

Abstract
Single-electron N-heterocyclic carbene (NHC) catalysis has gained attention recently for the synthesis of C–C bonds. Guided by density functional theory and mechanistic analyses, we report the light-driven synthesis of aliphatic and α-amino ketones using single-electron NHC operators. Computational and experimental results reveal that the reactivity of the key radical intermediate is substrate-dependent and can be modulated through steric and electronic parameters of the NHC. Catalyst potential is harnessed in the visible-light driven generation of an acyl azolium radical species that undergoes selective coupling with various radical partners to afford diverse ketone products. This methodology is showcased in the direct late-stage functionalization of amino acids and pharmaceutical compounds, highlighting the utility of single-electron NHC operators.
Keywords: carbene, catalysis, density functional theory, late-stage functionalization, photochemistry
Graphical Abstract
We developed a single-electron approach for the construction of aliphatic ketones from activated carboxylic acids using merged NHC/photoredox catalysis. Computational studies and mechanistic analysis revealed that reactivity is dependent on both the substrate and NHC catalyst. This methodology was showcased in the direct functionalization of amino acids and bioactive compounds.
There is an ongoing need for mild synthetic methods with broad functional group tolerance for the construction of key bonds in natural products, pharmaceutical compounds, and organic materials. In particular, the formation of carbonyl-containing scaffolds remains a priority due to their synthetic utility and high prevalence in medicinally active compounds (Figure 1A).[1] While many living organisms naturally synthesize ketones through ketogenesis, photosynthesis, etc., chemists have developed numerous approaches to generate ketones through the formation of C–C bonds, among other methods (Figure 1B).[2] Dating back to the 19th century, conventional two-electron methods to access ketones from various acyl electrophiles often require organometallic reagents (Grignard),[3] Lewis acids or strong protic acids (Friedel-Crafts acylation),[4] or transition-metal catalysts (cross-coupling, Figure 1B).[5] Although their utility in synthetic chemistry is undisputed, these protocols are limited in some cases by low regioselectivity, poor functional group tolerance, and generation of corrosive waste. Alternatively, umpolung reactivity offers a distinct alternative to standard two-electron chemistry for the synthesis of ketones as well as other C–C and C–X bonds that would not otherwise be possible (Figure 1B).[6] In recent decades, N-heterocyclic carbenes (NHCs) have transformed the field of umpolung chemistry and emerged as a versatile tool for the catalytic generation of acyl anions, enolates, and homoenolates.[7] The scope of NHC-catalyzed transformations, however, was limited by their inability to engage sp3 electrophiles until the merger of carbene catalysis with single-electron chemistry.
Figure 1.
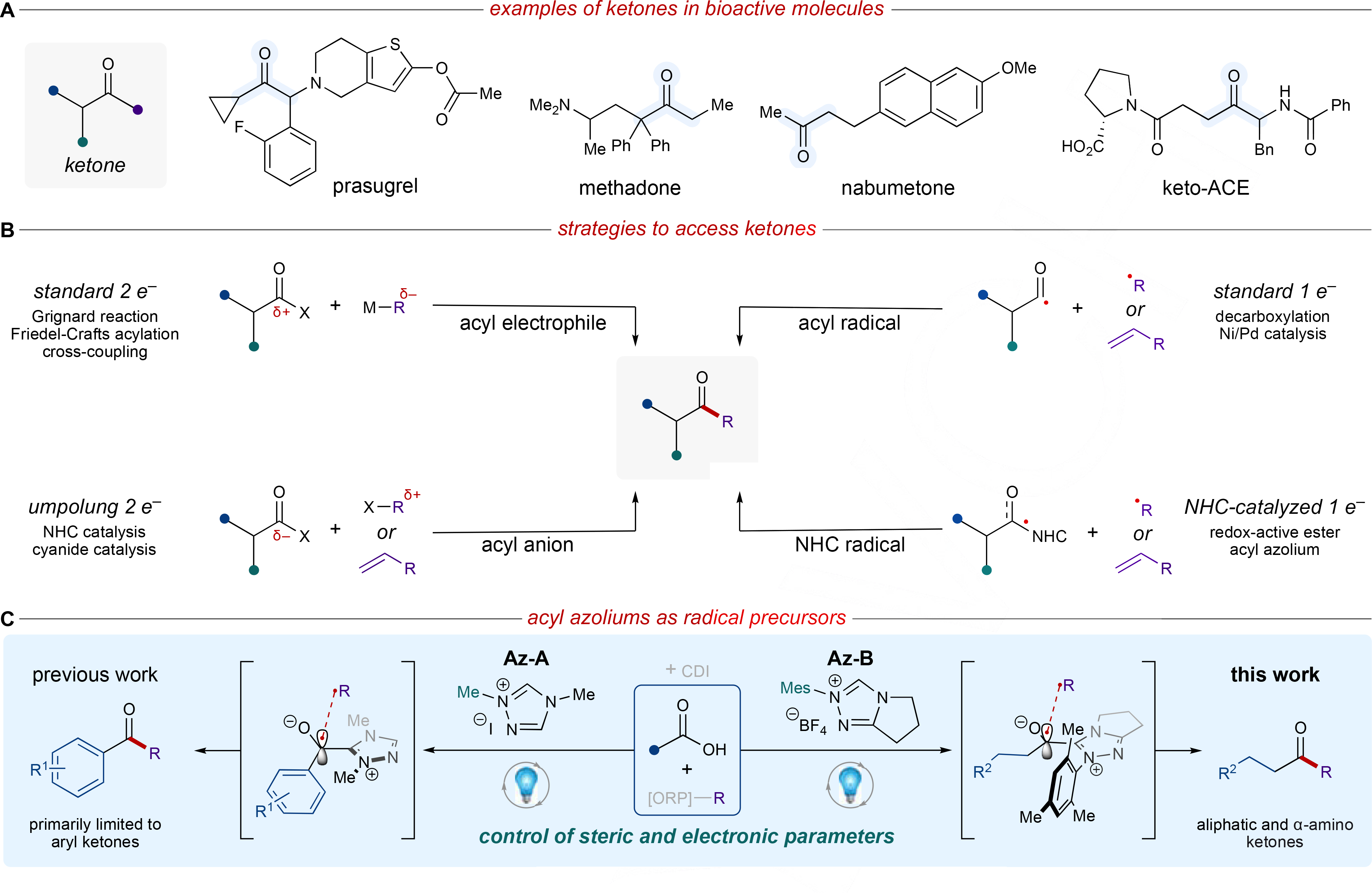
(A) Selected examples of ketones in pharmaceutically relevant compounds. (B) Standard approaches for ketone synthesis. (C) Acyl azolium radicals for the construction of aryl, alkyl, and α-amino ketones; ORP = oxidatively generated radical precursor, CDI = carbonyldiimidazole.
The recent renaissance of single-electron chemistry via photoredox catalysis and electrocatalysis has further enabled bond connections that were previously inaccessible using traditional pathways. While radical chemistry is well-established, the revival of photocatalysis and electrosynthesis has accelerated modern synthetic chemistry.[8] These advances have furnished numerous strategies for the synthesis of ketones (Figure 1B).[9] For example, photoredox/nickel dual catalysis has gained attention for its ability to synthesize ketone moieties via cross-coupling between nucleophilic acyl radicals and other radical species (e.g., aryl radicals from aryl halides).[10] New modes of reactivity have stemmed from these advances in radical chemistry, and the merging of single-electron chemistry with umpolung reactivity has facilitated even more opportunities.
While NHC-derived radical reactivity dates back to the early 2000s,[11] there has been a marked increase in single-electron NHC-catalyzed methods for the construction of C–C bonds reported in recent years (Figure 1B).[12] In 2019, Ohmiya and coworkers described the coupling of a persistent Breslow intermediate-derived radical with a transient alkyl radical.[13] This pioneering example of thermal single-electron NHC catalysis inspired the development of similar protocols by the groups of Li,[14] Wang,[15] and Hong,[16] among others.[17]
Concurrent with this work, light-driven carbene methods by our group,[18] Hopkinson,[19] Studer,[20] and others demonstrated complementary reactivity to established two-electron carbene catalysis.[20c, 21] In 2020, Hopkinson and coworkers reported a light-activated, NHC-catalyzed Diels-Alder reaction to construct isochromanone derivatives.[19a] Our group reported the first single-electron reduction of an acyl azolium for the formation of ketones from activated carboxylic acids.[18a] Radical-radical coupling of a Hantzsch ester-derived alkyl radical with an acyl azolium radical was achieved via a reductive quenching cycle. While this work highlighted the utility of reductively generated single-electron NHC operators, the scope was primarily limited to aryl substrates, a widespread limitation in the majority of recent radical NHC processes (Figure 1C).
The work described herein uses in-depth computational studies to shed light on key reactive intermediates, thus guiding hypothesis-driven experimentation to greatly expand this ketone synthesis platform (Figure 1C). In 2019, Bertrand and Martin disclosed a detailed study on the key radical intermediates in oxidative NHC processes.[22] Contrary to many mechanistic proposals in the literature, their studies suggest that these oxidative pathways proceed through single-electron transfer (SET) from the corresponding electron-rich enolate of the Breslow intermediate or through proton-coupled electron transfer (PCET) from the enol. They conclude that the relevant radical intermediates in these reactions have spin density concentrated primarily on the carbene carbon (C2, 40%) and not on the carbonyl carbon (C1, 10%).[22] While these results do not directly translate to our system due to substantial differences in radical generation (i.e., thermal vs. photochemical), their work provoked important mechanistic questions regarding the nature of single-electron NHC operators.
Based on Bertrand’s report,[22] our previous results,[18a] and a key report from Ohmiya,[23] we hypothesized that both electronic and steric alterations to the NHC could impact radical stability and accessibility, thus imparting control over reactivity. To explore this possibility, we performed density functional theory investigations with PBE[24]/6–31G*[25] & LANL2DZ[26] (for Cs) level of theory and SMD solvation corrections[27] in acetonitrile as implemented in Gaussian 16. Acyl azoliums derived from benzoic acid and hydrocinnamic acid were chosen as the model substrates to study the reactivity of aryl and aliphatic systems, respectively (Figure 1C, R1 = H and R2 = Ph, respectively). To analyze the steric and electronic impact of the NHC, our previously optimized NHC precursor (Az-A) and sterically encumbering N-mesityl pyrrolotriazolium (Az-B), which has been employed in other single-electron NHC reports,[20] were the primary focus of this study (Figure 1C).
An initial spin density analysis of the optimized aliphatic and aryl acyl azolium radical structures (III) with Az-A or Az-B revealed significant radical character on both C1 and C2 (Figure 2A, see SI, page S53). It is often assumed that the spin density population is concentrated on C1 and, as a result, selective coupling occurs with the C1-centered radical.[12] However, we propose that there are at least two operative radical-radical coupling pathways: 1) coupling at C1, which directly leads to product, and 2) coupling at C2, which may lead to unproductive off-cycle side reactivity.
Figure 2.
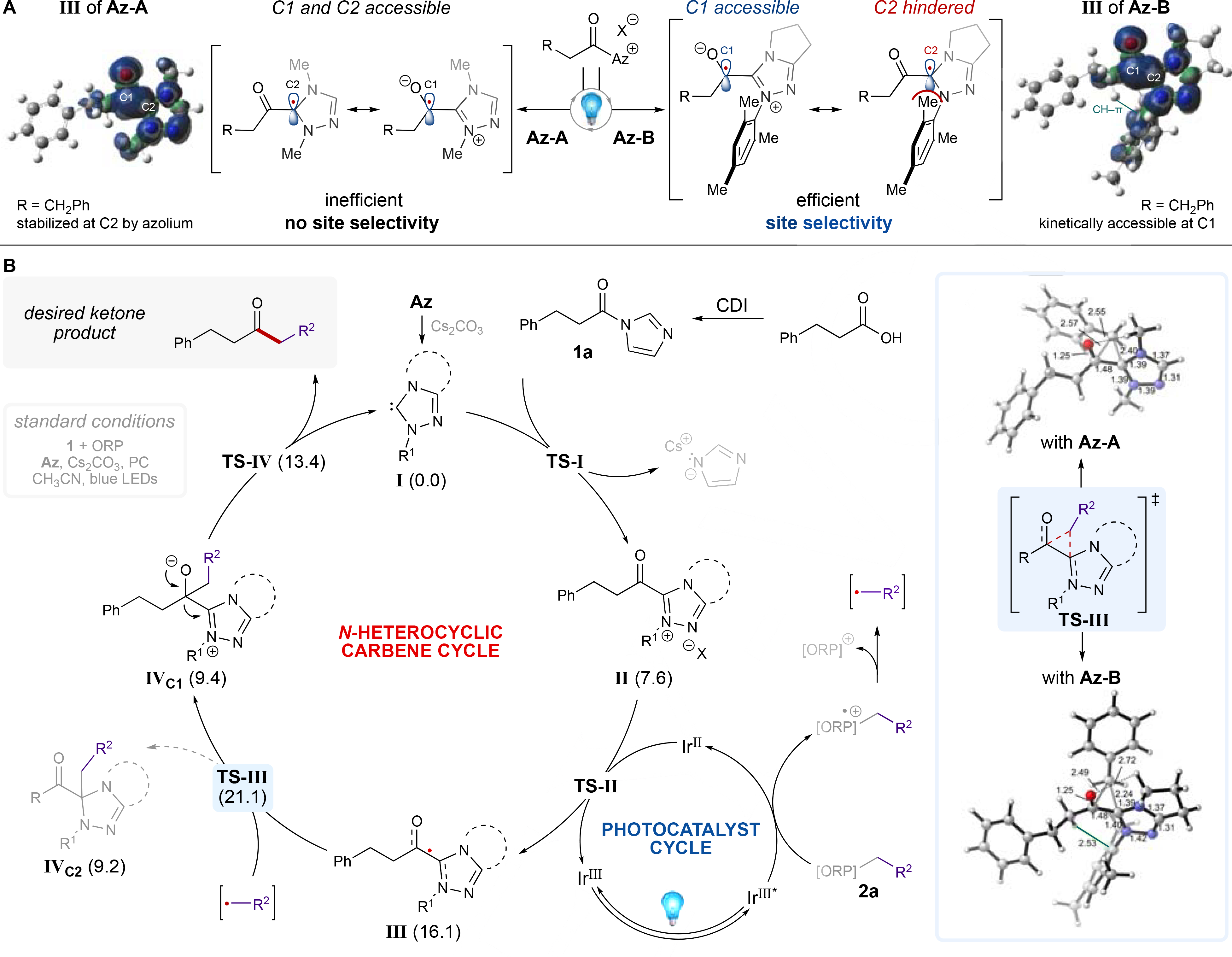
(A) Radical coupling accessibility model for aliphatic acyl azoliums with spin density distributions of optimized aliphatic acyl azolium structures (R = CH2Ph). (B) Proposed mechanism for the combined NHC/photoredox-catalyzed construction of aliphatic and α-amino ketones. Reported energies are for Az-B in kcal/mol.
Analysis of computed isodesmic reactions suggests that radical stability at C1 and C2 may be substrate dependent (see SI, page S56-S57). The extended conjugation in aryl substrates stabilizes the radical at C1, minimizing the effect that NHC structure has on reactivity. Alternatively, for aliphatic substrates, there is increased radical stability at C2 due to the lack of substrate conjugation, thus making reactivity more NHC-dependent. This C2 stability likely contributed to the limited reactivity of aliphatic substrates in our previous work.[18a]
We then investigated whether NHC steric parameters can influence productive coupling at C1 for aliphatic substrates. When we juxtaposed acyl azolium radicals III derived from Az-A and Az-B, different conformations were observed (see SI, page S58). Due to the torsional flexibility of the aliphatic substrate and the orthogonality of the mesityl group, Az-B rotates 180° with respect to the carbonyl to form a stabilizing CH-π interaction (1.5 kcal/mol more stable), potentially leading to efficacy of Az-B over Az-A (Figure 2A–B). Moreover, this interaction rigidifies the conformation, enforces the steric encumbrance imposed by the mesityl ring, and, thus, may facilitate favorable coupling at kinetically accessible C1 (Figure 2A).[28]
Given these results, we propose a combined NHC and photocatalytic mechanism for the synthesis of aliphatic ketones (Figure 2B). Operating through a reductive quenching photocatalytic cycle, the radical coupling partner is accessed via a single-electron oxidation event with the excited photocatalyst. Subsequent single-electron reduction of the acyl azolium (II, ΔG = 7.6 kcal/mol) furnishes an open-shell acyl azolium radical species (III, ΔG = 16.1 kcal/mol) with the radical distributed between C1 and C2. The radical coupling partner then approaches III to give IVC1 and IVC2 via a single three-membered transition state (TS-III, ΔG‡ = 21.1 kcal/mol for benzyl radical) with bonds forming between the benzyl radical center, C1 and C2. We computed the potential energy surfaces (PES) around TS-III for the aliphatic acyl azolium radicals III of Az-A and Az-B with the benzyl radical to further rationalize which carbon, C1 or C2, will undergo the radical-radical coupling (Figure 3, see SI, pages S60-S62). The Az-A PES reveals downhill energy paths leading to both C1- and C2-coupling products, suggesting a non-selective reaction wherein considerable IVC2 is formed. For Az-B, the PES features a downhill energy path to desired C1. In contrast, an uphill barrier exists on the path to undesired C2, which translates to a preference of 2.3 kcal/mol for C1 over C2, in line with our hypothesis that the mesityl group of Az-B may hinder coupling at the C2 center. These results suggest a greater likelihood for efficient C1-coupling over inefficient C2-coupling using Az-B as the NHC precursor (IVC1 and IVC2, ΔG = 9.4 and 9.2 kcal/mol, respectively). Finally, release of the NHC catalyst (TS-IV, ΔG‡ = 13.4 kcal/mol) affords the ketone product (ΔG = –16.3 kcal/mol).
Figure 3.
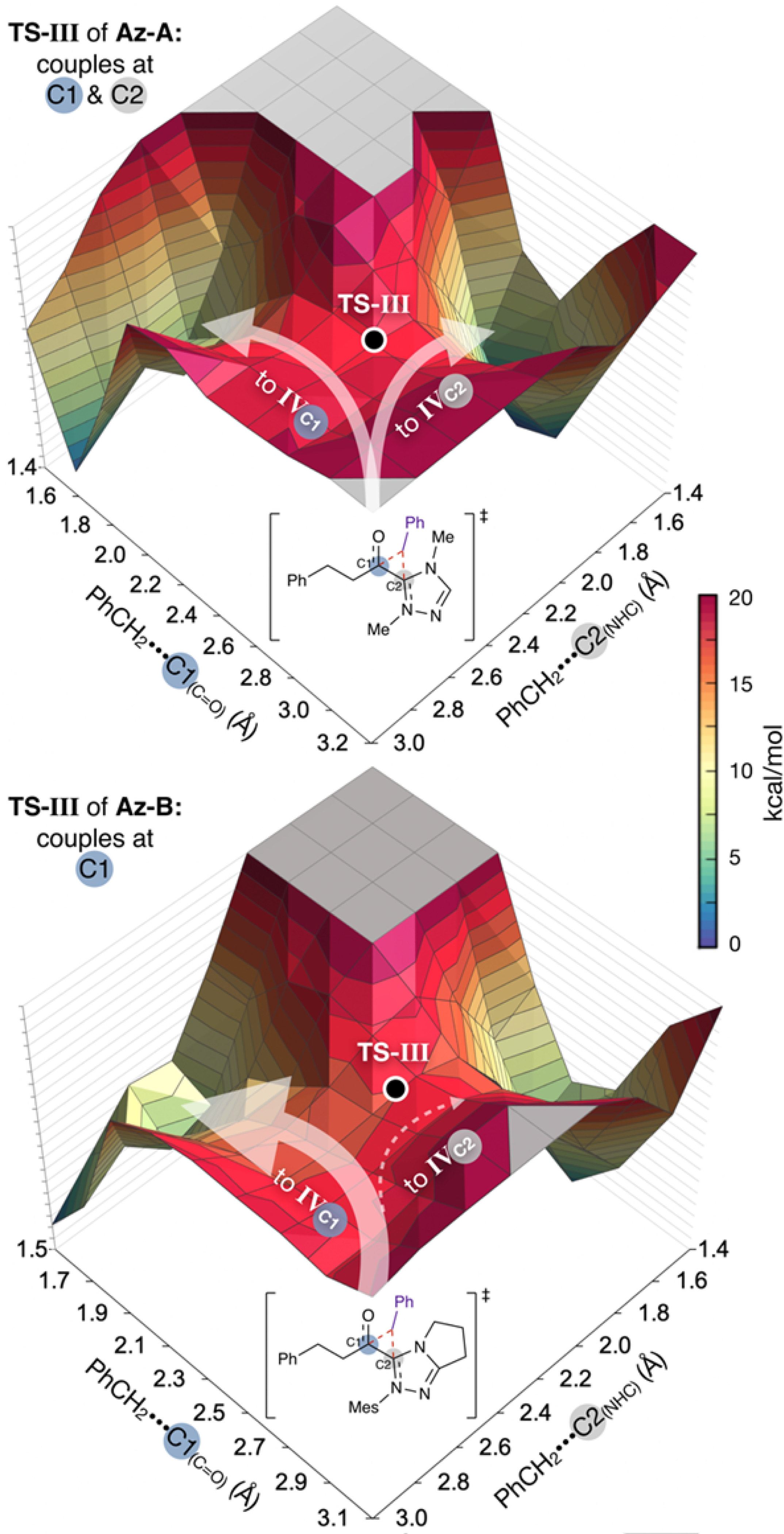
Computed potential energy surfaces for benzyl radical coordination to C1 or C2 of the aliphatic acyl azolium radical (III) with Az-A or Az-B.
This proposed mechanism thus directed experimental efforts to explore and validate our hypotheses (see SI, page S13-S16). A survey of selected azoliums revealed key trends in reactivity: yields decreased with electron withdrawing or highly conjugated NHC substituents and increased with sterically encumbering substituents. As predicted by our mechanistic analysis, the delicate balance of steric and electronic properties offered by Az-B enabled the benzylation of 1a to proceed with significantly less side reactivity compared to Az-A (see SI, page S15) and afforded the desired aliphatic ketone 3a in 72% yield (Table 1).
Table 1.
Substrate scope.[a]
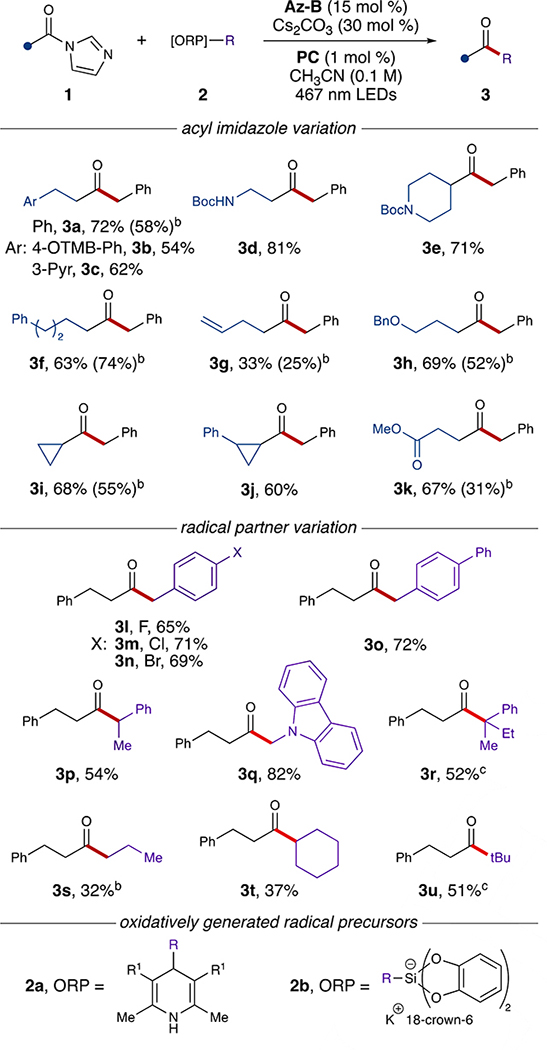 |
Reaction used 2a (R1 = CO2Et) and PC-1 ([Ir(dF[CF3]ppy)2(dtbpy)]PF6) unless otherwise noted.
Reaction used 2b and PC-2 (3DPAFIPN).
Reaction used 2a (R1 = CN) and PC-3 (4CzIPN). See SI for reaction details.
Given this result, the scope of aliphatic acyl imidazoles amenable to benzylation was investigated using Az-B as the NHC precursor (Table 1). Substituted hydrocinnamic acyl imidazoles were tolerated to provide the respective ketones in good yields (3b,c). The reaction proceeded with nitrogen- and ether-containing substrates, affording the desired products (3d,e,h) in good-to-excellent yields. Albeit low-yielding due to radical addition into the alkene, unsaturated 3g was successfully isolated. The reaction with cyclopropane acyl imidazoles furnished the respective ketone products (3i,j) in good yields, an unexpected result that provides additional mechanistic insight into the nature of III. Compared to traditional acyl radicals, which have demonstrated radical clock-like reactivity,[29] the distribution of radical density between C1 and C2 affords increased stability, thus illustrating a significant advantage of NHC-based radical chemistry. The chemoselectivity of this process was demonstrated in the isolation of ester-containing 3k, illustrating a bond construction that suffers from functional group incompatibility using conventional methods of ketone formation (e.g., Grignard reaction, see SI, page S17). In general, use of Az-B instead of Az-A with aliphatic substrates resulted in significant increases in yield (e.g., 32% increase for 3a and 64% increase for 3d) and enabled inclusion of substrates that demonstrated little-to-no reactivity using Az-A (e.g., 3g and 3k).
To further increase the practicality and overall utility of this method, the reaction with other radical coupling partners was also studied.[30] In addition to their ease of preparation, bis-catecholato silicates are bench stable and offer excellent substrate diversity. Minor modification of the reaction conditions enabled reactivity to be achieved using bis-catecholato silicates as an alternative radical precursor (2b), affording selected ketone products in comparable yields to those using Hantzsch esters (Table 1). In addition to tolerating different radical precursors as coupling partners, it was also determined that an organophotocatalyst (PC-2) could be substituted for PC-1, providing a more cost-effective variant for alkylation. A survey of Hantzsch esters (2a, R1 = CO2Et), Meyer nitriles (2a, R1 = CN), and silicates (2b) enabled addition of various functional groups while demonstrating the array of coupling partners viable in this reaction (Table 1). Benzyl Hantzsch esters containing electron-withdrawing and electron-donating groups afforded ketones (3l-3o) in good yields. Carbazole-containing 3q was isolated in high yield, offering a potential method to tag molecular probes for fluorescence studies.[31] The reaction proceeded smoothly with sterically congested radicals (3p,r) as well as primary, secondary, and tertiary alkyl radicals (3s-3u).
The direct functionalization of amino acids was achieved via in situ activation with carbonyldiimidazole (CDI). Various amino acids, including alkyl amino acids (e.g., N-Boc-L-valine), aromatic amino acids (e.g., N-Boc-L-tryptophan), and others were suitable for direct benzylation to afford α-amino ketones 4a-4g in good-to- excellent yields (Table 2A).[32] Even under the slightly basic conditions of this reaction, α-amino ketone 4b was prepared directly from N-Boc-L-valine with moderate stereoretention (87:13 er), suggesting that alkylation of chiral carboxylic acids is possible without complete erosion of stereochemistry.
Table 2.
(A) Synthesis of α-amino ketones via direct amino acid functionalization. (B) Late-stage functionalization of biologically-active compounds.[a]
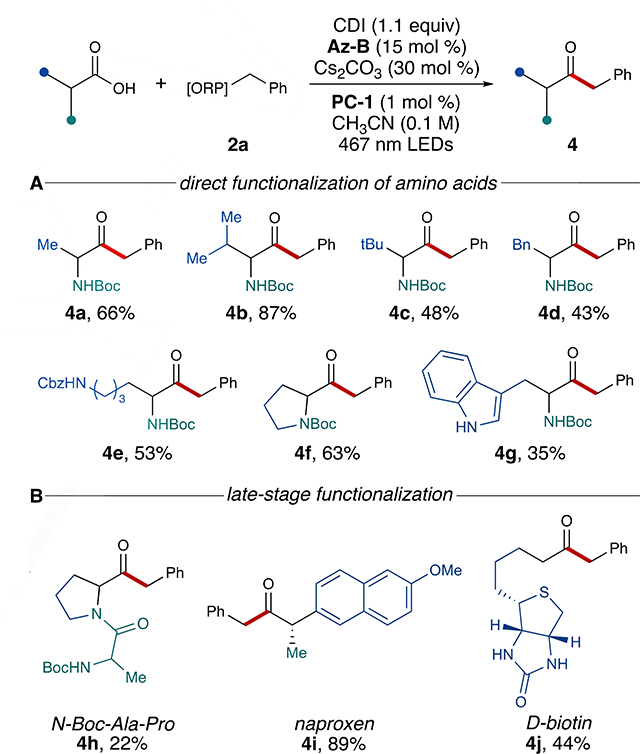 |
See SI for reaction details.
Lastly, the late-stage functionalization of bioactive compounds was accomplished using the same one-step protocol (Table 2B). The benzylation of a dipeptide was successful (4h), suggesting that this methodology may be suitable for converting short peptides into their ketone counterparts with additional optimization. Naproxen, an anti-inflammatory drug used to treat bone disorders, was directly benzylated to afford the corresponding ketone (4i) in excellent yield. The late-stage functionalization of heterobicyclic D-biotin, a vitamin essential to metabolism, was also achieved to give 4j in moderate yield, further highlighting the synthetic utility of this process.
The work described herein was developed via hypothesis-driven experimentation guided by computational analysis. In addressing a common limitation of single-electron NHC processes, we revealed mechanistic insights that may be applied to this catalysis platform to further advance the field, expand substrate scopes, and increase the utility of NHCs in synthesis. The modularity of these single-electron operators was highlighted in a combined NHC-catalyzed and photoredox protocol to synthesize aliphatic ketones from activated carboxylic acids; modulation of NHC steric and electronic parameters enabled increased yields of up to 65% compared to our previous results. In addition to significantly expanding the scope of this reaction, amino acids were directly functionalized to afford the corresponding α-amino ketones using a one-pot procedure, and the utility of this method was showcased in the late-state tailoring of bioactive compounds. Future correlation of computed results with ongoing experimental outcomes is expected to reveal additional mechanistic details regarding what factors control the PES to govern the selectivity of single-electron carbene-catalyzed methods.
Supplementary Material
Acknowledgements
The authors thank Northwestern University and the National Institute of General Medical Sciences (R35 GM136440) for support of this work. We also thank Ada Kwong (NU) and Saman Shafaie (NU) for assistance with HRMS. PHYC is the Bert and Emelyn Christensen Professor and gratefully acknowledges financial support from the Vicki & Patrick F. Stone family and the computing infrastructure in part provided by the National Science Foundation (CHE-1352663 and NSF Phase-2 CCI, Center for Sustainable Materials Chemistry CHE-1102637).
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].a) Chen KK, Ann. N.Y. Acad. Sci 1948, 51, 83–97; [DOI] [PubMed] [Google Scholar]; b) Boyle EA, Freemanm PC, Mangan FR, Thomson MJ, J. Pharm. Pharmacol 1982, 34, 562–569; [DOI] [PubMed] [Google Scholar]; c) Tanaka T, Kawase M, Tani S, Biorg. Med. Chem. 2004, 12, 501–505; [DOI] [PubMed] [Google Scholar]; d) Redelinghuys P, Nchinda AT, Chibale K, Sturrock ED, Biol. Chem 2006, 387, 461–466; [DOI] [PubMed] [Google Scholar]; e) Riley AB, Tafreshi MJ, Haber SL, Am. J. Health-Syst. Pharm 2008, 65, 1019–1028; [DOI] [PubMed] [Google Scholar]; f) Hoyos P, Sinisterra J-V, Molinari F, Alcántara AR, Domínguez de María P, Acc. Chem. Res 2010, 43, 288–299; [DOI] [PubMed] [Google Scholar]; g) Liu Y, Han S-J, Liu W-B, Stoltz BM, Acc. Chem. Res 2015, 48, 740–751; [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Allen LAT, Raclea R-C, Natho P, Parsons PJ, Org. Biomol. Chem 2021, 19, 498–513. [DOI] [PubMed] [Google Scholar]
- [2].a) Milstein D, Stille JK, J. Am. Chem. Soc 1978, 100, 3636–3638; [Google Scholar]; b) Richardson SK, in General and Synthetic Methods: Volume 14, Vol. 14 (Ed.: Pattenden G), The Royal Society of Chemistry, 1992, pp. 26–62; [Google Scholar]; c) Dieter RK, Tetrahedron 1999, 55, 4177–4236; [Google Scholar]; d) Zhou F, Li C-J, Nat. Commun 2014, 5, 4254; [DOI] [PubMed] [Google Scholar]; e) Miles DH, Guasch J, Toste FD, J. Am. Chem. Soc 2015, 137, 7632–7635; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Zhang M, Xie J, Zhu C, Nat. Commun 2018, 9, 3517; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Ruzi R, Liu K, Zhu C, Xie J, Nat. Commun 2020, 11, 3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].a) Grignard V, Hebd CR. Séances. Acad. Sci 1900, 130, 1322– 1325; [Google Scholar]; b) Nahm S, Weinreb SM, Tetrahedron Lett. 1981, 22, 3815–3818; [Google Scholar]; c) Alonso F, Lorenzo E, Yus M, J. Org. Chem 1996, 61, 6058–6059; [Google Scholar]; d) Knochel P, Dohle W, Gommermann N, Kneisel FF, Kopp F, Korn T, Sapountzis I, Vu VA, Angew. Chem. Int. Ed 2003, 42, 4302–4320; [DOI] [PubMed] [Google Scholar]; e) Kagan HB, Angew. Chem. Int. Ed 2012, 51, 7376–7382; [DOI] [PubMed] [Google Scholar]; f) Colas K, dos Santos ACVD, Mendoza A, Org. Lett 2019, 21, 7908–7913. [DOI] [PubMed] [Google Scholar]
- [4].a) Rueping M, Nachtsheim BJ, Beilstein J Org. Chem 2010, 6, 6; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Heravi MM, Zadsirjan V, Saedi P, Momeni T, RSC Adv. 2018, 8, 40061–40163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Yin H, Zhao C, You H, Lin K, Gong H, Chem. Commun 2012, 48, 7034–7036; [DOI] [PubMed] [Google Scholar]; b) Cherney AH, Kadunce NT, Reisman SE, J. Am. Chem. Soc 2013, 135, 7442–7445; [DOI] [PubMed] [Google Scholar]; c) Buchspies J, Szostak M, Catalysts 2019, 9, 53. [Google Scholar]
- [6].a) Johnson JS, Angew. Chem. Int. Ed 2004, 43, 1326–1328; [DOI] [PubMed] [Google Scholar]; b) Bugaut X, Glorius F, Chem. Soc. Rev 2012, 41, 3511–3522; [DOI] [PubMed] [Google Scholar]; c) Sanz-Marco A, Martinez-Erro S, Pauze M, Gómez-Bengoa E, Martín-Matute B, Nat. Commun 2019, 10, 5244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a) For reviews on NHC catalysis, see: Enders D, Niemeier O, Henseler A, Chem. Rev 2007, 107, 5606–5655; [DOI] [PubMed] [Google Scholar]; b) Biju AT, Kuhl N, Glorius F, Acc. Chem. Res 2011, 44, 1182–1195; [DOI] [PubMed] [Google Scholar]; c) Nair V, Menon RS, Biju AT, Sinu CR, Paul RR, Jose A, Sreekumar V, Chem. Soc. Rev 2011, 40, 5336–5346; [DOI] [PubMed] [Google Scholar]; d) Ryan SJ, Candish L, Lupton DW, Chem. Soc. Rev 2013, 42, 4906–4917; [DOI] [PubMed] [Google Scholar]; e) Hopkinson MN, Richter C, Schedler M, Glorius F, Nature 2014, 510, 485–496; [DOI] [PubMed] [Google Scholar]; f) Mahatthananchai J, Bode JW, Acc. Chem. Res 2014, 47, 696–707; [DOI] [PubMed] [Google Scholar]; g) Flanigan DM, Romanov-Michailidis F, White NA, Rovis T, Chem. Rev 2015, 115, 9307–9387; [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Wang MH, Scheidt KA, Angew. Chem. Int. Ed 2016, 55, 14912–14922; [DOI] [PubMed] [Google Scholar]; i) Chen X, Wang H, Jin Z, Chi YR, Chin. J. Chem 2020, 38, 1167–1202. [Google Scholar]
- [8].a) For reviews and selected work on photochemistry and electrochemistry, see: Yoon TP, Ischay MA, Du J, Nat. Chem 2010, 2, 527–532; [DOI] [PubMed] [Google Scholar]; b) Xuan J, Xiao W-J, Angew. Chem. Int. Ed 2012, 51, 6828–6838; [DOI] [PubMed] [Google Scholar]; c) Prier CK, Rankic DA, MacMillan DWC, Chem. Rev 2013, 113, 5322–5363; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Francke R, Little RD, Chem. Soc. Rev 2014, 43, 2492–2521; [DOI] [PubMed] [Google Scholar]; e) Romero NA, Nicewicz DA, Chem. Rev 2016, 116, 10075–10166; [DOI] [PubMed] [Google Scholar]; f) Skubi KL, Blum TR, Yoon TP, Chem. Rev 2016, 116, 10035–10074; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Shaw MH, Twilton J, MacMillan DWC, J. Org. Chem 2016, 81, 6898–6926; [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Yan M, Kawamata Y, Baran PS, Chem. Rev 2017, 117, 13230–13319; [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Huang H, Yu C, Zhang Y, Zhang Y, Mariano PS, Wang W, J. Am. Chem. Soc 2017, 139, 9799–9802; [DOI] [PubMed] [Google Scholar]; j) Zhang S, Li L, Li J, Shi J, Xu K, Gao W, Zong L, Li G, Findlater M, Angew. Chem. Int. Ed 2021, 7275. [DOI] [PubMed] [Google Scholar]
- [9].a) Deb A, Manna S, Modak A, Patra T, Maity S, Maiti D, Angew. Chem. Int. Ed 2013, 52, 9747–9750; [DOI] [PubMed] [Google Scholar]; b) Chen W, Liu Z, Tian J, Li J, Ma J, Cheng X, Li G, J. Am. Chem. Soc 2016, 138, 12312–12315; [DOI] [PubMed] [Google Scholar]; c) Raviola C, Protti S, Ravelli D, Fagnoni M, Green Chem. 2019, 21, 748–764; [Google Scholar]; d) Ni S, Padial NM, Kingston C, Vantourout JC, Schmitt DC, Edwards JT, Kruszyk MM, Merchant RR, Msykhailiuk PK, Sanchez BB, Yang S, Perry MA, Gallego GM, Mousseau JJ, Collins MR, Cherney RJ, Lebed PS, Chen JS, Qin T, Baran PS, J. Am. Chem. Soc 2019, 141, 6726–6739; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Chalotra N, Sultan S, Shah BA, Asian J. Org. Chem 2020, 9, 863–881. [Google Scholar]
- [10].a) Zuo Z, Ahneman DT, Chu L, Terrett JA, Doyle AG, MacMillan DWC, Science 2014, 345, 437–440; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chu L, Lipshultz JM, MacMillan DWC, Angew. Chem. Int. Ed 2015, 54, 7929–7933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].a) Chabrière E, Vernède X, Guigliarelli B, Charon M-H, Hatchikian EC, Fontecilla-Camps JC, Science 2001, 294, 2559–2563; [DOI] [PubMed] [Google Scholar]; b) Ragsdale SW, Chem. Rev 2003, 103, 2333; [DOI] [PubMed] [Google Scholar]; c) Guin J, De Sarkar S, Grimme S, Studer A, Angew. Chem. Int. Ed 2008, 47, 8727–8730; [DOI] [PubMed] [Google Scholar]; d) Kluger R, Tittmann K, Chem. Rev 2008, 108, 1797–1833; [DOI] [PubMed] [Google Scholar]; e) White NA, Rovis T, J. Am. Chem. Soc 2014, 136, 14674–14677; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Zhang Y, Du Y, Huang Z, Xu J, Wu X, Wang Y, Wang M, Yang S, Webster RD, Chi YR, J. Am. Chem. Soc 2015, 137, 2416–2419. [DOI] [PubMed] [Google Scholar]
- [12].a) For reviews on single-electron NHC catalysis, see: Ishii T, Nagao K, Ohmiya H, Chem. Sci 2020, 11, 5630–5636; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Li Q-Z, Zeng R, Han B, Li J-L, Chem. Eur. J 2021, 3238–3250; [DOI] [PubMed] [Google Scholar]; c) Liu Q, Chen X-Y, Org. Chem. Front 2020, 7, 2082–2087; [Google Scholar]; d) Mavroskoufis A, Jakob M, Hopkinson MN, ChemPhotoChem 2020, 4, 5147–5153; [Google Scholar]; e) Dai L, Ye S, Chin. Chem. Lett 2020, 32, 660–667. [Google Scholar]
- [13].Ishii T, Kakeno Y, Nagao K, Ohmiya H, J. Am. Chem. Soc 2019, 141, 3854–3858. [DOI] [PubMed] [Google Scholar]
- [14].Li J-L, Liu Y-Q, Zou W-L, Zeng R, Zhang X, Liu Y, Han B, He Y, Leng H-J, Li Q-Z, Angew. Chem. Int. Ed 2020, 59, 1863–1870. [DOI] [PubMed] [Google Scholar]
- [15].Zhang B, Peng Q, Guo D, Wang J, Org. Lett 2020, 22, 443–447. [DOI] [PubMed] [Google Scholar]
- [16].Kim I, Im H, Lee H, Hong S, Chem. Sci 2020, 11, 3192–3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].a) Ishii T, Ota K, Nagao K, Ohmiya H, J. Am. Chem. Soc 2019, 141, 14073–14077; [DOI] [PubMed] [Google Scholar]; b) Yang H-B, Wang Z-H, Li J-M, Wu C, Chem. Commun 2020, 56, 3801–3804. [DOI] [PubMed] [Google Scholar]
- [18].a) Bay AV, Fitzpatrick KP, Betori RC, Scheidt KA, Angew. Chem. Int. Ed. 2020, 59, 9143–9148; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Bayly AA, McDonald BR, Mrksich M, Scheidt KA, Proc. Natl. Acad. Sci. U. S. A 2020, 117, 13261–13266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].a) Mavroskoufis A, Rajes K, Golz P, Agrawal A, Ruß V, Götze JP, Hopkinson MN, Angew. Chem. Int. Ed. 2020, 59, 3190–3194; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hopkinson MN, Mavroskoufis A, Synlett 2021, 32, 95–101. [Google Scholar]
- [20].a) Liu K, Studer A, J. Am. Chem. Soc 2021, 143, 4903–4909; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Meng Q-Y, Lezius L, Studer A, Nat. Commun 2021, 12, 2068; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Meng Q-Y, Döben N, Studer A, Angew. Chem. Int. Ed 2020, 59, 19956–19960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].a) Dai L, Xia Z-H, Gao Y-Y, Gao Z-H, Ye S, Angew. Chem. Int. Ed 2019, 58, 18124–18130; [DOI] [PubMed] [Google Scholar]; b) Ren S-C, Lv W-X, Yang X, Yan J-L, Xu J, Wang F-X, Hao L, Chai H, Jin Z, Chi YR, ACS Catal. 2021, 11 2925–2934. [Google Scholar]
- [22].Regnier V, Romero EA, Molton F, Jazzar R, Bertrand G, Martin D, J. Am. Chem. Soc 2019, 141, 1109–1117. [DOI] [PubMed] [Google Scholar]
- [23].Kakeno Y, Kusakabe M, Nagao K, Ohmiya H, ACS Catal. 2020, 10, 8524–8529. [Google Scholar]
- [24].a) Perdew JP, Burke K, Ernzerhof M, Phys. Rev. Lett 1996, 77, 3865–3868; [DOI] [PubMed] [Google Scholar]; b) Perdew JP, Burke K, Ernzerhof M, Phys. Rev. Lett 1997, 78, 1396–1396. [DOI] [PubMed] [Google Scholar]
- [25].Hehre WJ, Ditchfield R, Pople JA, J. Chem. Phys 1972, 56, 2257–2261. [Google Scholar]
- [26].Hay PJ, Wadt WR, J. Chem. Phys 1985, 82, 270–283. [Google Scholar]
- [27].Marenich AV, Cramer CJ, Truhlar DG, J. Phys. Chem. B 2009, 113, 6378–6396. [DOI] [PubMed] [Google Scholar]
- [28].Clavier H, Nolan SP, Chem. Commun 2010, 46, 841–861. [DOI] [PubMed] [Google Scholar]
- [29].a) Liu J, Liu X-P, Wu H, Wei Y, Lu F-D, Guo K-R, Cheng Y, Xiao W-J, Chem. Commun 2020, 56, 11508–11511; [DOI] [PubMed] [Google Scholar]; b) Leifert D, Studer A, Angew. Chem. Int. Ed 2020, 59, 74–108. [DOI] [PubMed] [Google Scholar]
- [30].a) Nakajima K, Nojima S, Sakata K, Nishibayashi Y, ChemCatChem 2016, 8, 1028–1032; [Google Scholar]; b) Wang P-Z, Chen J-R, Xiao W-J, Org. Biomol. Chem 2019, 17, 6936–6951; [DOI] [PubMed] [Google Scholar]; c) Molander GA, J. Org. Chem 2015, 80, 7837–7848; [DOI] [PubMed] [Google Scholar]; d) Jouffroy M, Primer DN, Molander GA, J. Am. Chem. Soc 2016, 138, 475–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].a) Uoyama H, Goushi K, Shizu K, Nomura H, Adachi C, Nature 2012, 492, 234–238; [DOI] [PubMed] [Google Scholar]; b) Hörner A, Volz D, Hagendorn T, Fürniss D, Greb L, Rönicke F, Nieger M, Schepers U, Bräse S, RSC Adv. 2014, 4, 11528–11534; [Google Scholar]; c) Yu Y, Li N, Jin Q, Ji Z, Sun Z, Li G, Zhang S, You J, Microchem. J 2019, 145, 9–17. [Google Scholar]
- [32]. Due to the propsensity of free amines to be oxidized under photocatalytic conditions, this reaction currently necesitates the use of N-protecting groups.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.