5 μM final concentration) after 12, 24 or 48 hours of co-culture of tumor cells with T cells, and positive tumor cells were counted using the image cytometer.
T cell cytotoxicity assay using human tumor cells was performed using NY-ESO-1 transduced human CD8 T cells and BT549 human triple negative breast cancer cells which were HLA-A02*01 positive and endogenously expressed the NY-ESO-1 antigen. BT549 cells were co-cultured with human CD8 T cells that expressed a NY-ESO-1 TCR at increasing effector to target (E:T) ratios for 12–72 hours. Cytotoxicity was quantified by flow cytometry. All in vitro cytotoxicity assays were performed in human or murine T cell media (without addition of IL-2).
RNA extraction and RT-qPCR
Total RNA was extracted using the RNeasy Mini Kit (Qiagen) according to the manufacturer’s protocol. Extracted RNA (1 μg) was transcribed into cDNA using SuperScript IV VILO master mix with ezDNase enzyme according to the manufacturer’s protocol (ThermoFisher). The cDNA samples were diluted and used for real-time quantitative PCR (RT-qPCR). Taqman master mix (ThermoFisher) and gene-specific primers (sequences listed in Supplementary Table S6) were used for PCR amplification and detection using a QuantStudio 6 Flex Real-Time PCR System (ThermoFisher). The RT-qPCR data were normalized to GAPDH and HPRT (housekeeping genes) and presented as fold change of gene expression in the test sample compared to the control sample.
Protein extraction and immunoblotting
Whole cell extracts were prepared by lysis and sonication of cells in RIPA buffer [20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% NP-40, 1% sodium deoxycholate, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 μg/ml leupeptin] supplemented with Halt Protease and Phosphatase Inhibitor Cocktail (ThermoFisher). Protein concentrations were determined using a Bradford Protein Assay (Bio-Rad). Protein samples (20 μg) were resolved by SDS-PAGE using 4–12% NuPAGE Novex Bis-Tris mini gels (ThermoFisher) and transferred onto a PVDF membrane (Bio-Rad). Blots were blocked in 5% Blocker powder (Bio-Rad), 0.2% Tween in PBS and then incubated overnight with primary antibodies. Following incubation with secondary detection reagents and subsequent washing, blots were incubated in Western Lightening Plus-ECL substrate (PerkinElmer). Luminescence was captured using a ChemiDoc MP system (Bio-Rad Laboratories).
Tumor cell colony formation assay
B16F10, 4T1 and MC38 cells were trypsinized and transferred into fresh media, counted and diluted appropriately for seeding into 6-well plates at a density of 500 cells/well. Cells were allowed to grow for 5–6 days, with fresh media added at day 3. Cells were washed with PBS and stained with crystal violet solution (0.5% w/v crystal violet powder, 80% v/v H2O and 20% v/v methanol). Number of colonies and colony areas were quantified using ImageJ software based on the user’s manual.
Immunofluorescence imaging
Cells were grown on glass coverslips, washed with PBS and fixed in 4% paraformaldehyde in PBS for 20 min at room temperature. Cells were permeabilized in 0.2% Triton X-100 for 15 min before blocking for 30 min with 10% serum-containing blocker (ThermoFisher). Coverslips were then incubated with primary antibody overnight at 4°C in a humidified chamber, followed by incubation with a secondary antibody for 1 hour. Primary and secondary antibodies were diluted in blocking buffer, and all incubations were performed at room temperature. Coverslips were mounted using Prolong Gold Antifade Mounting Medium with DAPI (ThermoFisher). Imaging was performed using a Hamamatsu ORCA-Flash4.0 V3 Digital CMOS camera and Nikon Ti-E Motorized Microscope 2000U microscope with Plan Apo Lamda 60x/1.40 Oil Ph3 DM objectives. Images were captured with Nikon Elements Acquisition Software. All scoring was performed under blinded conditions. Three independent experiments with three biological replicates per group were performed.
Quantification of micronuclei in tumor cells
Tumor cells were stained with DAPI, and the percentage of cells that were positive for cytoplasmic micronuclei was determined using Nikon Ti inverted microscope using Plan Apo λ 100x/1.45 Oil DIC objective lens. Micronuclei were defined as discrete DNA aggregates separate from the primary nucleus for cells in which the morphology of the primary nucleus was normal. Cells with apoptotic appearance were excluded from the analysis. All scoring was performed under blinded conditions. Three independent experiments with three biological replicates per group (>100 cells counted per replicate) were performed.
Mouse tumor models
Female BALB/c (Jackson Laboratory #000651) or C57BL/6J (Jackson Laboratory #000664) mice of 4–6 weeks of age were purchased from The Jackson Laboratory. B16F10 or MC38 tumor cells (2×105) were injected subcutaneously in 50 μL of PBS into syngeneic C57BL/6J mice. 4T1 TNBC cells (1×105) were injected in 100μl of PBS supplemented with Matrigel orthotopically into the mammary fat pads of syngeneic BALB/c mice. Mice with similar tumor burden were randomized into treatment groups. Depletion of CD8 T cells in BALB/c and C57BL/6J mice was achieved by IP injection of 100 μg of CD8β mAb (Bio X Cell, Clone 53–5.8 #BE0223) in 100 μL of PBS on days −1, day 0, and then every 3rd day post tumor inoculation. Mice receiving an isotype control mAb (Bio X Cell, clone HRPN #BE0088) at the same dose in PBS were used as controls. CD8 T cell depletion was confirmed by labeling of CD8 T cells from spleens with a CD8 mAb (Biolegend #100741) followed by flow cytometric analysis (BD Fortessa, BD Biosciences). CD8 T cells were significantly depleted within 24 hours following administration of CD8β antibody and at the experimental endpoint.
For checkpoint blockade experiments, tumor-bearing mice were administered with anti-CTLA4 mAb (clone 9H10, #BP0131, 100 μg/injection) or corresponding isotype control Ab (polyclonal Syrian hamster IgG, 100μg/mouse). Alternatively, mice received anti-PD1 (clone 29F.1A12, #BE0273, 200μg/injection) or rat IgG2a isotype control Ab, anti-trinitrophenol (Clone: 2A3, 200 μg/injection) starting on day 7 post tumor inoculation and then every 3rd day. The specific endpoint for each experiment is indicated in the figure legends.
For CARM1 inhibitor experiments, mice received CARM1 inhibitor EZM2302 (dose of 150mg/kg in 100μL) or vehicle (5% Dextrose) twice daily via oral gavage. Inhibitor treatment was performed for 14 days because limited quantities of the compound were available.
4T1 metastasis assay
Carm1-KO or control-KO 4T1 cells (105 cells) were injected into the mammary fat pad of 6-week old BALB/c mice. Three weeks later, lung tissue was washed three times with PBS and fixed in Bouin’s solution (10 mL per lung) for 4–5 days. Visible metastatic nodules were counted under a stereomicroscope (Leica).
Doxycycline- inducible expression of Carm1 in vivo
C57Bl/6 mice bearing Carm1-KO B16F10 tumor cells (transduced with Dox-Carm1 or empty vector constructs) were fed a doxycycline containing diet (625ppm, Envigo Teclad) until the experimental endpoint (18 days). Mice receiving the regular feed were used as controls. Intra-tumoral expression of Carm1 was confirmed by Western blotting at the experimental endpoint.
Flow cytometry analysis of tumor-infiltrating immune cells
Tumors were excised on day 18 following tumor cell inoculation and cut into small pieces using sterile scalpels in serum-free RPMI 1640 media (ThermoFisher #11875093). Tissue was dissociated in 1 mg/ml Collagenase Type IV (Sigma-Aldrich #C5138), 20 units/ml DNAse Type IV (Sigma-Aldrich #D5205), 0.1 mg/ml Hyaluronidase Type V (Sigma-Aldrich #H6254) using GentleMACS C or M tubes on the GentleMACS™ Dissociator (Miltenyi Biotec #130-093-235) followed by incubation at 37°C for 20 min. The resulting cell suspension was passed through a 70 μm filter and pelleted by centrifugation at 300 x g for 5 min. To remove red blood cells, ACK lysis buffer (3x by volume) was added for 45–60 seconds followed by 2 volumes of RPMI to stop red cell lysis. Pelleted cells from pooled supernatants (>300 x g or 1500 rpm, 5 min) were resuspend in the appropriate buffer for flow cytometric analysis of tumors.
Single cell suspensions were stained with 5 μg/mL of Fc receptor blocking anti-mouse CD16/CD32 antibody (clone 2.4G2, BD PharMingen) at 4°C for 5 min before staining of surface proteins with an antibody cocktail at 4°C for 30min in a volume of 100 μL. Cells were then washed twice with PBS, stained with LIVE/DEAD Fixable Dead Cell Stain Kit (Molecular Probes) at 4°C for 15 min and washed twice with staining buffer (PBS supplemented with 1% BSA and 2 mM EDTA). Finally, cells were fixed by incubation in BD Cytofix Fixation Buffer (BD Biosciences) at 4°C for 30min. Samples were analyzed using a BD LSR Fortessa X-20 cell analyzer and BD FACSDiva Software version 8.0. For intracellular staining, cells were stained with surface markers, fixed in Fix/Perm buffer (eBioscience) for 15min, washed in permeabilization buffer (eBioscience) twice and stained with primary antibodies targeting intracellular proteins in permeabilization buffer for 30min at 4°C. Data analysis was performed on FlowJo 10.
Bulk RNA-seq analysis
B16F10 tumor cells or OT-I CD8 T cells were edited with Carm1 or control gRNAs, and loss of Carm1 protein expression was confirmed by Western blot analysis. Three biological replicates were used to extract total RNA using the RNeasy Plus Mini Kit (Qiagen #74134) according to the manufacturer’s protocol. RNA quality was checked using an Agilent BioAnalyzer 2000 instrument. RNA with an integrity number of greater than 9.5 was used for subsequent analyses. RNA-seq analysis was performed by GeneWiz. The standard mRNA library preparation TruSeq RNA Sample Prep Kit v2 (Illumina) was used for library preparation. DNA concentration of libraries was quantified by Qubit (Invitrogen), and equal quantities of DNA were mixed for sequencing. Single-end 75bp sequencing was performed for edited tumor cells, whereas paired-end 150bp sequencing was performed for edited CD8 T cells on an Illumina NextSeq 500 instrument. Gene count quantification was performed with RSEM (41). In the Carm1 KO tumors, we derived a list of genes from the tumor Carm1 KO RNA-seq study, where differential expression study (DES) was performed between the Carm1 KO group and the control group. Using the cutoff of “abs(og2FC)>0.05 & FDR<0.05 & TMP>1”, we obtained 146 upregulated genes and 18 downregulated genes (Table S5). Together, these 164 genes represent a gene signature of ‘Carm1 knockout’ or ‘Carm1 inhibition’, such that the derived gene signature is enriched in Carm1 deficient cells. Statistics for differentially expressed genes were calculated by DESeq2 (42). Criteria of log2FC>1, FDR<0.05, and average TPM >1 were utilized to call significantly differentially expressed genes for the CD8 T cell KO analysis and criteria of FDR<0.01 was used for tumor cell KO experiment.
Mammalian native elongating transcript sequencing (mNET-Seq)
The samples were processed as recommended previously (29). Briefly, three sets of 8×106 cells per experimental condition were processed separately and pooled after MNase treatment. Protein G Dynabeads (ThermoFisher, 10004D) were coupled with the following antibodies: RNAPII Ser2-P (Abcam, Rabbit Poly Ab #5095, 5μg/IP), RNAPII CTD (clone 8WG16, Biolegend #664912, 5μg/IP). NEBNext Small RNA-seq Library Prep kit (#E7300S) was used for library synthesis. Prior to library preparation, sample quality assessed using the Agilent RNA 6000 Pico Kit (#5067–1513). Samples were processed per manufacturer’s protocol for Next-Generation Sequencing (NGS) with the exception of the size selection step, which was performed as described previously (29). We selected barcoded fragments of 150–250bp, which correspond to RNA fragments of 23bp-123bp. Samples were sequenced on the Illumina Nextseq500 platform using a PE75 flow cell. Analysis was performed by the Molecular Biology Core Facility at Dana Farber Cancer Institute as described previously (29).
Computational analyses
mNET-Seq data analysis
mNET-Seq reads were processed as described in (43). Briefly, adapters were removed using software tool Cutadapt (version 1.18), and the trimmed reads were aligned to the mouse genome (mm10) using TopHat (version 2.1.1). Location and strandness of the 5’-end of the second read in each concordant read pair were identified and used for further analysis. R coding environment (r-project.org) was used to compute read frequencies and perform statistical analyses.
Cancer Cell Line Encyclopedia (CCLE) analysis
To evaluate CARM1 expression and its association with different molecular phenotypes in human cancer cell lines, we collected and curated RNA-seq and mutational profiles of 1,208 cell lines from Depmap (44). To investigate associations between CARM1 expression and biomarkers and pathways, we fit linear regression models with biomarker and pathway score as output variable and CARM1 mRNA level as input variable, and with cancer type adjusted. Adjusted p-values were retrieved and reported for each biomarker and pathway.
TCGA data analysis
We collected cancer data sets with both patient survival duration and tumor gene expression profiles from the TCGA database (45). When clinical information was available, we separated the breast cancer data sets into the PAM50 (Prediction Analysis of Microarray 50) subtypes (46) of luminal A, luminal B, Her2 positive, and basal. Each PAM50 subtype is known to have a distinct genomics profile (47) and degree of cytotoxic T cell infiltration (48). Among all TCGA cancers, melanoma is annotated in terms of primary and metastatic sites; Head and neck cancer (HNSC) is annotated corresponding to its HPV status. The CARM1 expression level was compared between cancer tissues and their matched normal tissue when both datasets were available. For each sample, the transcriptomic profile was log2(1+TPM) transformed. We standardized the log scale transcriptome data across patients by quantile-normalization, and further normalized the expression of each gene by subtracting the average of all samples, where a zero value indicated the average expression. The correlations between CARM1 expression and pathway scores were assessed by Spearman correlations. To further assess the clinical relevance of CARM1 expression in cancer, we examined whether CARM1 expression and MED12 were linked to survival benefits in multiple cancer types using Cox regression model.
Human single-cell RNA-seq data analysis
CARM1 expression in human malignant cells was evaluated at single cell resolution. Human malignant cell scRNA-seq data from basal cell carcinoma (GSE123813) (3,551 cells) (49), head and neck squamous cell carcinoma (GSE103322) (2,488 cells) (50), acute myeloid leukemia (GSE116256) (12,489 cells) (51), acute lymphoblastic leukemia (GSE132509) (21,370 cells), multiple myeloma (GSE141299) (16,840 cells), non-small cell lung cancer (GSE143423) (9,237 cells) were retrieved and processed. For each collected dataset, quality control, clustering, and cell-type annotation was uniformly performed. The annotated malignant cells were confirmed by inferCNV (52) based on their copy number variations (CNVs). The pathway scores for the single cell data were derived using AddModuleScore module from Seurat package (53). Statistical comparisons were made with two-sided unpaired Mann-Whitney tests and Spearman correlations.
Gene Ontology and Pathway enrichment analysis
Gene ontology and pathway enrichment were performed with Metascape (54) or GSEA/mSigDB (55). For the bulk tumor Carm1 knockout in our studies, differentially expressed genes were selected for pathway enrichment studies. Pathway signature genes were obtained from the GSEA/mSigDB hallmark gene set collection. In bulk RNA data, pathway signature scores were calculated with mean normalized mRNA expression in each data set. Spearman correlations were calculated for the expression patterns of the pathway signatures in the CCLE, TCGA, and scRNA-seq data. The correlations were adjusted with estimated purity score where both data were available in the TCGA data.
Statistical analyses
Statistical analyses were performed using R3.6 and GraphPad Prism 6 software. Each experiment was performed 2–3 times as indicated. Unpaired Student’s t-test, 2-way ANOVA or unpaired two-sided Mann-Whitney test were used as indicated for comparisons between two groups, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns (non-significant). For in vivo studies, sample size was determined as a function of effect size ((difference in means)/(SD) = 2.0) for a two-sample t-test comparison assuming a significance level of 5%, a power of 90%, and a two-sided t-test. Normal distribution was confirmed using normal probability plot (GraphPad Prism 6.0, GraphPad Software, San Diego, CA), variance was assessed within and between groups. The means of groups were compared using Student’s t-test. The growth of primary tumors over time was analyzed using two-way ANOVA with multiple comparisons. For comparing mouse survival curves, a Log-rank (Mantel-Cox) test was used. All p-values are two-sided, and statistical significance was evaluated at the 0.05 level.
Data and materials availability
All transcriptomics data generated during this study (RNA-seq, mNET-seq and DRIP-seq) have been deposited at NCBI GEO (Gene Expression Omnibus) with accession numbers GSE144917, GSE149139 and GSE148905.
Supplementary Material
Significance.
Resistance to cancer immunotherapy remains a major challenge. Targeting of CARM1 enables immunotherapy of resistant tumors by enhancing T cell functionality and preserving memory-like T cell populations within tumors. CARM1 inhibition also sensitizes resistant tumor cells to immune attack by inducing a tumor cell-intrinsic type I interferon response.
Acknowledgments
We thank Qiakai Xu (Elledge lab) for help in selecting gRNAs for the epigenetic CRISPR library. Paula Montero Llopis and Ryan Stephansky (Microscopy Resources, Harvard Medical School) for technical support in imaging data analysis, Zach Herbert (Genomics core facility, DFCI) for providing expertise in NGS projects and data analysis. This work was supported by the Parker Institute for Cancer Immunotherapy (PICI to K.W.W.), NIH grants R01 CA238039, R01 CA251599 and P01 CA163222 (to K.W.W.), R01 CA234018 (to X.S.L. and K.W.W), a Department of Defense Distinguished Investigator Award (W81XWH-18-1-0469 to S.J.E.) and a 2018 AACR-AstraZeneca Lung Cancer Research Fellowship, Grant Number 18-40-12-KUMA (to S.K.). Work in the Chedin lab is supported by NIH grant R01 GM120607. A.B. was supported by a T32 training grant (T32 CA207021). R.E.T. was supported by a graduate fellowship from A*STAR (Singapore). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health/NCI.
Footnotes
Declaration of Interests: K.W.W. serves on the scientific advisory board of TCR2 Therapeutics, TScan Therapeutics and Nextechinvest and receives sponsored research funding from Bristol-Myers Squibb and Novartis. He is a co-founder of Immunitas Therapeutics, a biotech company. S.J.E. is a co-founder of TScan Therapeutics, a biotech company.
References
- 1.Bouwman P, Jonkers J. The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nat Rev Cancer 2012;12(9):587–98 doi 10.1038/nrc3342. [DOI] [PubMed] [Google Scholar]
- 2.Naito Y, Saito K, Shiiba K, Ohuchi A, Saigenji K, Nagura H, et al. CD8+ T cells infiltrated within cancer cell nests as a prognostic factor in human colorectal cancer. Cancer Res 1998;58(16):3491–4. [PubMed] [Google Scholar]
- 3.Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F, et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci U S A 2005;102(51):18538–43 10.1073/pnas.0509182102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen Q, Sun L, Chen ZJ. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat Immunol 2016;17(10):1142–9 doi 10.1038/ni.3558. [DOI] [PubMed] [Google Scholar]
- 5.Hervas-Stubbs S, Perez-Gracia JL, Rouzaut A, Sanmamed MF, Le Bon A, Melero I. Direct effects of type I interferons on cells of the immune system. Clin Cancer Res 2011;17(9):2619–27 doi 10.1158/1078-0432.Ccr-10-1114. [DOI] [PubMed] [Google Scholar]
- 6.Schmid P, Chui SY, Emens LA. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. Reply. N Engl J Med 2019;380(10):987–8 doi 10.1056/NEJMc1900150. [DOI] [PubMed] [Google Scholar]
- 7.Wysocka J, Allis CD, Coonrod S. Histone arginine methylation and its dynamic regulation. Frontiers in bioscience : a journal and virtual library 2006;11:344–55. [DOI] [PubMed] [Google Scholar]
- 8.Daujat S, Bauer UM, Shah V, Turner B, Berger S, Kouzarides T. Crosstalk between CARM1 methylation and CBP acetylation on histone H3. Curr Biol 2002;12(24):2090–7 doi 10.1016/s0960-9822(02)01387-8. [DOI] [PubMed] [Google Scholar]
- 9.Chen D, Ma H, Hong H, Koh SS, Huang SM, Schurter BT, et al. Regulation of transcription by a protein methyltransferase. Science 1999;284(5423):2174–7 doi 10.1126/science.284.5423.2174. [DOI] [PubMed] [Google Scholar]
- 10.Chen D, Huang SM, Stallcup MR. Synergistic, p160 coactivator-dependent enhancement of estrogen receptor function by CARM1 and p300. The Journal of biological chemistry 2000;275(52):40810–6 doi 10.1074/jbc.M005459200. [DOI] [PubMed] [Google Scholar]
- 11.Kim YR, Lee BK, Park RY, Nguyen NT, Bae JA, Kwon DD, et al. Differential CARM1 expression in prostate and colorectal cancers. BMC Cancer 2010;10:197 doi 10.1186/1471-2407-10-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Al-Dhaheri M, Wu J, Skliris GP, Li J, Higashimato K, Wang Y, et al. CARM1 is an important determinant of ERalpha-dependent breast cancer cell differentiation and proliferation in breast cancer cells. Cancer Res 2011;71(6):2118–28 doi 10.1158/0008-5472.CAN-10-2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jansen CS, Prokhnevska N, Master VA, Sanda MG, Carlisle JW, Bilen MA, et al. An intra-tumoral niche maintains and differentiates stem-like CD8 T cells. Nature 2019;576(7787):465–70 doi 10.1038/s41586-019-1836-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Siddiqui I, Schaeuble K, Chennupati V, Fuertes Marraco SA, Calderon-Copete S, Pais Ferreira D, et al. Intratumoral Tcf1(+)PD-1(+)CD8(+) T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity 2019;50(1):195–211 e10 doi 10.1016/j.immuni.2018.12.021. [DOI] [PubMed] [Google Scholar]
- 15.Drew AE, Moradei O, Jacques SL, Rioux N, Boriack-Sjodin AP, Allain C, et al. Identification of a CARM1 Inhibitor with Potent In Vitro and In Vivo Activity in Preclinical Models of Multiple Myeloma. Sci Rep 2017;7(1):17993 doi 10.1038/s41598-017-18446-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gajewski TF, Fuertes MB, Woo SR. Innate immune sensing of cancer: clues from an identified role for type I IFNs. Cancer Immunol Immunother 2012;61(8):1343–7 doi 10.1007/s00262-012-1305-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dunn GP, Ikeda H, Bruce AT, Koebel C, Uppaluri R, Bui J, et al. Interferon-gamma and cancer immunoediting. Immunol Res 2005;32(1–3):231–45. [DOI] [PubMed] [Google Scholar]
- 18.Kinner A, Wu W, Staudt C, Iliakis G. Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res 2008;36(17):5678–94 doi 10.1093/nar/gkn550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fenech M, Kirsch-Volders M, Natarajan AT, Surralles J, Crott JW, Parry J, et al. Molecular mechanisms of micronucleus, nucleoplasmic bridge and nuclear bud formation in mammalian and human cells. Mutagenesis 2011;26(1):125–32 doi 10.1093/mutage/geq052. [DOI] [PubMed] [Google Scholar]
- 20.Mackenzie KJ, Carroll P, Martin CA, Murina O, Fluteau A, Simpson DJ, et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 2017;548(7668):461–5 doi 10.1038/nature23449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee YH, Bedford MT, Stallcup MR. Regulated recruitment of tumor suppressor BRCA1 to the p21 gene by coactivator methylation. Genes Dev 2011;25(2):176–88 doi 10.1101/gad.1975811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bakhoum SF, Cantley LC. The Multifaceted Role of Chromosomal Instability in Cancer and Its Microenvironment. Cell 2018;174(6):1347–60 doi 10.1016/j.cell.2018.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, et al. Loss of IFN-gamma Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell 2016;167(2):397–404.e9 doi 10.1016/j.cell.2016.08.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang TH, et al. The Immune Landscape of Cancer. Immunity 2019;51(2):411–2 doi 10.1016/j.immuni.2019.08.004. [DOI] [PubMed] [Google Scholar]
- 25.Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A 2010;107(9):4275–80 doi 10.1073/pnas.0915174107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang Y, Lu Y, Espejo A, Wu J, Xu W, Liang S, et al. TDRD3 is an effector molecule for arginine-methylated histone marks. Molecular cell 2010;40(6):1016–23 doi 10.1016/j.molcel.2010.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheng D, Vemulapalli V, Lu Y, Shen J, Aoyagi S, Fry CJ, et al. CARM1 methylates MED12 to regulate its RNA-binding ability. Life Sci Alliance 2018;1(5):e201800117 doi 10.26508/lsa.201800117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Buratowski S. Progression through the RNA polymerase II CTD cycle. Mol Cell 2009;36(4):541–6 doi 10.1016/j.molcel.2009.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nojima T, Gomes T, Carmo-Fonseca M, Proudfoot NJ. Mammalian NET-seq analysis defines nascent RNA profiles and associated RNA processing genome-wide. Nat Protoc 2016;11(3):413–28 doi 10.1038/nprot.2016.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang Y, McBride KM, Hensley S, Lu Y, Chedin F, Bedford MT. Arginine methylation facilitates the recruitment of TOP3B to chromatin to prevent R loop accumulation. Molecular cell 2014;53(3):484–97 doi 10.1016/j.molcel.2014.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sanz LA, Chedin F. High-resolution, strand-specific R-loop mapping via S9.6-based DNA-RNA immunoprecipitation and high-throughput sequencing. Nat Protoc 2019;14(6):1734–55 doi 10.1038/s41596-019-0159-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol 2013;14(10):1014–22 doi 10.1038/ni.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, Sancho D. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol 2020;20(1):7–24 doi 10.1038/s41577-019-0210-z. [DOI] [PubMed] [Google Scholar]
- 34.Raghu D, Xue HH, Mielke LA. Control of Lymphocyte Fate, Infection, and Tumor Immunity by TCF-1. Trends in immunology 2019;40(12):1149–62 doi 10.1016/j.it.2019.10.006. [DOI] [PubMed] [Google Scholar]
- 35.Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, et al. Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell 2018;175(4):998–1013 e20 doi 10.1016/j.cell.2018.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gautam S, Fioravanti J, Zhu W, Le Gall JB, Brohawn P, Lacey NE, et al. The transcription factor c-Myb regulates CD8(+) T cell stemness and antitumor immunity. Nat Immunol 2019;20(3):337–49 doi 10.1038/s41590-018-0311-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 2017;548(7668):466–70 doi 10.1038/nature23470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, et al. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell reports 2015;11(7):1018–30 doi 10.1016/j.celrep.2015.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Busch DH, Frassle SP, Sommermeyer D, Buchholz VR, Riddell SR. Role of memory T cell subsets for adoptive immunotherapy. Semin Immunol 2016;28(1):28–34 doi 10.1016/j.smim.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, et al. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol 2014;15(12):554 doi 10.1186/s13059-014-0554-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 2011;12:323 doi 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15(12):550 doi 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nojima T, Gomes T, Grosso ARF, Kimura H, Dye MJ, Dhir S, et al. Mammalian NET-Seq Reveals Genome-wide Nascent Transcription Coupled to RNA Processing. Cell 2015;161(3):526–40 doi 10.1016/j.cell.2015.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ghandi M, Huang FW, Jane-Valbuena J, Kryukov GV, Lo CC, McDonald ER 3rd, et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 2019;569(7757):503–8 doi 10.1038/s41586-019-1186-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, Ellrott K, et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet 2013;45(10):1113–20 doi 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol 2009;27(8):1160–7 doi 10.1200/jco.2008.18.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hoadley KA, Yau C, Wolf DM, Cherniack AD, Tamborero D, Ng S, et al. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell 2014;158(4):929–44 doi 10.1016/j.cell.2014.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miyan M, Schmidt-Mende J, Kiessling R, Poschke I, de Boniface J. Differential tumor infiltration by T-cells characterizes intrinsic molecular subtypes in breast cancer. J Transl Med 2016;14(1):227 doi 10.1186/s12967-016-0983-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tirosh I, Izar B, Prakadan SM, Wadsworth MH 2nd, Treacy D, Trombetta JJ, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016;352(6282):189–96 doi 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang Z, Liu R, Jin R, Fan Y, Li T, Shuai Y, et al. Integrating Clinical and Genetic Analysis of Perineural Invasion in Head and Neck Squamous Cell Carcinoma. Front Oncol 2019;9:434 doi 10.3389/fonc.2019.00434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.van Galen P, Hovestadt V, Wadsworth Ii MH, Hughes TK, Griffin GK, Battaglia S, et al. Single-Cell RNA-Seq Reveals AML Hierarchies Relevant to Disease Progression and Immunity. Cell 2019;176(6):1265–81.e24 doi 10.1016/j.cell.2019.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014;344(6190):1396–401 doi 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nature biotechnology 2018;36(5):411–20 doi 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun 2019;10(1):1523 doi 10.1038/s41467-019-09234-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 2015;1(6):417–25 doi 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.