

Abstract
The rapidly acting antidepressants ketamine and scopolamine exert behavioral effects that can last several days to weeks in some patients. The molecular mechanisms underlying the maintenance of these antidepressant effects are unknown. Here, we show that methyl-CpG-binding protein 2 (MeCP2) phosphorylation at Ser421 (pMeCP2) is essential for the sustained, but not the rapid, antidepressant effects of ketamine and scopolamine in mice. Our results reveal that pMeCP2 is downstream of BDNF, a critical factor in ketamine and scopolamine antidepressant action. In addition, we show that pMeCP2 is required for the long-term regulation of synaptic strength following ketamine or scopolamine administration. These results demonstrate that pMeCP2 and associated synaptic plasticity are essential determinants of sustained antidepressant effects.
Main
Major depressive disorder (MDD) is a prevalent mental disorder and a leading cause of morbidity worldwide. Traditional antidepressants are an effective treatment for some patients. However, while these drugs rapidly change monoaminergic transmission, their antidepressant effect typically takes several weeks to emerge1. In contrast, ketamine, a noncompetitive N-methyl-D-aspartate (NMDA) receptor antagonist, exerts rapid and sustained antidepressant effects and represents a new class of antidepressants2, 3. The mood-elevating effects of a single subanesthetic dose of ketamine are observed within a few hours and can be maintained for days, and in some patients for more than a week2, 3. Scopolamine, a muscarinic acetylcholine receptor antagonist, has also been shown to produce an antidepressant effect within one day, with beneficial effects maintained for several days in some patients4, 5. Despite its clinical importance, the mechanism by which the rapid action of ketamine and scopolamine transitions to longer-lasting antidepressant effects is unclear.
Ketamine’s antidepressant effects have been attributed to a range of targets6–8. One well-supported theory posits that it is due to its effects as an NMDA receptor antagonist, resulting in the inactivation of the calcium/calmodulin-dependent eukaryotic elongation factor 2 kinase (eEF2K)6, 9. Inactivation of eEF2K leads to rapid desuppression of protein synthesis, including brain-derived neurotrophic factor (BDNF), which increases the insertion of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors on the postsynaptic membrane and results in a rapid and persistent potentiation of AMPA receptor-mediated Schaffer collateral (SC) to CA1 field excitatory postsynaptic potentials (fEPSPs)6, 9, 10. This ketamine-mediated synaptic potentiation, similar to the antidepressant effects, is dependent on BDNF6, 9. Scopolamine also increases BDNF release and activates its high-affinity receptor, tropomyosin receptor kinase B (TrkB), with recent data showing BDNF is necessary for scopolamine’s antidepressant effects11, 12. This BDNF requirement for the rapid antidepressant action of ketamine and scopolamine suggests an initial point of convergence that can be triggered by different mechanisms, such as blockade of the NMDA receptor or muscarinic acetylcholine receptor antagonism.
Ketamine and scopolamine have rapid effects on neuronal function, BDNF expression, and behavior6, 9, 11, 13; however, it is unclear how these drugs give rise to long-lasting changes that persist for days or weeks. Transcription-dependent mechanisms play a critical role in persistent synaptic and behavioral changes14. Methyl-CpG-binding protein 2 (MeCP2), cAMP response element-binding protein (CREB), and myocyte enhancer factor 2C (MEF2C) are three well-documented transcription factors that transduce alterations in synaptic activity and Ca2+ signaling into sustained changes in synaptic efficacy. MeCP2 is a transcriptional regulator originally identified as a protein that binds to methylated CpG sites and interacts with other transcriptional complexes15–17. CREB is a transcription activator that regulates transcriptional responses to Ca2+ influx by changes in neural activity18, 19 and plays a critical role in long-term memory and synaptic plasticity20. Existing literature shows that in response to BDNF upregulation as well as neuronal activation, MeCP2 is activated by phosphorylation at S421 and CREB by phosphorylation at S13317-19, 21. MEF2C is a critical regulator of the number of spine and synapse and synaptic function and is responsive to long-term changes in activity22, 23. In this study, we examined whether the sustained effects of ketamine and scopolamine are dependent on these factors.
Results
Ketamine’s sustained antidepressant effects require pMeCP2
Ketamine exerts antidepressant-like effects within 30 min that can persist for more than 7 days in mice6, 24, similar to the long-lasting antidepressant effects seen in some patients3. In particular, ketamine is known to reduce immobility in the forced swim test (FST) up to 7 days following drug treatment, and the FST has been used in numerous studies to assess ketamine’s mechanism of action6, 24–26. While the rapid antidepressant effects are translation-dependent6, we surmised that the sustained antidepressant effects may involve transcription mechanisms. To test this premise, we focused on three key transcription factors, MeCP2, CREB, and MEF2C, to investigate whether they are involved in the sustained antidepressant effects of ketamine in mice. To test whether ketamine activates MeCP2 or CREB, we quantified phosphorylation of MeCP2 (S421, pMeCP2) and CREB (S133, pCREB) in the hippocampus at 30 min (when the ketamine’s rapid effect in the FST can be detected6) and 7 days after subanesthetic ketamine administration. Additionally, we examined BDNF protein levels; in agreement with previous work6, BDNF levels were increased 30 min after ketamine treatment but by 7 days had returned to baseline levels (Fig. 1a,b). In contrast, pMeCP2 levels were unchanged 30 min after acute ketamine treatment but increased at 7 days, with no change in total MeCP2 expression (Fig. 1a,b).
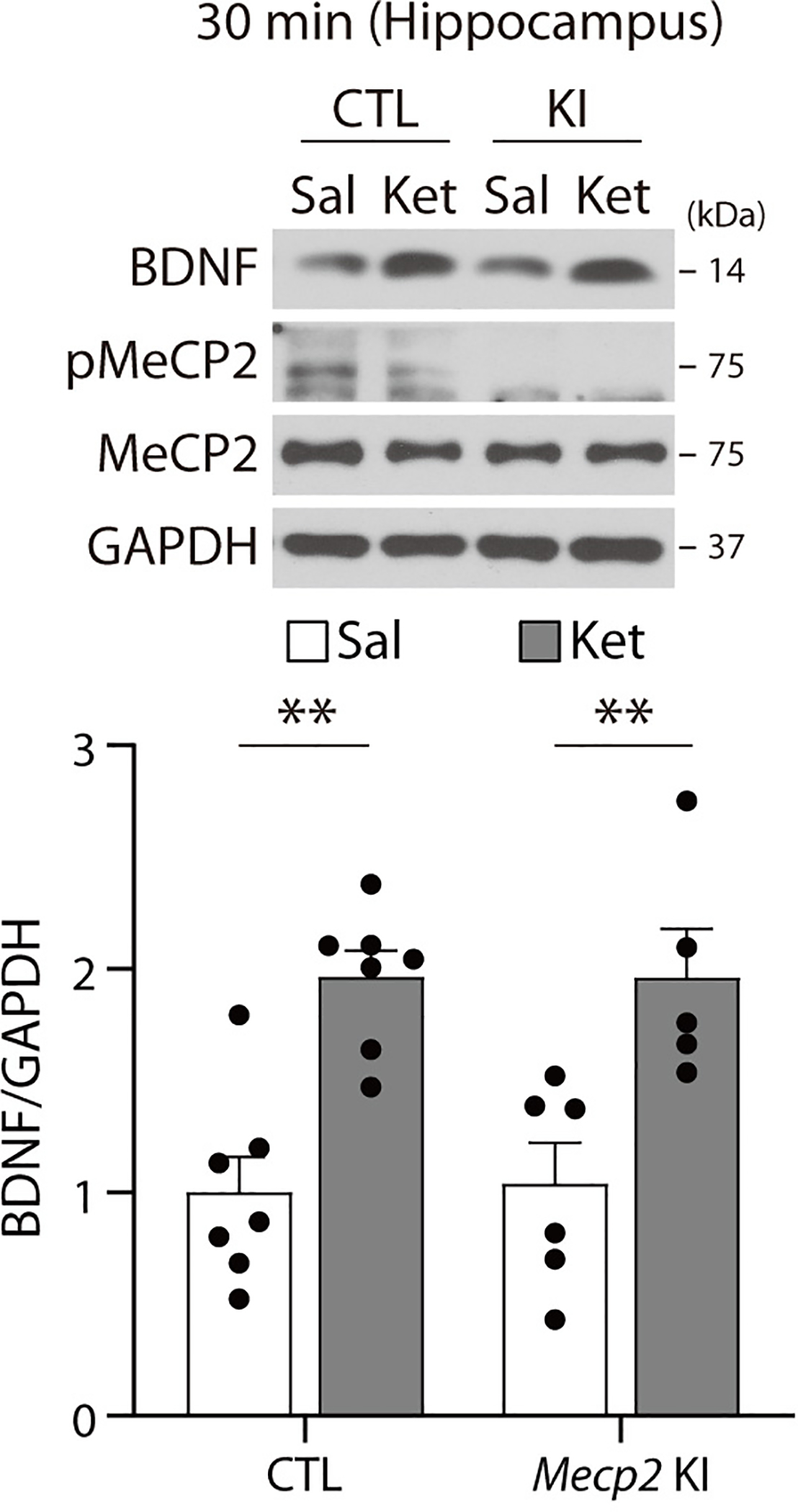
Fig. 1. BDNF-dependent pMeCP2 is essential for sustained antidepressant response of ketamine.
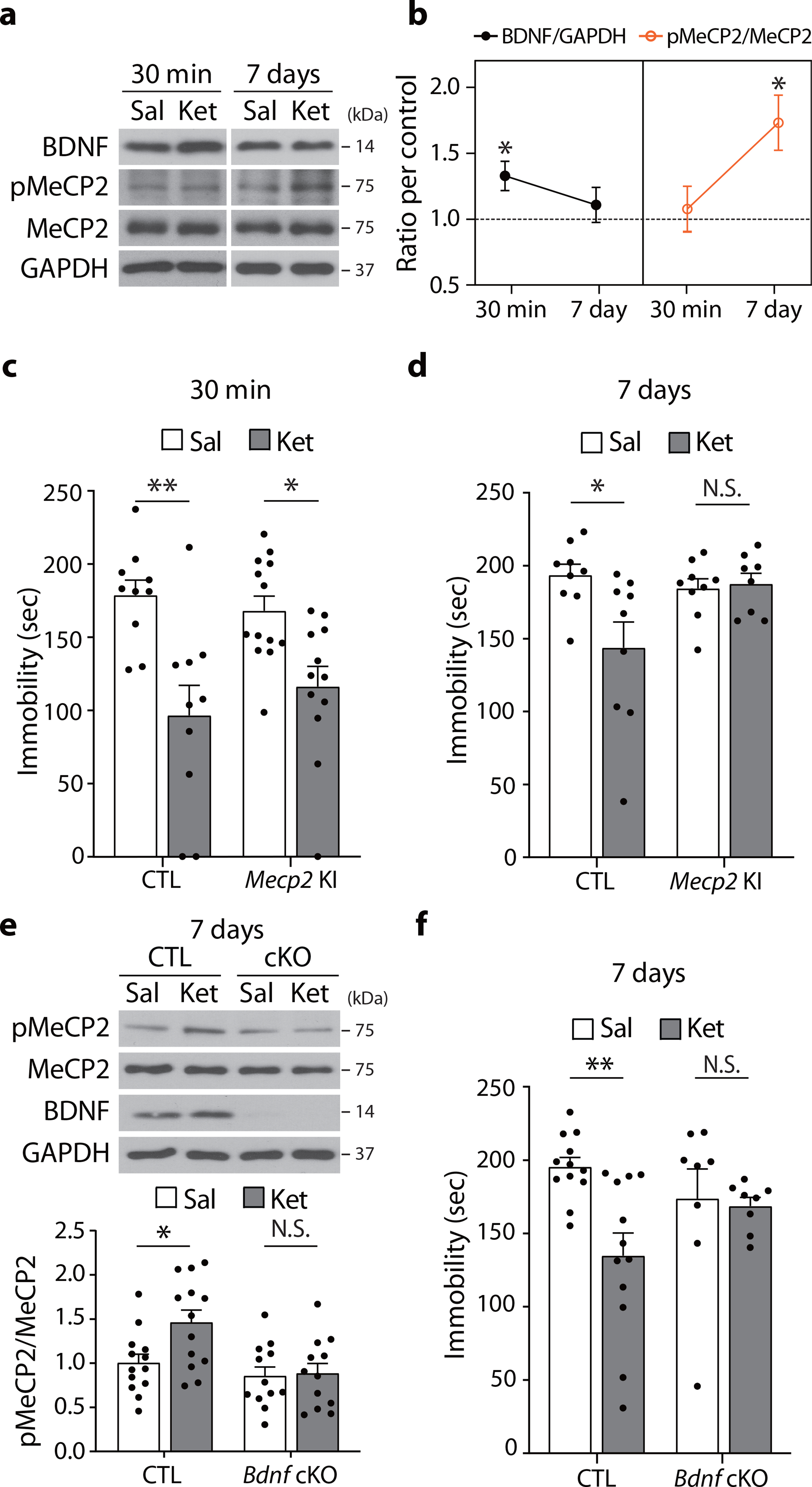
a,b, BDNF and pMeCP2 levels were measured in the hippocampus of C57BL/6J mice 30 min and 7 days after ketamine treatment. BDNF levels were increased at 30 min and returned to baseline at 7 days after treatment (30 min - P = 0.0189, Sal, Ket: n = 8, 7 mice, 7 days - P = 0.5872, Sal, Ket: n = 9, 9 mice). pMeCP2 levels were increased 7 days but not 30 min after injection (30 min - P = 0.7126, Sal, Ket: n = 8, 8 mice, 7 days - Sal, Ket: n = 9, 11 mice). c, In the FST, Mecp2 CTL and KI mice showed a significant reduction in time spent immobile 30 min after ketamine, compared to the respective saline-treated control groups (Genotype x Drug: P = 0.2875, Drug: P < 0.0001, CTL -Sal, CTL -Ket, KI-Sal, KI-Ket: n = 10, 10, 13, 12 mice). d, In the FST, Mecp2 CTL, but not KI, mice showed a significant reduction in time spent immobile 7 days after ketamine compared to the respective saline-treated control groups (Genotype x Drug: P = 0.0222, CTL-Sal, CTL-Ket, KI-Sal, KI-Ket: n = 9, 9, 9, 8 mice). e, Western blot analysis of hippocampi from Bdnf cKO and CTL mice 7 days after ketamine treatment. Ketamine increased pMeCP2 levels in the CTL but not in cKO mice (Genotype x Drug: P = 0.0465, CTL-Sal, CTL-Ket, cKO-Sal, cKO-Ket: n = 13, 13, 11, 12 mice). f, In the FST, Ketamine reduced time spent immobile in CTL mice but not in Bdnf cKO mice 7 days after injection (Genotype x Drug: P = 0.0456, CTL-Sal, CTL-Ket, cKO-Sal, cKO-Ket: n = 12, 12, 8, 8 mice). b: two-sided unpaired t-test. c-f: two-way ANOVA with Tukey’s multiple comparisons. Graphs represent mean ± S.E.M.; N.S.: not significant, *, P < 0.05, **, P < 0.01. Sal: saline, Ket: ketamine, CTL: littermate control. For detailed statistical information, see Supplementary Table 1.
The medial prefrontal cortex (mPFC) has been implicated in mediating the antidepressant action of ketamine7, 25. However, in contrast to the hippocampus, no significant change in pMeCP2 levels was observed in the mPFC 7 days after ketamine treatment (Extended Data Fig.1). This indicates ketamine-mediated changes in pMeCP2 expression are region-specific rather than global.
We next determined whether pMeCP2 is required for ketamine’s sustained antidepressant effects. We used Mecp2 S421A knock-in (Mecp2 KI) mice in which phosphorylation at S421 is ablated by substituting the serine to alanine27. A single subanesthetic dose of ketamine reduced immobility time in the FST at 30 min post-injection in both wild-type littermate controls and Mecp2 KI mice, indicating that the rapid antidepressant-like responses are not pMeCP2-dependent (Fig. 1c). In a separate cohort of mice, ketamine-injected littermate controls also exhibited reduced immobility 7 days post-injection, whereas Mecp2 KI mice did not (Fig. 1d). These data show that pMeCP2 is essential for ketamine’s persistent, but not rapid, antidepressant action.
We next tested whether the ketamine-mediated increase in pMeCP2 levels is dependent on BDNF. We utilized inducible Bdnf conditional KO (Bdnf cKO) mice in which BDNF is selectively deleted in the forebrain28. A single ketamine injection increased hippocampal pMeCP2 levels 7 days later in littermate controls but not in Bdnf cKO mice (Fig. 1e). Conversely, we examined whether the ketamine-induced increase in BDNF levels was dependent on pMeCP2, and observed that Mecp2 S421A KI mice still showed a rapid upregulation of BDNF protein expression 30 min following ketamine (Extended Data Fig. 2). We then examined whether ketamine’s prolonged antidepressant effects require BDNF, and found that, unlike littermate controls, Bdnf cKO mice did not show reduced immobility in the FST 7 days post-ketamine (Fig. 1f).
Next, to validate whether transcriptional mechanisms are indeed essential for ketamine’s long-term effect, we treated C57BL/6J mice with actinomycin D, a transcription inhibitor, 30 min before ketamine injection and assessed their behavior in the FST 7 days later (Fig. 2a). We observed that actinomycin D pretreatment prevented the ketamine-mediated reduction of immobility in the FST at 7 days, validating the premise that transcription-dependent processes mediate the longer-term antidepressant effects of ketamine.
Fig. 2. CREB and MEF2C are not involved in the sustained effects of ketamine.
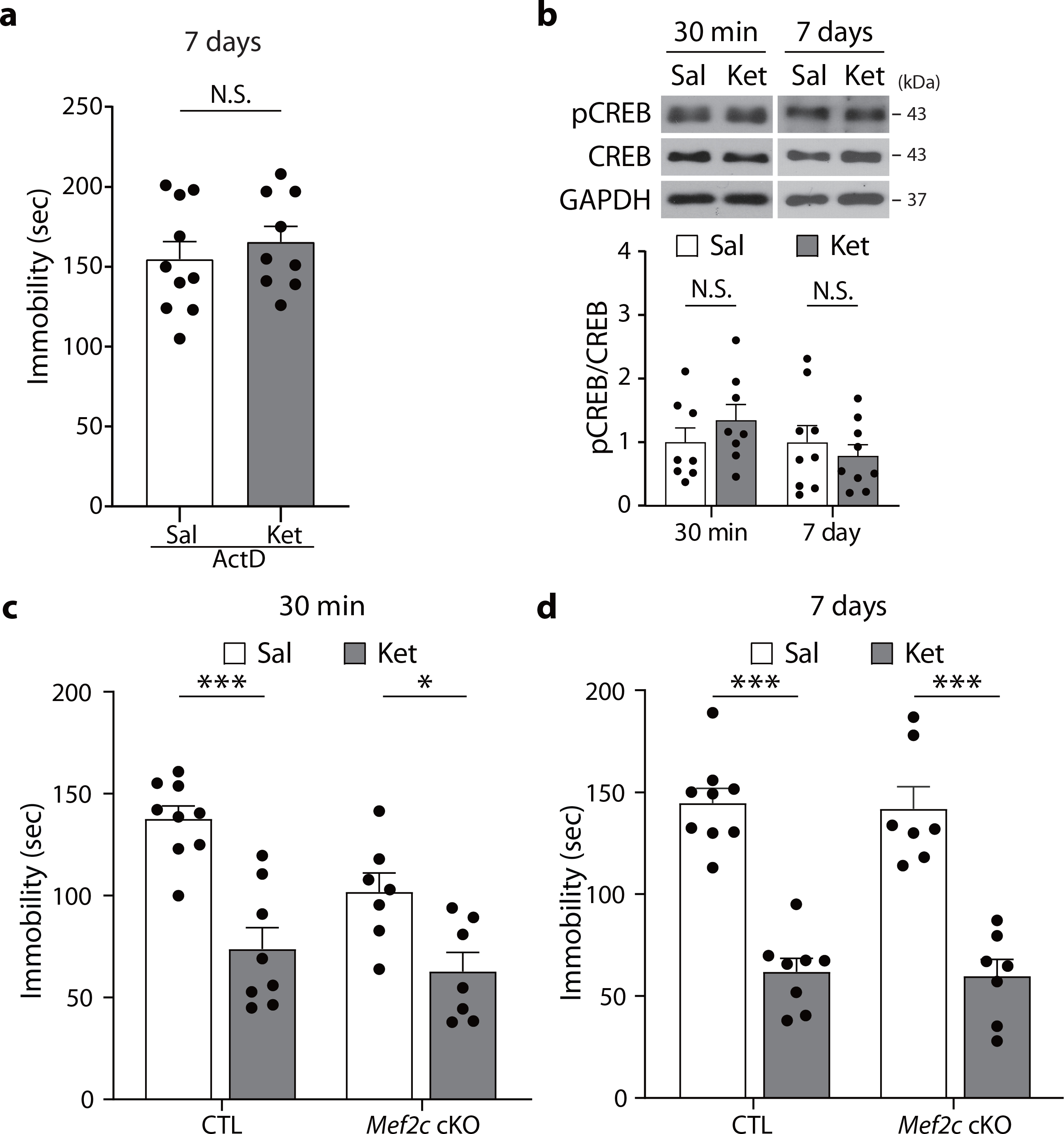
a, Actinomycin D was administered to C57BL/6J 30 min prior to the saline or ketamine injection, and mice were tested 7 days later in the FST. Actinomycin D pretreatment abolished the ketamine-mediated reduction of immobility (P = 0.4826, ActD-Sal, ActD-Ket: n = 10, 9 mice). b, pCREB levels were measured 30 min and 7 days after ketamine treatment with Western blot analysis. No significant differences were observed between saline- and ketamine-treated groups (30 min - P = 0.3165, Sal, Ket: n = 8, 8 mice, 7 days - P = 0.4968, Sal, Ket: n = 9, 9 mice). c,d, Data showing FST with postnatal Mef2c cKO and CTL mice. The same cohorts were tested in the FST 30 min and 7 days after ketamine injection. Ketamine decreased immobility in the FST 30 min (c) and 7 days (d) after injection, in both CTL and cKO mice (30 min - Genotype x Drug: P = 0.1735, Drug: P < 0.0001, 7 days - Genotype x Drug: P = 0.9693, Drug: P < 0.0001, CTL-Sal, CTL-Ket, cKO-Sal, cKO-Ket: n = 9, 8, 7, 7 mice per group). a,b: two-sided unpaired t-test. c,d: two-way ANOVA with Tukey’s multiple comparisons. Graphs represent mean ± S.E.M., N.S.: not significant, *, P < 0.05, ***, P < 0.001. Sal: saline, Ket: ketamine, CTL: littermate control. For detailed statistical information, see Supplementary Table 1.
In contrast, under the same conditions, we did not observe any changes in pCREB or CREB levels following ketamine treatment at either time point (Fig. 2b). To assess whether the transcription factor MEF2C was required for ketamine’s antidepressant effects, we used conditional Mef2c knockout (cKO) mice, in which forebrain Mef2c is deleted postnatally, resulting in a significant increase in the number of excitatory synapses23. A single subanesthetic dose of ketamine decreased immobility in the FST to a similar extent in Mef2c cKO mice and littermate controls at both 30 min and 7 days post-injection, suggesting that MEF2C was not required for ketamine’s rapid or long-term effects (Fig. 2c,d). Collectively, these data demonstrate that pMeCP2 and associated transcriptional mechanisms — but not pCREB or MEF2C — are specifically required for the sustained antidepressant effects of ketamine, likely by acting as a downstream effector of BDNF.
Ketamine induces pMeCP2-dependent metaplasticity
A critical area of study is how to sustain ketamine’s antidepressant response in patients. Electrophysiological studies have shown ketamine rapidly potentiates AMPA receptor-mediated SC–CA1 fEPSPs in the hippocampus of naïve (unstressed) mice; this ketamine-mediated synaptic potentiation has been postulated to be a neural substrate of rapid antidepressant effects6, 9, 29, 30. Therefore, we performed experiments to determine whether this potentiation persists for 7 days post-ketamine injection. We obtained field recordings to assess ketamine’s effects on fEPSPs in the SC–CA1 synapses of hippocampal slices from C57BL/6J mice 7 days after intraperitoneal ketamine injection. There were no significant shifts in input-output curves compared to saline-injected C57BL/6J mice, showing hippocampal synaptic potentiation is not detectable 7 days following acute ketamine (Extended Data Fig. 3a).
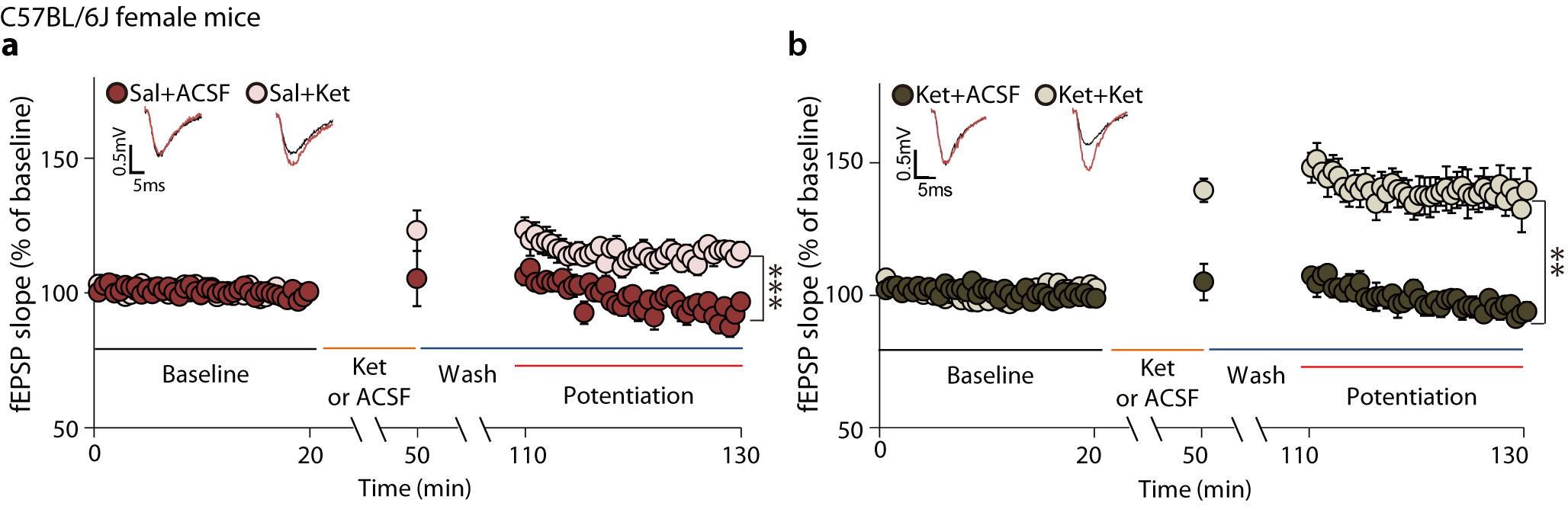
Clinical studies suggest that repeated ketamine infusions may be more effective than a single infusion in patients with MDD31, 32. Reductions in depressive symptoms have been reported to be prolonged in patient responders given repeated infusions33. Since ketamine’s clinical effects may be cumulative31–33, we examined whether previous ketamine exposure impacts the AMPA receptor-mediated synaptic potentiation in hippocampal slices treated with ketamine (Fig. 3a). C57BL/6J mice were injected with ketamine or saline; 7 days later the animals were sacrificed, and ketamine or ACSF was perfused onto hippocampal slices. In slices from mice injected with saline 7 days prior, ketamine perfusion induced rapid synaptic potentiation (Fig. 3b: 116 ± 1.994%). This ketamine-induced synaptic potentiation was > two-fold higher in slices from mice that had received ketamine 7 days earlier (Fig. 3c: 140.7 ± 10.57%, unpaired t-test, Sal-Ket in Fig. 3b vs Ket-Ket in Fig. 3c, P = 0.0266). ACSF perfusion onto hippocampal slices did not produce any significant changes compared to baseline (Sal+ACSF in Fig. 3b and Ket+ACSF in Fig. 3c), indicating that synaptic potentiation only occurs following ketamine perfusion. Taken together, these results suggest that ketamine administration may produce a priming effect, such that subsequent ketamine treatment causes a form of metaplasticity, as indicated by the augmented synaptic potentiation.
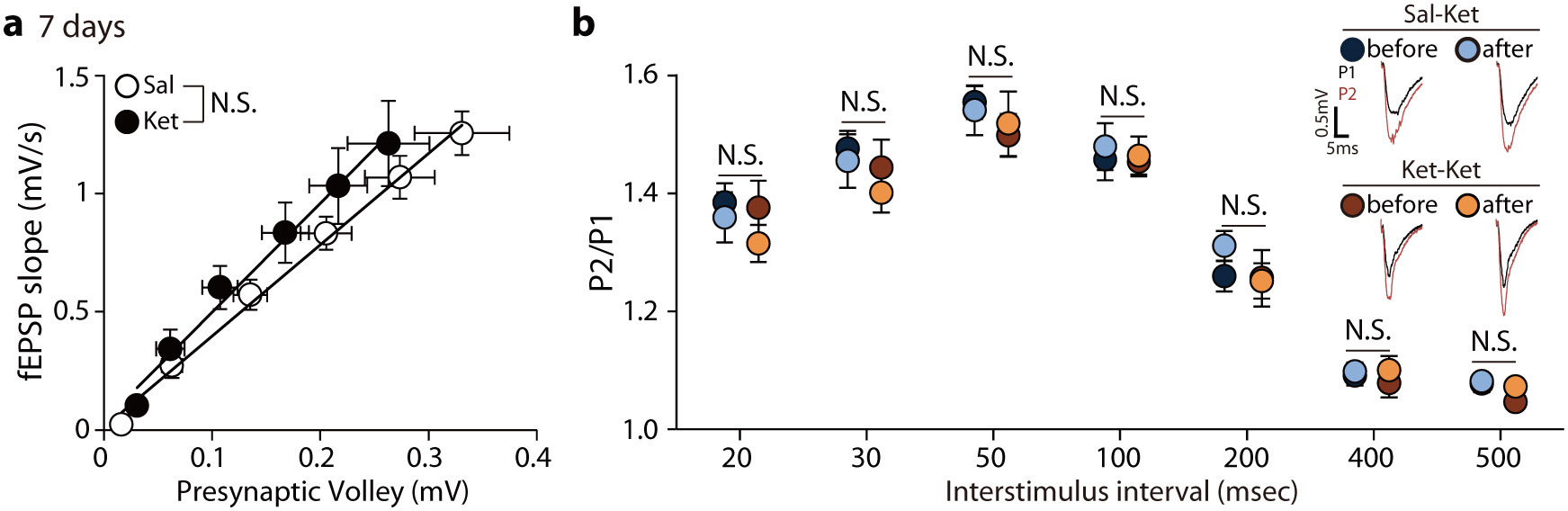
Fig. 3. pMeCP2 is essential for ketamine-induced metaplasticity in SC–CA1 synapses.
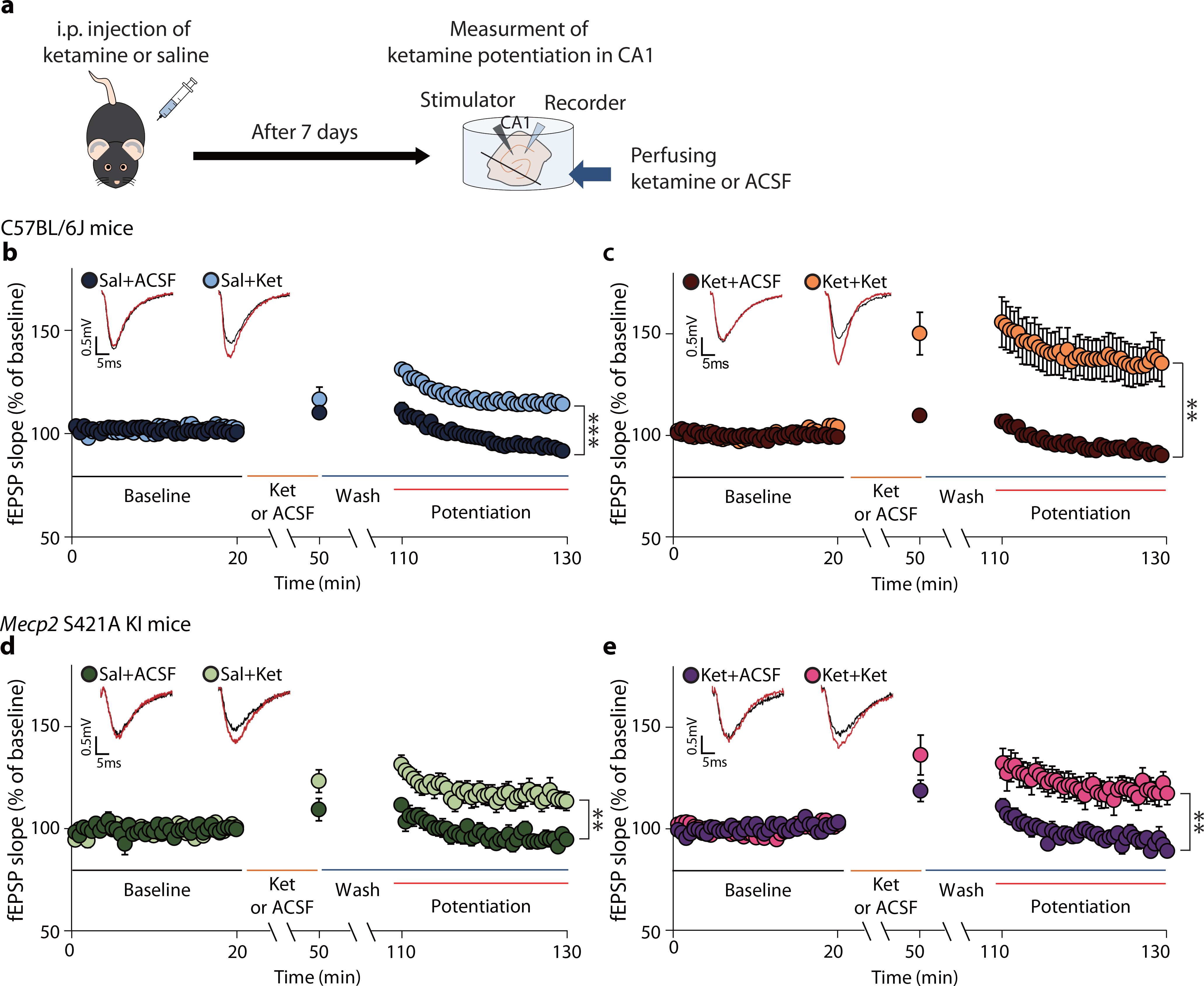
a, The experimental design for measurement of ketamine-potentiation. Mice were treated with saline or ketamine 7 days later hippocampal slices were prepared and fEPSPs recorded in the CA1 region. b,c, Ketamine-potentiation was measured in hippocampal CA1 from C57BL/6J mice previously given either a saline (b) or ketamine (c) injection. Ketamine perfusion induced significant potentiation in both groups, and augmented potentiation was observed in the group previously treated with the ketamine (c: Ket+Ket, 140.7 ± 10.57%), compared to the group previously treated with saline (b: Sal+Ket, 116 ± 1.994%), (two-sided unpaired t-test, b - Sal+ACSF vs Sal+Ket: P < 0.0001, Sal+ACSF, Sal+Ket: n = 6, 11 slices, c - Ket+ACSF vs Ket+Ket: P = 0.0019, Ket+ACSF, Ket+Ket: n = 8, 10 slices, Sal+Ket in b vs Ket+Ket in c: P = 0.0266). d, e, Ketamine-potentiation was measured in the hippocampal CA1 from Mecp2 KI mice previously given either a saline (d) or ketamine injection (e). Ketamine-potentiation was intact in Mecp2 KI mice (d) but the augmented potentiation was not observed in the KI group previously treated with ketamine (e: Ket+Ket, 121.6 ± 6.139%), compared to the group previously treated with saline (d: Sal+Ket, 118.7 ± 4.103%), (two-sided unpaired t-test, d - Sal+ACSF vs Sal+Ket: P = 0.0017, Sal+ACSF, Sal+Ket: n = 7, 7 slices, e - Ket+ACSF vs Ket+Ket:, P = 0.0017, Ket+ACSF, Ket+Ket: n = 8, 9 slices, Sal+Ket in d vs Ket+Ket in e: P = 0.7249). Representative traces are from recorded results during the measurement of baseline and potentiation. Graphs represent mean ± S.E.M., **, P < 0.01, ***, P < 0.001. Ket: ketamine, Sal: saline, Sal+ACSF: ACSF perfusion onto hippocampal slices from saline-injected mice, Sal+Ket: ketamine perfusion onto hippocampal slices from saline-injected mice, Ket+ACSF: ACSF perfusion onto hippocampal slices from ketamine-injected mice, Ket+Ket: ketamine perfusion onto hippocampal slices from ketamine-injected mice. For detailed statistical information, see Supplementary Table 1.
Since the experiments above involved male mice, we next used the same protocol to determine whether the enhanced ketamine-mediated synaptic potentiation also occurs in female C57BL/6J mice. Indeed, slices from female ketamine-injected mice showed higher synaptic potentiation after ketamine perfusion than slices from saline-treated female mice, indicating this metaplasticity is similar in both sexes (Extended Data Fig. 4a,b).
The metaplasticity observed after prior ketamine administration may underlie ketamine’s cumulative effects seen in patients31–33. To better understand what may be driving this plasticity, we assessed paired-pulse ratios (PPRs), an index of presynaptic release probability, by perfusing ketamine onto hippocampal slices of C57BL/6J mice treated 7 days earlier with ketamine or saline. There were no significant changes in PPRs in the SC–CA1 synapses of the ketamine-pretreatment group, compared to the saline-pretreatment group, following ketamine perfusion, suggesting the priming effect that produces the metaplasticity is not presynaptic (Extended Data Fig. 3b).
Our initial experiments showed that the increase in pMeCP2 7 days post-ketamine was required for behavioral effects at that time point (Fig. 1b,d). We, therefore, tested if pMeCP2 is also required for the metaplasticity. Mecp2 KI mice were injected with acute ketamine or saline and sacrificed 7 days later; ketamine or ACSF was then perfused onto hippocampal slices, and synaptic responses were recorded. Saline-treated Mecp2 KI mice had a rapid ketamine-induced synaptic potentiation (Fig. 3d, 118.7 ± 4.103%) similar to littermate controls (Fig. 3b, 116 ± 1.994%), demonstrating that loss of pMeCP2 does not interfere with the induction of this type of synaptic plasticity. However, slices from ketamine-injected Mecp2 KI mice did not show this augmented plasticity, revealing a requirement for pMeCP2 in ketamine-induced metaplasticity (unpaired t-test, Sal-Ket in Fig. 3d: 118.7 ± 4.103% vs Ket-Ket in Fig. 3e: 121.6 ± 6.139%, P = 0.7249). ACSF perfusion did not produce significant synaptic response changes in any of the experiments (Sal+ACSF in Fig. 3d and Ket+ACSF in Fig. 3e). Collectively, these data show that a single ketamine injection increases hippocampal pMeCP2 levels 7 days later, and this is necessary for synaptic priming and metaplasticity.
So far, our data show that ketamine produces a rapid increase in BDNF protein levels at 30 min post-injection that is required for the increase in pMeCP2 levels 7 days later. This increase in pMeCP2 levels is necessary for the ketamine-mediated metaplasticity and the sustained behavioral effects. However, it is unclear which molecular mechanism links the increased BDNF expression to the increase in pMeCP2 following ketamine treatment. Previous studies have reported that Ca2+/calmodulin-dependent protein kinase II (CaMKII) may be an upstream regulator for phosphorylation of MeCP2 induced by BDNF or neural activity changes17, 34. By responding to Ca2+ influx and binding to the Ca2+/calmodulin complex, CaMKII is autophosphorylated at threonine 286/287 residues, which in turn mediates phosphorylation of downstream targets in an activity-dependent manner35. Therefore, we examined phosphorylation of CaMKIIα (T286, pCaMKIIα) and CaMKIIβ (T287, pCaMKIIβ), two major isoforms in the brain36, 3 and 7 days after ketamine treatment in the hippocampal CA1 region. While ketamine did not change pCaMKIIα levels, pCaMKIIβ levels were increased at 3 days post-ketamine and had returned to baseline at 7 days (Extended Data Fig. 5a,b). Increased pMeCP2 levels were observed at 3 and 7 days post-ketamine, whereas BDNF protein levels were rapidly increased 30 min post-ketamine but had returned to baseline by 3 days post-injection (Fig. 1a,b and Extended Data Fig. 5a,b). Collectively, these results suggest that pMeCP2 is not a direct substrate of BDNF-TrkB signaling. Rather, the increased pCaMKIIβ suggests that Ca2+ signaling changes may be an intermediate mechanism to induce pMeCP2 in the hippocampal CA1 region, although further studies will be necessary to test this premise.
Scopolamine’s sustained antidepressant effects require pMeCP2
Scopolamine has antidepressant effects as early as the evening of the administered day (~several hours) and can last in some patients for several days4, 5. Previous work showed that scopolamine increases BDNF release and induces TrkB phosphorylation in the rat prefrontal cortex11. BDNF appears to be required for scopolamine’s effects, as the BDNF Val66Met mutation, which impairs BDNF processing and release37, or infusion of an anti-BDNF neutralizing antibody into the cortex of rats attenuates scopolamine’s antidepressant-related responses11. However, the targets downstream of BDNF involved in mediating the longer-term effects are unknown.
To address this question, we first examined whether a single scopolamine injection induces an antidepressant-like effect in C57BL/6J mice and whether it was sustained for 24 hrs, similarly to patients. Mice were tested in the FST at 8 hrs or (in a separate cohort) 24 hrs post-injection. Scopolamine decreased immobility time at 8 hrs and 24 hrs post-injection compared to vehicle-treated mice (Fig. 4a). We observed that the sustained effects of scopolamine in the FST were maintained at 7 days after injection (Extended Data Fig. 6a). To examine whether this effect was mediated by a transcriptional mechanism, we pretreated C57BL/6J mice with actinomycin D prior to scopolamine and tested them 24 hours later in the FST. Actinomycin D prevented the reduction of immobility by scopolamine (Extended Data Fig. 7), indicating that blocking transcription prevents scopolamine’s antidepressant-like effects, similar to the earlier observation with ketamine.
Fig. 4. BDNF-dependent pMeCP2 mediates the sustained antidepressant response of scopolamine.
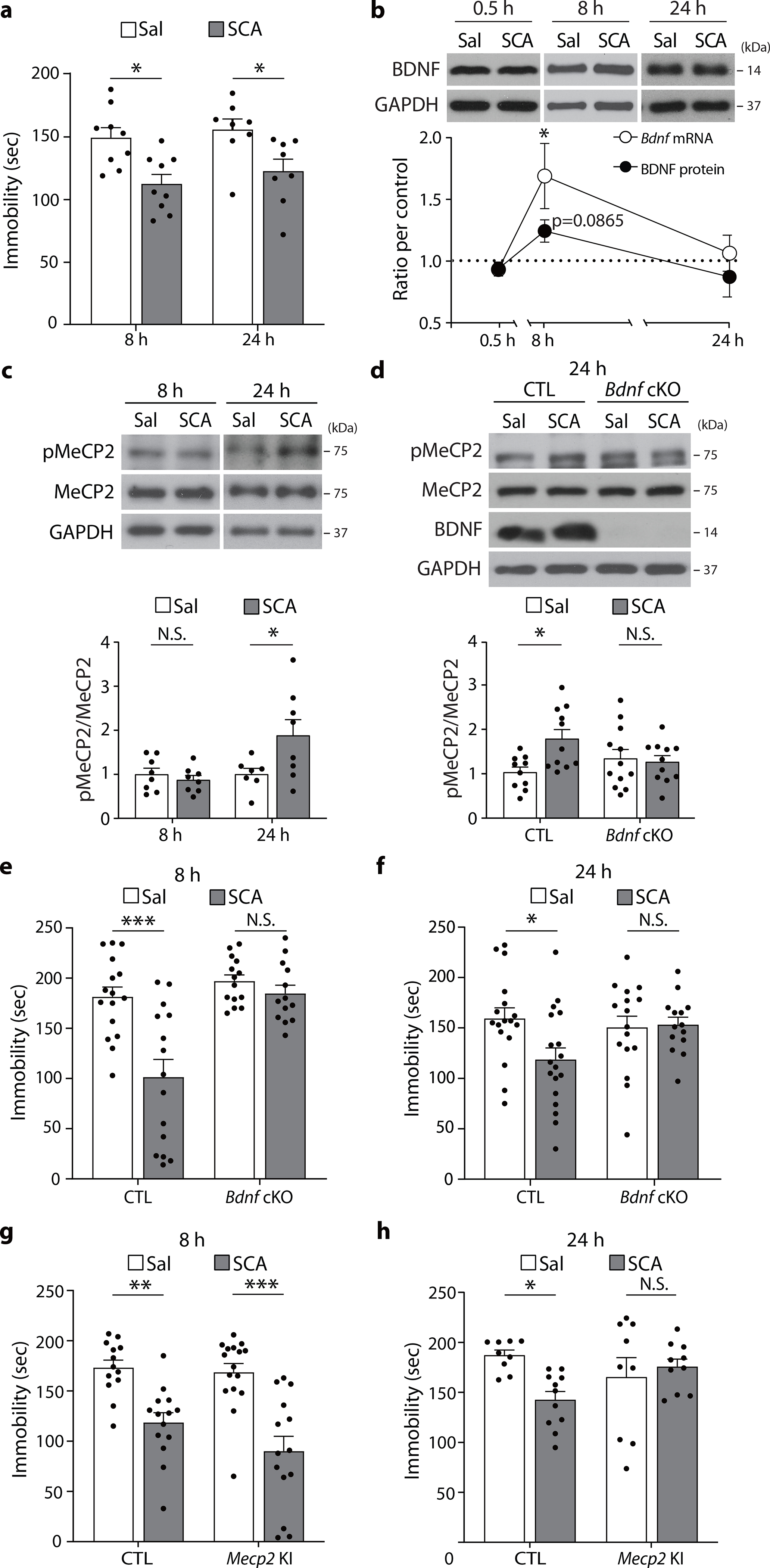
a, Scopolamine decreased time spent immobile in the FST compared to the saline-treated group, both 8 hrs and 24 hrs after treatment (8 hr - Sal, SCA: n = 9, 9 mice, 24 hr - Sal, SCA: n = 8, 8 mice). b, Eight hours after scopolamine treatment, Bdnf mRNA levels were significantly increased (Sal, SCA: n = 7, 7 mice), and BDNF protein levels showed an increased tendency (Sal, SCA: n = 15, 15 mice). Both mRNA and protein levels returned to baseline by 24 hrs (Sal, SCA: n = 8, 8 mice). c, pMeCP2 levels were increased 24 hrs after scopolamine treatment in the hippocampus (8 hrs - Sal, SCA: n = 8, 8 mice, 24 hrs - Sal, SCA: n = 7, 8 mice). d, Scopolamine increased hippocampal pMeCP2 levels in the CTL mice but not in the Bdnf cKO mice 24 hrs after injection (CTL-Sal, CTL-SCA, cKO-Sal, cKO-SCA: n = 10, 12, 11, 11 mice). e, Time spent immobile was reduced in the CTL mice but not in the Bdnf cKO mice 8 hrs after scopolamine treatment (CTL-Sal, CTL-SCA, cKO-Sal, cKO-SCA: n = 16, 14, 15, 13 mice). f, Scopolamine decreased immobility time in the CTL mice but not in the Bdnf cKO mice 8 hrs after treatment (CTL-Sal, CTL-SCA, cKO-Sal, cKO-SCA: n = 17, 18, 16, 14 mice). g, Scopoloamine reduced time spent immobile in both the CTL mice and the Mecp2 KI mice 8 hrs after treatment (CTL-Sal, CTL-SCA, KI-Sal, KI-SCA: n = 13, 16, 14, 14 mice). h, Scopolamine decreased immobility time in the CTL mice but not in the Mecp2 KI mice at 24 hrs after treatment (CTL-Sal, CTL-SCA, KI-Sal, KI-SCA: n = 9, 11, 9, 10 mice). a,d,e-h: two-way ANOVA with Tukey’s multiple comparisons. b,c: two-sided unpaired t-test or Welch’s correction. Graphs represent mean ± S.E.M., N.S.: not significant, *, P < 0.05, **, P < 0.01, ***, P < 0.001. Sal: saline, SCA: scopolamine, CTL: littermate control. For detailed statistical information, see Supplementary Table 1.
We next determined hippocampal BDNF mRNA and protein expression and pMeCP2 levels in separate cohorts of C57BL/6J mice 8 or 24 hrs after scopolamine treatment. Eight hours after a single scopolamine injection, Bdnf mRNA levels were significantly increased with a trend towards an increase in protein expression (Fig. 4b); however, there was no change in pMeCP2 or total MeCP2 levels compared to saline-treated mice (Fig. 4c). BDNF mRNA and protein levels had returned to baseline levels by 24h after scopolamine treatment (Fig. 4b), whereas at that time point there was a significant increase in hippocampal pMeCP2 levels relative to MeCP2 (Fig. 4c). The latter change was hippocampus-specific since no significant change was observed in the mPFC (Extended Data Fig. 8). The scopolamine-mediated increase in phosphorylation levels of MeCP2 did not extend to pCREB, as neither pCREB nor total CREB levels were altered 8 and 24 hours after scopolamine (Extended Data Fig. 9).
Next, we investigated whether BDNF was required for the scopolamine-induced increase in hippocampal pMeCP2 expression. Consistent with our earlier data (Fig. 4c), pMeCP2 levels were increased 24 hours after scopolamine injection in littermate control mice (Fig. 4d). However, there was no such increase in scopolamine-treated Bdnf cKO mice (Fig. 4d). We also quantified Bdnf mRNA levels in Mecp2 KI mice 8 hrs after scopolamine treatment, to examine the involvement of pMeCP2 in regulating Bdnf mRNA at this early time point. We observed a significant increase in Bdnf mRNA levels in both littermate control and Mecp2 KI mice (Extended Data Fig. 10), showing that pMeCP2 is not regulating BDNF expression at this early time point when scopolamine already has a behavioral effect.
To examine the requirement of BDNF and pMeCP2 in the rapid and/or sustained antidepressant action of scopolamine, separate cohorts of Bdnf cKO and Mecp2 KI mice and littermate controls were tested in the FST 8 or 24 hrs after a single scopolamine or saline injection. Scopolamine significantly decreased the immobility time in littermate controls at both 8 hrs and 24 hrs, but not in the Bdnf cKO mice (Fig. 4e,f). Scopolamine did reduce immobility of Mecp2 KI mice at 8 hrs, but not at 24 hrs post-injection (Fig. 4g,h). These results suggest that, similar to ketamine, scopolamine requires BDNF-dependent MeCP2 phosphorylation to induce a sustained antidepressant response.
Scopolamine-induced synaptic plasticity requires pMeCP2
We perfused scopolamine onto hippocampal slices and recorded fEPSPs and PPRs at SC–CA1 synapses to assess synaptic efficacy. A 1 hr perfusion of scopolamine did not elicit detectable changes in fEPSP responses (Fig. 5a) or PPRs (Fig. 5d), presumably due to suppression of cholinergic inputs in acute slices. We, therefore, perfused the acetylcholinesterase inhibitor physostigmine onto hippocampal slices for 20 min and then measured fEPSPs and PPRs in SC–CA1 synapses. Increasing cholinergic tone by application of an acetylcholinesterase inhibitor or a cholinergic receptor agonist induces long-term depression in the SC–CA1 synapses of the hippocampus38, 39, which has been suggested to be involved in the pathophysiology in depression40, 41. Physostigmine application reduced fEPSP responses, and the reduction reached static levels within 20 min and was maintained for at least 1 hr (Fig. 5b), similar to a previous report39. The PPRs were increased at 20 to 100 msec interstimulus interval (Fig. 5e), suggesting that increased cholinergic tone suppresses synaptic responses by decreasing presynaptic release probability.
Fig. 5. Phospho-MeCP2 is essential for sustained changes in presynaptic release probability by scopolamine.
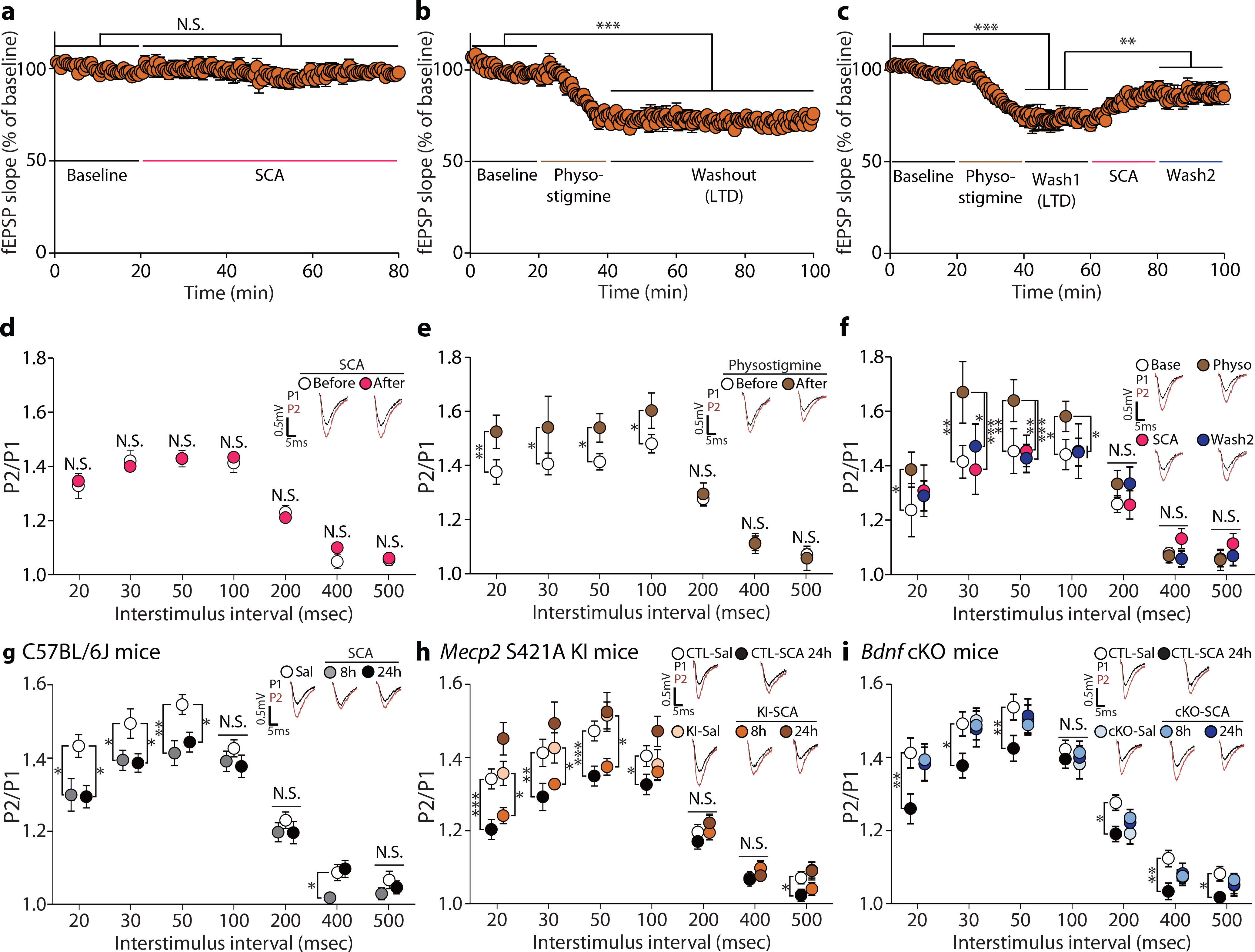
a-f, fEPSPs and PPRs were recorded in SC–CA1 synapses before and after perfusion of scopolamine or physostigmine onto hippocampal slices. Scopolamine application did not affect the fEPSP response (a) and PPRs (d) [All P > 0.05, n = 8 slices]. Physostigmine induced long-term depression for an hour (b, n = 7 slices) and increased PPRs at 20 to 100 msec interstimulus interval (e, n = 7 slices). Scopolamine treatment following physostigmine-induced long-term-depression rescued the depressed fEPSP (c, n = 11 slices) and normalized the increased PPRs at 30 to 100 msec interstimulus interval to control levels (f, n = 11 slices). g, PPRs were significantly decreased at 8 hrs and the reduction was still observed at 24 hrs at 20, 30, and 50 msec interstimulus interval [Sal, SCA (8 h), SCA (24 h): n = 20, 24, 20 slices]. h, Scopolamine decreased PPRs at 20, 30, 50, and 100 msec interstimulus intervals in slices from Mecp2 CTL mice 24hs after the injection. In hippocampal slices from Mecp2 KI mice, PPRs were decreased at 20, 30, and 50 msec interstimulus intervals 8 hr but not 24 hrs after scopolamine treatment [CTL-Sal, CTL-SCA (24 h), KI-Sal, KI-SCA (8 h), KI-SCA (24 h) : n = 20, 20, 17, 15, 14 slices ]. i, Scopolamine decreased PPRs in slices from Bdnf CTL mice 24 hrs after treatment. However, PPRs were not significantly changed in slices from Bdnf cKO mice at 8 or 24 hrs after scopolamine treatment [CTL-Sal, CTL-SCA (24 h), cKO-Sal, cKO-SCA (8 h), cKO-SCA (24 h): n = 31, 27, 21, 11, 26 slices]. Representative traces are from results recorded at 30 msec interstimulus interval in their respective experiments. a,b,d,e: two-sided paired t-test. c,f: One-way RM ANOVA with Tukey’s multiple comparison. g,h,i: One-Way ANOVA with Tukey’s multiple comparison. h,i: two-sided unpaired t-test, Mann-Whitney test, Welch’s correction. Graphs represent mean ± S.E.M., N.S.: not significant, *, P < 0.05, **, P < 0.01, ***, P < 0.001. Sal: saline, SCA: scopolamine, CTL: littermate control. For detailed statistical information, see Supplementary Table 1.
To investigate whether scopolamine rescues these suppressed synaptic responses, we applied scopolamine for 20 min onto hippocampal slices following the physostigmine-induced long-term depression. Scopolamine significantly restored the suppressed fEPSP response (Fig. 5c) and the increased PPRs to control levels at 30 to 100 msec interstimulus interval conditions (Fig. 5f). These recovered changes were maintained even after the washout of scopolamine (Wash2 in Fig. 5c,f). These results suggest scopolamine rescues suppressed hippocampal responses by countering the increased cholinergic tone and increasing presynaptic release probability. These findings also demonstrate that scopolamine-induced synaptic effects can be maintained even after removal of scopolamine, suggesting a possible sustained mechanism.
Since our behavioral and molecular data show that scopolamine induces antidepressant effects and changes in BDNF or pMeCP2 levels at 8 hrs and 24 hrs after treatment, we investigated whether presynaptic functional changes are observed in the hippocampus at these time points. We recorded fEPSPs in the SC–CA1 synapses of C57BL/6J mice at 8 or 24 hrs after scopolamine. PPRs at 20 to 50 ms interstimulus intervals were significantly decreased at 8 hrs and 24 hrs post-scopolamine (Fig. 5g). The reduction in PPRs was also observed at 20 and 30 msec interstimulus interval conditions 7 days after scopolamine treatment along with the behavioral effect (Extended Data Fig. 6a,b), suggesting a sustained increase in presynaptic release probability after scopolamine administration.
We next examined whether pMeCP2 was required for the sustained presynaptic functional changes induced by scopolamine. We assessed PPRs in the SC–CA1 synapses of Mecp2 KI mice 8 or 24 hrs after intraperitoneal scopolamine injection. Rather unexpectedly, in Mecp2 KI mice the PPRs were significantly reduced 8 hrs but not 24 hrs after scopolamine treatment compared to saline (Fig. 5h). These results suggest a key role for pMeCP2 selectively in the late phase of presynaptic efficacy changes, consistent with the requirement for pMeCP2 in the sustained (but not the rapid) antidepressant effects of scopolamine. We also assessed PPRs in hippocampal slices from Bdnf cKO mice treated with scopolamine. We observed no change in PPRs in Bdnf cKO mice at 8 and 24 hrs after scopolamine, demonstrating an essential role for BDNF in both the rapid and sustained synaptic effects (Fig. 5i). Collectively, these data show that BDNF plays a key role in presynaptic function in the early stages of antidepressant action, while pMeCP2 is required to maintain these presynaptic changes induced by scopolamine.
M1-antagonism increases pMeCP2 and presynaptic release probability
Scopolamine is a muscarinic acetylcholine receptor antagonist and a previous study suggested antagonism of muscarinic acetylcholine receptor M1 mediates scopolamine’s antidepressant effects42. Therefore, we treated C57BL/6J mice with pirenzepine, a blood-brain barrier permeable muscarinic acetylcholine receptor M1 selective antagonist. Pirenzepine produced an increase in pMeCP2, but not in pCREB (Fig. 6a), as well as an increase in the presynaptic release probability of SC–CA1 synapses 24 hrs after treatment, similar to scopolamine (Fig. 6b).
Fig. 6. Pirenzepine up-regulates MeCP2 phosphorylation and increases release probability.
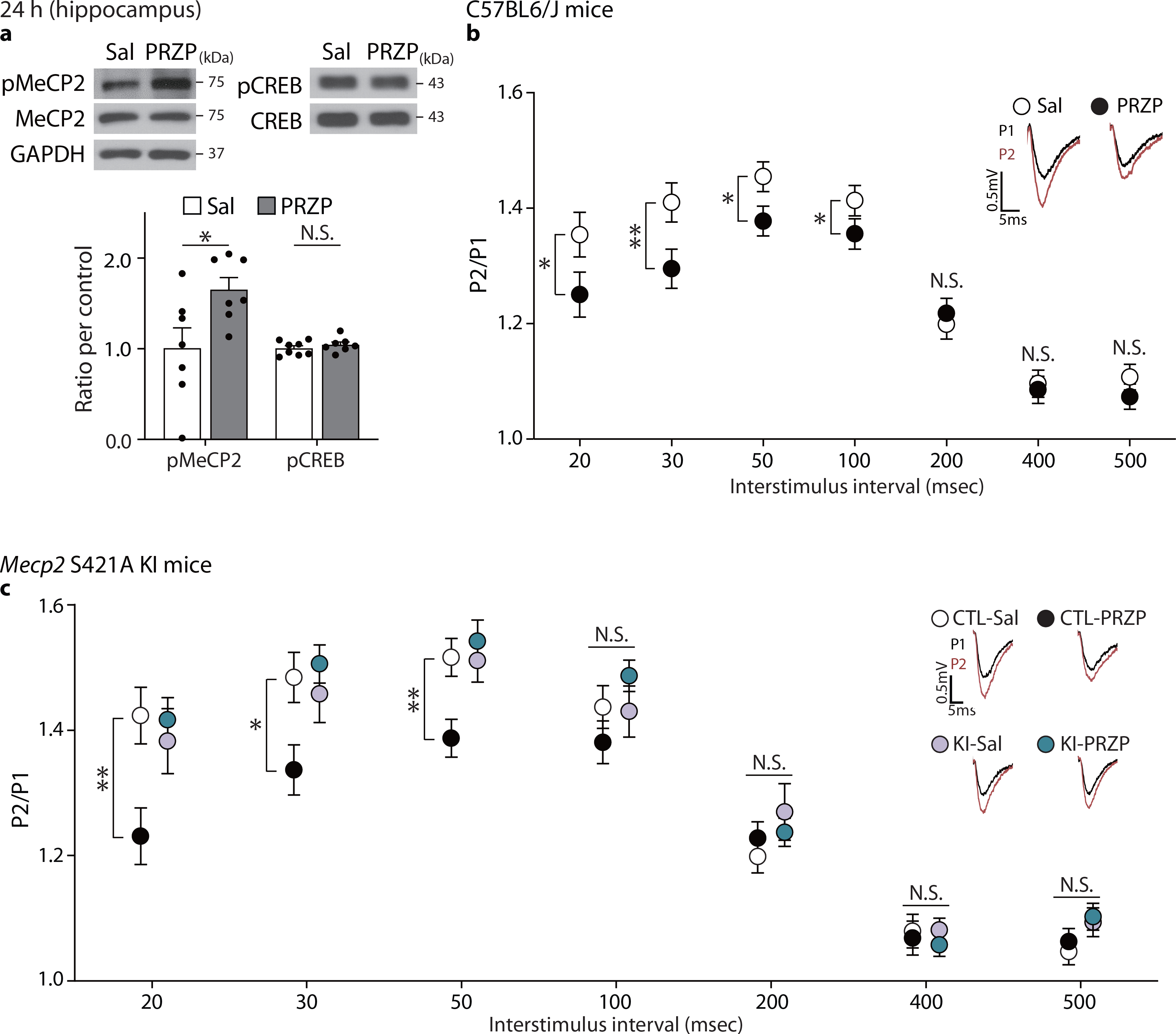
a, pMeCP2 levels were measured in the hippocampus of C57BL/6J mice 24 hrs after pirenzepine treatment (5mg/kg) with Western blot analysis. Pirenzepine significantly increased pMeCP2 levels but not pCREB levels (pMeCP2/MeCP - P = 0.0298, n = 7 mice per group, pCREB/CREB - P = 0.3604, Sal, PRZP: n = 8, 7 mice). b, To investigate whether the scopolamine-induced reduction of PPRs results from M1 antagonism, PPRs were measured in CA1 hippocampal slices from C57BL/6J mice treated with saline or pirenzepine 24 hrs before the hippocampal slice preparation. PPRs were significantly decreased in the pirenzepine-treated group at the 20 msec, 30 msec, 50 msec, and 100 msec interstimulus interval conditions (20, 30, 50, and 100 msec P < 0.05, Sal, PRZP: n = 20, 28 slices). c, To confirm whether the sustained reduction of PPRs by M1-receptor specific antagonism requires pMeCP2, PPRs were measured in CA1 hippocampal slices from Mecp2 KI or CTL mice 24 hrs after pirenzepine treatment. Pirenzepine-treated CTL group showed decreased PPRs compared to saline-treated CTL group, but the effects of pirenzepine were impaired in the Mecp2 KI mice [Genotype x Drug : 20 msec - P = 0.0049, 30 msec - P = 0.0069, 50 msec - P = 0.0083, CTL-Sal, CTL-PRZP, KI-Sal, KI-PRZP: n = 18, 18, 16, 18 slices]. Representative traces are from results recorded at 30 msec interstimulus interval in the respective groups. a,b: two-sided unpaired t-test or Mann-Whitney test, c: two-way ANOVA with Tukey’s multiple comparisons. Graphs represent mean ± S.E.M., N.S.: not significant, *, P < 0.05, **, P < 0.01, Sal: saline, PRZP: pirenzepine, CTL: littermate control. For detailed statistical information, see Supplementary Table 1.
We next determined whether muscarinic acetylcholine receptor M1 antagonism also requires pMeCP2 to elicit sustained presynaptic changes in the hippocampus. We observed the effect of pirenzepine on presynaptic release probability was abolished in slices from Mecp2 KI mice 24 hrs after scopolamine (Fig. 6c). Taken together, these findings suggest that scopolamine-mediated MeCP2 phosphorylation and sustained presynaptic functional changes in SC–CA1 synapses are due to muscarinic acetylcholine receptor M1 antagonism on presynaptic terminals rather than postsynaptic changes in synaptic efficacy.
Sustained effects of ketamine and scopolamine in stressed mice
To further validate the long-term behavioral effects of ketamine and scopolamine, C57BL/6J mice were subjected to the corticosterone-induced animal model of depression43 and then tested in the tail suspension test (TST), a behavioral paradigm that has been shown to detect sustained antidepressant effects25. As expected, mice treated with corticosterone for 21 days showed an increase in immobility compared to vehicle-treatment (No Cort). A single ketamine injection reduced immobility at 7 days post-injection to control (No Cort) levels (Fig. 7a). Similarly, scopolamine reduced immobility in corticosterone-treated mice at 24 hrs post-injection, and the effects were maintained for 7 days (Fig. 7b).
Fig. 7. Sustained antidepressant effects of ketamine and scopolamine in a corticosterone-induced animal model of depression.
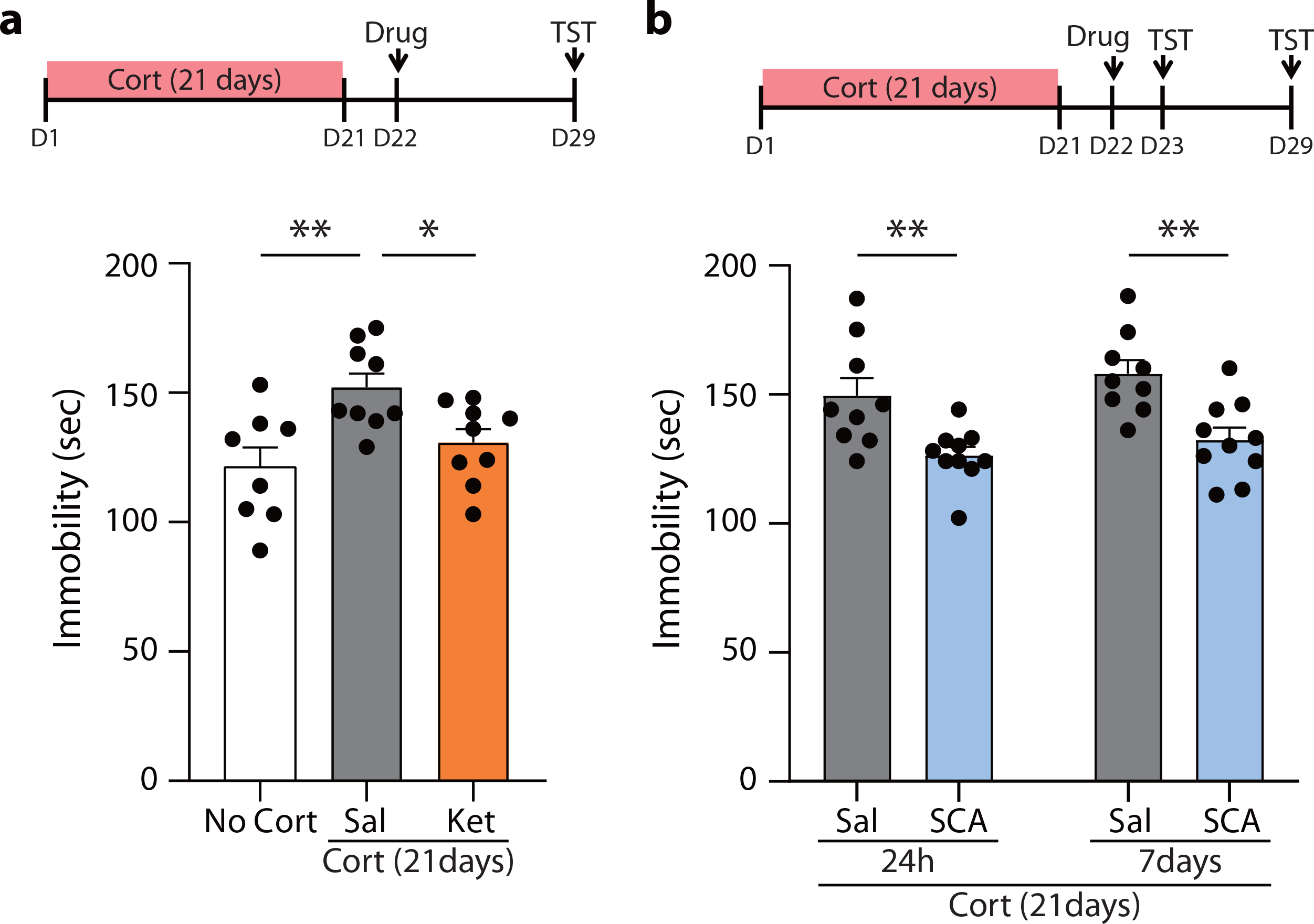
a,b, Corticosterone was dissolved in ethanol and diluted in tap water (100ng/ml, 1% Ethanol). Corticosterone was given in water bottles with free access for 21 days. For a vehicle control group, 1% ethanol was given in the water bottle for 21 days. a, One day after the last corticosterone treatment, ketamine was administered to C57BL/6J mice, and the mice were tested 7 days later in the tail suspension test (TST). The saline-treated corticosterone group showed a significant increase in the time spent immobile during the TST compared to the saline-treated vehicle control group. The ketamine-treated corticosterone group showed a significant decrease in the immobile duration compared to the saline-treated corticosterone group (one-way ANOVA with Tukey’s multiple comparisons, P = 0.0054, No Cort, Cort-Sal, Cort-Ket: n = 8, 9, 9 mice). b, One day after the termination of the corticosterone treatment, scopolamine was given, and the TST was conducted with the same cohort 24 hrs and 7 days after the drug treatment. Scopolamine significantly reduced the time spent immobile at 24 hrs and its effects were maintained for 7 days (two-way RM ANOVA, Time x Drug: P = 0.7989, Time: P = 0.1408, Drug: P = 0.0004, Sal, SCA: n = 9, 10 mice). Graphs represent mean ± S.E.M., *, P < 0.05, **, P < 0.01, Sal: saline, Ket: ketamine, SCA: scopolamine. For detailed statistical information, see Supplementary Table 1.
Discussion
We have demonstrated a crucial role for pMeCP2 in the prolonged antidepressant and synaptic effects of ketamine and scopolamine, the actions of which are dependent on BDNF. Ketamine and scopolamine induce distinct types of synaptic plasticity that nevertheless result in a similar increase in glutamatergic synaptic efficacy, suggesting a common synaptic endpoint. The sustained behavioral and synaptic effects of ketamine and scopolamine require BDNF-dependent phosphorylation of MeCP2 at S421. Of note, Bdnf cKO mice had similar baseline levels of pMeCP2 to littermate controls, and it was only after antidepressant treatment that differences in pMeCP2 were observed. These results suggest that rapid antidepressant treatment induces the initial elevations in BDNF signaling and then the subsequent increase in pMeCP2 which leads to these downstream events.
Our data show ketamine and scopolamine regulate MeCP2 phosphorylation, which is required for the persistent behavioral and synaptic effects, without affecting levels of pCREB. The transcription factor MEF2C – a key regulator of synapse numbers and structural plasticity22, 23 – also was not required for the antidepressant effects of ketamine, suggesting that synaptic functional changes rather than sole alterations in synapse structure are critical for the maintenance of the antidepressant effect. Treatment with a transcriptional inhibitor also prevented the sustained antidepressant effects of ketamine and scopolamine, indicating the involvement of a transcription-dependent mechanism in sustained antidepressant action, presumably through pMeCP2. Our results are consistent with earlier findings that pMeCP2 is required for the long-term, but not acute, effects of imipramine44. Taken together, these data suggest that pMeCP2 is a key consolidating factor for persistent antidepressant effects. Notably, BDNF is necessary to exert an antidepressant-like effect6, 11, 28, and the downstream target of pMeCP2 in the sustained antidepressant and synaptic effects. These results suggest that antidepressant drugs, including those that increase monoaminergic transmission or block NMDA receptors or muscarinic acetylcholine receptors, may have a mechanistic point of convergence in antidepressant action.
The requirement for pMeCP2 in long-lasting antidepressant effects may suggest transcriptional regulation of downstream targets that underlie long-term plasticity and behavioral changes, although specific targets of MeCP2 have been difficult to identify given the generalized binding of MeCP2 to chromatin45. Previous studies have shown that Bdnf transcription can be regulated by pMeCP217, 46. However, we did not observe changes in BDNF expression at the later time points in which pMeCP2 levels were increased. Moreover, the ketamine- and scopolamine-mediated rapid increases in BDNF levels were intact in Mecp2 KI mice. These results suggest that the ketamine and scopolamine-induced increase in BDNF levels are not regulated by pMeCP2, at least in the hippocampus at the time points examined. It is possible that target genes regulated by pMeCP2 may be involved in the sustained behavioral and synaptic effects. Future studies will be needed to identify these targets and determine their role in sustained antidepressant action.
BDNF levels at 30 min were required for the changes in pMeCP2 at 7 days following ketamine treatment; the temporal gap between these two molecular changes indicates the possibility of an intermediate process. We observed that BDNF levels were at baseline levels at 3 days post-ketamine, whereas pCaMKIIβ and pMeCP2 levels were increased at this time point in the CA1, where ketamine-mediated potentiation occurs. These data suggest that increased pMeCP2 levels are not directly induced from activated BDNF–TrkB signaling. Presumably, given that CaMKIIβ has higher sensitivity to Ca2+/CaM than CaMKIIα47, the increase in pCaMKIIβ levels may indicate an increase in activity-dependent Ca2+ influx, which is not high enough to induce pCaMKIIα, at this time point. Therefore, one could speculate that the activated Ca2+ signaling may be an intermediate process to link BDNF and pMeCP2. In support of this hypothesis, previous studies demonstrated that CaMKII is an upstream molecule that can mediate phosphorylation of MeCP217, 34. Ketamine has been shown to increase phosphorylation of CaMKIIα following eEF2 dephosphorylation48, which is required for rapid translation of BDNF and synaptic potentiation in the hippocampus6. Moreover, the blockade of CaMKII activity has been shown to prevent the antidepressant-like effects of ketamine 24 hrs after treatment48. Additional studies are necessary to further investigate this hypothesis and determine the role of Ca2+-dependent signaling as an intermediate process between BDNF and pMeCP2 following antidepressant action.
A rather unexpected result of our current work is that repeated ketamine induces a type of metaplasticity through postsynaptic functional changes. Initial ketamine administration rapidly augments potentiation of AMPA receptor-mediated SC–CA1 fEPSPs, although this potentiation is not maintained for 7 days. Instead, the initial dose of ketamine appears to produce a priming effect such that subsequent ketamine administration further augments this plasticity. pMeCP2 is necessary for this ketamine-induced metaplasticity as well as the sustained antidepressant action, but not for the rapid synaptic or behavioral effects. This temporally specific requirement of pMeCP2 highlights its role as a downstream effector for long-term antidepressant action. These data also suggest a potential framework for ketamine’s rapid and persistent action. Ketamine has rapid effects in the hippocampus, leading to the potentiation of AMPA receptor-mediated SC–CA1 fEPSPs. However, by 7 days this potentiation in the hippocampus is no longer present (or relatively undetectable). It is possible that any remaining synaptic changes at this time point are below the resolution of our current measurements, or that synaptic changes may be ‘transferred’ to other brain regions for the long-term antidepressant effects. In support of the latter premise, previous work has shown that ketamine acutely induces BDNF–TrkB signaling in the ventral hippocampus, which was required for its sustained antidepressant effects, and that inactivation of the ventral hippocampus–mPFC pathway prevented the sustained, but not the acute antidepressant effect of ketamine49. Taken together, these data suggest that the synaptic basis for the rapid antidepressant effects of ketamine is initiated in the hippocampus and then may be transferred to other brain regions such as the prefrontal cortex region. A subsequent ketamine treatment then re-engages the hippocampus to produce the metaplasticity. Clinical data suggest that repeated-ketamine-infusions have cumulative and sustained antidepressant effects31–33. This synaptic metaplasticity of ketamine may explain why repeated ketamine treatment shows more enhanced and prolonged antidepressant effects compared to a single injection31–33. Further experiments focusing on the downstream mechanisms of this metaplasticity may provide critical insights into how repeated ketamine treatment produces more potent and prolonged antidepressant effects.
Increased cholinergic tone has been suggested to be an important pathogenic factor in MDD41, 50. Increased cholinergic tone suppresses hippocampal synaptic responses by inhibiting presynaptic release probability, whereas scopolamine rescues the suppressed synaptic response by increasing release probability. This increased presynaptic release probability is maintained even after the removal of scopolamine suggesting a possible sustained mechanism is involved. Similar to ketamine, scopolamine requires BDNF to exert its rapid antidepressant and synaptic effects. The scopolamine-mediated increase in pMeCP2 is BDNF-dependent and, based on our current data, pMeCP2 is required for the sustained antidepressant and synaptic effects. In addition, our results demonstrate that pirenzepine induces similar molecular and synaptic effects as scopolamine and also requires pMeCP2 for its synaptic effects. This supports the premise that scopolamine’s antidepressant and synaptic effects are mediated by muscarinic acetylcholine receptor M1 antagonism. Collectively, these results suggest that scopolamine induces sustained presynaptic release and an increase in glutamatergic synaptic efficacy in BDNF-dependent effect on MeCP2 phosphorylation, which may be a key mechanism underlying its sustained antidepressant effects.
Ketamine rapidly increases BDNF protein expression and augments postsynaptic activity by rapid potentiation of AMPA receptor-mediated SC–CA1 fEPSPs6, 9, whereas scopolamine increases BDNF mRNA and protein over a longer time course and reduces paired-pulse ratios, suggesting an increase in neurotransmitter release probability. While ketamine and scopolamine work through differing mechanisms, including distinct manners in the regulation of BDNF expression, the changes in synaptic plasticity results in a similar increase in synaptic efficacy.
Based on the results of this study, we propose that BDNF-dependent pMeCP2 is a convergent target for sustained antidepressant effects. In addition to mediating the behavioral effects of ketamine and scopolamine, the BDNF-dependent pMeCP2 signaling also mediates distinct forms of synaptic plasticity in the presynaptic and postsynaptic compartments by scopolamine and ketamine, respectively. These findings elucidate the key role played by MeCP2 in the maintenance of rapid antidepressant responses, and thus highlight potential therapeutic targets for novel antidepressants that produce both rapid and long-lasting effects. Identification of mechanisms downstream of BDNF-dependent pMeCP2 signaling may provide novel targets for treating symptoms of MDD.
Methods
Mice.
C57BL/6J male and female mice aged 2 to 6 months were used for this study. Animals were maintained on a 12h-light/12h-dark cycle, at ambient temperature (23 ± 3°C) and humidity (50 ± 20%) with ad libitum access to chow pellets and water. All animal procedures were performed in accordance with the guide for the care and use of laboratory animals and were approved by the Institutional Animal Care and Use Committee (IACUC) at UT Southwestern Medical Center and Vanderbilt University.
Mecp2 S421A knock-in (KI) mice.
Mecp2 S421A KI mice were generated as previously described27. Heterozygous Mecp2 S421A female mice were mated with wild-type male mice. Hemizygous KI male and littermate controls (2 to 9 months old) were used for the current study.
Bdnf cKO mice.
Bdnf cKO mice were generated by crossing mice carrying neuron-specific enolase (NSE)-tetracycline transactivator (tTA), tetracycline operator sequences (TetOp)-Cre recombinase, with floxed BDNF mice as previously described28. We bred the inducible Bdnf cKO mice without doxycycline administration. These Bdnf cKO mice have a deletion of BDNF in the late embryonic stage and no defects in general growth and neuronal structure28, 51, 52. Bdnf cKO male and littermate controls (2 to 9 months old) were used for the current study.
Mef2c conditional knock-out (cKO) mice.
Conditional Mef2 cKO mice were produced as previously described22, 23. Floxed Mef2c mice were mated with transgenic mice expressing Cre recombinase under the control of CaMKⅡ promoter (CaMKⅡ-Cre93). Mef2c KO male and littermate controls (2 to 6 months old) were used for the current study.
Drugs.
Drug doses were used as follows: ketamine (Hospira) 5 mg/kg, scopolamine (Sigma) 0.35mg/kg, pirenzepine (Tocris) 5 mg/kg. Drugs were dissolved in 0.9% saline just before use and injected intraperitoneally.
Forced swim test.
The forced swim test was performed as previously reported6. Briefly, mice were placed in a 4L Pyrex glass beaker containing 3L of water (23–24°C) for 6 min. The time mice spent immobile was measured during the last 4 min of the test. Water was changed between each animal.
Corticosterone-induced animal model of depression.
Corticosterone (Sigma Aldrich) was dissolved in 100% ethanol to make a final concentration of 100 μg/ml and 1% ethanol. The corticosterone solution was given to mice with the drinking water bottle ad libitum for 21 days. For the vehicle control group, 1% of ethanol in drinking water was provided. Drug treatment or brain preparation was conducted 24hrs after the termination of the corticosterone treatment.
Tail suspension test.
Each mouse was suspended within a tail suspension test chamber (Med Associates) for 6 min. The tail of each mouse was inserted into the clear hollow cylindrical climb stopper (4 cm length, 1.5 g) and then was attached to a vertical aluminum bar with adhesive tape. The time mice spent immobile was measured during the last 4 min of the test. The chamber was cleaned with an antiseptic solution after each trial.
Quantitative RT-PCR.
Hippocampal slices (2 per mouse, ~ 1 mm thick) were removed from C57BL/6J mice at designated time points after drug treatment. The slices were snap-frozen and RNA was extracted with Trizol reagent (Invitrogen), and reverse transcribed with SuperScript™ III reverse transcriptase (Invitrogen). PCR reaction mixture was made with Power SYBR® Green PCR master mix (Applied Biosystems). PCR reaction was processed with ABI7500 fast (Applied Biosystems) and fold changes were calculated by using the comparative threshold cycle (Ct) method. Primer sequences are as follows; for Bdnf, forward: ccataaggacgcggacttgtaca, reverse: agacatgtttgcggcatccag, for Gapdh, forward: aggtcggtgtgaacggatt, reverse: tgtagaccatgtagttgaggtca.
Brain preparation and western blot analysis.
Hippocampal slices (~1 mm thick) were collected from C57BL/6J mice at designated time points after drug treatment. The slices were immediately snap-frozen and lysed using RIPA buffer [50mM Tris, pH7.4, 1% Igepal, 0.1% SDS, 0.5% Na deoxycholate, 4mM EDTA, 150mM NaCl, protease inhibitors (Roche) and phosphatase inhibitors (2mM sodium orthovanadate, 50mM sodium fluoride, 10mM sodium pyrophosphate)]. The total protein amount was quantified by bicinchoninic acid assay (Thermo Fisher Scientific). An equal amount of protein was loaded on the respective wells of SDS-PAGE gels and transferred to the nitrocellulose membrane. After blocking the membranes for 1 hr at room temperature in blocking solution, blots were washed three times with 0.1% TBS-Tween and incubated overnight at 4°C with the following primary antibodies: MeCP2 (1:4,000, Thermo Fisher, PA1–888), phospho-MeCP2 (S421, 1:1,000, a generous gift from Dr. Anne West53 and ECM Biosciences, MP46111), GAPDH (1:100,000, Cell Signaling Technology, 2118), CREB (1:5,000, Cell Signaling Technology, 9197), phospho-CREB (Ser133, 1:5,000, Merck Millipore, 06–519), BDNF (1:1,500, Abcam, ab108319), CaMKIIα (1:5,000, Cell Signaling Technology, 3357S), CaMKIIβ (1:500,000, Abcam, ab34703), phospho-CaMKII (D21E4, 1:50,000, Cell Signaling Technology, 12716S). After washing three times with 0.1% TBS-Tween solution, blots were incubated with peroxidase-labeled secondary antibody [Goat anti-rabbit IgG (H+L) conjugated to peroxidase, 1:5000, Vector Laboratories, PI-1000–1] at room temperature for 2hrs. For blocking and incubating the blots, 5% skim milk in 0.1% TBS-Tween solution was used. The protein bands were detected using enhanced chemiluminescence (Biorad) and exposed to x-ray film.
Hippocampal slice field recording.
Mice 2 months old or older were anesthetized using isoflurane and decapitated. The brain was immediately removed and sliced using a vibratome (VT 1000S, Leica) in ice-cold dissection buffer containing the following (in mM): 2.6 KCl, 1.25 NaH2PO4, 26 NaHCO3, 0.5 CaCl2, 5 MgCl2, 212 sucrose, and 10 D-glucose. To record field potentiation in the SC–CA1 synapses, the CA3 region was surgically removed from each slice after sectioning. The slices were transferred into a reservoir chamber filled with ACSF containing the following (in mM): 124 NaCl, 5 KCl, 1.25 NaH2PO4, 26 NaHCO3, 2 CaCl2, 2 MgCl2, and 10 D-glucose. Slices were incubated at 30°C for 2–3 hrs of a recovery period. ACSF and dissection buffer were equilibrated with 95% O2 and 5% CO2.
For recording, slices were transferred to a submerged recording chamber, maintained at 30°C, and perfused continuously with ACSF at a rate of 3 ml/min. fEPSPs were recorded with extracellular recording electrodes filled with ACSF (resistance, 1 – 2MΩ) and placed in CA1 area stratum radiatum. fEPSPs were evoked by monophasic stimulation (duration, 200 μs) of Schaffer collateral/commissural afferents with a concentric bipolar microelectrode (FHC). Stable baseline responses were collected every 30 sec using a stimulus yielding 50 – 75% of the maximum peak. fEPSPs were filtered at 2 kHz and digitized at 10 kHz on a personal computer with a customized program (LabVIEW 8.5, National Instruments). The response was measured as the initial slope (10 – 40% of the rising phase) of the fEPSPs.
Ketamine potentiation was measured as previously described with a slight modification9. Hippocampal slices were prepared from mice pretreated with ketamine (5 mg/kg, i.p.) or saline 7 days before. After measuring the baseline response for 20 min, ketamine (20μM) or ACSF was perfused at 3ml/min without recording for 30 min. After perfusion of drugs, a single response was measured, and then recording was stopped. After an hour, the response was recorded every 30 sec for an additional 20 min. The response recorded during this period is designated as a ketamine-potentiation. Potentiation magnitude is an averaged value of ketamine-potentiation responses. All responses were normalized to the baseline responses. Paired-pulse ratio (PPR) was measured with interstimulus intervals (ISIs) of 20, 30, 50, 100, 200, 400, and 500 ms before and after ketamine treatment. PPR was calculated by the slope ratio of the second to the first response (P2/P1).
For scopolamine field recordings, hippocampal slices were prepared from mice 24 hrs or 7 days after intraperitoneal injection of scopolamine or saline. After confirming a stable baseline, the input/output response was measured to determine the response intensity using 4, 8, 12, 16, 20, and 24 μA stimulation. PPR was also measured as mentioned above.
For physostigmine-induced LTD measurement, hippocampal slices were recovered for 3 hrs after slice preparation. Physostigmine (10μM) or scopolamine (10μM) was perfused at the designated time points, and the slope of fEPSP and PPRs were measured as described above.
Statistics.
All statistical analyses were performed using GraphPad Prism 7.03 software. The normality of data was checked by the Shapiro-Wilk test. Homogeneity of variance was checked using Bartlett’s test. Outliers were determined by Grubb’s outlier test (alpha = 0.05) and excluded from the analyses. For comparison between two groups, two-tailed paired or unpaired t-test, Welch’s correction t-test, or Mann-Whitney test was performed. For comparisons of three or more groups, one-way repeated measures (RM) analysis of variance (ANOVA), one-way ANOVA, two-way RM ANOVA, or two-way ANOVA was performed. Tukey’s multiple comparisons test was used for post-hoc comparisons after the ANOVA tests. No statistical methods were applied to predetermine sample sizes, but our sample sizes are similar to those of previous publications6, 9, 23, 25, 26. Mice were randomly assigned to experimental groups. The experimenters were not blinded to experimental group assignment, as the same experimenter habituated mice and conducted tests. Data analyses were not performed blind to the experimental conditions, except the analysis for behavioral experiments. For detailed statistics information, see Supplementary Table 1. Statistical significance was designated as asterisks depending on P-value. *, P < 0.05, **, P < 0.01, ***, P < 0.001.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The raw data that supports the findings of the current study are available from the corresponding author upon reasonable request.
Extended Data
Extended Data Fig. 1. pMeCP2 is not changed in the medial prefrontal cortex 7 days after ketamine treatment.
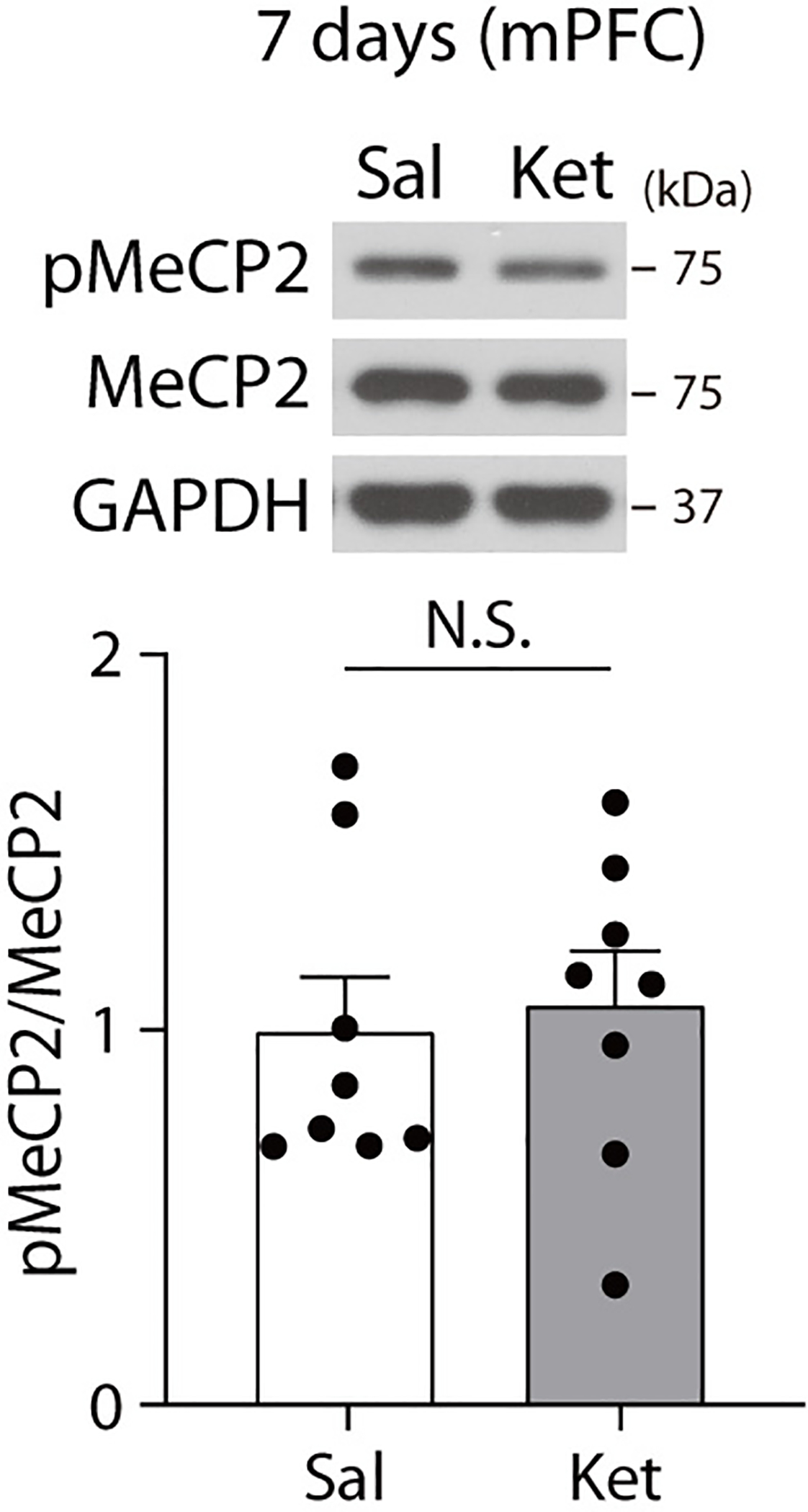
pMeCP2 levels were measured with Western blot analysis in the medial prefrontal cortex of C57BL/6J mice 7 days after ketamine treatment. No significant changes in pMeCP2 levels were observed between saline- and ketamine-treated groups. (two-sided unpaired t-test, ketamine: t(14) = 0.3298, P = 0.7464, n = 8 mice per group). In Western blot analysis for pMeCP2, membrane above about 70 KDa was cropped and used for immunoblotting. The graph represents mean ± S.E.M., N.S.: not significant, Sal: saline, Ket: ketamine. For detailed statistical information, see Supplementary Table 1.
Extended Data Fig. 2. Ketamine rapidly increases BDNF protein levels in the hippocampus of Mecp2 S421A KI mice.
BDNF protein levels were measured 30 min after ketamine treatment in Mecp2 KI and CTL mice by Western blot analysis. BDNF levels were significantly increased by ketamine treatment in both the CTL and KI mice (two-way ANOVA with Tukey’s multiple comparisons, Genotype x Drug: F(1, 21) = 0.0157, P = 0.9016, Genotype: F(1, 21) = 0.0115, P = 0.9158, Drug: F(1, 21) = 31.93, P < 0.0001, CTL-Sal, CTL-Ket, KI-Sal, KI-Ket: n = 7, 7, 6, 5 mice). In Western blot analysis for pMeCP2, membrane above about 70 KDa was cropped and used for immunoblotting. The graph represents mean ± S.E.M., **, P < 0.01, Sal: saline, Ket: ketamine, CTL: littermate control. For detailed statistical information, see Supplementary Table 1.
Extended Data Fig. 3. Effects of ketamine on pre-and post-synaptic function at 7 days after treatment.
a, Saline or ketamine was administered to C57BL/6J mice. The mice were sacrificed 7 days later, and slices were prepared for recordings. I-O curves were measured during baseline recording in the SC-CA1 synapses. I-O curves of Sal and Ket groups were from baseline recording in the Sal-Ket group of Fig. 3b and Ket-Ket group of Fig. 3c, respectively. The slope of I-O curves in the previous ketamine-treated group was not significantly different compared to the saline-treated group (two-sided unpaired t-test, t(17) = 1.448, P = 0.1659, Sal, Ket: n = 9, 10 slices). b, PPRs were measured before and after ketamine perfusion onto hippocampal slices of mice given either saline or ketamine and sacrificed 7 days later for slice electrophysiology. PPRs were not significantly changed by either previous ketamine injection or subsequent ketamine perfusion (two-way ANOVA with Tukey’s multiple comparisons, all P-values > 0.05, Sal-Ket, Ket-Ket: n = 11, 10 slices). Representative traces are from data recorded at 30 msec interstimulus interval in the respective treatment groups. Graphs represent mean ± S.E.M., N.S.: not significant, Sal: saline, Ket: ketamine. Sal+Ket: ketamine perfusion onto hippocampal slices from saline-injected mice, Ket+Ket: ketamine perfusion onto hippocampal slices from ketamine-injected mice. For detailed statistical information, see Supplementary Table 1.
Extended Data Fig. 4. Ketamine-induced metaplasticity is observed in female mice.
a,b, Ketamine-potentiation was measured in the Schaffer collateral to CA1 synapses of C57BL/6J female mice given either saline (a) or ketamine (b) injection 7 days before slice preparation. Augmented potentiation was observed in the group previously treated with ketamine (b: Ket+Ket, 138.5 ± 6.717%), compared to the group previously treated with saline (a: Sal+Ket, 114 ± 3.158%), (two-sided unpaired t-test, a - Sal+ACSF vs Sal+Ket: t(11) = 4.207, P = 0.0015, Sal+ACSF, Sal+Ket: n = 6, 7 slices, b - Ket+ACSF vs Ket+Ket: t(10) = 5.528, P = 0.0003, Ket+ACSF, Ket+Ket: n = 6, 6 slices, Sal+Ket in a vs Ket+Ket in b: t(11) = 3.465, P = 0.0053). Graphs represent mean ± S.E.M., **, P < 0.01, ***, P < 0.001, Sal: saline, Ket: ketamine. Sal+ACSF: ACSF perfusion onto hippocampal slices from saline-injected mice, Sal+Ket: ketamine perfusion onto hippocampal slices from saline-injected mice, Ket+ACSF: ACSF perfusion onto hippocampal slices from ketamine-injected mice, Ket+Ket: ketamine perfusion onto hippocampal slices from ketamine-injected mice. For detailed statistical information, see Supplementary Table 1.
Extended Data Fig. 5. Molecular changes in the CA1 hippocampal region at 3 or 7 days after ketamine treatment.
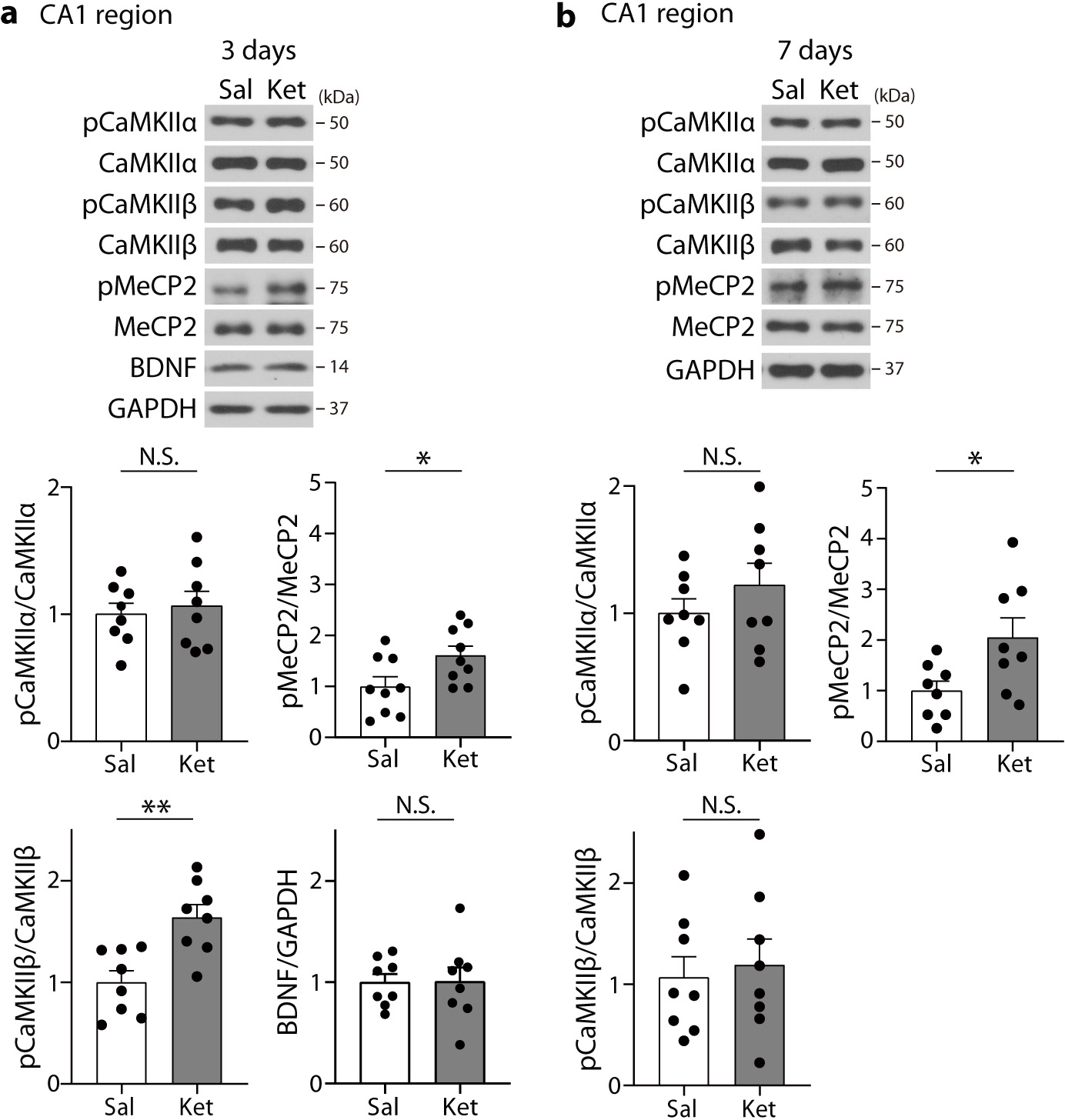
a,b, The hippocampal CA1 area was collected in C57BL6/J mice 3 or 7 days after ketamine treatment. Phosphorylation of CaMKIIβ, but not CaMKIIα, was significantly increased by ketamine at 3 days compared to the saline group, and the increased phosphorylation returned to the levels of the saline-treated group at 7 days (two-sided unpaired t-test, 3 days, CaMKIIα: t(14) = 0.4369, P = 0.6688, CaMKIIβ: t(14) = 3.756, P = 0.0021, n = 8 mice per group, 7 days, CaMKIIα: t(14) = 1.052, P = 0.3107, CaMKIIβ: t(14) = 0.3796, P = 0.7100, n = 8 mice per group). pMeCP2 levels were significantly increased at both 3 and 7 days (two-sided unpaired t-test, 3 days: t(16) = 2.332, P = 0.0331, n = 9 mice per group, 7 days: t(14) = 2.440, P = 0.0286, n = 8 mice per group). BDNF levels were not changed at 3 days (two-sided unpaired t-test, t(14) = 0.0433, P = 0.9661, n = 8 mice per group). In Western blot analysis for pMeCP2, membrane above about 70 KDa was cropped and used for immunoblotting. Graphs represent mean ± S.E.M., N.S.: not significant, *, P < 0.05, **, P < 0.01, Sal: saline, Ket: ketamine. For detailed statistical information, see Supplementary Table 1.
Extended Data Fig. 6. Effects of scopolamine at 7 days after injection.
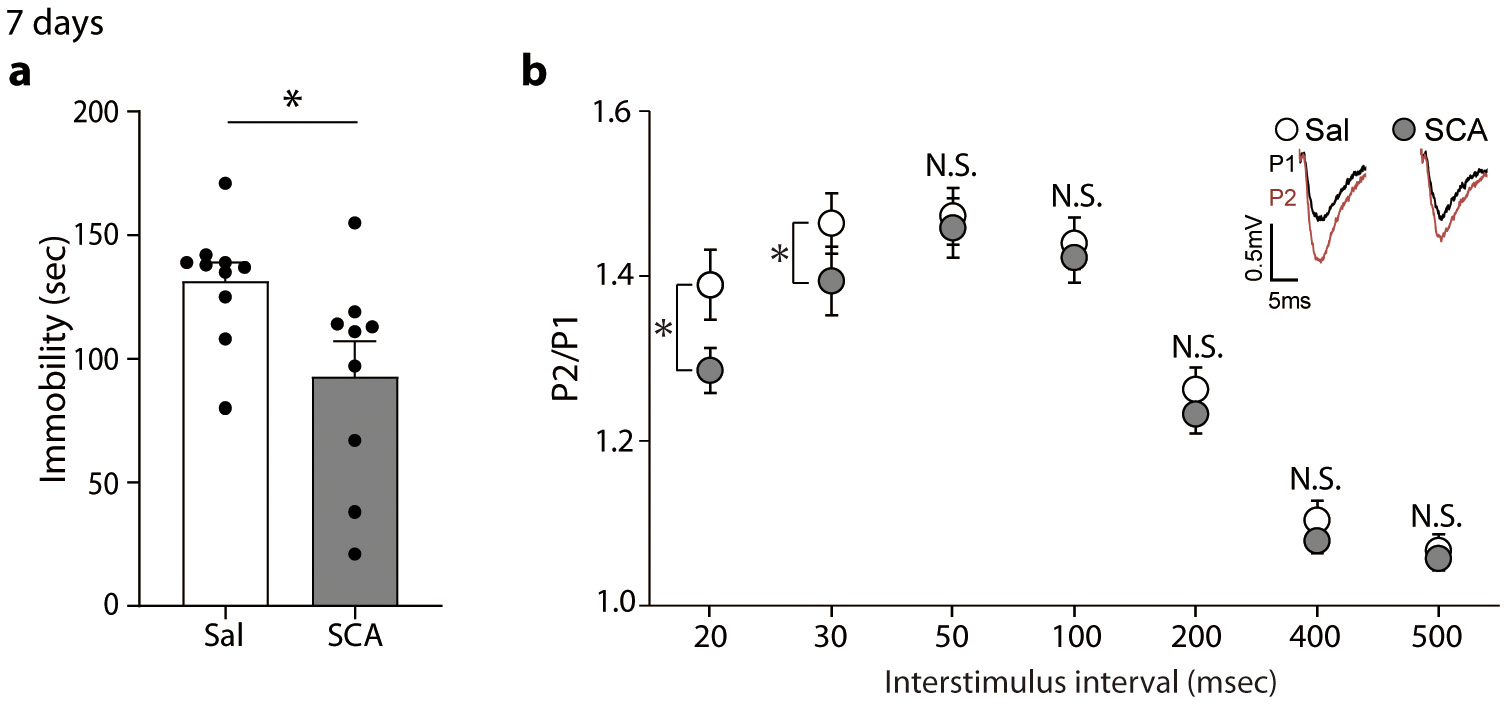
a, Immobility was measured in the FST 7 days after scopolamine treatment. The scopolamine-treated group showed a significant reduction in time spent immobile compared to the saline-treated group (two-sided unpaired t-test, t(17) = 2.469, P = 0.0244, Sal, SCA: n = 9, 10 mice). b, PPRs were analyzed in the hippocampal CA1 area of C57BL/6J mice given scopolamine 7 days prior. The PPRs were significantly reduced in the scopolamine-treated group compared to the saline-treated group at 20 and 30 msec interstimulus interval condition (two-sided Mann-Whitney test, 20 msec: U = 210, P = 0.0314, 30 msec: U = 207, P = 0.0270, Sal, SCA: n = 24, 27 slices). Graphs represent mean ± S.E.M., N.S.: not significant, *, P < 0.05, Sal: saline, SCA: scopolamine. For detailed statistical information, see Supplementary Table 1.
Extended Data Fig. 7. Actinomycin D prevents scopolamine-mediated sustained behavioral effects.
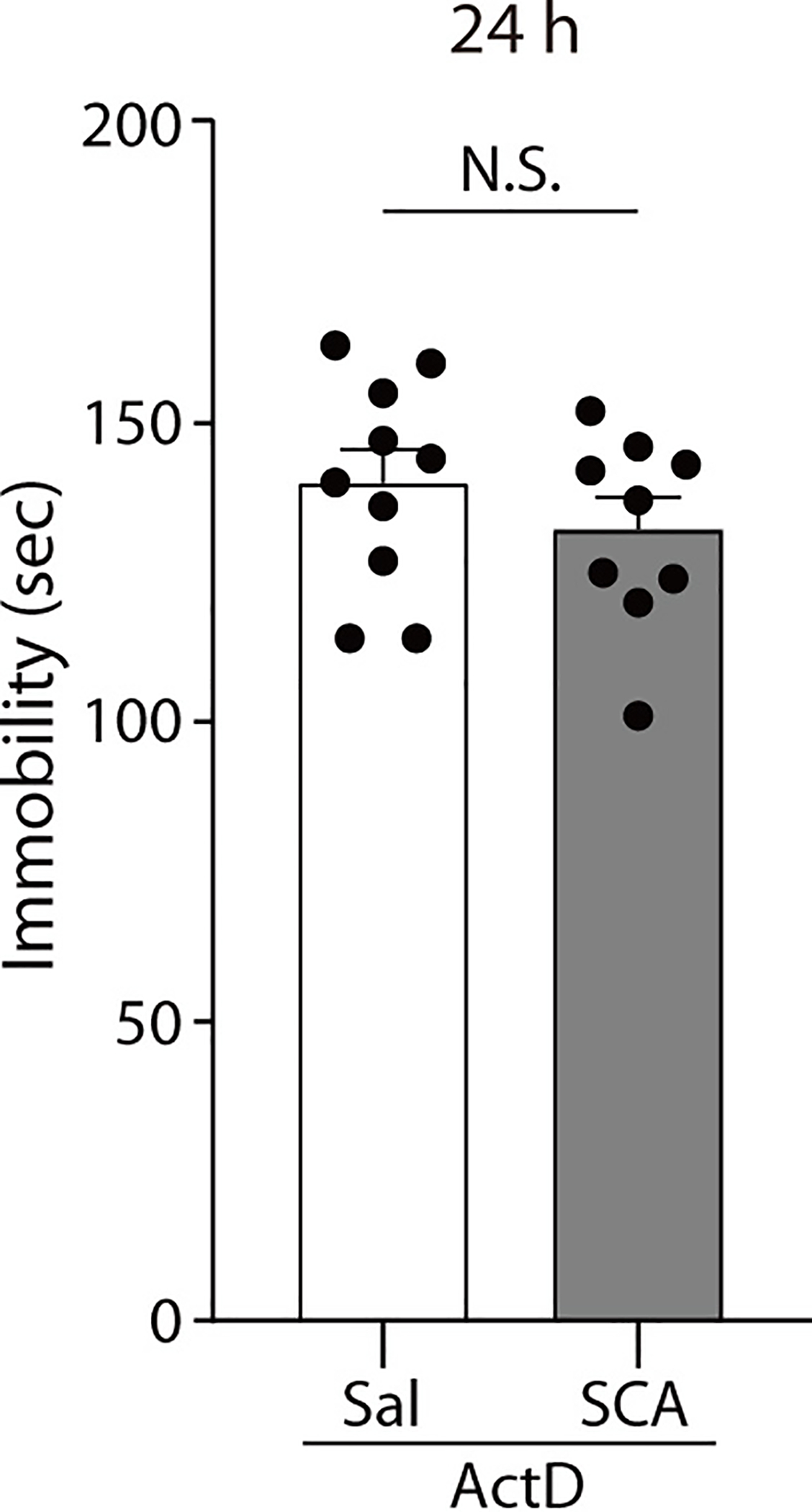
Actinomycin D was administered to C57BL/6J mice prior to the saline or scopolamine treatment, and the mice were tested 24 hrs later in the FST. Scopolamine did not significantly reduce the duration of immobility of the mice pretreated with actinomycin D (two-sided unpaired t-test, t(17) = 1.006, P = 0.3286, ActD-Sal, ActD-SCA : n = 10, 9 mice). The Graph represents mean ± S.E.M., N.S.: not significant, Sal: saline, SCA: scopolamine. For detailed statistical information, see Supplementary Table 1.
Extended Data Fig. 8. pMeCP2 is not changed in the medial prefrontal cortex 24 hrs after scopolamine treatment.
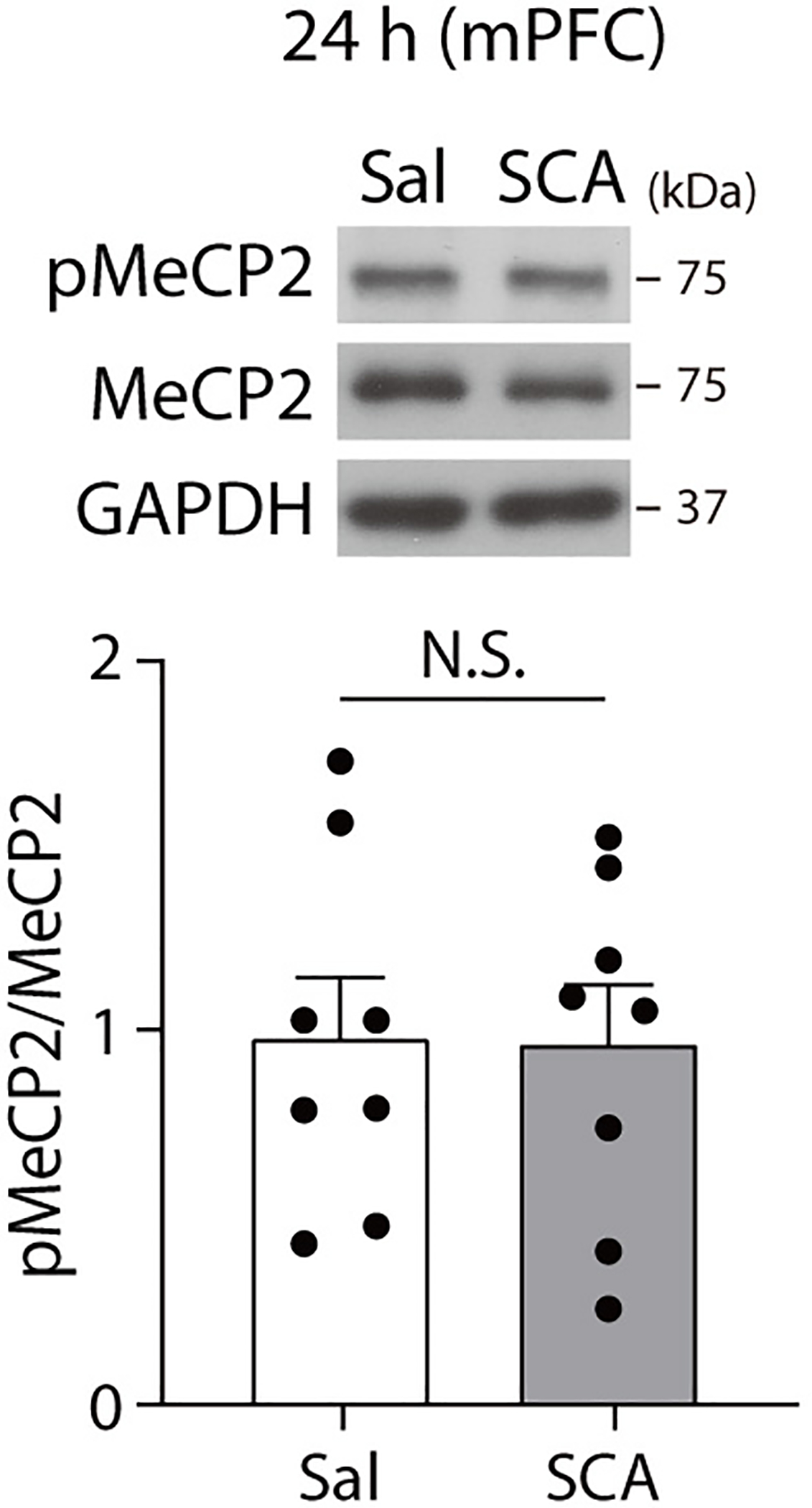
pMeCP2 levels were measured in the medial prefrontal cortex of C57BL/6J mice 24 hrs after scopolamine treatment with Western blot analysis. No significant differences in pMeCP2 levels were observed between saline- and scopolamine-treated groups (two-sided unpaired t-test, t(14) = 0.0705, P = 0.9448, n = 8 mice per group). In Western blot analysis for pMeCP2, membrane above about 70 KDa was cropped and used for immunoblotting. The Graph represents mean ± S.E.M., N.S.: not significant, Sal: saline, SCA: scopolamine. For detailed statistical information, see Supplementary Table 1.
Extended Data Fig. 9. Scopolamine does not affect CREB phosphorylation.
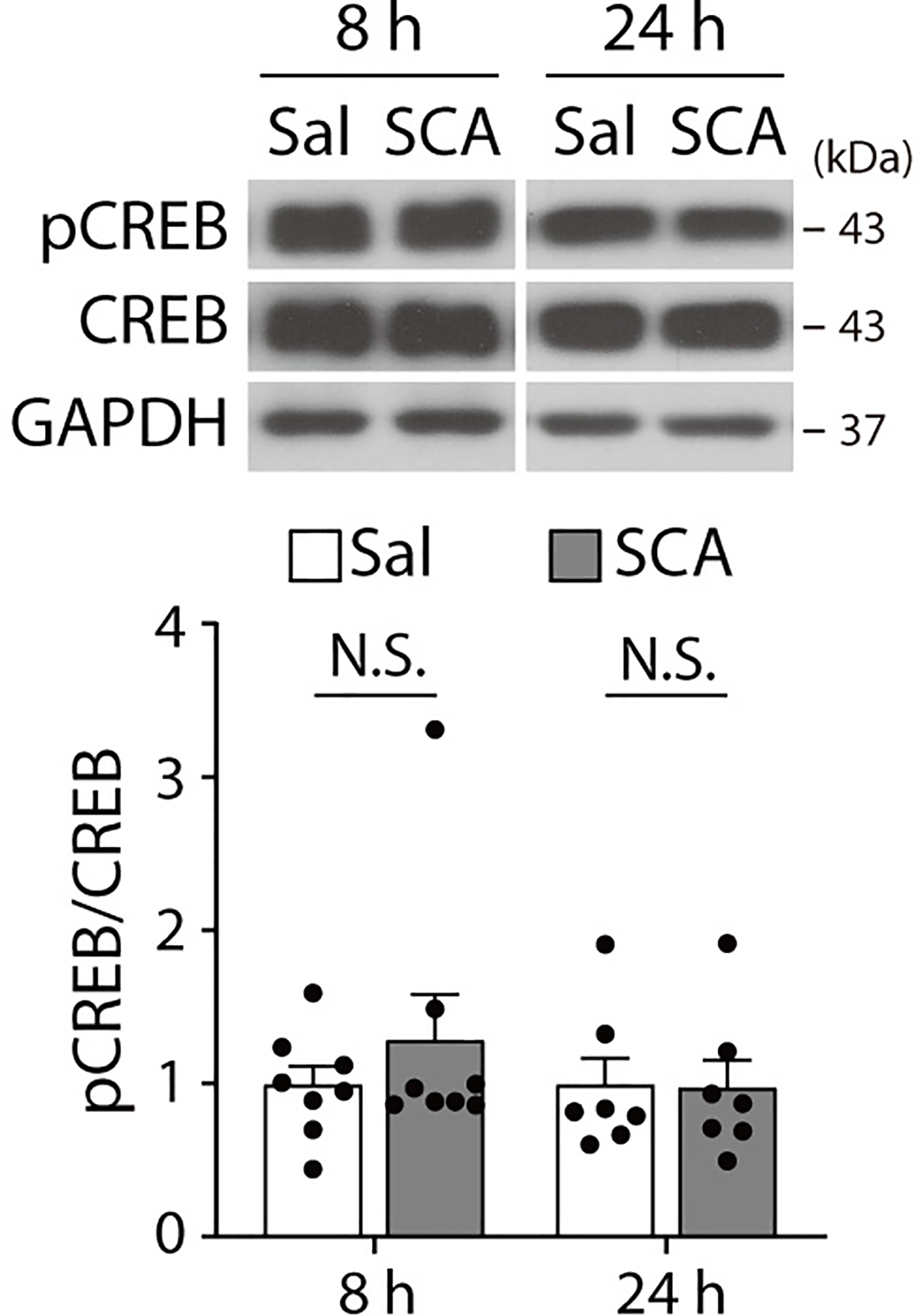
pCREB levels were measured in the hippocampus of C57BL/6J mice with Western blot analysis 8 hrs and 24 hrs after scopolamine treatment. No significant differences were observed between Sal- and SCA-treated groups at either time point (two-sided unpaired t-test or Welch’s correct t-test, 8 hrs: t(9.272) = 0.9004, P = 0.3907, n = 8 mice per group, 24 hrs: t(12) = 0.0677, P = 0.9472, n = 7 mice per group). The Graph represents mean ± S.E.M., N.S.: not significant, Sal: Saline, SCA: Scopolamine. For detailed statistical information, see Supplementary Table 1.
Extended Data Fig. 10. Scopolamine increases Bdnf mRNA levels in the hippocampus of Mecp2 S421A KI mice 8 hours after injection.
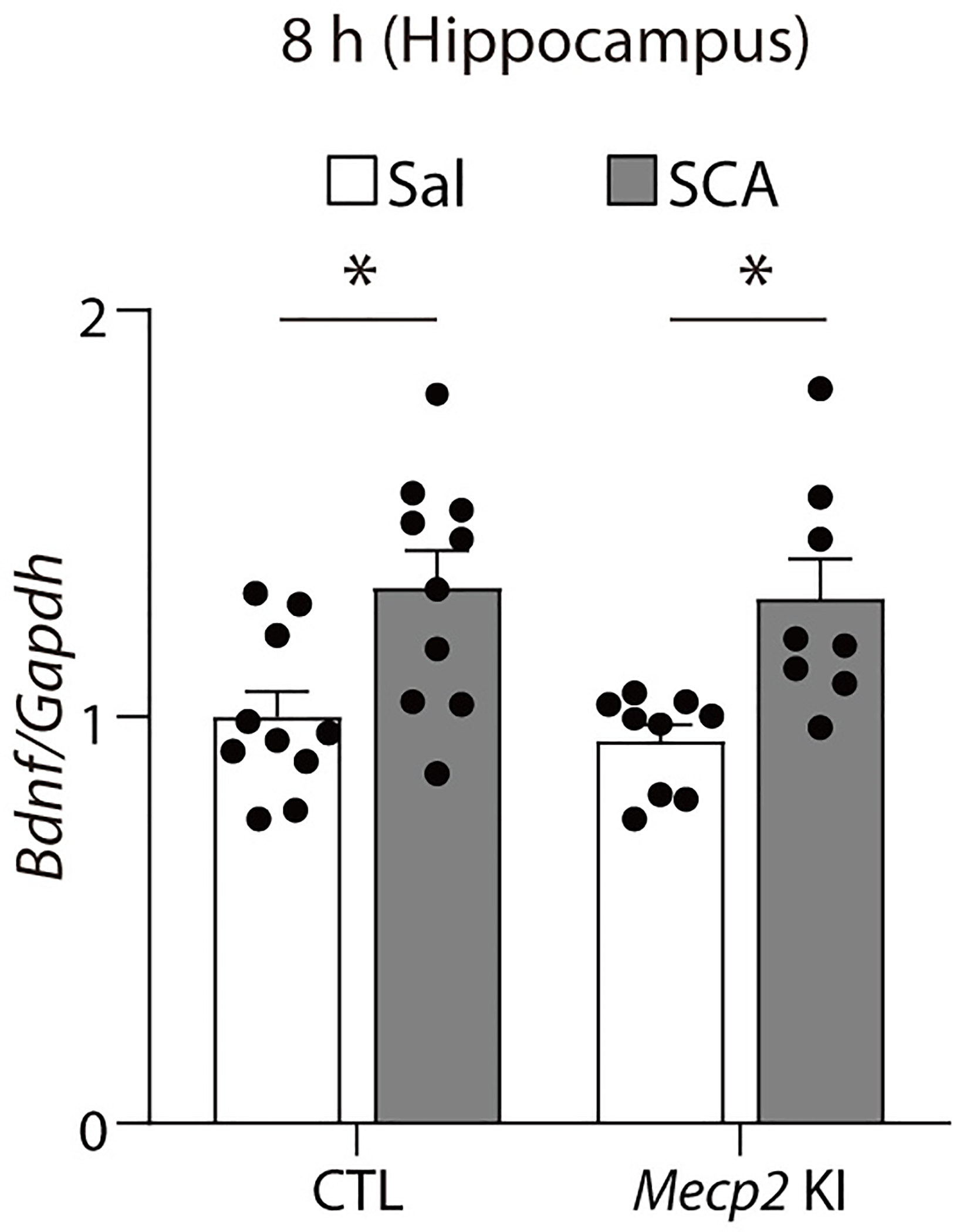
Bdnf mRNA levels were measured with quantitative real-time PCR in CTL and Mecp2 KI mice 8 hrs after scopolamine. Scopolamine treatment significantly increased Bdnf mRNA levels in the hippocampus of both the CTL and KI mice (two-way ANOVA with Tukey’s multiple comparisons, Genotype x Drug: F(1, 33) = 0.0467, P = 0.8302, Genotype: F(1, 33) = 0.3223, P = 0.5741, Drug: F(1, 33) = 19.22, P = 0.0001, CTL-Sal, CTL-SCA, KI-Sal, KI-SCA: n = 10, 9, 10, 8 mice). The Graph represents mean ± S.E.M., *, P < 0.05, Sal: saline, SCA: scopolamine, CTL: littermate control. For detailed statistical information, see Supplementary Table 1.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank members of the Monteggia and Kavalali laboratory for helpful advice and discussions on the manuscript. We thank Dr. Kristen Szabla for the initial insight into this project. We thank Dr. Anne West for providing the initial pMeCP2 antibody for these studies. This work was supported by National Institutes of Health Grants MH070727 and MH081060 (to L.M.M.) and MH066198 (to E.T.K.), the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2016R1A6A3A03008533, J.K.), and post-doctoral fellowships from the Elisabeth and Alfred Ahlqvist foundation within the Swedish Pharmaceutical Society and the Swedish Society for Medical Research (to C.B.).
Footnotes
Competing Interests
The authors declare no competing interests.
References
- 1.Quitkin FM, et al. Use of pattern analysis to identify true drug response. A replication. Arch Gen Psychiatry 44, 259–264 (1987). [DOI] [PubMed] [Google Scholar]
- 2.Berman RM, et al. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry 47, 351–354 (2000). [DOI] [PubMed] [Google Scholar]
- 3.Zarate CA Jr., et al. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry 63, 856–864 (2006). [DOI] [PubMed] [Google Scholar]
- 4.Furey ML & Drevets WC Antidepressant efficacy of the antimuscarinic drug scopolamine: a randomized, placebo-controlled clinical trial. Arch Gen Psychiatry 63, 1121–1129 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Drevets WC & Furey ML Replication of scopolamine’s antidepressant efficacy in major depressive disorder: a randomized, placebo-controlled clinical trial. Biol Psychiatry 67, 432–438 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Autry AE, et al. NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature 475, 91–95 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li N, et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 329, 959–964 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beurel E, Song L & Jope RS Inhibition of glycogen synthase kinase-3 is necessary for the rapid antidepressant effect of ketamine in mice. Mol Psychiatry 16, 1068–1070 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nosyreva E, et al. Acute suppression of spontaneous neurotransmission drives synaptic potentiation. J Neurosci 33, 6990–7002 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Suzuki K, Nosyreva E, Hunt KW, Kavalali ET & Monteggia LM Effects of a ketamine metabolite on synaptic NMDAR function. Nature 546, E1–E3 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Ghosal S, et al. Activity-Dependent Brain-Derived Neurotrophic Factor Release Is Required for the Rapid Antidepressant Actions of Scopolamine. Biol Psychiatry 83, 29–37 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Monteggia LM & Kavalali ET Scopolamine and ketamine: evidence of convergence? Biol Psychiatry 74, 712–713 (2013). [DOI] [PubMed] [Google Scholar]
- 13.Voleti B, et al. Scopolamine rapidly increases mammalian target of rapamycin complex 1 signaling, synaptogenesis, and antidepressant behavioral responses. Biol Psychiatry 74, 742–749 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alberini CM Transcription factors in long-term memory and synaptic plasticity. Physiol Rev 89, 121–145 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nan X, Campoy FJ & Bird A MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell 88, 471–481 (1997). [DOI] [PubMed] [Google Scholar]
- 16.Guy J, Cheval H, Selfridge J & Bird A The role of MeCP2 in the brain. Annu Rev Cell Dev Biol 27, 631–652 (2011). [DOI] [PubMed] [Google Scholar]
- 17.Zhou Z, et al. Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron 52, 255–269 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sheng M, Thompson MA & Greenberg ME CREB: a Ca(2+)-regulated transcription factor phosphorylated by calmodulin-dependent kinases. Science 252, 1427–1430 (1991). [DOI] [PubMed] [Google Scholar]
- 19.Tao X, Finkbeiner S, Arnold DB, Shaywitz AJ & Greenberg ME Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron 20, 709–726 (1998). [DOI] [PubMed] [Google Scholar]
- 20.Bourtchuladze R, et al. Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell 79, 59–68 (1994). [DOI] [PubMed] [Google Scholar]
- 21.Finkbeiner S, et al. CREB: a major mediator of neuronal neurotrophin responses. Neuron 19, 1031–1047 (1997). [DOI] [PubMed] [Google Scholar]
- 22.Barbosa AC, et al. MEF2C, a transcription factor that facilitates learning and memory by negative regulation of synapse numbers and function. Proc Natl Acad Sci U S A 105, 9391–9396 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adachi M, Lin PY, Pranav H & Monteggia LM Postnatal Loss of Mef2c Results in Dissociation of Effects on Synapse Number and Learning and Memory. Biol Psychiatry 80, 140–148 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maeng S, et al. Cellular mechanisms underlying the antidepressant effects of ketamine: role of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors. Biol Psychiatry 63, 349–352 (2008). [DOI] [PubMed] [Google Scholar]
- 25.Moda-Sava RN, et al. Sustained rescue of prefrontal circuit dysfunction by antidepressant-induced spine formation. Science 364 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zanos P, et al. NMDAR inhibition-independent antidepressant actions of ketamine metabolites. Nature 533, 481–486 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cohen S, et al. Genome-wide activity-dependent MeCP2 phosphorylation regulates nervous system development and function. Neuron 72, 72–85 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Monteggia LM, et al. Essential role of brain-derived neurotrophic factor in adult hippocampal function. Proc Natl Acad Sci U S A 101, 10827–10832 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim JW & Monteggia LM Increasing doses of ketamine curtail antidepressant responses and suppress associated synaptic signaling pathways. Behav Brain Res 380, 112378 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang K, et al. Essential roles of AMPA receptor GluA1 phosphorylation and presynaptic HCN channels in fast-acting antidepressant responses of ketamine. Sci Signal 9, ra123 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.aan het Rot M, et al. Safety and efficacy of repeated-dose intravenous ketamine for treatment-resistant depression. Biol Psychiatry 67, 139–145 (2010). [DOI] [PubMed] [Google Scholar]
- 32.Murrough JW, et al. Rapid and longer-term antidepressant effects of repeated ketamine infusions in treatment-resistant major depression. Biol Psychiatry 74, 250–256 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Phillips JL, et al. Single, Repeated, and Maintenance Ketamine Infusions for Treatment-Resistant Depression: A Randomized Controlled Trial. Am J Psychiatry 176, 401–409 (2019). [DOI] [PubMed] [Google Scholar]
- 34.Buchthal B, Lau D, Weiss U, Weislogel JM & Bading H Nuclear calcium signaling controls methyl-CpG-binding protein 2 (MeCP2) phosphorylation on serine 421 following synaptic activity. J Biol Chem 287, 30967–30974 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bayer KU & Schulman H CaM Kinase: Still Inspiring at 40. Neuron 103, 380–394 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tobimatsu T & Fujisawa H Tissue-specific expression of four types of rat calmodulin-dependent protein kinase II mRNAs. J Biol Chem 264, 17907–17912 (1989). [PubMed] [Google Scholar]
- 37.Egan MF, et al. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell 112, 257–269 (2003). [DOI] [PubMed] [Google Scholar]
- 38.Scheiderer CL, et al. Sympathetic sprouting drives hippocampal cholinergic reinnervation that prevents loss of a muscarinic receptor-dependent long-term depression at CA3-CA1 synapses. J Neurosci 26, 3745–3756 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mans RA, Warmus BA, Smith CC & McMahon LL An acetylcholinesterase inhibitor, eserine, induces long-term depression at CA3-CA1 synapses in the hippocampus of adult rats. J Neurophysiol 112, 2388–2397 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Holderbach R, Clark K, Moreau JL, Bischofberger J & Normann C Enhanced long-term synaptic depression in an animal model of depression. Biol Psychiatry 62, 92–100 (2007). [DOI] [PubMed] [Google Scholar]
- 41.Mineur YS, et al. Cholinergic signaling in the hippocampus regulates social stress resilience and anxiety- and depression-like behavior. Proc Natl Acad Sci U S A 110, 3573–3578 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Navarria A, et al. Rapid antidepressant actions of scopolamine: Role of medial prefrontal cortex and M1-subtype muscarinic acetylcholine receptors. Neurobiol Dis 82, 254–261 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gourley SL & Taylor JR Recapitulation and reversal of a persistent depression-like syndrome in rodents. Curr Protoc Neurosci Chapter 9, Unit 9 32 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hutchinson AN, Deng JV, Cohen S & West AE Phosphorylation of MeCP2 at Ser421 contributes to chronic antidepressant action. J Neurosci 32, 14355–14363 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Skene PJ, et al. Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol Cell 37, 457–468 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen WG, et al. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science 302, 885–889 (2003). [DOI] [PubMed] [Google Scholar]
- 47.Brocke L, Chiang LW, Wagner PD & Schulman H Functional implications of the subunit composition of neuronal CaM kinase II. J Biol Chem 274, 22713–22722 (1999). [DOI] [PubMed] [Google Scholar]
- 48.Adaikkan C, Taha E, Barrera I, David O & Rosenblum K Calcium/Calmodulin-Dependent Protein Kinase II and Eukaryotic Elongation Factor 2 Kinase Pathways Mediate the Antidepressant Action of Ketamine. Biol Psychiatry 84, 65–75 (2018). [DOI] [PubMed] [Google Scholar]
- 49.Carreno FR, et al. Activation of a ventral hippocampus-medial prefrontal cortex pathway is both necessary and sufficient for an antidepressant response to ketamine. Mol Psychiatry 21, 1298–1308 (2016). [DOI] [PubMed] [Google Scholar]
- 50.Dulawa SC & Janowsky DS Cholinergic regulation of mood: from basic and clinical studies to emerging therapeutics. Mol Psychiatry 24, 694–709 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Reference for Methods
- 51.Chen J, et al. Transgenic animals with inducible, targeted gene expression in brain. Mol Pharmacol 54, 495–503 (1998). [DOI] [PubMed] [Google Scholar]
- 52.Hill JJ, et al. Analysis of pyramidal neuron morphology in an inducible knockout of brain-derived neurotrophic factor. Biol Psychiatry 57, 932–934 (2005). [DOI] [PubMed] [Google Scholar]
- 53.Deng JV, et al. MeCP2 in the nucleus accumbens contributes to neural and behavioral responses to psychostimulants. Nat Neurosci 13, 1128–1136 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw data that supports the findings of the current study are available from the corresponding author upon reasonable request.