

Abstract
Purpose:
To define dominant molecular and cellular features associated with PD-1/PD-L1 blockade resistance in metastatic urothelial cancer.
Experimental Design:
We pursued an unbiased approach using bulk RNA sequencing data from two clinical trials to discover (IMvigor 210) and validate (CheckMate 275) pre-treatment molecular features associated with resistance to PD-1/PD-L1 blockade in metastatic urothelial cancer. We then generated single-cell RNA sequencing data from muscle-invasive bladder cancer specimens to dissect the cellular composition underlying the identified gene signatures.
Results:
We identified an adaptive immune response gene signature associated with response and a pro-tumorigenic inflammation gene signature associated with resistance to PD-1/PD-L1 blockade. The adaptive immune response:pro-tumorigenic inflammation signature expression ratio, coined the 2IR score, best correlated with clinical outcomes and was externally validated. Mapping these bulk gene signatures onto single-cell RNA sequencing data uncovered their underlying cellular diversity with prominent expression of the pro-tumorigenic inflammation signature by myeloid phagocytic cells. However, heterogeneity in expression of adaptive immune and pro-tumorigenic inflammation genes was observed among single myeloid phagocytic cells quantified as the Msc2IR score. Single myeloid phagocytic cells with low Msc2IR scores demonstrated upregulation of proinflammatory cytokines/chemokines and downregulation of antigen presentation genes, were unrelated to M1 versus M2 polarization, and were enriched in pre-treatment blood from patients with PD-L1 blockade-resistant metastatic urothelial cancer.
Conclusions:
The balance of adaptive immunity and pro-tumorigenic inflammation in individual tumor microenvironments is associated with PD-1/PD-L1 resistance in urothelial cancer with the latter linked to a proinflammatory cellular state of myeloid phagocytic cells detectable in tumor and blood.
Introduction
Standard treatment for metastatic urothelial cancer (UC) of the bladder has historically been limited to platinum-based chemotherapy. However, the treatment landscape has experienced a major shift with the introduction of several PD-1/PD-L1 immune checkpoint inhibitors (CPI) into the armamentarium.1–5 These therapies are characterized by durable responses, often measured in years, but achieved in only a subset of ~15–25% of patients. This therapeutic profile has led to intensive investigation into dominant mechanisms of intrinsic resistance in pursuit of biomarkers and combination strategies to extend the benefits of CPI to additional patients.
Responses to CPI are thought to be predicated on a pre-existing anti-tumor T cell response restrained due to adaptive immune resistance.6 Measures of T cell infiltration, IFNγ-related gene signatures, and PD-L1 expression, colloquially referred to as reflecting “hot” or “inflamed” tumors, have all been correlated with response to CPI in patients with UC and other cancers.1,3 While classifying tumors as “inflamed” versus “non-inflamed” provides a convenient framework for conceptualizing the immunobiology underlying sensitivity and resistance to CPI, this nomenclature fails to distinguish anti-tumor versus pro-tumorigenic inflammation. Pro-tumorigenic inflammation, a “Hallmark of Cancer” pathogenesis7, involves a TME shaped by activated fibroblasts, endothelial cells and innate immune cells, particularly myeloid phagocytic cells, which promote cancer growth and progression at least in part by impairing antitumor immunity.7–9 Antitumor immunity and pro-tumorigenic inflammation coexist in delicate spatiotemporal balance in individual TMEs complicating identification of tumors in the clinic which are resistant to CPI due to the latter and obfuscating identification of cellular populations or signaling interactions for prioritization as therapeutic targets to overcome such resistance.
To identify dominant molecular and cellular interactions in the UC TME associated with CPI resistance, we pursued an unbiased approach (Figure 1). We first used pre-CPI treatment bulk RNA sequencing data from a large clinical trial cohort and identified two gene signatures independently associated with CPI outcomes beyond tumor mutational burden alone: 1) a signature enriched in adaptive immune response genes and associated with better CPI outcomes dubbed the adaptive immune response signature, and 2) a signature enriched in inflammation and innate immune genes and associated with worse CPI outcomes dubbed the pro-tumorigenic inflammation signature. Consistent with the notion that antitumor immunity and pro-tumorigenic inflammation coexist within individual TMEs, the adaptive immune response:pro-tumorigenic inflammation gene expression ratio, coined the 2IR score (adaptive Immune:pro-tumorigenic Inflammation Ratio), had the largest effect on clinical outcomes and was validated in an independent UC clinical trial cohort. We then generated single-cell RNA sequencing (scRNA-seq) data from UC bladder specimens to uncover the cellular composition underlying the gene signatures revealing diverse cellular populations contributed to both the adaptive immune response and pro-tumorigenic inflammation gene signatures. Though the pro-tumorigenic inflammation signature was expressed prominently by myeloid phagocytic cells as a whole, diverse expression of the adaptive immune response and pro-tumorigenic signature genes were observed across individual macrophages/monocytes and neutrophils leading to application of the 2IR score to each individual cell (Myeloid Single Cell Immune:protumorigenic Inflammation Ratio or Msc2IR score). Myeloid phagocytic cells with low Msc2IR scores demonstrated upregulation of proinflammatory cytokines (e.g, IL1B) and chemokines (e.g., CCL20) and downregulation of antigen presentation genes, could not be discerned based on M1 versus M2 classification, and were enriched in the pretreatment blood of patients with CPI-resistant metastatic UC. Thus, the balance of adaptive immunity and pro-tumorigenic inflammation is associated with CPI outcomes in UC and resistance associated with the latter may be driven by interactions among diverse cell types in the TME and linked to a proinflammatory cellular state of myeloid phagocytic cells detectable in both the TME and peripheral blood.
Figure 1. Cohorts and workflow for discovery of gene signatures associated with sensitivity and resistance to anti-PD-1/PD-L1 treatment in metastatic urothelial cancer.
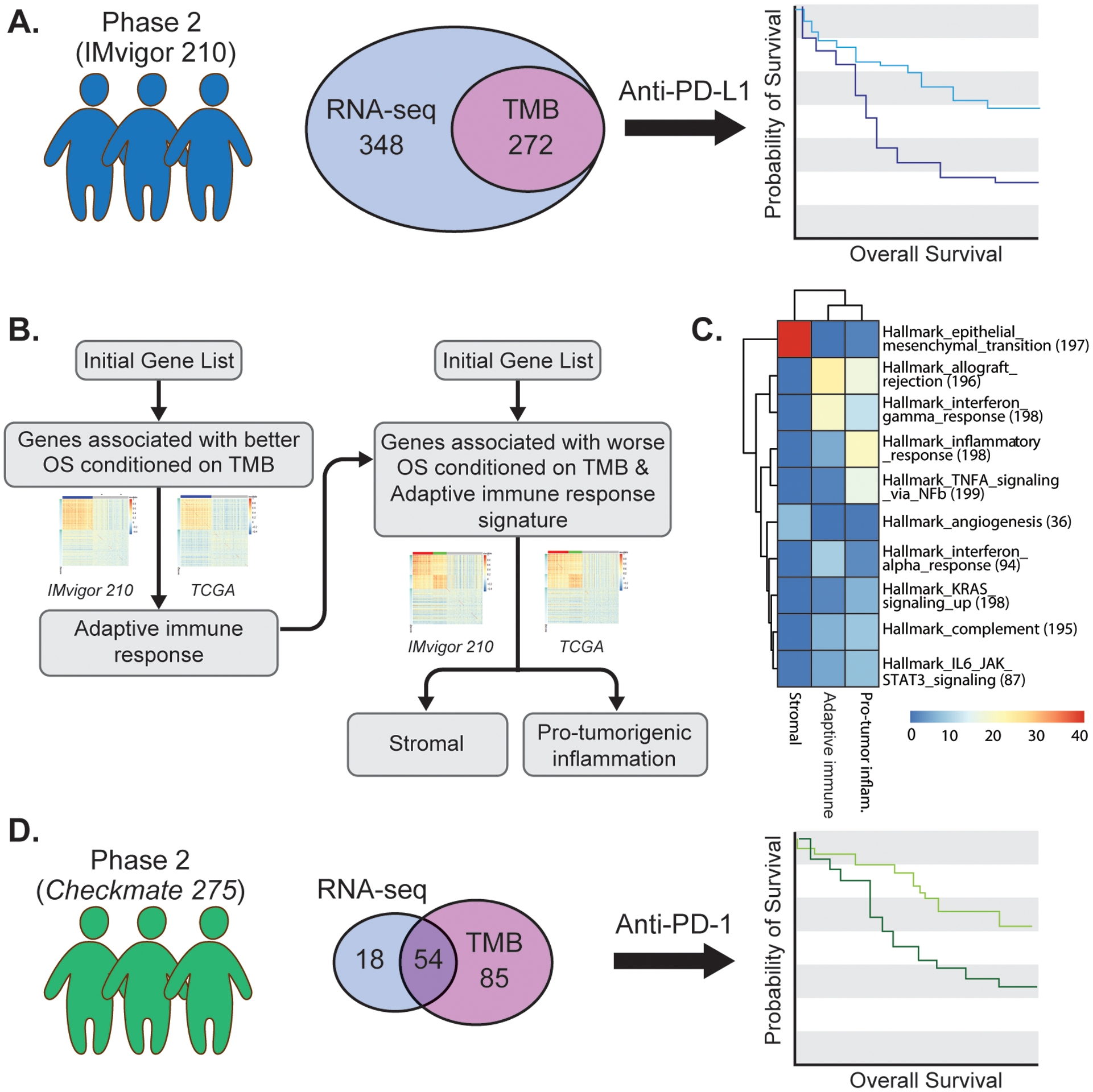
A. IMvigor 210 was a single-arm phase 2 study investigating PD-L1 inhibition with atezolizumab in patients with metastatic urothelial cancer. The illustration depicts the numbers of patients with available pre-PD-L1 inhibition RNA-sequencing (RNA-seq) data, tumor mutational burden (TMB) data, or both, derived from archival tumor specimens available for the current analysis. B. Step-wise approach to the identification of consistently co-expressed gene modules, conditioned on TMB, associated with better overall survival or worse overall survival with PD-L1 blockade treatment in patients with metastatic urothelial cancer. Data from The Cancer Genome Atlas (TCGA) urothelial bladder cancer dataset was used to identify consistently co-expressed gene modules (see Methods). C. Hallmark pathways enriched in the adaptive immune response, pro-tumorigenic inflammation, and stromal gene signatures using Fisher’s exact test (nominal two-sided p-value <1e-5). Color corresponds to the −log10 of the p-value. D. Checkmate 275 was a single-arm phase 2 study investigating PD-1 inhibition with nivolumab in patients with metastatic urothelial cancer. The illustration depicts the number of patients with available pre-PD-1 inhibition RNA-sequencing data, TMB data, or both derived from archival tumor specimens used for validation of the association between the adaptive immune response, pro-tumorigenic inflammation, and stromal gene signatures and outcomes with PD-1/PD-L1 blockade in metastatic urothelial cancer.
Materials and Methods
Identification and validation of gene signatures associated with CPI outcomes from bulk RNA sequencing data
Patient cohorts with tumor mutational burden data and/or bulk RNA sequencing data
Three datasets including bulk RNA sequencing (RNAseq) data from patients with urothelial cancer were analyzed in this study (Figure 1 and Supplemental Table S1): IMvigor 210, The Cancer Genome Atlas (TCGA) urothelial bladder cancer dataset, and the Checkmate 275 study.
IMvigor 210 was a single arm phase 2 study investigating PD-L1 inhibition with atezolizumab (1200 mg intravenously every 3 weeks) in patients with metastatic urothelial cancer (NCT01208652, NCT02951767). The primary endpoint of the trial was the objective response rate according to Response Evaluation Criteria In Solid Tumors v1.1. Patients with metastatic urothelial cancer progressing despite prior platinum-based chemotherapy, or chemotherapy-naïve patients who were not eligible for cisplatin-based chemotherapy, were eligible. The results of IMvigor 210 have previously been reported.1,11 Patients enrolled on IMvigor 210 were required to have archival tumor tissue obtained within two years of study entry submitted for analysis which including bulk RNAseq as well as targeted next-generation sequencing-based genomic profiling for 395 cancer-related genes (FoundationOne, Foundation Medicine, Cambridge MA). For the current study, RNAseq data, TMB (“FMOne mutation burden per MB”), objective response rate, and survival data for 348 unique patients were extracted using the R package IMvigor210CoreBiologies (http://research-pub.gene.com/IMvigor210CoreBiologies/)
The Cancer Genome Atlas bladder cancer dataset includes patients with clinically localized muscle-invasive urothelial cancer of the bladder who underwent radical cystectomy. This cohort has previously been described in detail45 and RNAseq data (“Level_3_RSEM_genes_normalized”) for 408 unique patients was downloaded from Firehose (2016_01_28) at the Broad Institute (https://confluence.broadinstitute.org/display/GDAC/Home/). The updated clinical data were downloaded from an integrated TCGA pan-cancer clinical data resource.46
Checkmate 275 was a single arm phase 2 study investigating PD-1 inhibition with nivolumab (3 mg/kg intravenously every 3 weeks) in patients with metastatic urothelial cancer (NCT02387996). The primary endpoint of the trial was the objective response rate according to Response Evaluation Criteria In Solid Tumors v1.1. Patients with metastatic urothelial cancer progressing despite prior platinum-based chemotherapy were eligible. The results of Checkmate 275 have previously been reported.3 Patients enrolled on Checkmate 275 were required to have archival tumor tissue submitted for analysis which included bulk RNAseq and whole exome sequencing. Patients who were consented for genomic studies and had tumor material that passed quality control were included in the current analysis. RNAseq and tumor mutational burden data was provided by Bristol Myers-Squibb and the latter was calculated as the missense mutation count. TMB (n=139) and/or RNAseq (n=72) data, objective response rate, and survival data was available with both TMB and RNAseq data available for 54 patients.
Preprocessing of bulk RNAseq expression datasets
For the IMvigor 210 dataset, only genes with a read count >1 in more than 10% of the samples were considered. The raw read count data from the IMvigor and Checkmate 275 datasets were first transformed to RPKM and then scaled patient-wise such that the 75% quantile of each sample was equal to 1000 (similar to the RSEM normalization47). To facilitate analysis across datasets, only 16339 genes common among the three datasets were analyzed in this study. Batch effects were removed across the three datasets using R package ComBat.48
Step-wise identification of consistently co-expressed gene modules (CCGMs)
With the goal of identifying consistently coexpressed gene modules associated with survival, we first identified genes nominally associated with better overall survival outcomes in the IMvigor 210 dataset. A bivariable Cox regression model was used to estimate the association between the expression of each gene, Genei, with the overall survival conditional on TMB: Surv(Event, Time) ~ Genei + log(TMB). We identified 1193 genes for which higher expression was associated with better survival outcomes (nominal P-value of two-sided Wald’s test <0.05). We employed a lenient P-value cutoff to be as inclusive as possible at this initial gene selection step, and then identified consistently co-expressed gene modules (CCGMs) to enrich for true signals and filter out possible noise. A CCGM is defined as a list of genes that are co-regulated in multiple datasets. Using weighted correlation network analysis49 among these 1193 genes, we identified one co-expression module in the IMvigor 210 dataset (735 genes) and one co-expression module in the TCGA dataset (600 genes). Significant overlap (575 genes) existed between the modules identified in the IMvigor 210 and TCGA datasets ( p<1e-16 by two-sided Fisher’s exact test) and these 575 overlapping genes were considered a CCGM, referred to as the “adaptive immune response” signature.
Next, we identified genes associated with worse survival outcomes conditioned on both TMB and the adaptive immune response signature genes (Madaptive immune). Specifically, we assessed the association of each gene (Genei) with overall survival using a multivariable Cox regression model Surv(Event, Time) ~ Genei + Madaptive immune + log(TMB)), where Madaptive immune was calculated for each sample by averaging expression of the adaptive immune response signature genes. A total of 1498 genes were associated with worse survival outcomes (nominal P-value of two-sided Wald’s test <0.05). The weighted correlation network analysis was conducted for the 1498 genes in both the IMvigor 210 and TCGA datasets, followed by the overlapping analysis analogous to the methodology described for derivation of the adaptive immune response signature which resulted in two CCGMs, i.e. the “pro-tumorigenic inflammation” signature (437 genes) and stromal signature (287 genes). A third CCGM (50 genes) enriched with HALLMARK_MYC_targets was not further pursued in this study given its small size. We further updated the “adaptive immune response” signature by excluding genes associated with worse survival in this three variate Cox regression model (Z>1.5), resulting in 483 genes in the module.
To investigate the pathways enriched in each CCGM, we compared the signature genes with the HALLMARK and canonical gene sets in the Molecular Signatures Database (MSigDB, software.broadinstitute.org/gsea/msigdb)50 using Fisher’s exact test (nominal p-value of two-sided test <1e-5).
Calculation of the 2IR score, identification of top-ranked genes, comparison with other biomarkers, and multiplex immunohistochemistry is detailed in the Supplemental Methods.
Univariable and multivariable models
Cox proportional hazard regression models (coxph() function) were performed using the R package survival to evaluate the association between the gene signatures and TMB with overall survival (OS). When signature expression and TMB were treated as continuous variables, they were standardized to N(0,1) before entering the Cox regression model to estimate hazard ratio and confidence interval, and the significance testing was performed by Wald’s test. When the 2IR score was discretized into tertiles, the R package survminor was used to plot the Kaplan Meier curve, and the significance testing for differences in OS was performed using the log-rank test. Logistic regression models were performed to evaluate the association between the gene modules and TMB with objective response. In the logistic regression, a complete response or partial response were treated as 1, and stable disease or progressive disease were treated as 0. The signature expression and TMB were similarly standardized before entering the logistic regression model to estimate the coefficient, and the significance testing was performed by Wald’s test. All statistical analyses and figures were generated in R version 3.6.3.
Single-cell RNA Sequencing of Urothelial Cancer Specimens
Sample collection and specimen processing
Primary urothelial bladder cancer tumor tissue was obtained after obtaining informed consent in the context of an institutional review board approved genitourinary cancer clinical database and specimen collection protocol (IRB #10–1180) at the Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai. Patients undergoing transurethral resection of bladder tumor had a portion of their tumor placed immediately into RPMI medium in the operating room. The specimen was then transferred to the laboratory for further processing. Patients undergoing radical cystectomy and lymph node dissection had their bladder and lymph nodes sent directly to the pathology suite upon completion of the lymph node dissection. The bladder was bivalved and a portion of visible tumor was then placed in media as above. Adjacent normal tissue was identified in a subset of specimens based on visual inspection. The specimen was then transferred to the laboratory for further processing.
Tissue specimens were processed immediately upon receipt and dissociated into single cell suspensions using the GentleMACS Octodissociator with kit matched to the tissue type (Miltenyi Biotech) following the manufacturer’s instructions. Single-cell RNA sequencing was performed on these samples using the Chromium platform (10x Genomics, Pleasanton, CA) with the 3’ gene expression (3’ GEX) V3 kit, using an input of ~10,000 cells. Briefly, Gel-Bead in Emulsions (GEMs) were generated on the sample chip in the Chromium controller. Barcoded cDNA was extracted from the GEMs by Post-GEM RT-cleanup and amplified for 12 cycles. Amplified cDNA was fragmented and subjected to end-repair, poly A-tailing, adapter ligation, and 10X-specific sample indexing following the manufacturer’s protocol. Libraries were quantified using Bioanalyzer (Agilent) and QuBit (Thermofisher) analysis. Libraries were sequenced in paired end mode on a NovaSeq instrument (Illumina, San Diego, CA) targeting a depth of 50,000–100,000 reads per cell. Sequencing data was aligned and quantified using the Cell Ranger Single-Cell Software Suite (version 3.0, 10x Genomics) against the provided GRCh38 human reference genome.
Preprocessing
Seurat26 (version 3.0) was used to process the single-cell RNA sequencing data. After filtering cells with a high percentage (>20%) of mitochondrial reads and cells with <200 or >6000 genes detected, as well as potential doublets uncovered during subsequent analysis steps, 19,708 cells from 10 samples and 22,175 genes with nonzero read counts in > 5 cells were included for further analysis.
Identification of major cell clusters
After the read count data was log-normalized, the most variable 2000 genes were selected. Then the effect of the unique molecular identifier (UMI) count and percentage of mitochondria per cell was regressed out, followed by dimensionality reduction using principle component analysis (PCA). Finally, the cells were clustered using the K-nearest neighbors graph-based methods as implemented in Seurat (with the top 20 PC and resolution = 0.5). Cells were grouped into 9 major cell clusters based on the canonical cell-type-specific markers: T/NK (“CD3E”), B/plasma (“MS4A1”,”MZB1”,”CD79A”), DC (“HLA-DQA1”, “HLA-DQB1”), Mast (“MS4A2”), Macrophage/Monocyte (“C1QA”,”LYZ”), Endothelial (“PLVAP”), Fibroblast-related (“DCN”, “ACTA2”), Epithelial (“KRT19”) and Neuronal cells (NNAT). The identification of minor cell clusters, Msc2IR score calculation, and Nichenet analysis are detailed in the Supplemental Methods.
Association between cell subsets and adaptive immune response, pro-tumorigenic inflammation, and stromal signatures.
For each of the 9 major cell clusters, we identified cell type-overexpressed genes using FindAllMarker() function in Seurat package (with default parameters). The overlap between each of the adaptive immune response, pro-tumorigenic inflammation, and stromal signature genes and overexpressed genes among the major cell clusters was assessed using odds ratio and p-value (two-sided Fisher’s exact test).
Peripheral blood mononuclear cell single-cell RNA sequencing cohort
Single-cell RNA sequencing data for 10 frozen PBMC samples derived from pre-treatment peripheral blood of 5 patients with metastatic UC who achieved an objective response to treatment with atezolizumab and 5 patients with metastatic urothelial cancer who did not achieve an objective response to treatment with atezolizumab in the setting of the IMvigor 210 study were downloaded from GEO (GSE145281). The peripheral blood single-cell RNA sequencing cohort and analysis are detailed in the Supplemental Methods.
Results
Gene signatures independently associated with CPI outcomes in patients with UC.
To identify molecular features associated with survival in CPI-treated patients with metastatic UC, we utilized bulk RNA sequencing and TMB data from the IMvigor 210 study, a large single arm phase 2 trial testing the PD-L1 inhibitor, atezolizumab (Figure 1a).1,10,11 This cohort has been previously described and additional details are provided in Table S1; RNA sequencing and TMB data were available for 348 and 272 patients, respectively.10 We pursued step-wise identification of consistently co-expressed gene modules, which focused on identifying gene modules associated with overall survival (OS) and utilizing gene modularity to enrich for true signals (Figure 1b, Figure S1; see Methods). Given the correlation between TMB and response to CPI in UC10,12, we explored genes associated with OS conditioning on TMB (see Methods) and identified a signature consisting of 1193 genes associated with longer OS. To refine this signature, we performed meta-analysis of co-expression patterns13,14 using both the IMvigor 210 and The Cancer Genome Atlas (TCGA) UC datasets and identified a consistently co-expressed gene module comprised of 483 genes (see Methods and Supplementary Data S1). Gene set enrichment analysis revealed that this module was highly enriched in adaptive immune response-related genes (Figure 1c and Figure S2) and was therefore labeled the adaptive immune response signature.
In the second step, we further analyzed the IMvigor 210 dataset to identify genes associated with survival conditioning on both TMB and the adaptive immune response signature (Figure 1b). We identified 1498 genes associated with shorter OS. We again applied meta-analysis of co-expression patterns15,16 in the IMvigor 210 and TCGA UC datasets and identified two consistently co-expressed gene modules for further analysis. The first module consisting of 437 genes, was enriched in inflammation and innate immune genes (Figure 1c, Figure S2, and Supplementary Data S1) and associated with shorter OS and was therefore labeled the pro-tumorigenic inflammation signature. The second module associated with shorter OS, consisting of 287 genes, was enriched in epithelial mesenchymal transition (EMT)- and extracellular matrix (ECM)-related genes (Figure 1c and Supplementary Data S1) and consistent with our prior work17 was named the stromal signature. Importantly, expression of the adaptive immune response and stromal signatures were both positively correlated with the adaptive immune response module (Figure S3) such that their disparate impact on OS was only revealed using our stepwise approach (Figure S4) and suggesting that the balance of these features in individual tumors may impact CPI outcomes.
We next sought to define the independent contribution of the three gene signatures to outcomes with CPI in the IMvigor 210 cohort. Models combining both the adaptive immune response and pro-tumorigenic inflammation signatures (see Methods), particularly the adaptive Immune response:pro-tumorigenic inflammation signature expression Ratio (hereafter referred to as the 2IR score), demonstrated the largest effect size on OS and similar findings were observed with objective response rate (Figure 2a–c and Table S2). Importantly, when both the pro-tumorigenic inflammation and stromal signatures were entered into a multivariable model along with the adaptive immune response signature and TMB, the stromal module was no longer independently associated with OS (Figure 2a and Table S2). These findings indicated that (a) the balance of cellular and molecular events underlying the adaptive immune response versus pro-tumorigenic inflammation signatures within an individual UC TME may dictate outcomes with CPI and (b) the negative impact of the stromal signature on outcomes may be more indirect and mediated via the events underlying the pro-tumorigenic inflammation signature (Figure 2a).
Figure 2. The adaptive immune response and pro-tumorigenic inflammation gene signatures, and the ratio of signature expression termed the 2IR score, are associated with clinical outcomes with PD-1/PD-L1 blockade in patients with metastatic urothelial cancer.
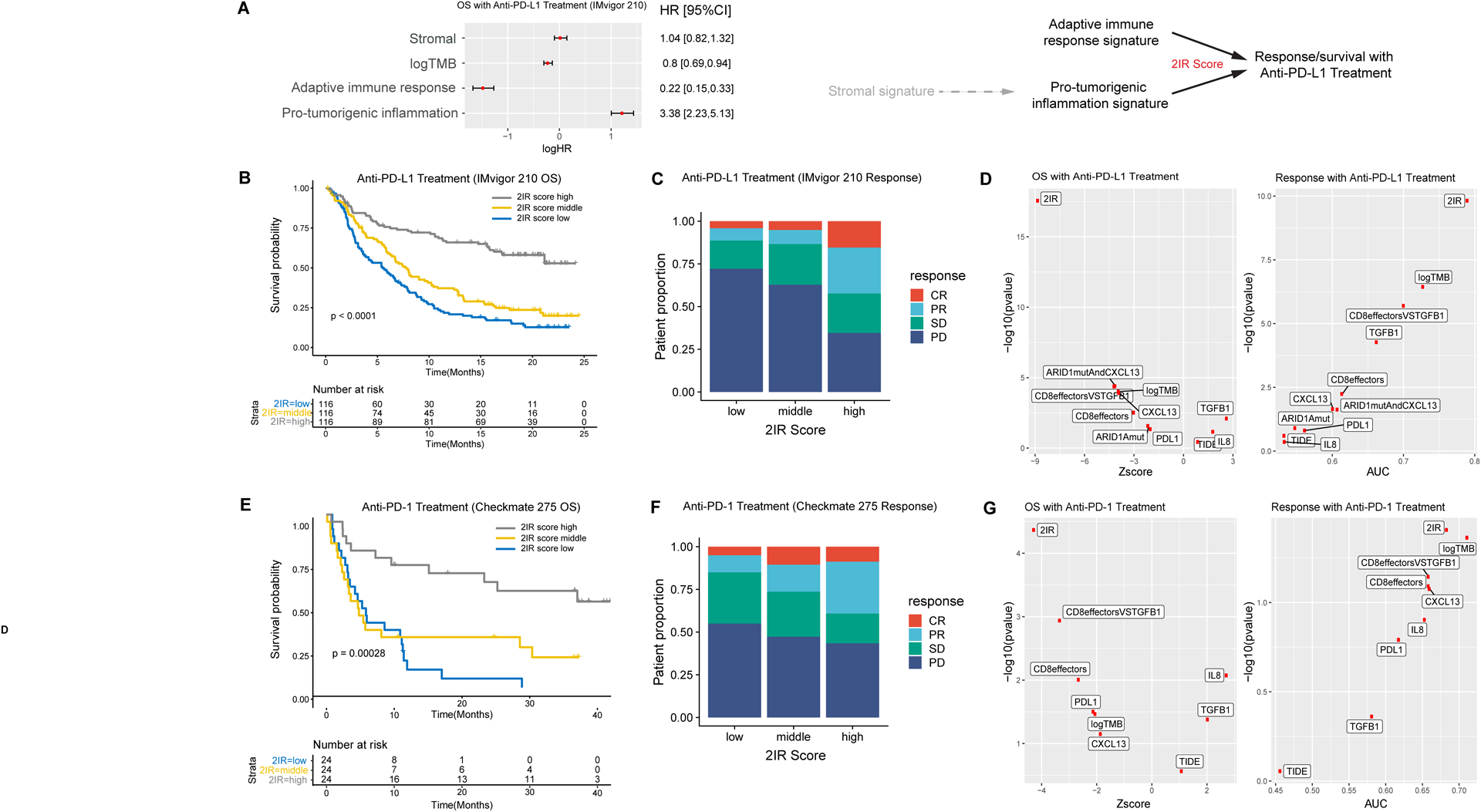
A. Multivariable Cox regression model for overall survival (OS; n=272 patients with RNA sequencing and tumor mutational burden (TMB) data) including adaptive immune response, pro-tumorigenic inflammation, and stromal gene signature expression, as well as TMB from the IMvigor 210 cohort (HR, hazard ratio; 95% CI, 95% confidence interval; error bars represent 95% CI of the HRs). Gene signature expression and TMB were standardized before entering the Cox regression model. The plot indicates log HRs while annotation provides HRs. Schematic representation of the relationship of the adaptive immune response, pro-tumorigenic inflammation, and stromal gene signatures and outcomes with atezolizumab indicating potential indirect role of the stromal signature on resistance mediated more directly through the pro-tumorigenic inflammation signature and the 2IR score representing the adaptive Immune response:pro-tumorigenic Inflammation gene signature expression Ratio. B. Kaplan-Meier curve for overall survival (OS) stratified by the 2IR score cut at tertiles in the IMvigor 210 cohort (n=348 patients with RNA sequencing data; log-rank p value shown). C. Objective response rate with PD-L1 blockade in the IMvigor 210 cohort according to the 2IR score (cut at tertiles). For each 2IR score tertile, bar graphs depict the percentage of patients achieving a complete response (CR), partial response (PR), stable disease (SD), or progressive disease (PD) as the best objective response with PD-L1 blockade. D. The association between each biomarker (or biomarker combination) and overall survival (OS) in the IMvigor 210 cohort was evaluated using the Z-score by univariate Cox regression analysis and the p-value by log likelihood ratio test (left). The association between each biomarker and response to PD-L1 blockade (CR/PR versus SD/PD) was evaluated using the area under curve (AUC) score and the p-value by the Wald’s test in univariate logistic regression (right). E. Kaplan-Meier curves for overall survival (OS) stratified by the 2IR score (cut at tertiles) in the Checkmate 275 cohort (n=72 patients with RNA sequencing data; log rank p value shown). F. Objective response rate with PD-1 blockade in the Checkmate 275 cohort according to the 2IR score (cut at tertiles). For each 2IR score tertile, bar graphs depict the percentage of patients achieving a complete response (CR), partial response (PR), stable disease (SD), or progressive disease (PD) as the best objective response with PD-1 blockade. G. The association between each biomarker (or biomarker combination) and overall survival (OS) in the Checkmate 275 cohort was evaluated using the Z-score by univariate Cox regression analysis and the p-value by log likelihood ratio test (left). The association between each biomarker and response to PD-1 blockade (CR/PR versus SD/PD) was evaluated using the area under curve (AUC) score and the p-value by the Wald’s test in univariate logistic regression (right).
Given the potential practical advantages of smaller sets of genes for validation and clinical biomarker development, we identified the top-ranked genes within the three signatures (see Methods, Table S3, and Figure S5). Signature scores derived from these smaller gene sets demonstrated similar associations with OS compared with scores derived from the full gene sets (Table S4).
The 2IR score was validated in an independent cohort of patients with metastatic UC treated with PD-1 blockade, conveyed information beyond previously identified features, and was associated with the cellular organization of the TME.
For validation, we utilized TMB and RNA sequencing data from the Checkmate 275 study, a single-arm phase 2 trial evaluating the PD-1 inhibitor, nivolumab, in patients with metastatic UC (Figure 1d).3 This cohort has been previously described, with further detail provided in Table S1; RNA sequencing and TMB data were available for 72 and 139 patients, respectively.3 The adaptive immune response, pro-tumorigenic inflammation, and stromal gene signatures demonstrated similar associations with OS, progression free survival (PFS) and response rate in the Checkmate 275 cohort (Table S5). As observed in the IMvigor 210 cohort, the 2IR score in the Checkmate 275 cohort demonstrated the largest effect on CPI outcomes (Figure 2e,f). Furthermore, the 2IR score remained significantly associated with overall survival after several clinical prognostic factors were taken into consideration (supplementary results and Table S6).
Other cancer cell-intrinsic and TME-related features have been associated with CPI outcomes in UC and other tumors.10,18–20 The 2IR score demonstrated favorable performance characteristics relative to such features including PD-L1 protein expression, the tumor immune dysfunction and exclusion (TIDE) and CD8 effector T cell gene signatures, ARID1A mutation status, and CXCL13, TGFB1, or CXCL8 (IL8) gene expression (Figure 2d,g; see Figure S7 for correlation between these features and the 2IR score) in both the IMvigor 210 and Checkmate 275 cohorts. Less dramatic findings were observed applying the 2IR score to the TCGA UC dataset, a cohort of patients with muscle-invasive UC of the bladder treated with curative-intent cystectomy (Figure S6), suggesting that the 2IR score may impart predictive rather than solely prognostic information. Taken together, the 2IR score, representing the balance of expression of the adaptive immune response and pro-tumorigenic inflammation gene signatures within individual TMEs, is associated with objective response and OS in CPI-treated patients with metastatic UC in two clinical trial cohorts and conveys information beyond that achieved with previously identified features.
We next sought to characterize the relationship between the 2IR score and the cellular organization of the UC TME, particularly the spatial localization of T cells, based on prior work from our group and others linking T cell spatial localization with CPI resistance in UC.10,17 We employed a tissue profiling approach known as multiplexed immunohistochemical consecutive staining on a single slide (MICSSS)21,22 on a subset of 19 specimens from the Checkmate 275 cohort with matched RNA sequencing data. Notably, MICSSS revealed that specimens with higher 2IR scores exhibited occasional tertiary lymphoid-like structures (Figure 3a,b and Figure S8) consistent with prior findings linking such structures with improved CPI outcomes.23,24 To quantify the localization of T cells according to gene signature expression, we defined cancer cell and stromal zones based on pan-cytokeratin staining using a machine learning segmentation tool and examined CD8+ expression in 76 regions of interest across the 19 specimens (see Methods and Figure 3e). Lower 2IR scores correlated with decreased CD8+ T cell enumeration in cancer cell nests, with T cells more prominently localized to the stromal regions, suggestive of a T cell “excluded” phenotype (Figure 3c–g).10,17 These findings suggested that CPI resistance associated with lower 2IR scores may be related to impairment of T cell trafficking and/or function prompting us to further probe the cellular populations and interactions underlying the gene signatures.
Figure 3. The adaptive immune response and pro-tumorigenic inflammation gene signatures are associated with spatial organization of immune cells in the tumor microenvironment.
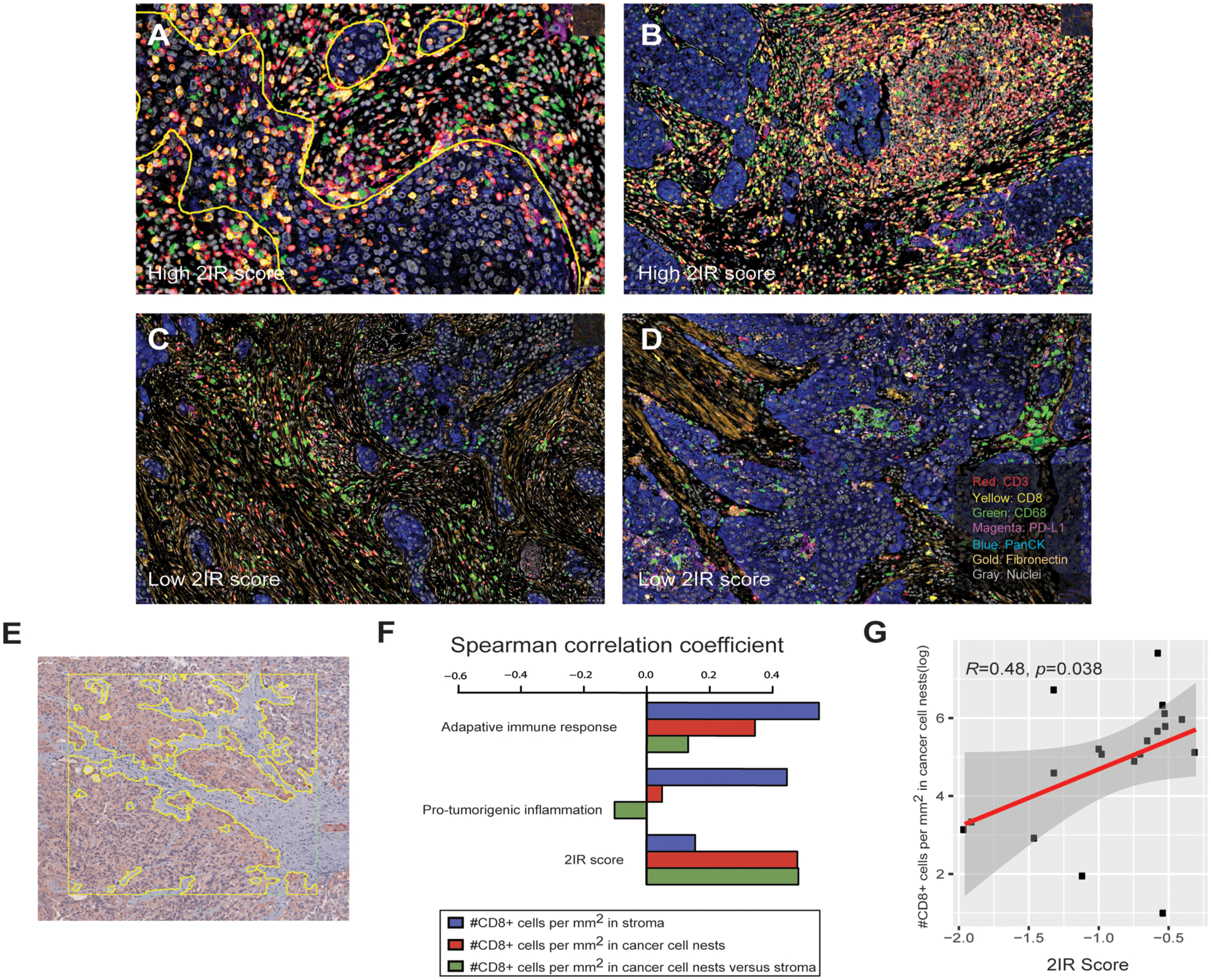
A-D. Representative images of multiplexed immunohistochemical consecutive staining on a single slide (MICSSS) demonstrating abundance of CD8+ T cells (A, B) and tertiary lymphoid-like structures (B) in specimens with high 2IR scores and a paucity of CD8+ T cells and prominent macrophages and stroma (C, D) in specimens with a low 2IR scores. Yellow outline in panel A represents demarcation of cancer cell nests. All slides were initially scanned at 20x magnification. E. Representative image of urothelial cancer specimen demonstrating region of interest (ROI), designated by the square, and machine learning-based segmentation of cancer cell nest and stromal zones to define T cell localization in the tumor microenvironment using pancytokeratin immunohistochemical staining, designated by the yellow outline bordering cytokeratin-expressing cells. F. Spearman’s correlation between enumeration of CD8+ T cells localized to cancer cell nests or stromal zones and adaptive immune response gene signature, pro-tumorigenic inflammation gene signature, or 2IR score. The results are based on analysis of 76 ROIs across 19 specimens with both immunohistochemistry and RNA sequencing data from the Checkmate 275 cohort. G. Correlation between enumeration of CD8+ T cells localized to cancer cell nests and the 2IR score. The results are based on analysis of 76 ROIs across 19 specimens with both immunohistochemistry and RNA sequencing data from the Checkmate 275 cohort. Spearman’s correlation was used to determine the correlation coefficient R and p value.
Diverse cellular populations underlie the adaptive immune response, pro-tumorigenic inflammation, and stromal gene signatures.
Our gene signatures were derived from bulk RNA sequencing data from archival UC specimens obtained pre-treatment with CPI, the vast majority of which represented invasive primary tumors (Table S1). Therefore, to map the cellular origins of the three gene signatures (Figure 4a), we performed droplet-based encapsulation single cell RNA sequencing (scRNA-seq) on an analogous set of eight freshly resected invasive UC bladder specimens as well as two specimens derived from adjacent grossly normal urothelium using the 10x Genomics Chromium system (see Methods and cohort characteristics in Table S7). The characteristics of the cohort are detailed in Table S7. After excluding cells not passing quality control (see Figure S9 for QC plots), 19,708 cells from the 10 samples were analyzed. A median of 1456 genes were detected per cell. We performed graph-based clustering as implemented in the Seurat package.26 A two-stage clustering approach was employed in which cells were first grouped into major populations and subsequently further partitioned into minor populations (see Methods).
Figure 4. Defining the cellular origins of adaptive immune response, pro-tumorigenic inflammation, and stromal gene signature expression using single-cell RNA sequencing.
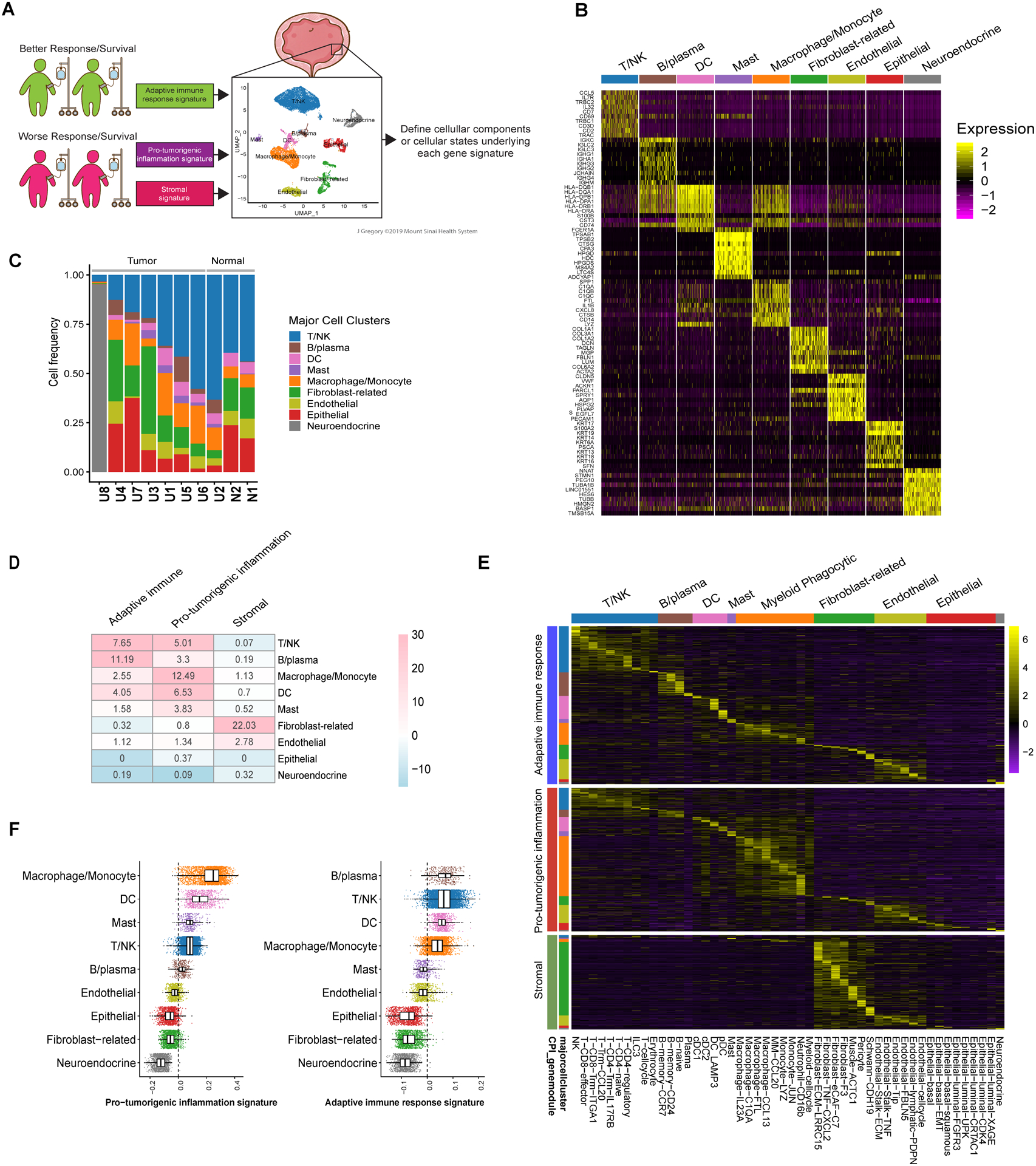
A. Schematic representation of projection of gene signatures identified using bulk RNA sequencing data linked to outcomes with anti-PD-1/PD-L1 treatment onto single-cell RNA sequencing data generated from a separate cohort of invasive urothelial bladder cancer specimens. The illustration depicts nine major cell clusters visualized using Uniform Manifold Approximation and Projection (UMAP) across eight urothelial cancer specimens and two adjacent normal urothelial cancer specimens profiled using droplet-based encapsulation single-cell RNA sequencing. The adaptive immune response, pro-tumorigenic inflammation, and stromal gene signatures identified using bulk RNA sequencing data from clinical trial cohorts were projected onto the single-cell RNA sequencing data to define the predominant cellular sources of the respective signature gene expression. B. Single-cell expression of top 10 overexpressed genes in each major cell cluster. Heatmap visualization color-coding the scaled gene expression level for selected marker genes (rows). Visualized are 500 randomly selected cells per cluster. C. Frequency of cell populations in individual samples included in the single-cell RNA sequencing cohort. For each sample, bar graphs depict the percentage of cells in clusters associated with each population. Samples were ranked according to T/NK cell frequency. Normal indicates samples obtained for urothelial tissue that was considered grossly normal by visual inspection adjacent to site of harvested tumor tissue. D. Heatmap of overlap between genes comprising the adaptive immune response, pro-tumorigenic inflammation, and stromal gene signatures and genes overexpressed in each of the major cell clusters in the single-cell RNA sequencing cohort. The number in each cell corresponds to the odds ratio for the corresponding overlap between genes, the color corresponds to the −log10 p-value (for enrichment) or log10 p value (for depletion) by two-sided Fisher’s exact test. E. Heatmap visualizing the expression of adaptive immune response, pro-tumorigenic, and stromal signature genes across each of the major and minor cell clusters in the single-cell RNA sequencing cohort. F. Expression level of pro-tumorigenic inflammation signature genes per cell (left) and adaptive immune response signature genes per cell (right) as assessed by the AddModuleScore() function in the Seurat package across major cell populations.
Canonical marker genes revealed nine major cell populations identified by scRNA-seq including T- and NK cells, B-cells, myeloid-lineage cells, non-hematopoietic stromal cells, and epithelial cells (Figure 4b,c, Supplementary Data S2). To determine the origins of the adaptive immune response, pro-tumorigenic inflammation, and stromal signatures, the expression pattern of the signature genes was assessed among these major cell populations (Figure 4d). While adaptive immune response gene expression was prominent among T and B cells [(odds ratio (OR) = 7.65 and 11.19, respectively] and pro-tumorigenic inflammation gene expression was prominent among monocytes/macrophages (OR = 12.49), diversity in expression of signature genes among cell populations was observed (Figure 4d). For example, there were pro-tumorigenic inflammation signature genes overexpressed in T cells (OR = 5.01) and adaptive immune response signature genes overexpressed in monocytes/macrophages (OR = 2.55). Notably, expression of stromal signature genes demonstrated much less cellular diversity and was detected predominantly from cancer associated fibroblast (CAF) (OR = 22.03) and endothelial (OR = 2.78) cell populations.
To determine if the adaptive immune response and pro-tumorigenic inflammation signature genes expressed by a given major cell type were arising from discrete cellular subpopulations (e.g., adaptive immune response genes from one subset of macrophages and pro-tumorigenic inflammation genes from another subset of macrophages), we subjected each major cluster to a second round of partitioning revealing a total of 50 minor cell clusters (described in detail in Supplemental Results and Figures S10–16). Unexpectedly, we observed expression of both adaptive immune response and pro-tumorigenic signature genes within most minor cell populations (Figure 4e). Hence, the adaptive immune response and pro-tumorigenic inflammation gene signatures are contributed to by diverse cell types within the TME and may be linked to underlying cellular states rather than discrete cellular subpopulations.
Individual myeloid phagocytic cells demonstrate heterogeneous expression of adaptive immune response and pro-tumorigenic inflammation signature genes.
Myeloid phagocytic cells demonstrated the most prominent expression of the pro-tumorigenic inflammation signature genes that were associated with CPI resistance in our clinical trial cohorts, yet also expressed some adaptive immune response signature genes (Figure 4e). Consequently, the expression level of pro-tumorigenic inflammation signature per cell (as assessed by the AddModuleScore() function in the Seurat package) was highest in myeloid phagocytic cells (Figure 4f). In addition, compared with other types of cells in the scRNAseq dataset, myeloid phagocytic cells had much higher variance in the expression of the pro-tumorigenic inflammation signature genes (aka. heterogeneity of molecular state, Figure 4f) yet comparable variance in adaptive immune signature genes (Figure S4f). Thus, we turned further attention to the myeloid phagocytic cells. We identified seven minor monocyte/macrophage populations and one neutrophil population by scRNA-seq analysis (Figure 5a and b and Figure S17a). While some of these minor cell populations demonstrated higher expression of M1 versus M2 signature genes (Figure 5c), or vice versa, heterogeneity of monocyte/macrophage minor populations was observed beyond classical M1 and M2 polarization as has been documented in prior analyses.27,28 The macrophage populations resembled previously described “TAM-like macrophages” with increased expression of APOE, C1QA, C1QB, SLC40A1 and TREM2.27,29 Two populations demonstrated higher expression of S100A family genes, but lower M1 and M2 signature gene expression, and were annotated as monocyte-Jun and monocyte-LYZ. These clusters resembled previously described “MDSC-like macrophages” with overexpression of THBS1, S100A8, FCN1 and VCAN.27 A population annotated as MM-CCL2 shared marker genes with both “TAM-like” and “MDSC-like” macrophages.
Figure 5. The pro-tumorigenic inflammation gene signature is expressed prominently by myeloid phagocytic cells and low Msc2IR score myeloid phagocytic cells are characterized by increased expression of proinflammatory genes and decreased expression of antigen presentation genes.
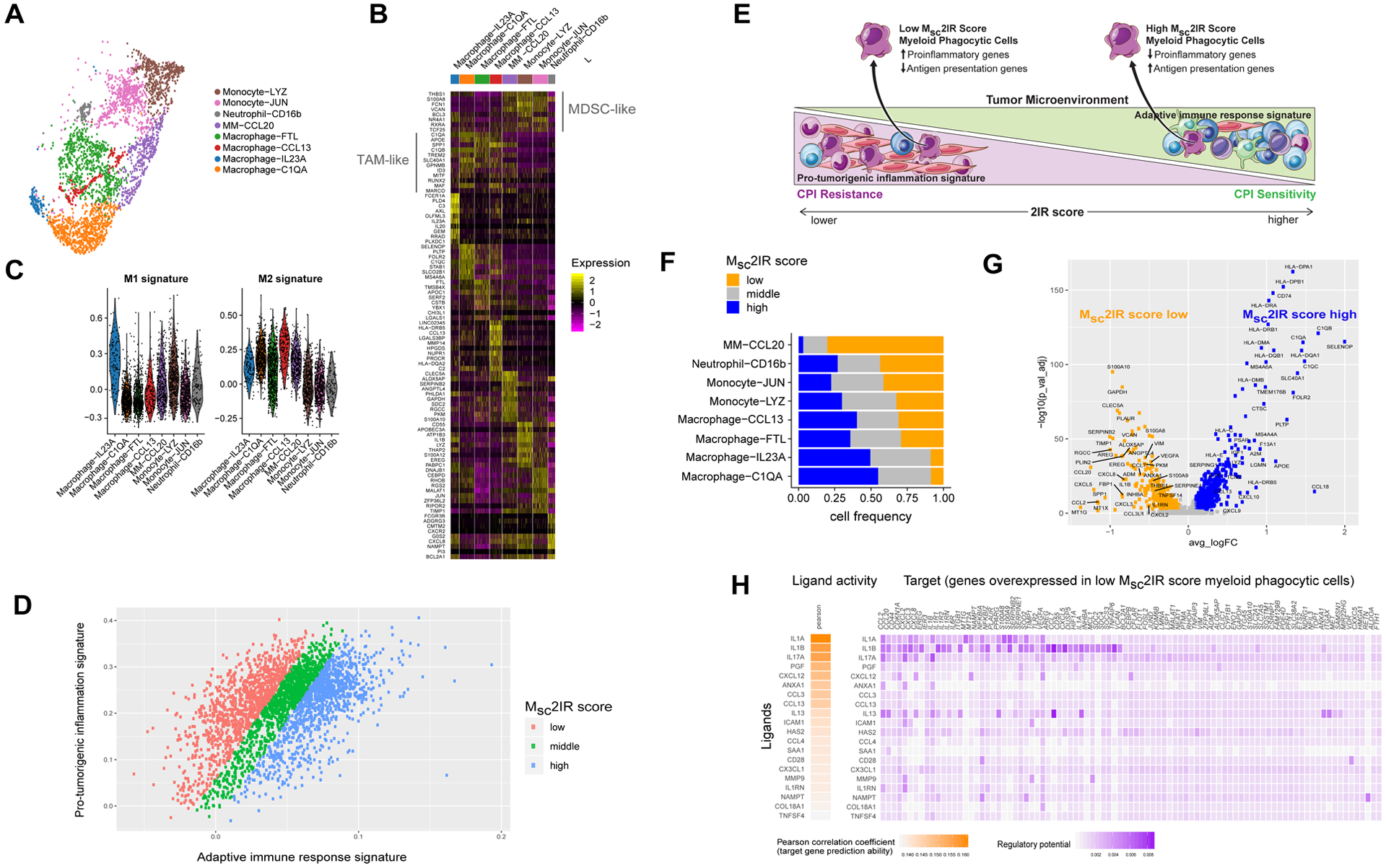
A. Eight minor myeloid phagocytic cell clusters visualized using Uniform Manifold Approximation and Projection (UMAP) across eight urothelial cancer specimens and two adjacent normal urothelial cancer specimens profiled using droplet-based encapsulation single-cell RNA sequencing. B. Myeloid phagocytic cell populations in the single-cell RNA sequencing cohort. Heatmap visualization color-coding the scaled gene expression level for selected marker genes (rows). Visualized are 200 randomly selected cells per cluster or all cells when the cell cluster contained <200 cells. C. Expression level of M1 and M2 macrophage polarization signature genes in the myeloid phagocytic cell populations as assessed by the AddModuleScore() function in the Seurat package. D. Expression of pro-tumorigenic inflammation signature genes versus adaptive immune response genes in single myeloid phagocytic cells in the urothelial cancer tumor microenvironment and classification of single myeloid phagocytic cells by myeloid single cell 2IR (Msc2IR) score. E. Schematic representation of the relationship between the 2IR score in the urothelial cancer tumor microenvironment based on bulk RNA sequencing and the Msc2IR score in individual myeloid phagocytic cells based on single cell RNA sequencing. F. The frequency of cells with low, intermediate and high Msc2IR score within each myeloid phagocytic cell minor population. G. Volcano plot of genes differentially expressed between myeloid phagocytic cells with high versus low Msc2IR scores. P-value was calculated by Wilcoxon rank-sum test and then adjusted by Bonferroni correction. Genes with log fold change (FC) >0.1 and adjusted p-value <0.05 were considered as significant. H. Top-ranking ligands inferred to regulate genes upregulated in low Msc2IR score myeloid phagocytic cells according to NicheNet. Heatmap visualization of ligand activity and downstream target genes inferred to be regulated by each respective ligand.
We next sought to better characterize the myeloid cells that might be linked to CPI resistance. Myeloid phagocytic cells are highly plastic, educated by cellular and signaling interactions in the TME, and play diverse roles in promoting and restraining anticancer immunity.30 Our single cell characterization of UC specimens revealed diversity in expression of the pro-tumorigenic inflammation and adaptive immune response signature genes across individual macrophages/monocytes and neutrophils (Figure 5d). Intrigued by the observation that the balance of pro-tumorigenic inflammation and adaptive immunity plays a key role in bulk UC specimens, and reasoning that such balance might be relevant at the level of individual cells, we extended this concept to the monocyte/macrophage and neutrophil population and calculated a 2IR score for each individual cell termed the myeloid single cell 2IR score (Msc2IR score; see Figure 5d,e and Methods). While low Msc2IR score cells were observed across all minor myeloid phagocytic cell populations and were not correlated with M1 or M2 signatures (Figure S17b), these cells were highly enriched in the MM-CCL20 minor population (OR=11.0, p-value<1e-16 by fisher’s exact test) and under-represented in the Macrophage-C1QA minor population (OR=0.14, p-value<1e-16 by fisher’s exact test; Figure 5f).
Differential gene expression and gene set enrichment analysis of myeloid phagocytic cells with low versus high Msc2IR scores revealed upregulation of proinflammatory pathways and top-ranking genes such as IL1B, CXCL8 (IL8), SPP1, and CCL20 in the former while the latter demonstrated upregulation of genes and pathways related to antigen presentation and the T cell recruiting chemokines CXCL9 and CXCL10 (Figure 5e,g and S18).
Pro-tumorigenic monocytes derived from patients with renal cancer have previously been shown to express proinflammatory cytokines and chemokines, including IL1B, CCL20, and CXCL8 (IL8), via an IL-1β-dependent mechanism.31 We sought to define putative therapeutic targets implicated in polarizing myeloid phagocytic cells with low Msc2IR scores and not restrict our analysis to genes overexpressed in low Msc2IR score cells but rather seek upstream ligands implicated in driving the expression of such genes. We therefore used NicheNet32, an approach that predicts ligands that modulate target gene expression by leveraging prior knowledge of signaling pathways and transcriptional regulatory networks (see Methods). Indeed, this analysis revealed that IL-1α and IL-1β were the top-ranked ligands inferred to regulate genes overexpressed in low Msc2IR score cells (Figure 5h). Both IL1A and IL1B were also predominantly expressed by myeloid phagocytic cells in our single cell cohort (Figure S19).
Thus, the Msc2IR score, reflecting the balance of adaptive immune response and pro-tumorigenic inflammation gene expression in individual myeloid phagocytic cells, may reflect the plasticity of these cells in the TME (Figure 5e). Low Msc2IR score monocytes/macrophages and neutrophils, with upregulation of proinflammatory genes and downregulation of antigen presentation genes and not delineated by classical M1 versus M2 polarization or graph-based unsupervised cell clustering, may define a cellular state of myeloid phagocytic cells contributing to CPI resistance.
Monocytes with low Msc2IR scores are enriched in the pre-treatment peripheral blood of patients with CPI-resistant metastatic UC.
We next asked whether similar heterogeneity in Msc2IR scores was present in monocytes in the peripheral blood of patients with metastatic UC and whether these populations were associated with CPI resistance. Single-cell RNA sequencing data from peripheral blood mononuclear cells collected prior to the initiation of treatment with anti-PD-L1 CPI from five patients who achieved an objective response, and five patients who did not achieve an objective response, were utilized (see Methods and Figure S20). We calculated Msc2IR scores in individual monocytes identifying low, intermediate, and high Msc2IR score populations. Monocytes with low Msc2IR scores were significantly enriched in the peripheral blood of patients with CPI-resistant versus CPI-responsive metastatic UC (Figure 6a; p value =0.0048 by two-sided t-test). Alternatively, the five patients who responded to CPI could not be readily distinguished from the five patients with CPI resistant metastatic UC using monocyte minor populations identified by graph-based unsupervised cell clustering (Figure 6b), individual genes such as CXCL8 (IL8) (Figure 6c) or M1 and M2 signatures (Figure S21). Similar to our findings in the UC TME, low Msc2IR score monocytes in the pre-treatment peripheral blood of patients with metastatic UC demonstrated upregulation of proinflammatory genes and downregulation of antigen presentation genes (Figure 6d) and IL-1α and IL-1β were the top ranked ligands inferred to regulate this gene expression program. Therefore, low Msc2IR score myeloid cells are present in both the TME and peripheral blood of patients with UC and are associated with CPI resistance.
Figure 6. Low Msc2IR score monocytes are enriched in the pre-treatment peripheral blood of patients with metastatic urothelial cancer resistant to anti-PD-L1 treatment.
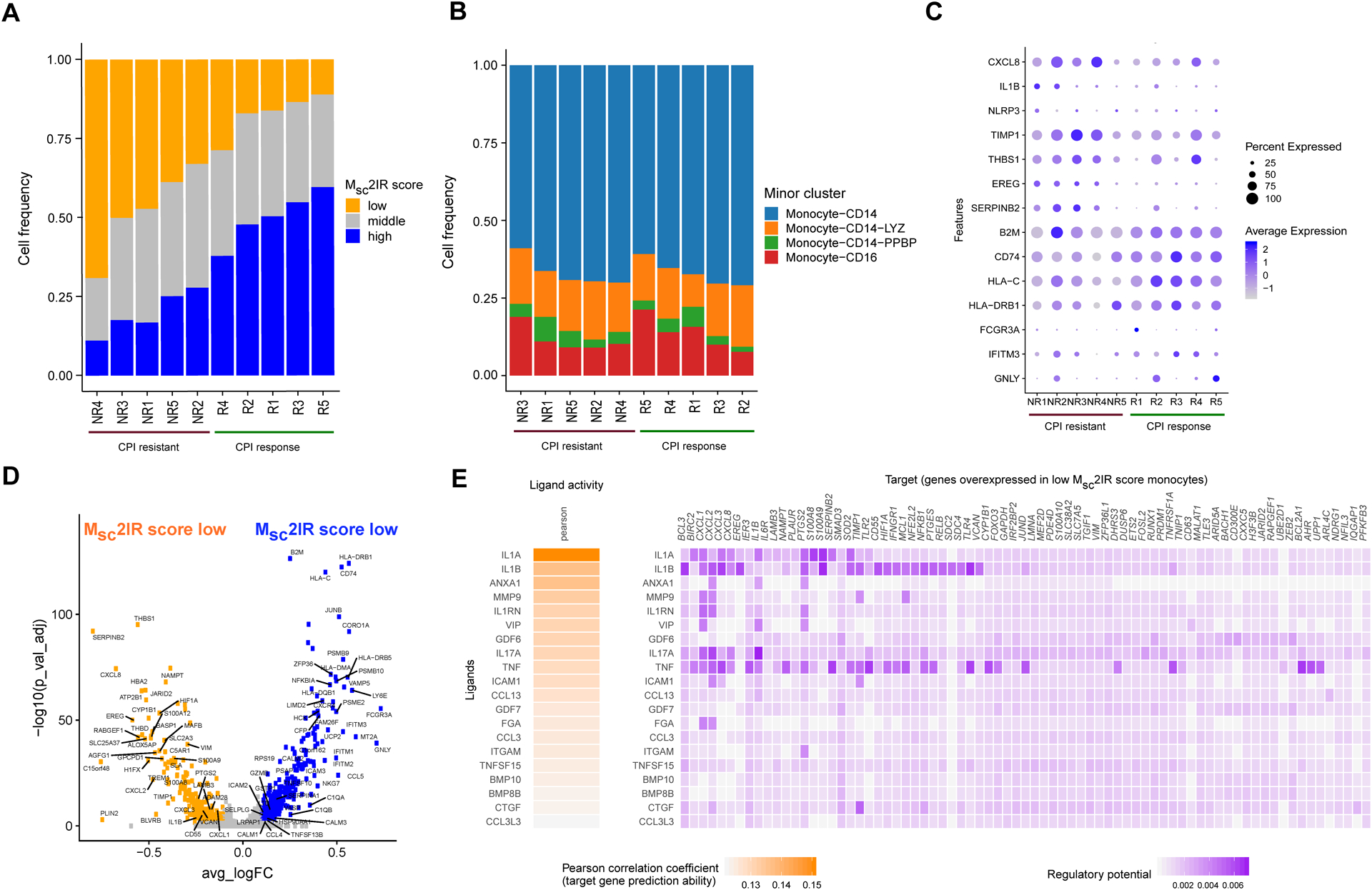
Single-cell RNA sequencing data from peripheral blood mononuclear cells collected prior to the initiation of treatment from five patients with metastatic urothelial cancer who achieved an objective response, and five patients with metastatic urothelial cancer who did not achieve an objective response, to anti-PD-L1 immune checkpoint inhibition (CPI). A. The frequency of monocytes with low, intermediate and high Msc2IR scores in the pre-treatment peripheral blood of patients (n=10 patients) resistant or sensitive to anti-PD-L1 CPI. B. The frequency of monocyte minor cell populations in the pre-treatment peripheral blood of patients (n=10 patients) resistant or sensitive to anti-PD-L1 CPI. C. Dot plot of expression of select genes in monocytes from pre-treatment peripheral blood of patients (n=10 patients) resistant or sensitive to anti-PD-L1 CPI. D. Volcano plot of genes differentially expressed between peripheral blood monocytes with high and low 2IR score. P-value was calculated by Wilcoxon rank-sum test and then adjusted by Bonferroni correction. Genes with log fold change (FC) >0.1 and adjusted p-value <0.05 were considered as significant. E. Top-ranking ligands inferred to regulate genes upregulated in low Msc2IR score peripheral blood monocytes according to NicheNet. Heatmap visualization of ligand activity and downstream target genes inferred to be regulated by each respective ligand.
Discussion
Pro-tumorigenic inflammation is recognized as a “Hallmark of Cancer” pathogenesis.7,33,34 However, antitumor immunity and tumor-promoting inflammation coexist in delicate balance complicating dissecting the role of the latter in mediating CPI resistance in studies using human specimens. Using an unbiased approach, we identified an adaptive immune response gene signature associated with better CPI outcomes and pro-tumorigenic inflammation and stromal gene signatures associated with worse CPI outcomes in patients with metastatic UC. We further demonstrated that: 1) expression of the three gene signatures were positively correlated with one another, consistent with the coexistence and balance between antitumor immunity and tumor-promoting inflammation, 2) the stromal gene signature, linked to activated CAFs, did not convey independent information related to CPI outcomes beyond the pro-tumorigenic inflammation signature suggesting a more indirect role (e.g., recruitment and education of myeloid cells), 3) the 2IR score, reflecting the balance of antitumor immunity and tumor-promoting inflammation in individual TMEs, best correlated with CPI outcomes and was reflective of diverse cell types in the TME and 4) low Msc2IR score myeloid phagocytic cells were characterized by increased expression of proinflammatory genes and decreased expression of antigen presentation genes, could not be discerned based on classical M1 versus M2 polarization, and were enriched in the pre-treatment blood of patients with metastatic UC resistant to CPI. Together, our findings define a myeloid phagocytic cell state associated with CPI resistance, highlight potential approaches to identify patients potentially best suited for therapies seeking to overcome pro-tumorigenic inflammation-related CPI-resistance, and delineate putative therapeutic targets for further study.
Our overarching goal was to identify dominant clinically relevant features correlated with CPI resistance that might be linked to underlying immunobiology and associated therapeutic targets for prioritization for further preclinical and clinical testing as CPI-based combination strategies. Additionally, with further refinement and validation, the identified tissue and blood-based features could prove valuable in establishing proof-of-concept in early phase clinical development of combination regimens targeting myeloid-related CPI resistance, through associations with clinical outcomes and/or pharmacodynamic monitoring (e.g., serial changes in low Msc2IR score monocytes in peripheral blood).
Other studies have linked aspects of the TME with response/resistance to CPI in UC and other malignancies. Features reflecting an activated tumor stroma, including EMT- and TGFβ-related gene signatures have been correlated with poor outcomes with CPI treatment.10,17 In our current study, our stromal gene signature was no longer independently associated with CPI outcomes when the pro-tumorigenic inflammation signature was included in multivariable models suggesting the former may play a more indirect role. Multiple studies have correlated T cell gene signatures, or related measures of adaptive immune resistance, with sensitivity to CPI.1,3,35 However, analyses utilizing human tumor specimens demonstrating an independent association between gene signatures reflecting pro-tumorigenic inflammation, or related myeloid cells, and CPI resistance have been much more limited. This disconnect is likely in part related to the positive correlation between gene signatures reflecting the presence of T cells, and other stromal and immune cells, despite a disparate impact on outcomes complicating dissecting the role of the latter and due to the plasticity of immune cells in the TME. Recently, elevated levels of serum/plasma IL-8, traced primarily to the myeloid cell compartment, was associated with decreased efficacy of CPI across several tumor types.19,36 These studies likely relate to similar biology exposed in our analysis, though we extend these findings by: (1) contextualizing the importance of pro-tumorigenic inflammation in contributing to CPI resistance in UC relative to other features defined through an unbiased approach, (2) providing a refined understanding of the myeloid cellular state associated with pro-tumorigenic inflammation characterized by expression of a number of inflammatory cytokines and chemokines, beyond IL-8 alone, suggesting that targeting upstream regulators may be required for optimal therapeutic modulation, and (3) defining tumor-tissue and blood-based measures using bulk or scRNA sequencing to identify tumors for which pro-tumorigenic inflammation may be contributing to CPI resistance in UC.
Myeloid phagocytic cells have been linked to suppression of antitumor immunity across a range of malignancies via a variety of mechanisms though clinically tractable approaches to target myeloid cell-related CPI resistance have remained elusive.37–39 IL-1 was among the top-ranked ligands inferred to regulate the low Msc2IR score myeloid phagocytic cell gene program in line with prior experimental data demonstrating that inflammatory cytokine and chemokine production from pro-tumorigenic monocytes in patients with renal carcinoma was IL-1β-dependent.31 IL-1β has been considered a “master regulator” of inflammation involved in the tumor-promoting and immune suppressive function of myeloid cells, anti-IL-1β combined with anti-PD-1 therapy abrogated tumor growth in model systems, and anti-IL-1β has been associated with lower cancer mortality in human studies.31,40–42 Building on this collective work, our findings raise the hypothesis that targeting IL-1 may reverse the inflammatory phenotype of low Msc2IR score myeloid phagocytic cells and may represent a rational combination strategy to overcome CPI resistance in a defined subset of patients with UC. Additional studies are required to refine the role of IL-1α versus IL-1β in this context though IL-1β is not present in cells from healthy individuals, and is a product of limited cell types such as myeloid-phagocytic cells, whereas IL-1α is more ubiquitously expressed. Clinical trials combining CPI and anti-IL-1 therapies have already been initiated (NCT03631199, NCT03742349). Further, the gene expression program of low Msc2IR score myeloid phagocytic cells in the TME reveals several additional putative targets previously linked to inflammatory disorders worthy of further investigation.43,44
There are potential limitations to our study. While our study is among the first to characterize the UC TME at the single cell RNA level, the quantity of single cells from each specimen was variable with two specimens contributing a large proportion of cells; a larger cohort is required to establish a definitive cellular atlas of UC specimens. Still, the main goal of our scRNA-seq cohort in this study was to uncover the cellular origins our gene signatures derived from bulk RNA sequencing data. Features of urothelial cancer cells are likely associated with sensitivity and resistance to CPI. However, beyond TMB, on which our three gene signatures were conditioned, our current analysis was focused on the TME given the expression of the module genes when projected onto our scRNA-seq data. Cancer cell-intrinsic features that contribute to immune escape and ultimately shape the pro-tumorigenic inflamed TME require further study. Though we linked low Msc2IR score monocytes in peripheral blood with resistance to CPI in patients with metastatic UC, we did not have paired scRNA-seq data from the matched primary tumors to directly explore the association between the TME and circulating immune cells. Together, these considerations underscore the need for additional studies of UC specimens profiled at single cell resolution and linked to CPI treatment outcomes.
Our study identified and validated key gene signatures associated with sensitivity or resistance to CPI in patients with metastatic UC related to adaptive immunity and pro-tumorigenic inflammation, defined the 2IR score as reflecting such balance in individual UC TMEs, established the Msc2IR score as reflecting the cellular state of myeloid phagocytic cells linked to CPI resistance, and identified putative therapeutic targets to overcome resistance. Future work exploring the 2IR and Msc2IR scores in clinical trials seeking to overcome myeloid-related CPI resistance, further defining and credentialing “master regulators” of low Msc2IR score myeloid cell polarization as putative therapeutic targets, and dissecting the dominant mechanisms of immune suppression related to these cells may help facilitate extension of the benefits of CPI to additional patients with UC.
Supplementary Material
Statement of translational relevance.
Using an unbiased approach, we identified and validated gene signatures related to adaptive immunity and pro-tumorigenic inflammation associated with sensitivity or resistance to PD-1/PD-L1 blockade in patients with metastatic urothelial cancer. We defined the balance of these signatures, coined the 2IR score, in individual urothelial cancer tumor microenvironments best correlated with clinical outcomes and defined cellular states of single myeloid cells linked to these microenvironments and PD-1/PD-L1 blockade resistance. Integrating these bulk and single cell RNA signatures into clinical trials seeking to overcome myeloid cell-related PD-1/PD-L1 blockade resistance, and further credentialing therapeutic targets linked pro-tumorigenic inflammation, may help facilitate extension of the benefits of PD-1/PD-L1 blockade to additional patients with urothelial cancer.
Acknowledgements
The results shown here are in part based upon data generated by the TCGA Research Network: http://cancergenome.nih.gov/.
We thank Jill Gregory at the Icahn School of Medicine Instructional Technology Group for assistance with graphic design.
Funding:
CA196521 (MDG, NB, WKO), NIH Loan Repayment Program (SI)
Competing interests:
MDG has served as a consultant or advisor to BioMotiv, Janssen, Dendreon, Merck, Glaxo Smith Kline, Lilly, Astellas, Genentech, Bristol-Myers Squibb, Novartis, Pfizer, EMD Serono, Astra Zeneca, Seattle Genetics, Incyte, Aileron, Dracen, Inovio, NuMab, Dragonfly Therapeutics. He has received research funding from Janssen, Dendreon, Novartis, Bristol-Myers Squibb, Merck, Astra Zeneca, Genentech/Roche.
NB has served as a consultant or advisor to Neon, Tempest, Checkpoint Sciences, Curevac, Primevax, Novartis, Array BioPharma, Roche, Avidea, Boehringer Ingelheim, Rome Therapeutics, Roswell Park, Parker Institute for Cancer Immunotherapy. She has received research funding from Novocure, Ludwg Institute, Celldex, Genentech, Oncovir, Melanoma Research Alliance, Leukemia & Lymphoma Society, NSTEM, Regeneron.
WKO has served as a consultant to Astellas, Astra Zeneca, Bayer, Janssen, Sanofi, Sema4 and TeneoBio.
SG reports consultancy and/or advisory roles for Merck, Neon Therapeutics and OncoMed and research funding from Bristol-Myers Squibb, Genentech, Immune Design, Agenus, Janssen R&D, Pfizer, Takeda, and Regeneron. S.G. is a named co-inventor on an issued patent for multiplex immunohistochemistry to characterize tumors and treatment responses. The technology is filed through Icahn School of Medicine at Mount Sinai (ISMMS) and non-exclusively licensed to Caprion. Mount Sinai has received payments associated with this technology and Dr. Gnjatic is entitled to future payments.
PS reports consultantships for Oncolytics, Jounce, BioAtla, Forty-Seven, Polaris, Marker, Codiak, ImaginAB, Hummingbird, Dragonfly, Lytix, Lava Therapeutics, Infinity and Achelois. Dr. Sharma reports ownership interests in Constellation, Oncolytics, Apricity Health, Hummingbird, Dragonfly, Lytix, Lava Therapeutics, Achelois, Jounce, Neon, BioAtla, Forty-Seven, Polaris, Marker, Codiak, and ImaginAb. PS also owns a patent licensed to Jounce.
JS has served as a speaker for Pfizer.
AH has served as a consultant or advisor to HTG Molecular.
JZ, LW, and ES are employees of SEMA4
References
- 1.Rosenberg JE, Hoffman-Censits J, Powles T, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: A single-arm, multicentre, phase 2 trial. Lancet 2016;387(10031). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Powles T, O’Donnell PH, Massard C, et al. Efficacy and Safety of Durvalumab in Locally Advanced or Metastatic Urothelial Carcinoma. JAMA Oncol 2017;45(2):e172411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sharma P, Retz M, Siefker-Radtke A, et al. Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): a multicentre, single-arm, phase 2 trial. Lancet Oncol 2017;18(3). [DOI] [PubMed] [Google Scholar]
- 4.Bellmunt J, de Wit R, Vaughn DJ, et al. Pembrolizumab as Second-Line Therapy for Advanced Urothelial Carcinoma. N Engl J Med 2017;NEJMoa1613683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Patel MR, Ellerton J, Infante JR, et al. Avelumab in metastatic urothelial carcinoma after platinum failure (JAVELIN Solid Tumor): pooled results from two expansion cohorts of an open-label, phase 1 trial. Lancet Oncol 2018;19(1):51–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tumeh PC, Harview CL, Yearley JH, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014;515(7528):568–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144(5):646–74. [DOI] [PubMed] [Google Scholar]
- 8.Veglia F, Perego M, Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nat Immunol 2018;19(2):108–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Noy R, Pollard JW. Tumor-Associated Macrophages: From Mechanisms to Therapy. Immunity. 2014;41(1):49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mariathasan S, Turley SJ, Nickles D, et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018;554(7693):544–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Balar AV, Galsky MD, Rosenberg JE, et al. Atezolizumab as first-line treatment in cisplatin-ineligible patients with locally advanced and metastatic urothelial carcinoma: a single-arm, multicentre, phase 2 trial. Lancet 2017;389(10064):67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Galsky MD, Saci A, Szabo PM, et al. Impact of tumor mutation burden on nivolumab efficacy in second-line urothelial carcinoma patients: Exploratory analysis of the phase ii checkmate 275 study. Ann Oncol 2017;28(suppl_5). [Google Scholar]
- 13.Chen Y, Zhu J, Lum PY, et al. Variations in DNA elucidate molecular networks that cause disease. Nature 2008;452(7186):429–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Emilsson V, Thorleifsson G, Zhang B, et al. Genetics of gene expression and its effect on disease. Nature 2008;452(7186):423–8. [DOI] [PubMed] [Google Scholar]
- 15.Wang K, Narayanan M, Zhong H, Tompa M, Schadt EE, Zhu J. Meta-analysis of Inter-species Liver Co-expression Networks Elucidates Traits Associated with Common Human Diseases. PLoS Comput Biol 2009;5(12):e1000616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Narayanan M, Huynh JL, Wang K, et al. Common dysregulation network in the human prefrontal cortex underlies two neurodegenerative diseases. Mol Syst Biol 2014;10(7):743–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang L, Saci A, Szabo PM, et al. EMT- and stroma-related gene expression and resistance to PD-1 blockade in urothelial cancer. Nat Commun 2018;9(1):3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jiang P, Gu S, Pan D, et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med 2018;24(10):1550–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yuen KC, Liu L-F, Gupta V, et al. High systemic and tumor-associated IL-8 correlates with reduced clinical benefit of PD-L1 blockade. Nat Med [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goswami S, Chen Y, Anandhan S, et al. ARID1A mutation plus CXCL13 expression act as combinatorial biomarkers to predict responses to immune checkpoint therapy in mUCC. Sci Transl Med 2020;12(548). [DOI] [PubMed] [Google Scholar]
- 21.Remark R, Merghoub T, Grabe N, et al. In-depth tissue profiling using multiplexed immunohistochemical consecutive staining on single slide. Sci Immunol 2016;1(1):aaf6925–aaf6925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Akturk G, Sweeney R, Remark R, Merad M, Gnjatic S. Multiplexed Immunohistochemical Consecutive Staining on Single Slide (MICSSS): Multiplexed Chromogenic IHC Assay for High-Dimensional Tissue Analysis. In: Methods in Molecular Biology. Humana Press Inc; 2020. p. 497–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Helmink BA, Reddy SM, Gao J, et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 2020;577(7791):549–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cabrita R, Lauss M, Sanna A, et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 2020;577(7791):561–5. [DOI] [PubMed] [Google Scholar]
- 25.Newman AM, Liu CL, Green MR, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 2015;12(5):453–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stuart T, Butler A, Hoffman P, et al. Comprehensive Integration of Single-Cell Data. Cell 2019;177(7):1888–1902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Q, He Y, Luo N, et al. Landscape and Dynamics of Single Immune Cells in Hepatocellular Carcinoma. Cell 2019;179(4):829–845.e20. [DOI] [PubMed] [Google Scholar]
- 28.Azizi E, Carr AJ, Plitas G, et al. Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell 2018;174(5):1293–1308.e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lavin Y, Kobayashi S, Leader A, et al. Innate Immune Landscape in Early Lung Adenocarcinoma by Paired Single-Cell Analyses. Cell 2017;169(4):750–765.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Locati M, Curtale G, Mantovani A. Diversity, Mechanisms, and Significance of Macrophage Plasticity. Annu Rev Pathol Mech Dis 2020;15:123–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chittezhath M, Dhillon MK, Lim JY, et al. Molecular Profiling Reveals a Tumor-Promoting Phenotype of Monocytes and Macrophages in Human Cancer Progression. Immunity 2014;41(5):815–29. [DOI] [PubMed] [Google Scholar]
- 32.Browaeys R, Saelens W, Saeys Y. NicheNet: modeling intercellular communication by linking ligands to target genes. Nat Methods 2020;17(2):159–62. [DOI] [PubMed] [Google Scholar]
- 33.Coussens LM, Zitvogel L, Palucka AK. Neutralizing tumor-promoting chronic inflammation: A magic bullet? Science (80-. ). 2013;339(6117):286–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shalapour S, Karin M. Pas de Deux: Control of Anti-tumor Immunity by Cancer-Associated Inflammation. Immunity. 2019;51(1):15–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cristescu R, Mogg R, Ayers M, et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 2018;362(6411):eaar3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schalper KA, Carleton M, Zhou M, et al. Elevated serum interleukin-8 is associated with enhanced intratumor neutrophils and reduced clinical benefit of immune-checkpoint inhibitors. Nat Med 2020;26(5):688–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol 2017;14(7):399–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gabrilovich DI. Myeloid-derived suppressor cells. Cancer Immunol Res 2017;5(1):3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peranzoni E, Lemoine J, Vimeux L, et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. Proc Natl Acad Sci U S A 2018;115(17):E4041–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Condamine T, Gabrilovich DI. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol. 2011;32(1):19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kaplanov I, Carmi Y, Kornetsky R, et al. Blocking IL-1β reverses the immunosuppression in mouse breast cancer and synergizes with anti–PD-1 for tumor abrogation. Proc Natl Acad Sci U S A 2019;116(4):1361–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ridker PM, MacFadyen JG, Thuren T, et al. Effect of interleukin-1β inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet 2017;390(10105):1833–42. [DOI] [PubMed] [Google Scholar]
- 43.Ranasinghe R, Eri R. Modulation of the CCR6-CCl20 axis: A potential therapeutic target in inflammation and cancer. Med. 2018;54(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang W, Wang H, Sun M, et al. CXCL5/CXCR2 axis in tumor microenvironment as potential diagnostic biomarker and therapeutic target. Cancer Commun. 2020;40(2–3):69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Robertson AG, Kim J, Al-Ahmadie H, et al. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell 2017;171(3):540–556.e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu J, Lichtenberg T, Hoadley KA, et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018;173(2):400–416.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li B, Dewey CN. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 2011;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2007;8(1):118–27. [DOI] [PubMed] [Google Scholar]
- 49.Langfelder P, Horvath S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinformatics 2008;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011;27(12):1739–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bankhead P, Loughrey MB, Fernández JA, et al. QuPath: Open source software for digital pathology image analysis. Sci Rep 2017;7(1):1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Puram SV, Tirosh I, Parikh AS, et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 2017;171(7):1611–1624.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Aran D, Looney AP, Liu L, et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol 2019;20(2):163–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Monaco G, Lee B, Xu W, et al. RNA-Seq Signatures Normalized by mRNA Abundance Allow Absolute Deconvolution of Human Immune Cell Types. Cell Rep 2019;26(6):1627–1640.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.