

Abstract
Mutant-selective KRASG12C inhibitors, such as MRTX849 (adagrasib) and AMG 510 (sotorasib), have demonstrated efficacy in KRASG12C-mutant cancers including non-small cell lung cancer (NSCLC). However, mechanisms underlying clinical acquired resistance to KRASG12C inhibitors remain undetermined. To begin to define the mechanistic spectrum of acquired resistance, we describe a KRASG12C NSCLC patient who developed polyclonal acquired resistance to MRTX849 with the emergence of 10 heterogeneous resistance alterations in serial cell-free DNA spanning four genes (KRAS, NRAS, BRAF, MAP2K1), all of which converge to reactivate RAS-MAPK signaling. Notably, a novel KRASY96D mutation affecting the switch-II pocket, to which MRTX849 and other inactive-state inhibitors bind, was identified that interferes with key protein-drug interactions and confers resistance to these inhibitors in engineered and patient-derived KRASG12C cancer models. Interestingly, a novel, functionally distinct tri-complex KRASG12C active-state inhibitor RM-018 retained the ability to bind and inhibit KRASG12C/Y96D and could overcome resistance.
Keywords: KRAS G12C, acquired resistance, MRTX849, NSCLC
INTRODUCTION
The development of compounds that bind covalently to cysteine 12 in KRASG12C cancers has ushered in a new era in efforts to target KRAS directly. Biochemically these agents lock KRAS in its inactive GDP-bound conformation, thereby inhibiting downstream signaling leading to preclinical anti-tumor responses (1–3). The lead clinical compounds sotorasib (AMG 510) and adagrasib (MRTX849) have advanced rapidly and demonstrated tolerability and single-agent activity across KRASG12C-mutant cancers (4,5). In patients with advanced non-small cell lung cancer (NSCLC) harboring KRASG12C (which comprises approximately 13% of all lung adenocarcinoma), AMG 510 and MRTX849 have demonstrated meaningful efficacy with objective response rates of 37% and 45%, and disease control rates of 81% and 96%, respectively (6,7). AMG 510 has recently received the Breakthrough Therapy designation from the U. S. Food and Drug Administration for the treatment of patients with advanced KRASG12C-mutant NSCLC following at least one prior systemic therapy. Multiple ongoing trials seek to augment responses to KRASG12C inhibitors through combination strategies.
Preclinical studies with MRTX849 and other KRASG12C inhibitors have suggested several mechanisms of upfront resistance, including reactivation of ERK-dependent signaling to bypass KRASG12C blockade (4). Prior work by our group and others has identified adaptive RAS pathway feedback reactivation as a key mechanism of primary resistance to KRASG12C inhibition (8–13). However, the key mechanisms of clinical acquired resistance to KRASG12C inhibitors are currently unknown. Here, as an initial effort to characterize the clinical landscape of potential acquired resistance mechanisms to KRASG12C inhibitors, we present a KRASG12C-mutant NSCLC patient who developed acquired resistance to MRTX849, characterized by the emergence of 10 individual resistance alterations involving four RAS-MAPK genes. All of these resistance alterations converge to reactivate RAS-MAPK signaling, implicating this as a potential central mechanism of acquired resistance. We also identify a novel KRASY96D resistance mutation in the switch-II pocket of KRAS through serial cell-free DNA analysis. Through structural modeling and in vitro functional studies, we find that KRASY96D confers resistance to multiple KRASG12C inhibitors currently in clinical development, but identify a novel active-state KRASG12C inhibitor RM-018 which is able to overcome KRASG12C/Y96D-mediated resistance.
RESULTS
Heterogeneous acquired resistance alterations converge on RAS-MAPK reactivation
A 67-year-old female with metastatic KRASG12C-mutant NSCLC was treated on the dose expansion cohort of the phase 1 study of MRTX849 (NCT03785249) (Methods; further detailed in Supplementary Methods). Initial scans showed a 32% reduction in tumor size (by RECIST v1.1), but after approximately 4 months of treatment the patient developed progressive disease, and the patient discontinued therapy at 5.5 months (Fig. 1A). In order to identify putative mechanisms of acquired resistance to MRTX849 in this patient, we assessed serial cell-free DNA (cfDNA) using a targeted next-generation sequencing assay (Guardant360, Guardant Health) and droplet digital PCR (ddPCR). Upon development of acquired resistance, the original KRASG12C and TP53F338fs variants present in pre-treatment tumor and cfDNA were again detected in cfDNA, but were accompanied by the emergence of 10 distinct mutations affecting RAS-MAPK components KRAS, NRAS, BRAF, and MAP2K1 (which encodes the MEK1 protein) identified across cfDNA specimens obtained after disease progression (Fig. 1B, Supplementary Table S1). The lower allele frequencies of these alterations relative to the truncal KRASG12C and TP53 mutations are consistent with the emergence of these mutations in heterogeneous subclonal populations. These included three activating NRAS mutations (NRASQ61L, NRASQ61K, NRASQ61R), which can drive active RAS signaling in a KRAS-independent manner, and BRAFV600E, which can maintain MAPK signaling downstream of KRASG12C in the presence of MRTX849 (Supplementary Fig. S1). Three MAP2K1 mutations (MAP2K1K57N, MAP2K1Q56P, MAP2K1E102-I103del) previously demonstrated to be activating and known to be involved in resistance to upstream MAPK pathway inhibitors (i.e. BRAF inhibitors) were also identified (14,15).
Figure 1. Acquired resistance to KRASG12C inhibitor MRTX849 (adagrasib).
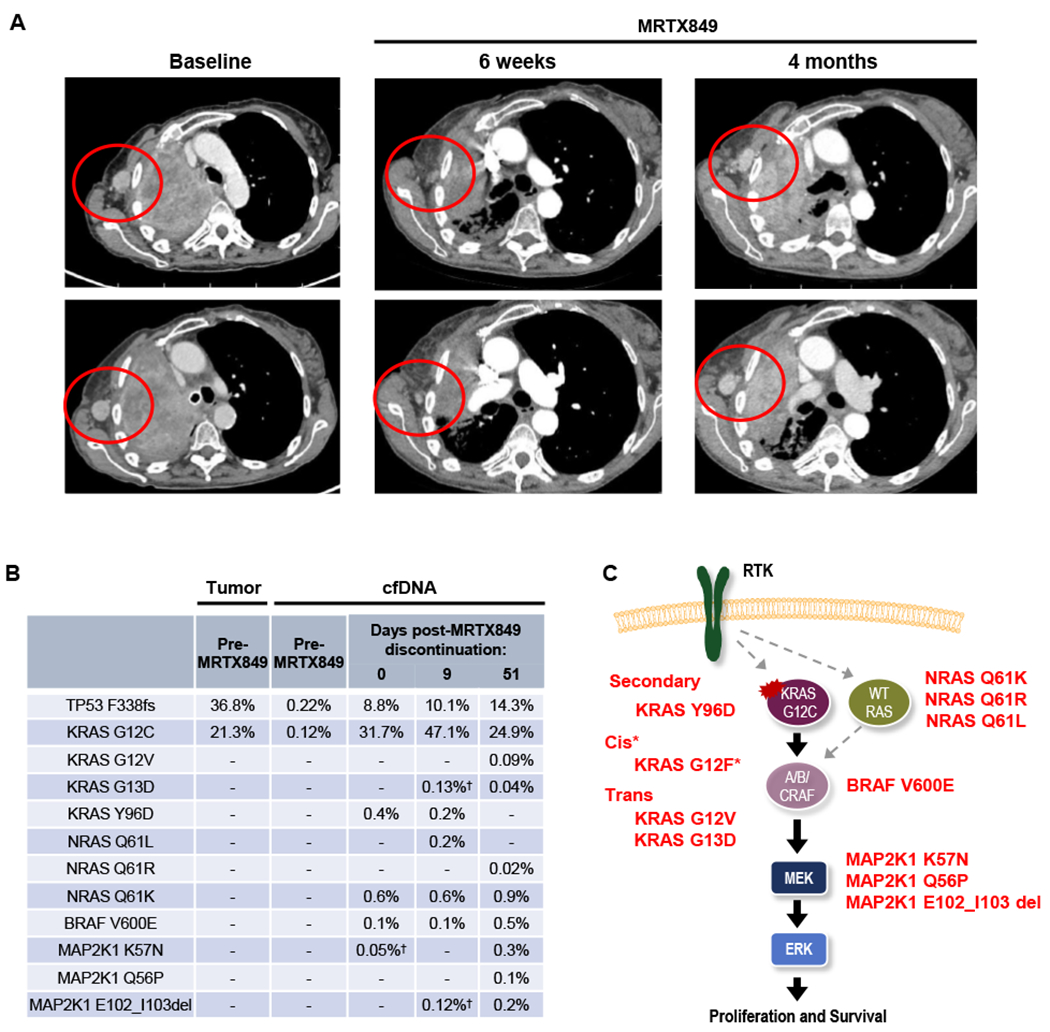
A, Computed tomography (CT) images of the patient’s axillary lymph node metastasis at baseline, during response to MRTX849, and at progression on MRTX849. B, Variant allele fractions (VAFs) of mutations detected in the patient’s serial plasma samples. † indicates the mutations were detected by digital droplet PCR but not by plasma NGS. C, Alterations detected in post-MRTX849 cfDNA include acquired mutations in KRAS as well as multiple components of the MAPK signaling cascade. *KRASG12F represents a potential resistance mechanism supported by limited sequencing reads, as shown in Supplementary Figure S2.
Additionally, three KRAS mutations emerged in the post-progression cfDNA. Two of these mutations are known activating mutations KRASG13D and KRASG12V, and mutant-selective KRASG12C inhibitors have previously been shown to be ineffective against these mutations (4,16). A deeper analysis of individual sequencing reads from cfDNA suggested that these mutations seemed to occur in trans to the original KRASG12C mutation (Supplementary Figs. S2A and B), likely arising in the remaining wild type copy of KRAS, which appeared to be retained based on pre-treatment tumor sequencing (Supplementary Table S2). However, it is not possible from the cfDNA data to confirm that these mutations co-exist in cells that also harbor the original KRASG12C mutation. Notably, a single, well-supported family of sequencing reads from the same original template molecule showed the concurrent presence of both nucleotide changes corresponding to KRASG12C and KRASG12V in cis on the same strand, which would encode for a KRASG12F mutation. While it is not possible to confirm the presence of this mutation based on a single read family, this finding raises the possibility that cis mutations resulting in “loss” of the original KRASG12C mutation and conversion to a different KRAS mutation might be another potential mechanism of resistance. Notably, all putative resistance mutations identified are predicted to converge on reactivation of RAS-MAPK pathway signaling, suggesting that this may represent a common primary mechanism of acquired resistance to KRASG12C inhibitors (Fig. 1C).
Interestingly, the third KRAS mutation identified, KRASY96D, represents a novel mutation that is not known to be activating. Notably, while KRAS is the most commonly mutated oncogene in human cancer, a search of two large tumor mutational databases—COSMIC and GENIE, which collectively contain >450,000 molecularly characterized cancers (17,18)—did not reveal a single previously identified mutation at the KRASY96 locus among >75,000 cases with documented KRAS mutations (Supplementary Table S3). However, the Y96 residue is associated with the Switch-II pocket to which MRTX849 and other inactive-state KRASG12C inhibitors bind, suggesting that the previously undescribed Y96D mutation may have a novel and specific role in driving resistance to KRASG12C inhibitors.
Structural Modeling of KRASG12C/Y96D
To understand the significance of the acquired KRASY96D mutation, we performed structural modeling of the G12C-mutant and G12C/Y96D-double mutant KRAS proteins bound to the KRASG12C inhibitors MRTX849, AMG 510, and ARS-1620 (Fig. 2). These three inhibitors bind the GDP-state of KRASG12C and exploit a cryptic pocket formed by the central beta sheet of RAS and Switch II [first identified by Ostrem et al. (3)]. To determine the effects of the amino acid substitution at the Y96 locus, crystal structures of MRTX849, AMG 510, and ARS-1620 bound to KRASG12C were modeled for interactions with the Y96 residue within the Switch II pocket. (16,19–21). The hydroxyl group of Y96 forms a direct hydrogen bond with the pyrimidine ring of MTRX849, which is abolished with the Y96D mutation. Y96D also disrupts the water mediated hydrogen bond between Y96 and a carboxyl group on AMG 510. Finally, while Y96 does not form a direct hydrogen bond with ARS-1620, it stabilizes the interaction with ARS-1620 through pi-stacking with the phenyl ring of Y96, which is disrupted with the Y96D mutation. Additionally, by introducing a negatively charged amino acid, the Y96D mutation changes the hydrophobic nature of the binding pocket for all three compounds to a substantially more hydrophilic pocket, which may further destabilize binding.
Figure 2. Structural basis for resistance to KRAS G12C inhibition conferred by KRASY96D.
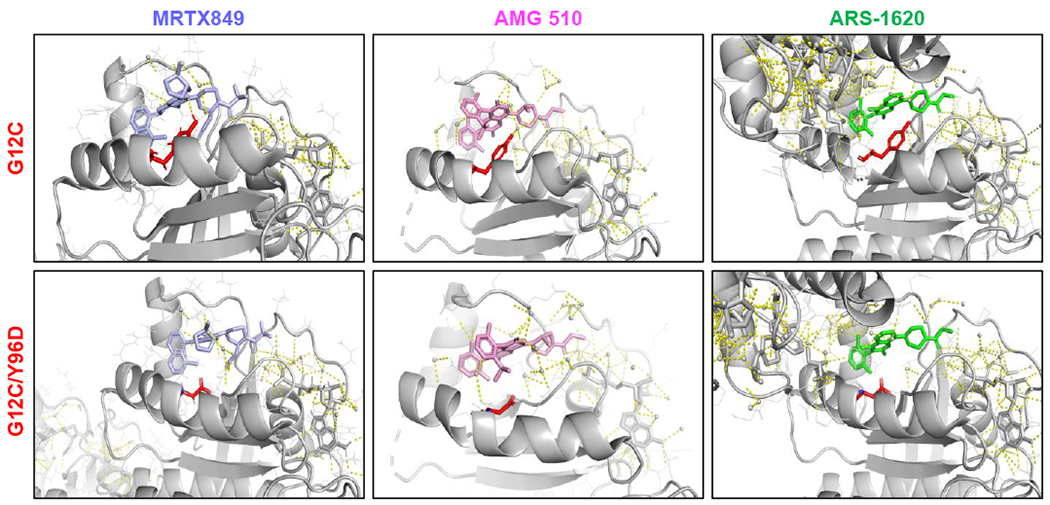
Shown are the modeled crystal structures of MRTX849 (6UT0), AMG 510 (6OIM), and ARS-1620 (5V9U) bound to KRASG12C (top panels) and KRASG12C/Y96D (bottom panels), highlighting the loss of the hydrogen bonds between MRTX849 or AMG 510 and the Y96 residue and the disruption of the switch II pocket dynamics between ARS-1620 and KRASG12C/Y96D.
Functional characterization of KRASY96D
To assess whether KRASY96D can mediate resistance to MRTX849 and other inactive-state KRASG12C inhibitors, we expressed KRASG12C or the KRASG12C/Y96D double mutant in NCI-H358 (KRASG12C-mutant NSCLC), MIA PaCa-2 (KRASG12C-mutant pancreatic ductal adenocarcinoma), as well as Ba/F3 cells, which lack endogenous KRASG12C, but become oncogene-dependent upon withdrawal of IL-3. In cell viability assays, relative to KRASG12C-expressing controls, cells expressing KRASG12C/Y96D showed marked resistance to three KRASG12C inhibitors, with IC50 shifts of >100-fold for MRTX849 and AMG 510 and ~20-fold for ARS-1620 (Fig. 3A, Supplementary Table S4).
Figure 3. Cellular characterization of KRASY96D in KRASG12C-mutant models.
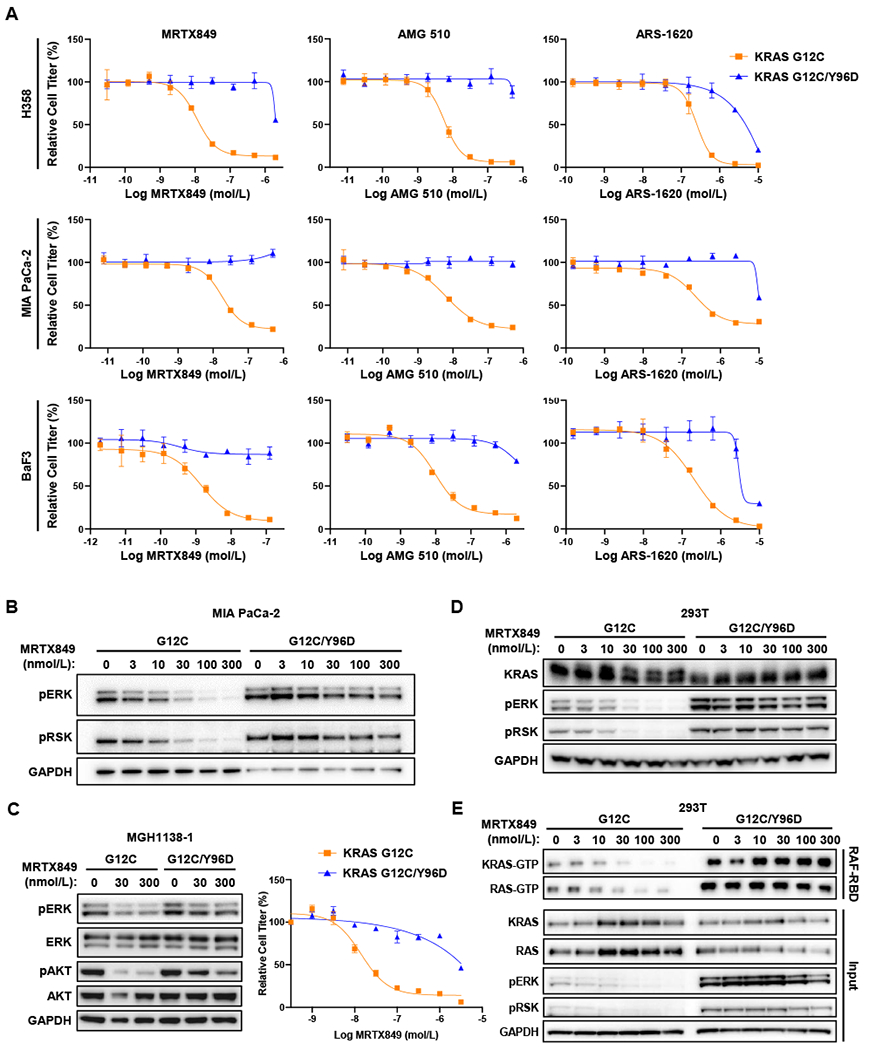
A, Cell viability assays performed with NCI-H358, MIA PaCa-2 and Ba/F3 cells infected with retrovirus packaging KRAS (G12C or G12C/Y96D). Cell lines were treated with indicated drugs for 72 hours and the viabilities were measured with CellTiter-Glo. B, Western blot analysis was performed after treating MIA PaCa-2 cells stably expressing KRASG12C or KRASG12C/Y96D with MRTX849 for 4 hours. C, MGH1138-1 cells expressing KRASG12C or KRASG12C/Y96D were treated with MRTX849 for 4 hours and subjected to western blot analysis (left) and cell viability assay following 72 hours of treatment with the indicated concentrations of MRTX849 (right). D, Western blot analysis of HEK293T cells transiently expressing KRAS mutants after treatment with MRTX849 for 4 hours. E, RAS-GTP pulldown was performed after treating HEK293T stably expressing KRAS mutants with MRTX849.
Consistent with the effects on cell viability, RAS-MAPK pathway activity, as measured by levels of phosphorylated (p)ERK and pRSK were sustained in KRASG12C/Y96D-expressing MIA PaCa-2 cells even at high concentrations of MRTX849, relative to cells expressing KRASG12C alone (Fig. 3B). Similarly, in KRASG12C-mutant NSCLC cells in which PI3K signaling is driven by mutant KRAS, including an existing patient-derived model MGH1138-1, persistent pERK and pAKT levels were observed with KRASG12C/Y96D in the presence of MRTX849, relative to KRASG12C expression alone (Fig. 3C; Supplementary Fig. S3). KRASG12C/Y96D also drove marked resistance to MRTX849 in the patient-derived MGH1138-1 model. Furthermore, in 293T cells, which lack endogenous KRASG12C expression, MRTX849 was unable to inhibit pERK levels driven by KRASG12C/Y96D (Fig. 3D). Since MRTX849 and other inactive-state KRASG12C inhibitors bind covalently to KRASG12C, an electrophoretic mobility shift of drug-adducted KRASG12C can be observed upon drug binding due to increased molecular weight. However, this mobility shift was no longer observed when 293T cells expressing KRASG12C/Y96D were treated with MRTX849, suggesting that the Y96D mutation may abrogate inhibitor binding. Notably, KRASG12C/Y96D appeared to have higher basal activation than KRASG12C, as measured by a higher proportion of the active GTP-bound form of KRAS, though activation still appeared to be partly dependent on upstream pathway input (Supplementary Figs. S4A and B). Finally, while a decrease in guanosine triphosphate (GTP)-bound KRAS (representing the active state) was observed in KRASG12C-expressing cells treated with MRTX849, levels of active GTP-bound KRAS were maintained in KRASG12C/Y96D expressing cells (Fig. 3E) (22). These results suggest that the KRASY96D mutation disrupts KRASG12C inhibitor binding, leading to sustained KRAS signaling and therapeutic resistance.
The active state KRASG12C inhibitor RM-018 overcomes KRASG12C/Y96D
As KRASG12C/Y96D conferred resistance to multiple KRASG12C inhibitors currently in clinical development, suggestive of shared vulnerability for this class of inhibitors, we sought to identify whether a structurally and functionally distinct KRASG12C inhibitor might retain potency against this resistance mutation. RM-018 is a novel KRASG12C inhibitor which binds specifically to the GTP-bound, active (“RAS(ON)”) state of KRASG12C. RM-018 is a “tri-complex” KRAS inhibitor, which exploits a highly abundant chaperone protein, cyclophilin A, to bind and inhibit KRASG12C, as previously described (Fig. 4A; structure shown in Supplementary Fig. S5) (23,24). Briefly, upon entering the cell, RM-018 forms a “binary complex” with cyclophilin A. This binary complex can associate with the active state of KRASG12C, aided by protein-protein surface interactions between cyclophilin A and KRAS, and forms a covalent bond with KRASG12C in a mutant-selective manner. This resultant “tri-complex” inhibits KRASG12C through binding of cyclophilin A leading to steric occlusion preventing association of downstream effector proteins. Given the markedly different mechanism of action of this class of inhibitor, we hypothesized that the inhibitory activity of RM-018 might be differentially affected by KRASY96D compared to inactive-state KRASG12C inhibitors.
Figure 4. Novel KRAS inhibitor RM-018 overcomes KRASG12C/Y96D.
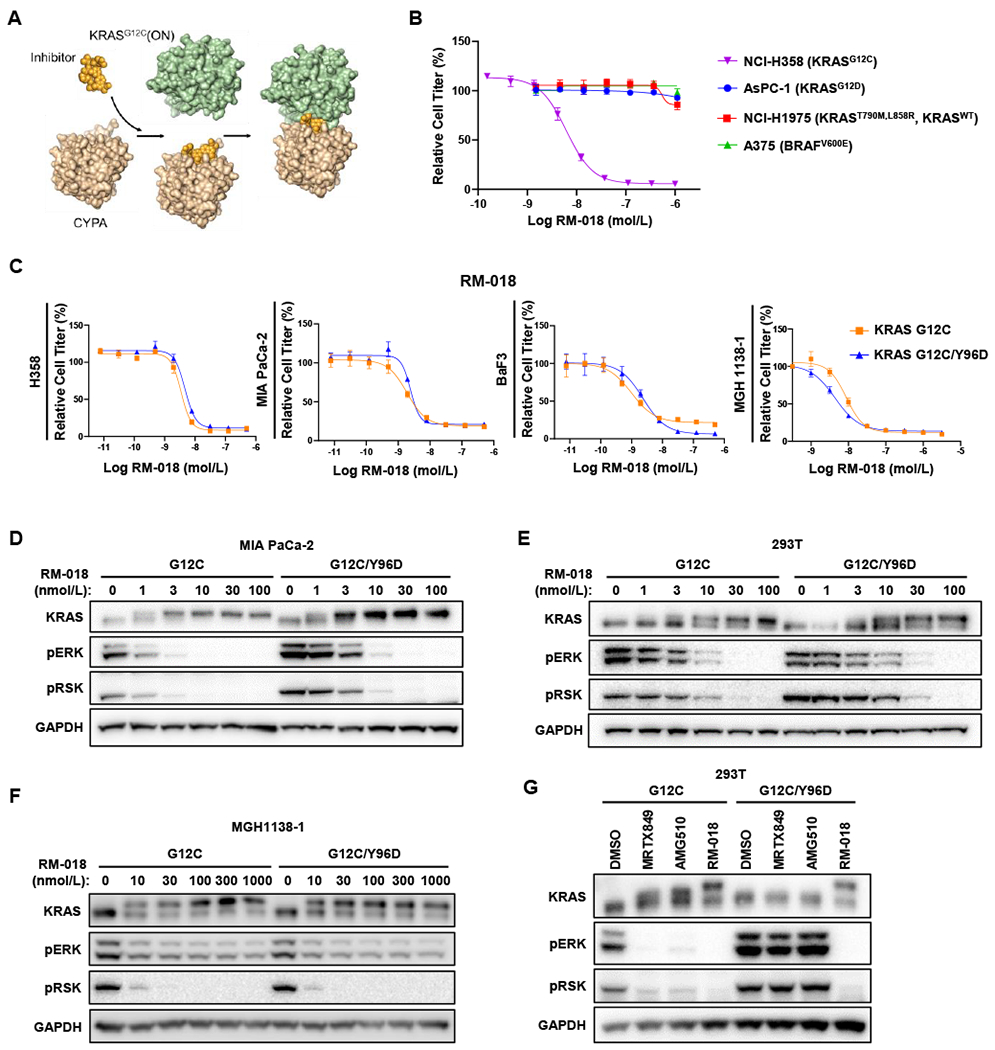
A, Mechanism of action of RM-018. B, RM-018 selectively inhibits cell viability in cells harboring KRASG12C. C, Cell viability assays performed with NCI-H358, MIA PaCa-2, Ba/F3 and MGH1138-1 cells stably infected with KRASG12C or KRASG12C/Y96D treated for 72 hours with RM-018. D and E, Western blot analysis performed in MIA PaCa-2 stably expressing KRASG12C or KRASG12C/Y96D (D) and HEK293T cells transiently expressing KRAS mutant (E) after treatment of RM-018 for 4 hours. F, Western blot analysis of MGH1138-1 cells transiently expressing KRASG12C or KRASG12C/Y96D) after treatment with RM-018 for 4 hours. G, HEK293T cells transiently expressing KRAS mutant were treated with the indicated drug at 100 nmol/L each for 4 hours and then subjected to western blot analysis.
RM-018 demonstrated selectivity for KRASG12C-driven cells, exhibiting low nanomolar potency in KRASG12C-mutant H358 cells, while not impairing the viability of cells driven by KRASG12D, BRAFV600E, or RTK-driven signaling through wild-type RAS (Fig. 4B). Interestingly, while KRASG12C/Y96D expression led to marked IC50 shifts of >100-fold for MRTX849 and AMG 510 and ~20-fold for ARS-1620 (Fig. 3A) relative to KRASG12C expression alone, the efficacy of RM-018 on cell viability was largely unaffected by KRASG12C/Y96D expression with IC50 shifts of only ~2-fold (Fig. 4C, Supplementary Table S4). In addition, RM-018 was able to inhibit pERK and pRSK levels with similar potency in the presence of KRASG12C or KRASG12C/Y96D expression in both MIA PaCa-2, 293T cells, and the patient-derived KRASG12C-mutant NSCLC cell line MGH1138-1 (Figs. 4D–F). Inhibition of cell viability by RM-018 was also unaffected by KRASG12C/Y96D expression in the patient-derived MGH1138-1 model. Furthermore, the KRAS mobility shift induced by covalent binding of RM-018 was observed in both cell lines in the presence of either KRASG12C or KRASG12C/Y96D expression, suggesting that binding of RM-018 to KRAS is not abrogated by the KRASY96D mutation. Indeed, while a KRAS mobility shift due to covalent drug binding was observed in 293T cells expressing KRASG12C for MRTX849, AMG 510, and RM-018, only RM-018 exhibited this same mobility shift and was able to inhibit downstream signaling in the presence of the KRASG12C/Y96D mutation (Fig. 4G). Taken together, these data suggest that RM-018 retains the ability to bind and inhibit KRASG12C/Y96D and may represent a potential therapeutic strategy to overcome this acquired resistance mechanism.
DISCUSSION
The arrival of covalent KRASG12C-selective inhibitors in the clinic and early signs of activity demonstrated by MRTX849 and AMG 510 have generated great enthusiasm (4,5). However, our experiences across targeted therapies in lung and other cancers collectively demonstrate that acquired resistance to the KRASG12C inhibitors will represent an inevitable challenge going forward. While preclinical studies have nominated putative mechanisms of upfront resistance including RAS-MAPK pathway reactivation (4,8), mechanisms of acquired resistance to MRTX849 or AMG 510 causing disease relapse in patients remain unknown.
The acquired resistance demonstrated in this patient is instructive in highlighting several points. First, ten distinct resistance alterations arose in this patient, all converging on the reactivation of RAS-MAPK signaling, suggesting that this may be a central common mechanism of acquired resistance. RAS reactivation occurred by multiple different mechanisms, including (1) activation of another RAS isoform (in this case, NRAS); (2) other KRAS activating mutations in trans (G13D, G12V); (3) potential loss of KRAS G12C through a mutational switch to a different KRAS mutation in cis, though supported by limited sequencing reads; and (4) a novel secondary alteration in KRAS (i.e., Y96D) which alters inhibitor binding.
Structural modeling predicted that this KRASY96D mutation disrupts critical hydrogen bonding between the Y96 residue of KRAS and MRTX849. Importantly, we found that this KRASY96D mutation conferred resistance not only to MRTX849 but also to additional KRASG12C-selective inhibitors in clinical development, AMG 510 and ARS-1620, highlighting that mutations affecting the Y96 residue of KRAS may represent a shared vulnerability for the currently available KRASG12C inhibitors. Continued investigation into clinical mechanisms of resistance to KRASG12C inhibitors in larger cohorts of patients will be required to define the spectrum and frequency of KRASY96 (as well as other on-target) mutations. Nonetheless, in light of our observations, it may be necessary to develop novel compounds that are able to target KRASG12C/Y96D in order to overcome resistance to KRASG12C inhibitors in clinic. RM-018 was identified as a novel KRASG12C inhibitor with a distinct mechanism of action for targeting KRAS, which was uniquely able to overcome KRASG12C/Y96D across multiple models. These results suggest that a novel, distinct KRAS inhibitor could theoretically be utilized to target resistance after an acquired KRAS resistance mutation emerges on the initial KRASG12C inhibitor, and support efforts towards rational design of next-generation inhibitors.
Of note, however, the abundance of mutations downstream of RAS suggest that MAPK reactivation alone may be sufficient to drive resistance in at least some KRASG12C cancers. At the time of disease progression on MRTX849, this patient’s plasma biopsy revealed a low allele fraction of KRASY96D particularly when compared to the significantly higher allele fraction of KRASG12C (0.2% vs 9.9%, respectively), suggestive of the presence of polyclonal resistance to MRTX849 (with KRASG12C/Y96D-harboring tumor cells representing a minor subclone). Concordant with this notion and prior reports based on preclinical models, subsequent post-treatment plasma samples in this case revealed numerous mutations affecting various nodes of the RAS-MAPK pathway (including NRAS, BRAF, and MAP2K1 mutations). Thus, rational combinations of KRASG12C inhibitors with downstream MAPK pathway inhibitors may be needed to successfully prevent or overcome resistance. While analysis of additional patients is clearly needed to more fully define the spectrum of potential acquired resistance mechanisms to KRASG12C inhibitors, this study begins to delineate some of the potential resistance alterations that may be observed clinically. Ultimately, an iterative discovery process of identifying mechanisms of resistance and validating in vitro with a detailed structural and molecular understanding should help advance the development of novel strategies for therapeutic targeting of KRAS-mutant cancers.
METHODS
Patient treatment and specimen collection
The patient was treated with MRTX849 dosed 600 mg twice daily on the phase 1 study (KRYSTAL-1) after providing written informed consent (ClinicalTrials.gov identifier: NCT03785249). She had received two prior lines of therapy. All pre- and post-treatment biopsies and genotyping were performed in accordance with the Massachusetts General Hospital (MGH) institutional review board-approved protocol and in accordance with the Declaration of Helsinki. The pre-treatment tumor specimen was analyzed using the MGH SNaPshot next-generation sequencing assay (25). All cfDNA samples were sequenced using the commercially available Guardant360 assay (Guardant Health; Redwood City, CA). More detailed patient history is available in the Supplement.
Cell Lines and Reagents
Ba/F3 cells were obtained from the RIKEN BRC Cell Bank (RIKEN BioResource Center). MGH1138-1 cells were generated from a KRASG12C-mutant NSCLC patient using methods that have been previously described (26). Prior to cell line generation, the patient provided written informed consent to participate in a Dana Farber/Harvard Cancer Center Institutional Review Board-approved protocol giving permission for research to be performed on their sample. The remaining cell lines were obtained from ATCC or the Center for Molecular Therapeutics at the MGH Cancer Center (Boston, MA) which routinely performs cell line authentication testing by SNP and short-tandem repeat analysis. HEK293T cells were maintained in DMEM supplemented with 10% FBS. MIA PaCa-2 and NCI-H358 cells were maintained in DMEM/F12 supplemented with 10% FBS. LU-65 and MGH1138-1 cells were maintained in RPMI supplemented with 10% FBS. Ba/F3 cells were maintained in DMEM supplemented with 10% FBS and 10 ng/mL interleukin-3 (IL-3). KRAS (G12C or G12C/Y96D) gene was inserted in pMXs-Puro Retroviral Expression Vector, which was purchased from Cell Biolabs. Retrovirus packaging mutated KRAS genes were produced with HEK293T cells. After concentration of virus with Retro-Concentin Retro Concentration Reagent (System Biosciences), MIA PaCa-2, NCI-H358 and Ba/F3 cells were infected with the virus packaging either KRAS G12C or G12C/Y96D gene. After 48 hours of incubation, the cells were treated with puromycin (1-2 μ;g/mL) for another 48 hours. IL-3 was withdrawn to select for Ba/F3 cells dependent on mutant KRAS signaling after 48 hours of puromycin treatment. The remaining cells were maintained in media supplemented with puromycin. For transient expression experiments, a day after seeding the cells, pMXs-Puro-KRASG12C or pMXs-Puro-KRASG12C/Y96D vectors were induced with Lipofectamine 2000 Transfection Reagent (ThermoFisher Scientific) following manufacturer’s protocol. After 16-24 hours of incubation, cells were treated with inhibitors for 4 hours. AMG 510 was purchased from MedChemExpress. MRTX849 and ARS-1620 were purchased from Selleck Chemicals. RM-018 was provided by Revolution Medicines (Redwood City, CA, USA), and details of the chemical synthesis can be found in International Patent Application No. PCT/US2020/058841
Cell Viability Assays
Cells lines were seeded in 96-well plate at 2-10 × 103 cells/well depending on cell lines and after 24 treated with a serial dilution of drugs and incubated for 72 hours. Cell viability was measured with CellTiter-Glo (Promega).
Western Blot Analysis
Cell lines were treated with MRTX849, AMG 510 or RM-018 for 4 hours and lysates were prepared as described previously (27). All antibodies were diluted in 5% bovine serum albumin as follows: KRAS (Sigma), phospho-ERK (Thr202/Tyr204,1:1,000, Cell Signaling Technology), p44/42 MAPK (Erk1/2) (1:1000, Cell signaling Technology), phospho-RSK1 (T359+S363, 1:1,000, Abcam), phospho-Akt (Ser473, 1:1000, Cell Signaling Technology), AKT (1:1000, Cell Signaling Technology) and GAPDH (1:1,000, MilliporeSigma).
RAS-GTP pulldown
After indicated inhibitor treatment, RAS activity was assessed by GST-RAF-RBD pulldown (Cell Signaling Technology), followed by western blot analysis with pan-RAS or RAS isoform–specific antibodies. Pulldown samples and whole-cell lysates were resolved on 4-12% Bis-Tris Gels and western blotting was performed using antibodies against KRAS (Sigma) and pan-RAS (Cell Signaling Technology).
Structural Modeling
Publicly available crystal structures of KRASG12C in complex with MRTX849 (PDB:6UT0), AMG 510 (PDB:6OIM), and ARS-1620 (PDB:5V9U) were downloaded from the RCSB Protein Data Bank (PDB)(28). Structures were rendered in PyMol (The PyMOL Molecular Graphics System) and analyzed for hydrogen bonds and other molecular interactions between the KRASG12C inhibitors and the KRAS protein. Structures of Y96 amino acid mutation were generated by Protein Mutagenesis Wizard implemented in PyMol, with one of the backbone dependent rotamers manually selected.
ctDNA extraction and digital droplet PCR
Whole blood was collected by routine phlebotomy in two 10 mL Streck tubes. Plasma was separated within 1-4 days of collection through two different centrifugation steps (the first at room temperature for 10 minutes at 1,600 × g and the second at 3,000 × g for the same time and temperature). Plasma was stored at −80°C until ctDNA extraction. ctDNA was extracted from plasma using the QIAamp Circulating Nucleic Acid Kit (QIAGEN) with 60 min of proteinase K incubation at 60 degrees Celsius. All other steps were performed according to the manufacturer’s instructions. For droplet digital PCR (ddPCR) experiments, DNA template (up to 10 μL, with a total of 20 ng) was added to 12.5 μL of ddPCR Supermix for Probes (Bio-Rad) and 1.25 μL of the custom primer/probe mixture. This reaction mix was added to a DG8 cartridge together with 60 μL of Droplet Generation Oil for Probes (Bio-Rad) and used for droplet generation. Droplets were then transferred to a 96-well plate (Eppendorf) and then thermal cycled with the following conditions: 5 minutes at 95°C, 40 cycles of 94°C for 30 seconds, 55°C (with a few grades difference among assays) for 1 minute followed by 98°C for 10 minutes (Ramp Rate 2°C/sec). Droplets were analyzed with the QX200 Droplet Reader (Bio-Rad) for fluorescent measurement of FAM and HEX probes. Gating was performed based on positive and negative controls, and mutant populations were identified. The ddPCR data were analyzed with QuantaSoft analysis software (Bio-Rad) to obtain Fractional Abundance of the mutant DNA alleles in the wild-type/normal background. The quantification of the target molecule was presented as the number of total copies (mutant plus wild-type) per sample in each reaction. Allelic fraction is calculated as follows: AF % = (Nmut/(Nmut+Nwt))*100), where Nmut is the number of mutant alleles and Nwt is the number of wild-type alleles per reaction. ddPCR analysis of normal control plasma DNA (from cell lines) and no DNA template controls were always included. Probe and primer sequences are available upon request.
Supplementary Material
STATEMENT OF SIGNIFICANCE.
In one of the first reports of clinical acquired resistance to KRASG12C inhibitors, our data suggest polyclonal RAS-MAPK reactivation as a central resistance mechanism. We also identify a novel KRAS switch-II pocket mutation that impairs binding and drives resistance to inactive-state inhibitors but is surmountable by a functionally-distinct KRASG12C inhibitor.
ACKNOWLEDGEMENTS:
RM-018 was kindly provided by Revolution Medicines (Redwood City, CA, USA).
Financial Support:
This research was supported in part by a Mark Foundation for Cancer Research EXTOL Project Grant (JFG, ANH) and a Stand Up To Cancer – American Cancer Society Lung Cancer Dream Team Translational Research Grant (JFG, ANH; grant number: SU2C-AACR-DT17-15). Stand Up To Cancer (SU2C) is a division of the Entertainment Industry Foundation. The indicated SU2C research grant is administered by the American Association for Cancer Research, a scientific partner of SU2C. JJL is partially supported by funding from NIH R01-CA164273. CL is supported by funding from NIH 1F32CA250231-01. NT is partially supported by funding from US-JFCR Scientist Exchange Program. L.B-P is supported by AACR (19-20-45-BARP), Damon Runyon (DRR-62-20), and NIH/NCI (R00 CA215249).
Footnotes
Conflicts of Interest:
JJL has served as a compensated consultant for Genentech, C4 Therapeutics, Blueprint Medicines, Nuvalent, Turning Point Therapeutics, and Elevation Oncology; received honorarium and travel support from Pfizer; received institutional research funds from Hengrui Therapeutics, Turning Point Therapeutics, Neon Therapeutics, Relay Therapeutics, Bayer, Elevation Oncology, Roche, and Novartis; received CME funding from OncLive, MedStar Health, and Northwell Health.
LK is an employee and shareholder of Guardant Health.
DJ has received scientific advisory board fee from Eisai, EMD Serono, Genentech, Ipsen, Novartis, Guardant, Petra Pharma, Mapkure, Vibliome Therapeutics, and Relay Therapeutics, and institutional research funds from Novartis, Genentech, EMD Serono, Eisai, Takeda, Placon Therapeutics, Takeda, Syros, Ribon Therapeutics, Infinity Pharmaceuticals, InventisBio and Amgen.
JFG has served as a compensated consultant or received honoraria from Bristol-Myers Squibb, Genentech, Ariad/Takeda, Loxo/Lilly, Blueprint, Oncorus, Regeneron, Gilead, AstraZeneca, Pfizer, Incyte, Novartis, Merck, Agios, Amgen, and Array; research support from Novartis, Genentech/Roche, and Ariad/Takeda; institutional research support from Bristol-Myers Squibb, Tesaro, Moderna, Blueprint, Jounce, Array Biopharma, Merck, Adaptimmune, Novartis, and Alexo; and has an immediate family member who is an employee with equity at Ironwood Pharmaceuticals.
SJK has served as a compensated consultant for Merck, BMS, Eli Lilly, Natera, Daiichi Sankyo and Pieris Oncology; owns stock/equity in Turning Point Therapeutics, Inc..
LBP is a co-founder/owns stock/equity and consults for Scorpion Therapeutics.
ANH has served as a compensated consultant for Nuvalent, Inc.; received research support from Amgen, Eli Lilly, Roche/Genentech, Relay Therapeutics, Blueprint Medicines, Nuvalent Inc., and Novartis.
RSH has received consulting honoraria from Novartis, Daichii Sankyo, EMD Serono, Boehringer Ingelheim, Tarveda, Apollomics, and has research funding (to institution, not to self) from Agios, Abbvie, Daichii Sankyo, Novartis, Lilly, Mirati, Corvus, Genentech Roche, Exelixis, Turning Point.
RBC has received consulting or speaking fees from Abbvie, Amgen, Array Biopharma/Pfizer, Asana Biosciences, Astex Pharmaceuticals, AstraZeneca, Avidity Biosciences, BMS, C4 Therapeutics, Chugai, Elicio, Erasca, Fog Pharma, Genentech, Guardant Health, Ipsen, Kinnate Biopharma, LOXO, Merrimack, Mirati Therapeutics, Natera, Navire, N-of-one/Qiagen, Novartis, nRichDx, Remix Therapeutics, Revolution Medicines, Roche, Roivant, Shionogi, Shire, Spectrum Pharmaceuticals, Symphogen, Tango Therapeutics, Taiho, Warp Drive Bio, Zikani Therapeutics; holds equity in Avidity Biosciences, C4 Therapeutics, Erasca, Kinnate Biopharma, nRichDx, Remix Therapeutics, and Revolution Medicines; and has received research funding from Asana, AstraZeneca, Lilly, Novartis, and Sanofi.
The remaining authors have no conflicts to disclose.
REFERENCES
- 1.Fell JB, Fischer JP, Baer BR, Blake JF, Bouhana K, Briere DM, et al. Identification of the Clinical Development Candidate MRTX849, a Covalent KRAS(G12C) Inhibitor for the Treatment of Cancer. J Med Chem 2020;63(13):6679–93 doi 10.1021/acs.jmedchem.9b02052. [DOI] [PubMed] [Google Scholar]
- 2.Lanman BA, Allen JR, Allen JG, Amegadzie AK, Ashton KS, Booker SK, et al. Discovery of a Covalent Inhibitor of KRAS(G12C) (AMG 510) for the Treatment of Solid Tumors. J Med Chem 2020;63(1):52–65 doi 10.1021/acs.jmedchem.9b01180. [DOI] [PubMed] [Google Scholar]
- 3.Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013;503(7477):548–51 doi 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hallin J, Engstrom LD, Hargis L, Calinisan A, Aranda R, Briere DM, et al. The KRAS(G12C) Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov 2020;10(1):54–71 doi 10.1158/2159-8290.CD-19-1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, et al. KRAS(G12C) Inhibition with Sotorasib in Advanced Solid Tumors. N Engl J Med 2020;383(13):1207–17 doi 10.1056/NEJMoa1917239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li B, Skoulidis F, Falchook G, Sacher A, Velcheti V, Dy G, et al. PS01.07 Registrational Phase 2 Trial of Sotorasib in KRAS p.G12C Mutant NSCLC: First Disclosure of the Codebreak 100 Primary Analysis. Journal of Thoracic Oncology 2021;16(3):S61 doi 10.1016/j.jtho.2021.01.321. [DOI] [Google Scholar]
- 7.Jänne PA, Rybkin II, Spira AI, Riely GJ, Papadopoulos KP, Sabari JK, et al. KRYSTAL-1: Activity and Safety of Adagrasib (MRTX849) in Advanced/ Metastatic Non–Small-Cell Lung Cancer (NSCLC) Harboring KRAS G12C Mutation. European Journal of Cancer 2020;138:S1–S2 doi 10.1016/S0959-8049(20)31076-5. [DOI] [Google Scholar]
- 8.Ryan MB, Fece de la Cruz F, Phat S, Myers DT, Wong E, Shahzade HA, et al. Vertical Pathway Inhibition Overcomes Adaptive Feedback Resistance to KRAS(G12C) Inhibition. Clin Cancer Res 2020;26(7):1633–43 doi 10.1158/1078-0432.CCR-19-3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Misale S, Fatherree JP, Cortez E, Li C, Bilton S, Timonina D, et al. KRAS G12C NSCLC Models Are Sensitive to Direct Targeting of KRAS in Combination with PI3K Inhibition. Clin Cancer Res 2019;25(2):796–807 doi 10.1158/1078-0432.CCR-18-0368. [DOI] [PubMed] [Google Scholar]
- 10.Santana-Codina N, Chandhoke AS, Yu Q, Malachowska B, Kuljanin M, Gikandi A, et al. Defining and Targeting Adaptations to Oncogenic KRAS(G12C) Inhibition Using Quantitative Temporal Proteomics. Cell Rep 2020;30(13):4584–99 e4 doi 10.1016/j.celrep.2020.03.021. [DOI] [PubMed] [Google Scholar]
- 11.Solanki HS, Welsh EA, Fang B, Izumi V, Darville L, Stone B, et al. Cell Type-specific Adaptive Signaling Responses to KRAS(G12C) Inhibition. Clin Cancer Res 2021. doi 10.1158/1078-0432.CCR-20-3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xue JY, Zhao Y, Aronowitz J, Mai TT, Vides A, Qeriqi B, et al. Rapid non-uniform adaptation to conformation-specific KRAS(G12C) inhibition. Nature 2020;577(7790):421–5 doi 10.1038/s41586-019-1884-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fedele C, Li S, Teng KW, Foster CJR, Peng D, Ran H, et al. SHP2 inhibition diminishes KRASG12C cycling and promotes tumor microenvironment remodeling. J Exp Med 2021;218(1) doi 10.1084/jem.20201414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kinoshita-Kikuta E, Kinoshita E, Ueda S, Ino Y, Kimura Y, Hirano H, et al. Increase in constitutively active MEK1 species by introduction of MEK1 mutations identified in cancers. Biochim Biophys Acta Proteins Proteom 2019;1867(1):62–70 doi 10.1016/j.bbapap.2018.05.004. [DOI] [PubMed] [Google Scholar]
- 15.Gao Y, Chang MT, McKay D, Na N, Zhou B, Yaeger R, et al. Allele-Specific Mechanisms of Activation of MEK1 Mutants Determine Their Properties. Cancer Discov 2018;8(5):648–61 doi 10.1158/2159-8290.CD-17-1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019;575(7781):217–23 doi 10.1038/s41586-019-1694-1. [DOI] [PubMed] [Google Scholar]
- 17.Sondka Z, Bamford S, Cole CG, Ward SA, Dunham I, Forbes SA. The COSMIC Cancer Gene Census: describing genetic dysfunction across all human cancers. Nat Rev Cancer 2018;18(11):696–705 doi 10.1038/s41568-018-0060-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Consortium APG. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov 2017;7(8):818–31 doi 10.1158/2159-8290.CD-17-0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fell JB, Fischer JP, Baer BR, Ballard J, Blake JF, Bouhana K, et al. Discovery of Tetrahydropyridopyrimidines as Irreversible Covalent Inhibitors of KRAS-G12C with In Vivo Activity. ACS medicinal chemistry letters 2018;9(12):1230–4 doi 10.1021/acsmedchemlett.8b00382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Janes MR, Zhang J, Li LS, Hansen R, Peters U, Guo X, et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 2018;172(3):578–89.e17 doi 10.1016/j.cell.2018.01.006. [DOI] [PubMed] [Google Scholar]
- 21.Chen H, Smaill JB, Liu T, Ding K, Lu X. Small-Molecule Inhibitors Directly Targeting KRAS as Anticancer Therapeutics. Journal of medicinal chemistry 2020;63(23):14404–24 doi 10.1021/acs.jmedchem.0c01312. [DOI] [PubMed] [Google Scholar]
- 22.Zeng M, Lu J, Li L, Feru F, Quan C, Gero TW, et al. Potent and Selective Covalent Quinazoline Inhibitors of KRAS G12 C. Cell Chem Biol 2017;24(8):1005–16 e3 doi 10.1016/j.chembiol.2017.06.017. [DOI] [PubMed] [Google Scholar]
- 23.Schulze CJ, Bermingham A, Choy TJ, Cregg JJ, Kiss G, Marquez A, et al. Abstract PR10: Tri-complex inhibitors of the oncogenic, GTP-bound form of KRASG12C overcome RTK-mediated escape mechanisms and drive tumor regressions in vivo. Molecular Cancer Therapeutics 2019;18(12 Supplement):PR10–PR doi 10.1158/1535-7163.Targ-19-pr10. [DOI] [Google Scholar]
- 24.Nichols R, Schulze C, Bermingham A, Choy T, Cregg J, Kiss G, et al. A06 Tri-complex Inhibitors of the Oncogenic, GTP-Bound Form of KRASG12C Overcome RTK-Mediated Escape Mechanisms and Drive Tumor Regressions in Preclinical Models of NSCLC. Journal of Thoracic Oncology 2020;15(2):S13–S4 doi 10.1016/j.jtho.2019.12.035. [DOI] [Google Scholar]
- 25.Zheng Z, Liebers M, Zhelyazkova B, Cao Y, Panditi D, Lynch KD, et al. Anchored multiplex PCR for targeted next-generation sequencing. Nat Med 2014;20(12):1479–84 doi 10.1038/nm.3729. [DOI] [PubMed] [Google Scholar]
- 26.Crystal AS, Shaw AT, Sequist LV, Friboulet L, Niederst MJ, Lockerman EL, et al. Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science 2014;346(6216):1480–6 doi 10.1126/science.1254721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ahronian LG, Sennott EM, Van Allen EM, Wagle N, Kwak EL, Faris JE, et al. Clinical Acquired Resistance to RAF Inhibitor Combinations in BRAF-Mutant Colorectal Cancer through MAPK Pathway Alterations. Cancer Discov 2015;5(4):358–67 doi 10.1158/2159-8290.CD-14-1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The Protein Data Bank. Nucleic Acids Research 2000;28(1):235–42 doi 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.