

Abstract
The human neonatal cerebellum is a fourth of its adult size, yet contains the blueprint required to integrate environmental cues with developing motor, cognitive, and emotional skills into adulthood. Although mature cerebellar neuroanatomy is well studied, understanding its developmental origins is limited. Here, we systematically mapped the molecular, cellular, and spatial composition of human fetal cerebellum by combining laser capture microscopy and SPLiT-seq single-nucleus transcriptomics. We profiled functionally distinct regions and gene expression dynamics within cell types and across development. The resulting cell atlas demonstrates that the molecular organization of the cerebellar anlage recapitulates cytoarchitecturally distinct regions and developmentally transient cell types that are distinct from the mouse cerebellum. By mapping genes dominant for pediatric and adult neurological disorders onto our dataset, we identify relevant cell types underlying disease mechanisms. These data provide a resource for probing the cellular basis of human cerebellar development and disease.
The cerebellum, identified in classic neuroanatomy studies over a hundred years ago, integrates neuronal networks that couple motor function with cognition, emotional regulation, and language.1, 2 Its unique structure and function depend on the precise developmental coordination of molecular and cellular programs among multiple cell types3, 4, and when these go awry, result in disease. Cerebellar diseases include congenital structural abnormalities in children and cerebellar ataxias in adults that have specific cerebellar pathology, but also disorders that impact multiple brain regions including the cerebellum, such as autism spectrum disorders (ASD) and Alzheimer’s disease.1, 5–7 Although cerebellar development is relatively well understood in mice,8, 9 we have only begun to unravel the complexity and dynamics of human cerebellar development.
Cerebellar spatial organization results from timed cellular proliferation and differentiation within distinct progenitor niches and coordinated cell migration during development.10–12 A unique feature of the human cerebellum is that the progenitor zones have extended proliferative activity, suggesting human-specific expansion of these zones.10 Emerging from the dorsal hindbrain, the cerebellar anlage contains two separate proliferative niches; the ventricular zone (VZ) which lines the fourth ventricle, and the rhombic lip (RL) which is dorsal to the VZ and adjacent to the developing choroid plexus. The VZ, which gives rise to GABAergic neurons, is active during early embryonic stages and dominates the cerebellar anlage with nascent VZ-derived Purkinje cells by 10 post conceptional weeks (PCW) (Fig. 1a). After 10 PCW, the RL expands to generate glutamatergic neurons, the majority of which are granule cell progenitors, which migrate rostrally to form the external granule cell layer (EGL) on the dorsal surface. By mid-gestation, Purkinje cells reorganize into a single cell layer (PCL) under the EGL. At the same time, granule cell progenitors in the EGL proliferate, differentiate, and migrate inward, to form the internal granule cell layer located just below the PCL.
Fig. 1 |. Overview of prenatal cerebellar development and the data generated in this study.
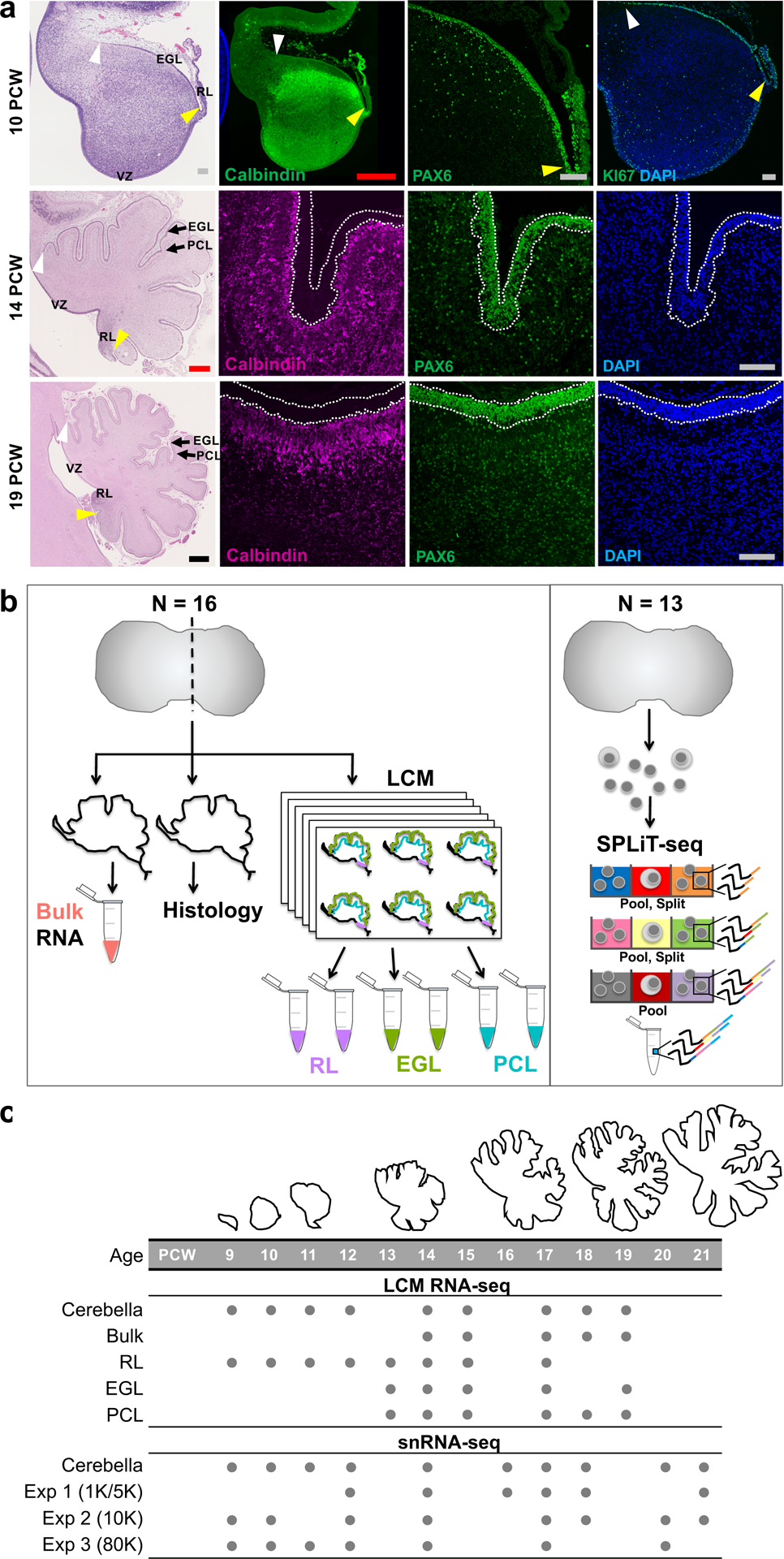
a, Midsagittal sections of the human fetal cerebellum stained with hematoxylin and eosin (H&E) or markers for Purkinje cells (Calbindin) or rhombic lip (RL) and external granule cell layer (EGL; PAX6 and KI67). A minimum of 2 samples per age were stained with adjacent sections used for histology and immunocytochemistry. The ventricular zone (VZ), RL, EGL, and Purkinje cell layer (PCL) are shown. Arrowheads mark the anterior (yellow) and posterior (white) EGL across the dorsal surface of the cerebellar anlage. At 9 PCW, the cerebellar anlage is dominated by Purkinje cells, with a thin nascent EGL extending from the RL. By 19 PCW, Purkinje cells have migrated radially to establish a multicellular layer (PCL) beneath the EGL. Scale bars: 100 um (grey), 500 um (red), 1 mm (blue). 10 PCW H&E section was used previously in Fig. 1 of Haldipur et al., 2019. b, Schematic illustrating LCM (left) and SPLiT-seq (right) experimental workflows. c, The time span of fetal cerebellar development represented by line drawings of midsagittal sections of the cerebellum (to-scale) showing a dramatic change in size and foliation from 9 to 19 PCW. Below is the distribution of cerebella in this study. Biological and technical replicate samples are not shown (RNA-seq sample numbers: n = 13 for bulk; 9 for RL; 17 for EGL; 18 for PCL; snRNA-seq sample numbers: n = 6 for Exp 1; 11 for Exp 2; 9 for Exp 3).
Many core molecular programs and cellular cues that guide the extensive growth and coordinated reorganization during cerebellar development are known.3, 8, 9 However, cerebellum is not well represented in previous bulk and single-cell transcriptomic studies of the developing human brain.13–17 In the BrainSpan atlas of the developing human brain, samples from neocortical regions account for 68% (415/607) of the samples.15 In contrast, cerebellar samples account for only 6% (35/607) of the total samples (Supplementary Fig. 1). Prenatal development of the cerebellum is even less well represented in BrainSpan, with cerebellum comprising only 5% (13/261) of the prenatal samples [8–37 postconceptional weeks (PCW)]. Further, the available data are predominantly derived from bulk transcriptomic analysis. As it stands, the available human brain transcriptomic data are not likely to capture the depth and breadth of the molecular repertoire in the cerebellum, especially during early development.
Here, we characterize the transcriptional and cellular landscape of the developing human cerebellum by combining laser capture microdissection (LCM) of spatially defined progenitor and neuronal populations with single-nucleus transcriptomic sequencing. We report single-nucleus combinatorial indexing that profiles the transcriptomes of 70,000 cells across prenatal cerebellar development from 9–21 PCW. We cross-compare this data with the BrainSpan dataset and with a published mouse dataset. Our work establishes a ‘Developmental Cell Atlas of the Human Cerebellum’ as a solid foundation enabling novel discoveries related to cerebellar development and origin of disease.
RESULTS
Study design and data generation
To characterize the transcriptional landscape of the prenatal human cerebellum, we generated and analyzed transcriptomic data using direct and inferred approaches to define cell populations. We performed bulk RNA-seq from spatially demarcated progenitor and neuronal regions isolated by LCM (57 samples from 16 cerebella) and single cell RNA-seq (69,174 cells/nuclei from 13 cerebella) from 29 postmortem cerebella obtained from clinically and histopathologically unremarkable donors of both sexes across fetal development (Fig. 1b and Supplementary Tables 1–2).
To obtain populations preferentially enriched for primary progenitors and neurons, we devised a consistent experimental workflow to isolate cells occupying RL, EGL, and PCL (Fig. 1b, and Extended Data Fig. 1a). Specifically, we dissected whole cerebella from fetal specimens with intact calvaria (the top part of the skull) to ensure correct orientation for each sample, and sectioned frozen cerebella in the sagittal plane through the cerebellar midline lobe (vermis). We then isolated RNA from one section for each specimen (referred to from here on as ‘bulk’) and assessed RNA quality [RNA integrity number (RIN), 7.7±0.95 (mean±s.d.)] (Supplementary Table 1). For our LCM sample collection, we visually localized the EGL, which is easily identifiable in sagittal sections as a cell-dense layer on the dorsal surface of the developing cerebellar anlage, and attained adjacent sections using an anti-Calbindin antibody, a well-known Purkinje cell marker, to identify the PCL. Finally, we isolated total RNA from our 57 samples. We had previously performed LCM and RNA-seq of the RL10 and included this dataset in our analysis.
For our sequencing libraries, we selected the Illumina TruSeq RNA Access Library Prep Kit because it requires low total RNA input, yet maintains high sensitivity. We then performed paired-end Illumina high-quality sequencing on bulk cerebellum (N=13), RL (N=9), EGL (N=17), and PCL (N=18) from 16 mid-gestation (9–21 PCW) fetal specimens (Fig. 1b and Supplementary Table 1). By comparing gene expression of established RL (LMX1A, BARHL1), EGL (ATOH1, PAX6), and PCL (CALB1, SKOR2) markers between the RNA-seq dataset from LCM-isolated samples and bulk-isolated cerebellum, we validated the technical quality of our LCM-isolation; expression of these six neuron-specific markers confirmed the specificity of our enrichment, with the highest expression detected in the appropriate samples (Extended Data Fig. 1b).
To complement our spatially defined analyses, we performed three single-nucleus RNA-seq (snRNA-seq) experiments using 26 samples from an independent set of 13 cerebella ranging in age from 9 to 21 PCW (Fig. 1c and Supplementary Table 2). We used split-pool ligation-based transcriptome sequencing (SPLiT-seq), a multi-step barcoding strategy combined with RNA-seq which increases throughput by enabling simultaneously interrogation of thousands of cells/nuclei in multiplexed samples.18 Single-cell (8 samples) and single-nucleus (12 samples) level transcriptomic data were generated in technical replicates for 10 cerebella across two experiments; data for the remaining 3 cerebella were generated in a single experiment.
Transcriptional analysis of spatially defined neural zones
To characterize the global transcriptional landscapes in RL, EGL, and PCL, and RL, we applied principal component analysis (PCA) to the expression profiles of LCM-isolated regionally distinct regions and from bulk cerebellum. PCA visualized sample clustering corresponding to neuronal region, with PC1 distinguishing RL and EGL from PCL and bulk cerebellum and PC2 distinguishing RL from EGL (Fig. 2a). To identify spatially regulated genes, we evaluated differential gene expression between each LCM-isolated zone and bulk cerebellum using a modest threshold (false discovery rate, FDR <0.05 and log2-transformed fold change >1.5) and including library prep batch, age, and region as covariates (Fig. 2b–d). This analysis identified 1,111 differentially expressed genes (1.5-fold, FDR<0.05) between the neuronal zones: 627 genes showed increased expression in RL, 612 genes in EGL, and 168 genes in PCL compared to bulk cerebellum (Fig. 2e and Supplementary Table 3). The RL genes were enriched for cell cycle (hsa04110, FDR = 1.2×10−18) and p53 signaling pathways (hsa04115, FDR = 6.3×10−6), as were the EGL genes (hsa04110, FDR = 1.1×10−14 and hsa04115, FDR = 0.0007) (Supplementary Table 4). The PCL genes showed little pathway enrichment. Subsets of genes were specifically expressed in each captured region (Supplementary Fig. 2): 184 RL-specific genes, 176 EGL-specific genes, and 142 PCL-specific genes. The RL genes were enriched in Hippo signaling, stem cell pluripotency regulation, and TGFβ signaling (Fig. 2f), with expression increasing across mid-gestation (Supplementary Fig. 2d). EGL genes were enriched in MAPK, Ras, and Rap1 signaling (Fig. 2g and Supplementary Fig. 2e). Again, we detected little pathway enrichment among PCL genes (Supplementary Table 4).
Fig. 2 |. Spatial transcriptional analysis of the developing human cerebellum.

a, Principal component analysis indicates that the largest source of variation among RNA-seq samples was spatial location, accounting for 57% of the variance, and verifying LCM successfully captured these regions. b-d, Volcano plots illustrating differential expression of genes for each spatial region versus bulk cerebellum. Colored dots represent genes with significant expression [FDR<0.05; Log2(FC)>1.5]. Selected canonical genes with significant expression are labeled. Significance was determined by the Wald test and adjusted using FDR. Statistics are presented in Supplementary Table 3. e, Heatmap of the top 10 genes with significant expression per spatial region (RL, EGL, PCL) are shown for each sample. Samples are ordered by region [RL (purple), EGL (green), PCL (turquoise), bulk (salmon)], then by ascending age (9 to 19 PCW). High expression is in red and low expression is in blue. f,g, Heatmaps of genes and pathways expressed in RL (f) and EGL (g) identified by gene ontology analysis. High expression is in red and low expression is in blue; Z-score legend as in e. Colored boxes indicate genes represented in enriched pathways: Hippo signaling (green), signaling pathways regulating pluripotency of stem cells (magenta), and Tgfβ signaling (blue) in f; MAPK signaling (orange), Rap1 (green), Ras (pink) in g. Statistics are presented in Supplementary Table 4. h, Heatmap of genes expressed in each WGCNA module enriched in a. High expression is in red and low expression is in blue; Z-score legend as in e. Colored boxes represent the cerebellar region interpretation for each WGCNA module, as in Supplementary Table 5.
To identify cellular components of the spatial cerebellar transcriptome, we performed weighted gene co-expression network analysis (WGCNA)19 on all 57 LCM samples and identified 21 modules of co-expressed genes (Extended Data Fig. 2–4, and Supplementary Tables 5–6). We curated 21 gene co-expression modules according to spatial relationships between enriched regions and shared gene expression between regions within the RL lineage. Of these, 9 modules showed expression differences among cerebellar regions (spatial), 8 modules showed expression differences in both RL and EGL (RL lineage), one module was enriched in bulk cerebellum, and three modules did not show differential expression among the regions captured.
When we compared our 21 gene coexpression modules to the 73 modules generated in the most recent BrainSpan analysis of human neurodevelopment15, which comprises 16 anatomical brain regions including cerebellar cortex, we found that 26 of the 73 BrainSpan modules were correlated with the modules derived from our data (Fig. 2h and Supplementary Fig. 3). We found that genes in 14 BrainSpan modules were enriched among genes with spatial expression in prenatal cerebellum, 8 of which were enriched in the RL lineage (RL and EGL), 2 of which were enriched bulk cerebellum, and 2 of which were correlated with modules that were not differentially expressed in the prenatal cerebellum. Overall, the majority of these 14 BrainSpan modules were highly expressed prenatally in all brain regions and contained multiple neural and non-neural cell types (Supplementary Table 3). Among the 15 cerebellar-specific BrainSpan modules, only one (M11) was shared with our data (M9). The M11 module is highly expressed in postnatal cerebellum and includes granule cell markers such as PAX6 and GABRA6. This result was expected given that our data are exclusively prenatal, when Purkinje cells dominate, while BrainSpan contains a small number of primarily postnatal cerebellum samples, when the granule cell population is vastly dominant relative to all other cell types in the cerebellum20. In our data, PAX6 is found in M14, which is highly expressed in both RL and EGL, consistent with the granule neuron lineage, and enriched in processes regulating DNA (Supplementary Table 6).
Cell types in the developing human cerebellum
We performed snRNA-seq to define cell types and assemble cell-type specific transcriptomes in the developing human cerebellum from 9 to 21 PCW (Supplementary Table 2). Using SPLiT-seq,18 we sequenced 92,314 nuclei (~21,000 raw reads per nucleus) with a median transcript capture of 1,214 unique molecular identifiers (UMIs) per nucleus (Supplementary Table 7). We removed outlier cells with too few (<200) or too many (dataset specific cutoffs) genes detected. We used DoubletFinder21 to detect and discard 5% likely doublets. The remaining 69,174 nuclei had an average of 3,626 transcripts/UMIs per nucleus from 1,332 genes. We merged a total of four datasets; two datasets generated previously,22 and two datasets generated in the present study. Each dataset was filtered independently, after which we used Seurat v323 to integrate all four datasets. We applied Louvain clustering and UMAP visualization to all cells in the integrated dataset (Fig. 3a). Nuclei from replicate samples processed in separate experiments were similarly distributed while nuclei from different developmental stages were not (Extended Data Fig. 5). We used known marker genes to manually annotate 21 distinct cell types, then validated the expression of selected marker genes using immunohistochemistry or in situ hybridization (Fig. 3b,c and Supplementary Table 8).
Fig. 3 |. Identifying the major cell types of the developing human cerebellum.
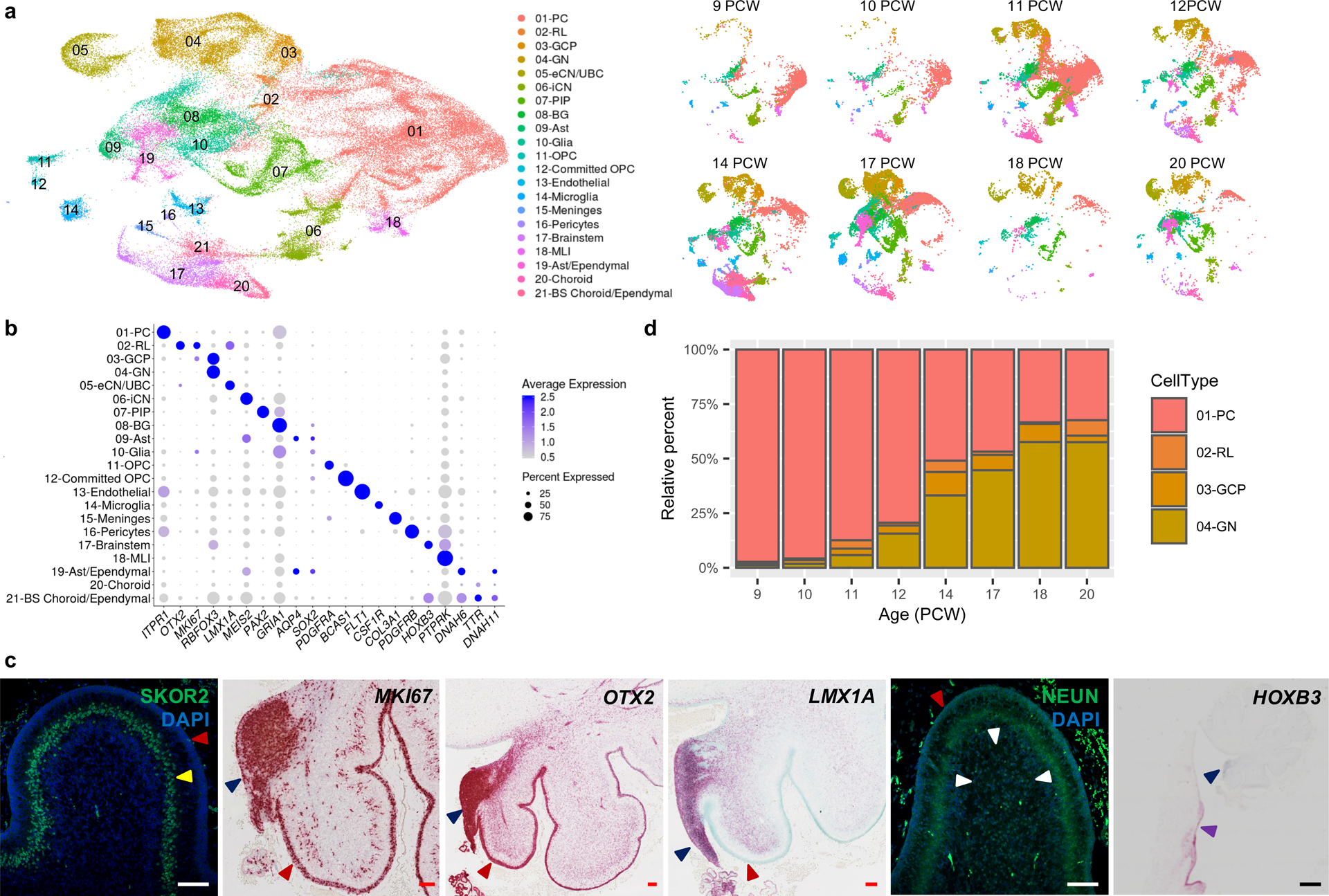
a, UMAP visualization of 67,174 human cerebellar nuclei colored by cluster identity from Louvain clustering and annotated on the basis of marker genes. The same UMAP is plotted at right, showing only nuclei from each age (nuclei numbers from left to right: n = 5,003 for 9 PCW; 2,329 for 10 PCW; 20,364 for 11 PCW; 7,119 for 12 PCW; 11,213 for 14 PCW; 15,556 for 17 PCW; 1,617 for 18 PCW; 5,177 for 20 PCW). b, Dot plot showing the expression of one selected marker gene per cell type. The size of the dot represents the percentage of nuclei within a cell type in which that marker was detected and its color represents the average expression level. Statistics are presented in Supplementary Table 9. c, Midsagittal sections of the human fetal cerebellum at 18 PCW stained with selected marker genes for Purkinje cells (SKOR2), proliferation (MKI67), RL (OTX2 and LMX1A), granule neurons (NEUN), and brainstem (HOXB3). Adjacent sections from one sample were stained for OTX2 and HOXB3; a minimum of 3 sections from each of 3 samples were stained for the other markers. The EGL, PCL, internal granule cell layer, RL and brainstem are indicated by red, yellow, white, blue and purple arrowheads, respectively. Sections are counterstained using DAPI for immunohistochemistry (SKOR2, NEUN) or Fast Green for in situ hybridization (MKI67, OTX2, LMX1A, HOXB3). Scale bar = 100 um and 1 mm (HOXB3). LMX1A was used previously in Fig. 3G of Haldipur et al., 2019. d, Stacked bar charts show the percentage of the four major cell types from each age sampled. Bar colors represent Purkinje cells (PC), rhombic lip (RL), granule cell precursors (GCP), or granule neurons (GN).
Across the 21 major cell types, 4,443 genes (FDR < 0.05) were differentially expressed (Supplementary Table 9). We identified 239 cell-type specific marker genes (average LogFC>1.5; Extended Data Fig. 6), many that were previously characterized as markers of the respective cell types. For example, we detected CA8, ITPR1, DAB1, and RORA in Purkinje cells, SLIT2 in RL, and RELN and RBFOX3 in granule neurons.
The 21 cell types as a group are represented by a median of 1,659 nuclei (ranging from 25,724 Purkinje cells to 189 pericytes). Across developmental time points, our analysis mirrored known changes in the cellular composition of the four major cerebellar cell types (Purkinje cells, RL, granule cell progenitors, granule neurons) (Fig. 3d). For instance, at 9 PCW, Purkinje cells comprised 97% (3,736/3,839) of the total nuclei recovered from the major cell types present, then gradually declined to 32% (371/1,145) at 20 PCW. Conversely, granule neurons in the cerebellar anlage at 9 PCW comprised 1% (44/3,839) of the total nuclei recovered, then increased across development to reach 58% (659/1,145) at 20 PCW. Cell type composition among samples was most consistent in our largest dataset (Extended Data Fig. 7). Overall, RL comprised only 1% (1,018/69,174) of the total nuclei recovered from the cerebellum across development, with 822 (81%) RL nuclei detected among 59,608 total nuclei recovered in our largest dataset (Extended Data Fig. 5a and Supplementary Table 7).
Molecular distinction between RL compartments
We recently demonstrated that the human RL has unique cytoarchitectural features that are not shared with other vertebrates, including the non-human primate, macaque.10 Specifically, in human fetal brain development, the RL begins as a simple proliferative progenitor niche, but then becomes compartmentalized into ventricular (RLVZ) and subventricular zones (RLSVZ) which persist until birth. To identify molecular characteristics of the uniquely human RL progenitor subsets, we selected and subclustered cells in the RL population and examined the molecular correlates that define the RLVZ and RLSVZ compartments (Fig. 4). To annotate the subclusters, we first examined the expression of classic RL markers. Indeed, MKI67, PAX6, and LMX1A were expressed throughout the subclusters, consistent with their known expression as RL markers. WLS, SOX2, and CRYAB were restricted to one subcluster, identifying it as the RLVZ (Fig. 4b and Supplementary Table 10). Another subcluster expressed CA8, suggesting they are likely Purkinje cells originating from the intermediate zone, and another expressed LMX1B, consistent with choroid plexus epithelium. We observed marked changes in the proportions of cells within RL compartments during development, with the proportion of cells occupying the RLVZ generally decreasing across development and cells in the RLSVZ increasing. Next, we identified additional genes with RL spatially-restricted expression. We selected the top RL markers defined by our spatial RNA-seq analysis (Fig. 2f) and examined expression at the single-cell level within the RL subclusters (Fig. 4e). OLIG3, RSPO3, and SLFN13 were expressed throughout the RL while WNT2B, CALCB, ATP6V1C2, and CALCA were expressed in the RLVZ, and DPYD expression was enriched in the RLSVZ.
Fig. 4 |. Analysis of RL compartments at single-cell resolution.
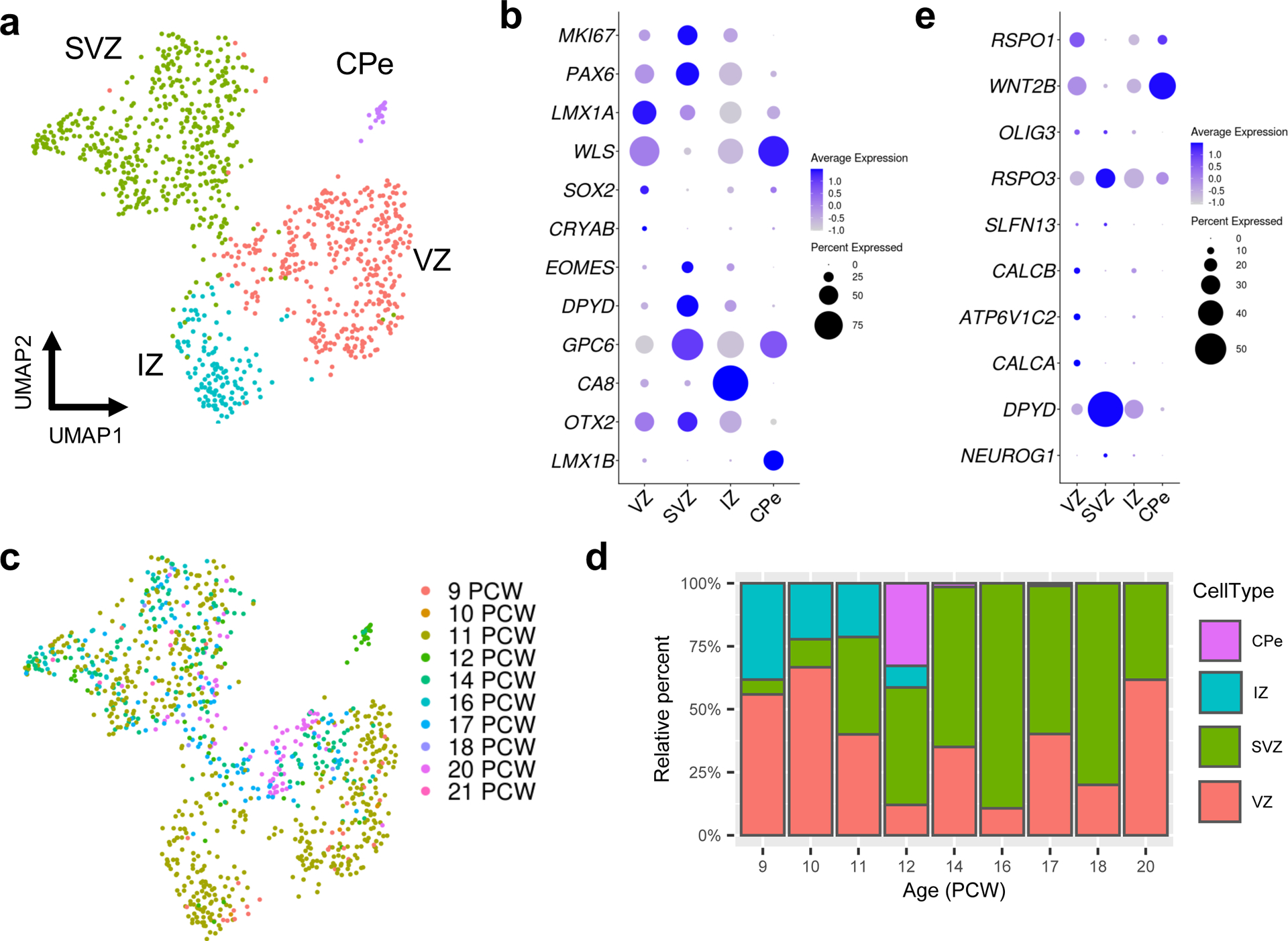
a, UMAP visualization and marker-based annotation of the RL subclusters (n = 1,018 nuclei; 466 for SVZ; 390 for VZ; 135 for IZ; 21 for CPe). CPe, choroid plexus epithelium; IZ, intermediate zone; SVZ, subventricular zone; VZ ventricular zone. b, Dot plot showing the expression of selected marker genes in subclusters. c, The same UMAP as in a with nuclei colored by sample age (n = 34 for 9 PCW; 9 for 10 PCW, 535 for 11 PCW; 58 for 12 PCW; 137 for 14 PCW; 56 for 16 PCW; 97 for 17 PCW, 5 for 18 PCW; 81 for 20 PCW; 6 for 21 PCW). d, Stacked bar charts show the percentage of the RL subclusters by sample age. e, Dot plot showing the expression of the top 10 most differentially expressed genes from the spatial transcriptional analysis of the RL (Fig. 2e and Supplementary Table 3).
Developmental trajectory of the RL lineage
The RL gives rise to all glutamatergic neuronal subtypes of the developing cerebellum in a sequential manner.24, 25 First, glutamatergic neurons destined to become cerebellar nuclei neurons (eCN), which integrate GABAergic Purkinje cell-mediated and excitatory mossy fiber/climbing inputs to serve as major output tracts, are generated. Second, granule cell progenitors (GCP) that proliferate, differentiate, and migrate to form the internal granule layer arise, and lastly, unipolar brush cell (UBC) interneurons that make presynaptic connections with vestibular ganglia and nuclei are formed.24, 26 To resolve lineage trajectories of the RL, GCP, GN, and eCN/UBC subpopulations, we subclustered the cells, and ordered them according to pseudotime using Monocle 327 (Fig. 5a–b). We confirmed predicted developmental trajectories, including temporal progression and expression of classic markers, with one branch of the RL trajectory giving rise to GCP, then granule neurons, and the second branch giving rise to eCN/UBC (Fig. 5b). As progenitors differentiate into eCN/UBC, canonical RL gene expression (MKI67, OTX2, LMX1A, EOMES) declines, and as GCP differentiate into GN, MIK67 and DCC expression in GCP declines concurrent with increased expression of RELN and RBFOX3. As we had done for the RL compartments, we selected the top markers for RL and EGL defined by our spatial RNA-seq analysis (Fig. 2e) and examined expression of these marker genes at the single-cell level within the RL trajectory (Fig. 5c). Among the top 10 RL markers, RSPO1, WNT2B, OLIG3, SLFN13, CALCB, CALCA, and ATP6V1C2 expression was largely confined to the RL lineage, whereas RSPO3 and DPYD expression was highest in both RL and eCN/UBC. Among the top 10 EGL markers, expression was largely confined to the GCP, though the overall magnitude of expression was low.
Fig. 5 |. Characterization of the RL trajectory.
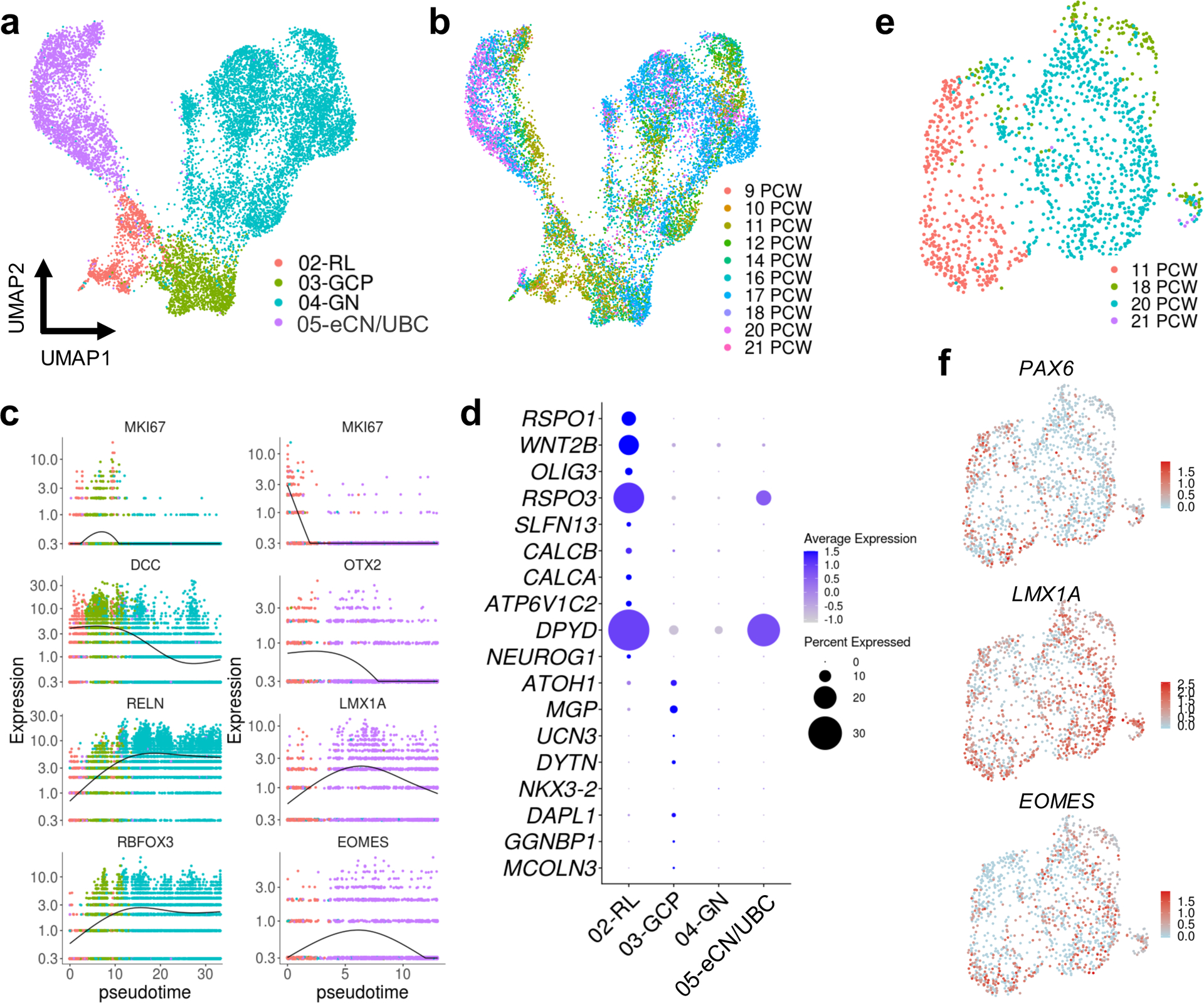
a, UMAP visualization and marker-based annotation of cell types that originate from the RL (n = 12,243; 1,018 for RL; 1,659 for GCP; 6,727 for GN; 2,839 for eCN/UBC). eCN/UBC, excitatory cerebellar nuclei/unipolar brush cells; GCP, granule cell progenitors; GN, granule neurons; RL, rhombic lip. b, The same UMAP as in a with nuclei colored by sample age (n = 120 for 9 PCW; 61 for 10 PCW; 2,190 for 11 PCW; 1,053 for 12 PCW; 1,663 for 14 PCW; 432 for 16 PCW; 4,410 for 17 PCW; 627 for 18 PCW; 1,626 for 20 PCW; 89 for 21 PCW). c, Kinetics plot showing the relative expression of RL trajectory marker genes across developmental pseudotime. Dots are colored according to cell types as in a. d, Dot plot showing the expression of the top 10 most differentially expressed genes from the spatial transcriptional analysis of RL and EGL (Fig. 2e and Supplementary Table 3). e, UMAP visualization of the eCN/UBC cluster including 11, 18, 20, 21 PCW samples. Nuclei are colored by sample age (n = 1,424; 436 for 11 PCW; 138 for 18 PCW; 842 for 20 PCW; 8 for 21 PCW). f, The same UMAP as in e with nuclei colored by expression level for the indicated gene.
Consistent with their RL origin, eCN and UBC express classic RL markers PAX6, LMX1A, and EOMES.26, 28 We identified eCN/UBC on the basis of these markers and the absence of MIK67 expression (since eCN and UBC are non-proliferative at the ages sampled) (Fig. 3a–b, Supplementary Tables 8 and 9). The cells within this cluster were present in all ages sampled (9–21 PCW) and were distinct from other glutamatergic neurons (GCP and GN) that also originate from RL neural progenitors (Fig. 5). To more clearly delineate the different developmental origins of eCN and UBC, we selected cells in the eCN/UBC population from 11 PCW or 18–21 PCW, subclustered the cells, and examined the molecular correlates that define eCN and UBC (Fig. 5). We found that PAX6, LMX1A, and EOMES were expressed throughout this cluster. Although we attempted to distinguish eCN and UBC by examining LMX1A/EOMES co-expression in the eDCN/UBC cluster, only a few co-expressing cells were detectable (Extended Data Fig. 8), limiting this analysis.
Purkinje cells dominate the developing cerebellar anlage
In the adult brain, Purkinje neurons form a single layer with extensive dendritic arborization in the molecular layer and axons projecting to the deep cerebellar nuclei to coordinate all motor output. By 10 PCW, inhibitory Purkinje neurons dominate the cerebellar anlage29 and, as expected, represented the cell type with the most nuclei recovered in our dataset (Fig. 3). Nuclei within this cluster were present in all ages sampled (9–21 PCW) and were distinct from other GABAergic neurons (inhibitory cerebellar nuclei and PAX2+ interneuron progenitors) that also originate from VZ neural progenitors. To examine early markers of human Purkinje cell subtypes, we selected and subclustered nuclei in the Purkinje cell cluster, then used Monocle 327 to order them in pseudotime (Fig. 6). By labeling the cells by sample age, we detected a temporal progression (Fig. 6a). Plotting relative gene expression in pseudotime and then coloring cells by sample age demonstrated little fluctuation in canonical Purkinje cell marker gene expression, with the exception of CALB1 and SKOR2 (Fig. 6b). CALB1 was expressed at higher levels in later samples, while SKOR2 expression declined with increasing gestational age. RORA was expressed throughout the Purkinje cell cluster, as were markers that in mouse display parasagittal banding patterns of alternating Purkinje cells in mouse,30 including PLCB4 and EBF2 (Fig. 6c). Few Purkinje cells expressed more mature markers, ALDOC and PCP2.
Fig. 6 |. Purkinje cells.
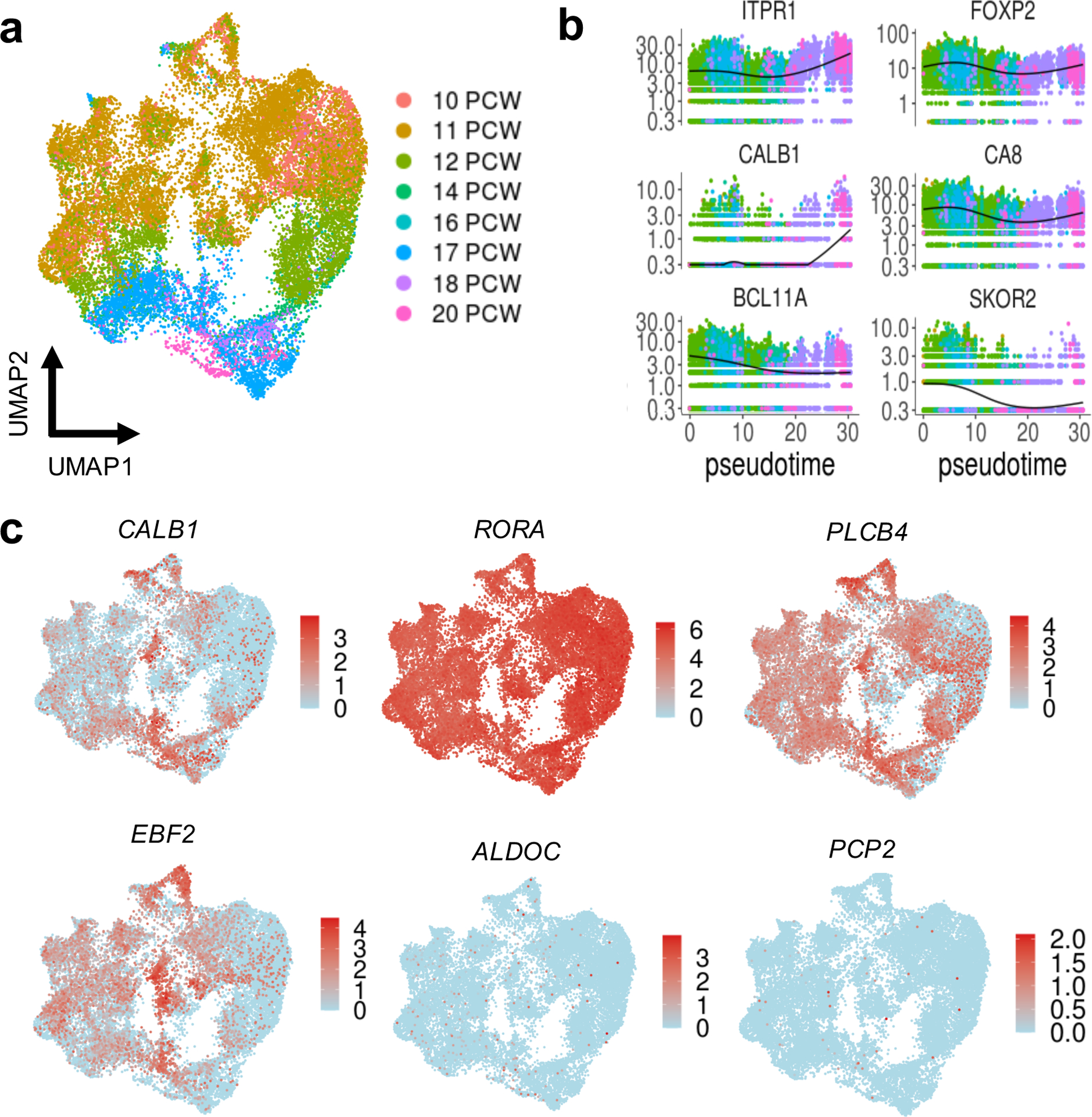
a, UMAP visualization of the PC cluster. Nuclei are colored by sample age (n = 25,724; 3,736 for 9 PCW; 1,131 for 10 PCW; 12,182 for 11 PCW; 3,543 for 12 PCW; 1,346 for 14 PCW; 26 for 16 PCW; 3,144 for 17 PCW; 245 for 18 PCW; 371 for 20 PCW). b, Kinetics plot showing the relative expression of PC marker genes across developmental pseudotime. Dots are colored by sample age as in a. c, The same UMAP as in a with nuclei colored by expression level for the indicated gene.
Deconvolution of LCM and BrainSpan
LCM is a technique used to harvest subpopulations of cells from precise anatomical regions of a heterogeneous tissue sample.31, 32 However, such samples can be contaminated with cell types in adjacent tissues. Therefore, we sought to directly investigate the cell type composition of our LCM samples by using the reference gene expression profiles from our snRNA-seq dataset. We used CIBERSORTx,33 a machine learning method for inferring cell-type-specific gene expression profiles, to establish a transcriptional signature for each of the 21 cell types detected. This approach allowed us to estimate the relative proportions of each cell type present in every sample of our spatial transcriptional dataset. Overall, we found that the expected cell type had the highest relative abundance in each LCM sample (Extended Data Fig. 9). Cells corresponding to the RL (02-RL) cluster were most abundant in the LCM RL samples (median 52%, range 35–57%), while they represented only 7% of the cells in the EGL samples, and were absent from PCL and bulk cerebellum samples. GCP (03-GCP) were the most abundant cell type present in LCM EGL samples (median 49%, range 40–54%), while they represented 6.5% of RL, 2% of PCL, and 6% of bulk cerebellum. Purkinje cells (01-PC) were the most abundant cell type present in the LCM PCL samples (median 43%, range 32–57%) while they represented 16% of bulk cerebellum and were absent from RL and EGL samples.
We also estimated the cell type composition of bulk cerebellar samples from BrainSpan using our fetal cerebellar transcriptional signatures. Overall, Purkinje cells were more abundant in bulk cerebellar samples from fetal development (Supplementary Fig. 4). Deconvolution using our fetal transcriptional signatures estimated that Purkinje cells made up a median of 23% in BrainSpan fetal samples and a median of 13% in postnatal samples. Endothelial cells (median 21%, range 15–55%), iCN (median 10%, range 4–22%), and glia (median 10%, range 5–14%) were also detectable in BrainSpan fetal samples. However, several low abundance cell types were not detectable (median 0%) in the majority of BrainSpan fetal samples, including RL, PIP, Bergmann glia, OPC, microglia, meninges, pericytes, molecular layer interneurons, astrocytes and ependymal cells, and choroid plexus.
Human-mouse cell-type homology
To examine conservation of cellular architecture in the developing cerebellum between human and mouse, we used LIGER34, 35 to align the transcriptomic cell types in our human fetal cerebellum with the cell types in a published dataset from the mouse developing cerebellum.36 Overall, the joint analysis identified strong concordance between human and mouse cluster assignments for the individual datasets (Fig. 7). Shared metagene factors corresponded to the genes that define particular cell types in both species. First, we examined human RL metagenes. Factor 10 showed high loading values for RRM2, PCNA, and LIG1. These genes were enriched in DNA replication (hsa03030, FDR = 1.04×10–12) and cell cycle (hsa04110, FDR = 4.55×10–6) pathways consistent with their identity as neural progenitors. In mice, factor 10 corresponded to neural stem cells and GCP and UBC progenitors rather than to cells from the RL. Next, we examined human PC metagenes. Factors 13 and 15 showed high loading values for ITPR1, EBF1, PDE1C and RORA, DAB1, FOXP2, respectively, indicating they were PC specific. Notably, ventricular zone progenitors that express PTF1A were present in the mouse, but not the human data. These factors corresponded to two subpopulations in mice, PC and differentiating PC.
Fig. 7 |. Human-mouse cross-species analysis.

a, UMAP plot of nuclei from human cerebellum and cells from mouse cerebellum following LIGER analysis, showing only nuclei from human cerebellum (n = 69,174) and colored by cell type from the original analysis. b, Riverplot showing the relationship between original cluster assignments from our human cerebellum and a published mouse cerebellum dataset. c, UMAP plot of nuclei from human cerebellum and cells from mouse cerebellum following LIGER analysis, showing only cells from mouse cerebellum (n = 39,130) and colored by cell type from the original analysis. d-f, UMAP plots showing cell factor loading values and gene loading plots for factors corresponding to RL (d), and PC (e, f). g, UMAP plots show the human (n = 1,018) and mouse (7,034) cell types contributing to factor 10. Dot plot shows expression of canonical RL genes delineated in human and mouse clusters. h, UMAP plots show the human and mouse PC clusters. Dot plot shows expression of canonical PC genes delineated in human and mouse PC clusters.
Cellular convergence of disease
Cerebellar dysfunction underlies major childhood neurodevelopmental and adult-onset neurodegenerative disorders.1, 2 As a framework for understanding these complex disorders, we used our atlas of developing human cerebellum to identify the cell types in which mutations can act to cause pediatric and adult diseases (Fig. 8 and Supplementary Table 11 and 12). We first examined the enrichment of genes implicated in structural cerebellar malformations, namely cerebellar hypoplasia and Dandy-Walker malformation, that are commonly diagnosed prenatally.5, 22 We found that 72% of genes associated with these common cerebellar malformations were expressed in the fetal cerebellum (Fig. 8a). These genes were significantly enriched in Purkinje cells, with prominent expression of AUTS2, BCL11A, EBF2, and EBF3, endothelial cells (MACF1, SHANK3), and pericytes (LAMC1, NID1, PDGFRB). Next, we examined the enrichment of genes that cause Joubert syndrome, a recessive neurodevelopmental ciliopathy defined by a distinctive hindbrain malformation.37 None of the Joubert syndrome genes showed significant enrichment in cerebellar cell types (Extended Fig. 10a). Then, we examined the enrichment of high-confidence ASD risk genes.38–42 Gene expression varied substantially across cell types with significant enrichment of gene expression in multiple cell types: Purkinje cells, granule neurons, eCN/UBC, inhibitory cerebellar nuclei (iCN), Pax2+ interneuron progenitors (PIP), committed OPC, endothelial cells, pericytes, brainstem, molecular layer interneurons (MLI), choroid plexus, and brainstem choroid plexus/ependemyl cells (Fig. 8b). ASD genes were most prominently expressed in Purkinje cells (ASXL3, BCL11A, CTTNBP2, SHANK2, SUV420H1), eCN/UBC (CCSER1, DIP2C, FXBO11, NRXN1, NUAK1, PCM1), committed OPC (DSCAM, MYO5A, NCKAP1, PCDH11X, PRKAR1B, TCF7L2), and pericytes (INTS6, MED13, PTEN, SMURF1, SYNGAP1, ZC3H11A). We extended this analysis to examine high-confidence ID genes41 and found prominent expression with significant enrichment in Purkinje cells (ASXL3, AUTS2, BCL11A, CHD3, EBF3, FOXP2, GABRB2, PRKG1, TCF20), RL (ASXL1, CTCF, HIST1H1E, HNRNPK, HNRNPU, MSI1, NSD1, SMC1A, SYNCRIP, WHSC1), GCP (ADNP, CHD4, CSNK2A1, HNRNPK, HNRNPU, KAT6B, KDM6A, PRPF40A, SETBP1, SYNCRIP), and microglia (ANKRD11, CLTC, COL4A3BP, EHMT1, HIST1H2AC, MECP2, MED13L, MEF2C, USP9X, WDR26) (Fig. 8c). Lastly, we examined the expression of genes associated with two adult-onset neurodegenerative disorders: spinocerebellar ataxias (SCA) and Alzheimer’s disease (AD). SCA are progressive disorders with autosomal dominant inheritance that lead to irreversible Purkinje cell loss.7 SCA genes were significantly enriched in Purkinje cells, driven by DAB1 and ITPR1 expression (Fig. 8d). Alzheimer’s disease (AD) is a progressive disease associated with age-related cognitive decline and aberrant neuron-glial interactions.43 We examined the enrichment of AD risk genes identified in a recent case-control exome sequencing study.44 Though none of the AD genes showed significant enrichment in cerebellar cell types (Extended Fig. 10b), several genes were prominently expressed in microglia, consistent with emerging evidence.6, 43, 44 Taken together these findings demonstrate the value of our cerebellar developmental atlas as a rich resource for probing the cellular biology underlying complex disease.
Fig. 8 |. Cerebellar cell type enrichment in pediatric and adult diseases.
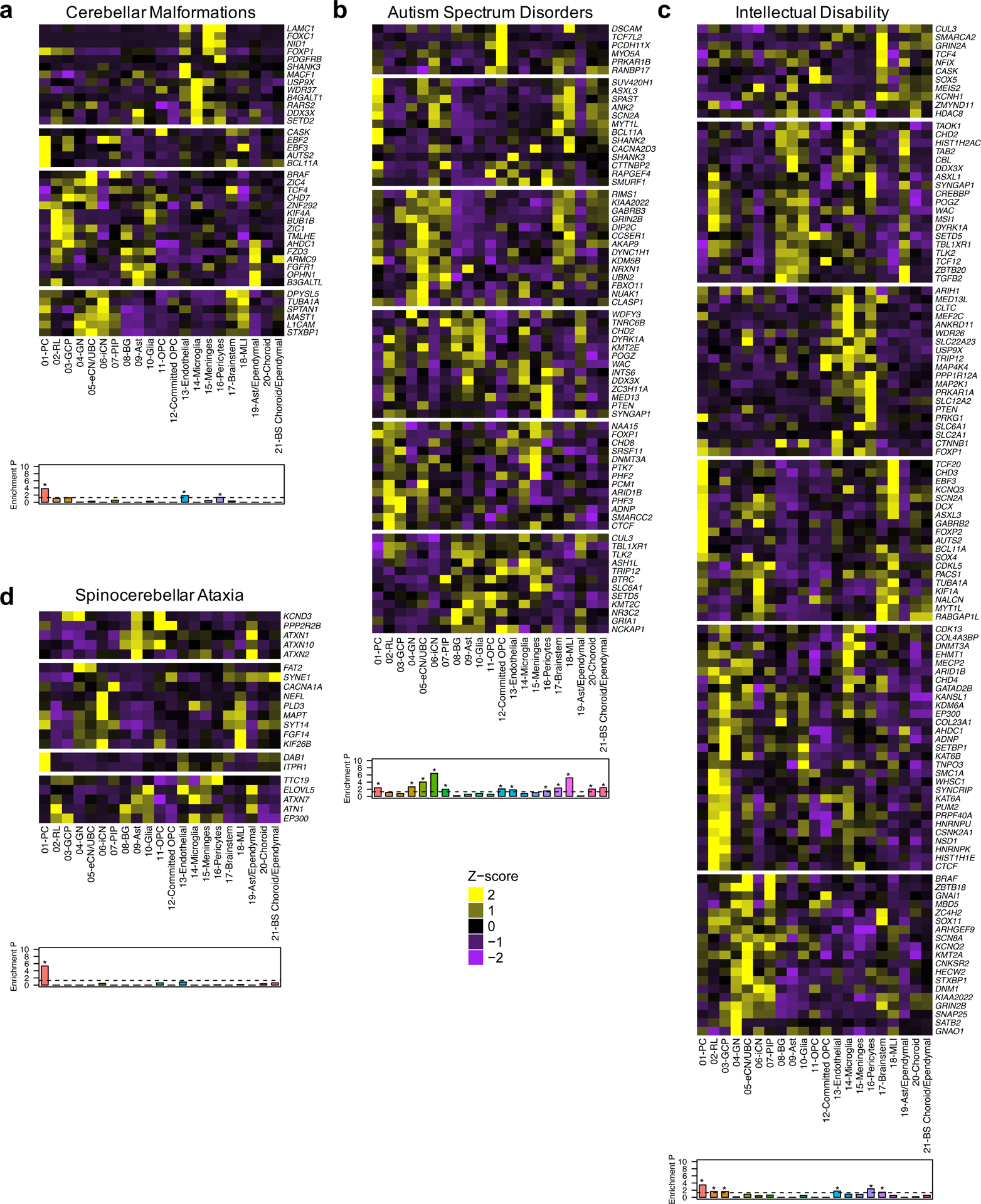
a-d, Heatmaps of mean expression per fetal cerebellar cell type for genes associated with pediatric (a, cerebellar malformations; b, autism spectrum disorders; c, intellectual disability) or adult (d, spinocerebellar ataxia) diseases. Color scheme is based on Z-score distribution. In the heatmaps, each row represents one gene and each column represents a single cell type. Gene expression was clustered by row. Horizontal white lines indicate branch divisions in row dendrograms (not shown). The full list of genes is provided in Supplementary Table 11. Enrichment P values (-Log10 P value) for each cell type are shown in the bottom bar plots. Significance was determined by one-sample Z-test, two-tailed P value. The dashed line is the significance threshold. Asterisk (*) indicates significance (P < 0.05) after Bonferroni correction: cerebellar malformations (P = 1.63 × 10−4 for 01-PC; 0.01 for 13-Endothelial; < 0.05 for 16-Pericytes), autism spectrum disorders (P = 0.004 for 01-PC; 0.002 for 04-GN; 9.43 × 10−5 for 05-eCN/UBC; 3.78 × 10−7 for 06-iCN; 0.01 for 07-PIP; 0.008 for 12-Committed OPC; 0.02 for 13-Endothelial; 0.03 for 16-Pericytes; 0.005 for 17-Brainstem; 6.74 × 10−6 for 18-MLI; 0.009 for 20-Choroid; 0.004 for 21-BS Choroid/ependymal), intellectual disability (P = 3.30 × 10−4 for 01-PC; 0.02 for 02-RL; 0.02 for 03-GCP; 0.02 for 13-Endothelial; 0.004 for 16-Pericytes; 0.04 for 17-Brainstem), spinocerebellar ataxia (P = 4.56 × 10−6 for 01-PC).
DISCUSSION
By combining microdissection and single-nucleus capture methods we provide a map of expression profiles for the major cell types present in the human cerebellum from 9 to 21 PCW. This ‘Developmental Cell Atlas of the Human Cerebellum’ provides molecular context for comparative evolution, benchmarking ex vivo model systems, and investigating disease cell type origins.
The RL is a transient stem cell reservoir for glutamatergic neuron progenitors in the developing cerebellum.24, 25 We recently reported that the human RL is composed of an inner RLVZ and an outer RLSVZ, a feature that appears to be unique to humans and might explain the evolutionary expansion of the human cerebellum.10 Here, we performed both subcluster and trajectory analyses of the small recovered RL population to confirm that we can readily distinguish the RLVZ and outer RLSVZ compartments. In addition, we putatively identified the intermediate zone,45 another transient progenitor region adjacent to the ventricular zone and the RL in early development, though this warrants further investigation given the few nuclei represented. Our human mouse comparisons indicate the human RL does not directly map onto the mouse RL. Additional cross species analysis that retain spatial localization of the human and mouse RL is necessary.
During mid-gestation, the RL produces cells that migrate to become excitatory granule neurons in the EGL and UBC, which are excitatory glutamatergic interneurons.3 Additional excitatory cerebellar interneurons (eCN) are also generated from the RL, but their formation is completed prior to 8 PCW.10 Distinguishing all of these closely related cell types is important because they have been implicated as the origin for group IV medulloblastoma, a poorly understood and aggressive subtype of childhood cerebellar tumor subtype.36, 46 Proliferative human RLSVZ progenitors are known to express both LMX1A and EOMES.10 Although we recovered a cell population that expresses LMX1A and EOMES, this subset did not express the proliferative marker MKI67, leading us to identify this cluster as eCN/UBC. While eCN express LMX1A but not EOMES, UBC express both LMX1A and EOMES. This cell cluster contained few nuclei (6 total), and we were unable to discriminate eCN and UBC.
Neurogenesis in the VZ concludes between 8 and 10 PCW, whereas extensive migration of all VZ derivatives, including Purkinje cells and PAX2+ interneuron progenitors, occurs during early and mid-fetal development.29 In the mouse, Purkinje cell morphology and circuitry appears nearly identical, however, at late gestational stages up to 50 molecularly heterogeneous Purkinje cell clusters, partly related to cell birthdates, are present.47 These Purkinje cell clusters are subsequently transformed into longitudinal stripes along the mediolateral axis in a way that correlates with function.30, 48 In our data set, Purkinje cells were the most frequent cell type recovered, but we did not readily detect Purkinje cell clusters with distinct transcriptional profiles. This is likely because Purkinje cell maturation begins during late gestation and peaks only after birth - once all GCPs in the EGL have differentiated and migrated inward to establish the IGL. Consistently, expression of canonical mature Purkinje cell markers (ALDOC, PCP2) was low, whereas expression of early Purkinje cell differentiation marker49 SKOR2 declined with increasing gestational age, demonstrating that we recovered immature Purkinje cells in our dataset (Fig. 6c). Indeed, human Purkinje cells around 20 PCW display a nascent dendritic arbor, which expands considerably during late gestation and continues after birth.29, 50 Human-mouse comparisons reveal substantial similarities in Purkinje cells across species. Additional sampling from earlier timepoints that capture more immature Purkinje cells and their progenitors and from later timepoints that capture Purkinje cell maturation are required to investigate differences in Purkinje cell trajectories between species.
Thus far, bulk RNA-seq data available from a limited number of fetal cerebellar samples has been reported.15, 16 Our data significantly augment prior work by providing 3-fold more spatially resolved bulk transcriptional data and adding 70,000 single-nucleus transcriptomes. By directly comparing our spatial RNA-seq data with bulk RNA-seq data, we show that similarities are scarce; only one co-expression module from BrainSpan cerebellum correlated with our fetal EGL data. Bulk cerebellar data primarily emphasize Purkinje cells in prenatal samples and granule neurons in postnatal samples, obscuring detection of rare and transient cell types. We applied transcriptional signatures for cell types detected in our ‘Developmental Cell Atlas of the Human Cerebellum’ to infer cell type composition of our LCM and BrainSpan bulk cerebellum RNA-seq datasets. Our spatially captured data show a 40% abundance of the targeted cell type, with <20% abundances for other cell types (Extended Data Fig. 9). When we applied our fetal cell type signatures to the cerebellum data in BrainSpan, only half of the cell types identified in our snRNA-seq dataset were detected in the BrainSpan fetal samples (Supplementary Fig. 4). Notably, RL was one of the cell types that was not detectable in bulk cerebellum.
Human cerebellar development is protracted – extending from 30 days post conception through the second postnatal year of life – and after birth is influenced by environmental and sensory cues that shape maturing brain circuitry.4, 50 The 17 week window of cerebellar development profiled here represents only a small slice of human cerebellar development. Yet, this time period instructs developmental processes that are fundamental for establishing the stereotypical lamination of the cerebellum that begins to emerge during this time,4, 10 and we find commonalities between human and mice. Importantly, we used our dataset to map genes associated with neurodevelopmental and adult onset neurodegenerative disorders to relevant cell types. Although future studies are required to complete the cellular and transcriptional characterization of the human cerebellum across the complete human lifespan, our unique dataset serves as a framework with which to identify cell types, verify lineage relationships, and establish the stoichiometry of cerebellar cell types across development.
METHODS
Cerebellum samples
Acquisition of human tissue samples was approved by the Seattle Children’s Hospital (SCH) Institutional Review Board. Experiments were performed in accordance with SCH ethical and legal guidelines. Specimens from fetal (9–21 PCW) human cerebellum were obtained from the Birth Defects Research Laboratory at the University of Washington or the Joint MRC/Wellcome (MR/R006237/1) Human Developmental Biology Resource51 (www.hdbr.org) with ethics board approval and maternal written consent obtained prior to specimen collection.
Histology, immunohistochemistry, and in situ hybridization analyses
Fixation, tissue processing, and immunohistochemistry were performed as previously described50 using the following primary antibodies: Calbindin (Swant, CB38, rabbit, 1:3000), PAX6 (Biolegend, 901301, rabbit, 1:300), SKOR2 (Novus, NBP2–14565, rabbit, 1:100), and NEUN (Millipore, MAB377, mouse, 1:100). All sections were counterstained using Vectashield DAPI (H1000, Vector labs) which marks all nuclei.
In situ hybridization was performed using commercially available probes from Advanced Cell Diagnostics. Manufacturer recommended protocols available on the ACD webpage were used without modification. Probes used include: LMX1A (#540661), MKI67 (#591771), ATOH1 (#417861), OTX2 (#484581), and HOXB3 (custom-made). All sections were counterstained using Fast Green.
Slides processed for fluorescent IHC were imaged using Zeiss LSM Meta confocal microscope and ZEN 2009 software (Zeiss). Brightfield imaging was performed using a Nanozoomer Digital Pathology slide scanner (Hamamatsu; Bridgewater, New Jersey). Barring minor adjustments limited to contrast and brightness to the entire image, no additional image alteration was performed.
Laser capture microdissection
Whole cerebellum was dissected from 16 fetal specimens that had intact calvaria to ensure correct orientation of the cerebellum. Intact cerebella were embedded in OCT, frozen at −80°C, and cryosectioned at 16-μm in the sagittal plane through the cerebellar vermis onto PEN Membrane Glass Slides (Applied Biosystems, USA). Total RNA was isolated from one whole section using the Qiagen RNeasy Micro Kit and RNA quality was assessed using the Agilent Bioanalyzer 6000 Pico Kit before proceeding with LCM. LCM was performed using the Leica DM LMD-6000 Laser Microdissection system to capture tissue containing PCL and EGL from each of 6–8 sections per slide into separate collection tubes. Total RNA was then isolated from LCM-enriched samples pooled across 9 slides using the Qiagen RNeasy Micro Kit. LCM was previously performed to capture RL ventricular (RLVZ) and subventricular zones (RLSVZ), then total RNA was isolated from RLVZ and RLSVZ resulting in two RNA samples per specimen.10
RNA-seq and analysis
Sequencing libraries were prepared using the Illumina TruSeq RNA Access Prep Kit (Illumina, USA) and 25 ng of total RNA per sample, according to the manufacturer’s protocol. RNA libraries were barcoded and sequenced including 6–8 samples per lane on an Illumina HiSeq 2000. FASTQ files for RLVZ and RLSVZ samples from the same specimen (phs001908.v1.p1) were merged to generate the RL dataset and analyzed together with data for the other samples. Paired-end reads (100 bp) were aligned to the Human reference genome (NCBI build 37/hg19) using STAR,52 genes counts were summarized using HTSeq,53 and gene-level differential expression was analyzed using DESeq254 specifying ~ batch + age + region as the experimental design. Six samples were deemed to be outliers because principal component analysis separated these samples from all others; these samples were removed from additional analyses. Sample sex was confirmed by comparing expression of the female-specific non-coding RNA XIST and the chromosome Y specific gene DDX3Y. Significant results are reported as Benjamini-Hochberg adjusted P values. Pathway enrichment was performed using String v11.0.55
Gene co-expression network analyses
Weighted gene co-expression network analysis (WGCNA) was performed using the R package.19 Summarized gene counts were converted to RPKM using RNA-SeQC56 v1.1.8. Log2-transformed RPKM values were used for this analysis as described previously.15
BrainSpan RNA-seq data
Gene-level expression data in counts and RPKM for the BrainSpan RNA-seq dataset generated from post-mortem human brain were downloaded (http://www.development.psychencode.org). We restricted our analysis to the cerebellum, selecting data from 35 individuals and including 3 brain regions: CBC (cerebellar cortex), CB (cerebellum), and URL (upper rhombic lip).
SPLiT-seq method
Specimens were flash frozen in liquid nitrogen and stored at −80°C until use. Frozen tissue samples (whole or half cerebellum) were pulverized on dry ice using a ceramic mortar and pestle. Pulverized samples were transferred to chilled microcentrifuge tubes and stored at −80°C until use. Nuclei were isolated from either 150 mg of pulverized tissue or an entire amount of dissected cerebellum using a published protocol.57 Nuclei were fixed according to the SPLiT-seq protocol.18 The SPLiT-seq method was performed in an initial experiment as previously described (experiment 1).22 Two additional SPLiT-seq experiments were performed using nuclei isolated from 13 cerebellar specimens using the published detailed experimental protocol.18 Libraries were first sequenced on an Illumina NextSeq using 150 nucleotide kits and paired-end sequencing. Libraries were then sequenced on an Illumina NovaSeq S2 flow cell by SeqMatic (experiment 2) or the Northwest Genomics Center at the University of Washington (experiment 3). We used the SPLiT-seq pipeline to convert FASTQ files into digital gene expression matrices from each sequencing run: https://github.com/yjzhang/split-seq-pipeline.
snRNA-seq analysis
Deep and shallow sequencing runs from experiment 1,22 and shallow sequencing runs from experiment 2 and experiment 3 were filtered independently (datasets 1K, 5K, 10K, and 80K, respectively). Nuclei with <200 genes, >4 standard deviations above the median number of genes or UMIs, or >1–5% mitochondrial genes were removed from the analysis (Supplementary Table 7). DoubletFinder21 was used to detect likely doublets assuming a rate of 5%, that were discarded from analysis. Sample sex was confirmed by counting reads mapped to the female-specific non-coding RNA XIST and the chromosome Y specific gene DDX3Y. We used Seurat23 v3 for downstream analysis. The four filtered datasets (1K, 5K, 10K, and 80K) were combined into a single dataset using canonical correlation analysis with anchors (‘FindIntegrationAnchors’) to correct for batch effects.58 The top 2,000 most variable genes were used to find individual cells in each sequencing run that originate from the same biological state, which became the anchors to merge runs together. The resulting dataset was then scaled and centered as well as regressed out cell cycle difference (S.score – G2M.score). Data dimensionality of the integrated dataset was reduced by principal component analysis (‘RunPCA’), then uniform manifold approximation and projection (‘RunUMAP’), then Shared Nearest Neighbor (SNN) Graph construction (‘FindNeighbors’), and finally Louvain clustering (‘FindClusters’) using the first 75 principal components and a resolution of 1.5 to determine cluster assignment. A Wilcoxon rank sum test was performed to identify differentially expressed genes for each cluster (‘FindAllMarkers’) and compare them to known gene markers for cell type assignment. One cluster with no significant differentially expressed genes and another cluster with an enrichment of mitochondrial genes were removed.
Subcluster analysis was performed by subsetting populations of interest from the overall dataset. Clustering and differential gene tests were repeated with a subpopulation specific number of principal components determined by ‘ElbowPlot’. Then, pseudotime analysis was performed using Monocle 3.27 Subsets were normalized (‘preprocess_cds’), dimension reduction was applied (‘reduce_dimension’), followed by clustering (‘cluster_cells’) and visualization (‘learn_graph’). Pseudotemporal ordering of cells was performed (‘order_cells’) by selecting a biologically relevant starting point. Genes of interest were used to construct the pseudotime trajectory (‘plot_genes_in_pseudotime’).
Cell type deconvolution
We used CIBERSORTx33 to estimate the cell type composition in the LCM-isolated and BrainSpan RNA-seq samples. We down sampled our integrated snRNA-seq dataset to 100 cells per cell type, built a cell type signature matrix with this digital expression matrix, and imputed cell fractions for each of the 57 LCM RNA-seq samples and 35 BrainSpan cerebellar samples.
Cross-species analysis
To analyze the developing cerebellum between human and mouse, we selected high-confidence human to mouse (one-to-one) orthologs from Ensembl release 101 (http://www.ensembl.org/biomart/martview). We downloaded single-cell RNA-sequencing data from 9 cerebellum samples across mouse embryonic and postnatal development from the Gene Expression Omnibus (GSE118068)36 and restricted our analysis to the most relevant period (E10, E12, E14, E16, E18, P0). We selected the union of orthologs present in our human dataset and in the downloaded mouse dataset (13,182 genes), then used LIGER34, 35 to integrate the filtered datasets and identify shared cell types in the cerebellum across these two species. Variable gene selection, normalization, and scaling of individual genes was performed on the combined dataset using integrative non-negative matrix factorization with k = 20 to define dataset-specific and shared metagenes, which correspond to genes that define particular cell types.
Gene set curation
Disease gene lists are provided in Supplementary Table 11. The cerebellar malformation gene list was obtained from exome sequencing analysis and published Dandy-Walker malformation and cerebellar hypoplasia genes.5, 22, 59 The CBLM list included 54 genes. The Joubert syndrome gene list was compiled from published Joubert syndrome genes.37, 60–62 The JS list included 42 genes. The ASD gene set was compiled by selecting high-confidence ASD genes identified through exome and genome sequencing.38–42, 63 The final ASD list included 108 genes. The intellectual disability (ID) gene list was compiled by selecting genes identified through exome sequencing.41, 64, 65 The final ID list included 186 genes. The spinocerebellar ataxia (SCA) gene set was compiled by selecting genes from OMIM phenotype PS164400. The SCA list included 44 genes. The Alzheimer’s disease gene list was compiled by selecting genes identified through exome sequencing.44 The ALZ list included 120 genes.
Cell type enrichment analysis
We used a one sample Z-test66 to identify cell types that showed enriched gene expression associated with particular gene sets. We calculated the average expression for each gene per cell type, then removed genes with expression values < 1 for more than one cell type to define a population size of 4,457 genes (Supplementary Table 12). Enrichment P values were corrected for multiple testing using the Bonferroni method.
Statistical Tests
No statistical methods were used to predetermine sample sizes. No randomization was used in this study. Distributions of the data were not tested. Statistical tests were performed using R 3.3.3 and RStudio 1.0.143. The Wald test was used to calculate differential gene expression and P values were adjusted using the FDR approach within DESeq254. A loess regression was used to estimate gene expression across time. Fisher’s exact test was used for gene ontology, pathway, and WGCNA module enrichment. The Wilcoxon rank sum test was used to calculate cluster markers within Seurat23. Three independent snRNA-seq experiments were performed. Gene set enrichment analysis was performed using a one-sided Z-test and P values were adjusted using the Bonferroni method.
Extended Data
Extended Data Fig. 1. Quality control related analyses of LCM RNA-seq data.
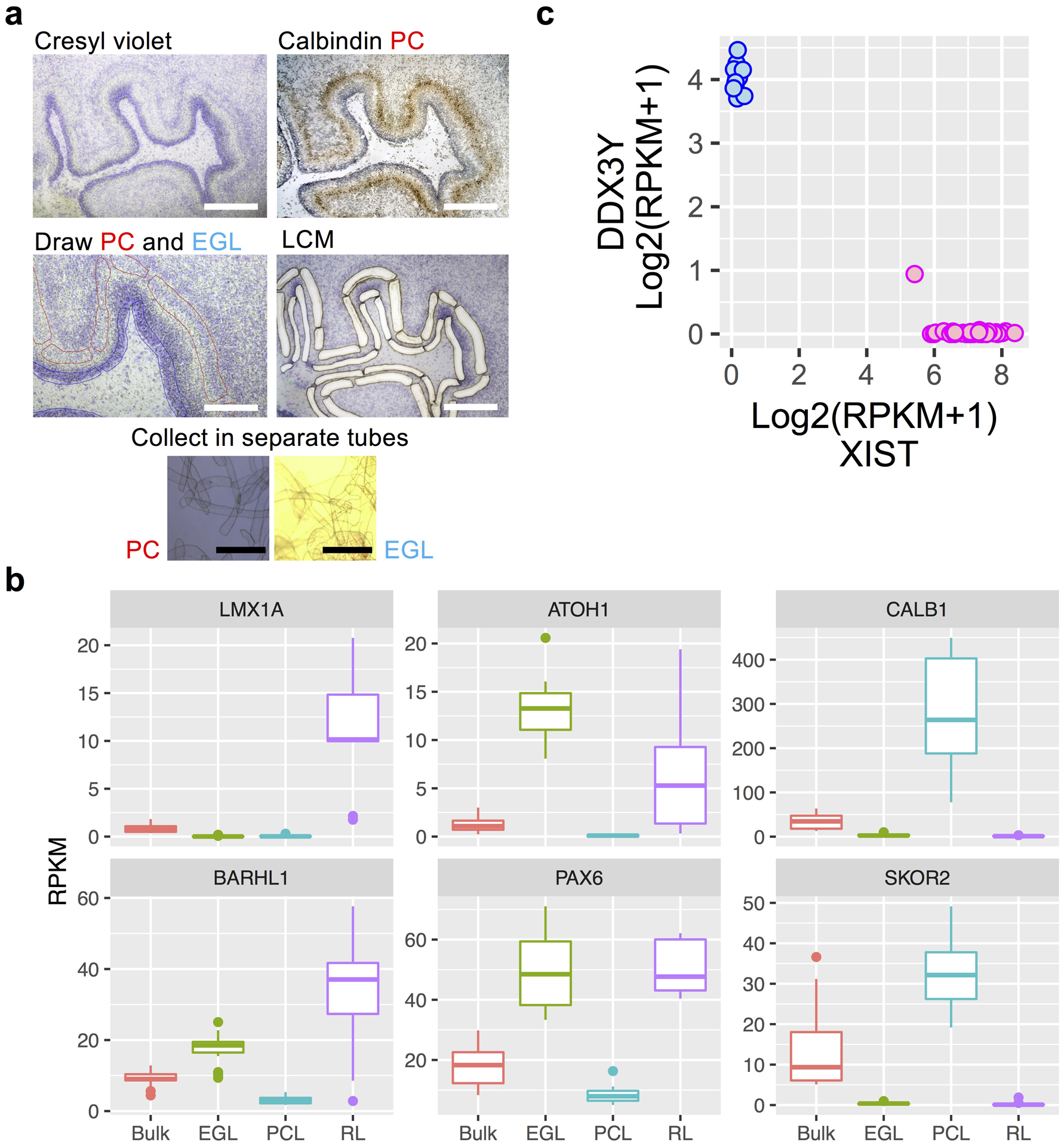
a, Example of cerebellum section stained with cresyl violet (purple) and anti-calbindin antibody (brown). Section before and after LCM and images of Purkinje cell (PC) and external granule cell layer (EGL) tissue captured into collection tubes are shown. Example shown is representative of 11 specimens. Scale bars: 200 um (white), 400 um (black). b, Boxplots of gene expression for established markers showing highest expression in the expected samples (box: 25–75th percentiles, whiskers: 10–90th percentiles, horizontal line in box: median). Dots indicate outliers. RNA-seq sample numbers per region: n = 13 for bulk; 17 for EGL; 18 for PCL; 9 for RL. c, Expression of the female-specific non-coding RNA XIST and the chromosome Y specific gene DDX3Y show correct sex assignment for female (pink) and male (blue) samples. RNA-seq sample numbers: n = 13 for bulk; 17 for EGL; 18 for PCL; 9 for RL. RNA-seq sample numbers per sex: n = 44 female; 13 male.
Extended Data Fig. 2. Co-expression modules in the developing human cerebellum.
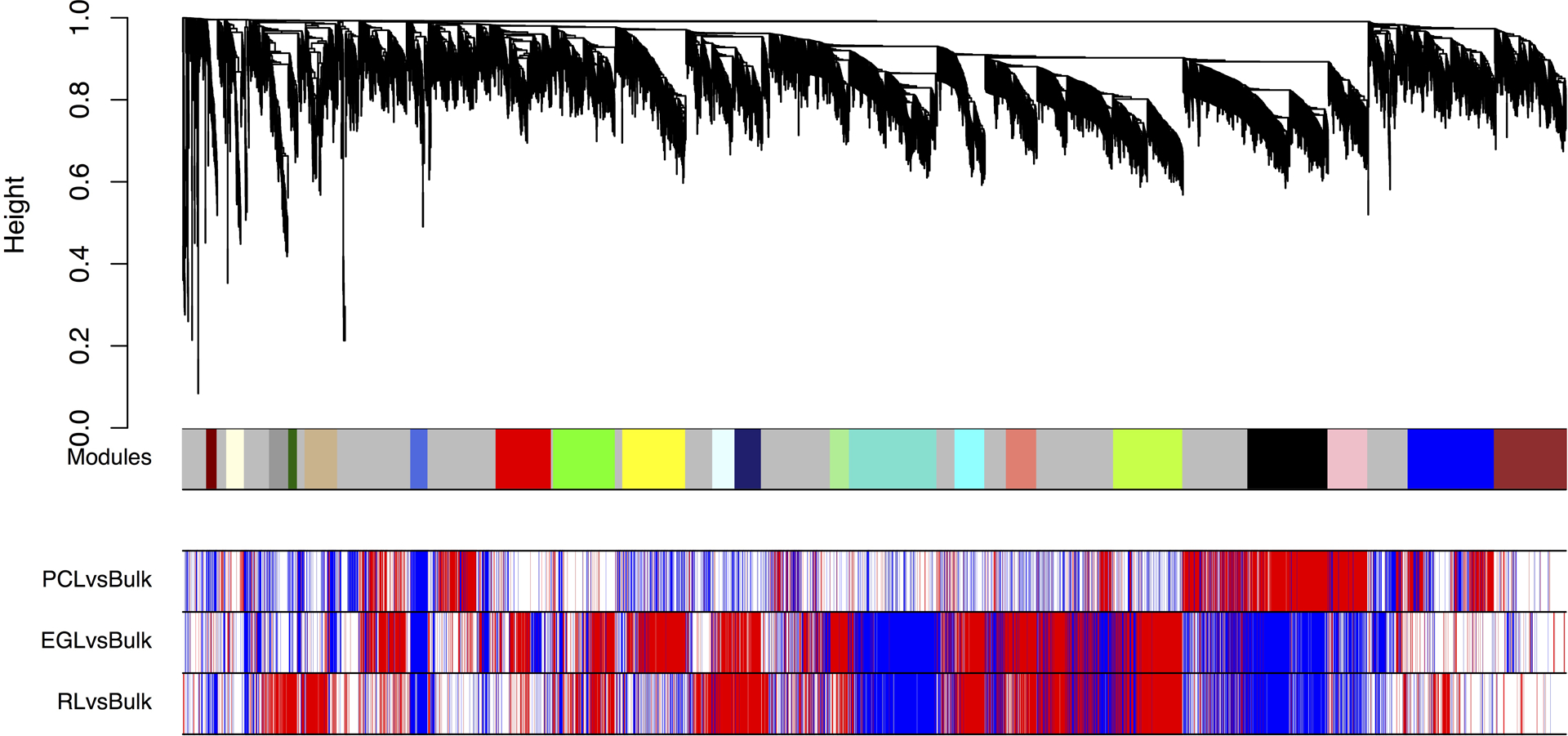
Weighted gene co-expression network analysis (WGCNA) dendrogram identified 21 modules comprised of 6,336 expressed genes (row 1). M0 (grey) comprised of nonclustered genes was not analyzed further. Rows 2–4 show differential expression relationships between module genes and LCM-enriched region compared to bulk expression. EGL, external granule cell layer; PCL, Purkinje cell layer; RL, rhombic lip.
Extended Data Fig. 3. Co-expression modules in the developing human cerebellum by region.
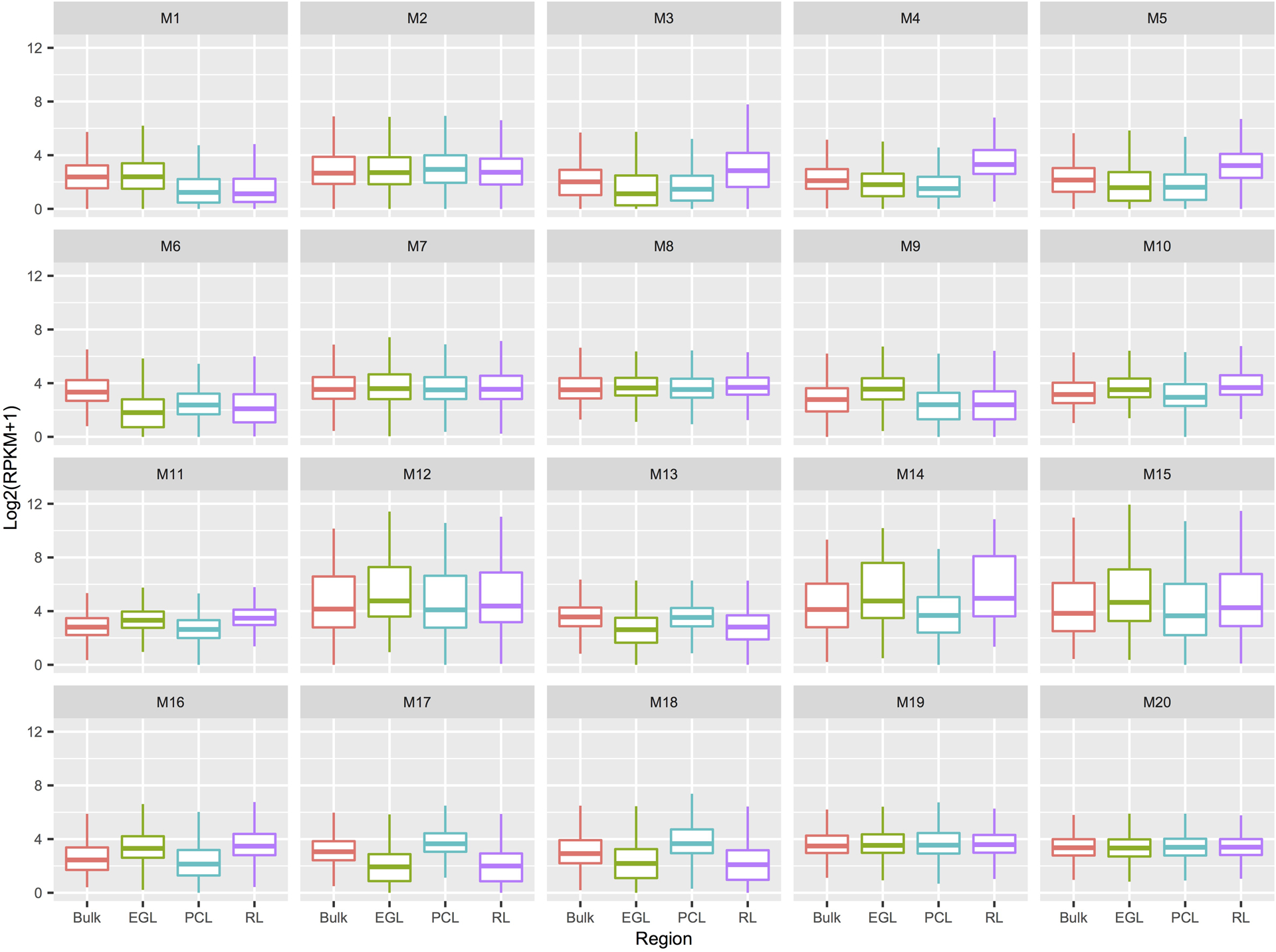
Boxplots of gene expression per WGCNA module for bulk and spatial regions (box: 25–75th percentiles, whiskers: 10–90th percentiles, horizontal line in box: median). Number of genes per module: n = 48 for M1; 81 for M2; 88 for M3; 40 for M4; 149 for M5; 79 for M6; 253 for M7; 283 for M8; 288 for M9; 102 for M10; 121 for M11; 87 for M12; 401 for M13; 136 for M14; 139 for M15; 317 for M16; 367 for M17; 182 for M18; 395 for M19; 327 for M20. EGL, external granule cell layer; PCL, Purkinje cell layer; RPKM, reads per kilobase of transcript per million mapped reads; RL, rhombic lip.
Extended Data Fig. 4. Co-expression modules in the developing human cerebellum by age.
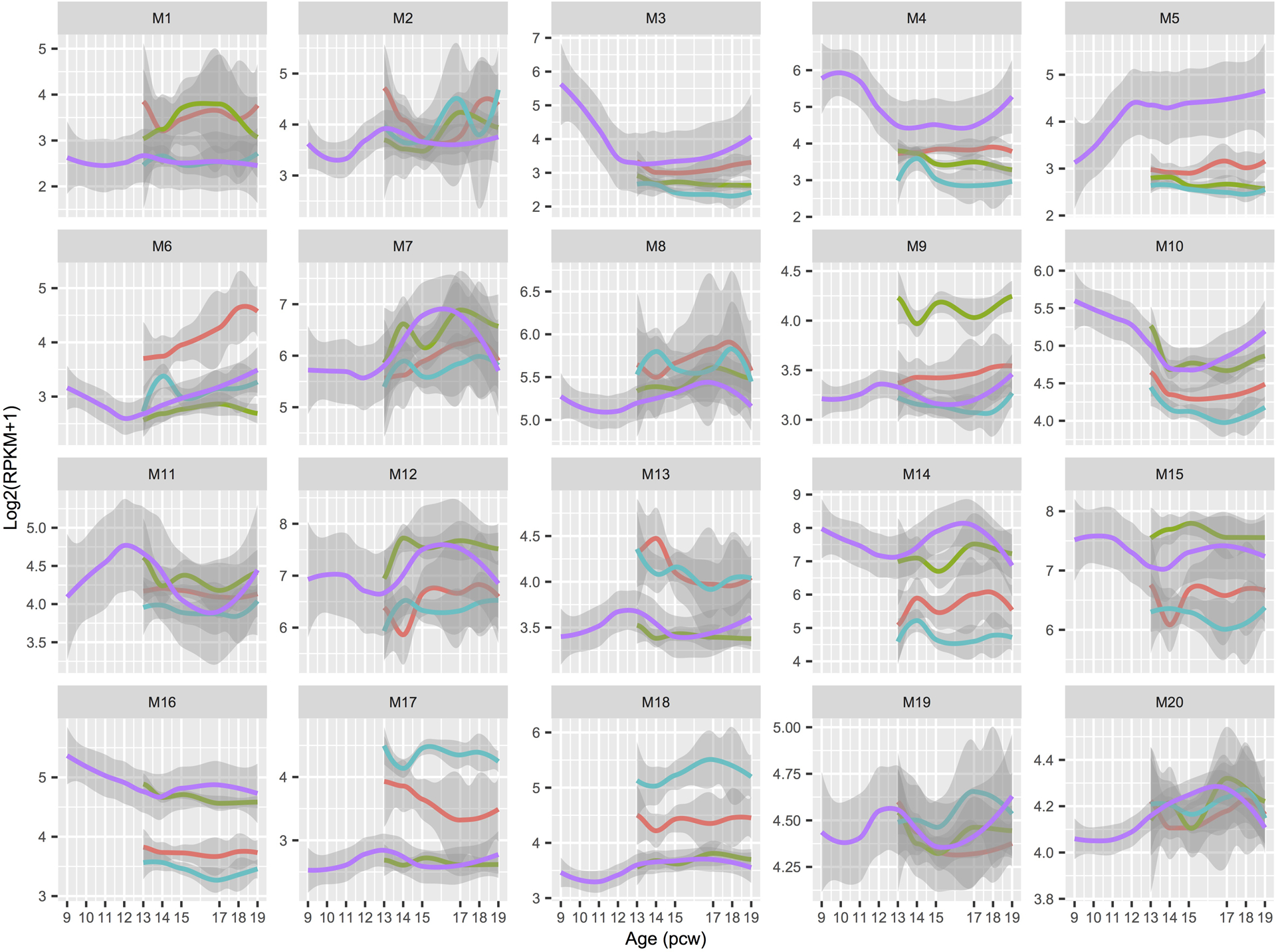
LOESS expression values across development are shown with 95% CIs per module. Spatial regions are distinguished by colors: bulk (salmon); EGL (green); PCL (turquoise); RL (purple). EGL, external granule cell layer; PCW, postconceptional week; PCL, Purkinje cell layer; RPKM, reads per kilobase of transcript per million mapped reads; RL, rhombic lip.
Extended Data Fig. 5. Quality control related analyses of snRNA-seq data.
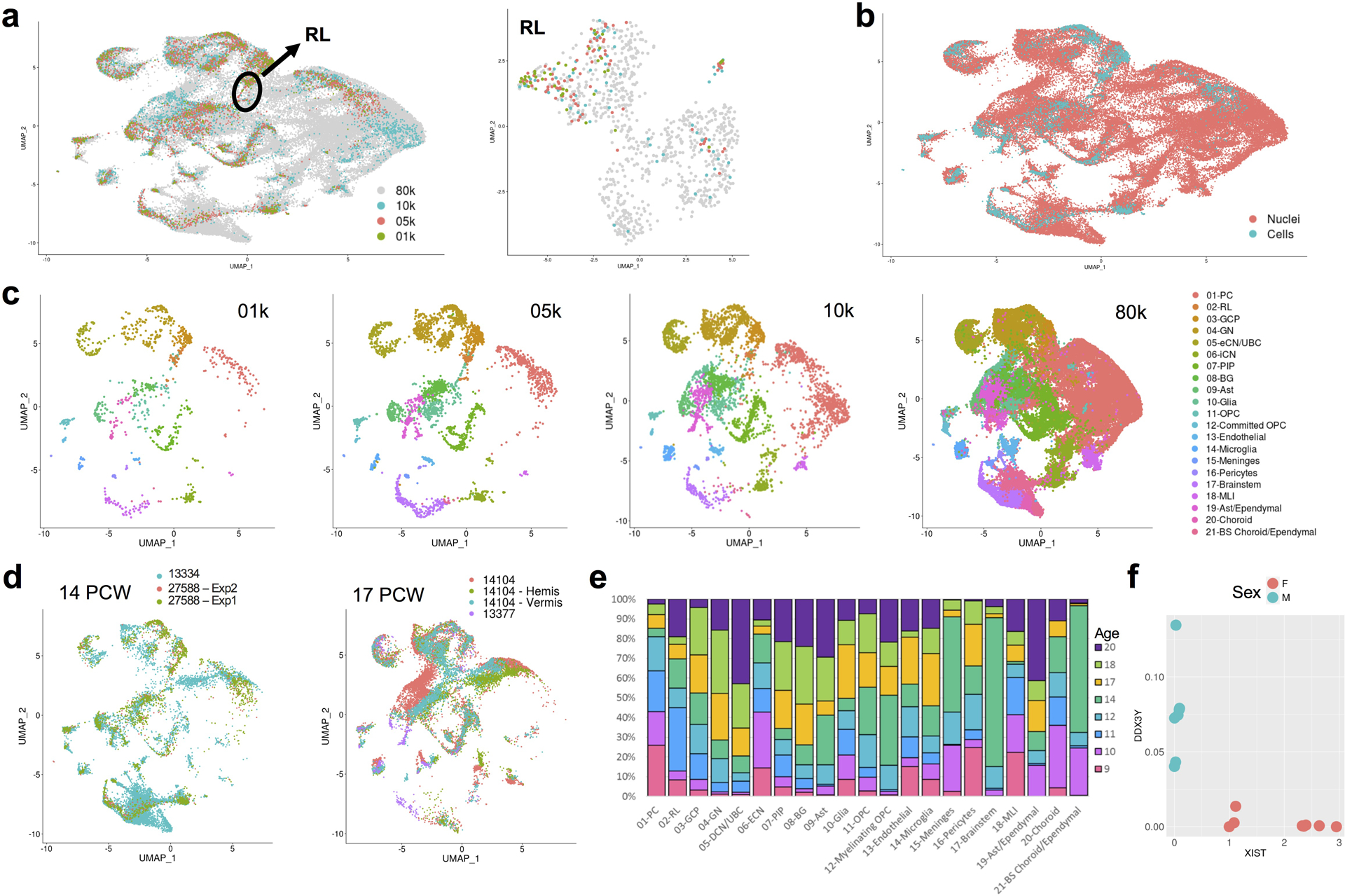
a, UMAP visualization of 69,174 human cerebellar nuclei colored by dataset (n = 1,076 for 01k; 3,530 for 05k; 4,960 for 10k; 59,608 for 80k). Rhombic lip (RL) is circled. UMAP visualization of 1,018 RL nuclei colored by dataset at right (nuclei numbers: n = 41 for 01k; 88 for 05k; 67 for 10k; 822 for 80k). b, The same UMAP as in a with nuclei colored by type (n = 4,462 cells; 64,712 nuclei). c, The same UMAP as in a and b showing nuclei from each dataset. Nuclei are colored by cell type. d, The same UMAP as in a-c showing nuclei sampled from same age biological and technical replicates (n = 11,213 for 14 PCW; 8,453 nuclei for 13334; 2,098 cells for 27588 Exp1; 662 cells for 27588 Exp2; n = 15,556 for 17 PCW; 524 cells for 13377; 8,540 nuclei for 14104; 3,364 nuclei for 14104h; 3,128 nuclei for 14104v). e, Stacked bar chart shows the percentage of age sampled in each of the 21 cell types. Bar colors represent age sampled in postconceptional weeks (9–20 PCW). f, Expression of the female-specific non-coding RNA XIST and the chromosome Y specific gene DDX3Y show correct sex assignment for female (salmon) and male (turquoise) samples (n = 14 female; 12 male).
Extended Data Fig. 6. Cell-type-specific marker genes.
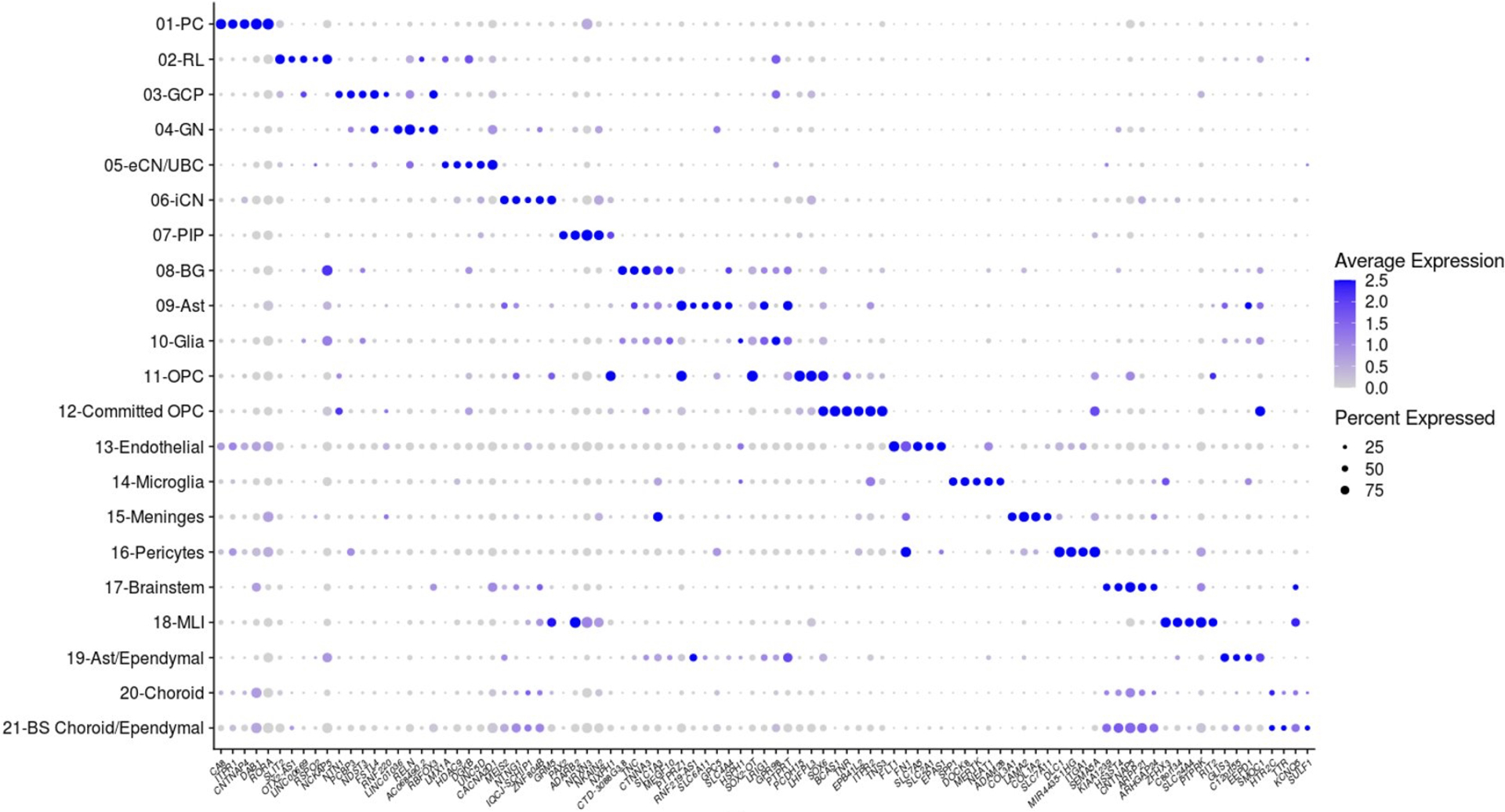
Dot plot showing expression of the top 5 most differentially expressed genes for each of the 21 cell types identified in early and mid-gestation fetal cerebellum. The size of the dot represents the percentage of cells within a cell type in which that gene was detected and its color represents the average expression level. Statistics are presented in Supplementary Table 9.
Extended Data Fig. 7. Distribution of major cell types.
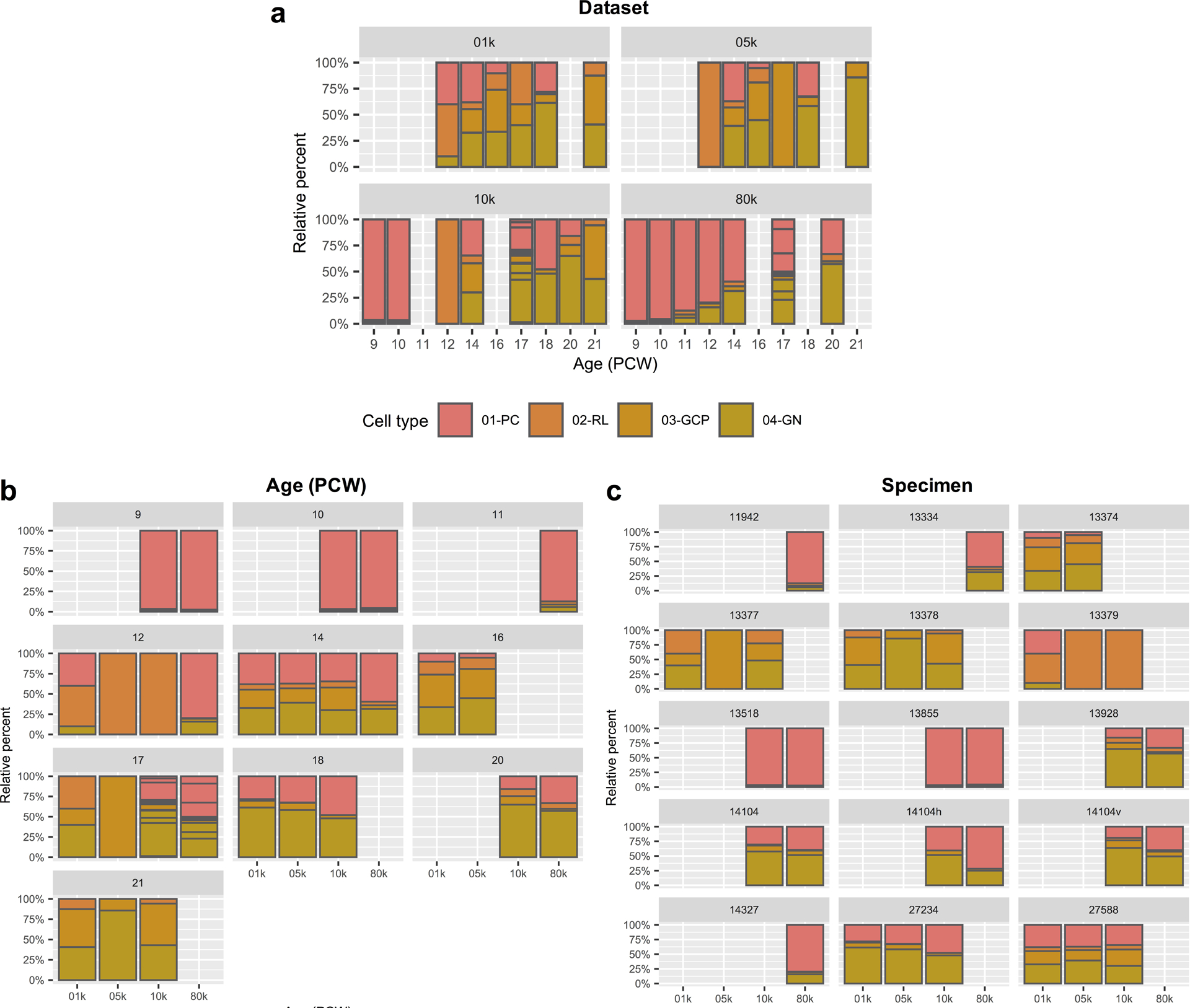
a-c, Stacked bar charts show the percentage of the four major cell types from each dataset (a), developmental age (b), and specimen (c). Dataset 01k and 05k from experiment (Exp) 1 represent deep and shallow sequencing runs, respectively, from the same 6 samples (one per age). Dataset 10k from Exp 2 represents 11 samples (7 for a single age and 4 for 17 PCW), including 5 replicates from Exp 1. Dataset 80k from Exp 3 represents 9 samples (6 for a single age and 3 for 17 PCW), including 6 replicates from Exp 2. Sample and experiment characteristics are presented in Supplementary Table 2 and 7.
Extended Data Fig. 8. Co-expression of marker genes in eCN/UBC.
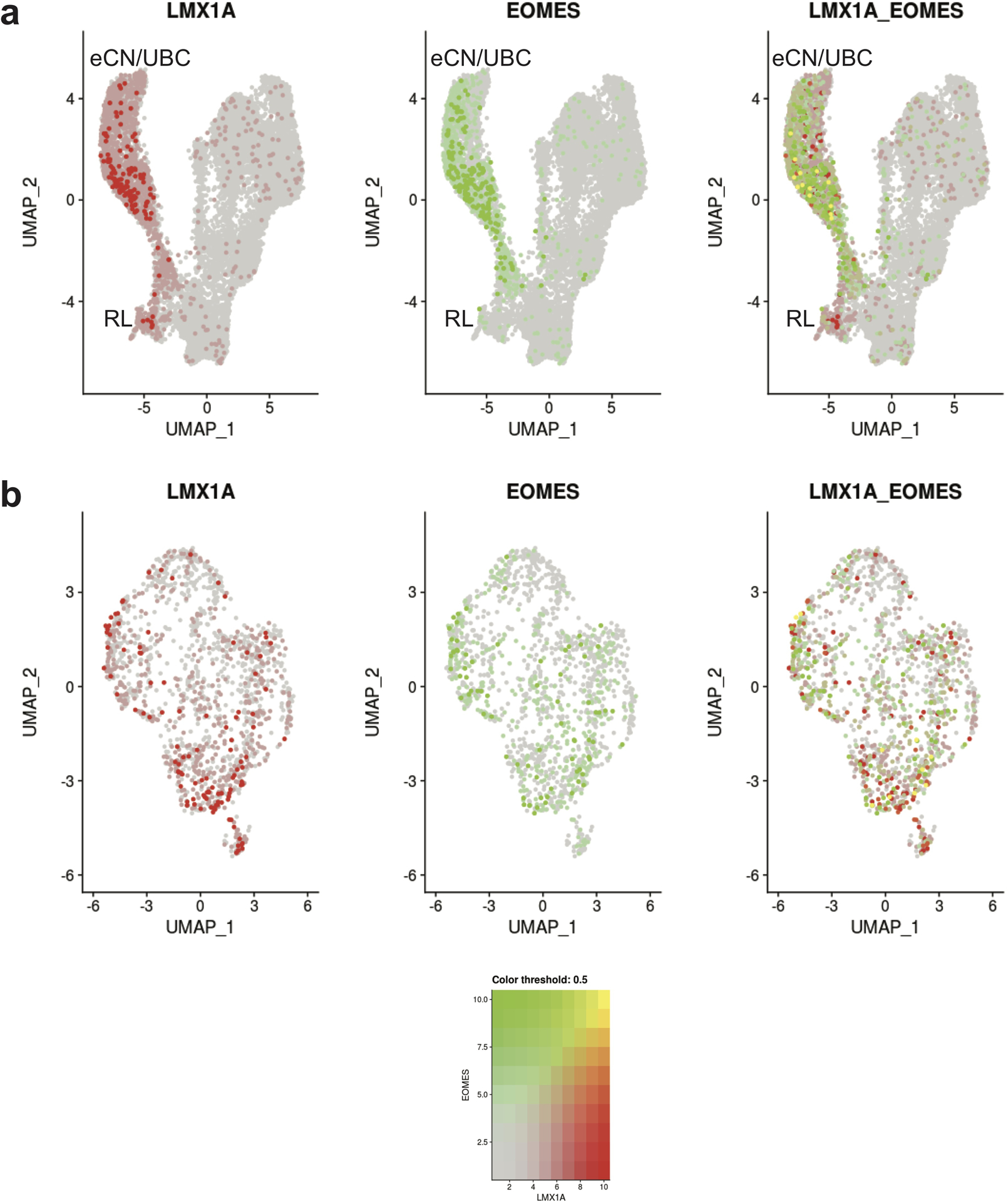
a, The same UMAP visualization of cell types that originate from the RL as in Fig. 5a with nuclei colored by expression level for LMX1A (red), EOMES (green), and co-expression (yellow). b, The same UMAP visualization the eCN/UBC subcluster as in Fig. 5e with nuclei colored by expression level for LMX1A (red), EOMES (green), and co-expression (yellow).
Extended Data Fig. 9. Cell type heterogeneity in LCM-isolated regions of the cerebellum.
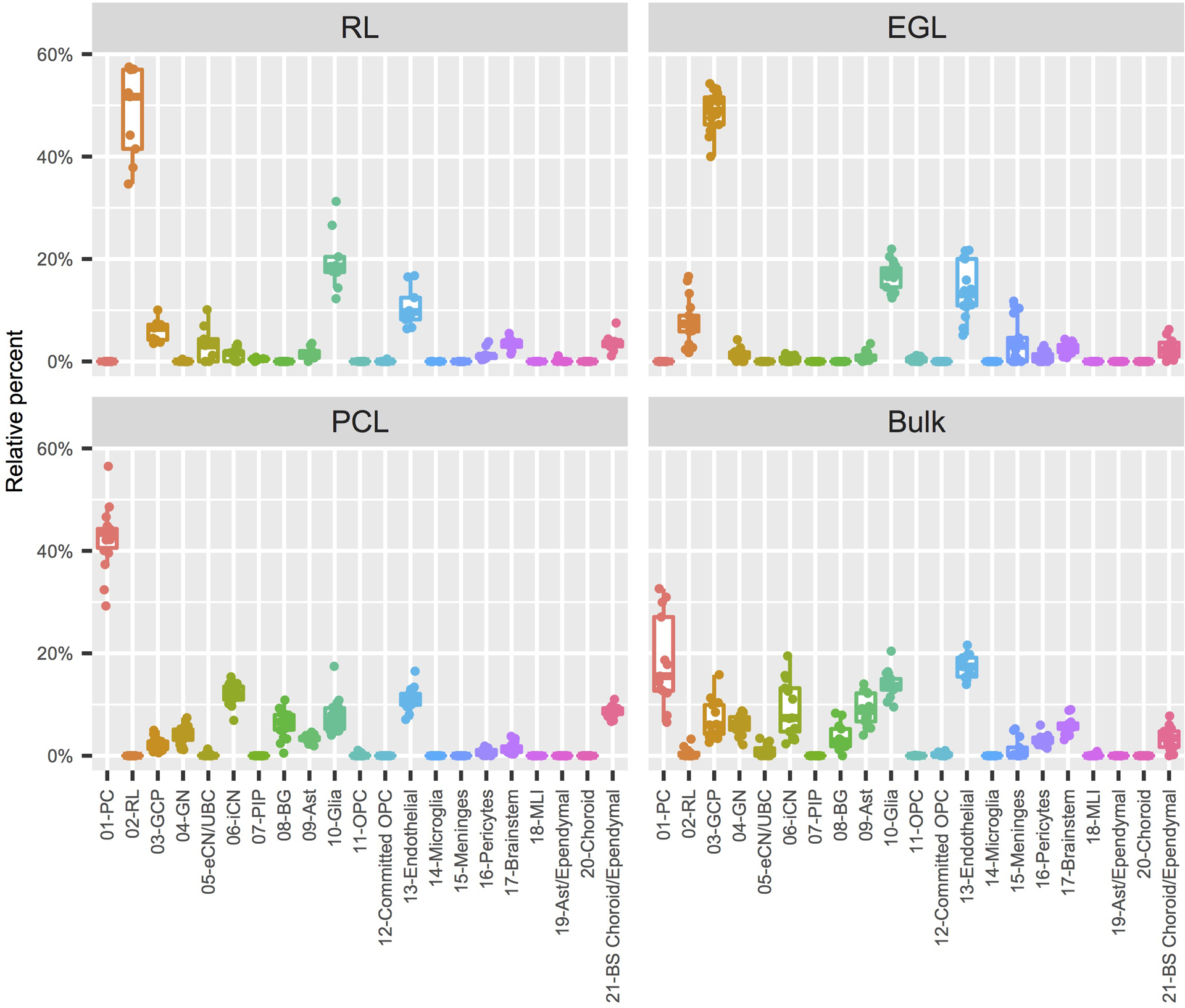
Box plots (box: 25–75th percentiles, whiskers: 10–90th percentiles, horizontal line in box: median) with data points (dots) showing the proportion of each of the 21 cell types from the Developmental Cell Atlas of the Human Cerebellum represented in the LCM RNA-seq data, grouped by LCM-isolated region. RL, rhombic lip; EGL, external granule cell layer; PCL, Purkinje cell layer.
Extended Data Fig. 10. Cerebellar cell type enrichment in Joubert syndrome and Alzheimer’s disease.

Heatmaps of mean expression per fetal cerebellar cell type for genes associated with Joubert syndrome (a) or Alzheimer’s disease (b). Color scheme is based on Z-score distribution. In the heatmaps, each row represents one gene and each column represents a single cell type. Horizontal white lines indicate branch divisions in the clustering dendrograms (not shown). The full list of genes is provided in Supplementary Table 11. Enrichment P values (-Log10 P value) for each cell type are shown in the bottom bar plots. Significance determined by one-sample Z-test, two-tailed P value. The dashed line is the Bonferroni significance threshold (P<0.05); no gene enrichment was detected among the 21 cerebellar cell types.
Supplementary Material
Acknowledgements
This study was funded by the National Institutes of Health under National Institute of Neurological Disorders and Stroke (NINDS), National Institute of Child Health and Human Development (NICHD), and National Institute of Mental Health (NIMH) grant numbers NS095733 to K.J.M., HD000836 to I.A.G., NS050375 to W.B.D, and MH110926, MH116488, and MH106934 to N.S. The project that gave rise to these results received the support of a fellowship from “la Caixa” Foundation (ID 100010434) to G. Santpere. The fellowship code is LCF/BQ/PI19/11690010. K.A.A. received a Parental Leave Grant from Life Science Editors and would like to thank Dr. Christina Lilliehook for editorial assistance. This publication is part of the Human Cell Atlas -www.humancellatlas.org/publications
Footnotes
Competing interests
Charles Roco, Alexander B. Rosenberg, and Georg Seelig are shareholders of Parse Biosciences. The remaining authors declare no competing interests.
Data availability
Processed data are available through the Human Cell Atlas (https://www.covid19cellatlas.org/aldinger20), the UCSC Cell Browser (https://cbl-dev.cells.ucsc.edu), and upon request. Sequence data were deposited into the Database of Genotypes and Phenotypes (dbGaP), under accession number phs001908.v2.p1, and available upon request.
Code availability
No custom code was used in this study. Open source algorithms were used as detailed in analysis methods. Details on how these algorithms were used are available from the corresponding author upon request.
REFERENCES
- 1.Sathyanesan A, et al. Emerging connections between cerebellar development, behaviour and complex brain disorders. Nat Rev Neurosci 20, 298–313 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schmahmann JD The cerebellum and cognition. Neurosci Lett 688, 62–75 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Leto K, et al. Consensus Paper: Cerebellar Development. Cerebellum 15, 789–828 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rakic P & Sidman RL Histogenesis of cortical layers in human cerebellum, particularly the lamina dissecans. J Comp Neurol 139, 473–500 (1970). [DOI] [PubMed] [Google Scholar]
- 5.Aldinger KA & Doherty D The genetics of cerebellar malformations. Semin Fetal Neonatal Med 21, 321–332 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoxha E, et al. The Emerging Role of Altered Cerebellar Synaptic Processing in Alzheimer’s Disease. Front Aging Neurosci 10, 396 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klockgether T, Mariotti C & Paulson HL Spinocerebellar ataxia. Nat Rev Dis Primers 5, 24 (2019). [DOI] [PubMed] [Google Scholar]
- 8.Corrales JD, Rocco GL, Blaess S, Guo Q & Joyner AL Spatial pattern of sonic hedgehog signaling through Gli genes during cerebellum development. Development 131, 5581–5590 (2004). [DOI] [PubMed] [Google Scholar]
- 9.Dahmane N & Ruiz i Altaba A Sonic hedgehog regulates the growth and patterning of the cerebellum. Development 126, 3089–3100 (1999). [DOI] [PubMed] [Google Scholar]
- 10.Haldipur P, et al. Spatiotemporal expansion of primary progenitor zones in the developing human cerebellum. Science 366, 454–460 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Holgado BL, Guerreiro Stucklin A, Garzia L, Daniels C & Taylor MD Tailoring Medulloblastoma Treatment Through Genomics: Making a Change, One Subgroup at a Time. Annu Rev Genomics Hum Genet 18, 143–166 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Volpe JJ Cerebellum of the premature infant: rapidly developing, vulnerable, clinically important. J Child Neurol 24, 1085–1104 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson MB, et al. Functional and evolutionary insights into human brain development through global transcriptome analysis. Neuron 62, 494–509 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kang HJ, et al. Spatio-temporal transcriptome of the human brain. Nature 478, 483–489 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li M, et al. Integrative functional genomic analysis of human brain development and neuropsychiatric risks. Science 362 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miller JA, et al. Transcriptional landscape of the prenatal human brain. Nature 508, 199–206 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mu Q, Chen Y & Wang J Deciphering Brain Complexity Using Single-cell Sequencing. Genomics Proteomics Bioinformatics 17, 344–366 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rosenberg AB, et al. Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science 360, 176–182 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang B & Horvath S A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol 4, Article17 (2005). [DOI] [PubMed] [Google Scholar]
- 20.Lange W Cell number and cell density in the cerebellar cortex of man and some other mammals. Cell Tissue Res 157, 115–124 (1975). [DOI] [PubMed] [Google Scholar]
- 21.McGinnis CS, Murrow LM & Gartner ZJ DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Syst 8, 329–337 e324 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aldinger KA, et al. Redefining the Etiologic Landscape of Cerebellar Malformations. Am J Hum Genet 105, 606–615 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Butler A, Hoffman P, Smibert P, Papalexi E & Satija R Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36, 411–420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Machold R & Fishell G Math1 is expressed in temporally discrete pools of cerebellar rhombic-lip neural progenitors. Neuron 48, 17–24 (2005). [DOI] [PubMed] [Google Scholar]
- 25.Wang VY, Rose MF & Zoghbi HY Math1 expression redefines the rhombic lip derivatives and reveals novel lineages within the brainstem and cerebellum. Neuron 48, 31–43 (2005). [DOI] [PubMed] [Google Scholar]
- 26.Englund C, et al. Unipolar brush cells of the cerebellum are produced in the rhombic lip and migrate through developing white matter. J Neurosci 26, 9184–9195 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cao J, et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fink AJ, et al. Development of the deep cerebellar nuclei: transcription factors and cell migration from the rhombic lip. J Neurosci 26, 3066–3076 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zecevic N & Rakic P Differentiation of Purkinje cells and their relationship to other components of developing cerebellar cortex in man. J Comp Neurol 167, 27–47 (1976). [DOI] [PubMed] [Google Scholar]
- 30.Dastjerdi FV, Consalez GG & Hawkes R Pattern formation during development of the embryonic cerebellum. Front Neuroanat 6, 10 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Emmert-Buck MR, et al. Laser capture microdissection. Science 274, 998–1001 (1996). [DOI] [PubMed] [Google Scholar]
- 32.Espina V, et al. Laser-capture microdissection. Nat Protoc 1, 586–603 (2006). [DOI] [PubMed] [Google Scholar]
- 33.Newman AM, et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol 37, 773–782 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu J, et al. Jointly defining cell types from multiple single-cell datasets using LIGER. Nat Protoc 15, 3632–3662 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Welch JD, et al. Single-Cell Multi-omic Integration Compares and Contrasts Features of Brain Cell Identity. Cell 177, 1873–1887 e1817 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vladoiu MC, et al. Childhood cerebellar tumours mirror conserved fetal transcriptional programs. Nature 572, 67–73 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Van De Weghe JC, et al. Mutations in ARMC9, which Encodes a Basal Body Protein, Cause Joubert Syndrome in Humans and Ciliopathy Phenotypes in Zebrafish. Am J Hum Genet 101, 23–36 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Feliciano P, et al. Exome sequencing of 457 autism families recruited online provides evidence for autism risk genes. NPJ Genom Med 4, 19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.RK CY, et al. Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat Neurosci 20, 602–611 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ruzzo EK, et al. Inherited and De Novo Genetic Risk for Autism Impacts Shared Networks. Cell 178, 850–866 e826 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Willsey AJ, et al. The Psychiatric Cell Map Initiative: A Convergent Systems Biological Approach to Illuminating Key Molecular Pathways in Neuropsychiatric Disorders. Cell 174, 505–520 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yuen RK, et al. Whole-genome sequencing of quartet families with autism spectrum disorder. Nat Med 21, 185–191 (2015). [DOI] [PubMed] [Google Scholar]
- 43.De Strooper B & Karran E The Cellular Phase of Alzheimer’s Disease. Cell 164, 603–615 (2016). [DOI] [PubMed] [Google Scholar]
- 44.Bis JC, et al. Whole exome sequencing study identifies novel rare and common Alzheimer’s-Associated variants involved in immune response and transcriptional regulation. Mol Psychiatry (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wizeman JW, Guo Q, Wilion EM & Li JY Specification of diverse cell types during early neurogenesis of the mouse cerebellum. Elife 8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hovestadt V, et al. Resolving medulloblastoma cellular architecture by single-cell genomics. Nature 572, 74–79 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carter RA, et al. A Single-Cell Transcriptional Atlas of the Developing Murine Cerebellum. Curr Biol 28, 2910–2920 e2912 (2018). [DOI] [PubMed] [Google Scholar]
- 48.Sillitoe RV & Joyner AL Morphology, molecular codes, and circuitry produce the three-dimensional complexity of the cerebellum. Annu Rev Cell Dev Biol 23, 549–577 (2007). [DOI] [PubMed] [Google Scholar]
- 49.Nakatani T, Minaki Y, Kumai M, Nitta C & Ono Y The c-Ski family member and transcriptional regulator Corl2/Skor2 promotes early differentiation of cerebellar Purkinje cells. Dev Biol 388, 68–80 (2014). [DOI] [PubMed] [Google Scholar]
- 50.Haldipur P, et al. Preterm delivery disrupts the developmental program of the cerebellum. PLoS One 6, e23449 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gerrelli D, Lisgo S, Copp AJ & Lindsay S Enabling research with human embryonic and fetal tissue resources. Development 142, 3073–3076 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Anders S, Pyl PT & Huber W HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Szklarczyk D, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res 47, D607–D613 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.DeLuca DS, et al. RNA-SeQC: RNA-seq metrics for quality control and process optimization. Bioinformatics 28, 1530–1532 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hodge RD, et al. Conserved cell types with divergent features in human versus mouse cortex. Nature 573, 61–68 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stuart T, et al. Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902 e1821 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mirzaa GM, et al. De novo and inherited variants in ZNF292 underlie a neurodevelopmental disorder with features of autism spectrum disorder. Genet Med 22, 538–546 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Epting D, et al. Loss of CBY1 results in a ciliopathy characterized by features of Joubert syndrome. Hum Mutat 41, 2179–2194 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Latour BL, et al. Dysfunction of the ciliary ARMC9/TOGARAM1 protein module causes Joubert syndrome. J Clin Invest 130, 4423–4439 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Luo M, et al. Disrupted intraflagellar transport due to IFT74 variants causes Joubert syndrome. Genet Med (2021). [DOI] [PubMed] [Google Scholar]
- 63.Sanders SJ, et al. Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron 87, 1215–1233 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Deciphering Developmental Disorders S Prevalence and architecture of de novo mutations in developmental disorders. Nature 542, 433–438 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rauch A, et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet 380, 1674–1682 (2012). [DOI] [PubMed] [Google Scholar]
- 66.Irizarry RA, Wang C, Zhou Y & Speed TP Gene set enrichment analysis made simple. Stat Methods Med Res 18, 565–575 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.