

Abstract
Purpose
Neoadjuvant immunotherapy may improve the clinical outcome of regionally advanced operable melanoma and allows for rapid clinical and pathologic assessment of response. We examined neoadjuvant pembrolizumab and high dose interferon alfa-2b (HDI) therapy in patients with resectable advanced melanoma.
Patients and Methods
Patients with resectable stage III/IV melanoma were treated with concurrent pembrolizumab 200 mg intravenously (IV) every 3 weeks and HDI 20 MU/m2/day IV, 5 days/week for 4 weeks, then 10 MU/m2/day subcutaneously 3 days/week for 2 weeks. Definitive surgery followed, and adjuvant combination immunotherapy completing a year of treatment. Primary endpoint was safety of the combination. Secondary endpoints included overall response rate (ORR), pathologic complete response (pCR), recurrence-free survival (RFS), and overall survival (OS). Blood samples for correlative studies were collected throughout. Tumor tissue was assessed by immunohistochemistry (IHC) and flow cytometry at baseline and at surgery.
Results
31 patients were enrolled, and 30 were evaluable. At data cutoff (October 2, 2019), median follow-up for OS was 37.87 months (range; 33.2–43.47). Median OS and RFS were not reached. Radiographic ORR was 73.3% (95% CI: 55.5%−85.8%), with a 43% (95% CI: 27.3%−60.1%) pathologic complete response (pCR) rate. None of the patients with a pCR have recurred. HDI and pembrolizumab were discontinued in 73% and 43% of patients, respectively. Correlative analyses suggested that intratumoral PD-1/PD-L1 interaction and HLA-DR expression are associated with pCR (p=0.002 and p=0.008, respectively).
Conclusions
Neoadjuvant concurrent HDI and pembrolizumab demonstrated promising clinical activity despite high rates of treatment discontinuation. pCR is a prognostic indicator.
Introduction
Adjuvant therapy with immune checkpoint inhibitors (ICI) is now standard of care in resected stage III or IV melanoma, and targeted therapy (TT) is an alternative option for patients with a BRAF V600E/K mutation.1,2 Neoadjuvant therapy may provide several advantages: debulking of disease prior to surgery, earlier treatment of micrometastases, and tissue analysis at time of surgery to correlate treatment response with potential biomarkers. Furthermore, phase I and II studies suggest that neoadjuvant therapy is safe, does not lead to development of unresectable disease or delays in definitive surgical management, and is potentially superior or at least non-inferior to adjuvant therapy in regards to relapse free survival (RFS), including in patients with in-transit metastases.3–14
Pathologic complete response (pCR), defined as the absence of viable tumor cells by histologic examination, is associated with reduced recurrence and increased overall survival (OS) in breast cancer.15 Furthermore, it is increasingly accepted as a significant prognostic marker in melanoma.6,16–22 In a pooled analysis of 184 patients treated with neoadjuvant ICI (n=133) or TT (n=51), none of the patients with a pCR following ICI recurred at a median follow up of 10 months.5
Multiple immunotherapy agents, including combinations, have been evaluated in the neoadjuvant setting. Neoadjuvant pembrolizumab resulted in a 30% complete or near complete (<10% viable tumor) pathologic response rate after a single treatment dose.8 Paired administration of PD-1/PD-L1 axis blockers with CTLA-4 inhibitors has been shown to improve pCR (45% vs 25%) but at the cost of significantly increased toxicity (73% vs 8% treatment related adverse events (trAE).12 Alternative ICI neoadjuvant dosing regimens adopting a reduced dose of ipilimumab while increasing the dose of nivolumab have sought to minimize toxicity while maintaining pathologic response.4
There is evidence that ICI and interferon-alpha (IFN-α), which engage multiple immune compartments, may also improve response rates.9,23–25 In the adjuvant setting, high dose IFN-α has been shown to improve both relapse-free and overall survival.26,27 IFN-α enhances class I expression28 and directly activates innate and adaptive immune responses, including CD4+ T, CD8+ T-cells and natural killer cells.29,30 In experimental models, it is required by dendritic cells to mediate tumor rejection.31–33 Furthermore, the combination of PD-1/PD-L1 blockade and IFN-α has been shown to be superior to either alone in B16 melanoma-bearing mice, and in vitro IFN-α exposure has been shown to increase PD-L1 expression in human melanoma cells.34 In patients with unresectable melanoma, the combination of pembrolizumab and pegylated IFN-α2b resulted in a 60.5% objective response rate.25
In this pilot phase Ib/II study (ClinicalTrials.gov identifier: NCT02339324) of neoadjuvant pembrolizumab and high dose IFNα−2b (IFN), we evaluated the safety, efficacy, and immune correlates of combination therapy in high-risk surgically resectable stage III or IV melanoma patients.
Patients and Methods
Patients
This multi-center, open-label, phase Ib/II trial enrolled patients with resectable stage III/IV PD-1-naive melanoma (Tx-4 N1b-3M0–1;AJCC 7th edition). Key eligibility criteria included age ≥18 years with histologically confirmed mucosal or cutaneous melanoma, measurable disease per Response Evaluation Criteria in Solid Tumors version 1.1 (RECIST v1.1), surgically resectable disease (confirmed by surgical oncology prior to treatment start), and an Eastern Cooperative Oncology Group performance status of 0 or 1. Patients who relapsed following adjuvant ipilimumab and had prior immune-related adverse events (irAEs) could enroll if these had resolved ≤ grade 1. Patients requiring prednisone ≤ 10 mg/day or equivalent were eligible. Patients were excluded if they had active autoimmune disease requiring systemic immune suppression (excluding asthma, atopy, type I diabetes, hypothyroidism and vitiligo). Approval to treat patients was obtained from the UPMC-Hillman Cancer Center Institutional Review Board (No. 19020212), Roswell Park Cancer Institute (No. 00005890) and Hershey Penn State IRB (No. 00002662). The authors attest that written informed consent was obtained from all patients involved in the study.
Study Design and Treatment
This clinical trial enrolled 31 patients, of whom 30 were evaluable. The treatment plan consisted of 3 phases: induction, definitive surgery and maintenance. During the induction portion, patients received pembrolizumab 200 mg intravenous (IV) every 3 weeks for 2 doses, starting the first week of IFN administration, given concurrently with IFN at 20 MU/m2/d IV for 5 of 7 consecutive days for 4 weeks, followed by 10 MU/m2/d subcutaneously (SC) every other day, 3 times per week for 2 weeks. The maintenance phase (following recovery from surgery) consisted of pembrolizumab 200 mg IV every 3 weeks given concurrently with IFN at 10 MU/m2/d SC every other day, 3 times a week for 46 additional weeks. Dose delays of both agents were permitted. Dose reductions were permitted for IFN only. Pembrolizumab was either given or skipped. Dose-limiting toxicities (DLT) were defined as grade 3 or greater AEs within 28 days of the first treatment cycle. Grade 3 fatigue was excluded from the DLT definition. Patients were treated until disease recurrence, intolerable toxicity or for up to 1 year, whichever occurred first.
Objectives
The primary objective of this study was to assess the safety profile of combination therapy with HDI and pembrolizumab. Secondary objectives included investigator-assessed radiologic objective response rate (ORR) by RECIST v1.1, pathologic complete response rate (pCR), relapse-free survival (RFS), overall survival (OS), and correlative analyses.
Assessments
Radiographic imaging (positron emission tomography or CT) was performed at baseline, at week 6 (after completion of the induction phase and before definitive surgery), and every 12 weeks during the maintenance phase. Baseline MRI brain was required at enrollment. During follow-up, patients underwent imaging every 3 months up until year 2, every 6 months years 2–5, and planned for every year thereafter for up to 15 years. Patients who developed recurrent melanoma were followed for survival and subsequent treatment. Response assessments followed RECIST v1.1. Treatment related AEs (trAEs) were evaluated using the National Cancer Institute Common Terminology Criteria for Adverse Events version 4.0. Pathologic response was evaluated by 3 board certified pathologists who were blinded to the assessment of the other pathologists. If there was a discrepancy, a 4th pathologist reviewed the case and the majority evaluation was utilized. Complete pathologic response (pCR) was defined as no evidence of viable tumor by histologic assessment. A major pathologic response was defined as <10% residual melanoma in the surgical specimen.
Statistical Analysis
R version 3.6.1 was used for the analysis of demographic, safety and efficacy data. Toxicities that were possibly, probably or definitely related to the study regimen are included in the analysis. Kaplan-Meier’s method was used for the analysis of time-to-event endpoint. Fisher exact tests were used to assess the association between response and exposure to prior adjuvant therapy. Cohen’s κ statistics was used to measure the agreement between radiographic response and pathological response.
When evaluating the fluorescence immunohistochemistry (FIHC) data, pre-therapy included baseline data for all samples (n = 11). A Wilcoxon test was used to determine statistical significance for baseline vs. surgery paired samples (n=8) and unpaired T test evaluating pCR and non-pCR samples (n = 14) using GraphPad Prism v 8.1.2. Flow cytometry assessments employed paired t-test for evaluating baseline and post-treatment samples from TILs and ANOVA in addition to paired student’s t-test for evaluating PBMCs where baseline, 6-weeks and 12-week biomarker assessments were possible, with p-values <0.05 considered significant. To evaluate significant biomarker changes in OS, patients were classified into a low OS group and a high OS group using the median of the OS. To assess statistical significance of different biomarkers to patient response, t-test was applied to compare the two groups and those biomarkers with a two-tailed p-value below 0.05 were considered to be of statistical significance.
Tumor Biopsies and Blood Collection
Tumor surgical, core or punch biopsies were obtained pre-treatment and tissue was obtained at the time of definitive surgery. Blood was collected at baseline, at weeks 6 and 12, at 6 and 12 months, and at time of disease recurrence. The tumor samples were formalin fixed and paraffin-embedded, and portions were prepared for single cell suspensions of tumor and tumor infiltrating lymphocytes (TIL) by enzymatic digestion. For peripheral blood mononuclear cells (PBMC), blood was drawn into heparin-containing tubes and for serum into tubes without anticoagulant at baseline and at 6 weeks, 12 weeks, 6 and 12 months, and at disease recurrence. The samples were processed by the UPMC-Hillman Cancer Center Immunologic Monitoring Lab and PBMC were isolated by Ficoll gradient and cryopreserved for batched testing according to standard operating procedures. The liquid nitrogen (PBMC) and −80C (serum) freezers were continuously monitored for temperature.
Flow Cytometry
Flow cytometry was performed on PBMC and on TIL. Briefly, up to 4×106 cryopreserved PBMCs or tumor tissue cells were thawed quickly in a 37ºC water bath and washed with pre-warmed RPMI medium containing 10% FBS. Cells were stained with fixable viability dye eFluor™506 (Thermo Fisher Scientific, San Diego, CA) at a dilution of 1:400 in wash buffer (phosphate-buffered saline containing 0.1% sodium azide and 2% fetal bovine serum) followed by staining with fluorochrome tagged antibodies for approximately 30 minutes at room temperature in the dark, as previously described35. After incubation, cells were washed once in wash buffer and fixed using 0.5% formalin buffer or subjected to intra-cellular staining. For intra-cellular staining to detect Ki-67 expression, PBMCs were first stained with surface antibody cocktails followed by permeabilization and staining for Ki-67 according to the manufacturer’s instructions (Foxp3 / Transcription Factor Staining Buffer Set, Thermofisher, Carlsbad, CA). For characterization of different immune cell subsets, the following antibodies were used CD3 (UCHT1), CD4 (RPA-T4), CD8 (SK1), CD5 (L17F21), CD19 (SJ25C1), CD25 (M-A251), CD28 (CD28.2), CD38 (HIT2), CD45 (HI30), CD45RA (HI100), CD45RO (UCHL1), CD183 (1C6/CXCR3), CD194 (1G1), CD197 (150503) CD279 (EH12.1), CD366 (7D3), HLA-DR (G46–6), and Ki-67 (B56) from (BD Biosciences, San Diego, CA). Additional antibodies were obtained from Biolegend (San Diego, CA), including CD27 (O323), CD134 (ACT35), CD196 (G034E3), or from ThermoFisher (Carlsbad, CA), CD127 (eBioRDR5), CD223 (3DS223H), CD278 (ISA-3) and fixable viability dye (FVD-eF506). Stained Samples were acquired in a BD LSRFortessa X-20 equipped with 5 lasers (BD Biosciences, San Jose, CA) and data was analyzed using FlowJo software (FlowJo LLC, Ashland, OR).
For data analysis, after exclusion of debris, doublets and dead cells, B cells were gated based on the expression of CD19 and T cells were gated based on the expression of CD3. T cells were further gated for CD4+ and CD8+ subsets. Both CD4+ and CD8+ cells were subsequently analyzed for the expression of various markers to define helper T cell subsets, memory T cell subsets and checkpoint inhibitor expression. Gate placement for all checkpoint molecules was set based on naïve T cells with a summary of gating strategy for different populations shown in Supplementary Figure 1.
Fluorescence Immunohistochemistry
Multiplexed FIHC analyses were performed to detect PD-1 and PD-L1 expression, lymphoid infiltrates (CD3, CD4, CD8, Foxp3) and CD11b, IDO1, HLA-DR in the context of tumor. PD-1/PD-L1/S100 and CD11b/HLA-DR/IDO1/S100 staining was performed as previously described36. Image analysis was performed using AQUA (Navigate BioPharma, Inc.) to determine the percent of all DAPI cells PD-1+, PD-L1+, or the proportion of PD-1+ cells in close proximity to PD-L1+ cells, defined as the Interaction Score, and CD11b+, HLA-DR+, IDO1+, or IDO1+HLA-DR+S100+ cells.
The CD4, CD8, CD25, FoxP3, Ki67, cytokeratin panel was performed using a fully automated staining protocol on the Bond Rx (Leica). Slides were dewaxed using the Bond Rx followed by antigen retrieval in ER2 (Leica) buffer at 95°C for 20 minutes. Primary antibodies were incubated for 1 hour, detected with either EnVision+ HRP Mouse or EnVision+ HRP Rabbit for 30 minutes and slides were heat cycled in ER1 (Leica) buffer for 20 minutes at 95°C after each round of primary, secondary and Opal fluorophore staining. Slides were stained with 1:100 dilution of mouse anti-CD4 (4B12, Agilent), detected with Opal 520 (Akoya Biosciences), 1:400 dilution of mouse anti-CD8 (C8/144B, Agilent), detected with Opal 620 (Akoya Biosciences), 1:100 dilution of rabbit anti-FoxP3 (D2W8E, Cell Signaling Technologies), detected with Opal 540 (Akoya Biosciences), 1:400 dilution of rabbit anti-CD25 (SP176, Sigma Aldrich), detected with Opal 570 (Akoya Biosciences), 1:1000 dilution of mouse anti-Ki67 (MIB-1, Agilent), detected with Opal 650 (Akoya Biosciences), and 1:400 dilution of mouse anti-cytokeratin (AE1/AE3, Agilent), detected with Opal 690, and finally incubated for 10 minutes with spectral DAPI (Akoya Biosciences). AQUAnalysis was used to determine the percent of all DAPI cells CD4+, CD8+, CD25+, FoxP3+ or Ki67+. T cells were identified as either CD4+ or CD8+. Regulatory T cells were defined as CD25+FoxP3+CD4+ cell populations.
Results
Patient Population
31 patients were enrolled from March 2015 to June 2018. Baseline characteristics are listed in Table 1. On central pathology review, 1 patient was found to have sarcoma (rather than melanoma) and was thus taken off study and excluded from analysis. An additional melanoma patient was enrolled, resulting in 30 eligible patients. The median age was 59 years (range: 26 to 83 years). 14 patients (47%) had stage IIIB, 9 (30%) had stage IIIC, and 7 patients had stage IV disease (23%) by AJCC 7th edition. 4 patients (13%) had in-transit disease.17 patients (53%) had recurrent disease after prior definitive surgery. 6 patients (20%) had received prior adjuvant therapy, 4 (13%) with IFN, 1 with ipilimumab (3%), and 1 with IFN and ipilimumab (3%). 3 of these patients recurred at an interval of at least 3 years, and 2 patients recurred at an interval of 1 year from completion of prior adjuvant therapy. All patients were anti-PD-1 naïve.
Table 1:
Patient Demographics and Clinical Characteristics
| Variable | No. of Patients (%) | |
|---|---|---|
| Age (years; Median (range) | 59 (26–83) | |
| Cutaneous primary | 27 (90) | |
| Mucosal | 3 (10) | |
| Sex | Female | 8 (27) |
| Male | 22 (73) | |
| Performance Status |
0 | 25 (83) |
| 1 | 5 (17) | |
| Recurrent disease after prior surgery | 17 (57) | |
| Presence of in-transit metastases | 4 (13) | |
| Stage (AJCC 7) | IIIB | 14 (47) |
| IIIC | 9 (30) | |
| M1a, M1b | 7 (23) | |
Treatment
A median of 18 doses of IFN (range; min 8-max 20) were given during the 4-week induction period. 12 patients (40%) required HDI dose reduction during induction. 22 patients (73%) had HDI discontinued (either during induction or maintenance); 21 for toxicity and 1 for PD. During IFN maintenance, a median of 44.5 doses were given (range; min 0-max 139), and 16 (53%) required dose reduction (12 had 1 dose reduction and 4 had 2 dose reductions). A median of 16 doses of pembrolizumab were completed (range; min 2-max 18). 9 (30%) discontinued pembrolizumab due to toxicity, and 4 (13%) for recurrence of disease. All patients underwent definitive surgical resection as planned and on schedule.
Treatment-Related Toxicities
All patients experienced at least one treatment-related AE (trRAEs) of any grade, as detailed in Table 2. The most common trAEs of any grade were fatigue in 30 (100%), elevated aspartate aminotransferase (AST) in 24 (80%), hypophosphatemia in 23 (77%) and fever in 19 (63%). The most common grade 3 TRAEs were hypophosphatemia in 13 (43%), fatigue in 11 (37%), and elevated creatine kinase (CPK) in 6 (20%). Grade 4 toxicities included hyperglycemia, elevated lipase, elevated CPK, and lymphopenia (1 each, 3%). One grade 5 event occurred following a viral prodrome, 6 months after discontinuing study therapy. Autopsy revealed pneumonia and myocarditis, and this event was not attributed to treatment.
Table 2:
Select adverse events possibly, probably or definitely related to Pembrolizumab and/or HDI (N=30)*
| Type | Any Grade | Grade 3 | Grade 4 | |||
|---|---|---|---|---|---|---|
| No. Patients | % | No. Pts. | % | No. Pts. | % | |
| Rash maculopapular | 11 | 37 | - | - | - | - |
| Diarrhea/Colitis | 16 | 53 | 1 | 3 | - | - |
| AST elevation | 24 | 80 | - | - | - | - |
| ALT elevation | 17 | 57 | 2 | 7 | ||
| Adrenal insufficiency | 3 | 10 | - | - | - | - |
| Hypothyroidism | 9 | 30 | - | - | - | - |
| Lipase increased | 10 | 33 | 4 | 13 | - | - |
| Hyponatremia | 19 | 63 | 1 | 3 | - | - |
| Hypophosphatemia | 23 | 77 | 13 | 43 | - | - |
| Depression | 6 | 20 | - | - | - | - |
| Arthralgia | 5 | 17 | 1 | 3 | - | - |
| CPK elevation | 17 | 57 | 6 | 20 | 1 | 3 |
| Fatigue | 30 | 100 | 11 | 37 | - | - |
| Fever | 19 | 63 | 1 | 3 | - | - |
| Lymphopenia | 15 | 50 | 4 | 13 | 1 | 3 |
| Myalgia | 16 | 53 | - | - | - | - |
One grade 5 event occurred 6 months after completion of therapy with autopsy evidence of pneumonia and myocarditis.
Antitumor Activity
All 30 patients were evaluated for efficacy, and all 30 patients underwent surgery as planned. On restaging scans at 6 weeks (before surgery), the radiographic objective response rate (ORR) per RECIST 1.1 was 73.3% (95% CI 55.5%−85.8%). 7 (23%) had a complete radiographic response (CR),15 (50%) had a partial response (PR), 7 (23%) had stable disease (SD) and 1 (3%) had progressive disease (PD). There were no differences in response rate by exposure to prior adjuvant therapy (p=0.3). All patients underwent restaging with CT (rather than PET/CT) scans.
Pathologic examination at time of surgery confirmed complete pathologic response in 13 patients (43%; 95% CI 27%−61%) (Figure 1). Four additional patients (13%) had a major pathologic response, with less than 10% residual disease. Thirteen patients were pathologic non-responders (>10% residual pathologic disease), including the 3 patients with mucosal melanoma. There were no differences in pCR seen when analyzed by prior exposure to adjuvant therapy. There was little association between ORR and pCR (Cohen’s κ = 0.024).
Figure 1:
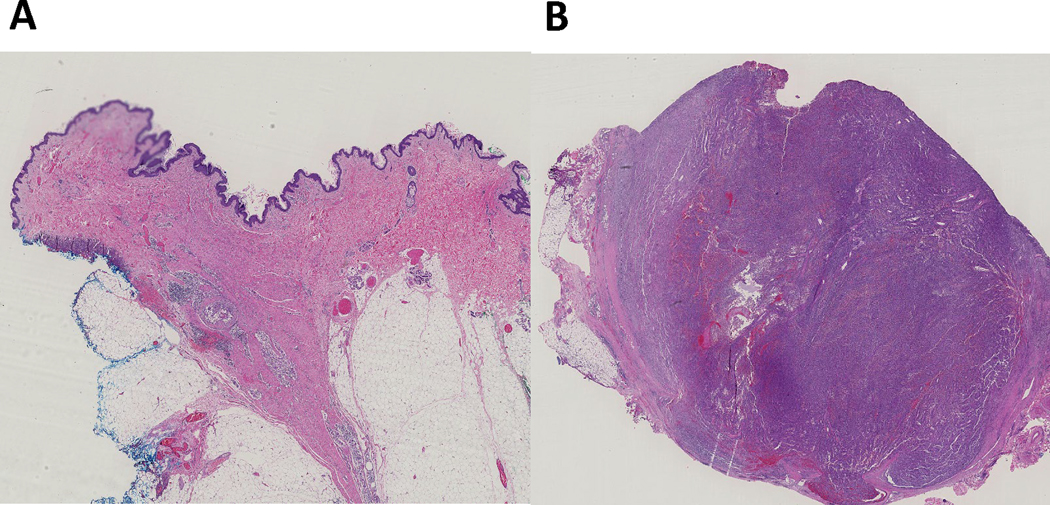
Representative H&E slide images of a pathologic complete response (A) and pathologic non-response (B).
The median follow-up time for RFS was 29.17 months (17.27–36.8), and median follow-up time for OS was 37.87 months (33.2–43.47). Neither median OS (Figure 2A) or RFS (Figure 2B) have been reached. Five patients (17%) had recurrent disease, with a median time to recurrence of 14.5 months. None of the patients achieving a pCR have recurred. Of the four patients with a major pathologic response, 1 patient had recurrent disease and has died, and 3 patients remain without evidence of disease. At data cut-off, 5 patients had died. One patient died without evidence of disease at age 85, of unknown etiology. One patient died of pneumonia following a viral prodrome 6 months after completion of study therapy. Autopsy was performed and showed evidence of myocarditis, which was attributed to viral pneumonia and was not attributed to treatment. Three patients died of progressive disease after treatment on subsequent systemic therapy. Of the 3 patients with mucosal melanoma, all remain without evidence of disease.
Figure 2: Kaplan-Meier plots of overall survival (A) and recurrence free survival (B).

A. The median follow-up time for OS is 37.87 months (33.2–43.47). Median OS has not been reached. B. The median follow-up time for RFS is 29.17 months (17.27–36.8). Median RFS has not been reached.
Neoadjuvant treatment positively modulates the tumor microenvironment
In patients with residual pathologic disease at time of surgery, IHC was performed on 13 paired samples, at baseline and from the surgical specimen. Treatment was associated with a significant increase in the % of CD8 T cells (p=0.04) in the tumor microenvironment (TME; Figure 3A). There was also an increase in the % of T cells (p=0.1), including CD4+ (p=0.3), though this did not reach statistical significance. No significant differences were observed when separating the paired data according to recurrence status. The % of Treg cells (p=0.03) and CD11b + myeloid cells (p=0.047) were also significantly increased (Figure 3B and 3C). No significant differences were observed when separating the paired data according to recurrence status. At the time of surgery, PD-1 expressing non-tumor cells were increased (p=0.04), and PD-L1 expressing non-tumor cells were also increased (p=0.02) (Figure 4A and 4B). The PD-1/PD-L1 interaction score was significantly increased (p=0.008) with treatment (Figure 3C). Similar trends were noted when evaluating the paired patient samples, regardless of recurrence. Tumor cells expressing IDO1 and HLA-DR+ did not change significantly after treatment (p=0.2). We evaluated T-cell exhaustion markers on tumor infiltrating lymphocytes (TIL) by flow cytometry both pre- and post-neoadjuvant therapy. CD8+ T cells expressing both PD-1 and TIM-3 were significantly decreased with treatment (p=0.014), as were CD4+ PD1+TIM-3+ cells (p=0.001) (Figure 5A and 5B). No significant changes in LAG-3 expression were noted.
Figure 3:

Treatment is associated with a significant increase in CD8+ T cells (A), T regulatory (B) and myeloid cells (C) in the TME.
Figure 4:

Treatment is associated with a profound increase in PD-1 (A) and PD-L1 expression (B) in non-tumor cells, and an increased PD-1/PD-L1 interaction score (C).
Figure 5:

Treatment is associated with decreased PD-1+TIM-3+ CD8 (A) and CD8 T cells (B).
pCR is associated with intratumoral PD-1/PDL-1 and HLA-DR expression at baseline
We sought to determine the biologic correlates of pCR. Immunohistochemistry (IHC) and flow cytometry (FC) on tumor samples were performed. In 14 samples (5 with pCR, and 9 without pCR) higher baseline levels of non-tumor cell PD-1 (Figure 6A) and a higher PD-1/PD-L1 interaction score (Figure 6B) were associated with pCR (p= 0.01 and 0.002, respectively). Higher baseline values of HLA-DR on non-tumor cells was also associated with pCR (p=0.008) (Figure 6C).
Figure 6:

pCR is associated with increased PD-1+ expression (A), a higher PD-1/PD-L1 interaction score (B) and higher HLA-DR expression (C) in non-tumor cells at baseline.
Neoadjuvant treatment impacts the circulating immune system
We investigated the impact of treatment on the immune compartment in peripheral blood at baseline and on therapy (at 6 weeks and at 12 weeks). Treatment was associated with decreased CD4+PD1+ cells (p=0.0003) at weeks 6 and 12, and a dramatic increase in CD8+Ki-67+ (p=0.0006) and CD8+PD1+Ki-67+ (p=0.002) cells at week 6. Marked increases in other activated T-cell phenotypes were seen with treatment, including CD4+ICOS+ (p=0.0054), and CD8+CD38+HLA-DR+ (p=0.0005) cells (Supplementary Figure 2). We did not note any association of circulating parameters measured and pCR.
Discussion
The results of this phase Ib/II trial demonstrate significant clinical activity of the combination of pembrolizumab and HDI in the neoadjuvant setting. The ORR of 73.3% and 43% pCR rate compare favorably with the results of previously reported neoadjuvant trials of HDI in melanoma as monotherapy, and in combination with ipilimumab, as well as neoadjuvant studies that have tested ipilimumab, anti-PD1 antibodies and other combinations.9,14,37–39 Neoadjuvant HDI monotherapy using the standard FDA-approved regimen given intravenously for 4 weeks (as tested here) had an ORR of 55% and pCR of 15%.37 Neoadjuvant ipilimumab monotherapy showed an ORR of 9%, with 0% pCR in a subsequent trial.14 Combining ipilimumab and HDI resulted in a higher preoperative radiological response rate of 36%, and pCR of 32%.9 With the advent of anti-PD1, pembrolizumab was evaluated in the neoadjuvant setting, showing a 30% complete or near complete (<10% viable tumor) pathologic response rate after a single treatment dose.8 A phase II study compared neoadjuvant nivolumab (3 mg/kg) (Arm A) to neoadjuvant ipilimumab (3 mg/kg) plus nivolumab (1 mg/kg) (Arm B).12 Arm A was associated with pCR and ORR both of 25%, compared to pCR 45% and ORR 73% in Arm B.12 Combination neoadjuvant ipilimumab (3 mg/kg) and nivolumab (1 mg/kg) was tested in a second study (OpAcin) and was also associated with a high pathologic response rate, though 90% of patients experienced one or more grade 3/4 adverse events, illustrating the need to evaluate alternative less toxic combination regimens in the neoadjuvant setting.11 More recently, the phase II OPACIN-NEO trial determined that the regimen consisting of 2 doses of ipilimumab 1 mg/kg and nivolumab 3 mg/kg was favored for further evaluation based on lower toxicity and comparable efficacy to the more toxic dosing regimen utilizing a higher ipilimumab and a lower nivolumab dose, with a 20% grade 3/4 irAE rate and a pathologic response rate of 77%.4 Importantly, none of the 13 patients with a pCR had recurred after a median follow up of 17.6 months, confirming the importance of pCR as a potential predictive biomarker of durable clinical benefit in patients with melanoma, also supported by other reports.5 It is also interesting to note that of the four patients with a major pathologic response, 3 patients remain without evidence of disease, suggesting that a major pathologic response may also have predictive significance.
The median RFS in this study has not been reached at a median follow-up of 29.17 months. While this compares favorably to other reported adjuvant studies, it is difficult to draw any conclusions given our small sample size. At a minimum follow-up of 18 months, the 12-month RFS in Checkmate-238 was 70.5% in the nivolumab group and 60.8% in the ipilimumab group (HR 0.65; 97.56% CI: 0.51–0.83; p<0.001),40 with the RFS benefit sustained at 2441 and 48 months.42 In Keynote-054, at 3-year median follow-up, RFS was 64% in the pembrolizumab arm and 44% in the placebo group (HR 0.56; 95% CI: 0.47–0.68)43.
In this study, a high proportion of patients required HDI dose reduction, both during induction and maintenance therapy. Indeed, this regimen was associated with high rates of AEs, the most common grade 3 AEs being hypophosphatemia and fatigue. Hypophosphatemia has been reported in p to 29% of patients treated with pembrolizumab, and fatigue is frequently reported with HDI. We did not observe any DLTs. Only 4 grade 4 adverse events were reported. One patient died 6 months after completing therapy, following a viral illness. Autopsy revealed pneumonia and myocarditis, and the patient’s death was not attributed to treatment related toxicity. However, given other reports in the literature of immune related cardiotoxicity with PD1 and/or CTLA4 blockade we cannot rule out the possibility of a late immune related toxicity, which is rare but possible with immune checkpoint inhibitors.44 Overall, though this regimen is associated with significant fatigue and constitutional symptoms, as would be expected with HDI, the combination does not appear to have unexpected toxicities not known with HDI or pembrolizumab. On the other hand, our combination demonstrated high ORR and pCR rates (surpassing that of HDI monotherapy37 or anti-PD1 monotherapy8,12), in spite of HDI dose reductions and a significant portion of patients (43%) eventually discontinuing pembrolizumab prior to the one year limit. The rate of pembrolizumab discontinuation was higher in this study than would be expected. This may be due to significant constitutional symptoms with HDI, lowering each patient’s threshold to withstand further toxicity, and the difficulty of assigning attribution with combination therapy.
Mechanistically, treatment significantly increased CD8+ T cells, Treg and myeloid cells in the TME, in line with prior reports.8,12,37 The correlation between proliferation of TIL with Treg and myeloid cells suggests early and rapid upregulation of immunoregulatory feedback loops within the TME. Interestingly, intratumoral Ki-67+ by IHC was not significantly changed, whereas in the peripheral blood, a dramatic increase in CD4+PD1+, CD8+Ki-67+ and CD8+PD1+Ki-67+ was noted. This is similar to observations in prior reports of increased T cell activation in the periphery at 3 weeks following a dose of neoadjuvant pembrolizumab, without a consistent increase in Ki-67 in the intratumoral compartment.8 These observations may indicate that reinvigoration of T cells in the tumor occurs early, with rapid feedback mechanisms to dampen the immune response by week 6 (including increased Treg and myeloid cells). It is also possible that CD4+ and CD8+ T cells (with or without PD-1) are reinvigorated in the periphery with systemic neoadjuvant therapy, before infiltrating the tumor. Intratumoral PD-1 and PD-L1 expression were increased with treatment, as was the PD-1/PDL1 interaction score, which has been shown to be associated with improved response to anti-PD1 immunotherapy.36 Similarly, in this study, an increased PD-L1/PD-L1 interaction score correlates with pCR. Furthermore, increased HLA-DR+ expression is associated with a higher likelihood with pCR, suggesting that higher T cell activation at baseline is associated with improved clinical outcomes.
In conclusion, the combination of neoadjuvant pembrolizumab and HDI appears to have a manageable safety profile and is active in patients with resectable melanoma, with evidence of robust immune modulation within the tumor and in the peripheral blood. An increased PD-1/PD-L1 interaction score and increased HLA-DR expression are associated with pCR, which appears to be a robust prognostic biomarker.
Supplementary Material
Statement of Translational Relevance.
Patients with regionally advanced melanoma continue to have a poor prognosis, and treatment with neoadjuvant therapy is of increasing interest. Furthermore, neoadjuvant treatment allows for rapid assessment of clinical and pathologic response. This study evaluates the efficacy of neoadjuvant pembrolizumab and high dose interferon in patients with regionally advanced, resectable melanoma. Collection of tumor samples at baseline and after 6 weeks of combination treatment, and collection of blood throughout, allows for evaluation of mechanisms of response. Here, we evaluate the impact that combination treatment has on both the circulating immune component, and within the tumor microenvironment, and show that combination treatment modules both. Our correlative analyses show that intratumoral PD-1/PD-L1 interaction and HLA-DR expression are associated with pCR, and that pCR is associated with improved clinical outcomes. These findings may be of translational relevance in further neoadjuvant studies.
Acknowledgments
Funding acknowledgements:
Clinical trial funding:
Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA provided drug and financial support for the study
YGN: Hillman Fellows for Innovative Cancer Research Program funded by the Henry L. Hillman Foundation; Cancer Center Support Grant P30CA04790
LB: Skin SPORE CCSG for IMCPL
Disclosures
YGN: Merck, Pfizer, BMS (research support). Array BioPharma (Advisory Board 2018)
DD:
Merck, Bristol-Myers Squibb, Checkmate Pharmaceuticals, CellSight Technologies, Mecdpacto, Tesaro/GlaxoSmithKline (research support); and Array Biopharma, Checkmate Pharmaceuticals, Incyte, Immunocore, Merck, Shionogi (consulting).
LHB:
StemImmune/Calidi Scientific and Medical Advisory Board, April 6, 2017-present
SapVax Advisory Board meetings 2017–2020
NextCure, Scientific Advisory Board, 2018-present
Western Oncolytics, Scientific Advisory Board, 2018-present
Torque Therapeutics/Repertoire Immune Medicines, Scientific Advisory Board, 2018–2020
Khloris, Scientific Advisory Board, 2020-present
Pyxis, Scientific Advisory Board, 2019-present
Cytomix, Scientific Advisory Board, 2019-present
Vir, Scientific Advisory Board meeting, Feb. 2020
Adaptimmune, Advisory Board meeting, July 2020
RAPT Scientific Advisory Board, 2020-present
Igor Puzanov:
Consulting/advisory relationship for Roche, AbbVie, Inovio, 4SC, Amgen, Amgen (travel and accommodation).
AP, BC, CL, GS, IS, JB, JT, JB, JYK, ZA: employees of Navigate BioPharma Services
Hassane Zarour, MD: Conflict of Interest as of April 2020
Bristol Myers Squibb, Inc., Checkmate, MERCK SHARP & DOHME, Tesaro, GlaxoSmith Kline, Eli Lilly
JMK:
Consulting:
Amgen, Inc., BMS, Checkmate, Novartis
Research:
Amgen, Inc., BMS, Castle Biosciences, Inc., Checkmate, Immunocore LLC, Iovance, Novartis
AAT:
Merck, OncoSec, Genentech-Roche, Bristol Myers Squibb, Clinigen (research support)
Consulting/Advisory Board participation
Merck, Bristol Myers Squibb, Novartis, Genentech-Roche, Array Biopharma, Partner Therapeutics, Sanofi-Genzyme/Regeneron, Pfizer, EMD Serono, NewLink Genetics, BioNTech, Immunocore, Eisai
Footnotes
The remaining authors declare no conflicts of interest.
References
- 1.National Comprehensive Cancer Network: Cutaneous Melanoma (Version 1.2020), 2020 [Google Scholar]
- 2.Long GV, Hauschild A, Santinami M, et al. : Adjuvant Dabrafenib plus Trametinib in Stage III BRAF-Mutated Melanoma. N Engl J Med 377:1813–1823, 2017 [DOI] [PubMed] [Google Scholar]
- 3.Weber J, Glutsch V, Geissinger E, et al. : Neoadjuvant immunotherapy with combined ipilimumab and nivolumab in patients with melanoma with primary or in transit disease. Br J Dermatol, 2019 [DOI] [PubMed] [Google Scholar]
- 4.Rozeman EA, Menzies AM, van Akkooi ACJ, et al. : Identification of the optimal combination dosing schedule of neoadjuvant ipilimumab plus nivolumab in macroscopic stage III melanoma (OpACIN-neo): a multicentre, phase 2, randomised, controlled trial. Lancet Oncol 20:948–960, 2019 [DOI] [PubMed] [Google Scholar]
- 5.Menzies AM, Rozeman EA, Amaria RN, et al. : Pathological response and survival with neoadjuvant therapy in melanoma: A pooled analysis from the International Neoadjuvant Melanoma Consortium (INMC). Journal of Clinical Oncology 37:9503–9503, 2019 [DOI] [PubMed] [Google Scholar]
- 6.Mahuron K LL, Daud A, Alvarado M: Treatment outcomes of neoadjuvant immunotherapy in patients with locally advanced melanoma. Annals of Surgical Oncology 26:S7, 2019 [Google Scholar]
- 7.Long GV, Saw RPM, Lo S, et al. : Neoadjuvant dabrafenib combined with trametinib for resectable, stage IIIB-C, BRAF(V600) mutation-positive melanoma (NeoCombi): a single-arm, open-label, single-centre, phase 2 trial. Lancet Oncol 20:961–971, 2019 [DOI] [PubMed] [Google Scholar]
- 8.Huang AC, Orlowski RJ, Xu X, et al. : A single dose of neoadjuvant PD-1 blockade predicts clinical outcomes in resectable melanoma. Nat Med 25:454–461, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tarhini A, Lin Y, Lin H, et al. : Neoadjuvant ipilimumab (3 mg/kg or 10 mg/kg) and high dose IFN-alpha2b in locally/regionally advanced melanoma: safety, efficacy and impact on T-cell repertoire. J Immunother Cancer 6:112, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jakub JW, Racz JM, Hieken TJ, et al. : Neoadjuvant systemic therapy for regionally advanced melanoma. J Surg Oncol 117:1164–1169, 2018 [DOI] [PubMed] [Google Scholar]
- 11.Blank CU, Rozeman EA, Fanchi LF, et al. : Neoadjuvant versus adjuvant ipilimumab plus nivolumab in macroscopic stage III melanoma. Nat Med 24:1655–1661, 2018 [DOI] [PubMed] [Google Scholar]
- 12.Amaria RN, Reddy SM, Tawbi HA, et al. : Neoadjuvant immune checkpoint blockade in high-risk resectable melanoma. Nat Med 24:1649–1654, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Amaria RN, Prieto PA, Tetzlaff MT, et al. : Neoadjuvant plus adjuvant dabrafenib and trametinib versus standard of care in patients with high-risk, surgically resectable melanoma: a single-centre, open-label, randomised, phase 2 trial. Lancet Oncol 19:181–193, 2018 [DOI] [PubMed] [Google Scholar]
- 14.Tarhini AA, Edington H, Butterfield LH, et al. : Immune monitoring of the circulation and the tumor microenvironment in patients with regionally advanced melanoma receiving neoadjuvant ipilimumab. PLoS One 9:e87705, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spring L, Fell G, Arfe A, et al. : Abstract GS2–03: Pathological complete response after neoadjuvant chemotherapy and impact on breast cancer recurrence and mortality, stratified by breast cancer subtypes and adjuvant chemotherapy usage: Individual patient-level meta-analyses of over 27,000 patients. Cancer Research 79:GS2–03-GS2–03, 2019 [Google Scholar]
- 16.Spagnolo F, Croce E, Boutros A, et al. : Neoadjuvant treatments in patients with high-risk resectable stage III/IV melanoma. Expert Rev Anticancer Ther:1–11, 2020 [DOI] [PubMed] [Google Scholar]
- 17.Pelster MS, Amaria RN: Neoadjuvant Immunotherapy for Locally Advanced Melanoma. Curr Treat Options Oncol 21:10, 2020 [DOI] [PubMed] [Google Scholar]
- 18.Sun J, Kirichenko DA, Zager JS, et al. : The emergence of neoadjuvant therapy in advanced melanoma. Melanoma Manag 6:MMT27, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O’Donnell JS, Hoefsmit EP, Smyth MJ, et al. : The Promise of Neoadjuvant Immunotherapy and Surgery for Cancer Treatment. Clin Cancer Res 25:5743–5751, 2019 [DOI] [PubMed] [Google Scholar]
- 20.Liu JY, Lowe M: Neoadjuvant Treatments for Advanced Resectable Melanoma. J Surg Oncol 119:216–221, 2019 [DOI] [PubMed] [Google Scholar]
- 21.Khunger A, Buchwald ZS, Lowe M, et al. : Neoadjuvant therapy of locally/regionally advanced melanoma. Ther Adv Med Oncol 11:1758835919866959, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Amaria RN, Menzies AM, Burton EM, et al. : Neoadjuvant systemic therapy in melanoma: recommendations of the International Neoadjuvant Melanoma Consortium. Lancet Oncol 20:e378–e389, 2019 [DOI] [PubMed] [Google Scholar]
- 23.Tarhini AA, Lee SJ, Hodi FS, et al. : Phase III Study of Adjuvant Ipilimumab (3 or 10 mg/kg) Versus High-Dose Interferon Alfa-2b for Resected High-Risk Melanoma: North American Intergroup E1609. J Clin Oncol 38:567–575, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tarhini AA, Lee SJ, Li X, et al. : E3611-A Randomized Phase II Study of Ipilimumab at 3 or 10 mg/kg Alone or in Combination with High-Dose Interferon-alpha2b in Advanced Melanoma. Clin Cancer Res 25:524–532, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Davar D, Wang H, Chauvin JM, et al. : Phase Ib/II Study of Pembrolizumab and Pegylated-Interferon Alfa-2b in Advanced Melanoma. J Clin Oncol:JCO1800632, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Najjar YG, Puligandla M, Lee SJ, et al. : An updated analysis of 4 randomized ECOG trials of high-dose interferon in the adjuvant treatment of melanoma. Cancer 125:3013–3024, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tarhini AA, Gogas H, Kirkwood JM: IFN-alpha in the treatment of melanoma. J Immunol 189:3789–93, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ferrantini M, Capone I, Belardelli F: Interferon-alpha and cancer: mechanisms of action and new perspectives of clinical use. Biochimie 89:884–93, 2007 [DOI] [PubMed] [Google Scholar]
- 29.Zitvogel L, Galluzzi L, Kepp O, et al. : Type I interferons in anticancer immunity. Nat Rev Immunol 15:405–14, 2015 [DOI] [PubMed] [Google Scholar]
- 30.Curtsinger JM, Valenzuela JO, Agarwal P, et al. : Type I IFNs provide a third signal to CD8 T cells to stimulate clonal expansion and differentiation. J Immunol 174:4465–9, 2005 [DOI] [PubMed] [Google Scholar]
- 31.Spaapen RM, Leung MY, Fuertes MB, et al. : Therapeutic activity of high-dose intratumoral IFN-beta requires direct effect on the tumor vasculature. J Immunol 193:4254–60, 2014 [DOI] [PubMed] [Google Scholar]
- 32.Fuertes MB, Kacha AK, Kline J, et al. : Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med 208:2005–16, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Diamond MS, Kinder M, Matsushita H, et al. : Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med 208:1989–2003, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iwai Y, Ishida M, Tanaka Y, et al. : Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A 99:12293–7, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khunger A, Sarikonda G, Tsau J, et al. : Multimarker scores of Th1 and Th2 immune cellular profiles in peripheral blood predict response and immune related toxicity with CTLA4 blockade and IFNalpha in melanoma. Transl Oncol 14:101014, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johnson DB, Bordeaux J, Kim JY, et al. : Quantitative Spatial Profiling of PD-1/PD-L1 Interaction and HLA-DR/IDO-1 Predicts Improved Outcomes of Anti-PD-1 Therapies in Metastatic Melanoma. Clin Cancer Res 24:5250–5260, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moschos SJ, Edington HD, Land SR, et al. : Neoadjuvant treatment of regional stage IIIB melanoma with high-dose interferon alfa-2b induces objective tumor regression in association with modulation of tumor infiltrating host cellular immune responses. J Clin Oncol 24:3164–71, 2006 [DOI] [PubMed] [Google Scholar]
- 38.Huang AC, Orlowski RJ, Xu X, et al. : A single dose of neoadjuvant PD-1 blockade predicts clinical outcomes in resectable melanoma. Nature Medicine:1, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Blank CU, Rozeman EA, Fanchi LF, et al. : Neoadjuvant versus adjuvant ipilimumab plus nivolumab in macroscopic stage III melanoma. Nature medicine:1, 2018 [DOI] [PubMed] [Google Scholar]
- 40.Weber J, Mandala M, Del Vecchio M, et al. : Adjuvant Nivolumab versus Ipilimumab in Resected Stage III or IV Melanoma. N Engl J Med 377:1824–1835, 2017 [DOI] [PubMed] [Google Scholar]
- 41.Weber JAoOsv-vam:
- 42.Ascierto PA, Del Vecchio M, Mandala M, et al. : Adjuvant nivolumab versus ipilimumab in resected stage IIIB-C and stage IV melanoma (CheckMate 238): 4-year results from a multicentre, double-blind, randomised, controlled, phase 3 trial. Lancet Oncol 21:1465–1477, 2020 [DOI] [PubMed] [Google Scholar]
- 43.Eggermont AMJCOS:
- 44.Johnson DB, Balko JM, Compton ML, et al. : Fulminant Myocarditis with Combination Immune Checkpoint Blockade. N Engl J Med 375:1749–1755, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.