

Abstract
Background and Aims.
Epstein-Barr Virus (EBV)-associated Gastric Carcinoma (EBVaGC) is the most common EBV-associated cancer and accounts for ~10% of all gastric cancers (GC). Epstein-Barr virus Nuclear Antigen 1 (EBNA1), which is critical for the replication and maintenance of the EBV latent genome, is consistently expressed in all EBVaGC tumors. We previously developed small molecule inhibitors of EBNA1. In this study, we investigated the efficacy and selectivity of an EBNA1 inhibitor in cell-based and animal xenograft models of EBV-positive and EBV-negative gastric carcinoma.
Methods.
We tested the potency of an EBNA1 inhibitor, VK-1727, in vitro and in xenograft studies, using EBV-positive (SNU-719 and YCCEL1) and EBV-negative (AGS and MKN74) GC cell lines. After treatment, we analyzed cell viability, proliferation, and RNA expression of EBV genes by RT-qPCR.
Results.
Treatment with VK-1727 selectively inhibits cell cycle progression and proliferation in vitro. In animal studies, treatment with an EBNA1 inhibitor resulted in a significant dose-dependent decrease in tumor growth in EBVaGC xenograft models, but not in EBV-negative GC xenograft studies. Gene expression analysis revealed that short term treatment in cell culture tended towards viral gene activation, while long-term treatment in animal xenografts showed a significant decrease in viral gene expression.
Conclusions.
EBNA1 inhibitors are potent and selective inhibitors of cell growth in tissue culture and animal models of EBV-positive GC. Long-term treatment with EBNA1 inhibitors may lead to loss of EBV in mouse xenografts. These results suggest that pharmacological targeting of EBNA1 may be an effective strategy to treat patients with EBVaGC.
Keywords: Epstein-Barr Virus, Epstein-Barr nuclear antigen 1, Gastric Cancer, Xenograft Antitumor Assays
Mini-Abstract:
Treatment with an EBNA1 inhibitor resulted in a significant decrease in tumor growth in animal models of EBV-positive, but not EBV-negative, gastric carcinoma.
Introduction:
Gastric cancer (GC) is an aggressive and heterogenous disease and is a leading cause of cancer mortality in the world. In the United States, GC makes up 6.8% of total cancer cases and 8.8% of total cancer-associated deaths (1). There are four types of GC diagnosed based on the appearance of cancerous cells in different cell types and layers of the stomach with adenocarcinomas accounting for 90% of GC; adenocarcinomas are further divided into cardia (the top part of the stomach) and noncardia (the lower part of the stomach) cancers. Several environmental risk factors, including the infectious agents Helicobacter pylori and Epstein-Barr Virus (EBV) are highly associated with subtypes of GC. It is estimated that EBV associated gastric carcinomas (EBVaGC) comprise approximately 10% (11% of males and 6% of females) of all gastric cancers and represent the most common malignancy caused by EBV infection (2). The majority of EBVaGCs are lymphoepithelial-like carcinomas with a distinct molecular and clinical profile compared to EBV negative GCs. Moreover, EBVaGCs respond differently to antineoplastic therapy than to non-EBV GC and have different mean survival times, indicating EBVaGC is a distinct subtype that requires specific therapeutic strategies (3). Many EBVaGCs acquire resistance to standard of care chemotherapeutic agents and there are currently no EBV specific therapeutics available for treating EBVaGC.
EBV is a ubiquitous human γ-herpesvirus virus that is associated with diverse lymphoid and epithelial malignancies (4). EBV typically infects and establishes long-term latent infection in B-lymphocytes, but also replicates in epithelial cells in the oropharynx. EBV epithelial malignancies include endemic forms of nasopharyngeal carcinoma (NPC) and more recently discovered in subtypes of gastric carcinomas. Viral DNA structural analysis indicates that EBV positive cells in NPC and EBVaGC are monoclonal indicating that EBV infection precedes the clonal expansion of the carcinoma (5, 6). EBV maintains a stable latent infection in EBVaGC with viral gene expression limited to Epstein-Barr Nuclear Antigen 1 (EBNA1), BamHI fragment A rightward transcript (BART), and Epstein-Barr virus-encoded small RNAs (EBER). However, in about 40% of cases, latent membrane protein 2A (LMP2A) is also expressed (5). With respect to histopathology, lymphoepithelioma (LE)-like GC has been characterized as an undifferentiated carcinoma with high lymphocytic infiltration. It is also worth noting that LEGC resembles NPC histologically and shares some common genetic and epigenetic alterations, including DNA hypermethylation (7, 8).
EBNA1 is the only virally encoded protein consistently expressed in all EBV-positive cancers (9, 10). Experiments with siRNA knockdown of EBNA1 demonstrate that it is required for persistence of the EBV genome and continued proliferation of EBV-positive cells both in vitro and in vivo (11–13). EBNA1 binds to the viral origin of replication (OriP) and recruits cellular replication machinery to promote viral genome replication and episomal maintenance (10, 14). EBNA1 promotes immortalization and host cell survival by manipulating several cellular processes including p53 stabilization and pro-apoptotic pathways (15, 16). In addition to regulating the transcription of several viral latency genes, EBNA1 also binds directly to promoter regions and regulates the expression of several cellular genes, including the GC-specific tumor suppressor genes gastrokine 1 and 2 (17).
We previously reported the discovery of a series of small molecule inhibitors that block EBNA1-DNA binding (18, 19). EBNA1 inhibitors show activity in several biochemical and cell-based assays, including chromatin immunoprecipitation (ChIP) and DNA replication assays. In addition, EBNA1 inhibitors suppress tumor growth in several EBV-dependent xenograft models, including patient-derived xenografts for NPC. In this paper, we extend previous studies to investigate the efficacy and selectivity of EBNA1 inhibitors in tissue culture and mouse xenograft models of EBV-positive and negative GC. We observe significant tumor growth inhibition in EBV-positive GC xenografts, but not in EBV-negative xenografts. We also characterize viral gene expression after treatment.
Materials and Methods:
Cell lines and reagents
The identity of EBV-positive and negative cell lines [LCL352 (B-cell/LCL, EBV+), BJAB (B-cell, EBV−), C666-1 (NPC, EBV+), HK-1 (NPC, EBV−), YCCEL1 (GC, EBV+), SNU719 (GC, EBV+), AGS (GS, EBV−), and MKN74 EBV−)] were confirmed by short tandem repeat microsatellite testing. Cell lines were grown in RPMI-1640 medium (Gibco, BRL) containing 10% fetal bovine serum (FBS; Gibco, Gaithersburg, MD, USA) penicillin and streptomycin (50 U/ml), and L-glutamine. All cells were incubated at 37°C under a 5% CO2 humidified atmosphere. For bioimaging studies, the cell lines were transduced with a lentivirus constitutively expressing green fluorescent protein (GFP)-Internal Ribosome Entry Site (IRES)-luciferase transgene.
Resazurin cell respiration assay
Cells were plated at 8 x 103 cells (in 100 μl) per well in a 96-well plate. Inhibitors were added 24 hours later over a 10-point concentration range with two-fold dilutions (0.1953 to 100 μM) in quadruplicate wells. As positive and negative controls, DMSO alone (0.4%) and puromycin (20 μg/ml) treated wells, respectively, were also plated in quadruplicate wells. After 72 hours, resazurin (20 μl, final 50 μM) was added to each well. After 6 hours, plates were read on a PerkinElmer EnVision XCite multilabel plate reader (excitation, 560 nm; emission, 590 nm).
BrdU proliferation assay
Cells were plated at 5 x 104 per well (in 200 μl) in a 96-well plate and treated with DMSO alone (0.4%) or VK-1727 (0.25 or 2.5 μM). At 24 and 48 hours after treatment, the plates were centrifuged at 1000 rpm for 5 min, and the medium was carefully aspirated and replaced with fresh medium containing DMSO alone or EBNA1 inhibitor at the appropriate concentration. After 72 hours, a BrdU cell proliferation assay was performed according to the manufacturer’s instructions (CytoSelect BrdU Cell Proliferation ELISA Kit, Cell Biolabs, Inc.). Briefly, 10 μl of a 10x BrdU solution (100 μM BrdU) was added to each well, and plates were incubated at 37°C. Three hours later, cells were centrifuged at 1000 rpm for 5 min, washed three times with 100 ul of phosphate-buffered saline (PBS), and treated with fix/denature solution. Thereafter, cells were incubated with 100 μl of anti-BrdU antibody for 1 hour. Cells were washed three times with 1x wash buffer, and after incubation with a secondary HRP-conjugated antibody, a substrate solution was applied. Plates were read on a PerkinElmer EnVision XCite multilabel plate reader (absorbance, 450 nm). Data are represented as a percentage decrease in BrdU compared to DMSO treated control.
Analysis of cell cycle kinetics
LCL352 (B-cell/LCL, EBV+), BJAB (B-cell, EBV−), C666-1 (NPC, EBV+), HK-1 (NPC, EBV−), YCCEL1 (GC, EBV+), SNU719 (GC, EBV+), AGS (GS, EBV−), and MKN74 EBV−), cells were seeded at 2.4 x 105 cells/well in 6-well plates and exposed to VK-1727 (10 μM and 25 μM) in biological triplicates per each condition. After 72 h, cells were permeabilized with cold, 70% ethanol and resuspended in PBS containing PI (10 mg/mL) and RNAse A solution (100 μg/mL). Flow cytometry was performed on a BD-LSR II (BD Biosciences; Bedford, MA) and data were analyzed using FloJo software (Ashland, OR).
Cell growth assay
Each cell line was seeded at 2.4 x 105 cells/well overnight in a 6-well plate and treated the next day with EBNA1 inhibitor (VK-1727) alone or in combination with ganciclovir (GCV). 72 hours later, cells were stained with trypan blue and cell counts were obtained using the Countess II cell counter.
Animals
All institutional and national guidelines for the care and use of laboratory animals were followed. All procedures were approved by The Wistar Institute Institutional Animal Care and Use Committee (IACUC) in accordance with the Animal Welfare Act (AWA) and PHS Policy on Humane Care and Use of Laboratory Animals. All mice in this study were managed in accordance with the NIH Office of Laboratory Animal Welfare: “PHS Policy on the Humane Care and Use of Research Animals”; the recommendations of the American Association for Accreditation of Laboratory Animal Care (AAALAC).
Xenograft studies were performed in NSG mice (NOD.Cg-Prkdcscid Il2rgtm1Wjl/ScJ;) bred in-house at The Wistar Institute under protocol number 112092. There is a preponderance of GC in males (2:1; male vs female) and in the developing world, especially in Eastern Asia (1). Therefore, both male and female mice were used in this study. All mice were enrolled in their respective studies at 8 weeks of age and housed in micro-isolator cages in a designated, specific pathogen-free facility at The Wistar Institute (Philadelphia, PA), where they were fed sterile food and water ad libitum. Mice were euthanized via CO2 administration according to the American Association for Accreditation of Laboratory Animal Care (AAALAC) euthanasia guidelines.
Tumor implantation, grouping, and equalization
Mice were anesthetized using isoflurane and engrafted with a cell suspension (>98% viability) of 5 x 106 cells resuspended in 1x PBS (pH 7.4) and mixed with 20% of cold Matrigel (Corning Life Sciences) and maintained on ice. Cells were injected subcutaneously into the flank of each animal. Animals were weighed three-times per week and monitored daily. All tumors were measured by caliper, and tumor volume was calculated as follows: tumor volume = [length (L) × width (W)2]/2. After the average tumor volume reached 100 mm3, mice were grouped (n = 8 per group; 4 male and 4 female); between 28-45 days post engraftment depending on the cell line. Groups were normalized by bioluminescent imaging using the Spectrum IVIS CT Bioluminescent Imaging System (Perkin-Elmer; Waltham, MA) ensure the average flux (photons/sec) was equivalent across groups and to monitor cell growth throughout the study.
After randomization, treatment (vehicle or 10 mg/kg VK-1727) was administered by intraperitoneal injection twice a day (bis in die, b.i.d.) in a dose volume of 10 ml/kg body weight. Tumors growth was measured by IVIS bioluminescent imaging.
Visualization and quantitation of tumor growth using the Xenogen IVIS Bioluminescent Imaging System
For imaging studies, mice were injected with D-luciferin (Gold Biotechnology), i.p. at a dose of 7.5 mg/kg in a dose volume of 10 ml/kg body weight 15 minutes prior to imaging; this was the optimal interval between luciferin injection and bioluminescent imaging as determined by an initial kinetic curve for these cell lines in mice. Mice were anesthetized using isoflurane prior to and during imaging.
Ki67 analysis by flow cytometry
Tumor sections were homogenized and filtered through a 45 μM filter, followed by filtration through a 35 μM filter. Cells were washed twice with PBS and then fixed with 70% cold ethanol for one hour at −20°C. Cells were then washed three times with cell staining buffer (PBS with 5% fetal bovine serum and 0.09% sodium azide). After decanting cell staining buffer 100 μl of cell suspension was mixed with an antibody to Ki67 conjugated with Alexa 594 (Biolegend, San Diego, CA) and incubated at room temperature for 30 minutes. Cells were washed three times with cell staining buffer and flow cytometry was performed on a BD-LSR II (BD Biosciences; Bedford, MA). Data were analyzed using FloJo software (Ashland, OR).
Compound formulation and treatment schedules
For cell-based assays, compounds were weighed and resuspended in DMSO at a concentration of 50 mM, prior to dilution to desired concentration while maintaining a final concentration of DMSO at 0.4%. For animal studies, compounds were weighed and transferred to graduated tubes, and formulation reagents were added slowly dropwise in the following order: 1.0% Tween 80 (Sigma), 5% dimethylacetamide (Sigma), 15% PEG400 (polyethylene glycol, molecular weight 400) (Sigma), 10% propylene glycol, 30% PBS, and 39% water; 0.01% of 1N NaOH was added to clarify final solution. The vehicle control contained formulation reagents without the compounds.
Gene expression analysis
Tumor sections were flash-frozen, and later, RNA was isolated using the RNeasy Mini Kit (Qiagen) and treated with deoxyribonuclease (Qiagen). Reverse transcription followed by real-time (or quantitative) PCR (RT-qPCR) was used to measure gene expression levels of EBV genes. Primers used for qPCR of EBV genes include: EBNA1 F (5’-GGTCGTGGACGTGGAGAAAA-3’), EBNA1 R (5’-GGTGGAGACCCGGATGAT G3’); EBER2 F (5’-TTGCCCTAGTGGTTTCGGACACA3’-), EBER1 F (5’-TTTGCTAGGGAGGAGACGTGTGT-3’), EBER1 R (5’-AAGCAGAGTCTGGGAAGACAACCA-3’); EBER2 R (5’-ACTTGCAAATGCTCTAGGCGGGAA-3’); ZTA F (5—TCTGAACTAGAAATAAAGCGATACAAGAA-3’), ZTA R (TTGGGCACATCTGCTTCAAC); LMP1 F (5’-TCCAGAATTGACGGAAGAGGTT-3’), LMP1 R (5’-GCCACCGTCTGTCATCGAA-3’); BARF0 F(5’-GGTACGCTGTAGAAGCTGTTGAAG-3’), BARF0 R (5’-TGATATGGCCAGGCATCGA-3’); BART Ex1 F (5’-GTAGGCACTAGCCTCTTCATGTGA-3’), BART Ex1 R (5’-GGTAAGGGCTACGTCCGAGTCT-3’); BART Ex5 F (5’-GCTTGATGACGATGCCACAT-3’), and BART Ex5 R (5’-AATGCGCCGTCATTCCAT-3’). GUSB was used as a cellular control: GUSB (5’-CGCCCTGCCTATCTGTATTC and 5’-TCCCCACAGGGAGTGTGTAG-3’).
The average cycle threshold (CT) was determined by three independent samples. Template-negative (quantitative PCR reaction mixtures without cDNA) and RT-negative (RNA after genomic DNA elimination) conditions were used as controls. All data were normalized to the housekeeping gene GUSB and quantitative PCR data for the relative quantification were calculated with the ΔΔCt method. The level of EBNA1 transcript in cells treated with vehicle control (DMSO; 0.4%, vol/vol) was set as 1.
EBV viral load
Tumor sections were flash-frozen, and later, homogenized by tissuelyser and DNA was isolated DNeasy lood and issue kit (Qiagen, Hilden, Germany). PCR was used to quantify EBV viral load using a standard curve generated with Namalwa cells (two copies per cell). Primers used include: EBNA1 F 5’- TCA TCA TCA TCC GGG TCT CC-3’; EBNA1 R 5’- CCT ACA GGG TGG AAA AAT GGC −3’; β-actin F 5’- GCC ATG GTT GTG CCA TTA CA-3’; β-actin R 5’- GGC CAG GTT CTC TTT TTA TTT CTG-3’
Dot blot for DNA methylation
YCCEL1 and SNU719 were plated on 150 mm plates at a seeding density of 3X106 cells per plate. Twenty-four hours later, EBNA1 inhibitor (VK-1727) was added at a concentration of 0, 10, or 20 μM to duplicate plates. The DNA-demethylating agent decitabine (5-Aza-2’deoxycytadine; Sigma, St. Louis, MO) was used at 7.5 μM as a positive control. All plates had a final DMSO concentration of 0.4%. Medium was refreshed with EBNA1 inhibitor, DMSO alone, or decitabine three days later. At day seven, cells were harvested, and genomic DNA was extracted from the cells using the DNeasy lood and issue kit (Qiagen, Hilden, Germany). 1 μg of genomic DNA was denatured in 0.1M NaOH for ten min at 95°C, neutralized with 1M and diluted by two-fold. 4 μl of serial diluted genomic DNA was spotted on Hybond-N+ membrane (PerkinElmer, MA, USA). ir-dried membrane w subjected to DNA crosslinking UV crosslinker (Spectronics Corporation, NY, USA). After blocking in 5% BSA in TBST for 1h, membrane w incubated with 1:1000 diluted anti-5-methylcytosine (5-mC) monoclonal antibody (Abcam, Cambridge, UK) at 4° C overnight and then secondary antibody for 1h at room temperature. , dot blot w developed by incubation with ECL chemiluminescence for 1 min mages were captured and analyzed by Li-Cor Fc platform.
Results:
EBNA1 inhibitor decrease the proliferation of EBV-positive, but not EBV-negative, GC cells
To determine the effect of the EBNA1 inhibitor VK-1727 (Figure 1A) treatment on GC cell line proliferation, SNU719 (GC, EBV+) YCCEL1 cells (GC, EBV+) AGS (GC, EBV−) MKN74 (GC, EBV−) were treated with vehicle control (0.4% DMSO) or EBNA1 inhibitor VK-1727 with a dose of 0.25 μM or 2.5 μM. We performed a cell proliferation assay to measure 5-bromo-2’-deoxyuridine (BrdU) incorporation at 3 concentrations, 0, 0.25 and 2.5 μM. We found that EBNA1 inhibitor selectively inhibits proliferation of EBV-positive SNU719 and YCCEL1 cells, but not similarly growing EBV-negative AGS and MKN74 cells (Figure 1B). These results are similar to results obtained using other EBV positive and negative cell lines, including LCL352 (LCL/B-cell, EBV+), BJAB (B-cell, EBV−), C666-1 (NPC, EBV+), and HK1 (NPC, EBV−). These results suggest that VK-1727 is a potent and selective inhibitor of cell proliferation of EBV-positive GC cells compared with EBV-negative GC cells. In addition, the inhibition of proliferation in EBV-positive B-cells and NPC cells confirms that VK-1727 inhibits growth of EBV positive cells, regardless of their cell lineage or latency type.
Figure 1: Selective inhibition of EBV-positive cell lines by EBNA1 inhibitor VK-1727.
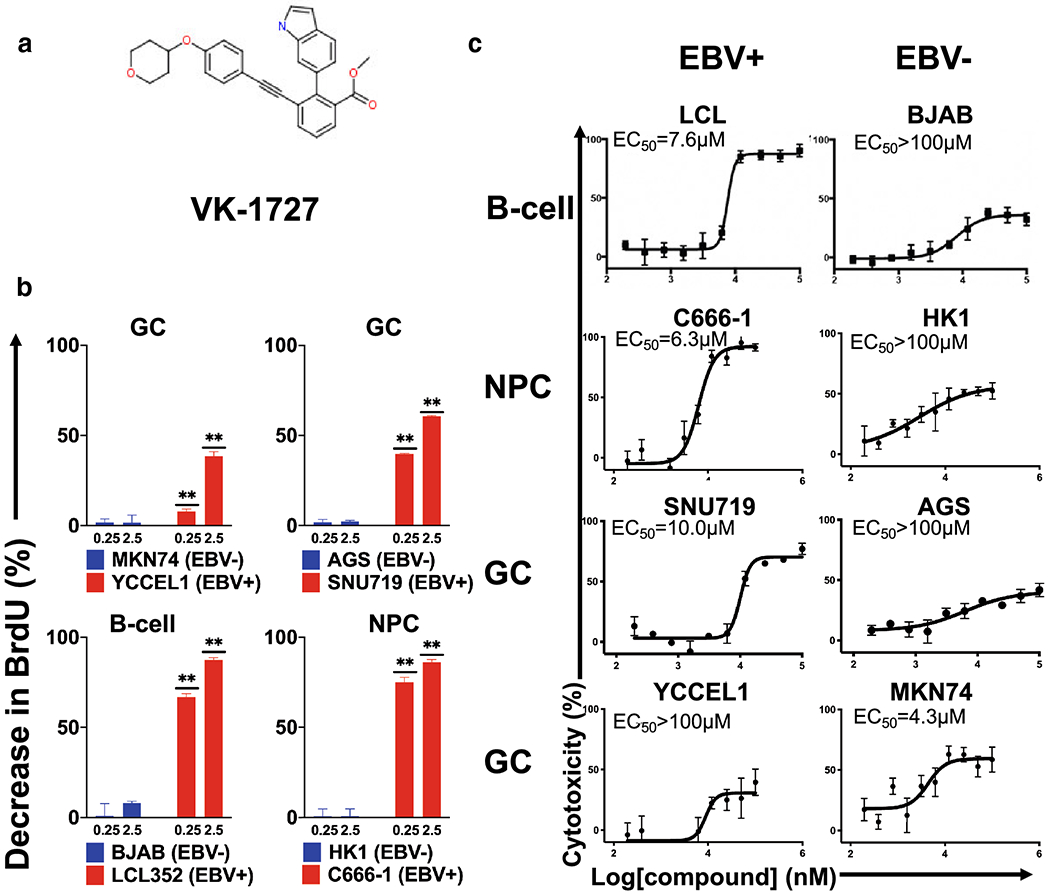
A. Structure of EBNA1 inhibitor VK-1727. B. Selective inhibition of cell proliferation as measured by significant decreases (Student T test; ** p < 0.0001) in BrdU compared to DMSO control in EBV-positive (red), but not EBV-negative (blue) cell lines at 0.25 and 2.5 μM. C. Resazurin-based cell metabolism assay using EBV-positive lymphoid cells LCL (EBV-positive) vs BJAB (EBV-negative) (top panels); NPC cells C666-1 (EBV+) vs HK-1 (EBV−) (middle panels); and gastric carcinoma cells SNU719 (EBV+) vs AGS (EBV−) (bottom panels) in a resazurin based cell viability assay, 72 hours after treatment.
As an additional measure of the effect of the EBNA1 inhibitor on cell viability, we used a resazurin assay to measure changes in metabolic activity of cells treated with VK-1727 (Figure 1C and S1). To determine the selectivity of EBNA1 inhibitor, the EBV-positive and negative cell lines were treated with VK-1727 and EC50 values calculated (Figure 1B). VK-1727 decreased the proliferation of most EBV-positive cells in a dose -dependent manner (Figure 1B). The EC50 value for the EBV-positive cell lines LCL352, C666-1, and SNU719 cells were 7.9 μM, 6.3 μM, and 10 μM, respectively), compared to the EBV-negative cell lines BJAB, HK1, and AGS, which were all had EC50s greater than 100 μM. Similar results were observed after 96 hours of incubation (Figure S1). Cell viability as measured by resazurin did not reach the 50% level at even the highest concentration of compound for the EBV-negative cell lines BJAB (B-cell), HK1 (NPC), and AGS (GC). The selective inhibition was consistent for all cells except for YCCEL1 (GC, EBV+) and MKN74 (GC, EBV−), where the EC50 for YCCEL1 was greater than 100 μM, in spite of the anti-proliferative effect of VK-1727 measured by other assays (Figure 1B and Figure 3B) and the decrease in cell number and cellular adherence observed in YCCEL1 treated with 10 μM or 25 μM VK-1727 by microscopy (Figure 2A and Figure S2).
Figure 3. EBV gene expression patterns in vitro and inhibition of proliferation in EBV-positive cell lines.
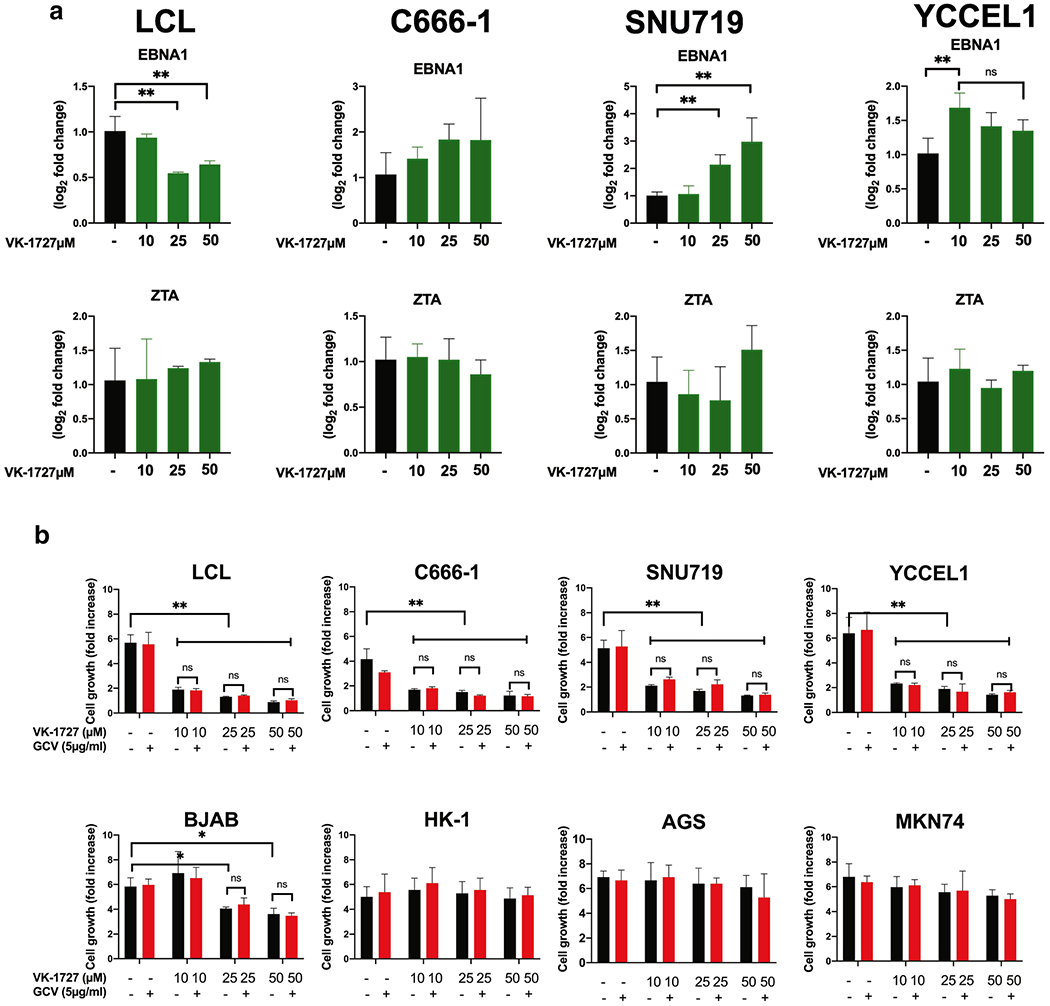
A. Expression of EBV genes EBNA1 and ZTA in EBV-positive cell lines measured by ΔΔCT method (Student T Test; ** p < 0.001, *** p < 0.0001). B. Cell counts are plotted as fold increase in cell number after 72 hours treatment. Each bar represents mean fold increase +/− SD of 3 replicates (EBV-positive lines: SNU719, C666-1, YCCEL1, and LCL352; EBV-negative lines: AGS, HK-1, MKN74, and BJAB). Cells were treated with DMSO, VK-127 (at 10, 25, or 50 μM), and VK-1727 with GCV (5 μg/ml).
Figure 2: EBNA1 inhibitor VK-1727 inhibits cell cycle progression.
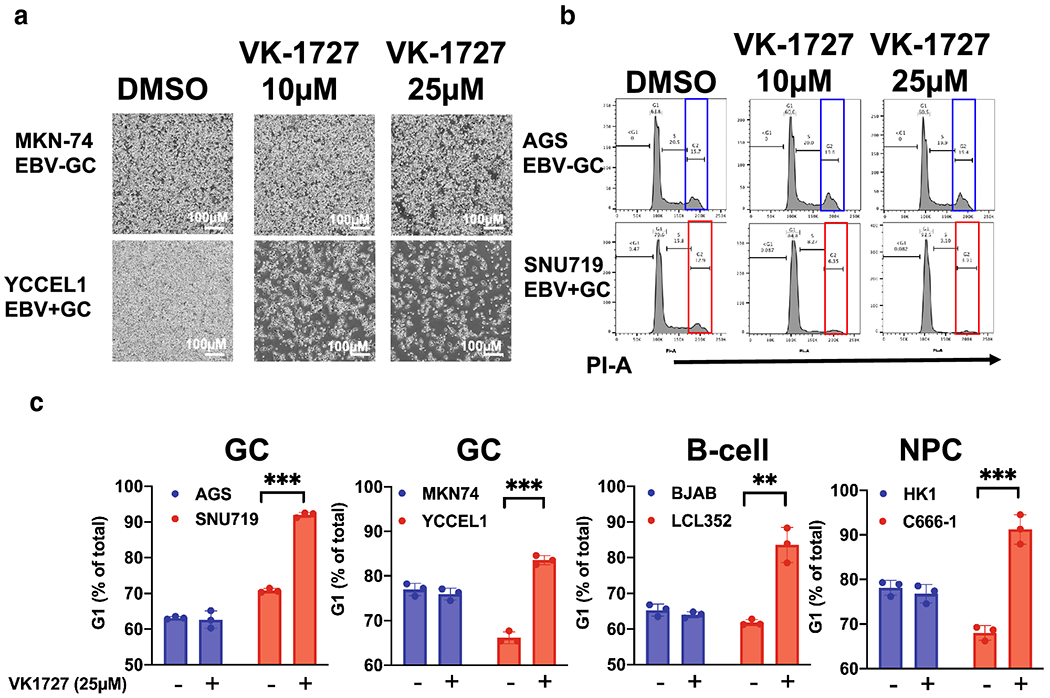
A. Images of EBV− (MKN74) and EBV+ (YCCEL1) GC cells with 4x objective using the Nikon TIE inverted microscope 72 hours after treatment with DMSO or VK-1727 (10 μM or 25 μM). Bar = 500 μm) B. Cell cycle profiles EBV− (AGS) and EBV+ (YCCEL1) cells treated with DMSO and 10 or 25 μM VK-1727 measured by FACS flow cytometry analysis of propidium iodide staining. Cell cycle changes, including a decrease in G2 (box) and an increase in G1, are observed in EBV-positive (SNU719), but not EBV-negative (AGS) gastric carcinoma cells. C. Cell cycle analysis of EBV-positive and EBV-negative GC, B-cells, and NPC cells treated with DMSO or VK-1727. G1 populations are represented as a percentage as total. (Student T test; ** p < 0.001, *** p < 0.0001). An increase in G1 as a percentage of the total population is increased in EBV-positive cells of B-cell, NPC, and GC origin (LCL, SNU719, YCCEL1, and C666-1), not in EBV-negative cells (Student T Test; ** p < 0.001).
EBNA1 inhibitor treatment perturbs the cell cycle in EBV-positive GC and other EBV-positive cells
To begin to understand the mechanism of action of VK-1727 in inhibiting cell proliferation, we first analyzed the cell cycle profiles for the EBV-positive GC line SNU719 and the EBV-negative GC line AGS using either 10 μM or 25 μM VK-1727 or DMSO as control for 72 hours (Figure 2B). Although cell cycle populations for AGS remained consistent during treatment with VK-1727, the cell cycle profile of EBV-positive SNU719 was clearly perturbed by VK-1727 treatment and a flattening of the G2 peak and increase in the G1 population was observed (Figure 2B). We followed this initial experiment to include two EBV-positive GC lines (SNU719 and YCCEL1), two EBV-negative GC lines (AGS and MKN74), two B-cell lymphomas lines (LCL352, EBV+ and BJAB, EBV−) and two NPC lines (C666-1, EBV+ and HK1, EBV−) after treatment with 25 μM VK-1727 (Figure 2C). EBNA1 inhibitor treatment altered the cell cycle profiles of all EBV-positive cell lines (SNU719, YCCEL1, LCL352, and C666-1) with a significant increase in the percentage of the population remaining in G1, while having no detectable effects on the cell cycle profiles of the EBV-negative cell lines (AGS, MKN74, BJAB, or HK1) (Figure 2B). These data suggest that the VK1727 inhibits cell cycle progression in EBV-positive cell types, again, regardless of their cell lineage or latency state. Notably, we did not see an increase in the <G1 population, suggesting that the EBNA1 inhibitor VK-1727 is cytostatic, rather than cytotoxic under these conditions.
EBNA1 inhibitors increase EBNA1 transcription and block cell proliferation of EBVaGC cell lines in vitro
EBV gene expression was measured after addition of VK-1727 to EBV positive B-cells (LCL352), NPC cells (C666-1), and EBVaGC cells (SNU719 and YCCEL1) (Figure 3). For these studies, we focused on the EBV latency gene EBNA1 and the EBV lytic gene BZLF/ZTA. In LCLs, EBNA1 gene expression was significantly reduced 72 hours after treatment with 25 μM and 50 μM of VK-1727; ZTA expression was unaffected (Figure 3A). In contrast, treatment with VK-1727 increased EBNA1 expression in both EBVaGC cell lines SNU719 and YCCEL1. These data suggest that VK-1727 is inhibiting EBNA1 in both LCLs and EBVaGC cells and indicates that the short-term effects of this treatment are different in cells of different lineages (B-cell compared with GC) and latency types (latency III vs latency I, respectively).
It was, therefore, of interest to determine whether EBNA1 inhibitor treatment induced lytic activation in EBV-positive cells, we treated EBV-positive cell lines (LCL, C666-1, SNU719 and YCCEL1) with 10-30 μM VK-1727 alone or in the presence of the antiviral drug ganciclovir (GCV), a synthetic analogue of 2’-deoxy-guanosine that is activated and phosphorylated in cells expressing lytic cycle enzymes of EBV. GCV would be expected to further inhibit viability of cells expressing EBV lytic enzymes. When we measure cell proliferation, here measured as fold increase in cell numbers 72 hours after treatment, we saw significant inhibition of cell proliferation in all EBV-positive cell lines (LCL352, C666-1, SNU719, and YCCEL1) treated with VK-1727, but not in EBV-negative cell lines (BJAB, HK1, AGS, and MKN74), with the exception of a decrease in BJAB proliferation at the highest concentrations of VK-1727 (Figure 3B). However, there was no additive effect of GCV observed, suggesting that VK-1727 does not induce lytic activation of EBVaGC or other EBV-positive cell lines.
EBNA1 inhibitors block EBV-driven tumor growth in vivo
To assess the efficacy of EBNA1 inhibitors in vivo, we engrafted the EBV-positive GC cell lines SNU714 and YCCEL1 into NSG mice and assessed tumor growth by bioluminescence (Figure 4A). After tumors were >100 mm3, mice were assigned to treatment groups to equalize tumor sizes by both size and bioluminescence as measured by Flux (Photons/sec). Eight mice per group (four male and four female) were assigned to be treated with vehicle, or 10 mg/kg VK-1727 Mice were administered compound or vehicle twice a day (b.i.d.) intraperitoneally for 24 days. We observed a significant decrease in tumor growth in EBV-positive tumors (YCCEL1 and SNU719) when treated with VK-1727. In the SNU719 and YCCEL1 xenograft studies, we observe that treatment results in tumor growth inhibition (TGI) of 61.2% and 67% at 10 mg/kg VK-1727 compared to vehicle control. To assess the selectivity of EBNA1 inhibitors, we performed similar xenograft experiments using the AGS and MKN74 cell lines (Figure 4B). A side-by-side comparison of bioluminescence showed that no growth inhibition was observed in EBV-negative GC tumors (AGS and MKN74) treated with VK-1727 (Figure 4C). Treatment with VK-1727 did not result in any appreciable weight loss compared to vehicle control (Figure 4D).
Figure 4. VK-1727 Inhibits EBV-positive GC tumor growth in xenografts.
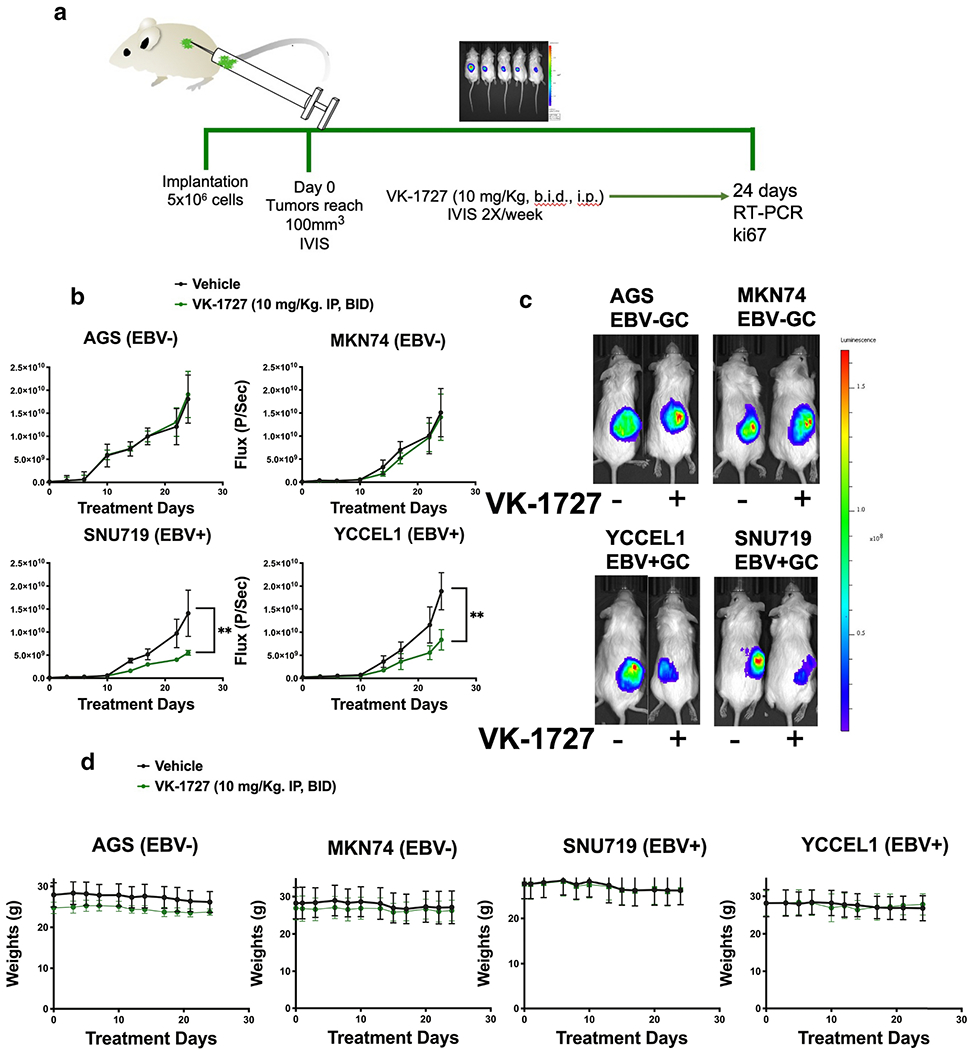
A. Schema illustrating key features of the in vivo experiments that were performed with VK-1727. NSG mice were implanted with 5 x 106 SNU719, YCCEL1, AGS, or MKN cells subcutaneously. When tumors reached 100 mm3, mice were randomized and treated daily with 0 (vehicle) or 10 mg/kg VK-1727, twice daily (b.i.d.). During treatment, the mice were continuously imaged using a Spectrum IVIS CT Bioluminescent Imaging System. At Day 24, the mice were sacrificed, weighed, and tumors were collected for further analyses. B. Graphs showing the changes in bioluminescence flux (photons/s) over time) (Student T test; ** p < 0.01). C. Images comparing the bioluminescence signals that were observed in mice implanted with GC cell lines treated with vehicle or VK-1727. For each pair, the left mouse was treated with vehicle and the right mouse was treated with VK-1727. D. Graphs comparing the changes in weight that were observed in mice engrafted with GC cell lines and treated with vehicle or VK-1727, twice daily i. p. Treatment with VK-1727 was not associated with weight loss or other clinical signs.
Treatment with EBNA1 inhibitors significantly reduces latent viral gene expression and EBV-viral load in EBV-positive GC xenografts
To assess the effect of treatment with EBNA1 inhibitors on viral gene expression, we isolated RNA from tumors at the end of the xenograft studies and performed quantitative RT-qPCR experiments for SNU719 (Figure 5A) and YCCEL1 (Figure 5B). We observe a significant (p<0.001) reduction in EBNA1, EBER1 and EBER2 in SNU719 and YCCEL1 tumors extracted from VK-1727 treated animals compared to vehicle. LMP1 mRNA expression was reduced in YCCEL1, but not SNU719, while BART miRNA exon 5 (BART Ex-5) was decreased in VK-1727 treated SNU179 tumors, but to a lesser extent in YCCEL1 (Figures 5a and 5b). We also observed that EBV viral loads were reduced after 24 days of treatment (Figure 5C). Others have shown that SNU-719 cells do not express EBNA2 or LMP1 protein (20) and YCCEL1 cells have been characterized as expressing a modified latency program I (21). GC cell lines AGS and MKN74 were confirmed to be EBV negative by qPCR (Figure S3).
Figure 5. EBV gene expression and cell proliferation is decreased in EBV-positive GC treated in vivo.
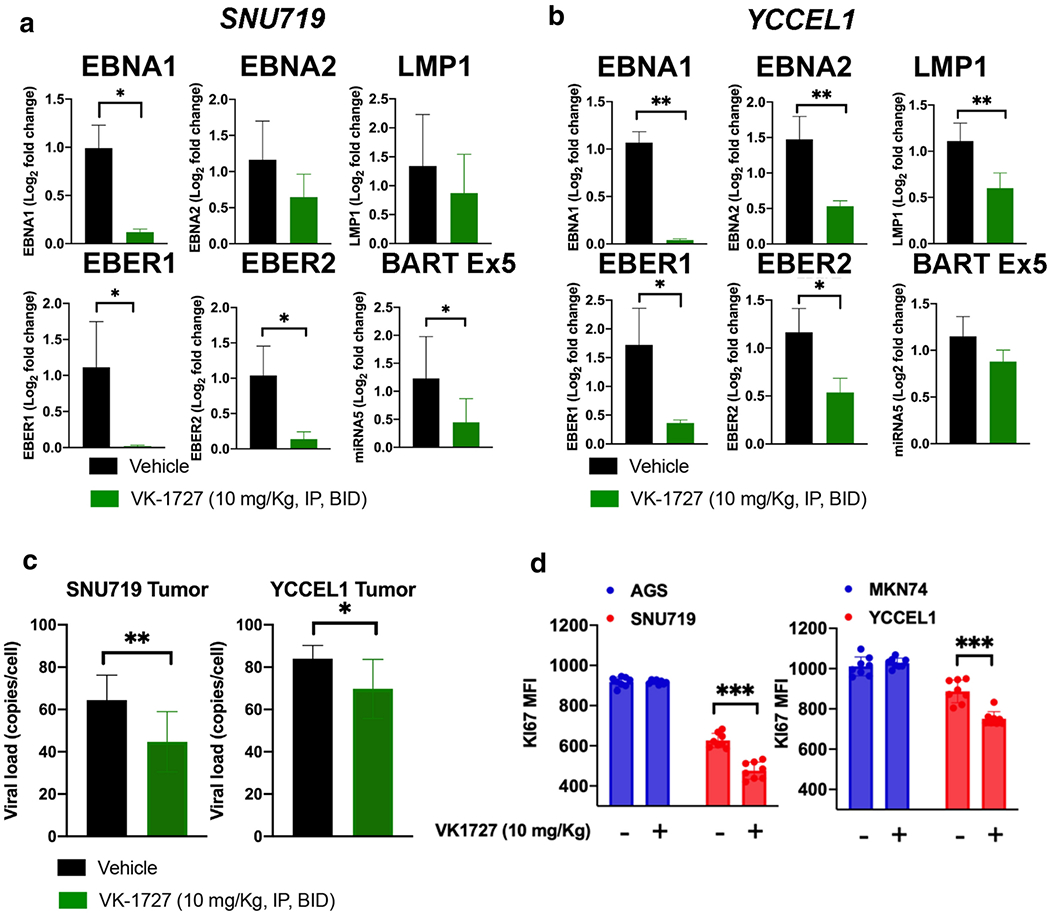
A. RT-PCR showing expression of EBNA1, EBNA2, LMP1, EBER1, EBER2, and BART Ex5 in SNU719 xenografts. B. RT-PCR showing expression of EBNA1, EBNA2, LMP1, EBER1, EBER2, and BART Ex5 in YCCEL1 xenografts. C. EBV viral loads in SNU719 and YCCEL1 tumor material from animals treated with VK-1727 or vehicle (Student T test; * p < 0.05, **p<0.01) D. Ki67 expression (mean fluorescence intensity as measured by flow cytometry).
Treatment with EBNA1 inhibitors significantly reduces proliferation in EBV-positive GC xenografts
To determine the effect of the EBNA1 inhibitor VK-1727 on proliferation in vivo, we measured Ki67 expression in tumors extracted from mice treated with VK-1727 (10 mg/kg) and vehicle by flow cytometry (Figure 5D). Ki67 was significantly reduced in EBV-positive GC tumors (SNU719 and YCCEL1), but not AGS or MKN74, consistent with the observation that VK-1727 decreases the proliferation of these cells.
Treatment with EBNA1 inhibitors demethylates DNA in EBVaGC cell lines.
To determine the effect of EBNA1 inhibition on the DNA hypermethylation profile of EBVaGC cell lines, we quantifiied by dot blot assay the global DNA methylation of EBVaGC cells treated with EBNA1 inhibitor or decitabine as a positive control (Figure 6). We observed a modest, but significant DNA decrease in global DNA methylation after seven days of treatment with the EBNA1 inhibitors in both SNU719 (Figure 6A and C) and YCCEL1 (Figure 6B and C).
Figure 6. DNA demethylation in YCCEL1 and SNU719 cells treated with EBNA1 inhibitors.
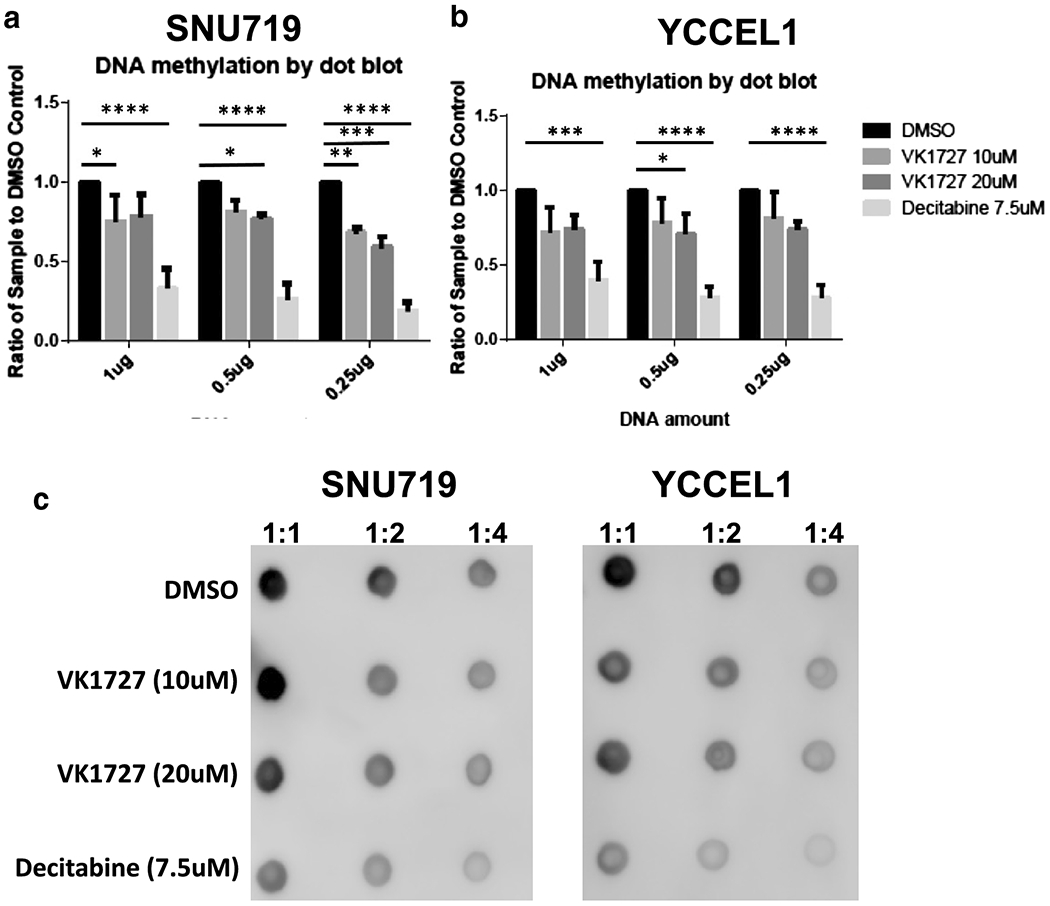
A. Quantification of DNA methylation by dot blot in SNU719 represented as ratio compared to DMSO control. B. quantification of DNA methylation by dot blot in YCCEL1 represented as ratio compared to DMSO control (Dunnett’s test). C. Representative dot blots for SNU719 and YCCEL1.
Discussion:
EBNA1 is critical for EBV episome maintenance and latent gene expression and provides a selection advantage to EBV positive tumors. We previously described the fragment-based development of EBNA1 inhibitors that block the DNA binding activity as demonstrated in Chromatin Immunoprecipitation (ChIP) assays and EBNA1-dependent replication assays (18, 19). We demonstrated that EBNA1 inhibitors have in vivo activity in B cell lymphoma and several NPC xenograft models. In this study, we sought to extend the applicability of EBNA1 inhibitors to EBV-associated GC xenograft models. We also wanted to demonstrate selectivity by comparing EBV-positive and EBV-negative GC cell lines in vitro and in vivo.
Here, we show that EBNA1 inhibitors significantly reduce the proliferation and tumor growth of EBV-positive cells in vitro and in vivo. Again, these results suggest that EBNA1 inhibitors are both EBV-specific and effective in vivo (Figure 4B). Similar to previous studies (18), no weight loss or other adverse effects were observed in treated animals, indicating the safety of these compounds in pre-clinical models (Figure 4D). EBNA1 inhibitors appear to be cytostatic in that tumors do not regress after initiation of treatment in immunocompromised mouse models and we have not observed clear evidence of apoptosis in treated cells (e.g. no increase in the <G1 population by cell cycle analysis).
We also demonstrate that EBNA1 inhibitors selective inhibit the growth of EBV-positive, but not EBV-negative cells. Previously, we demonstrated that EBNA1 does not affect the growth of the EBV-negative lung carcinoma cell line A549 (18). We now extend these studies to the EBV-negative GC lines AGS and MKN74. EBNA1 has structural similarities with other viral proteins (LANA from KSHV, E2 from HPV), but not other proteins in the human proteome (22). Therefore, we would not expect EBNA1 inhibitors to be associated with any on-target toxicities. One limitation of our study is that the EBV-positive/negative GC lines used here do not have the same genetic background. Therefore, selectivity may be idiosyncratic in this limited study with four GC cell lines.
Treatment with EBNA1 inhibitors significantly decreases the expression of viral genes and EBV viral load in vivo. This observation is consistent with previous observations in in vivo models of NPC and B-cell lymphomas. Expression levels of EBNA1, EBER1 and EBER2 are significantly reduced compared to vehicle controls in EBV-positive GC tumors from animals treated with VK-1727 at 10 mg/kg, twice per day. These results are expected given the critical role EBNA1 plays in the replication and maintenance of EBV. Of interest, decreased EBV expression is not observed during short-term (72 hour) treatment of EBV-positive GC cells in culture, which may either indicate that prolonged, consistent application of drug is required to decrease viral gene expression or may suggest differential responses of these tumors in vivo, within the tumor microenvironment. In addition, the short-term effects of viral mRNA expression were different in EBV-positive LCLs compared to EBV-positive GC or EBV-positive NPC cells, suggesting the initial effects of EBNA1 inhibition on viral and cellular gene expression may differ in EBV positive cells of different origin. Co-treatment of VK-1727 with GCV did not result in further decreases in cell proliferation, suggesting that there is VK-1727 does not induce full lytic activation in EBVaGC early in response to treatment with EBNA1 inhibitors (Figure 3B). In addition, EBNA1 inhibitors caused a slight reduction in levels of DNA methylation, suggesting that DNA demethylation may correlate with EBNA1 inhibition and tumor growth suppression in these EBVaGC models.
We previously performed gene expression analysis using the PanCancer Pathways Panel of 700 human genes supplemented with an analysis of ~30 EBV expressed genes in tumors from animals engrafted with an EBV-positive NPC-PDX (18). In those studies, we demonstrated that expression levels from EBV-encoded genes, including EBNA1, EBNA2, LMP1, and ZTA were significantly lower in tumors treated with an EBNA1 inhibitor. Cellular gene pathways for the cell cycle, PI3K, chromosomal modifications, TGF-β, STAT, RAS, NOTCH and MAPK were also significantly affected by EBNA1 inhibitor treatment. The PI3K/AKT pathway is of particular interest because it is induced by EBV-latent membrane proteins 1 and 2A (LMP1 and LMP2A) and is thought to play an important role in EBV-induced malignancies by affecting cell survival, apoptosis, proliferation, and genomic instability (23). It has also been suggested that the activation of the PI3K/Akt pathway can result in drug resistance to chemotherapy (23). In addition, recent studies have indicated that the expression of the AT-rich interaction domain 1A (ARID1A) and phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit α (PIK3CA) are associated with the depth of invasion of gastric cancer (24). Therefore, effective treatment of EBVaGCs is likely to be enhanced by combining EBNA1 inhibitors with inhibitors of other perturbed pathways (e.g. Idelalisib, a PI3K inhibitor that we have shown has activity against EBV-positive NPC tumors in vivo (18)) or ubiquitin conjugation inhibitors. In the future, we anticipate that combining EBNA1 inhibitors with inhibitors of other perturbed pathways may reveal functional crosstalk useful for developing a rationale for combinatorial therapies.
Collectively, these results suggest that pharmacological inhibition of EBNA1 has profound effects on EBV and cellular gene expression, correlating with suppression in EBV-driven tumor cell growth. EBNA1 is essential in promoting the continued proliferative capacity of latently infected cells. Our study demonstrates that EBNA1 inhibitors selectively suppress proliferation of EBV-driven tumor cells. This study may provide a rationale for the use EBNA1 inhibitors as a therapeutic option for patients with EBV-associated GC.
Supplementary Material
S1. Selective inhibition of EBV-positive cell lines by EBNA1 inhibitor VK-1727. EBNA1 Resazurin-based cell metabolism assay using EBV-positive lymphoid cells LCL (EBV+) vs BJAB (EBV−) (top panels); NPC cells C666-1 (EBV+) vs HK-1 (EBV−) (middle panels); and gastric carcinoma cells SNU719 (EBV+) vs AGS (EBV−) (bottom panels) in a resazurin based cell viability assay, 96 hours after treatment.
S2. EBNA1 inhibitor VK-1727 inhibits cell growth and cell adhesion. Images of EBV-negative(HK1, AGS, BJAB) and EBV-positive (YCCEL1, LCL352, C666-1) cells using a Nikon TIE inverted microscope with 4x objective at 72 hours after treatment with DMSO or VK-1727 (10 μM or 25 μM). Cell density and adhesion are decreased in VK-1727 treated EBV-positive cells compared to EBV-negative cells regardless of cell lineage or EBV latency type.
S3. EBV gene expression in GC cell lines. RT-PCR showing expression of EBNA1, EBNA2, BARF0, EBER1, EBER2, LMP1, ZTA, BART Ex 1/2 and BART Ex 5 in SNU719 and YCCEL1 cells. AGS and MKN74 cells are EBV-negative and, as expected, do not express EBV genes.
Acknowledgements:
We thank Frederick Kenney from The Wistar Imaging Facility for assistance with microscopy and the IVIS. We thank Jeffrey Faust and John Fundyga for sorting GFP-positive cells. The following research grants from NIH also supported this work S10OD021669-01A1 (Instrumentation grant for the IVIS Spectrum CT), NIH R01-CA093606 and R01-DE017336 to PML, and an NCI Cancer Center Support Grant Cancer Center Support Grant P30-CA010815 (Altieri). B.E.G. is supported by a Burroughs Wellcome Fund Career Award in Medical Sciences and NIH R01-AI137337 and R01-CA228700.
Competing Interests
This work was partially funded through a sponsored research agreement with Cullinan Apollo. The Wistar Institute on behalf of the authors T.E.M. and P.M.L. have filed patents covering composition of matter and their use on the small molecules disclosed here for the treatment of human cancer and other diseases (patent number WO2015073864, “EBNA1 Inhibitors and Their Method of Use”; WO2016183534, “EBNA1 Inhibitors and Methods using Same”). P.M.L. has an ownership interest in Vironika, LLC.
References:
- 1.Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–86. Epub 2014/09/16. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 2.Shibata D, Weiss LM. Epstein-Barr virus-associated gastric adenocarcinoma. Am J Pathol. 1992;140(4):769–74. Epub 1992/04/01. [PMC free article] [PubMed] [Google Scholar]
- 3.Camargo MC, Kim WH, Chiaravalli AM, Kim KM, Corvalan AH, Matsuo K, et al. Improved survival of gastric cancer with tumour Epstein-Barr virus positivity: an international pooled analysis. Gut. 2014;63(2):236–43. Epub 2013/04/13. doi: 10.1136/gutjnl-2013-304531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Young LS, Yap LF, Murray PG. Epstein-Barr virus: more than 50 years old and still providing surprises. Nat Rev Cancer. 2016;16(12):789–802. Epub 2016/11/04. doi: 10.1038/nrc.2016.92. [DOI] [PubMed] [Google Scholar]
- 5.Imai S, Koizumi S, Sugiura M, Tokunaga M, Uemura Y, Yamamoto N, et al. Gastric carcinoma: monoclonal epithelial malignant cells expressing Epstein-Barr virus latent infection protein. Proceedings of the National Academy of Sciences. 1994;91(19):9131. doi: 10.1073/pnas.91.19.9131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ott G, Kirchner T, Muller-Hermelink HK. Monoclonal Epstein-Barr virus genomes but lack of EBV-related protein expression in different types of gastric carcinoma. Histopathology. 1994;25(4):323–9. Epub 1994/10/01. doi: 10.1111/j.1365-2559.1994.tb01350.x. [DOI] [PubMed] [Google Scholar]
- 7.Fukayama M, Ushiku T. Epstein-Barr virus-associated gastric carcinoma. Pathol Res Pract. 2011. ;207(9):529–37. Epub 2011/09/29. doi: 10.1016/j.prp.2011.07.004. [DOI] [PubMed] [Google Scholar]
- 8.Oda K, Tamaru J, Takenouchi T, Mikata A, Nunomura M, Saitoh N, et al. Association of Epstein-Barr virus with gastric carcinoma with lymphoid stroma. Am J Pathol. 1993;143(4):1063–71. Epub 1993/10/01. [PMC free article] [PubMed] [Google Scholar]
- 9.Farrell PJ. Epstein-Barr Virus and Cancer. Annu Rev Pathol. 2019;14:29–53. Epub 2018/08/21. doi: 10.1146/annurev-pathmechdis-012418-013023. [DOI] [PubMed] [Google Scholar]
- 10.Frappier L EBNA1. Curr Top Microbiol Immunol. 2015;391:3–34. Epub 2015/10/03. doi: 10.1007/978-3-319-22834-1_1. [DOI] [PubMed] [Google Scholar]
- 11.Yin Q, Flemington EK. siRNAs against the Epstein Barr virus latency replication factor, EBNA1, inhibit its function and growth of EBV-dependent tumor cells. Virology. 2006;346(2):385–93. Epub 2005/12/14. doi: 10.1016/j.virol.2005.11.021. [DOI] [PubMed] [Google Scholar]
- 12.Wang J, Liang C, Meng F, Xu X, Wu Y, Lu L. Lentivirus-mediated RNA interference targeting EBNA1 gene inhibits the growth of GT-38 cells in vitro and in vivo. Oncol Lett. 2019; 18(3):2286–91. Epub 2019/06/28. doi: 10.3892/ol.2019.10543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dheekollu J, Malecka K, Wiedmer A, Delecluse HJ, Chiang AK, Altieri DC, et al. Carcinoma-risk variant of EBNA1 deregulates Epstein-Barr Virus episomal latency. Oncotarget. 2017;8(5):7248–64. Epub 2017/01/13. doi: 10.18632/oncotarget.14540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frappier L Contributions of Epstein-Barr nuclear antigen 1 (EBNA1) to cell immortalization and survival. Viruses. 2012;4(9):1537–47. Epub 2012/11/22. doi: 10.3390/v4091537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu J, Murakami M, Verma SC, Cai Q, Haldar S, Kaul R, et al. Epstein-Barr Virus nuclear antigen 1 (EBNA1) confers resistance to apoptosis in EBV-positive B-lymphoma cells through up-regulation of survivin. Virology. 2011;410(1):64–75. Epub 2010/11/26. doi: 10.1016/j.virol.2010.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sivachandran N, Dawson CW, Young LS, Liu FF, Middeldorp J, Frappier L. Contributions of the Epstein-Barr virus EBNA1 protein to gastric carcinoma. J Virol. 2012;86(1):60–8. Epub 2011/10/21. doi: 10.1128/jvi.05623-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu F, Tempera I, Lee HT, Dewispelaere K, Lieberman PM. EBNA1 binding and epigenetic regulation of gastrokine tumor suppressor genes in gastric carcinoma cells. Virol J. 2014; 11:12. Epub 2014/01/28. doi: 10.1186/1743-422x-11-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Messick TE, Smith GR, Soldan SS, McDonnell ME, Deakyne JS, Malecka KA, et al. Structure-based design of small-molecule inhibitors of EBNA1 DNA binding blocks Epstein-Barr virus latent infection and tumor growth. Sci Transl Med. 2019;11(482). Epub 2019/03/08. doi: 10.1126/scitranslmed.aau5612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Messick TE, Tolvinski L, Zartler ER, Moberg A, Frostell Å, Smith GR, et al. Biophysical Screens Identify Fragments That Bind to the Viral DNA-Binding Proteins EBNA1 and LANA. Molecules. 2020;25(7). Epub 2020/04/16. doi: 10.3390/molecules25071760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oh ST, Seo JS, Moon UY, Kang KH, Shin DJ, Yoon SK, et al. A naturally derived gastric cancer cell line shows latency I Epstein-Barr virus infection closely resembling EBV-associated gastric cancer. Virology. 2004;320(2):330–6. Epub 2004/03/16. doi: 10.1016/j.virol.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 21.Kim DN, Seo MK, Choi H, Kim SY, Shin HJ, Yoon AR, et al. Characterization of naturally Epstein-Barr virus-infected gastric carcinoma cell line YCCEL1. J Gen Virol. 2013;94(Pt 3):497–506. Epub 2012/11/24. doi: 10.1099/vir.0.045237-0. [DOI] [PubMed] [Google Scholar]
- 22.De Leo A, Calderon A, Lieberman PM. Control of Viral Latency by Episome Maintenance Proteins. Trends Microbiol. 2020;28(2):150–62. Epub 2019/10/19. doi: 10.1016/j.tim.2019.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen J Roles of the PI3K/Akt pathway in Epstein-Barr virus-induced cancers and therapeutic implications. World J Virol. 2012;1(6):154–61. Epub 2013/11/01. doi: 10.5501/wjv.v1.i6.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou H, Tan S, Li H, Lin X. Expression and significance of EBV, ARID1A and PIK3CA in gastric carcinoma. Mol Med Rep. 2019;19(3):2125–36. Epub 2019/02/13. doi: 10.3892/mmr.2019.9886. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
S1. Selective inhibition of EBV-positive cell lines by EBNA1 inhibitor VK-1727. EBNA1 Resazurin-based cell metabolism assay using EBV-positive lymphoid cells LCL (EBV+) vs BJAB (EBV−) (top panels); NPC cells C666-1 (EBV+) vs HK-1 (EBV−) (middle panels); and gastric carcinoma cells SNU719 (EBV+) vs AGS (EBV−) (bottom panels) in a resazurin based cell viability assay, 96 hours after treatment.
S2. EBNA1 inhibitor VK-1727 inhibits cell growth and cell adhesion. Images of EBV-negative(HK1, AGS, BJAB) and EBV-positive (YCCEL1, LCL352, C666-1) cells using a Nikon TIE inverted microscope with 4x objective at 72 hours after treatment with DMSO or VK-1727 (10 μM or 25 μM). Cell density and adhesion are decreased in VK-1727 treated EBV-positive cells compared to EBV-negative cells regardless of cell lineage or EBV latency type.
S3. EBV gene expression in GC cell lines. RT-PCR showing expression of EBNA1, EBNA2, BARF0, EBER1, EBER2, LMP1, ZTA, BART Ex 1/2 and BART Ex 5 in SNU719 and YCCEL1 cells. AGS and MKN74 cells are EBV-negative and, as expected, do not express EBV genes.