

Abstract
Immunotherapies targeting aspects of T cell functionality are efficacious in many solid tumors, but pancreatic ductal adenocarcinoma (PDAC) remains refractory to these treatments. Deeper understanding of the PDAC immune ecosystem is needed to identify additional therapeutic targets and predictive biomarkers for therapeutic response and resistance monitoring. To address these needs, we quantitatively evaluated leukocyte contexture in 135 human PDACs at single-cell resolution by profiling density and spatial distribution of myeloid and lymphoid cells within histopathologically-defined regions of surgical resections from treatment-naïve and presurgically (neoadjuvant)-treated patients and biopsies from metastatic PDAC. Resultant data establishes an immune atlas of PDAC heterogeneity, identifies leukocyte features correlating with clinical outcomes, and through an in silico study, provides guidance for use of PDAC tissue microarrays to optimally measure intratumoral immune heterogeneity. Atlas data has direct applicability as a reference for evaluating immune responses to investigational neoadjuvant PDAC therapeutics where pre-therapy baseline specimens are not available.
Keywords: pancreatic cancer, inflammation, prognostic biomarkers, tumor microenvironment, immune cells
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is a leading cause of cancer-related deaths in the United States, with a current 5-year survival rate of only 10% (1). Most patients present with late-stage metastatic disease, and even patients diagnosed at earlier stages who are eligible for potentially curative tumor resection have disease recurrence rates exceeding 80% (2). Standard-of-care cytotoxic therapies extend life expectancy only modestly (3,4), thus new therapeutic combinations are urgently needed.
Immunotherapies targeting immune checkpoint molecules have transformed clinical outcomes for patients with many types of solid tumors; however, these have yet to significantly impact outcomes for PDAC (5), except in a minority of patients harboring microsatellite instability-high (MSI-H) tumors (6). Poor immunogenicity of PDAC and its highly immunosuppressive tumor immune microenvironment (TiME) represent considerable hurdles to immunotherapeutic efficacy (7). A more nuanced understanding of human PDAC immune contexture and immune heterogeneity is needed to inform rational drug combinations and to effectively stratify patients for immunotherapies to which they are most likely to respond.
Recent development of innovative multiplexed imaging strategies now enables in situ phenotyping and spatial characterization of multiple cell populations simultaneously (8–11), thereby facilitating advances in our understanding of the cellular composition of tumors. For example, recent studies in PDAC have revealed extensive T cell heterogeneity where spatial localization holds prognostic significance (11–14). Despite these technological advances, many recent analyses have focused predominantly on features of adaptive immunity, particularly CD8+ T cells, without robust characterization of the full leukocyte milieu that also includes discrete subsets of B cells and myeloid cells, all of which harbor both pro- and anti-tumor functionality and likely influence patient outcome. Indeed, in a recent independent study, we profiled myeloid cells in 300+ human surgically resected PDACs and found significant associations between myeloid densities, spatial localization, and patient survival (15), thus supporting the need for further investigations integrating spatial analysis of both adaptive and innate components of the PDAC TiME. It is also incompletely understood how human PDAC immune contexture evolves with disease progression from early-stage to late-stage metastatic disease, or how various therapies impact the TiME. We previously reported that PDACs from patients who received neoadjuvant GVAX, a granulocyte-macrophage colony-stimulating factor (GM-CSF) tumor cell vaccine, clustered into low-myeloid inflamed and high-myeloid inflamed groups, where enhanced myeloid inflammation was associated with features of T cell suppression and shorter survival (8). In a subsequent study, we evaluated immune correlates of previously treated metastatic PDAC where patients received GVAX with cyclophosphamide (Cy) and CRS-207 (live, attenuated Listeria monocytogenes-expressing mesothelin), and we revealed that reduced myeloid to lymphoid ratios following therapy correlated with improved outcome (16). Together, these studies indicate that the PDAC TiME can be therapeutically impacted. Going forward, a deeper understanding of steady-state PDAC leukocyte ecosystems will aid in identification of immune features that underlie response and/or resistance to standard cytotoxic therapies as well as established and investigational immune therapy regimens. In addition, this will enable advanced clinical trial design for precision medicine approaches, similar to what we have recently reported in metastatic breast cancer (17).
To this end, we employed a chromogen-based multiplexed immunohistochemistry (mIHC) platform (8,18) to build a comprehensive immune atlas of 135 clinically- and genomically-annotated human PDAC specimens from a multi-institutional patient population. Through mIHC profiling of lymphoid and myeloid leukocyte lineages across distinct histopathological regions — including tumor, tumor adjacent stroma, tertiary lymphoid structures (TLS), and adjacent normal pancreas tissue — we report substantial inter- and intrapatient immune heterogeneity and identify leukocyte features associated with treatment status and clinical outcome. Collectively, this atlas will serve future studies by providing baseline steady-state characteristics of the PDAC TiME.
RESULTS
Evaluation of human PDAC immune contexture
To reveal the immune landscape of human PDAC, we used a robust mIHC platform designed to capture multiple leukocyte lineages and their functional status (8,18). As shown in Figure 1A, we evaluated primary PDAC from a multi-institutional population of treatment-naïve patients (n = 104; Cohorts 1 and 2) and patients who received physician’s choice chemotherapy and/or radiotherapy prior to surgical resection (n = 13; Cohort 1). In addition, we performed exploratory analysis of immune composition in primary PDACs versus distant PDAC metastases (n = 18; Cohort 3) and examined healthy normal (HN) pancreas from organ transplant donors (n = 6; Cohort 4) in order to compare neoplasia-associated leukocyte infiltration to steady-state healthy pancreatic tissue (Figure 1A, Tables S1–3).
Figure 1. Overview of human pancreas specimens and cell populations evaluated by mIHC.
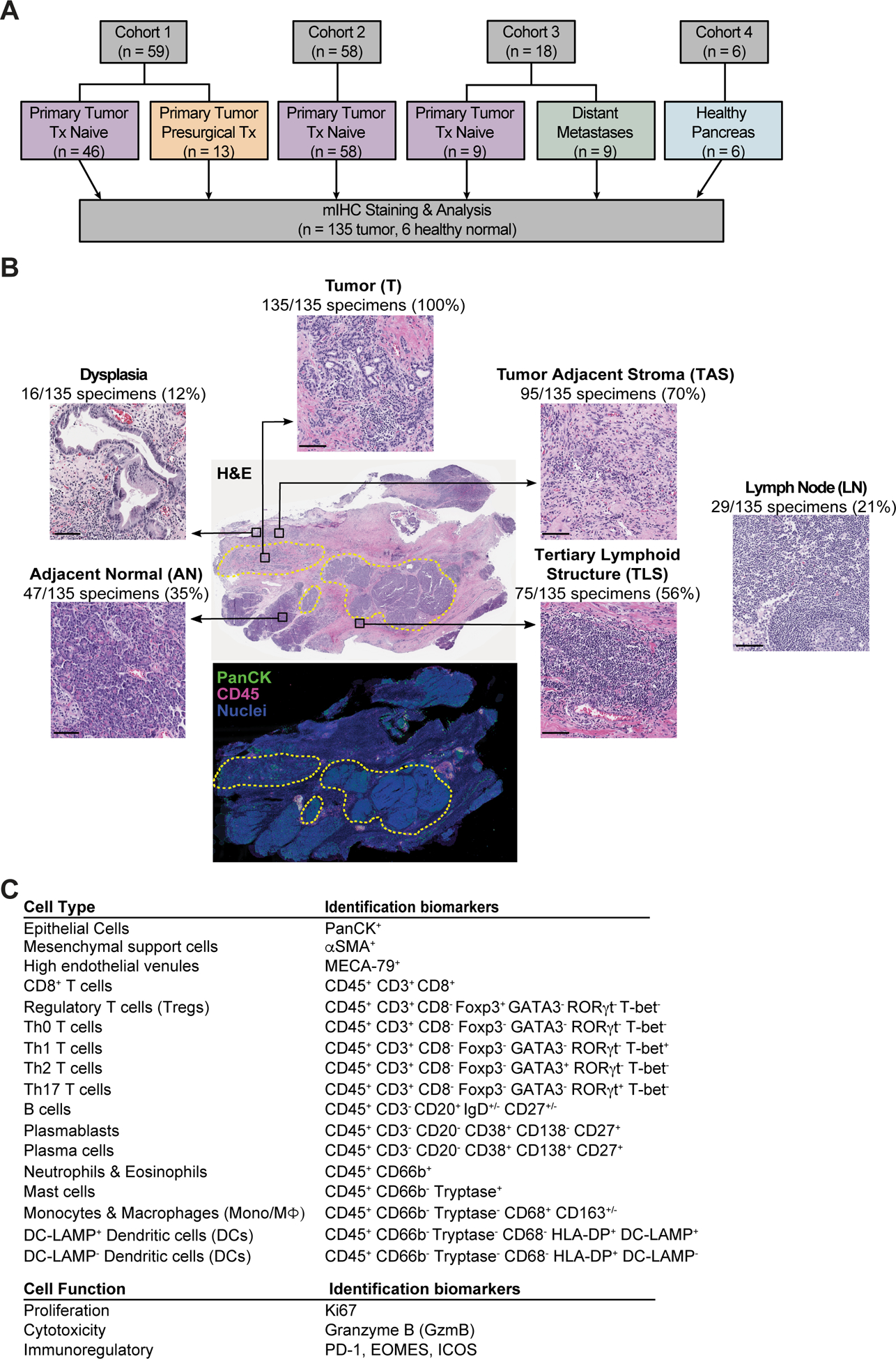
(A) Cohort diagram of human PDACs and healthy pancreas evaluated herein. (B) Representative PDAC tissue section stained with hematoxylin and eosin (H&E) (top) illustrating types of histopathologic regions analyzed, with corresponding serial section of mIHC-stained tissue (bottom) displayed in pseudocolor with pan-cytokeratin (PanCK; green), CD45 (pink), and nuclei (blue). Yellow dotted lines represent pathologist’s annotations of tumor (T) areas. Number of PDAC specimens containing each histopathologic region type are indicated. H&E image of LN at right is from a different treatment-naïve PDAC resection specimen within the cohort. Scale bars, 100 μm. (C) Cell lineages identified by hierarchical gating of lineage-selective and functional biomarkers during image cytometry analysis of mIHC staining.
Surgical resection specimens allowed evaluation of immune contexture from a “global” perspective in large tissue sections containing histopathologically distinct areas (Figure 1B). For all patient samples, we analyzed regions of interest (ROIs) within pathologist-annotated areas containing invasive tumor (T, 100% of specimens). Where possible, we also evaluated tumor adjacent stroma (TAS, contained in 70% of specimens) and adjacent ‘normal’ pancreas (AN; contained in 35% of specimens), each located outside of areas containing invasive carcinoma (Figure 1B). Dense lymphoid aggregates consistent with TLS were present in 56% of specimens (located within and/or outside of tumor areas), and a small fraction of samples contained lymph node (LN) and/or pre-invasive dysplasia (21% and 12% of specimens, respectively) that were also evaluated for leukocyte contexture (Figure 1B).
Three serial formalin-fixed paraffin-embedded (FFPE) tissue sections per patient were subjected to mIHC staining with antibody panels designed to identify lymphoid and myeloid populations (Figure 1C, Table S4), and digital images were quantitatively analyzed by image cytometry (8) (Figure S1A–C). Within the lymphoid lineage, we identified CD3+CD8+ T cells, CD3+CD8− T helper cells (Th0, Th1, Th2, Th17), regulatory T cells (Tregs), CD20+ B cells, and CD20− plasmablasts and plasma cells. T cells were further evaluated for expression of select immunoregulatory molecules (programmed cell death protein 1 [PD-1], inducible T cell costimulator [ICOS/CD278], Granzyme B [GzmB], Eomesodermin [EOMES]), and proliferation (Ki67). CD20+ B cells were categorized into memory and naïve populations based on CD27 and IgD expression. Myeloid subsets were identified as CD66b+ granulocytes (neutrophils and eosinophils), tryptase+ mast cells, dendritic cells (DCs), and CD68+ cells comprising both monocytes and macrophages. DC maturation status was assessed by expression of DC-LAMP, a lysosomal glycoprotein predominately expressed by mature DCs (19), while CD68+ cells were stratified by expression of the hemoglobin scavenger receptor CD163 to mark their alternative-activation status (20). Details on average tissue area and cell densities evaluated in each histopathologic region are provided in Table S5.
Treatment-naïve PDACs differentially cluster based on global immune phenotype
To evaluate immune complexity and interpatient heterogeneity of treatment-naïve PDACs, we performed unsupervised hierarchical clustering based on cell densities of CD45+ leukocyte subpopulations, pan-cytokeratin (PanCK)+ epithelial cells, and alpha smooth muscle actin (〈SMA)+ mesenchymal support cells (Figure 2A). We combined cell density data from all histopathological regions (Figure 1B), as we hypothesized that “global” quantitation, independent of histopathologic annotation, would yield data analogous to that obtained from tumor biopsies or tissue microarray (TMA) cores where histopathology of tissue available for analysis is often less prospectively defined. PDACs clustered into three groups based on leukocyte profiles: “myeloid-enriched”, “lymphoid-enriched”, and “hypo-inflamed” (Figure 2A). Principal component analysis (PCA) revealed two clusters were driven by relative densities of myeloid versus lymphoid cells, while the “hypo-inflamed’ cluster was not driven by either lineage and contained lowest abundance of overall leukocyte infiltrate (Figure 2B–C). Notably, densities of total lymphoid cells and lymphoid subpopulations did not significantly differ between the myeloid-enriched and lymphoid-enriched clusters, but lymphoid to myeloid ratio was elevated in the lymphoid-enriched group (Figure 2C, Figure S2A). In contrast, total myeloid cells, as well as select myeloid subpopulations — particularly neutrophils/eosinophils and monocytes/macrophages — were significantly elevated in the myeloid-enriched cluster as compared to others (Figure 2C, Figure S2A). We observed that these clusters arose independently of total area of tissue analyzed (Figure S2B), though there was some variability in the histopathologic regions represented in each cluster (Figure S2C). We next evaluated whether clusters were associated with patient survival and observed that patients in the lymphoid-enriched cluster had a modestly improved median overall survival (OS) as compared to patients in other clusters (24.3 months lymphoid-enriched, 20.7 months hypo-inflamed, 19.3 months myeloid-enriched); however, these global immune phenotypes were not significantly associated with outcome (Figure 2D).
Figure 2. Global analysis of treatment-naïve PDAC surgical resections reveals distinct immune phenotypes.
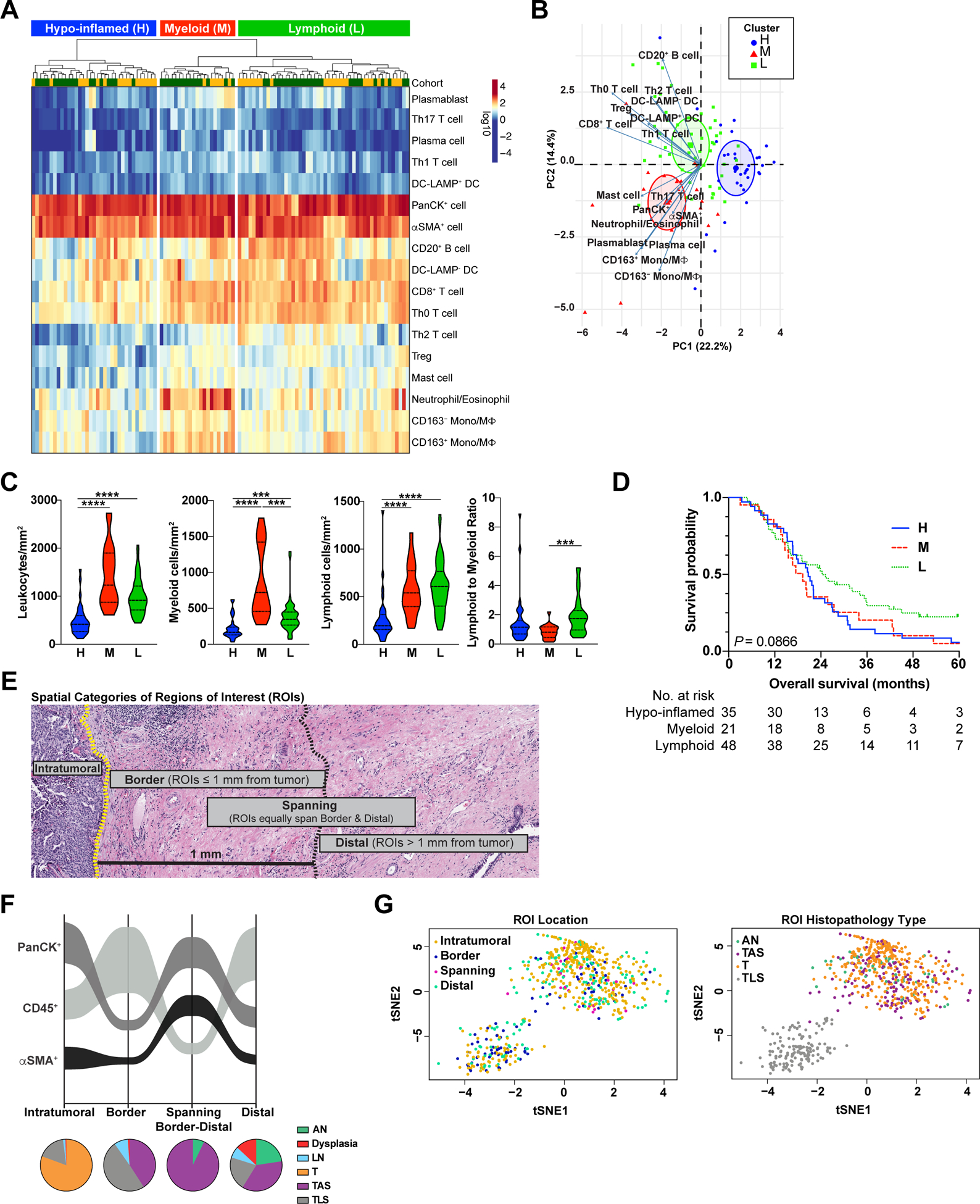
(A) Unsupervised hierarchical clustering of treatment-naive PDACs from Cohorts 1 (yellow, uppermost row) and 2 (green, uppermost row) showing cell densities of indicated cell subsets (rows). Cell densities for each patient (columns) reflect cumulative densities from all analyzed ROIs per patient. (B) PCA of cell population densities from ‘A’. Each symbol represents one patient. H, hypo-inflamed cluster, n = 35; M, myeloid-enriched cluster, n = 21; L, lymphoid-enriched cluster, n = 48. (C) Indicated cell densities and ratios based on clusters from ‘A’. Statistical differences between groups were determined by Kruskal-Wallis tests with Dunn’s correction for multiple comparisons. (D) Kaplan-Meier curves displaying OS of patients based on clusters defined in ‘A’. P-value was determined by log-rank test. (E) Representative H&E of PDAC depicting spatial categories (‘Intratumoral’, ‘Border’, ‘Spanning’, and ‘Distal’) assigned to each individual region of interest (ROI) analyzed. (F) Sankey flow diagram representing relative densities (cells/mm2) of PanCK+ epithelial cells, CD45+ leukocytes, and αSMA+ mesenchymal support cells within treatment-naïve PDAC specimens across each spatial category (X-axis). Populations are sorted on the Y-axis from highest (top) to lowest (bottom) density, where ribbon width is scaled to density. Pie charts below represent relative contribution of different histopathologic region types (AN, Dysplasia, LN, T, TAS, TLS) within each spatial category. Number of individual ROIs evaluated is listed in Table S6. (G) t-SNE representation of cell density within individual ROIs (dots) color coded by ROI spatial location (left) and histopathology type (right).
Spatial proximity of immune cells to invasive carcinoma holds prognostic significance for many cancer types (10,21,22). To investigate leukocyte distribution, we subclassified individual ROIs from all histopathologic regions into “intratumoral”, “border”, “spanning border-distal”, or “distal” spatial categories based on ROI proximity to nearest annotated area of invasive carcinoma (Figure 2E, Table S6). We then evaluated densities of total CD45+ leukocytes, PanCK+ epithelial cells, and 〈SMA+ cells in each spatial category and created corresponding spatial maps (Figure 2F). Of the cell types evaluated, PanCK+ cells were the densest population in intratumoral and spanning distal-border regions, whereas CD45+ leukocytes were densest in border and distal regions (Figure 2F). We used t-distributed stochastic neighbor embedding (t-SNE) to visualize relationships between ROI cell density, ROI location, and ROI histopathology type (Figure 2G). t-SNEs revealed considerable admixture of individual ROIs based on location and histopathology type, though TLS ROIs formed a distinct cluster. (Figure 2G, Figure S2D).
Total leukocyte density varies by histopathologic region
To further evaluate leukocyte composition and interpatient heterogeneity in distinct histopathologic regions, we quantified leukocyte density in each histopathologic region within treatment-naïve PDACs (Cohorts 1 and 2) and HN pancreas (Cohort 4). LNs and pre-invasive dysplasia were excluded from analyses given their low relative abundance in the dataset. Notably, leukocyte density was significantly higher in AN regions of PDAC as compared to HN pancreas (Figure 3A), although increased density in AN was not driven by influx of a particular leukocyte subset (Figure 3B). Thus, ostensibly “normal” pancreas tissue adjacent to invasive PDAC is not spared from inflammatory assault within the diseased organ.
Figure 3. Leukocyte density in treatment-naïve PDAC.
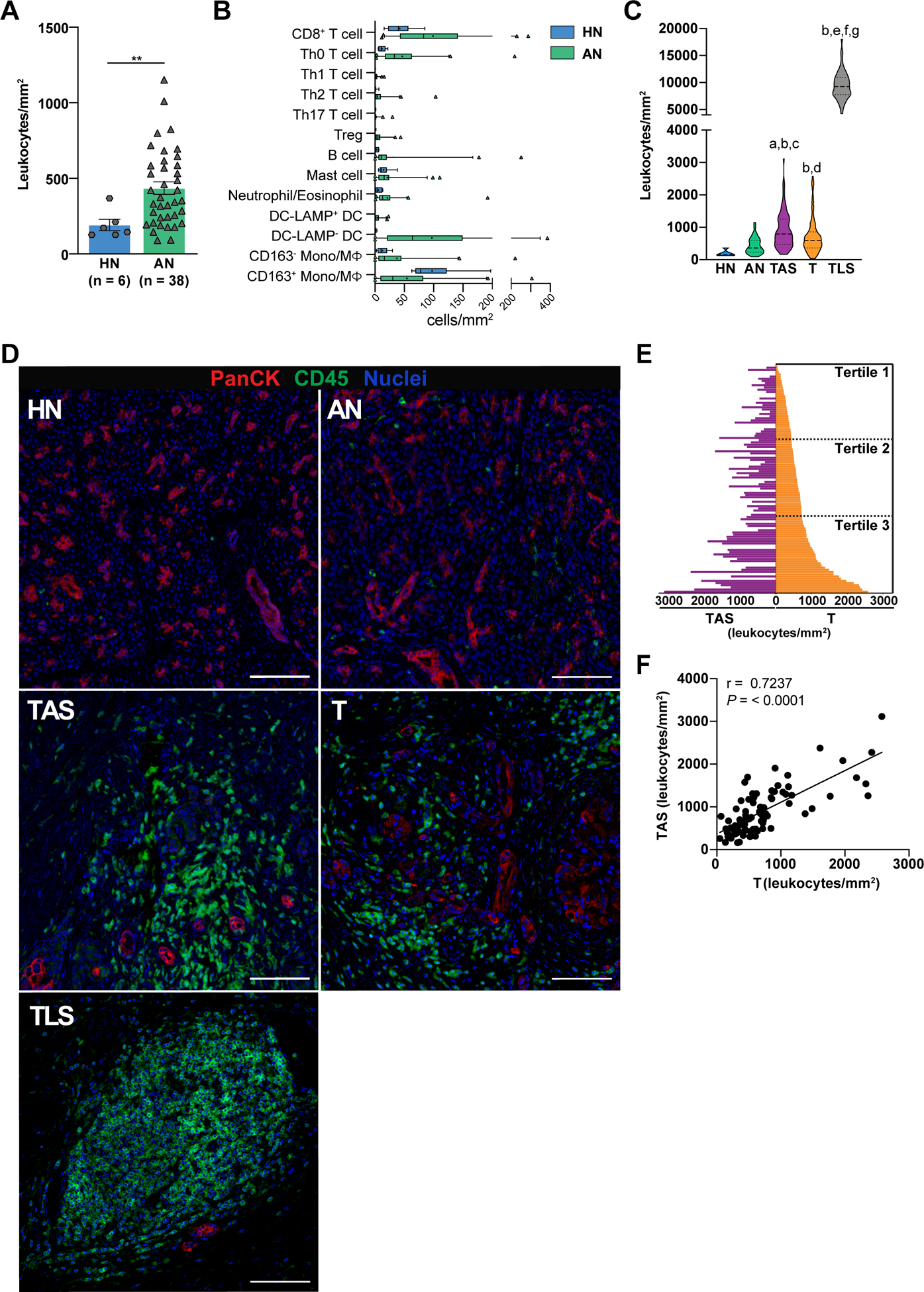
(A) Total leukocyte density in healthy normal (HN) pancreas from organ transplant donors versus PDAC adjacent normal (AN) pancreas. Each data point represents cumulative cell density from multiple ROIs in a single resection specimen. Statistical differences determined by two-tailed, unpaired Mann-Whitney U test. Data represented as mean ± SEM. (B) Leukocyte composition of HN compared to AN. Box plots show median and interquartile range with means indicated by (+) symbols. (C) Comparison of leukocyte density in HN and indicated treatment-naïve PDAC histopathology regions (from Cohorts 1 and 2). Statistical differences between histopathologic regions determined by mixed model repeated measures ANOVA on log-transformed data with heterogeneous compound symmetry (CSH) covariance structure to assess within-patient correlation. Tukey-Kramer post-hoc correction was applied, and adjusted P-values are reported: aP < 0.001 versus HN; bP < 0.0001 versus AN; cP <0.001 versus T; dP < 0.01 versus HN; eP < 0.0001 versus HN; fP < 0.0001 versus TAS; gP < 0.0001 versus T. (D) Representative pseudocolored images showing epithelial cells (PanCK+) and leukocytes (CD45+) in HN, AN, TAS, T, and TLS regions. Scale bars, 100 μm. (E) Leukocyte density within PDAC T regions (right) and patient-matched TAS (left) sorted low-to-high for intratumoral leukocyte density (T, n = 104; TAS, n = 81). Tumor leukocyte density tertiles are indicated. (F) Spearman correlation of leukocyte density in patient-matched T and TAS PDAC specimens (n = 81).
Evaluation of leukocyte density in other regions of PDAC revealed that TAS and T exhibited considerable interpatient heterogeneity, though average leukocyte density was highly similar between the two cohorts (Figure S3A–B). Comparison of all region types revealed that T, TAS, and TLS each contained significantly elevated leukocyte density as compared to AN. TAS regions as compared to T regions contained significantly higher leukocyte infiltrate, while TLS contained highest overall leukocyte density of any histopathology type evaluated (Figure 3C–D and Figure S3C).
Intratumoral density of leukocyte subpopulations has been associated with patient prognosis in PDAC and other malignancies (8,11,13,23–25), but it is unclear if total leukocyte density – as opposed to density or effector status of individual subpopulations – predicts outcome. When we examined matched pairs of TAS and T within the same patient, we found significant correlation in leukocyte density between the two sites (Figure 3E–F). Among leukocyte subpopulations, densities of CD8+ T cells, Th0 T cells, and Tregs were most significantly associated with total leukocyte density in both TAS and T regions (Figure S3D). However, regional leukocyte density in TAS or T was not independently predictive of disease-free survival (DFS), OS, or other evaluated clinical characteristics, including tumor stage and grade (Figure S3E, Tables S7–8), indicating that relative frequencies of leukocyte subpopulations as opposed to overall CD45+ leukocyte density may represent a critical metric.
Leukocyte subpopulations are differentially enriched between PDACs and across tissue regions
To investigate leukocyte identity within treatment-naïve PDACs, we first determined the degree of regional skewing present within myeloid and lymphoid infiltrates. AN possessed lowest myeloid and lymphoid density of all regions evaluated. TLS exhibited significantly increased lymphoid density as compared to all other regions and increased myeloid density as compared to T, likely reflecting TLS compactness and paucity of embedded epithelial cells. Myeloid density was similar across TAS and T regions, whereas lymphocytes were significantly enriched in TAS as compared to T (Figure 4A).
Figure 4. Regional characteristics of lymphoid and myeloid cell enrichment in PDAC.
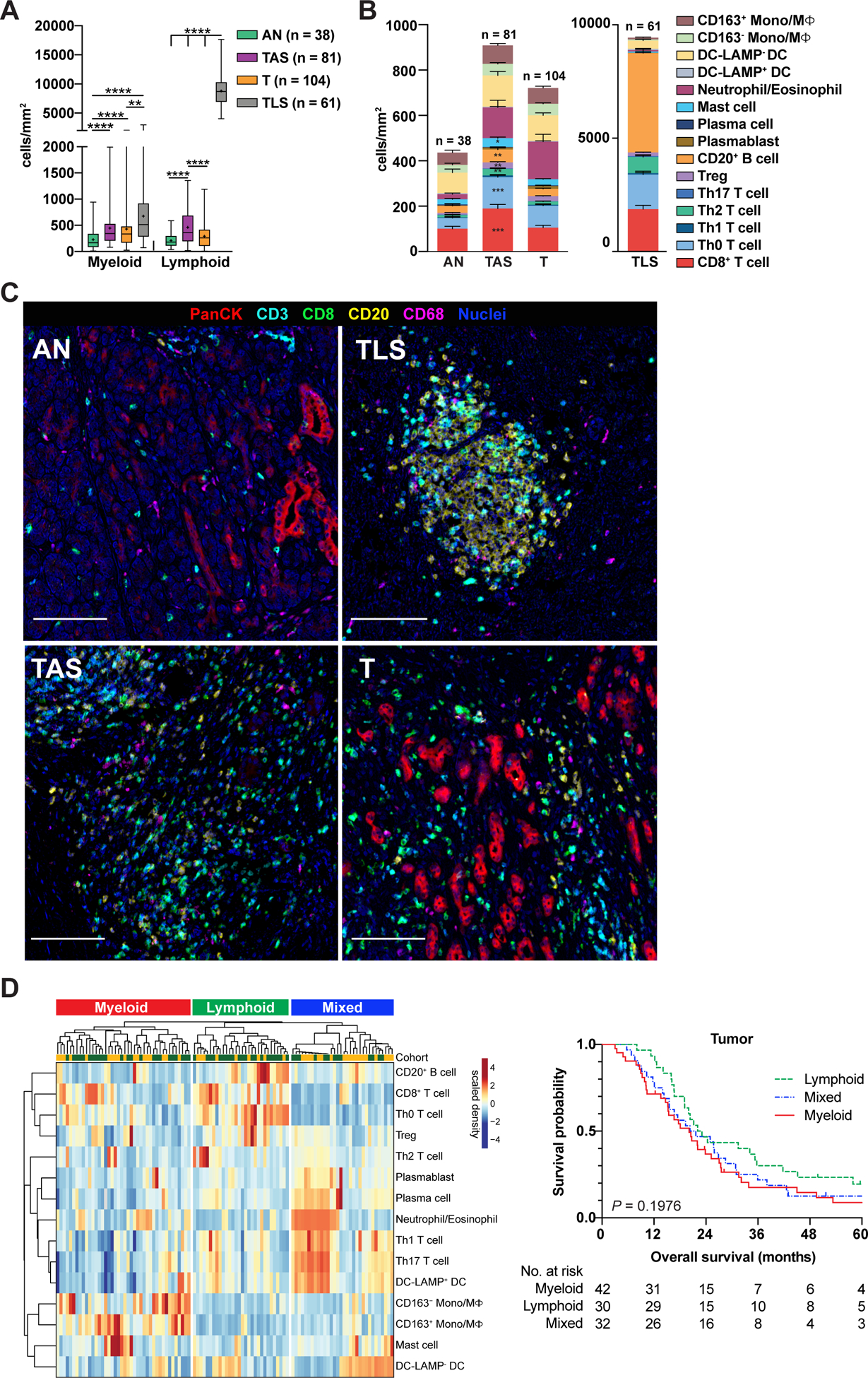
(A) Myeloid and lymphoid cell densities within indicated regions of treatment-naïve PDAC samples from Cohorts 1 and 2. ‘Myeloid’ reflects cumulative densities of mast cells, neutrophils/eosinophils, DCs, and Mono/MΦ. ‘Lymphoid’ reflects cumulative densities of CD3+ T cells and B cells, including plasma cells and plasmablasts. Statistical differences were determined by mixed model repeated measures ANOVA on log-transformed data with heterogeneous compound symmetry (CSH) covariance structure to assess within-patient correlation. Tukey-Kramer post-hoc correction was applied, and adjusted P-values are reported. (B) Immune composition of PDAC regions from surgical resection specimens shown in ‘A’. A mixed effects model was used to determine differences in cell population densities in T versus TAS. Data represented as mean ± SEM. (C) Representative pseudocolored images of regions quantitated in ‘B’ depicting PanCK+, CD3+, CD8+, CD20+, and CD68+ cell types. Scale bars, 100 μm. (D) Unsupervised hierarchical clustering (left) of treatment-naive PDACs from Cohorts 1 (yellow, uppermost row) and 2 (green, uppermost row) showing relative enrichment of indicated leukocyte subsets (rows) in tumor (T) regions. Data is patient-scaled and immune population z-scored for visualization. Each column represents one patient (n = 104) and reflects multiple tumor ROIs per specimen. Kaplan-Meier curve (right) displaying OS of patients based on clusters; P-value determined by log-rank test.
Deeper auditing revealed that discrete leukocyte subpopulations were present within each histopathologic region to varying degrees (Figure 4B–C, Figure S4A). Of lymphoid subsets, CD8+ T cell, Th0 and Th2 T helper cell, Treg, and B cell densities were significantly higher in TAS as compared to T regions (Figure 4B, left). Of myeloid populations, mast cells were significantly enriched in TAS as compared to T (Figure 4B, left). TLS were primarily composed of B cells, CD8+ T cells, and T helper cells, although DC-LAMP− DCs and mature DC-LAMP+ DCs were also present (Figure 4B, right), the latter of which are known to be critical within TLS for intra- or peri-tumoral antigen presentation and T cell priming (26). Further phenotyping of B cells and T helper cells revealed that CD27− IgD+ naïve B cells were most dense in AN, whereas expanded memory B cells (particularly CD27− IgD−) had highest density in TAS, T and TLS (Figure S4B). Tregs were least abundant in TLS as compared to other regions, and Th2 cells were significantly enriched in TLS as compared to other regions (Figure S4C).
Leukocyte milieu was independent of tumor histologic grade (Figure S4D). To determine if genomic alterations impacted PDAC leukocyte contexture, we next evaluated intratumoral leukocyte composition based on molecular status of the four main driver genes in PDAC (KRAS, CDK2NA, TP53, SMAD4) (27) in a subset of treatment-naïve samples. Leukocyte densities were generally consistent across tumors with distinct molecular profiles and, although sample sizes were small within molecular subgroups, we observed modest increases in neutrophil/eosinophil density in some PDACs, particularly those harboring TP53 alterations (Figure S4E).
To determine whether histologically discrete regions of treatment-naïve PDACs clustered into prognostically-relevant groups based on immune profile, we again performed unsupervised hierarchical clustering of patients, now considering leukocyte densities of either T regions or TAS regions only. Three clusters emerged in both T and TAS analyses (myeloid-enriched, lymphoid-enriched, and a mixed cluster with enrichment of both lymphocytes and myeloid cell types), though we did not observe associations between clusters and OS (Figure 4D, Figure S4F, Tables S9–S10).
High endothelial venule presence within TLS is associated with TLS cell density but not clinical outcome
TLS represent potential sites of T cell entry, priming, and clonal expansion within solid tumors and have been associated with improved survival and response to immunotherapy in several tumor contexts, including PDAC (28–32). To determine if TLS abundance represented a significant variable associated with survival in treatment-naïve PDACs, we quantified the number of total TLS present per single FFPE sample for each patient. 61 treatment-naïve PDACs in Cohorts 1 and 2 contained TLS, ranging in number from 1 to 46 (Figure S4G–H), where TLS quantity was modestly correlated with size of resection specimen (Figure S4I) but not associated with patient OS (Figure S4J).
High endothelial venules (HEVs) within TLS, identified by MECA-79 expression on lymphatic endothelial cells, facilitate trafficking of leukocytes and are associated with enhanced antitumoral immune responses (33). We observed that PDACs containing multiple TLS often exhibited heterogeneity in HEV presence. We detected MECA-79+ HEVs in 557 individual TLS (66% of total) (Figure S4K) and observed that HEVs were typically located within larger (by area) TLS containing higher densities of CD3+ T cells and CD20+ B cells, as compared to TLS lacking HEVs (Figure S4L). Higher overall T cell density in TLS with HEVs was driven by increased density of T helper cells, particularly Th0 and Th2 cells (Figure S4M). T and B cell functional state did not significantly differ based on HEV status (Figure S4N–P). We next examined whether HEV presence in TLS was associated with clinical features and revealed that HEV status was not associated with tumor histologic grade (Figure S4Q). Analysis of all treatment-naïve PDACs containing TLS (n = 61) revealed that proportion of HEV-containing TLS in a given specimen was not associated with patient survival (Figure S4R). Further, assessment of the longest-term and shortest-term treatment-naive survivors (1st and 4th quartile OS) that had TLS (n = 16 and 19, respectively) revealed that proportion of TLS containing HEVs was not correlated with exceptionally good or poor outcome (Figure S4S).
PDAC surgical resections exhibit extensive intrapatient immune heterogeneity.
We endeavored to understand degree of intrapatient heterogeneity in the current study by utilizing several approaches. First, we observed heterogeneity in leukocyte composition across individual ROIs from individual PDACs in T, TAS and TLS regions (Figure S5A–B). Next, we measured concordance of leukocyte population densities between two T ROIs from single patients and observed moderate correlations for many cell types (Figure S5C). Since studies investigating tumor immune contexture commonly utilize smaller tissue microarrays (TMAs) for uniform, high throughput analyses of large patient cohorts, we sought to determine how leukocyte composition in 1.0 mm diameter virtual TMA (vTMA) cores from T regions compared to composition within the multiple larger T ROIs evaluated per patient in the surgical resections studied herein. We first evaluated a random subset of 22 resection specimens from Cohorts 1 and 2 to determine the proportion of total pathologist-annotated invasive tumor on each slide that was captured in T ROIs analyzed by mIHC. Within these specimens, total tumor area ranged from 20–209 mm2 (average 94 mm2), and area within T ROIs was 12–31 mm2 (average 23 mm2) (Figure S5D). Five of these specimens representing a range of tumor sizes (Figure S5D, highlighted in red) and total leukocyte densities (Figure S5E) were selected for subsequent vTMA analysis. For each specimen, previously established T ROIs were further subdivided into 19–28 vTMA cores (Figure 5A). A variable abundance of CD3+ T cells, CD20+ B cells, CD68+ monocyte/macrophages, and other CD45+ leukocytes was documented across the vTMA cores, revealing that leukocyte composition of individual vTMA cores within an mIHC ROI varied as compared to the composition of the full mIHC ROI (Figure 5B, S5F), thereby identifying the spatial scale of leukocyte heterogeneity within PDAC.
Figure 5. Intrapatient leukocyte heterogeneity in treatment-naïve PDACs.
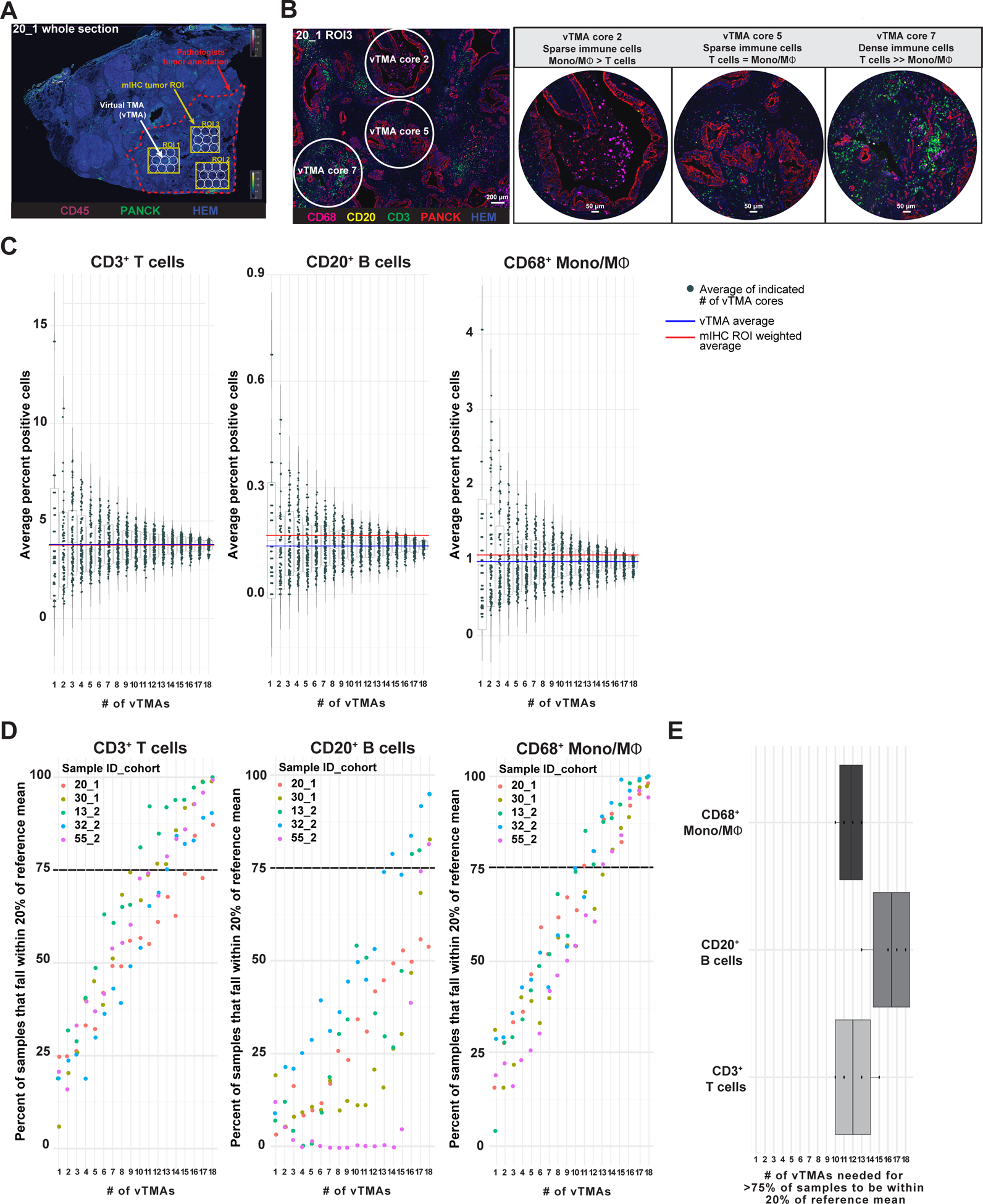
(A) Representative pseudocolored image depicting PanCK, CD45, and hematoxylin (nuclei) mIHC from one PDAC specimen (Sample 20, Cohort 1) with overlays indicating pathologist’s tumor annotation (red dashed line), T ROIs used in mIHC quantitative analysis (yellow boxes), and vTMA cores (white circles). (B) Pseudocolored image showing CD68, CD20, CD3, PanCK, and nuclei immunostaining of ROI3 from Sample 20 Cohort 1 (20_1), highlighting vTMA cores 2, 5, and 7. Scale bar equals 200 μm (left). Higher magnification images of vTMA cores (right). (C) CD3+ T cell (left), CD20+ B cell (middle), and CD68+ monocyte/macrophage (right) cell frequencies calculated from the average of 1–18 vTMA cores (x-axis) for 100 sample iterations. The vTMA averages (blue lines, sample reference) and mIHC ROI weighted averages (red line) are shown. (D) Percent of data from vTMA sample iterations that falls within 20% of the reference mean (vTMA mean, blue line in panel C) for 1–18 vTMA cores for 3 cell types and 5 patients. A cutoff of 75% of vTMA derived data falling within 20% of the reference mean was chosen as a relative confidence measure and is highlighted with a black dotted line. (E) Number of vTMA cores required to achieve 75% confidence level described in panel D for indicated cell types.
To capture intratumoral heterogeneity, it is standard practice to sample individual tumors using multiple TMA cores. Thus, we next investigated the effect of vTMA core number on the precision of leukocyte density estimates. To do this, we subsampled combinations of 1–18 vTMA cores from annotated T ROIs from each of five patient specimens 100 times each to generate a distribution of cellular abundance for CD3+ T cell, CD20+ B cell, and CD68+ monocytes/macrophages with ‘N’ number of vTMA cores (Figure 5C, S5G). As the number of vTMA cores increased, average cell abundance converged towards the average of total vTMA cores per patient (Figure 5C, S5G). To estimate how the number of vTMA cores reflected the average leukocyte abundance within the larger sample, we calculated the percent of vTMA subsamples that fell within 20% of the reference mean (average of all vTMA cores per specimen). To reach 75% probability of sampling within 20% of the reference mean, 10 or more vTMA cores were needed for the three cell populations evaluated (CD3+ T cells: 10–15 cores; CD20+ B cells: greater than 13 cores; CD68+ monocytes/macrophages: 11–14 cores) (Figure 5D–E). Although we evaluated only a small subset of patients and several T region ROIs, as opposed to the entire tumor area on a slide, this finding is consistent with analyses of similarly-sized TMA cores from different solid tumor types that concluded multiple TMA cores provide enhanced representation of immune contexture when compared to a larger tissue area or whole slide image (34,35).
Long-term and short-term survivors differ in ratio of T cells to CD68+ cells
Since unsupervised clustering of treatment-naive PDACs by global and intratumoral leukocyte profiles did not reveal associations between immune phenotype and OS (Figure 2D, Figure 4D), we hypothesized that evaluating the subset of patients with exceptionally poor versus exceptionally favorable OS might more clearly reveal distinct immune landscapes associated with clinical outcome. We thus identified patients as long-term and short-term survivors by stratifying treatment-naïve cases into quartiles and evaluating the 1st quartile (short-term survivors; median OS 9 months) and 4th quartile (long-term survivors; median OS 58 months) for leukocyte complexity (Figure 6A). Leukocyte subpopulation densities were similar in long-term and short-term survivors, with the exception of CD163− monocyte/macrophage density, which was significantly lower in PDACs from long-term survivors (Figure 6B, Figure S6A). Importantly, the ratio of CD8+ T cells to CD68+ monocytes/macrophages was significantly greater in T regions and modestly increased in TAS of long-term survivors (Figure 6C, Figure S6B).
Figure 6. CD8+ T cell to CD68+ cell ratios correlate with clinical outcome.
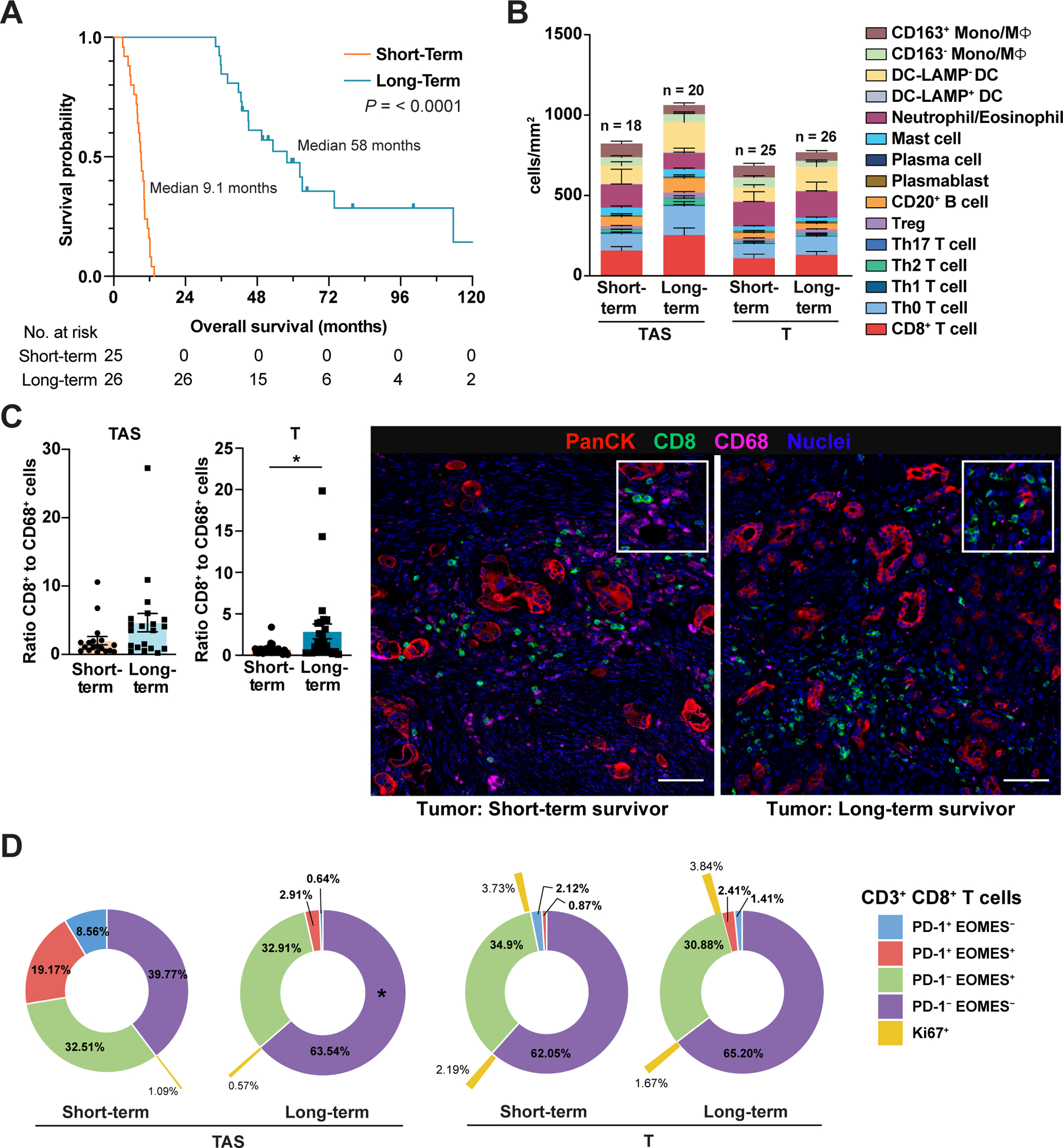
(A) Kaplan-Meier curves displaying OS of treatment-naïve short-term (1st quartile OS time) and long-term (4th quartile OS time) survivors from Cohorts 1 and 2. P-value determined by log-rank test. (B) Leukocyte composition in T and patient-matched TAS from short-term and long-term survivors. Data are represented as mean ± SEM. (C) Ratio of CD8+ T cells to total CD68+ Mono/MΦ in T (short-term survivor, n = 25; long-term survivor, n = 26) and patient-matched TAS (short-term survivor, n = 18; long-term survivor, n = 20) with corresponding pseudocolored images from representative tumor areas. Each data point represents a single patient. Statistical differences were determined by Mann-Whitney U test. Scale bars, 50 μm. Boxed insets show higher magnification. (D) Sunburst plots of patients shown in panels ‘A-C’ depicting average frequency of CD3+CD8+ T cells within T and TAS exhibiting PD-1 and/or EOMES expression. Percentage of PD-1/EOMES subpopulations positive for Ki67 are indicated in outermost ring (yellow). Statistical differences in T cell subpopulations in short-term versus long-term survivors were evaluated by a mixed-effects model with Sidak’s multiple comparisons test.
When measured as a frequency of total CD3+CD8− T cells, Th1 cells were significantly enriched in TAS of long-term survivors, but not within intratumoral regions (Figure S6C). B cell subsets were similar in both long-term and short-term survivors (Figure S6D). Interrogation of T cell functional status revealed no differences in cells singly positive for biomarkers indicative of activation (PD-1, ICOS), cytotoxicity (GzmB), or proliferation (Ki67) in long-term versus short-term survivors (Figure S6E). That said, T cells exist in tissues within a continuum of differentiation/activation/exhaustion states, thus we also evaluated CD8+ T cells for combined expression of PD-1 and EOMES to further inform on T cell functionality (36). PD-1 expression indicates T cell activation in response to antigen, while EOMES is expressed by effector cells and is required for conventional memory differentiation. Combined assessment of PD-1 and EOMES expression revealed no differences in intratumoral regions but an increased PD-1−EOMES− fraction in TAS of long-term survivors, likely representing a heterogeneous pool of naïve and effector cells (Figure 6D).
Presurgical cytotoxic therapy does not relieve indicators of intratumoral T cell dysfunction
Only 15% of PDAC patients are eligible for surgical resection at time of diagnosis (37), and treatment with chemotherapy and/or radiotherapy is one strategy for down-staging locally advanced or borderline resectable PDACs to resectable tumors. It is not yet fully understood how cytotoxic therapies impact the PDAC TiME. We thus investigated how presurgical treatment with chemo- and/or radiotherapy may impact leukocyte composition, spatial/regional distributions, and T cell functional status. Unsupervised hierarchical clustering of presurgically-treated PDACs revealed lymphoid-enriched, myeloid-enriched, and mixed tumors (Figure 7A), similar to results from treatment-naïve PDACs (Figure 4D). In contrast to treatment-naïve patients, survival analysis of presurgically-treated patients based on tumor clusters revealed a significant survival advantage for patients with lymphoid-enriched versus myeloid-enriched PDACs, although sample size was modest (Figure 7B). Moreover, presurgically-treated patients with highest intratumoral CD8+ T cell density, or high CD8+ T cell to CD68+ cell ratio trended toward enhanced OS (Figure S7A), analogous to our previous findings in PDAC patients treated with neoadjuvant GVAX (8) or GVAX/Cy/CRS-207 (16).
Figure 7. Presurgical therapy shapes immune contexture but does not relieve T cell dysfunction in primary PDAC.
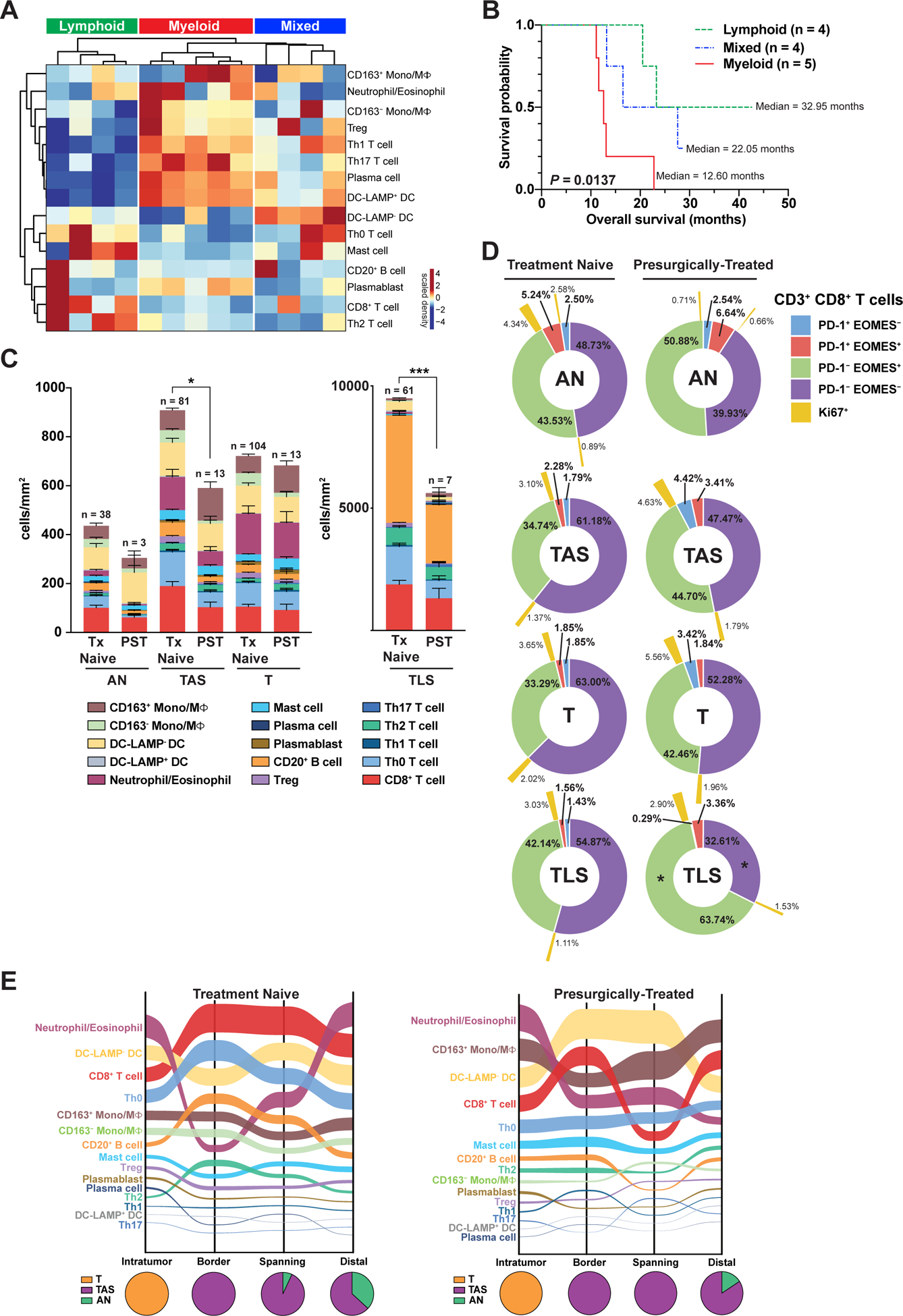
(A) Unsupervised clustering of ‘T’ regions of PDACs from patients who received chemotherapy and/or radiotherapy prior to surgical resection (n = 13; columns), showing relative intratumoral enrichment of indicated leukocyte subtypes (rows). (B) Kaplan-Meier curve of OS based on clusters in ‘A’ with ‘n’ indicating the number of individual PDACs per cluster. P-value determined by log-rank test. (C) Immune composition of indicated histopathologic regions (AN, TAS, T, TLS) from treatment-naïve and presurgically-treated (PST) specimens. Data presented as mean ± SEM. Differences in total leukocyte density in a given region type in treatment-naïve versus presurgically-treated PDAC determined by Mann-Whitney U test. (D) Sunburst plots depicting average frequency of CD3+CD8+ T cells exhibiting PD-1 and/or EOMES positivity in indicated histopathologic regions (AN, TAS, T, TLS) of the treatment-naïve (left) and presurgically-treated (right) PDACs depicted in ‘C’. Percentage of PD-1/EOMES subpopulations positive for Ki67 indicated in outermost ring (yellow). Statistical differences in T cell subpopulations comparing treatment-naïve and treated cases determined by a mixed-effects model with Sidak correction. (E) Sankey flow diagrams of treatment-naïve and presurgically-treated PDACs representing indicated leukocyte populations sorted on the Y-axis from highest (top) to lowest (bottom) cell density, where line width is scaled to cell density across four spatial categories. Pie charts below represent relative regional contribution of different PDAC histopathologic compartments (T, TAS, AN) within each spatial category. Number of individual ROIs evaluated in this analysis is summarized in Table S6.
To gain additional insight into how therapy potentially alters PDAC leukocyte composition, we evaluated leukocyte density and spatial distribution in presurgically-treated samples compared to unmatched treatment-naïve samples. While unmatched samples and small sample size precluded our ability to directly determine causal relationships between therapy and immune contexture, we observed several notable differences based on treatment status. Total leukocyte density was significantly reduced in TAS and TLS of presurgically-treated samples as compared to treatment-naïve, though similar leukocyte composition was observed within histopathologic regions (Figure 7C). To determine if presurgical therapy may have impacted T cell functional profiles, we evaluated CD3+CD8+ and CD3+CD8− T helper cells for expression of activation and effector biomarkers (Figure S7B–C). No differences in GzmB or ICOS were identified in T cells from treatment-naïve versus presurgically-treated PDACs, though increased frequency of PD-1+ CD8+ T cells and T helper cells was evident in TAS regions of presurgically-treated PDACs. T helper cell proliferation was also significantly elevated in TAS and T regions of pre-surgically treated versus treatment-naïve PDACs (Figure S7B–C), but this was not attributable to increases in any specific T helper subpopulation (Figure S7D). In addition, we evaluated CD8+ T cells for co-expression of PD-1 and EOMES; PD-1−EOMES− CD8+ T cells represented the majority of CD8+ T cells in AN, TAS, and T regions in both groups (Figure 7D). Within TLS, a significant decrease in PD-1−EOMES− cells and increase in PD-1−EOMES+ cells were observed in treated specimens as compared to treatment-naïve, indicating potential T cell skewing towards a late effector/memory phenotype following therapy; however, this change was not accompanied by changes in proliferation (Figure 7D).
To investigate whether therapy influences spatial distributions of leukocytes, we again subclassified individual ROIs into “border”, “spanning border-distal”, or “distal” spatial categories based on proximity to nearest area of invasive carcinoma, as in Figure 2. Because high leukocyte density in TLS substantially skewed spatial maps to reflect high B and T cell densities, we performed spatial analysis with (Figure S7E) and without (Figure 7E) TLS data to better appreciate their contribution to the PDAC TiME. Spatial maps omitting TLS revealed that densities of many leukocyte subpopulations were relatively constant across spatial categories independent of therapy. Spatial dynamics were most pronounced in neutrophils/eosinophils, DC-LAMP− DCs, and CD8+ T cells. CD8+ T cells were present at their highest density outside of tumor regions in treated and naïve tumors, while neutrophils/eosinophils were the most abundant population within intratumoral areas in both groups (Figure 7E). In contrast to treatment-naïve specimens, the second most abundant intratumoral leukocyte population in presurgically-treated PDACs was CD163+ monocytes/macrophages, perhaps correlating with their protumoral and T cell suppressive phenotype potentially elicited in response to cytotoxic therapy, and consistent with other reports revealing increased protumoral macrophage recruitment following neoadjuvant chemotherapy (38,39). Inclusion of TLS regions to these analyses revealed that CD20+ B cell density shifted to represent the most abundant leukocyte subtype in several spatial categories, particularly in border regions where TLS comprised approximately 50% of analyzed ROIs in both groups (Figure S7E). Collectively, spatial mapping revealed differences in treatment-naïve and treated PDACs, namely presurgically-treated tumors exhibited higher relative density of putatively immunosuppressive monocytes/macrophages than treatment-naïve tumors.
In addition to potential impact of therapy on the PDAC TiME, we hypothesized that leukocyte composition might also evolve with disease progression from primary tumor to distant metastases. We thus applied the mIHC antibody panels to a small independent cohort (Figure 1A, Cohort 3) of primary PDACs and PDAC metastases from unmatched patients. Unsupervised clustering of primary and metastatic tumors revealed that the majority of distant metastases derived from multiple anatomic sites clustered with other metastases as opposed to clustering with primary PDACs (Figure S7F), supporting the concept that immune contexture varies by disease site and highlighting this as an important consideration for applying immunotherapies.
DISCUSSION
A highly immunosuppressive TiME is thought to represent a major obstacle to effective PDAC therapy (40,41), but extent of immune heterogeneity and spatial distribution of leukocytes within human PDAC has not been fully investigated. In this study, we leveraged a quantitative mIHC approach to interrogate density, complexity, and spatial relationships of myeloid and lymphoid lineages, as well as functional and/or differentiation status of leukocyte subpopulations in multiple histopathological regions of a multi-institutional cohort of primary PDACs, consisting of 113 treatment-naïve primary PDACs, 13 presurgically-treated primary PDACs, and 9 PDAC distant metastases. Our results revealed that total leukocyte abundance and leukocyte subpopulation densities varied considerably across distinct histopathologic regions within single tumor resection specimens and also varied across patients.
Consistent with previous observations (12,14,41), CD8+ T cells and T helper cells together represented approximately one-third of all intratumoral leukocytes in treatment-naïve PDACs, with even higher densities found within stroma adjacent to invasive epithelium. A diverse assemblage of myeloid cells was present within tumor and adjacent stroma, with neutrophils/eosinophils being the predominant myeloid cell types in both, followed by CD68+ monocytes/macrophages, and DCs. Neutrophils, monocytes, and macrophages all possess potent T cell suppressive activities (42), consistent with data herein revealing paucity of T cells expressing indicators of activation, proliferation, or cytotoxicity across histopathologic regions. These observations support the assertion that, although human PDACs are not uniformly devoid of T cells, the myeloid microenvironment of PDAC is likely T cell suppressive, a point further bolstered by our observation that the majority of CD8+ T cells were PD-1−EOMES− or PD-1−EOMES+. PD-1 is thought to play a critical role in regulating entry of T cells into a differentiation program following antigen experience and T cell receptor activation (43,44), with PD-1+ T cells generally representing the target population for anti-PD-1 therapies (45). Thus, poor responses to PD-1 targeted immune therapies in PDAC observed thus far may be partly explained by the majority of CD8+ T cells lacking expression of PD-1 and/or the highly T cell suppressive TiME provided by myeloid subsets. These findings collectively support the notion that therapies aimed at relieving myeloid-based suppressive programs should be investigated.
In addition to high densities of putatively immunosuppressive myeloid subtypes, we also observed considerable abundance of DC-LAMP− DCs across histopathologic regions; these likely represent functionally immature cells with reduced capacity for T cell priming. However, cell surface markers evaluated herein could not further sub-stratify DCs into monocyte-derived, conventional, or plasmacytoid DC subtypes. Moreover, it is worth noting that distinct myeloid subpopulations share expression of many cell surface markers, making definitive identification of subsets challenging, even when applying multiple biomarkers. Just as CD68+ cells may be comprised of both monocytes and macrophages, the CD68−HLA+ DC-LAMP− cells identified as immature DCs could also reflect a fraction of monocytes. Despite the inability to fully resolve the complex identities and functional phenotypes of these populations, it is clear that as a whole, the treatment-naïve PDAC TiME is generally enriched in myeloid cell types commonly associated with T cell suppression and/or lack of T cell activating capability.
Importantly, we observed substantial intrapatient leukocyte heterogeneity within tumor ROIs, an essential aspect to consider when conducting TiME studies. By utilizing a vTMA approach, we found that greater numbers of vTMA cores led to a better representation of the average leukocyte abundance in larger ROIs. We also noted that analysis of more tissue area is especially important for accurate estimation of the abundance of low frequency populations, such as B cells, in our samples. In fact, highly variable B cell abundance across vTMA cores was evident in several cases where data appeared as segregated groups of averages, suggestive of clustered B cell organization across vTMAs, even though overt TLS structures were not present in vTMA ROIs. Altogether, the multiple approaches we took to describe inter- and intra-patient heterogeneity within the current study highlight the importance of histologic location and tissue area selection in bulk tumors for estimating abundance of multiple lineages of tumor-infiltrating leukocytes. Additional studies that continue to evaluate spatial distribution of cells across various tissue areas are warranted in order to further inform biological interpretation of human tissue analysis.
To interrogate potential impact of standard cytotoxic regimens on the PDAC TiME, we examined immune contexture and T cell functionality in presurgically-treated PDACs. We found that high intratumoral CD8+/CD68+ cell ratios correlated with longer median OS (25.4 months OS as compared to 14.8 months in patients with low CD8+/CD68+ cell ratios). Moreover, presurgically-treated patients whose tumors were lymphoid-enriched had significantly improved survival outcomes. These results are consistent with our prior evaluation of treated PDACs (8,16), thus underscoring the robustness of these metrics in predicting clinical outcome independent of treatment status or modality of presurgical therapy. Interestingly, lymphoid enrichment alone was not strongly correlated with survival in treatment-naïve patients as it was in presurgically-treated patients, thus highlighting this as a feature that could have unique utility as a predictive biomarker in the context of therapy. That said, relative distribution of CD163+ monocytes/macrophages skewed higher within tumor regions of pre-surgically treated specimens on average as compared to therapy-naïve, perhaps indicating the potential for sustained T cell suppression, even in the context of therapy. In agreement with this, we observed limited positivity of PD-1, EOMES, Ki67, and GzmB in CD8+ T cells in presurgically-treated samples. Notably, in TLS of presurgically-treated PDACs, PD-1−EOMES+ late effector/memory cells were the dominant CD8+ T cell population and PD-1+EOMES+ cells were modestly expanded compared to T cells in TLS of treatment-naïve patients, consistent with enhanced frequency of T cell activation events. However, CD8+ T cell proliferation was similarly low in TLS of treatment-naïve and presurgically-treated cohorts, consistent with recent reports revealing that T cell effector differentiation and proliferation can be unlinked (46). Taken together, our data indicate that conventional standard-of-care approaches may not substantially enhance intratumoral T cell activity in PDAC; however, it is important to acknowledge that this presurgically-treated population represents a small number of patients treated with a variety of therapeutic agents (Table S2). Future studies controlling for neoadjuvant therapy type and evaluating matched pre- and post-treatment samples will be needed to delineate drug-specific effects on the TiME.
Tumor cell intrinsic and extrinsic features evolve as tumor progression ensues and underlies the tremendous heterogeneity thought to limit success of cytotoxic, targeted, and immune therapy approaches. It is currently unclear how PDAC immune contexture evolves as tumors acquire additional genomic alterations, or how immune complexity varies in primary tumors versus distant metastases - both of these represent important considerations for patient stratification. Our data revealed modest enrichment in intratumoral neutrophils/eosinophils in PDACs harboring mutations in TP53, but we observed no other overt changes in immune complexity associated with molecular status, indicating perhaps that the TiME of PDACs is established early during neoplastic development. In an exploratory comparison of primary PDACs and unmatched metastases, we found that metastases clustered separately from primary tumors. Although sample number in this comparison was limited, and primary and metastatic tumors were not derived from matched patients, these data indicate that differences in immune complexity likely do exist between primary and metastatic lesions and may be significant for therapeutic responsiveness. Future studies with larger cohorts and paired primary and metastatic specimens are warranted to study these differences in greater detail and should also aim to investigate features of T cell phenotype. Consensus sets of protein biomarkers reflecting T cell functional state are now coming into focus (43,44,46,47); these will undoubtedly reveal novel mechanisms for inducing T cell memory, proliferation, and/or sustaining cytotoxicity, that surely will lead to new therapeutic opportunities.
Collectively, data presented herein provide a comprehensive atlas characterizing regional leukocyte contexture within treatment-naïve and presurgically-treated PDACs that can promote hypothesis generation, serve as a resource for future investigations, inform on the response to therapies when untreated baseline samples are not available, and provide metrics for stratification of patient therapy.
METHODS
Tissue Acquisition
Human PDAC specimens were obtained in accordance with the Declaration of Helsinki and were acquired with written informed consent and institutional review board (IRB) approval from Dana-Farber/Harvard Cancer Center, and the Oregon Pancreas Tissue Registry under Oregon Health & Science University (OHSU) IRB protocol #3609. Additional PDAC archival resection specimens and pre-study biopsies were collected from consented patients enrolled in the multi-center phase 1b PRINCE clinical trial (NCT03214250, sponsored by Parker Institute for Cancer Immunotherapy). Healthy normal pancreas was acquired through organ transplant programs at University of California San Francisco and OHSU. Clinical characteristics are provided in Tables S1–3.
mIHC Staining and Image Acquisition
mIHC was performed on 4–5 μm FFPE sections, as previously described (8,18). Briefly, tissues were fixed with 10% neutral buffered formalin, dehydrated in ethanol, and paraffin embedded using standard protocols. Slides were deparaffinized and stained with hematoxylin (S3301, Dako, Santa Clara, CA), followed by digital whole-slide scanning at 20X magnification on an Aperio AT2 scanner (Leica Biosystems, Wetzlar, Germany). Tissues were then subject to 20 minutes heat-mediated antigen retrieval in pH 6.0 Citra solution (HK080, BioGenex, Fremont, CA), followed by endogenous peroxidase blocking at room temperature (RT) in either 0.6% H2O2 for 20 minutes or Dako Dual Endogenous Enzyme Block (S2003, Dako, Santa Clara, CA) for 10 minutes. Protein blocking was performed for 10 minutes at RT with 5% normal goat serum and 2.5% bovine serum albumin in PBS. Tissues were incubated in primary antibodies for 30–60 minutes at RT, or overnight at 4°C; antibodies and staining conditions are listed in Table S4. Slides were then washed in TBST, and either anti-rat, anti-mouse, or anti-rabbit Histofine Simple Stain MAX PO horseradish peroxidase (HRP)- conjugated polymer (Nichirei Biosciences, Tokyo, Japan) was applied for 30 minutes at RT, followed by signal detection with aminoethyl carbazole (AEC) chromogen (Vector Laboratories, Burlingame, CA). Human tonsil was used in all rounds of mIHC as a staining control. Slides were digitally scanned after each chromogen development, followed by chromogen removal in 100% ethanol. For staining cycles with two rounds of antibody development, enzyme and protein blocking was repeated between rounds after chromogen removal, and tissue sections were taken through all steps described above from primary antibody application through slide scanning. All new staining cycles were started at the heat-mediated antigen retrieval step, in order to strip all antibodies from the previous cycle.
Statistical Analysis
For survival analyses, Kaplan-Meier curves were generated to demonstrate time to event and log-rank test was used to evaluate statistical significance. DFS was defined as time between surgery and disease recurrence. Cases with gross residual disease (R2 margin status) following surgery and cases with unknown date of disease recurrence were excluded from DFS analysis. OS was defined as time between date of surgical resection and date of death from any cause or date of last clinical contact. For presurgically-treated cases, OS was defined as time from start of therapy to date of death from any cause or last clinical contact. To compare leukocyte densities across samples and histopathologic region types, Mann-Whitney U tests, Kruskal-Wallis tests, mixed effects models, and multiple t-tests with adjustment for multiple comparisons were used. Specific tests are indicated in figure legends. Correlations between continuous variables were measured using Spearman rank correlation. P values < 0.05 were considered statistically significant, with * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001, **** P ≤ 0.0001 unless otherwise specified in legend. Statistical analyses were performed using GraphPad Prism 8 (GraphPad Software, San Diego, CA) and SAS 9.4 (SAS Institute, Cary, NC).
Supplementary Material
Significance:
We provide a phenotypic and spatial immune atlas of human PDAC identifying leukocyte composition at steady-state and following standard neoadjuvant therapies. These data have broad utility as a resource that can inform on leukocyte responses to emerging therapies where baseline tissues were not acquired.
Acknowledgements:
The authors thank members of the Coussens and Wolpin-Nowak laboratories for critical feedback, Mr. Justin Tibbitts, Ms. Teresa Beechwood, and Dr. Jacklyn Woods for laboratory management, and Ms. Cathy Love for administrative assistance. The authors thank the Parker Institute for Cancer Immunotherapy PRINCE trial translational team, the OHSU Histopathology Shared Resource, OHSU Surgical Pathology, the OHSU Cancer Registry, and the OHSU Knight Diagnostic Laboratories. The authors also acknowledge and thank all patients who donated tissue samples that made this study possible.
Funding:
The study and analyses were funded by a Stand Up To Cancer – Lustgarten Foundation Pancreatic Cancer Dream Team Translational Research Grant (Grant Number: SU2C-AACR-DT14–14) and the Brenden-Colson Center for Pancreatic Care at OHSU. Stand Up To Cancer is a division of the Entertainment Industry Foundation. The indicated grant is administered by the American Association for Cancer Research. L.M. Coussens acknowledges funding from the National Institutes of Health (1U01 CA224012, U2C CA233280, R01 CA223150, R01 CA226909, R21 HD099367), the Knight Cancer Institute, and the Brenden-Colson Center for Pancreatic Care at OHSU. Development of analytical methods used for image analysis at OHSU were developed and carried out with major support from the National Institutes of Health, National Cancer Institute Human Tumor Atlas Network (HTAN) Research Center (U2C CA233280), and the Prospect Creek Foundation to the OHSU SMMART (Serial Measurement of Molecular and Architectural Responses to Therapy) Program. B.M. Wolpin acknowledges funding from the Hale Family Center for Pancreatic Cancer Research, Lustgarten Foundation Dedicated Laboratory program, NIH grant U01 CA210171, NIH grant P50 CA127003, Stand Up To Cancer, Pancreatic Cancer Action Network, Noble Effort Fund, Wexler Family Fund, Promises for Purple and McCarthy Strong. S.A. Väyrynen is supported by the Finnish Cultural Foundation and Orion Research Foundation sr. RHV receives research funding from Apexigen, Fibrogen, Inovio, Janssen and Lilly. R.C. Sears acknowledges funding from the NIH (1U01 CA224012, U2C CA233280, U54 CA209988, R01 CA196228, and R01 CA186241) and the Brenden-Colson Center for Pancreatic Care at OHSU. This study was also made possible with support from the Oregon Clinical & Translational Research Institute (OCTRI), which is supported by the National Center for Advancing Translational Sciences, National Institutes of Health, through Grant Award Number UL1TR002369.
Footnotes
Conflict of Interests: R.H. Vonderheide reports having received consulting fees or honoraria from Celldex, Lilly, Medimmune, and Verastem. He is an inventor on a licensed patent relating to cancer cellular immunotherapy and receives royalties from Children’s Hospital Boston for a licensed research-only monoclonal antibody. E.M. Jaffee is a paid consultant for Adaptive Biotech, CSTONE, Achilles, DragonFly, and Genocea. She receives funding from Lustgarten Foundation and AduroBiotech and through a licensing agreement between AduroBiotech and JHU has the potential to receive royalties on GVAX. She is the Chief Medical Advisor for Lustgarten and serves on the National Cancer Advisory Board and as an Advisor to the Parker Institute for Cancer Immunotherapy (PICI). B.M. Wolpin reports research funding from Celgene and Eli Lilly and consulting for BioLineRx, Celgene, G1 Therapeutics, and GRAIL. L.M. Coussens is a paid consultant for Cell Signaling Technologies, AbbVie Inc., and Shasqi Inc., received reagent and/or research support from Plexxikon Inc., Pharmacyclics, Inc., Acerta Pharma, LLC, Deciphera Pharmaceuticals, LLC, Genentech, Inc., Roche Glycart AG, Syndax Pharmaceuticals Inc., Innate Pharma, NanoString Technologies, and Cell Signaling Technologies, is a member of the Scientific Advisory Boards of Syndax Pharmaceuticals, Carisma Therapeutics, Zymeworks, Inc, Verseau Therapeutics, Cytomix Therapeutics, Inc., and Kineta Inc, and is a member of the Lustgarten Therapeutics Advisory working group. No potential conflicts of interest were disclosed by the other authors.
Additional detailed methods can be found in the Supplementary Materials and Methods.
REFERENCES
- 1.Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA Cancer J Clin 2021;71(1):7–33 doi 10.3322/caac.21654. [DOI] [PubMed] [Google Scholar]
- 2.Garrido-Laguna I, Hidalgo M. Pancreatic cancer: from state-of-the-art treatments to promising novel therapies. Nat Rev Clin Oncol 2015;12(6):319–34 doi 10.1038/nrclinonc.2015.53. [DOI] [PubMed] [Google Scholar]
- 3.Conroy T, Hammel P, Hebbar M, Ben Abdelghani M, Wei AC, Raoul JL, et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. The New England journal of medicine 2018;379(25):2395–406 doi 10.1056/NEJMoa1809775. [DOI] [PubMed] [Google Scholar]
- 4.Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. The New England journal of medicine 2011;364(19):1817–25 doi 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- 5.Morrison AH, Byrne KT, Vonderheide RH. Immunotherapy and Prevention of Pancreatic Cancer. Trends Cancer 2018;4(6):418–28 doi 10.1016/j.trecan.2018.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017;357(6349):409–13 doi 10.1126/science.aan6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Karamitopoulou E Tumour microenvironment of pancreatic cancer: immune landscape is dictated by molecular and histopathological features. British journal of cancer 2019;121(1):5–14 doi 10.1038/s41416-019-0479-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsujikawa T, Kumar S, Borkar RN, Azimi V, Thibault G, Chang YH, et al. Quantitative Multiplex Immunohistochemistry Reveals Myeloid-Inflamed Tumor-Immune Complexity Associated with Poor Prognosis. Cell Rep 2017;19(1):203–17 doi 10.1016/j.celrep.2017.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin JR, Izar B, Wang S, Yapp C, Mei S, Shah PM, et al. Highly multiplexed immunofluorescence imaging of human tissues and tumors using t-CyCIF and conventional optical microscopes. Elife 2018;7 doi 10.7554/eLife.31657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Keren L, Bosse M, Marquez D, Angoshtari R, Jain S, Varma S, et al. A Structured Tumor-Immune Microenvironment in Triple Negative Breast Cancer Revealed by Multiplexed Ion Beam Imaging. Cell 2018;174(6):1373–87 e19 doi 10.1016/j.cell.2018.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carstens JL, Correa de Sampaio P, Yang D, Barua S, Wang H, Rao A, et al. Spatial computation of intratumoral T cells correlates with survival of patients with pancreatic cancer. Nature communications 2017;8:15095 doi 10.1038/ncomms15095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stromnes IM, Hulbert A, Pierce RH, Greenberg PD, Hingorani SR. T-cell Localization, Activation, and Clonal Expansion in Human Pancreatic Ductal Adenocarcinoma. Cancer Immunol Res 2017;5(11):978–91 doi 10.1158/2326-6066.CIR-16-0322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Masugi Y, Abe T, Ueno A, Fujii-Nishimura Y, Ojima H, Endo Y, et al. Characterization of spatial distribution of tumor-infiltrating CD8(+) T cells refines their prognostic utility for pancreatic cancer survival. Mod Pathol 2019;32(10):1495–507 doi 10.1038/s41379-019-0291-z. [DOI] [PubMed] [Google Scholar]
- 14.Steele NG, Carpenter ES, Kemp SB, Sirihorachai VR, The S, Delrosario L, et al. Multimodal mapping of the tumor and peripheral blood immune landscape in human pancreatic cancer. Nature Cancer 2020;1(11):1097–112 doi 10.1038/s43018-020-00121-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Väyrynen SA, Zhang J, Yuan C, Väyrynen JP, Dias Costa A, Williams H, et al. Composition, Spatial Characteristics, and Prognostic Significance of Myeloid Cell Infiltration in Pancreatic Cancer. Clinical Cancer Research 2021;27(4):1069–81 doi 10.1158/1078-0432.Ccr-20-3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsujikawa T, Crocenzi T, Durham JN, Sugar EA, Wu AA, Onners B, et al. Evaluation of Cyclophosphamide/GVAX Pancreas Followed by Listeria-Mesothelin (CRS-207) with or without Nivolumab in Patients with Pancreatic Cancer. Clinical Cancer Research 2020;26(14):3578–88 doi 10.1158/1078-0432.Ccr-19-3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnson BE, Creason AL, Stommel JM, Keck J, Parmar S, Betts CB, et al. An Integrated Clinical, Omic, and Image Atlas of an Evolving Metastatic Breast Cancer. bioRxiv 2020:2020.12.03.408500 doi 10.1101/2020.12.03.408500. [DOI] [Google Scholar]
- 18.Banik G, Betts CB, Liudahl SM, Sivagnanam S, Kawashima R, Cotechini T, et al. High-dimensional multiplexed immunohistochemical characterization of immune contexture in human cancers. Methods in enzymology 2020;635:1–20 doi 10.1016/bs.mie.2019.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Saint-Vis B, Vincent J, Vandenabeele S, Vanbervliet B, Pin JJ, Ait-Yahia S, et al. A novel lysosome-associated membrane glycoprotein, DC-LAMP, induced upon DC maturation, is transiently expressed in MHC class II compartment. Immunity 1998;9(3):325–36. [DOI] [PubMed] [Google Scholar]
- 20.DeNardo DG, Ruffell B. Macrophages as regulators of tumour immunity and immunotherapy. Nat Rev Immunol 2019;19(6):369–82 doi 10.1038/s41577-019-0127-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kather JN, Suarez-Carmona M, Charoentong P, Weis CA, Hirsch D, Bankhead P, et al. Topography of cancer-associated immune cells in human solid tumors. Elife 2018;7 doi 10.7554/eLife.36967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pages F, Mlecnik B, Marliot F, Bindea G, Ou FS, Bifulco C, et al. International validation of the consensus Immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet 2018;391(10135):2128–39 doi 10.1016/S0140-6736(18)30789-X. [DOI] [PubMed] [Google Scholar]
- 23.Fridman WH, Pages F, Sautes-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer 2012;12(4):298–306 doi 10.1038/nrc3245. [DOI] [PubMed] [Google Scholar]
- 24.Ino Y, Yamazaki-Itoh R, Shimada K, Iwasaki M, Kosuge T, Kanai Y, et al. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. British journal of cancer 2013;108(4):914–23 doi 10.1038/bjc.2013.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Balachandran VP, Luksza M, Zhao JN, Makarov V, Moral JA, Remark R, et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 2017;551(7681):512–6 doi 10.1038/nature24462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lutz ER, Wu AA, Bigelow E, Sharma R, Mo G, Soares K, et al. Immunotherapy converts nonimmunogenic pancreatic tumors into immunogenic foci of immune regulation. Cancer Immunol Res 2014;2(7):616–31 doi 10.1158/2326-6066.CIR-14-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Iacobuzio-Donahue CA. Genetic evolution of pancreatic cancer: lessons learnt from the pancreatic cancer genome sequencing project. Gut 2012;61(7):1085–94 doi 10.1136/gut.2010.236026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sautes-Fridman C, Petitprez F, Calderaro J, Fridman WH. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat Rev Cancer 2019;19(6):307–25 doi 10.1038/s41568-019-0144-6. [DOI] [PubMed] [Google Scholar]
- 29.Hiraoka N, Ino Y, Yamazaki-Itoh R, Kanai Y, Kosuge T, Shimada K. Intratumoral tertiary lymphoid organ is a favourable prognosticator in patients with pancreatic cancer. British journal of cancer 2015;112(11):1782–90 doi 10.1038/bjc.2015.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cabrita R, Lauss M, Sanna A, Donia M, Skaarup Larsen M, Mitra S, et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 2020;577(7791):561–5 doi 10.1038/s41586-019-1914-8. [DOI] [PubMed] [Google Scholar]
- 31.Petitprez F, de Reynies A, Keung EZ, Chen TW, Sun CM, Calderaro J, et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature 2020;577(7791):556–60 doi 10.1038/s41586-019-1906-8. [DOI] [PubMed] [Google Scholar]
- 32.Helmink BA, Reddy SM, Gao J, Zhang S, Basar R, Thakur R, et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 2020;577(7791):549–55 doi 10.1038/s41586-019-1922-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martinet L, Le Guellec S, Filleron T, Lamant L, Meyer N, Rochaix P, et al. High endothelial venules (HEVs) in human melanoma lesions: Major gateways for tumor-infiltrating lymphocytes. Oncoimmunology 2012;1(6):829–39 doi 10.4161/onci.20492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee ATJ, Chew W, Wilding CP, Guljar N, Smith MJ, Strauss DC, et al. The adequacy of tissue microarrays in the assessment of inter- and intra-tumoural heterogeneity of infiltrating lymphocyte burden in leiomyosarcoma. Sci Rep 2019;9(1):14602 doi 10.1038/s41598-019-50888-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gruber ES, Oberhuber G, Pils D, Stork T, Sinn K, Gruber S, et al. The Determination of Immunomodulation and Its Impact on Survival of Rectal Cancer Patients Depends on the Area Comprising a Tissue Microarray. Cancers (Basel) 2020;12(3) doi 10.3390/cancers12030563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol 2015;15(8):486–99 doi 10.1038/nri3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.White RR, Lowy AM. Clinical Management: Resectable Disease. Cancer J 2017;23(6):343–9 doi 10.1097/PPO.0000000000000291. [DOI] [PubMed] [Google Scholar]
- 38.DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov 2011;1(1):54–67 doi 10.1158/2159-8274.CD-10-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parra ER, Villalobos P, Behrens C, Jiang M, Pataer A, Swisher SG, et al. Effect of neoadjuvant chemotherapy on the immune microenvironment in non-small cell lung carcinomas as determined by multiplex immunofluorescence and image analysis approaches. J Immunother Cancer 2018;6(1):48 doi 10.1186/s40425-018-0368-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vonderheide RH. The Immune Revolution: A Case for Priming, Not Checkpoint. Cancer Cell 2018;33(4):563–9 doi 10.1016/j.ccell.2018.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blando J, Sharma A, Higa MG, Zhao H, Vence L, Yadav SS, et al. Comparison of immune infiltrates in melanoma and pancreatic cancer highlights VISTA as a potential target in pancreatic cancer. Proc Natl Acad Sci U S A 2019;116(5):1692–7 doi 10.1073/pnas.1811067116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med 2018;24(5):541–50 doi 10.1038/s41591-018-0014-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Blank CU, Haining WN, Held W, Hogan PG, Kallies A, Lugli E, et al. Defining ‘T cell exhaustion’. Nat Rev Immunol 2019;19(11):665–74 doi 10.1038/s41577-019-0221-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McLane LM, Abdel-Hakeem MS, Wherry EJ. CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annual review of immunology 2019;37:457–95 doi 10.1146/annurev-immunol-041015-055318. [DOI] [PubMed] [Google Scholar]
- 45.Ishida Y PD-1: Its Discovery, Involvement in Cancer Immunotherapy, and Beyond. Cells 2020;9(6) doi 10.3390/cells9061376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Beltra JC, Manne S, Abdel-Hakeem MS, Kurachi M, Giles JR, Chen Z, et al. Developmental Relationships of Four Exhausted CD8(+) T Cell Subsets Reveals Underlying Transcriptional and Epigenetic Landscape Control Mechanisms. Immunity 2020;52(5):825–41 e8 doi 10.1016/j.immuni.2020.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yu P, Lee Y, Liu W, Chin RK, Wang J, Wang Y, et al. Priming of naive T cells inside tumors leads to eradication of established tumors. Nat Immunol 2004;5(2):141–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.