

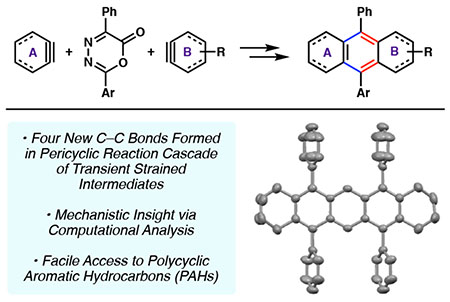
Abstract
We report a computational and experimental study of the reaction of oxadiazinones and strained alkynes to give polycyclic aromatic hydrocarbons (PAHs). The reaction proceeds by way of a pericyclic reaction cascade and leads to the formation of four new carbon–carbon bonds. Using M06-2X DFT calculations, we interrogate several mechanistic aspects of the reaction, such as why the use of non-aromatic strained alkynes can be used to access unsymmetrical PAHs, whereas the use of arynes in the methodology leads to symmetrical PAHs. In addition, experimental studies enable the rapid synthesis of new PAHs, including tetracene and pentacene scaffolds. These studies not only provide fundamental insight regarding the aforementioned cycloaddition cascades and synthetic access to PAH scaffolds, but are also expected to enable the synthesis of new materials.
Keywords: cyclic alkynes, arynes, cycloadditions, density functional theory, polycyclic aromatic hydrocarbons
Graphical Abstract
A computational and experimental study on the reaction of oxadiazinones and strained alkynes to give polycyclic aromatic hydrocarbons (PAHs) is reported. Several mechanistic aspects of the reaction, such as why the use of non-aromatic strained alkynes can be used to access unsymmetrical PAHs whereas the use of arynes in the methodology leads to symmetrical PAHs, are investigated. In addition, experimental studies enable the rapid synthesis of new PAHs, including tetracene and pentacene scaffolds.
Introduction
Strained cyclic alkynes and arynes were once avoided in organic synthesis because of their high reactivity and fleeting nature.[1] However, over the past two decades in particular, experimental and computational studies have expanded the utility of cyclic alkynes and arynes, while also enabling studies pertaining to reactivity and selectivity.[2,3,4] Notably, the use of Kobayashi silyl triflate precursors[2] to access these strained intermediates has paved the way for many synthetic advances. As such, strained cyclic alkynes and arynes have been employed in the synthesis of natural products,[5a] medicinally privileged scaffolds,[5b] ligands,[5c,5d] agrochemicals, and organic materials.[5e]
In one key area of interest to our laboratory, arynes have proven useful as building blocks in the synthesis of polycyclic aromatic hydrocarbons (PAHs). PAHs have had a remarkable impact on the field of materials science, as their unique electronic properties enable their use as intrinsic semiconductors, organic light-emitting diodes, organic field-effect transistors, and organic photovoltaics.[6] Traditional approaches to PAH synthesis typically involve aldol condensation, Friedel–Crafts acylation, aryne cycloadditions,[7] diyne polymerization/aromatization cascades,[8] radical alkyne annulations,[9] or sNar reactions to construct the desired C–C bonds in a stepwise manner and under harsh reaction conditions.[10]
In a seminal study reported in 1977, Steglich and coworkers demonstrated a promising alternative strategy to access PAHs that involved arynes.[11 ] As shown in Figure 1a, reaction of benzyne (1), generated from benzenediazonium-2-carboxylate, and oxadiazinone 2 provides 9,10-diphenylanthracene (4). The reaction involves an initial Diels–Alder/retro-Diels–Alder (DA/r-DA) reaction sequence to form benzopyrone 3 and a second DA/r-DA sequence to generate 4. Overall, this impressive reaction proceeds with extrusion of N2 and CO2 and the formation of four new C–C bonds. Decades later, Nuckolls and Wudl applied the Steglich chemistry to access pentacene and heptacene derivatives, respectively.[12,13,14,15] Notably, Christl demonstrated three examples of DA/r-DA cascades of oxadiazinones and benzyne leading directly to symmetric PAHS, including 6,13-diphenylpentacene and an anthracene dendrimer.[16] In these examples, benzopyrone intermediates are never observed or intercepted, thus only allowing for the formation of C2 symmetric products. With the aim of accessing a greater variety of scaffolds, our laboratory developed the modular synthetic platform shown in Figure 1b.[17] Using cyclic alkynes in the DA/r-DA cascade with unsymmetrically substituted oxadiazinones 6 allowed for access to stable and isolable pyrone 7. The ability to isolate pyrone 7 enabled the incorporation of an additional strained intermediate and, thus, for the generation of PAH scaffolds bearing four quadrants of differentiation. We also disclosed a one-pot variant wherein strained alkyne 5 and aryne 8 are cogenerated in the presence of oxadiazinone 6 to give tricyclic scaffolds 9 directly. Of note, Hosoya and coworkers recently demonstrated the use of 1,2-cyclohexyne in this methodology.[18] Altogether, the DA/r-DA cascades of cyclic alkynes and arynes allow rapid access to PAH scaffolds with promising applications. For example, compounds 10–13 demonstrate the promise of this methodology for accessing organic semiconductors,[13] stimuli-responsive materials,[17] biorthogonal labelling tools,[18] and donor-acceptor polymers (Figure 1c).[17]
Figure 1.
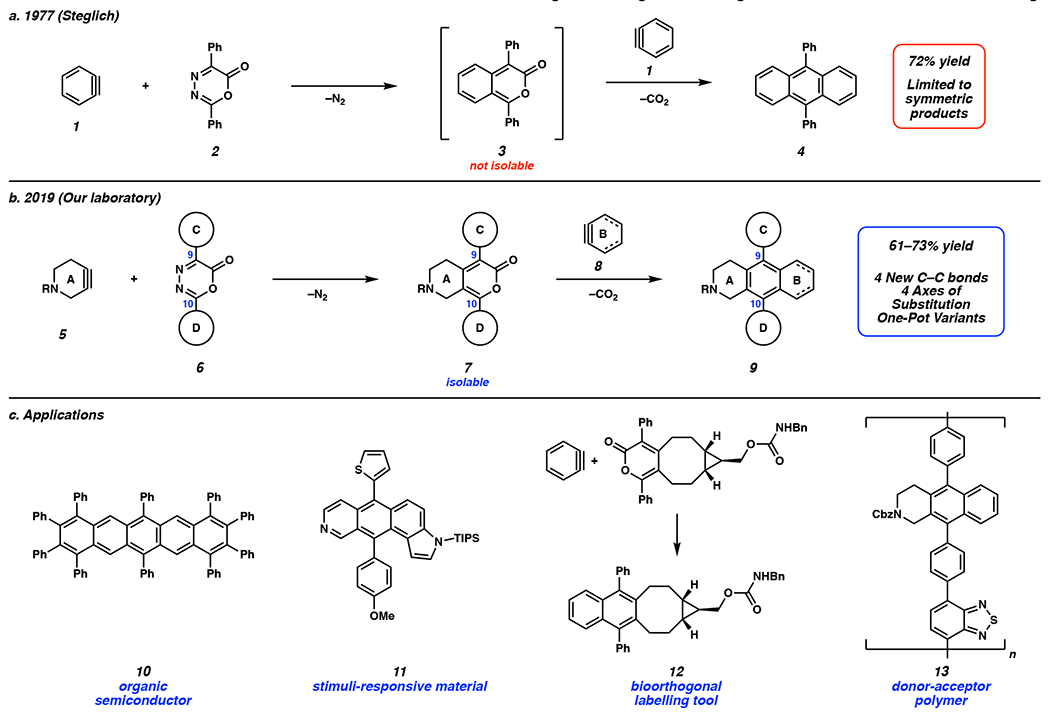
a) Seminal 1977 Steglich report of the DA/r-DA cascade of benzyne and an oxadiazinone. b) Recent 2019 study on the modular DA/r-DA cascade of strained alkynes and arynes with oxadiazinones. c) Applications of DA/r-DA reactions of strained cyclic alkynes and arynes with oxadiazinones. TIPS = triisopropyl silane, Bn = benzyl, Cbz = benzylcarbamate.
Despite that more than forty years have passed since Steglich’s pioneering study, key mechanistic features of the original DA/r-DA cascade and variants thereof have remained poorly understood. As such, we set out to shed light on mechanistic questions 1–4 highlighted in Figure 2. The cascade presumably involves DA cycloaddition of oxadiazinone 2 and strained cyclic intermediate 14 to form [2.2.2] bicycle 15 (DA1) and subsequent dinitrogen extrusion to afford pyrone 16 (r-DA1). Subsequently, pyrone 16 reacts with a second aryne or cyclic alkyne 8 to access bicyclic lactone 17 (DA2), which then expels carbon dioxide to afford PAH product 18 (r-DA2). We sought to understand (1) the reactivity differences between cyclic alkynes and arynes in the DA1, (2) the origins of selectivity in r-DA1, (3) the reactivity differences between alkyl- and benzopyrones in DA2, and (4) the steric and electron effects of rings A and B on r-DA2.
Figure 2.
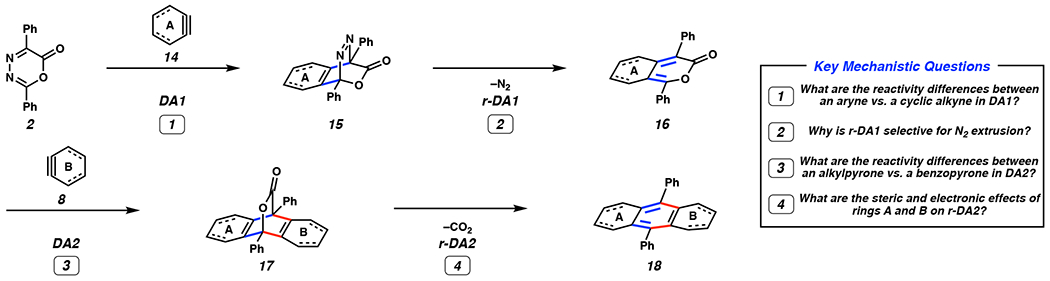
Overview of mechanism for the DA/r-DA cascade and key mechanistic questions. DA1 = Diels–Alder reaction 1, r-DA1 = retro-Diels–Alder reaction 1, DA2 = Diels–Alder reaction 2, r-DA2 = retro-Diels–Alder reaction 2.
Our study serves as the first computational analysis of the DA1 reactions of cyclic alkynes with oxadiazinones and the first comparative investigation of cyclic alkyne versus aryne reactivity in this reaction manifold. The origins of selective r-DA1 N2 extrusion, which has only been studied in the context of the DA/r-DA reactions of tetrazines with alkynes[19] and alkenes,[20],[21] is also elucidated. Furthermore, we provide the first theoretical examination of (1) DA2 reactions of cyclic alkynes with pyrones and (2) the role of aromaticity in r-DA2 reactions of bicyclic lactones. It is anticipated that examination of each of these 4 mechanistic steps will help build a foundational predictive model for synthetic planning of more complex PAH derivatives using the DA/r-DA cascades of cyclic alkynes and arynes with oxadiazinones. Additionally, we report an improved substrate scope for the DA/r-DA reactions of oxadiazinones and strained cyclic intermediates. We access 11 new anthracene analogues, in addition to tetracene and pentacene frameworks, via the DA/r-DA cascades of cyclohexyne and oxadiazinones. Altogether, our computational and experimental investigations are expected to provide a basis for expanding this modular synthetic strategy toward accessing isolable pyrones and, consequently, a greater variety of PAH scaffolds.
Results and Discussion
Experimental Comparison of Aryne vs. Cyclic Alkyne Reactivity.
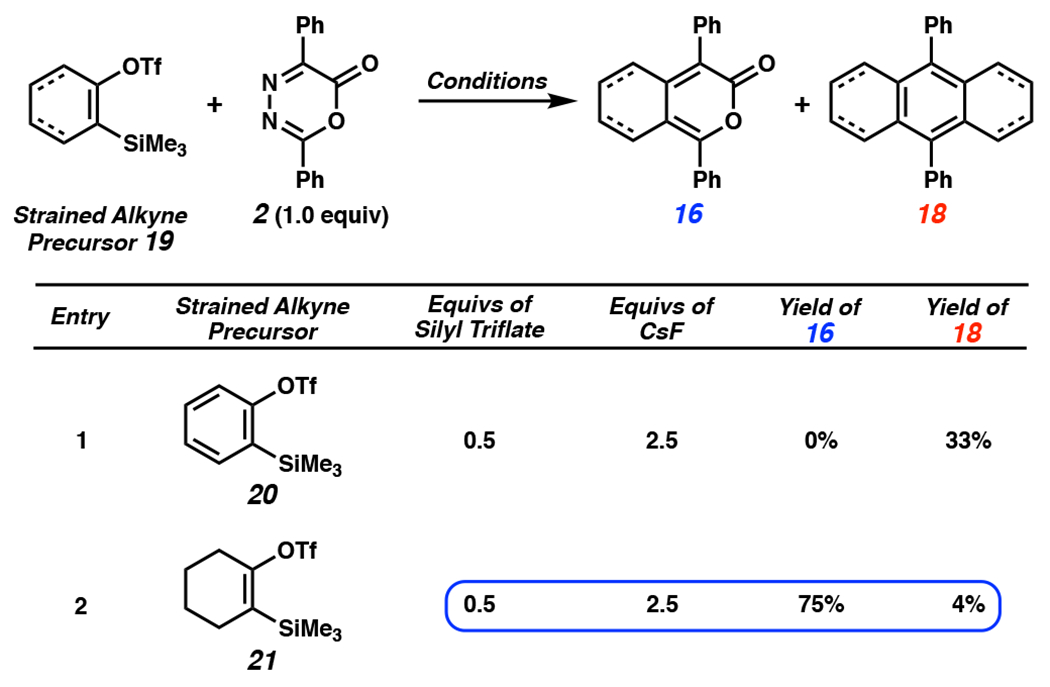
To initiate our studies, the reactivities of benzyne (1) and cyclohexyne in DA/r-DA cascades with oxadiazinone 2 were compared experimentally (Table 1). In an effort to favor the formation of benzopyrone 3, we carried out the reaction of benzyne precursor 20 with a two-fold excess of oxadiazinone 2. In this instance we only observed double addition product 18 in a 33% yield (entry 1) and no evidence of benzopyrone 3, consistent with previous observations.[11,15,17,18] In contrast, when cyclohexyne precursor 21 was employed in the presence of 2 equivalents of oxadiazinone 2, pyrone 16 was obtained in 75% yield (entry 2). This observation provides the basis for accessing nonsymmetric PAH scaffolds via a second DA/r-DA reaction sequence of pyrone 16 with a second, distinct aryne or cyclic alkyne. Recently, Hosoya and coworkers demonstrated that the reaction of a cyclohexyne precursor and oxadiazinone 2 can lead to selective formation of alkylpyrone 16,[18] albeit using slightly different reaction conditions than those identified herein. We hypothesized that benzopyrone 3 (Figure 1a) cannot be isolated in the reaction of benzyne (1) and oxadiazinone 2 because intermediate 3 is more reactive than oxadiazinone 2, preventing its isolation. These experimental results provided the framework for the mechanistic questions that we sought to answer using computational analyses (see Figure 2). Despite several decades passing since Steglich’s initial report[11] and recent accomplishments demonstrating the utility of cyclic alkynes for PAH synthesis,[17,18] these mechanistic questions have not been resolved.
Table 1.
Comparison of the reactivities of benzyne (derived from 20) and cyclohexyne (derived from 21) with oxadiazinone 2. Conditions: CsF, CH3CN (0.1 M), 23 °C, 12–14 h. OTf = trifluoromethanesulfonate.
 |
Computational Analysis of DA1.
To answer our first question regarding the reactivity differences between cyclic alkynes and arynes in DA1, we compared DA1 of oxadiazinone 2 with either cyclohexyne (23) or benzyne (1) (Figure 3a). Density functional theory (DFT) calculations were performed with Gaussian 16.22 The geometry of each species was optimized using the M06-2X functional and the 6-31G(d) basis set. Frequency calculations were performed at the same theoretical level as for geometry optimizations to verify the stationary points as either minima or first-order saddle points on the potential energy surface. Free energy corrections were calculated both with and without Truhlar’s quasiharmonic oscillator approximation. Single-point energy calculations were performed with the same functional using a 6-311+G(d,p) basis set and the SMD solvent model for acetonitrile to obtain more accurate energetics. HOMO and LUMO energies were computed using M06-2X/6-311+G(d,p). Optimized structures are presented using CYLview.
Figure 3.
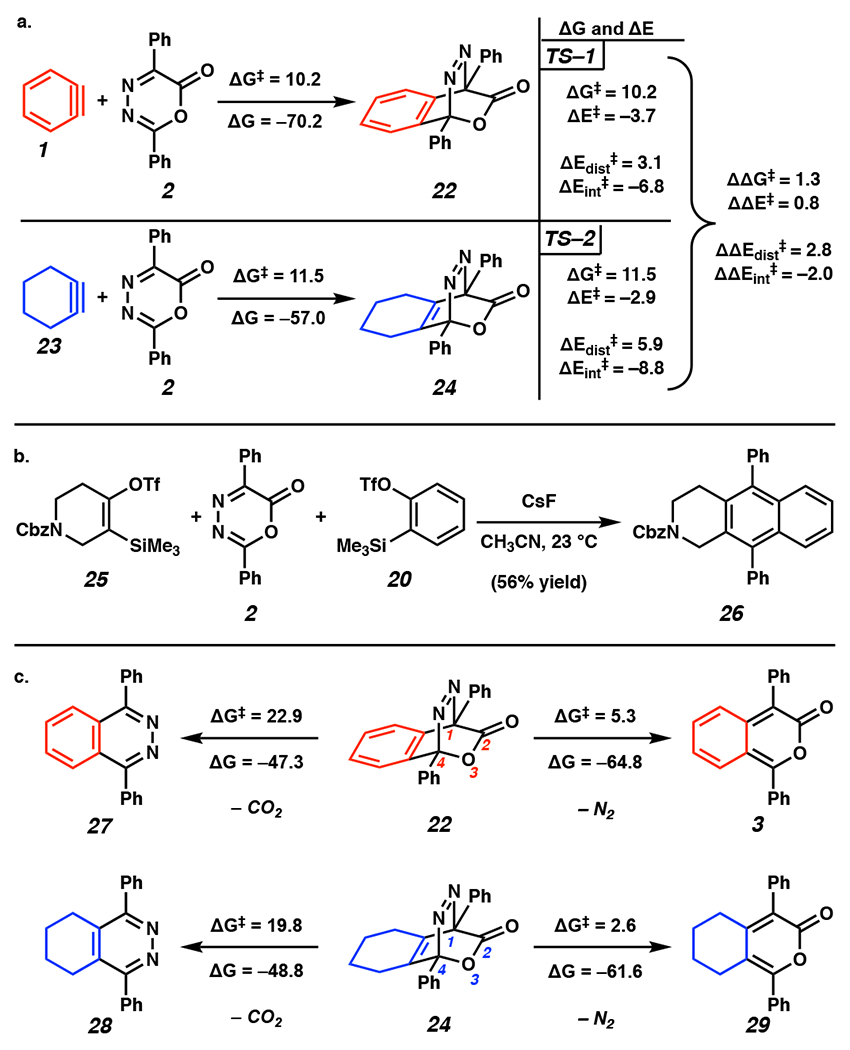
a) Computed ΔG and ΔG‡ values for DA1. b) Previously reported experimental results on the 1 pot reaction involving piperidyne precursor 25. c) Computed ΔG and ΔG‡ for selective N2 extrusion over CO2 release in r-DA1. Energies in kcal mol−1. OTf = trifluoromethanesulfonate, Cbz = benzylcarbamate.
Based on computed HOMO and LUMO energies (see the Supporting Information, Part II–B for MO energies), DA1 of benzyne (1) and oxadiazinone 2 is a neutral electron-demand DA reaction, involving either the HOMO or LUMO of the strained cyclic intermediate (i.e., the dienophile) and the HOMO or LUMO of the oxadiazinone (i.e., the diene). It has a Gibbs activation free energy (ΔG‡) of 10.1 kcal mol−1 and a Gibbs free energy (ΔG) of −70.2 kcal mol−1. In contrast, DA1 of cyclohexyne (23) and oxadiazinone 2 is an inverse electron-demand reaction, has a higher kinetic barrier (ΔG‡ = 11.5 kcal mol−1), and is less exothermic (ΔG = −57.0 kcal mol−1). A distortion/interaction activation strain (D/IAS) analysis[23] of DA1 was performed to understand the differences in reactivity of cyclohexyne (23) versus benzyne (1) with oxadiazinone 2. In a D/IAS analysis, activation potential energies (ΔE‡) are calculated for a transition state (TS) and further broken down into distortion energy (ΔEdist‡) and interaction energy (ΔEint‡). ΔEdist‡ is the energetic cost of deforming the ground states of the reactants into their TS geometries and can be roughly associated with the steric profile of a reaction. ΔEint‡ is the energetic benefit resulting from stabilizing electronic interactions between the reacting components at the TS and is, therefore, associated with the electronic profile of a reaction.
The D/IAS analysis was performed on the TSs for DA1 (i.e., TS–1 and TS–2). TS–1 corresponds to DA1 of benzyne (1) with oxadiazinone 2 and TS–2 corresponds to DA1 of cyclohexyne (23) with oxadiazinone 2. Based on D/IAS results, ΔEint‡ is more stabilizing in TS–2 than in TS–1 by 2.0 kcal mol−1.24 However, this effect is outcompeted by ΔEdist‡, which is 2.8 kcal mol−1 lower in TS–1 than in TS–2. This results in ΔΔE‡ of 0.8 kcal mol−1. The lower ΔEdist‡ in TS–2 indicates that predistortion of both reactants into their TS geometries in TS–1 results in a lower ΔE‡ (see the Supporting Information, Part II–C for analysis of TS geometries). The same degree of reactant pre-distortion is not observed in the reaction of cyclohexyne (23) and oxadiazinone 2. Resultantly, ΔE‡ of TS–1 is lower than that for TS–2.25 These results elucidate the differences in reactivity of benzyne (1) versus cyclohexyne (23) in DA1 with oxadiazinone 2. They are particularly interesting when considering one-pot reactions previously published by our group wherein a 1:1:1 ratio of an azacyclohexyne precursor 25, oxadiazinone 2, and benzyne precursor 20 are combined in the presence of a fluoride source and primarily give alkylpyrone and non-symmetric tricyclic scaffold 26 (56% yield).17 The aforementioned TS analysis would support the hypothesis that product distribution in the one-pot reaction sequence is governed by the rate of strained alkyne generation rather than the inherent reactivity differences between cyclic alkynes and arynes.
Computational Analysis of r-DA1.
We also examined r-DA1 toward answering mechanistic question 2: Why is r-DA1 selective for N2 extrusion? To our knowledge, computational analysis of r-DA N2 extrusion has only been studied in the context of DA/r-DA reactions of tetrazines with linear alkynes[26] and alkenes.[27] As such, we calculated ΔG‡ for r-DA1 of [2.2.2] bicycles 22 and 24 leading to pyrones 3 and 29, respectively, with expulsion of N2. As shown in Figure 3c, r-DA1 N2 extrusion occurs readily (ΔG2021; = 2.6 to 5.2 kcal mol−1). Interestingly, ΔG‡ for CO2 extrusion leading to tetrazines 27 and 28 is significantly higher (ΔG‡ = 19.8 to 22.9 kcal mol−1). We hypothesize that kinetic preference for N2 extrusion is due to predistortion of intermediates 22 and 24 into the corresponding TS geometries for N2 release. The change in C–N bond lengths that is required to arrive at the corresponding TSs for N2 release is smaller than the change in C–O and C–C bond lengths needed to arrive at TSs for CO2 extrusion (See the Supporting Information, Part II–C for TS geometries). Additionally, stabilizing hyperconjugative interactions between 1) the lone pair of O3 and the C4–N σ* orbital and 2) the C2=O π orbital and the adjacent C1–N σ* orbital lower ΔG‡ for r-DA1 (See the Supporting Information, Part II–C for computational support). N2 release is thermodynamically favored (ΔG = −61.6 kcal mol−1 to −64.8 kcal mol−1) over CO2 extrusion (ΔG = −47.3 kcal mol−1 to −48.8 kcal mol−1). This thermodynamic preference for release of N2 can be attributed to the relative bond strengths in starting material and products. More specifically, N2 release involves the cleavage of two weak C–N bonds in exchange for the formation of a strong N≡N bond.[28] Altogether, our DFT calculations reveal the thermodynamic and kinetic favorability of DA1 involving cyclohexyne (23) or benzyne (1) and oxadiazinone 2 followed by selective r-DA1 N2 release.
Computational Analysis of DA2.
Subsequently, we sought to answer mechanistic question 3: what are the reactivity differences between alkyl- and benzopyrones in DA2? To this end we studied four possible combinations for DA2 of benzopyrone 3 or alkylpyrone 29 with benzyne (1) or cyclohexyne (23) (Figure 4). Surprisingly, the HOMO and LUMO energies of pyrones 3 and 29 and strained cyclic intermediates 1 and 23 demonstrate that DA2 is a normal electron-demand reaction (see the Supporting Information, Part II–B for computed MO energies). This difference in the electronic profile of DA1 versus DA2 is due to the higher LUMO energy of pyrones 3 and 29 relative to oxadiazinone 2. [29] We first sought to compare the barrier differences between the reaction of benzyne (1) with benzopyrone 3 (Figure 4a) and cyclohexyne (23) with alkylpyrone 29 (Figure 4b).
Figure 4.
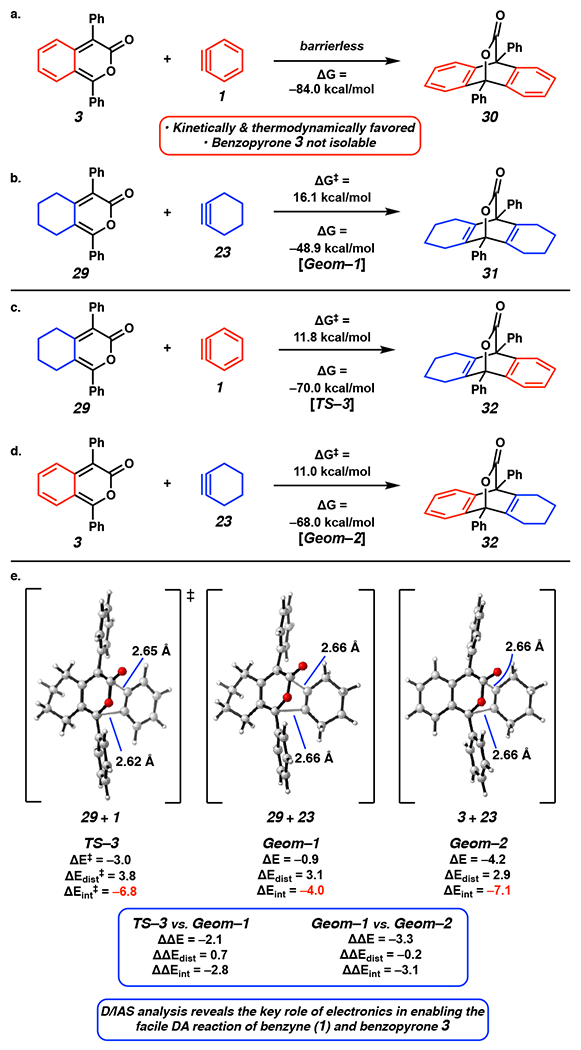
a) DA2 of benzopyrone 3 and benzyne (1). b) DA2 of alkylpyrone 29 and cyclohexyne (23). c) DA2 of alkylpyrone 29 with benzyne (1). d) DA2 of benzopyrone 3 and cyclohexyne (23). e) D/IAS analysis of analogous geometries for DA2 shown in Figure 4b–4d. Geom = analogous geometry.
Our computations demonstrate that DA2 of benzopyrone 3 and benzyne (1) is barrierless and highly exothermic (Δ = −84.0 kcal mol−1) (Figure 4a). [30] Compound 3 is an ortho-xylylene, which is highly reactive because reaction restores aromaticity of the benzene ring. By comparison, DA1 of cyclic alkyne 23 and oxadiazinone 2 has ΔG‡ of 11.5 kcal mol−1 and is exothermic by 57.0 kcal mol−1. These results give a clear picture as to why benzopyrone 3 cannot be isolated in the seminal studies by Steglich[11] as well as subsequent studies by Nuckolls[13] and Wudl.[12] In these cases, benzopyrone 3 is the more reactive diene and preferentially consumes any aryne generated before additional oxadiazinone 2 can react with aryne. This agrees with the experimental data shown in Table 1. In contrast, reaction of alkylpyrone 29 and cyclohexyne (23) (Figure 4b) has a ΔG‡ of 16.1 kcal mol−1 and is exothermic by 48.9 kcal mol−1. In this case, ΔG‡ for DA2 is significantly higher than that for DA1 between cycloalkyne 23 and oxadiazinone 2 (ΔΔG‡ = 4.6 kcal mol−1). This accounts for the experimental observation in Table 1, entry 2 where oxadiazinone 2 remains the most reactive diene throughout the progression of the reaction, allowing pyrone 29 to pool until oxadiazinone 2 is fully consumed.
To gain more insight into these reactivity differences, we sought to perform a D/IAS analysis. Unfortunately, a TS for the barrierless reaction between benzopyrone 3 and benzyne (1) (Figure 4a) could not be located. Two additional reactions were considered to gain insight on the barrierless process in Figure 4a. First, DA2 between alkylpyrone 29 and benzyne (1) (Figure 4c) was found to have ΔG‡ of 11.8 kcal mol−1 (TS–3) and to be exothermic by 70.0 kcal mol−1. Second, DA2 of benzopyrone 3 and cyclohexyne (23) (Figure 4d) was found to have ΔG‡ of 11.0 kcal mol−1 and to be exothermic by 68.0 kcal mol−1. These additional reactions allow us to further break down the steric and electronic factors contributing to the kinetic favorability of reaction between benzopyrone 3 and benzyne (1) (Figure 4a).
Because reaction of pyrone 29 with cyclohexyne (23) has a much later TS than do reactions of pyrone 29 with benzyne (23) and benzopyrone 29 with cyclohexyne (23), it was necessary to perform the D/IAS analysis at analogous geometries along each reaction coordinate.[31,32] The analogous geometries have similar bond forming lengths (Figure 4e). In particular, the TS for cycloaddition of pyrone 29 with benzyne (1), TS–3, was used directly for the D/IAS analysis. Analogous geometries Geom–1 and Geom–2 correspond to points along the reaction coordinates for DA2 of pyrone 29 with cyclohexyne (23) and benzopyrone 3 with cyclohexyne (23), respectively. Our computations reveal that TS–3 is energetically favored over Geom–1 by 2.1 kcal mol−1. Whereas ΔEdist was found to differ by only 0.7 kcal mol−1, ΔEint is more stabilizing in TS–3 by 2.8 kcal mol−1. This more stabilizing ΔEint results from a smaller HOMO/LUMO gap between the pyrone 29 HOMO and the benzyne (1) LUMO than between the pyrone 29 HOMO and the cyclohexyne (23) LUMO (see the Supporting Information, Part II–B for computed MO energies). Thus, the low-lying LUMO of benzyne (23) contributes to the kinetic favorability of DA2 involving benzopyrone 3 and benzyne (1) (Figure 4a).
A similar comparison was made between Geom–1 and Geom–2 to help elucidate the role of the pyrone’s steric and electronic profile on ΔG‡ for DA2 of benzopyrone 3 with benzyne (1) (Figure 4a). As shown in Figure 4e Geom–2 is energetically favored over Geom–1 by 3.3 kcal mol–1. D/IAS results demonstrate that ΔΔE‡ is largely attributed to ΔΔEint rather than ΔΔEdist. ΔEint is 3.1 kcal mol−1 lower in Geom–2, indicating that benzopyrone 3 has more favorable orbital interactions in DA2 with cyclohexyne (23) than does alkylpyrone 29. The high energy HOMO of benzopyrone 3 decreases the HOMO/LUMO gap for the reaction (see the Supporting Information, Part II–B for computed MO energies). This results in more stabilizing ΔEint in DA2 of benzopyrone 3 and cyclohexyne (23). Altogether, our D/IAS analysis of TS–3, Geom–1, and Geom–2 reveals the key role of electronics rather than sterics in enabling the facile DA2 of benzyne (1) and benzopyrone 3. As previously mentioned, benzopyrone 3 could not be isolated in experiments because it is more reactive than oxadiazinone 2 (Scheme 1a). The additional D/IAS results reveal that this is likely due to the presence of highly favorable electronic interactions in DA2 of benzopyrone 3 and benzyne (1). The absence of a barrier between benzyne (1) and benzopyrone 3 is the result of the very high reactivity of benzyne (1) and of the ortho-xylylene nature of benzopyrone 3. These combine to give high exothermicity and reactivity.
Computational Analysis of r-DA2.
Lastly, we aimed to answer mechanistic question 4: What are the steric and electronic effects of rings A and B on r-DA2? This mechanistic step starts from [2.2.2] bicyclic lactones 30–32, none of which have been isolated or observed due to their fleeting nature. Initially, we calculated ΔG‡ for r-DA2 of bicyclic lactones 30–32. Our computations demonstrate that CO2 extrusion from lactone 31 occurs most readily, having a ΔG‡ of 14.8 kcal mol−1 (Figure 5). For r-DA2 of substrate 32, ΔG‡ increases to 19.4 kcal mol−1. The r-DA2 of bicyclic lactone 30 leading to 9,10-diphenylanthracene (4) occurs with the highest energy barrier (ΔG‡ = 25.2 kcal mol−1). There is a good Evans-Polanyi relationship, that is as the reaction becomes less exothermic, ΔG‡ increases by about half the change in exothermicity.
Figure 5.
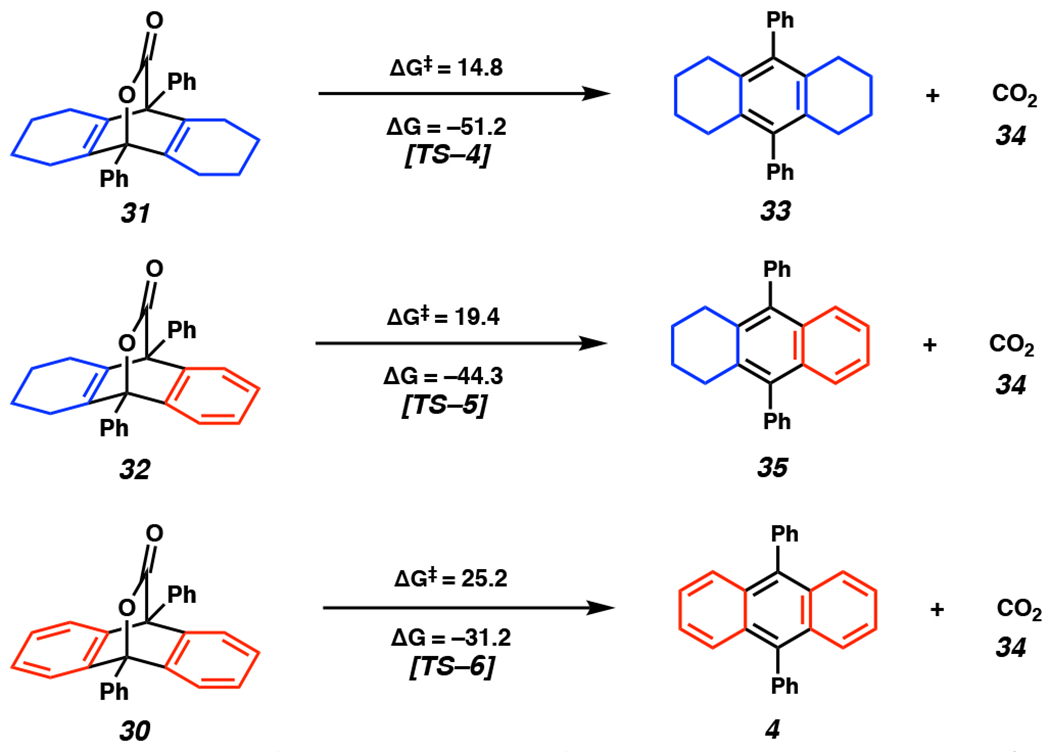
Computed ΔG and ΔG‡ values for r-DA2 of bicyclic lactones 30–32.
The ΔG values for the three cases are shown in Figure 5a and demonstrate distinct exergonicities. This mechanistic step had not been previously assessed in the context of PAH synthesis.[33] The r-DA2 of lactone 31 is exergonic by 51.2 kcal mol−1–reflective of the formation of a benzene ring [resonance energy (RE) of 36 kcal mol−1]. [34] The second case–reaction of substrate 32 to generate product 35–has a significantly lower thermodynamic driving force (ΔG = −44.3 kcal mol−1); the same strain is released, but one benzene is converted to naphthalene, which has an RE of 61 kcal mol−1[34] and is only a 25 kcal mol−1 increase in resonance stabilization. The third example is taken from the original Steglich work and corresponds to r-DA2 of lactone 30. It is least exergonic (ΔG = −31.2 kcal mol−1) since two benzenes are converted into an anthracene (RE = 83 kcal mol−1) and therefore involves an increase in RE of only 11 kcal mol−1. Notably, there is an approximate 0.5 correlation between ΔG and ΔG‡ for r-DA2 from bicyclic lactones 31 and 32 (Figure 5), which agrees well with Marcus theory.
Our studies on r-DA2 suggest the potential to modulate ΔG‡ and ΔG for r-DA2 via substrate design or careful selection of reaction conditions. Notably, our and Christl’s laboratories have encountered a common challenge with this methodology: subsequent aryne addition to the desired PAH product, resulting in triptycene scaffolds in low yields and as a complex mixture of products.[16,35] The high ΔG‡ for r-DA2 from tricycle 30 could potentially be leveraged to prevent undesired cycloaddition between electron-rich PAH products and a second equivalent of cyclic alkyne or aryne in the pursuit of higher order acenes.
Scope of Methodology.
Our mechanistic insight on the DA/r-DA cascades of oxadiazinones and strained cyclic intermediates motivated us to investigate the PAH scaffolds accessible via alkylpyrone 29. Having optimized reaction conditions for accessing pyrone 29, we focused our efforts on exploring the substrate scope for the addition of arynes and cyclic alkynes to 29 (Figure 6). Pyrone 29 successfully underwent the DA/r-DA reaction sequence with benzyne (1), generating tricycle 35 in 86% yield (entry 1). Alternate arynes were tolerated, as demonstrated by successful reactions of pyrone 29 with 1,2-naphthalyne and 4,5-indolyne, furnishing adducts 37 and 38, respectively (entries 2 and 3). An additional cyclohexyne (23) equivalent could be incorporated, generating adduct 33 (entry 4). A series of sp3-rich heterocyclic alkynes including 3,4-piperidynes[36] and 3,4-oxacyclohexyne[37] were also deemed suitable reaction partners, as demonstrated by formation of tricycles 39–41 (entries 5–7).
Figure 6.
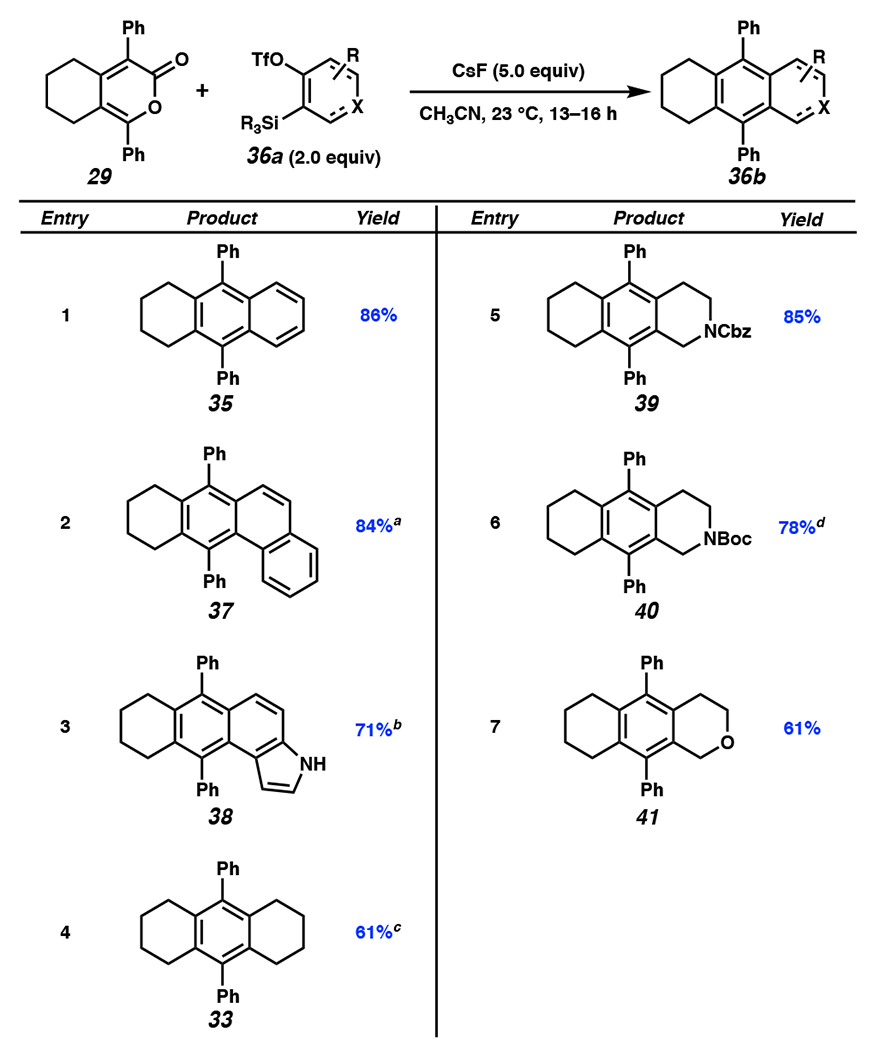
Substrate scope for the addition of arynes and cyclic alkynes to pyrone 29. aReaction run at 50 °C. bReaction run at 40 °C for 36 h. cReaction run for 36–39 h. dReaction run at 40 °C. OTf = trifluoromethanesulfonate, Cbz = benzylcarbamate, Boc = tert-butyloxycarbonyl.
DA/r-DA reactions of differentially-substituted oxadiazinones with silyl triflate 23 and subsequent addition of benzyne precursor 22 also provided access to non-symmetric PAHs (Figure 7). Nonsymmetric products bearing substituents commonly used in materials chemistry (e.g., thiophenes, cross-coupling handles, etc.) were well tolerated. Electron-donating and electron-withdrawing groups could also be incorporated (See the Supporting Information, Part II–D for computational analysis of substituents effects on DA1 and DA2). Well-established chemistry was used to prepare oxadiazinones 42a,[17,38,39] which were subjected to silyl triflate 23 under standard reaction conditions. The results shown in Figure 7 highlight the versatility of the methodology.
Figure 7.
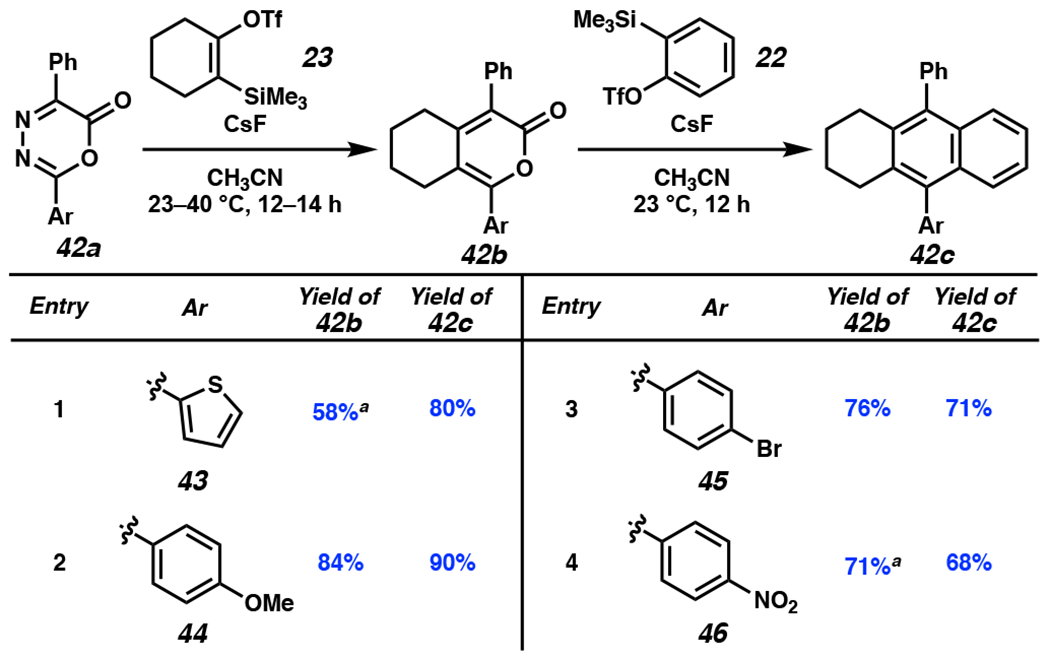
Variation of oxadiazinone 42a and reaction of the corresponding pyrone 42b with silyl triflate 22. aReaction run at 40 °C. OTf = trifluoromethanesulfonate.
Application of Methodology for the Synthesis of Extended PAH Carbon Frameworks.
We also sought to demonstrate the potential utility of our strategy for accessing the carbon frameworks of extended PAHs, such as tetracene and pentacene derivatives. Toward this goal, we demonstrated that pyrone 29 could undergo the DA/r-DA reaction sequence with silyl triflate precursor 47, which was synthesized in 2 steps from commercially available dibromohydroquinone (Figure 8). The resulting tricyclic silyl triflate 48 was then subjected to pyrone 49 or 29, providing access to tetracene and pentacene precursors 50 and 51, respectively (Figures 8a–8b). X-ray analysis of a single crystal of 51 was used to unambiguously assign the pentacyclic structure. Moreover, pentacycle 51 could be prepared in a direct fashion from pyrone 29 and silyl triflate 47 (Figure 8c). This notable transformation furnishes the desired pentacycle via sequential pericyclic reactions, namely two DA/r-DA sequences, and the assembly of four new C–C bonds in a single step.
Figure 8.
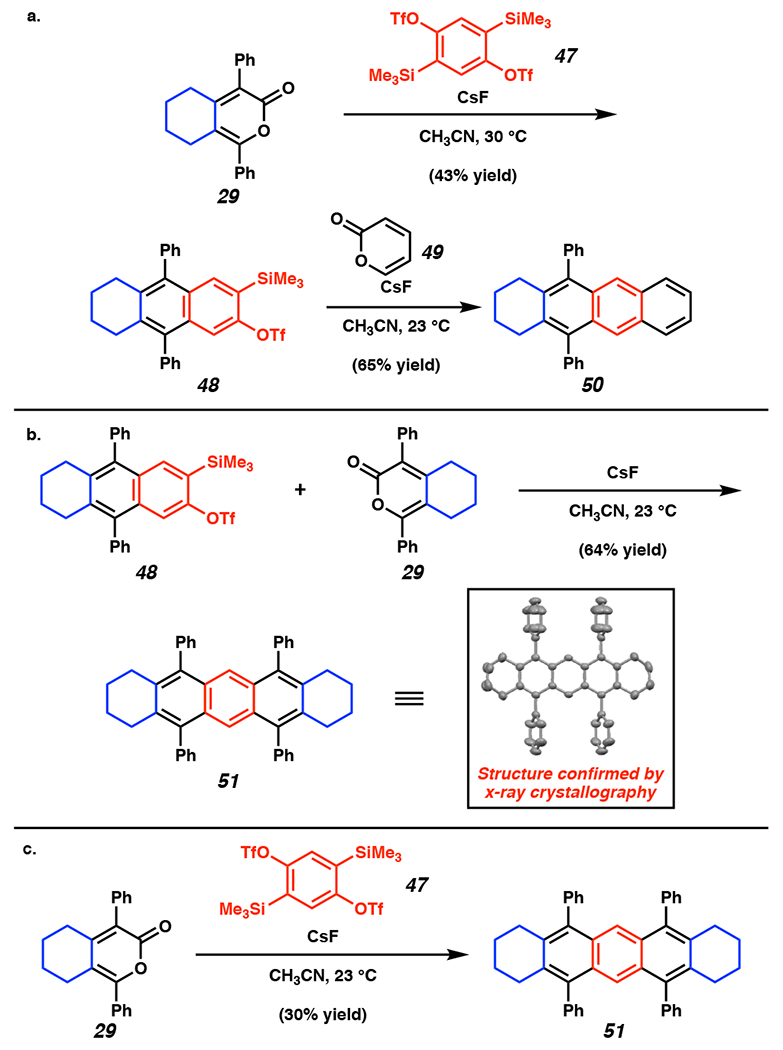
Synthetic application of the DA/r-DA reactions of alkylpyrone 29 for accessing extended PAH carbon frameworks 50 and 51. OTf = trifluoromethanesulfonate
Conclusion
We have performed a computational and experimental study of the reaction of strained alkynes with oxadiazinones to give PAH scaffolds, which are of great value in materials chemistry. The parent transformation was first reported in 1977 and used to access symmetrical products. Despite the intervening decades and recent versatile modifications to access unsymmetric PAH scaffolds, mechanistic studies on the transformation have been lacking. The DFT studies have answered several mechanistic questions about this pericyclic reaction cascade. Our findings help build a foundational predictive model for synthetic planning of more complex systems. In addition, our experimental studies have led to the efficient synthesis of new anthracene analogues, in addition to two extended frameworks. These studies not only provide fundamental insight on the cycloaddition cascade and synthetic access to PAH scaffolds but are also expected to enable the synthesis of new materials in future studies.
Supplementary Material
Acknowledgements
The authors are grateful to NIH-NIGMS (R35 GM139593 and R01 GM132432 to N.K.G., F31 GM130099-02 to M.R., and F32-GM122245 to E.R.D.), the National Science Foundation (CHE-1764328 to K.N.H.), the UCLA Graduate Division (J.S.D.), Bristol-Myers Squibb (J.S.D.), the Trueblood Family (N.K.G.), and the University of California, Los Angeles for financial support. Calculations were performed on the Hoffman2 cluster and the UCLA Institute of Digital Research and Education (IDRE) at UCLA and the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the National Science Foundation (OCI-1053575). These studies were supported by shared instrumentation grants from the NSF (CHE-1048804) and the National Center for Research Resources (S10RR025631).
References
- [1].For recent reviews on benzyne and related reactive intermediates, see:; (a) Bronner SM, Goetz AE, Garg NK, Synlett 2011, 2599–2604 [Google Scholar]; (b) Bhunia A, Yetra SR, Biju AT, Chem. Soc. Rev 2012, 41, 3140–3152 [DOI] [PubMed] [Google Scholar]; (c) Yoshida H, Takaki K, Synlett 2012, 23, 1725–1732 [Google Scholar]; (d) Dubrovskiy AV, Markina NA, Larock RC, Org. Biomol. Chem 2013, 11, 191–218 [DOI] [PubMed] [Google Scholar]; (e) Wu C, Shi F, Asian J Org. Chem 2013, 2, 116–125 [Google Scholar]; (f) Hoffmann RW, Suzuki K, Angew. Chem., Int. Ed 2013, 52, 2655–2656 [DOI] [PubMed] [Google Scholar]; (g) Yoshida S, Hosoya T, Chem. Lett 2015, 44, 1450–1460. [Google Scholar]
- [2].For a review on the synthetic utility of Kobayashi aryne precursors, see:; Shi J, Li L, Li Y, Chem. Rev 2021, 121, 7, 3892–4044. [DOI] [PubMed] [Google Scholar]
- [3].For a review on arynes in natural product synthesis, see:; Tadross PM, Stoltz BM, Chem. Rev 2012, 112, 3550–3557. [DOI] [PubMed] [Google Scholar]
- [4].For the aryne distortion/interaction model, see:; (a) Cheong PH-Y, Paton RS, Bronner SM, Im G-YJ, Garg NK, Houk KN, J. Am. Chem. Soc 2010, 132, 1267–1269 [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Im G-YJ, Bronner SM, Goetz AE, Paton RS, Cheong PH-Y, Houk KN, Garg NK, J. Am. Chem. Soc 2010, 132, 17933–17944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].(a) Corsello MA, Kim J, Garg NK, Nat. Chem 2017, 9, 944–949 [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ross SP, Hoye TR, Nat. Chem 2017, 9, 523–530 [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Surry DS, Buchwald SL, Angew. Chem., Int. Ed 2008, 47, 6338–6361 [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Mauger CC, Mignani GA, Org. Process Res. Dev 2004, 8, 1065–1071 [Google Scholar]; (e) Lin JB, Shah TK, Goetz AE, Garg NK, Houk KN, J. Am. Chem. Soc 2017, 139, 10447–10455. [DOI] [PubMed] [Google Scholar]
- [6].Anthony JE, Angew. Chem. Int. Ed 2008, 47, 452–483. [DOI] [PubMed] [Google Scholar]
- [7].Pérez D, Peña D, Guitián E, Eur. J. Org. Chem. 2013, 5981–6013. [Google Scholar]
- [8].For an example, see:; Jordan RS, Li YL, Lin C-W, McCurdy RD, Lin JB, Brosmer JL, Marsh KL, Khan SI, Houk KN, Kaner RB, Rubin Y. J. Am. Chem. Soc 2017, 139, 15878–15890. [DOI] [PubMed] [Google Scholar]
- [9].For an example, see:; Gonzalez-Rodriguez E, Abdo MA, dos Passos Gomes G, Ayad S, White FD, Tsvetkov NP, Hanson K, Alabugin IV. J. Am. Chem. Soc 2020, 142, 8352–8366. [DOI] [PubMed] [Google Scholar]
- [10].Stępień M, Gońka E, Żyła M, Sprutta N, Chem. Rev. 2017, 117, 3479–3716. [DOI] [PubMed] [Google Scholar]
- [11].Steglich W, Buschmann E, Gansen G, Wilschowitz L, Synthesis 1977, 252–253. [Google Scholar]
- [12].Chun D, Cheng Y, Wudl F, Angew. Chem., Int. Ed 2008, 47, 8380–8385.; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2008, 120, 8508–8513. [Google Scholar]
- [13].Miao Q, Chi X, Xiao S, Zeis R, Lefenfeld M, Siegrist T, Steigerwald ML, Nuckolls C, J. Am. Chem. Soc 2006, 128, 1340–1345. [DOI] [PubMed] [Google Scholar]
- [14].For additional examples of oxadiazinone and aryne DA/r-DA cascades, see:; Rickborn B, Org. React 1998, 53, 223–629. [Google Scholar]
- [15].Nuckolls’ application of the DA/r-DA cascade was low yielding (10–15% yield) and Wudl’s example required elevated temperature. However, both examples demonstrate the versatility of the Steglich methodology and provide access to coveted scaffolds.
- [16].Christl M, Gazz. Chim. Ital. 1986, 116, 1–17. [Google Scholar]
- [17].Darzi ER, Barber JS, Garg NK, Angew. Chem., Int. Ed 2019, 58, 9419–9424.; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2019, 131, 9519–9524. [Google Scholar]
- [18].Meguro T, Chen S, Kanemoto K, Yoshida S, Hosoya T, Chem. Lett 2019, 48, 582–585. [Google Scholar]
- [19].Sadasivam DV, Prasad E, Flowers RA, J. Phys. Chem. A 2006, 110, 1288–1294. [DOI] [PubMed] [Google Scholar]
- [20].Törk L, Jiménez-Osés G, Doubleday C, Liu F, Houk KN, J. Am. Chem. Soc 2015, 137, 4749–4758. [DOI] [PubMed] [Google Scholar]
- [21].Suh SE-, Chen S, Houk KN, Chenoweth DM, Chem. Sci 2018, 9, 7688–7693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].See the Supporting Information, Section XX for all computational references.
- [23].Houk KN, Bickelhaupt FM, Angew. Chem., Int. Ed 2017, 56, 10070–10086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].See the Supporting Information, Part II–C for a hypothesis regarding more stabilizing ΔEint‡ in TS–2 than in TS–1.
- [25].To confirm that lower ΔEdist‡ in DA1 of benzyne (1) with oxadiazinone 2 is not an artifact of the reaction being more exergonic than DA1 of cyclohexyne (23) with oxadiazinone 2, D/IAS analysis was performed along the reaction coordinate. These results are provided in the Supporting Information, Part II–C and confirm that ΔEdist is consistently lower in DA1 of benzyne (1) and oxadiazinone 2 as the reaction proceeds.
- [26].Sadasivam DV, Prasad E, Flowers RA, Birney DM, J. Phys. Chem 2006, 110, 1288–1294. [DOI] [PubMed] [Google Scholar]
- [27].(a) Törk L, Jiménez-Osés G, Doubleday C, Liu F, Houk KN, J. Am. Chem. Soc 2015, 137, 4749–4758 [DOI] [PubMed] [Google Scholar]; (b) Suh S-E, Chen S, Houk KN, Chenoweth DM, Chem. Sci 2018, 9, 7688–7693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Related bond dissociation energies: C–N 69 kcal mol−1, N=N 226 kcal mol−1, C–C 82 kcal mol−1, C–O 87 kcal mol−1, C=O 177 kcal mol−1. [Google Scholar]
- [29].In other words, pyrones 3 and 28 are less electron deficient than oxadiazinone 2. This means that the LUMO energies of pyrones 3 and 28 are higher than that of oxadiazinone 2.
- [30].No TS could be located on this reaction pathway and the high exothermicity of the transformation provides support for a barrierless process.
- [31].In a previous report, our lab explained the importance of performing the D/IAS analysis for TS structures and analogous geometries along the reaction coordinate; see reference 23.
- [32].See the Supporting Information, Part II for the TS structures of reactions involving addition of benzyne (1) to pyrone 29 and cyclohexyne (23) to pyrone 29.
- [33].Only one computational study on substituent effects in r-DA extrusion of CO2 (34) from pyrone cycloadducts has been reported, see:; Abdullahi MH, Thompson LM, Bearpark MJ, Vinader V, Afarinkia K, Tetrahedron 2016, 72, 6021–6024. [Google Scholar]
- [34].McMurry J Organic Chemistry; Cengage Learning, 2011. [Google Scholar]
- [35].See the Supporting Information, Part I for experimental results on aryne addition to PAH scaffolds.
- [36].McMahon TC, Medina JM, Yang Y-F, Simmons BJ, Houk KN, Garg NK, J. Am. Chem. Soc 2015, 137, 4082–4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Shah TK, Medina JM, Garg NK, J. Am. Chem. Soc 2016, 138, 4948–4954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Tîntas ML, Diac AP, Soran A, Terec A, Grosu I, Bogdan E, J. Mol. Struct 2014, 1058, 106–113. [Google Scholar]
- [39].For multigram synthesis of oxadiazinone 2, see:; Kelleghan AV, Spence KA, Garg NK, Org. Synth 2020, 97, 189–206. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.