

Abstract
Background:
Parental smoking is implicated in the etiology of acute lymphoblastic leukemia (ALL), the most common childhood cancer. We recently reported an association between an epigenetic biomarker of early-life tobacco smoke exposure at the AHRR gene and increased frequency of somatic gene deletions among ALL cases.
Methods:
Here, we further assess this association using two epigenetic biomarkers for maternal smoking during pregnancy — DNA methylation at AHRR CpG cg05575921 and a recently established polyepigenetic smoking score — in an expanded set of 482 B-cell ALL (B-ALL) cases in the California Childhood Leukemia Study with available Illumina 450K or MethylationEPIC array data. Multivariable Poisson regression models were used to test the associations between the epigenetic biomarkers and gene deletion numbers.
Results:
We found an association between DNA methylation at AHRR CpG cg05575921 and deletion number among 284 childhood B-ALL cases with MethylationEPIC array data, with a ratio of means (RM) of 1.31 (95% CI:1.02-1.69) for each 0.1 beta-value reduction in DNA methylation, an effect size similar to our previous report in an independent set of 198 B-ALL cases with 450K array data (meta-analysis summary RM [sRM]=1.32, 95% CI:1.10-1.57). The polyepigenetic smoking score was positively associated with gene deletion frequency among all 482 B-ALL cases (sRM=1.31 for each 4-unit increase in score; 95% CI:1.09-1.57).
Conclusions:
We provide further evidence that prenatal tobacco-smoke exposure may influence the generation of somatic copy-number deletions in childhood B-ALL.
Impact:
Analyses of deletion breakpoint sequences are required to further understand the mutagenic effects of tobacco smoke in childhood ALL.
Keywords: Prenatal tobacco smoking, childhood acute lymphoblastic leukemia, epigenetic biomarkers, somatic gene deletions, aryl hydrocarbon receptor repressor
Introduction
Acute lymphoblastic leukemia (ALL) is the most common childhood malignancy in the United States, with approximately 2,700 incident cases diagnosed under age 15 each year 1. Although survival rates for ALL have improved dramatically in recent decades, with overall 5-year survival now upwards of 90% 2, ALL remains a leading cause of disease-related mortality in children and current treatments still carry long-term health consequences 3–5. Therefore, prevention remains a top priority 6. In addition to known ALL risk factors with large effects, namely ionizing radiation and genetic predisposition syndromes 7–9, several environmental exposures have been associated with ALL etiology, including tobacco smoke, pesticides, paint, and air pollution 6,10; however, the causal mechanisms remain largely unclear 11.
Childhood ALL, in particular B-cell ALL (B-ALL), is thought to follow a “two-hit” model of leukemogenesis 12,13, with in utero development of a pre-leukemic clone 14,15 that progresses to overt leukemia following postnatal acquisition of secondary genetic changes 16. Deletions of genes involved in cell cycle control, and B-lymphocyte development and hematopoiesis 17–19 including, most commonly, CDKN2A, ETV6, PAX5, and IKZF1 18, comprise a large proportion of the secondary alterations in childhood B-ALL.
We recently reported a positive association between early-life tobacco smoke exposure and somatic gene deletions in childhood ALL cases, suggesting a potential etiologic role for parental and/or household smoking. In 559 childhood ALL cases in the California Childhood Leukemia Study (CCLS), self-reported maternal and paternal smoking were associated with an increased number of gene deletions 20. In a subset of 198 B-ALL cases for whom genome-wide DNA methylation data were available from Illumina® HumanMethylation450 BeadChip (450K) arrays, we validated this association using an epigenetic biomarker for maternal smoking during pregnancy at the AHRR gene 20–22.
In the current study, we examine the association between DNA methylation at the AHRR CpG cg05575921 and gene deletions in an expanded set of 482 B-ALL cases in the CCLS, including an additional 284 B-ALL cases with DNA methylation data now available from Illumina® Infinium MethylationEPIC BeadChip (EPIC) arrays. Further, we sought to expand our analysis of the impact of prenatal tobacco smoke exposure on gene deletion burden in childhood B-ALL by using a recently established polyepigenetic smoking score of in utero tobacco smoke exposure 23.
Materials and Methods
Ethics statement
This study was approved and reviewed by the Institutional Review Boards at the University of Southern California, the University of California, Berkeley, the California Department of Public Health, and all participating hospitals. Written informed consent was obtained from all study participants. This study was conducted in accordance with the Declaration of Helsinki.
Study population
The CCLS is a population-based case-control study conducted from 1995-2015 to examine the relationships between various environmental exposures, genetic factors, and childhood leukemia 10. Cases were identified within 72 hours after diagnosis at hospitals across California. Eligible criteria include (1) age under 15 years, (2) without prior cancer diagnosis, (3) residence in California at the time of diagnosis, and (4) having an English or Spanish-speaking biological parent available for interview. Controls were not included in the current case-only analysis. Newborn dried bloodspots (DBS) were obtained from the California Biobank Program Genetic Disease Screening Program. The current analysis included 482 B-ALL cases with available genome-wide DNA methylation array data and gene deletion frequency data (Figure 1). Of those with available race/ethnicity, 228 (58.9%) self-identified as Latino, 102 (26.4%) as non-Latino White, and 57 (14.7%) as other non-Latino races/ethnicities (including African American, Native American, Asian, and mixed/other groups) (Table 1).
Figure 1. Sample flowchart.
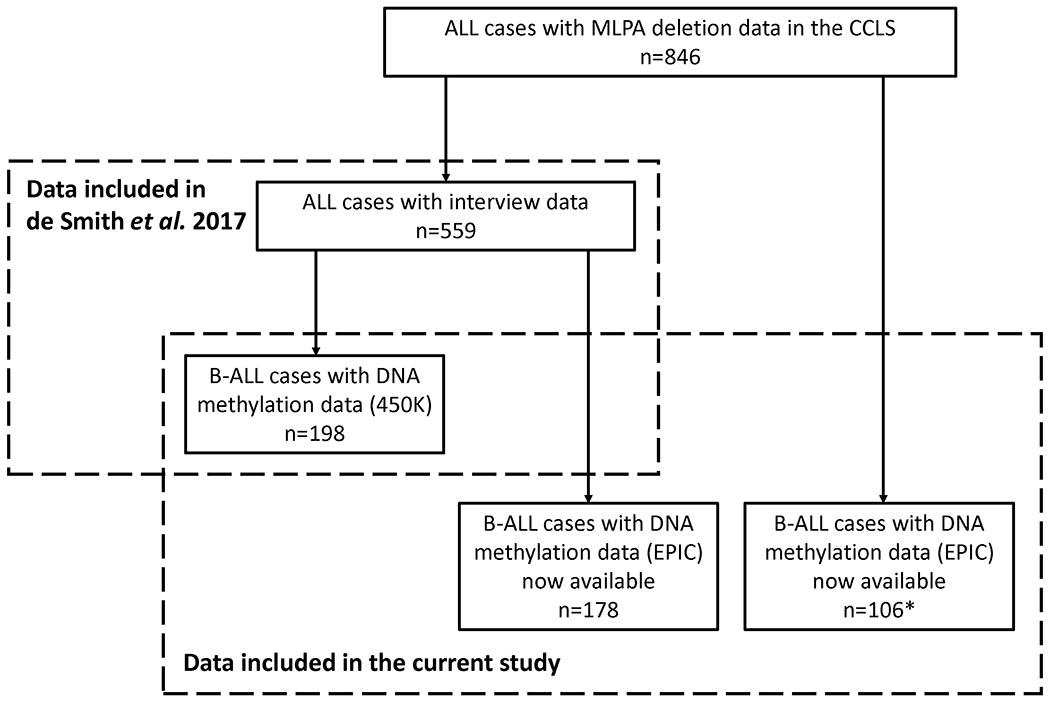
Left box: ALL cases included in the previous analysis for the association between early-life tobacco smoke and gene deletion frequencies (n=559) 20, in which 361 cases were analyzed only with interview data and 198 B-ALL cases had Illumina 450K genome-wide DNA methylation array data available and were thus included in the analyses of DNA methylation at the AHRR CpG cg05575921. Right box: samples included in the current study, including 198 B-ALL cases that were analyzed previously and 284 B-ALL cases now with available EPIC array DNA methylation data and MLPA gene deletion frequency data, of which 178 overlapped with the 361 cases that were analyzed previously only with interview data. In total, 482 B-ALL cases are included in our case-only analysis of prenatal tobacco smoke exposure and gene deletion frequency. *11 out of 106 B-ALL cases have interview data now available.
Table 1.
Characteristics of childhood B-ALL cases (n = 482) in the California Childhood Leukemia Study.
| Variables | Total (n = 482) | 450K (n = 198) | EPIC (n = 284) | P |
|---|---|---|---|---|
| Gestational age (weeks), mean (SD) | 39.31 (2.15) | 39.51 (1.92) | 39.06 (2.39) | 0.047 |
| Gestational age unknown (%) | 124 (25.7) | 124 (43.7) | ||
| Age at diagnosis (years), mean (SD) | 5.47 (3.44) | 5.36 (3.23) | 5.55 (3.58) | 0.550 |
| Sex (%) | ||||
| Females | 223 (46.3) | 91 (46.0) | 132 (46.5) | 0.984 |
| Males | 259 (53.7) | 107 (54.0) | 152 (53.5) | |
| Race/ethnicity (%) | ||||
| Latino | 228 (58.9) | 115 (58.1) | 113 (59.8) | 0.914 |
| Non-Latino White | 102 (26.4) | 54 (27.3) | 48 (25.4) | |
| Non-Latino Other | 57 (14.7) | 29 (14.6) | 28 (14.8) | |
| Race/ethnicity unknown (%) | 95 (19.7) | 95 (33.5) | ||
| Deletion number (%) | ||||
| 0 | 195 (40.5) | 73 (36.9) | 122 (43.0) | 0.459 |
| 1 | 151 (31.3) | 64 (32.3) | 87 (30.6) | |
| 2 | 88 (18.3) | 40 (20.2) | 48 (16.9) | |
| 3 | 35 (7.3) | 13 (6.6) | 22 (7.7) | |
| 4 | 9 (1.9) | 6 (3.0) | 3 (1.1) | |
| 5 | 4 (0.8) | 2 (1.0) | 2 (0.7) |
P-values comparing the characteristics of B-ALL cases in the 450K and EPIC array datasets were calculated using Studen’s t-tests for continuous variables (gestational age and age at diagnosis) and Chi-squared tests for categorical variables.
Somatic copy-number data
Copy-number at 8 commonly deleted gene regions (CDKN2A, ETV6, IKZF1, PAX5, BTG1, EBF1, RB1, and genes within the pseudoautosomal region [PAR1] of the sex chromosomes [CRLF2, CSF2RA, IL3RA]) was assayed in tumor DNA using multiplex ligation-dependent probe amplification (MLPA), as previously described 20,24.
Genome-wide DNA methylation arrays
For 198 B-ALL cases, genome-wide DNA methylation data were already available from Illumina® 450K arrays 20,25. For an additional 284 B-ALL cases, germline DNA was isolated from newborn DBS and bisulfite-treated as previously described 25, and subsequently assayed on Illumina® EPIC arrays. EPIC arrays include >850,000 CpG probes, comprising >90% of CpGs on 450K arrays plus an additional 413,743 CpGs. CpG beta values were normalized to remove batch effects according to the approach by Fortin et al. 26. Functional normalization was performed with noob background correction 27 by using the “preprocessFunnorm” function in the minfi package 28 through the Bioconductor project 29,30.
The AHRR CpG cg05575921 is included on both the 450K and EPIC arrays; we extracted beta values for this CpG for all 284 B-ALL cases assayed on the EPIC array to test for association with number of gene deletions, as previously performed for the 198 B-ALL cases in the 450K dataset 20.
We also calculated a new polyepigenetic smoking score for sustained maternal smoking during pregnancy, in both the 450K and EPIC datasets 23. In brief, for 450K data, we computed a DNA methylation-based smoking score as the linear combination of 28 previously selected maternal smoking-associated CpGs using their corresponding logistic LASSO regression coefficients (Supplementary Table S1) 23. For EPIC data, we calculated a score using 26 of the 28 CpGs that are included on the EPIC array, including the AHRR CpG cg05575921 23. Cases with missing data for any of these CpGs (due to detection P-values >0.01) were excluded from analyses involving polyepigenetic smoking scores.
AHRR DNA methylation quantitative trait locus (mQTL) genotype data
To account for potential genetic effects on DNA methylation at the AHRR CpG cg05575921, for 198 B-ALL cases (450K) we had available genotype data for SNP rs148405299, which was identified as an mQTL for cg05575921, as previously described 25. Additionally, for the new set of 284 B-ALL cases (EPIC) we genotyped SNP rs77111113, which is in perfect linkage disequilibrium with rs148405299 across all populations in LDlink (R2 = 1.0) 31, using a predesigned TaqMan SNP genotyping assay (ThermoFisher Scientific, Assay ID: C__25986435_10). Hereafter, we refer to either rs148405299 or rs77111113 as the AHRR-mQTL SNP.
Self-reported smoking exposures
Among B-ALL cases with available parent interview data, we tested the association between the DNA methylation-based biomarkers of maternal tobacco smoking in pregnancy and self-reported tobacco exposures as assessed by parent interviews 10,20. Dichotomous smoking variables (“yes” or “no”) included maternal/paternal ever smoking, maternal/paternal smoking 3 months before conception (preconception), maternal/paternal smoking at the time of the interview, maternal smoking during pregnancy, maternal smoking during breastfeeding, maternal prenatal smoking (during either preconception or pregnancy), maternal smoking during the year after birth, and child postnatal passive smoking. Continuous measures (number of cigarettes, pipes, or cigars per day) included maternal/paternal smoking preconception, maternal smoking during pregnancy, maternal smoking during breastfeeding, and maternal prenatal smoking (average of maternal smoking during preconception and during pregnancy). We also used combinations of responses from parental interviews to infer: 1) which mothers smoked throughout the entire duration of pregnancy, and 2) which mothers were never exposed to tobacco smoke from any source during pregnancy, allowing us to compare the DNA methylation-based biomarker levels of maternal smoking in pregnancy at these two extremes of self-reported smoking.
Statistical analyses
All statistical analyses were performed in R v 4.0.0 32. All 2-sided p-values below 0.05 indicate statistical significance. All analyses were performed separately in the 450K and EPIC datasets, including 198 and 284 B-ALL cases, respectively. Means and standard deviations were summarized to describe the distribution of continuous characteristics, and frequencies and proportions were computed for categorical characteristics.
We calculated Spearman rank correlation among self-reported tobacco smoking exposures, DNA methylation at the AHRR CpG cg05575921 and the polyepigenetic smoking scores. Linear regression models were additionally used to test for association between DNA methylation at the AHRR CpG cg05575921 or the polyepigenetic smoking scores and self-reported tobacco smoke exposures. To obtain independent effects of paternal smoking or maternal smoking on DNA methylation, maternal smoking was adjusted for paternal smoking in linear regression models, and vice versa (Supplementary Table S2). In addition, associations between the joint exposures of prenatal and postnatal tobacco smoking and DNA methylation were measured by fitting linear regression models for composite variables that were newly derived from paternal smoking preconception, maternal prenatal smoking and child postnatal passive smoking (Supplementary Table S3). Linear regression models were adjusted for cell type heterogeneity using principal components (PCs) derived from ReFACTor 33, and genetic ancestry using PCs derived from EPISTRUCTURE 34.
Linear regression models were used to assess whether the DNA methylation-based biomarkers had significant associations with child’s birth year, with the AHRR-mQTL SNP being additionally adjusted for DNA methylation at the AHRR CpG cg05575921 25.
Poisson regression models were used to test association between DNA methylation at the AHRR CpG cg05575921 and deletion numbers in 284 cases in the EPIC dataset, and between the polyepigenetic smoking scores and deletion numbers in both the 450K and EPIC datasets. Models were adjusted for ReFACTor and EPISTRUCTURE PCs and additionally adjusted for the AHRR-mQTL SNP to control for potential confounding 25. Models for ratios of means (RMs) were calculated for every 0.1 beta-value decrease 20 in AHRR cg05575921 methylation, and for every 4-unit increase in polyepigenetic scores. We also assessed the association between the polyepigenetic scores minus the AHRR CpG cg05575921 and deletion numbers. Sensitivity analysis was conducted in which the Poisson regression models were adjusted for self-reported race/ethnicity (i.e., Latino, non-Latino White, and non-Latino other), instead of EPISTRUCTURE PCs, in the subset of cases with available data (Table 1).
Fixed effect meta-analysis models were used to test for heterogeneity between 450K and EPIC datasets, and to generate summary effect estimates accounting for the variance of each dataset, using R packages tidymeta and metafor 35,36. Study heterogeneity was characterized with I2 statistics and their corresponding p-values 37.
Finally, we repeated the epigenetic biomarker and gene deletions analyses stratified by: 1) self-reported race/ethnicity, in the subset of B-ALL cases with available data (Table 1) and limited to Latinos and non-Latino Whites due to sample size; and 2) age of diagnosis, limited to ≥2 years of age, 0 to 5 years of age, and >5 years of age (as the number of ALL cases diagnosed <1 year of age in our study [n=8] was small).
Results
Demographic characteristics of the 482 B-ALL cases are summarized in Table 1, and the study design is illustrated in Figure 1. The distribution of deletions among 198 B-ALL cases in the 450K dataset and in the additional 284 B-ALL cases in the EPIC dataset were similar (Supplementary Fig. S1 and Supplementary Table S4). In the 450K dataset, 125/198 (63.1%) of cases harbored at least one gene deletion compared with 162/284 (57.0%) of cases in the EPIC dataset (Supplementary Fig. S1).
Association between self-reported smoking variables and DNA methylation-based biomarkers of maternal smoking in B-ALL cases
The median AHRR cg05575921 beta-value among B-ALL cases was 0.82 (interquartile range [IQR]: 0.79-0.85) in the 450K dataset and 0.81 (IQR: 0.78-0.84) in the EPIC dataset. The median polyepigenetic smoking score was −0.52 among 194 cases (4/198 cases excluded due to missing data) in the 450K dataset (IQR: −1.83-0.95) and 0.83 (IQR: −0.34-2.11) among 284 cases in the EPIC dataset (Supplementary Fig. S2). Mean methylation beta values of CpGs that were used to generate the polyepigenetic scores are summarized in Supplementary Table S1. Beta values of most of the CpGs were significantly different between B-ALL cases in the 450K dataset and the EPIC dataset (Supplementary Fig. S3), although a significant difference was not found for DNA methylation at the AHRR CpG cg05575921.
Self-reported tobacco smoking exposure data were available for all 198 B-ALL cases in the 450K dataset and 189 out of 284 cases in the EPIC dataset (Supplementary Table S5). The distributions of smoking variables were similar between cases in the 450K and EPIC datasets, although in general more cases in the 450K dataset were reported to be exposed to tobacco smoke. We did not find any evidence that DNA methylation at the AHRR CpG cg05575921 or the polyepigenetic smoking scores were associated with child’s birth year (Supplementary Fig. S4).
Maternal smoking variables were strongly correlated with each other (rho range: 0.36-1.00) and had relatively lower correlations with paternal smoking variables (rho range: 0.03-0.48) (Supplementary Fig. S5). The two DNA methylation-based biomarkers were significantly correlated (450K: rho = 0.54; EPIC: rho = 0.60). Decreased DNA methylation at the AHRR cg05575921 was correlated with maternal prenatal smoking exposures in both the 450K and EPIC datasets; it was additionally correlated with maternal smoking during breastfeeding and child passive smoking (via parental smoking) in the EPIC dataset. Increased polyepigenetic scores were significantly correlated with the majority of the self-reported parental smoking exposures in both 450K and EPIC data.
In both the 450K and EPIC datasets, polyepigenetic smoking scores were associated with nearly all of the self-reported smoking exposures in multivariable linear regression models (Figure 2). Decreased AHRR cg05575921 beta-value was mainly associated with maternal smoking exposures. Furthermore, joint exposures of maternal or paternal smoking and child postnatal passive smoking were significantly associated with the two epigenetic biomarkers.
Figure 2. Linear regression results for the associations of parental self-reported tobacco smoke exposures with DNA methylation at the AHRR CpG cg05575921 and with the polyepigenetic smoking score.
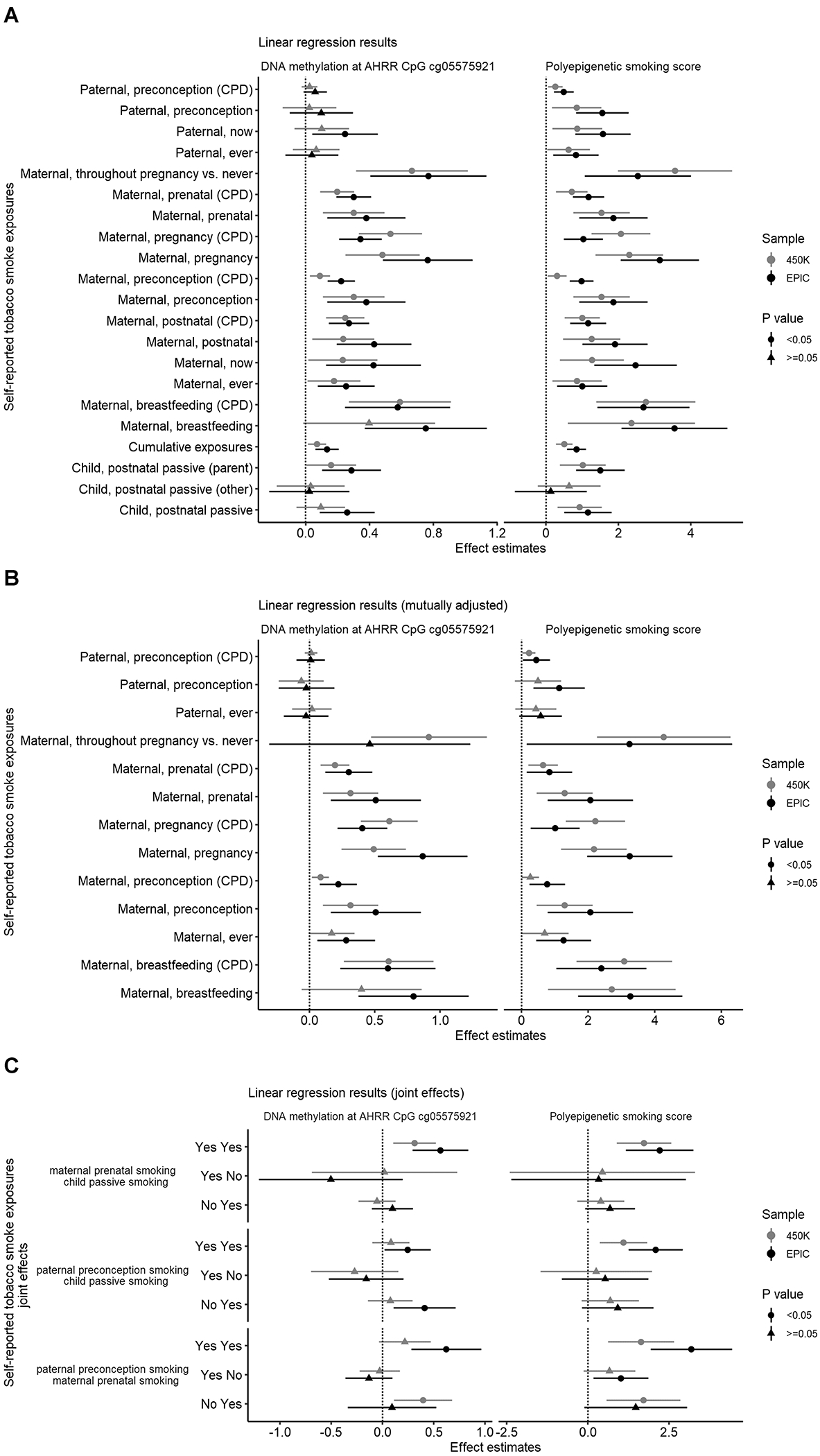
Paternal and maternal ever smoking were defined as having smoked at least 100 cigarettes, pipes, or cigars before the child’s diagnosis. Additional dichotomous variables only accounted for whether the mother or father smoked at all during the time period described. Child postnatal passive smoking was measured by child secondhand smoking from either parent, or from other persons aside from parents who smoked indoors, in order to show the presence of a regular smoker (e.g. the mother, father, or other individual) in the household up to the child’s third birthday or ALL diagnosis (whichever came first). Cumulative tobacco exposures were calculated from four binary exposures: paternal smoking during preconception, maternal smoking during preconception, maternal smoking during pregnancy, and child’s postnatal passive smoking. Parental continuous smoking exposures were measured by number of cigarettes, pipes, or cigars per day (CPD) in 5-unit increments. All linear regression models were adjusted for cell type heterogeneity and genetic ancestry. AHRR CpG cg05575921 beta value was multiplied by −10. Linear regression models were fitted for each smoking exposure variable in the 450K dataset and EPIC dataset. Panels show results from linear regression models for the outcome variable DNA methylation at the AHRR CpG cg05575921 (left) or for the outcome variable polyepigenetic smoking score (right). Centers of points and horizontal bars indicate point estimates and 95% confidence intervals. (A) Results from linear regression models adjusted for cell type heterogeneity and genetic ancestry only. (B) Independent effects from linear regression models additionally mutually adjusted for paternal and maternal smoking variables. (C) The joint exposures of prenatal and postnatal tobacco smoking from linear regression models for newly derived variables of paternal smoke preconception plus maternal prenatal smoking, maternal prenatal smoking plus child postnatal passive smoking, and paternal smoke preconception plus child postnatal passive smoking. Reference group: cases who were unexposed to both exposures that make up the joint effect.
Independent maternal and paternal smoking effects on DNA methylation were obtained from multivariable linear regression models (Figure 2). Maternal smoking exposures remained associated with polyepigenetic smoking scores and AHRR cg05575921 methylation while adjusting for paternal smoking preconception. In addition, paternal smoking during preconception remained associated with the polyepigenetic smoking score when controlling for maternal prenatal smoking. Notably, we found a −0.091 difference in the mean cg05575921 beta value and a ~4-unit difference for the polyepigenetic smoking score in the 450K dataset for mothers who smoked throughout pregnancy compared with mothers who were never exposed to tobacco smoke. The −0.091 difference is comparable to the previously reported −0.1 difference in AHRR cg05575921 beta-value of neonates of mothers with high cotinine levels versus mothers with undetectable cotinine levels 38. Therefore, we considered the corresponding 4-unit coefficient estimate in the same model for the polyepigenetic score to be biologically relevant, and subsequently computed RMs of deletion numbers for every 4-unit increase of the polyepigenetic score in both 450K and EPIC datasets.
DNA methylation-based biomarkers of tobacco smoke exposure are associated with gene deletion burden in childhood ALL
In the new EPIC dataset of 284 B-ALL cases, we found a 1.31-fold increase in the mean number of deletions with every 0.1 beta-value decrease in cg05575921 (95% CI, 1.02-1.69; Figure 3). After stratifying by sex, a stronger association presented in males (RM, 1.41; 95% CI, 0.99-2.02) compared to females (RM, 1.30; 95% CI, 0.87-1.95) (Supplementary Table S6), however, these differences were not significant in tests for heterogeneity (Phet = 0.599). In a meta-analysis of the 450K and EPIC datasets, the summary RM (sRM) was 1.32 (95% CI, 1.10-1.57; Figure 3).
Figure 3. Forest plots showing meta-analysis results of the association between epigenetic biomarkers of prenatal tobacco smoke exposure and gene deletion frequency in B-ALL cases.
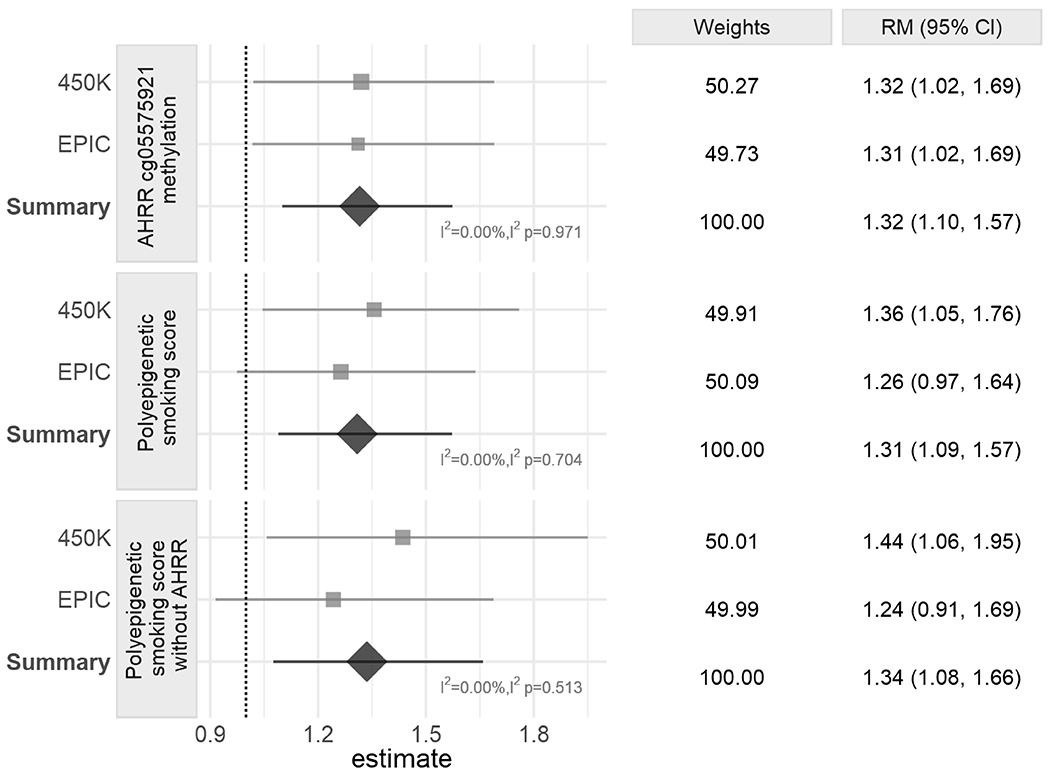
The panels include Poisson regression results for the association between deletion numbers and DNA methylation at the AHRR CpG cg05575921 (top), the polyepigenetic smoking score (middle), and the polyepigenetic smoking score excluding AHRR CpG cg05575921 (bottom). Ratio of means (RM) were calculated for every 0.1 beta value decrease of cg05575921 and every 4-unit increase of polyepigenetic smoking score. All Poisson regression models were adjusted for cell type heterogeneity and genetic ancestry. Models with exposure variable DNA methylation at the AHRR CpG cg05575921 were additionally adjusted for methyl-QTL SNP genotypes (rs148405299 in the 450K dataset and rs77111113 in the EPIC dataset). Centers of squares and horizontal bars through each indicate point estimates and 95% confidence intervals (CI) of individual set RM. Area of squares indicate relative weights of individual set. Vertical apices of diamonds and horizontal bars through each indicate summary RM and 95% CI. Relative weights (%) (proportional to the reciprocal of the sampling variance of the individual set) of two sets, RM, sRM, and 95% CI are summarized in the right panel.
We further extended our original analysis by constructing a DNA methylation-based smoking score, including the AHRR CpG and over 20 additional CpGs. In the meta-analysis of the 450K and EPIC datasets, the polyepigenetic score was also significantly associated with an increased number of deletions with a 1.31-fold increase in mean number of deletions for every 4-unit increase in the score (95% CI, 1.09-1.57; Figure 3). Similar effect sizes were seen for the association between the polyepigenetic score and the number of deletions in the 450K dataset (RM = 1.36; 95% CI, 1.05-1.76) and the EPIC dataset (RM = 1.26; 95% CI, 0.97-1.64), although the latter did not reach statistical significance (Figure 3).
We next explored whether removal of the AHRR CpG cg05575921 would impact the association between the polyepigenetic score and ALL patient gene deletion burden. A significant association between the modified polyepigenetic score and the number of deletions was still observed in the 450K dataset (RM = 1.44; 95% CI, 1.06-1.95), with a slightly attenuated effect in the EPIC dataset (RM = 1.24; 95% CI, 0.91-1.69; Figure 3). In the meta-analysis, the polyepigenetic smoking score excluding the AHRR CpG remained significantly positively associated with number of gene deletions (sRM = 1.34; 95% CI, 1.08-1.66; Figure 3).
No significant interaction effects were detected between the DNA methylation-based biomarkers and B-ALL cytogenetic subtypes (high-hyperdiploidy [HD-ALL] and ETV6-RUNX1 fusion) on deletion numbers (Supplementary Table S7).
Effect sizes from Poisson regression models adjusting for self-reported race/ethnicity were very similar to those adjusting for EPISTRUCTURE PCs (Supplementary Fig. S6). In analyses stratified by self-reported race/ethnicity, stronger associations between the DNA methylation-based biomarkers and gene deletions presented in non-Latino White compared to Latino B-ALL cases (Supplementary Fig. S7), although the differences were not significant in tests for heterogeneity (Phet >0.10). Finally, we assessed potential effects of patient age-at-diagnosis on our results, and observed similar associations between the epigenetic biomarkers and gene deletion burden after excluding cases diagnosed <2 years of age to those found in the overall B-ALL cases (Supplementary Fig. S8); the association between the polyepigenetic smoking score and gene deletions was slightly stronger among B-ALL cases diagnosed >5 years of age than those diagnosed ≤5 years of age (Phet >0.10).
Discussion
Somatic copy-number loss of lymphoid transcription factor and cell cycle control genes is an important driver of leukemogenesis in childhood ALL. Aberrant recombination-activating gene (RAG) activity, which normally drives antibody diversification as part of the adaptive immune system, is thought to underlie the formation of gene deletions in some ALL patients 39,40. However, few epidemiology studies have explored whether extrinsic factors influence the generation of somatic copy-number alterations in developing lymphocytes. Here, we provide further evidence that prenatal exposure to tobacco smoke may induce leukemia-causing gene deletions in ALL patients 20,41.
We recently reported that decreased DNA methylation at the AHRR CpG cg05575921, a biomarker for maternal smoking during pregnancy 38,42, was associated with an increased frequency of somatic gene deletions among childhood B-ALL cases 20. We have replicated this association in a larger, independent set of childhood B-ALL cases, assayed on Illumina® EPIC DNA methylation arrays, and found a remarkably similar effect size with a ratio of means of 1.31 in the current study compared with 1.32 in our previous report. Further, we found a similar positive association between gene deletion frequency and increased in utero tobacco smoke exposure in ALL cases as measured by a recently established polyepigenetic smoking score 23. This association remained after removal of the AHRR CpG from the smoking score and, thus, we were able to confirm our findings using an independent epigenetic biomarker.
Previous case-control studies based on questionnaire data reported significant association between paternal preconception smoking and childhood ALL risk, but no association between maternal smoking and childhood ALL risk 10,43,44. The discrepancy between our findings and the epidemiological literature on maternal smoking and childhood ALL risk could be due to several reasons. First, case-control studies limited to the use of self-reported data may be affected by recall bias, and may include potentially underreported smoking exposures 45 due to a perceived social stigma 46, in particular for maternal smoking. In addition, the two epigenetic biomarkers examined in this study can reflect particularly sustained maternal smoking throughout pregnancy 10,23,47, which is difficult to assess using single survey questions.
Second, we cannot rule out that these epigenetic biomarkers may also be proxies for non-maternal and/or postnatal tobacco smoke exposures that may impact the generation of gene deletions in childhood ALL. In our study, AHRR cg05575921 methylation was strongly associated with self-reported maternal prenatal smoking, consistent with previous findings that decreased methylation at cg05575921 was definitively associated with in utero exposure to maternal smoking 21,25, and not overtly connected to paternal smoking or secondhand smoke exposure 22. However, in contrast, the polyepigenetic smoking scores were associated with both self-reported maternal and paternal smoking exposures, suggesting that some CpGs included in this score may be associated with multiple sources of tobacco exposure. Moreover, both biomarkers were associated with the cumulative self-reported smoking exposures and the joint exposures of parental prenatal smoking with child postnatal passive smoking. These composite variables are indicative of smoking exposures in the household or residual prenatal smoking exposures that were not captured by single survey questions.
Third, the potential leukemogenic effects of tobacco smoke exposure on gene deletions in ALL patients may not translate to overall ALL risk in case-control studies, perhaps due to varying effects in different molecular subtypes. This is supported by the finding that the combination of paternal prenatal smoking with child postnatal passive smoking was significantly associated with ETV6-RUNX1 fusion ALL, but not with HD-ALL 10. HD-ALL is associated with a lower frequency of somatic gene deletions relative to other ALL subtypes 20 and, in our previous study, self-reported tobacco exposures were no longer associated with gene deletion frequency in ALL cases when restricted to HD-ALL 20, though we did not formally test for interaction. In the current study, we did not find significant interaction between ALL subtype and the DNA methylation-related biomarkers, but this may be due to a lack of power and warrants further investigation.
To our knowledge, this is the first study to (1) compute a polyepigenetic smoking score using neonatal DNA methylation data from EPIC arrays, and (2) test whether self-reported smoking exposures were associated with polyepigenetic scores derived from both 450K and EPIC data. The smoking score was developed by Reese et al. using 450K array data from newborn cord blood samples 23 and, in our study, we found largely consistent mean methylation beta values for the CpGs used to generate the score (Supplementary Table S1). Excluding the two CpGs present on 450K but not EPIC arrays caused little loss of performance in the 450K data; however, the predictive performance of the score using EPIC array data requires further investigation. We found that the majority of CpGs in the consensus smoking score showed significantly different average beta values between 450K and EPIC array data (21/26 CpGs in overall newborns; 19/26 CpGs in newborns not exposed to tobacco smoke during pregnancy) (Supplementary Fig. S3). Further, the consensus score was significantly lower in the 450K dataset (0.26 [IQR: −1.03-1.80]) than the EPIC dataset (0.83 [IQR: −0.34-2.11]; Wilcoxon test p = 0.003), despite more newborns in the 450K dataset being exposed to parental tobacco smoke according to interview data (Supplementary Table S5). The inter-array differences in the smoking score CpGs did not correspond consistently with their association with maternal smoking during pregnancy 38, nor were they likely explained by changes in individuals’ smoking behaviors over time (i.e., cohort effect) (Supplementary Fig. S4). They may instead be due to probe cross-reactivity or a shifted distribution of methylation values caused by increased Type II probe measurements on EPIC arrays 48,49.
The polyepigenetic smoking score was developed in a homogenous population from Norway 23, a country with different smoking habits (e.g. more prevalent use of hand-rolled cigarettes with higher nicotine and tar content) than the US 50. This may hamper the performance and generalizability of the score in our study, in which over 50% of cases were of non-white race/ethnicity, with a particularly large number of Latinos. In analyses stratified by self-reported race/ethnicity, both epigenetic biomarkers of tobacco smoke exposure showed a stronger association with the frequency of ALL gene deletions in non-Latino Whites than in Latinos. This might be attributable to a potentially superior performance of these biomarkers in predicting prenatal tobacco smoke exposure in non-Latino Whites compared to Latinos, but this warrants further evaluation as the number of non-Latino White cases in our study was relatively small. Nonetheless, the transferability of epigenetic biomarkers developed in largely European ancestry individuals across ancestrally diverse populations should be determined.
Our study does have several limitations that warrant consideration. Importantly, the DNA methylation-based biomarkers were derived from newborn DBS, thus we were not able to assess the potential effects of postnatal tobacco smoke exposure. Preleukemic clones may be present at birth, but at very low clonal frequencies in whole blood 51,52 and, thus, are unlikely to have influenced our DNA methylation results. This is supported by the minimal effects on our results after excluding B-ALL cases diagnosed <2 years of age. An additional limitation was our limited ability to study the effects of tobacco smoke exposure across different cytogenetic subtypes of ALL, due to sample size and a lack of information on subtypes beyond HD-ALL and ETV6-RUNX1 fusion. Further, our analyses were limited to the 8 commonly deleted genes targeted by the MLPA assays. These assays do not provide information on deletion breakpoint locations, hence we could not explore the molecular mechanisms underlying the formation of deletions in our ALL cases. Given that aberrant RAG-mediated V(D)J recombination underlies a large proportion of somatic gene deletions in ALL 39,40,53, it is compelling that cord blood lymphocytes in newborns of mothers exposed to tobacco smoke have been found to harbor a significantly increased frequency of off-target RAG recombination-mediated deletions than in newborns of mothers who were not exposed to tobacco 41,54, however, this remains to be examined in the setting of childhood ALL.
In summary, we provide further evidence that prenatal tobacco smoke exposure may influence the generation of somatic copy-number deletions in childhood B-ALL cases. Future epidemiological studies that incorporate both information on early-life exposure to tobacco smoke as well as whole-genome sequencing of ALL tumors and, in turn, analysis of mutational signatures and deletion breakpoint sequences are required to investigate the potential mutagenic effects of tobacco smoke in childhood ALL.
Supplementary Material
Acknowledgments:
The biospecimens and/or data used in this study were obtained from the California Biobank Program, (SIS request # 26), Section 6555(b), 17 CCR. The California Department of Public Health is not responsible for the results or conclusions drawn by the authors of this publication. For recruitment of subjects enrolled in the California Childhood Leukemia Study (CCLS), the authors gratefully acknowledge the families for their participation. We also thank the clinical investigators at the following collaborating hospitals for help in recruiting patients: University of California Davis Medical Center (Dr. Jonathan Ducore), University of California San Francisco (Drs. Mignon Loh and Katherine Matthay), Children’s Hospital of Central California (Dr. Vonda Crouse), Lucile Packard Children’s Hospital (Dr. Gary Dahl), Children’s Hospital Oakland (Drs. James Feusner and Carla Golden), Kaiser Permanente Roseville (formerly Sacramento) (Drs. Kent Jolly and Vincent Kiley), Kaiser Permanente Santa Clara (Drs. Carolyn Russo, Alan Wong, and Denah Taggart), Kaiser Permanente San Francisco (Dr. Kenneth Leung), Kaiser Permanente Oakland (Drs. Daniel Kronish and Stacy Month), California Pacific Medical Center (Dr. Louise Lo), Cedars-Sinai Medical Center (Dr. Fataneh Majlessipour), Children’s Hospital Los Angeles (Dr. Cecilia Fu), Children’s Hospital Orange County (Dr. Leonard Sender), Kaiser Permanente Los Angeles (Dr. Robert Cooper), Miller Children’s Hospital Long Beach (Dr. Amanda Termuhlen), University of California, San Diego Rady Children’s Hospital (Dr. William Roberts), and University of California, Los Angeles Mattel Children’s Hospital (Dr. Theodore Moore). We thank Robin Cooley and Steve Graham at the California Biobank Program, California Department of Public Health, for assistance in retrieval of neonatal dried bloodspot specimens for CCLS subjects.
Financial support, including the source and number of grants, for each author:
This research was supported by the California Tobacco-Related Disease Research Program, Grant No. 26IR-0005A (A.J.D., C.M., and A.Y.K.), by the National Institute of Environmental Health Sciences grants R01ES009137 and P42ES004705 (C.M., A.Y.K., J.L.W., and A.J.D.), R24ES028524 (C.M., A.Y.K., T.P.W., and L.M.), P01ES018172 (C.M., T.P.W., A.Y.K., L.M., J.L.W., and A.J.D.), and P50ES018172 (C.M., T.P.W., A.Y.K., L.M., J.L.W., S.C.K. and A.J.D.), by the United States Environmental Protection Agency under assistance agreement RD83451101 (C.M., T.P.W., A.Y.K., L.M., J.L.W., and A.J.D.) and RD83615901 (C.M., T.P.W., A.Y.K., L.M., J.L.W., S.C.K. and A.J.D.), and in part by the Children with Cancer UK (C.M.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or TRDRP. The contents of this document do not necessarily reflect the views and policies of the Environmental Protection Agency, nor does the EPA endorse trade names or recommend the use of commercial products mentioned in this document.
Footnotes
Conflict of interest: The authors declare no potential conflicts of interest.
References
- 1.Ward E, DeSantis C, Robbins A, Kohler B & Jemal A Childhood and adolescent cancer statistics, 2014. CA: A Cancer Journal for Clinicians 64, 83–103 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Cancer Facts & Figures. American Cancer Society; (2020). [Google Scholar]
- 3.Essig S et al. Estimating the risk for late effects of therapy in children newly diagnosed with standard risk acute lymphoblastic leukemia using an historical cohort: A report from the Childhood Cancer Survivor Study. Lancet Oncol 15, 841–851 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Winther JF & Schmiegelow K How safe is a standard-risk child with ALL? The Lancet Oncology 15, 782–783 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Mody R et al. Twenty-five—year follow-up among survivors of childhood acute lymphoblastic leukemia: a report from the Childhood Cancer Survivor Study. Blood 111, 5515–5523 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Whitehead TP, Metayer C, Wiemels JL, Singer AW & Miller MD Childhood Leukemia and Primary Prevention. Current Problems in Pediatric and Adolescent Health Care 46, 317–352 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Preston DL et al. Cancer incidence in atomic bomb survivors. Part III. Leukemia, lymphoma and multiple myeloma, 1950-1987. Radiat. Res. 137, S68–97 (1994). [PubMed] [Google Scholar]
- 8.Doll R & Wakeford R Risk of childhood cancer from fetal irradiation. BJR 70, 130–139 (1997). [DOI] [PubMed] [Google Scholar]
- 9.Pui C-H, Nichols KE & Yang JJ Somatic and germline genomics in paediatric acute lymphoblastic leukaemia. Nat Rev Clin Oncol 16, 227–240 (2019). [DOI] [PubMed] [Google Scholar]
- 10.Metayer C et al. Tobacco Smoke Exposure and the Risk of Childhood Acute Lymphoblastic and Myeloid Leukemias by Cytogenetic Subtype. Cancer Epidemiology Biomarkers & Prevention 22, 1600–1611 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Greaves M Infection, immune responses and the aetiology of childhood leukaemia. Nat Rev Cancer 6, 193–203 (2006). [DOI] [PubMed] [Google Scholar]
- 12.Greaves M Childhood leukaemia. BMJ 324, 283–287 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Greaves M A causal mechanism for childhood acute lymphoblastic leukaemia. Nat Rev Cancer 18, 471–484 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wiemels J et al. Prenatal origin of acute lymphoblastic leukaemia in children. The Lancet 354, 1499–1503 (1999). [DOI] [PubMed] [Google Scholar]
- 15.Greaves MF, Maia AT, Wiemels JL & Ford AM Leukemia in twins: lessons in natural history. Blood 102, 2321–2333 (2003). [DOI] [PubMed] [Google Scholar]
- 16.Bateman CM et al. Acquisition of genome-wide copy number alterations in monozygotic twins with acute lymphoblastic leukemia. Blood 115, 3553–3558 (2010). [DOI] [PubMed] [Google Scholar]
- 17.Mullighan CG et al. GENOMIC ANALYSIS OF THE CLONAL ORIGINS OF RELAPSED ACUTE LYMPHOBLASTIC LEUKEMIA. Science 322, 1377–1380 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schwab CJ et al. Genes commonly deleted in childhood B-cell precursor acute lymphoblastic leukemia: association with cytogenetics and clinical features. Haematologica 98, 1081–1088 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mullighan CG et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature 446, 758–764 (2007). [DOI] [PubMed] [Google Scholar]
- 20.de Smith AJ et al. Correlates of Prenatal and Early-Life Tobacco Smoke Exposure and Frequency of Common Gene Deletions in Childhood Acute Lymphoblastic Leukemia. Cancer Res 77, 1674–1683 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Joubert BR et al. DNA Methylation in Newborns and Maternal Smoking in Pregnancy: Genome-wide Consortium Meta-analysis. Am J Hum Genet 98, 680–696 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Joubert BR et al. Maternal Smoking and DNA Methylation in Newborns: In Utero Effect or Epigenetic Inheritance? Cancer Epidemiol Biomarkers Prev 23, 1007–1017 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reese SE et al. DNA Methylation Score as a Biomarker in Newborns for Sustained Maternal Smoking during Pregnancy. Environ Health Perspect 125, 760–766 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Walsh KM et al. Genomic ancestry and somatic alterations correlate with age at diagnosis in Hispanic children with B-cell ALL. Am J Hematol 89, 721–725 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gonseth S et al. Genetic contribution to variation in DNA methylation at maternal smoking-sensitive loci in exposed neonates. Epigenetics 11, 664–673 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fortin J-P et al. Functional normalization of 450k methylation array data improves replication in large cancer studies. Genome Biol 15, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Triche TJ, Weisenberger DJ, Van Den Berg D, Laird PW & Siegmund KD Low-level processing of Illumina Infinium DNA Methylation BeadArrays. Nucleic Acids Res 41, e90 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aryee MJ et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 30, 1363–1369 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huber W et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat Methods 12, 115–121 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gentleman RC et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biology 16 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Machiela MJ & Chanock SJ LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics 31, 3555–3557 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org/ (2020). [Google Scholar]
- 33.Rahmani E et al. Sparse PCA corrects for cell type heterogeneity in epigenome-wide association studies. Nat. Methods 13, 443–445 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rahmani E et al. Genome-wide methylation data mirror ancestry information. Epigenetics & Chromatin 10, 1 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barrett M tidymeta: Tidy and Plot Meta Analyses. R package version 0.1.0.9000. (2020). [Google Scholar]
- 36.Viechtbauer W Conducting meta-analyses in R with the metafor package. Journal of Statistical Software 36, 1–48 (2010). [Google Scholar]
- 37.Higgins JPT & Thompson SG Quantifying heterogeneity in a meta-analysis. Statistics in Medicine 21, 1539–1558 (2002). [DOI] [PubMed] [Google Scholar]
- 38.Joubert Bonnie R et al. 450K Epigenome-Wide Scan Identifies Differential DNA Methylation in Newborns Related to Maternal Smoking during Pregnancy. Environmental Health Perspectives 120, 1425–1431 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Papaemmanuil E et al. RAG-mediated recombination is the predominant driver of oncogenic rearrangement in ETV6-RUNX1 acute lymphoblastic leukemia. Nat Genet 46, 116–125 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mendes RD et al. PTEN microdeletions in T-cell acute lymphoblastic leukemia are caused by illegitimate RAG-mediated recombination events. Blood 124, 567–578 (2014). [DOI] [PubMed] [Google Scholar]
- 41.Finette BA, O’Neill JP, Vacek PM & Albertini RJ Gene mutations with characteristic deletions in cord blood T lymphocytes associated with passive maternal exposure to tobacco smoke. Nature Medicine 4, 1144–1151 (1998). [DOI] [PubMed] [Google Scholar]
- 42.Markunas CA et al. Identification of DNA Methylation Changes in Newborns Related to Maternal Smoking during Pregnancy. Environ Health Perspect 122, 1147–1153 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Orsi L et al. Parental smoking, maternal alcohol, coffee and tea consumption during pregnancy, and childhood acute leukemia: the ESTELLE study. Cancer Causes Control 26, 1003–1017 (2015). [DOI] [PubMed] [Google Scholar]
- 44.Milne E et al. Parental Prenatal Smoking and Risk of Childhood Acute Lymphoblastic Leukemia. Am J Epidemiol 175, 43–53 (2012). [DOI] [PubMed] [Google Scholar]
- 45.Rhomberg LR, Chandalia JK, Long CM & Goodman JE Measurement error in environmental epidemiology and the shape of exposure-response curves. Critical Reviews in Toxicology 41, 651–671 (2011). [DOI] [PubMed] [Google Scholar]
- 46.Rebagliato M Validation of self reported smoking. Journal of Epidemiology & Community Health 56, 163–164 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Klimentopoulou A et al. Maternal smoking during pregnancy and risk for childhood leukemia: a nationwide case-control study in Greece and meta-analysis. Pediatr Blood Cancer 58, 344–351 (2012). [DOI] [PubMed] [Google Scholar]
- 48.Pidsley R et al. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biology 17, 208 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Logue MW et al. The correlation of methylation levels measured using Illumina 450K and EPIC BeadChips in blood samples. Epigenomics 9, 1363–1371 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rolke HB, Bakke PS & Gallefoss F Relationships between hand-rolled cigarettes and primary lung cancer: A Norwegian experience. The Clinical Respiratory Journal 3, 152–160 (2009). [DOI] [PubMed] [Google Scholar]
- 51.Schäfer D et al. Five percent of healthy newborns have an ETV6-RUNX1 fusion as revealed by DNA-based GIPFEL screening. Blood 131, 821–826 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mori H et al. Chromosome translocations and covert leukemic clones are generated during normal fetal development. Proc Natl Acad Sci U S A 99, 8242–8247 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mullighan CG et al. BCR—ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature 453, 110–114 (2008). [DOI] [PubMed] [Google Scholar]
- 54.Grant SG Qualitatively and quantitatively similar effects of active and passive maternal tobacco smoke exposure on in utero mutagenesis at the HPRT locus. BMC Pediatr 5, 20 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.