

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is almost universally lethal. A critical unmet need exists to explore essential susceptibilities in PDAC and identify druggable targets to improve PDAC treatment. KRAS mutations dominate the genetic landscape of PDAC and lead to activation of multiple downstream pathways and cellular processes. Here, we investigated the requirement of these pathways for tumor maintenance using an inducible KrasG12D-driven PDAC mouse model (iKras model), identifying that RAF-MEK-MAPK signaling is the major effector for oncogenic KRAS-mediated tumor maintenance. However, consistent with previous studies, MEK inhibition had minimal therapeutic effect as a single agent for PDAC in vitro and in vivo. Although MEK inhibition partially downregulated transcription of glycolysis genes, it failed to suppress glycolytic flux in PDAC cells, which is a major metabolic effector of oncogenic KRAS. Accordingly, an in vivo genetic screen identified multiple glycolysis genes as potential targets that may sensitize tumor cells to MEK inhibition. Inhibition of glucose metabolism with low dose 2-deoxyglucose in combination with a MEK inhibitor induced apoptosis in KrasG12D-driven PDAC cells in vitro. The combination also inhibited xenograft PDAC tumor growth and prolonged overall survival in a genetically engineered PDAC mouse model. Molecular and metabolic analyses indicated that co-targeting glycolysis and MAPK signaling results in apoptosis via induction of lethal ER stress. Together, our work suggests that combined inhibition of glycolysis and the MAPK pathway may serve as an effective approach to target KRAS-driven PDAC.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is highly lethal with an overall 5-year survival of <10% and projected to be the second leading cause of cancer-related death by 2030 in the United States (1,2). PDAC is almost universally driven by mutant KRAS, which represents the earliest and the most frequent (>90%) genetic alteration. However, no effective inhibitors have been developed for mutant KRAS except for small molecule inhibitors targeting KRASG12C, which is present in just 1.5% human PDAC (3–5). To date, no targeted therapy has shown any major effect in PDAC patients yet.
KRAS mutations activate several downstream signaling pathways, including, but not limited to, the RAF/MEK/MAPK, phosphatidylinositol 3-kinase (PI3K)/AKT, and RalGDS pathways (6). Consequently, multiple cellular processes are activated such as proliferation, survival and KRAS-dependent metabolism pathways (7,8). However, it is likely that only a subset of KRAS surrogates play dominant roles during tumor maintenance. While several studies have established the roles of KRAS surrogates during PDAC initiation and development (5,9,10), the requirement of these pathways for tumor maintenance has not been thoroughly investigated. Recently, study using genetically engineered mouse models (GEMM) indicates that C-RAF is required for the maintenance of KRAS-driven lung adenocarcinomas (11).
Therapeutically, single agent targeting the MAPK or PI3K pathway has shown limited effects in PDAC (12,13). Although co-targeting MEK and PI3K pathways showed beneficial effect in preclinical models, the combination is too toxic to be used in clinic, indicating the need to identify alternative combination strategies. By using a PDAC GEMM driven by inducible KrasG12D (iKras model), we have established the critical roles of metabolism reprogramming in advanced tumors (14). Interestingly, recent studies have demonstrated the synergy between targeting autophagy (15,16), nucleoside metabolism (17) or mTORC1/2 (18) with MAPK inhibition in PDAC treatment, underscoring the therapeutic potential of co-targeting KRAS signaling and metabolism programs.
In this study, we conducted genetic studies to establish the dominant role of RAF-MAPK signaling for KRAS-dependent PDAC maintenance. However, MAPK inhibition alone fails to suppress the glycolysis flux induced by oncogenic KRAS. Accordingly, genetic or pharmacological inhibition of glycolysis synergizes with MAPK inhibition to suppress PDAC growth both in vivo and in vitro, largely due to the induction of lethal ER stress. Our study identified the co-inhibition of MAPK signaling and glycolysis flux as an alternative approach to target KRAS-driven PDAC.
Material and methods
Animals
All animal manipulations were approved by the Animal Care and Use Committees at The University of Texas, MD Anderson Cancer Center under protocol number 00001549. No patient samples were directly used in this study. TetO_Lox-Stop-Lox-KrasG12D (tetO_LKrasG12D), ROSA26-LSL-rtTA-IRES-GFP (ROSA_rtTA), p48-Cre and Trp53L strains were described previously (19). Mice were fed with doxy water (Dox 2g/L in sucrose 20g/L) starting at the 3-week-old to induce PDAC development. All the 11-week-old mice were randomized into four groups including control, 2DG treatment, Trametinib treatment and combination treatment. Trametinib was delivered by oral gavage (1mg/kg/day) and 2DG was by intraperitoneal injection (1000mg/kg/d). Drugs were given daily until the mice became visibly sick due to tumor burden which was confirmed by necropsy. The survival of the mice refers to weeks of treatment.
Cell culture and reagents
IMR90 and human PDAC cell lines HPAC, 8988T, PaTu8902, Miapaca2, DanG, S2013, and PANC1 were purchased from American Type Culture Collection (ATCC). PDX148 was established from PDX tumors (20). IMR90 was grown in Eagle’s Minimum Essential Medium with 10% FBS. The PDAC cell lines and PDX148 cells were cultured in RPMI1640 supplemented with 10% FBS. All the cell lines were tested negative for mycoplasma (LookOut Mycoplasma PCR Detection Kit) throughout the study. Cells were kept in culture for a maximum of 15 passages after recovery from frozen vials. Trametinib, SCH772984, BKM120, GDC-0623, gemcitabine, and paclitaxel were purchased from Selleckchem. 4-Phenylbutyric Acid (4-PBA) and 2DG were obtained from Sigma Aldrich. Heptelidic Acid was purchased from Cayman Chemical and R-GNE-140 was purchased from Medkoo Bioscience. POMHEX was provided by Dr. Florian L. Muller at MD Anderson Cancer Center.
Plasmids and reagents
The lentiviral shRNA clones were obtained from Sigma Aldrich (Supplementary Table S1). All ORFs were cloned into pHAGE-IRES-GFP using pENTR™/D-TOPO™ Cloning Kit (Thermofisher Scientific). The virus package plasmids psPAX2 (Addgene plasmid 12260) and pMD2.G (Addgene plasmid 12259) were purchased from Addgene.
Glucose consumption and lactate production by YSI
Cells were seeded in 12-well plate at ~30% confluence (blank wells without cells were used for a baseline reading of Glucose/lactate). Forty-eight hours later, the culture medium was collected and spinned at 1000g for 5min at 4°C. 250 μl supernatant was transferred into 96-well plate and read on YSI 2900 Biochemistry Analyzer (Agilent). The glucose consumption (Glcblank-Glctreated) and lactate production (Lactreated-Lacblank) was calculated and normalized by the cell number in each well.
Analysis of oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) by Seahorse XF Analyzers
The determination of OCR or ECAR values was performed on XF96 analyzers. 1×104 iKras cells were seeded each well in Seahorse XF96 Cell Culture Microplate. The cells were treated as Dox ON, Dox OFF or with Dox ON/Trametinib on the second day. After 24 h treatment, the cells were washed and the OCR or ECAR values were detected with Seahorse XF Cell Mito Stress Test following the manufacturer’s instruction. After seahorse analysis, the cells in 96-well plate were fixed with 4% paraformaldehyde for 15min, stained with DAPI and counted on Operetta High-Content Imaging System. The OCR and ECAR were normalized to the cell number in each well.
Quantitative RT-PCR
Total RNA was extracted by the Qiagen RNeasy kit following the manufacturer’s instruction. cDNA was generated by using the SuperScript IV First-Strand Synthesis System (Invitrogen). The qRT-PCR was performed with Fast SYBR Green Master Mix (Applied Biosystems) in StepOnePlus (Applied Biosystems). Relative expression of genes was calculated by 2−ΔΔCT method and normalized to beta-actin expression. All the primers are listed in Supplementary Table S2.
In vivo shRNA screen
The construction of customized shRNA library and method for shRNA screen in vivo were previously described (21,22). To perform the shRNA screen, the lentivirus package was prepared in 293FT cells using the second-generation packaging plasmids psPAX2 and pMD2.G. The lentivirus was concentrated using ultracentrifuge at 23,000 rpm for 3 h and the transducing unit was determined. The lentivirus infection was performed at ~0.3 transducing unit/cell with 10 µg/ml polybrene. After puromycin selection (2 µg/ml) for 48 h, cells were trypsinized, pooled together and 106 cells were washed with PBS and stored in −80°C as reference. The remaining cells were mixed with matrigel (1:1) and orthotopically injected at 106 cells/mouse pancreas. The screen was conducted in triplicates and an in vivo coverage of 1000 cells/barcode was guaranteed. The tumors were collected 10 days post injection and stored at −80°C.
Extraction of tumor DNA and NGS library preparation
The frozen tumors were minced to small pieces and dissociated with disposable gentleMACS M tubes (Miltenyi Biotech) with the gentleMACS dissociator (Miltenyi Biotec) in buffer P1 (QIAGEN, 1 mL Buffer/100 mg tumor) with 100 µg/mL RNase A. The cell pellet was lysed by adding 1/20 volume of 10% SDS (Promega). After incubating for 20 min at room temperature, the lysates were passed 10–15 times through a 22-gauge syringe needle to shear the genomic DNA. Then the genomic DNA were extracted using the Phenol-Chloroform solution. The DNA pellet was finally dissolved in UltraPure distilled water and DNA concentration was assessed by NanoDrop 2000 (Thermo Scientific). To prepare the NGS libraries, the barcodes were amplified from the equal amount of genomic DNA by 2 rounds of nested PCR. The primers are list in Supplementary Table S3. The required adapters for NGS were introduced in the second PCR reactions. Amplified PCR products were purified using QIAquick gel extraction kit (Qiagen) and quantified using High Sensitivity DNA Assay (Agilent Technologies) for the Agilent 2100 Bioanalyzer.
Screen data analysis
The shRNA screen data was analyzed as described previously (21,22). Illumina base calls were processed using CASAVA (v.1.8.2), and resulting reads were processed using our in-house pipeline. Following filtration and library-size normalization, reads counts in Vehicle or Trametinib samples were compared to the reference and a Log2 fold change was calculated.
TEM
Cells were cultured in 12-well plate and washed with PBS twice before fixed with a solution containing 3% glutaraldehyde and 2% paraformaldehyde in 0.1 M cacodylate buffer (pH 7.3). Samples were then washed in 0.1 M sodium cacodylate buffer, treated with 0.1% Millipore-filtered cacodylate buffered tannic acid, post-fixed with 1% buffered osmium and stained with 0.1% Millipore-filtered uranyl acetate. The samples were dehydrated in increasing concentrations of ethanol and then infiltrated and embedded in LX-112 medium. The samples were then polymerized in a 60°C oven for three days. Ultrathin sections were cut using a Leica Ultracut microtome (Leica, Deerfield, IL) and then stained with uranyl acetate and lead citrate in a Leica EM Stainer. The stained samples were examined in a JEM 1010 transmission electron microscope (JEOL USA, Inc., Peabody, MA) using an accelerating voltage of 80 kV. Digital images were obtained using an AMT imaging system (Advanced Microscopy Techniques Corp., Danvers, MA).
Cell Viability Assay
For the sphere formation, two thousand cells were seeded in the round bottom ultralow attached 96-well plates and cultured in medium with or without doxycycline for 1 week. The cell viability for each well was quantified using CellTiter-Glo® 2.0 Cell Viability Assay following the manufacturer’s instructions.
For the drug treatment, cells were plated in equal number in 24 well plates and treated with 2DG, Trametinib or combination. After 4 days’ treatment, cells were rinsed twice with PBS and stained by 0.25% Crystal violet in 20% methanol for 20 min and washed with water. Stained cells were scanned and destained with 10% Acetic acid. The absorbance was measured at 595 nm to quantify the relative cell numbers. The Bliss Score for the combination was calculated by online tools (https://synergyfinder.fimm.fi/).
Annexin V-PE and 7-AAD apoptosis assay
Induction of cell apoptosis was detected by PE Annexin V Apoptosis Detection Kit I (BD Pharmingen™) following the manufacturer’s instruction. Briefly, cells were seeded into 12-well plate at a density of 20000 cells/well and treated with the indicated concentration of 2DG, Trametinib or combination for 48 h. Then all the cells in each well were collected in 15ml tube and were pelleted by 200 g for 4 min. The cells were washed with PBS once and resuspended in the 100 µl 1×staining buffer in which 5 µl antibody and 5 µl 7-AAD were added. After staining at room temperature for 15 min, the samples were analyzed on Gallios Flow Cytometer (Beckman Counter). The FACS data were analyzed with FlowJo.
Xenograft
For orthotopic xenografts, 5×105 cells suspended in 10 µl 50% Corning Matrigel Matrix (Corning)/Opti-MEM media were injected into the pancreas of NCr nude mice.
For Sub-Q xenografts, 1×106 cells suspended in 100 µl Opti-MEM media were injected subcutaneously into the lower flank of NCr nude mice. Animals were fed with doxy water and treated with Trametinib (1mg/kg/d), 2DG (1000mg/kg/d) or combination. Tumor volumes and body weight were measured every four days and calculated using the formula (Volume =0.5×length×width2).
RNA sequencing
For the tumor samples, the mice were randomized into three groups (Kras-ON/Vehicle, Kras-OFF/Vehicle, Kras ON/Trametinib) on the 7th day after orthotopic injection. After 1 or 3 days of treatment, the tumors were collected for RNA extraction. For the cell samples, 5×105 iKras cell were seeded in the 10cm dish and started the treatment with vehicle, 1 mM 2DG, 25 nM Trametinib, or combination for 24 h. The RNA samples were collected in the TRIzol™ Reagent (1ml/10cm2).
Total RNA was extracted using the Qiagen RNeasy kit following the manufacturer’s instruction. The RNA samples with a RIN score >8 were used in further analysis. RNA library preparation, sequencing, raw data processing, and quality control were performed by Advanced Technology Genomics Core at MD Anderson Cancer Center. Reads were mapped using Tophat and FPKM values were generated with Cufflinks. The software package LIMMA (Linear Models for Microarray Data) was applied to detect significantly differentially expressed genes using Benjamini-Hochberg adjusted p-values. The raw or processed transcript sequencing data were submitted and are available through Gene Expression Omnibus database under the accession numbers GSE176241 and GSE176242.
Immunohistochemistry and Western Blot Analysis
For the IHC quantitation, xenograft tumors were collected from the mice treated with the drug for 12 days and fixed in 10% formalin overnight and embedded in paraffin. Immunohistochemical analysis was performed as described (23). To quantify of Ki67 and Caspase3 positive cells, a total of six views were taken from two separated tumors. In each view, the positive cells were counted and the percentage was calculated by the number of total cells. Antibodies used for immunohistochemistry and WB were listed in Supplementary Table S4 and Table S5.
Statistical analysis
To assess variance differences across various test groups, the data were analyzed using two-way ANOVA (multiple comparisons) in GraphPad Prism. Two-group comparisons were performed by using the unpaired 2-tailed t-test. For all figures with error bars, data are presented as mean ± SD unless otherwise stated. The mice survival was analyzed using Kaplan-Meier analysis. Tumor volume was analyzed using online Tumor Growth analysis software (https://kroemerlab.shinyapps.io/TumGrowth/). The level of significance was set at for p-value<0.01(**) or p-value <0.05 (*) in all figures.
Results
Active MAPK pathway is essential for PDAC maintenance
Our previous study has established the essential role of oncogenic KRAS for PDAC maintenance (14). To further dissect the respective contributions of KRAS downstream pathways, three effector domain missense mutants of KRAS, which preferentially activate RAF/MEK/ERK, PI3K or RalGDS pathway respectively (24,25), were ectopically expressed in the primary tumor cells derived from the iKras model (p48Cre; tetO_KrasG12D; Rosa_rtTAL/+; p53L/+) (Fig. 1A). Using GFP as a negative control, complete tumor regression of orthotopic xenografts upon KrasG12D inactivation following doxycycline (Dox) withdrawal (Fig. 1B). In contrast, the KRASG12V/T35S, which selectively activates the RAF/MAPK pathway (Fig. S1A), functions as potent as the endogenous KrasG12D, to maintain xenograft tumor growth following doxycycline withdrawal (Fig. 1B). Importantly, KRASG12V/Y40C (activates PI3K) or KRASG12V/E37G (activates RalGDS) (Fig. S1A) was less efficient to maintain the tumor growth upon extinction of the endogenous KrasG12D. In consistent with the in vivo observation, the induction of sphere formation by KRASG12V/T35S in the absence of endogenous KrasG12D is comparable with KRASG12V. However, the number of spheres was reduced by about 90% if KRASG12V/Y40C or KRASG12V/E37G was expressed in iKras cells in the absence of doxycycline (Fig. 1C–D). Therefore, our data implicates the requirement of RAF/MAPK signaling for KRAS-driven PDAC maintenance.
Figure 1. Active MAPK pathway is essential for PDAC maintenance.
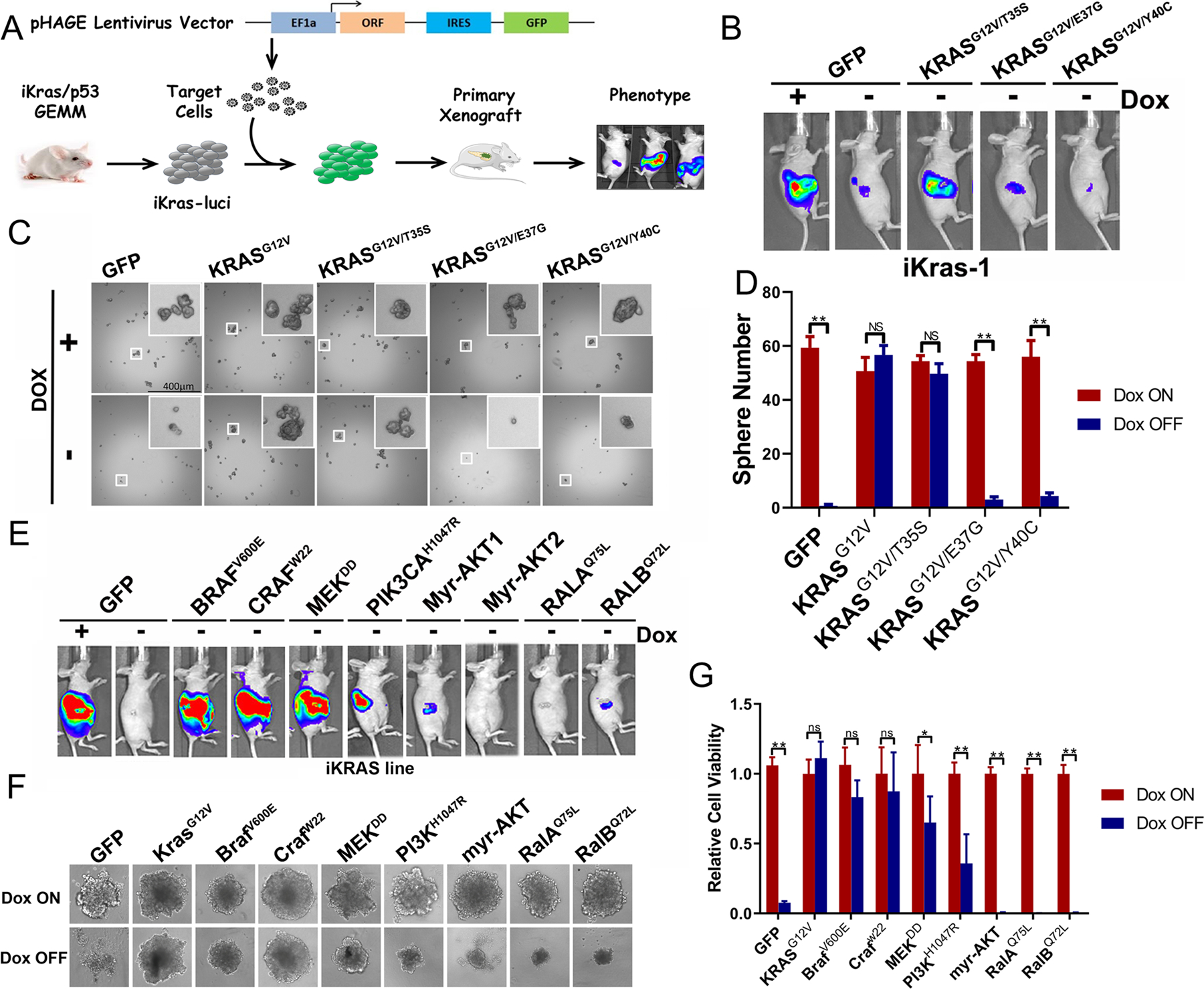
(A) Schematic diagram of investigating KRAS downstream surrogates in PDAC maintenance. The iKras cell was infected with lentivirus to overexpress the KRAS surrogates, sorted and orthotopically injected into nude mouse. Tumorigenesis was observed by bioluminescence imaging. (B) Tumorigenesis of iKras cells with KRASG12V/T35S, KRASG12V/E37G or KRASG12V/Y40C overexpression by bioluminescence imaging. (C) Sphere formation of iKras cell with KRASG12V/T35S, KRASG12V/E37G, KRASG12V/Y40C overexpression in the low-attached plate. (D) Quantification of sphere number in C (n=3, Mean ± SD). (E) Tumorigenesis of mouse PDAC cells with constitutively active KRAS downstream surrogates by bioluminescence imaging. (F) Sphere formation of iKras PDAC cells with constitutively active KRAS downstream surrogates in low attached plate. (G) Quantification of spheres in F by cell viability assay (n=3, Mean ± SD).
To further dissect the KRAS effector pathways in advanced tumors, we conducted gain of function experiments using several constitutively activated mutants for the key effectors in MAPK, PI3K or RalGDS pathway (Fig. S1B–C). Our data indicated that the constitutively activated C-RafW22, BRAFV600E, and MEKDD (26) were as efficient as mutant Kras to sustain tumor sphere formation in vitro and maintain tumor growth in vivo in the absence of doxycycline (Fig. 1E–G). In contrast, constitutive active PI3KH1047R was less competent to support tumor sphere growth and induced much delayed tumor formation (Fig. 1F–G and S1D). Additionally, in the absence of doxycycline, iKras cells expressing Myr-AKT or constitutive active RalGDS (RalAQ75L or RalBQ72L) failed to exhibit tumorigenic activity in vitro or in vivo (Fig. 1F–G and S1D). Together, our data indicates that RAF-MAPK signaling is the major surrogate for PDAC maintenance.
Inhibition of MAPK pathway fails to recapitulate glycolysis inhibition upon KRAS inactivation.
Despite the essential role of MAPK pathway in PDAC maintenance, blocking MAPK signaling with MEK inhibitors has been shown to exert marginal impact on tumor growth or overall survival in both preclinical models and PDAC patients (12,13,27–29). In an effort to further explore potential MAPK co-inhibition targets, we leveraged the unique ability of the iKras model to genetically extinct oncogenic Kras in advanced tumors and conducted global transcriptomic analysis to identify KRAS downstream effector pathways/activities that are not affected by MAPK inhibition. Specifically, RNAseq analysis was performed in orthotopic xenograft tumors established with primary tumor lines from the iKras/p53 model to compare the transcriptomic profiles following KRAS extinction or MEK inhibitor (MEKi) treatment for 1 or 3 days (Fig. 2A–B).
Figure 2. MEK inhibition fails to recapitulate glycolysis inhibition by Kras inactivation.
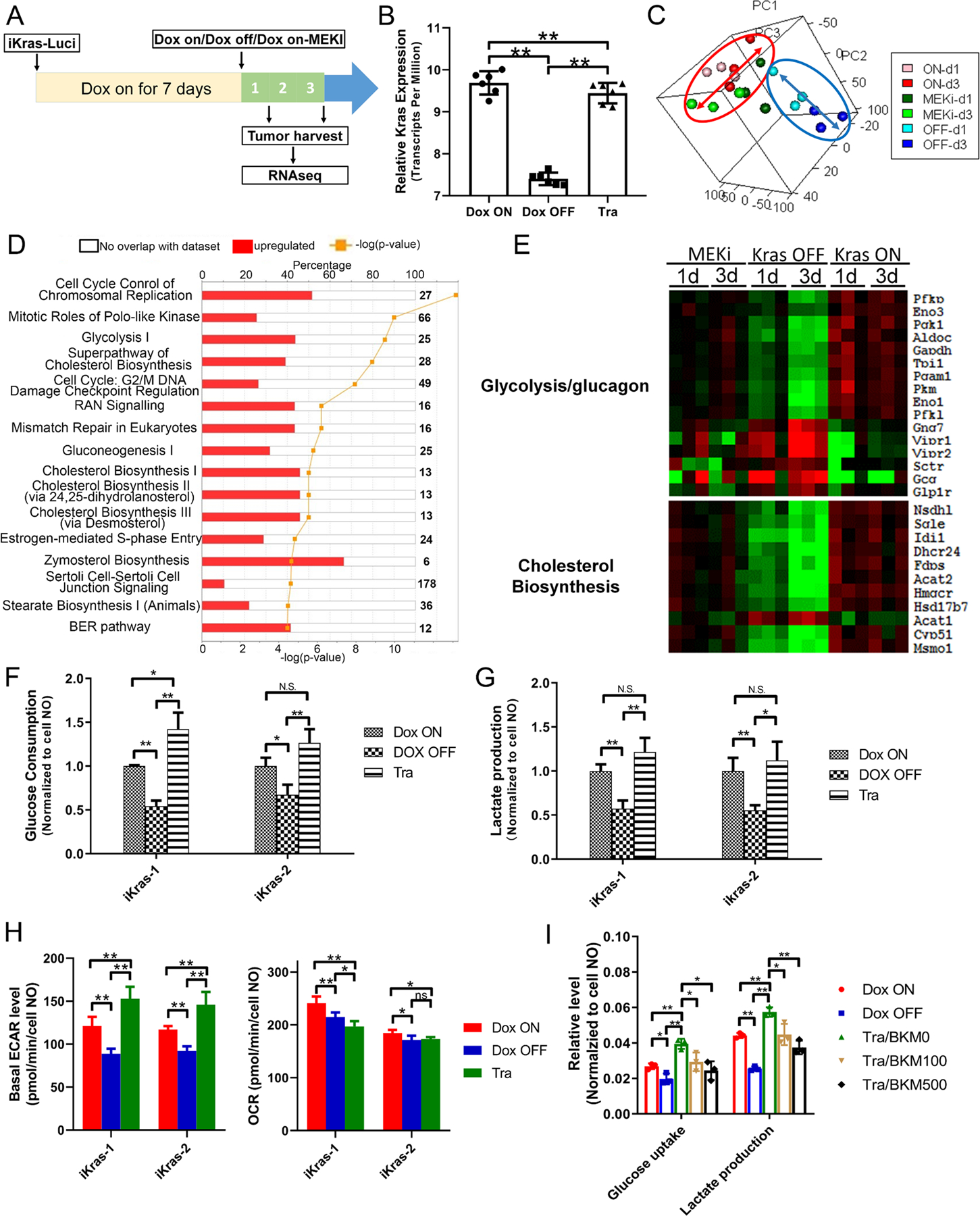
(A) Schematic illustration of transcriptomic analysis between xenograft upon Dox ON, Dox OFF or Tra treatment in vivo. Seven days post orthotopic injection of iKras cells, the mice were randomized into three group (Dox ON, Dox OFF or treated with 1 mg/kg/day Tra) and xenograft tumors were collected after 1- or 3-days treatment. (B) Kras expression in the xenograft upon Dox ON, Dox OFF or Tra treatment by RNA-seq (n=5, Mean ± SD). (C) PCA analysis of transcriptome changes between Dox ON, Dox OFF or Tra treatment. (D) IPA pathway analysis of differentially expressed genes between Dox OFF and Tra treatment. (E) Heatmap of the representative differentially expressed genes in respective pathway. (F-G) Glucose consumption and lactate production of iKras cells with Dox ON, Dox OFF or Tra treatment by YSI (n=3, Mean ± SD). Expression validation of genes involved in glycolysis by qPCR (n=3, Mean ± SD). (H) Basal ECAR or OCR value of iKras cells with Dox ON, Dox OFF or Tra treatment by Seahorse (n=4, Mean ± SD). (I) Glucose and lactate concentration in the medium of iKras cell was measured upon treatment (Tra: 25nM; BKM120: 100 nM or 500 nM) for 48h. The glucose consumption and lactate production were normalized based on cell number (n=3, Mean ± SD).
Compared to ON Dox tumors, a total of 206 genes were commonly suppressed in both Dox OFF and Trametinib treated group (Supplementary Table S6) which are enriched in secreted proteins and growth factors (Fig. S2A). Notably, a progressing transcriptomic change following KRAS extinction was observed in Dox OFF 1-day, and 3-day tumors compared to ON Dox tumors. However, such gradual expression changes were not observed in Trametinib-treated tumors (Fig. 2C), prompting the hypothesis that the pathways downstream of oncogenic KRAS that are not affected by treatment with MEKi could serve as coextinction targets that may cooperate with MEKi to recapitulate the impact of KRAS extinction. To this end, we conducted Ingenuity Pathway Analysis (IPA) on the differentially expressed genes in the Dox OFF group or the Trametinib treated group while compared with the Dox ON group. Half of the top-ten differentially expressed pathways preferentially enriched in OFF Dox tumors are associated with chromosol replication or cell cycle control (Fig. 2D and Fig. S2B), consistent with previous findings from an induced NRAS-driven melanoma mouse model (30). Interestingly, rest of the differentially enriched pathways are metabolism processes that we have previously shown to be driven by oncogenic KRAS (14), including glucose metabolism and cholesterol biosynthesis (Fig. 2D). As shown in the clustered heatmap, compared to the dramatic downregulation of genes in glucose metabolism and cholesterol biosynthesis pathways, Trametinib treatment partially decreases the expression of these metabolism genes (Fig. 2E). In addition, Trametinib treatment also failed to recapitulate the induction of amino acid and fatty acid degradation pathways following KRAS inactivation (Fig. S2C).
Biochemical analysis in iKras/p53 tumor cells upon KRAS inactivation indicated that glucose consumption or lactate production was dramatically downregulated, which is further supported by the decrease of ECAR as measured with Seahorse. Surprisingly, no significant decrease in glucose consumption or lactate production was detected when the cells were treated with Trametinib for 2 days (Fig. 2F–G). Moreover, Seahorse analysis showed that Trametinib treatment lead to a mild induction of ECAR compared with the iKras/p53 tumor cells grown in the presence of doxycycline (Fig. 2H and Fig. S2D–E). Accordingly, MEK inhibition also failed to suppress the glucose uptake and lactate production in human PDAC cell lines (Fig. S2F–G). BKM120, a selective PI3K inhibitor, was able to reverse the AKT activation induced by Trametinib treatment and impaired the glucose consumption and lactate production (Fig. 2I and S2H), indicating that the sustaining of glycolysis activity upon MEK inhibition is PI3K-AKT-dependent in iKras cell. Together, our transcriptomic analysis and biochemical analysis indicate that MEK inhibition alone is not sufficient to suppress the KRAS-mediated glucose flux in PDAC cells, likely due to the feedback activation of PI3K signaling.
Pooled shRNA library screening indicates glycolysis inhibition sensitizes iKras cells to MEK inhibition
To identify the metabolism genes that may sensitize Kras-driven PDAC cell to MEK inhibition upon depletion, we performed a pool-based in vivo loss-of-function screen in the orthotopic xenograft model as previously described (22). A customized bar-coded shRNA library comprising ∼3,400 shRNAs targeting ∼340 metabolism genes, including those KRAS-dependent metabolism genes (14) was packaged into lentivirus and infected iKras/p53 mouse PDAC cells. Orthotopic xenograft tumors were established in the nude mice and were treated with vehicle or Trametinib for 10 days before collection for next-generation sequencing. The log fold change (logFC) of bar-coded shRNA in the control or Trametinib-treated xenograft was calculated by comparison with reference group which was composed of library infected cells collected before orthotopic injection (Fig. 3A). In both control and Trametinib-treated xenograft, the positive control shRNA targeting Pmsa1 and Rpl30 are most depleted compared with the reference which indicates a high reliability of our screen system. Using logFC=−1 as a cut-off value, we obtained 36 candidate genes that were selectively depleted in MEKi-treated xenograft tumors compared to the control untreated ones (Supplementary Table S7). The depleted genes were most enriched in the glycolysis pathway (Fig. 3B), including Aldoa, Gapdhs, Hk2, Eno1 and PfkP (Fig. 3C–D).
Figure 3. Pooled shRNA library screening indicates glycolysis inhibition sensitizes iKras cells to MEK inhibition.
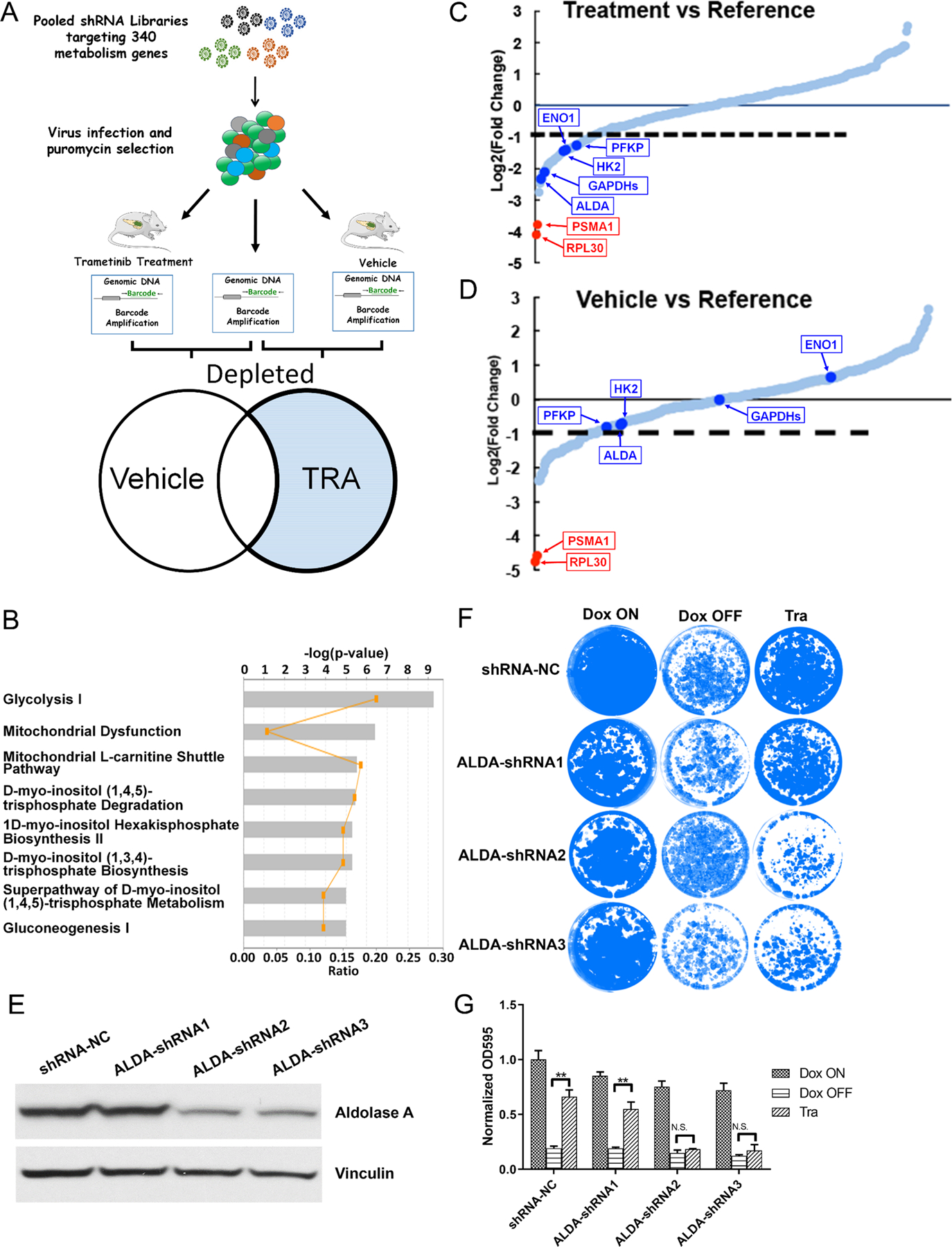
(A) Schematic illustration of pooled shRNA library screening in vivo. (B) IPA analysis of the depleted genes in shRNA library screen. (C-D) Relative abundance of each gene in the library in vehicle- or trametinib-treated tumors. Red dots: two positive control Rpl30 and Psma1; Blue Dots: five genes involved in glycolysis flux. (E) Knockdown of AldoA in iKras cell by shRNA. (F) Crystal violet staining of AldoA knockdown cell upon treatment of Dox ON, Dox OFF or 25 nM Tra for 4 days. (G) Quantification of crystal violet staining in F (n=3, Mean ± SD).
To validate the hits, we knocked down Aldoa in the iKras/p53 PDAC cells with shRNA (Fig. 3E). While knockdown of Aldoa itself has a limited effect on the cell growth, Aldoa-depleted cells are more sensitive to MEK inhibition compared with control cells, approximating the inhibitory effect on cell growth following KRAS inactivation (Fig. 3F–G). In addition, knocking down of Pfkp also sensitized the cell to Trametinib treatment while minimal effects were observed in Hk2 depleted cells (Fig. S3A–3C). Together, our data suggests that glycolysis inhibition may sensitize the KRAS-driven PDAC cells to MEK inhibition.
Glycolysis inhibition with 2DG synergizes with MEK inhibition to induce PDAC cell apoptosis
To test if pharmacological inhibition of glycolysis will synergize with MEK inhibition in KRAS-driven PDAC cells, we combined a well-known glycolysis inhibitor 2DG with Trametinib to treat iKras/p53 PDAC cells. Compared with cells with either 2DG or Trametinib single treatment, the combination shows a synergistic effect to decrease the proliferation of mouse PDAC cells from the iKras/p53 and LSL-Kras/p53 models, as well as human PDAC cell lines, including HPAC and Patu8902 cells (Fig. 4A–B). Treatment with additional glycolysis inhibitors, including GAPDH inhibitor Heptelidic acid (HA) or Enolase inhibitor POMHEX (31) also sensitized iKras cells to Trametinib treatment (Fig. S4A). Interestingly, no cooperative effect was observed in non-transformed human and mouse cell lines including IMR90, BJ, 293FT, HPNE, HPDE and 3T3 (Fig. 4A–B and S4B–C). To evaluate the synergy over a broad range of 2DG and Trametinib concentration, we computed the Bliss independence score for Trametinib and 2DG combination in PDAC cell lines. Compared to the low Bliss score observed in IMR90 cells, a strong synergistic effect was indicated in the iKras/p53 and LSL-Kras/p53 mouse PDAC cells (Fig. 4C–D), as well as human PDAC cells, including HPAC, and 8988T cells (Fig. 4E and Fig. S4D). Moreover, strong synergy was also observed between 2DG and additional MEK inhibitor GDC0623 or ERK inhibitor sch772984 (Fig. 4F and Fig. S4E). On the other hand, a low Bliss synergy score was observed when iKras cell line was treated with 2DG in combination with chemotherapy agents, such as gemcitabine or paclitaxel (Fig. 4G). Therefore, our data indicates that inhibition of glycolysis with 2DG specifically sensitizes PDAC cells to MAPK inhibition. Such synergy is due to the significant induction of apoptosis as shown by the increase in Annexin-V/7-AAD positive cells following the combination treatment, compared to single treatment alone groups. In contrast, the 2DG/Trametinib combination failed to induce apoptosis in IMR90 cells (Fig. 4H).
Figure 4. TRA and 2DG has a synergistic effect to induce apoptosis in mouse or human PDAC cell lines.
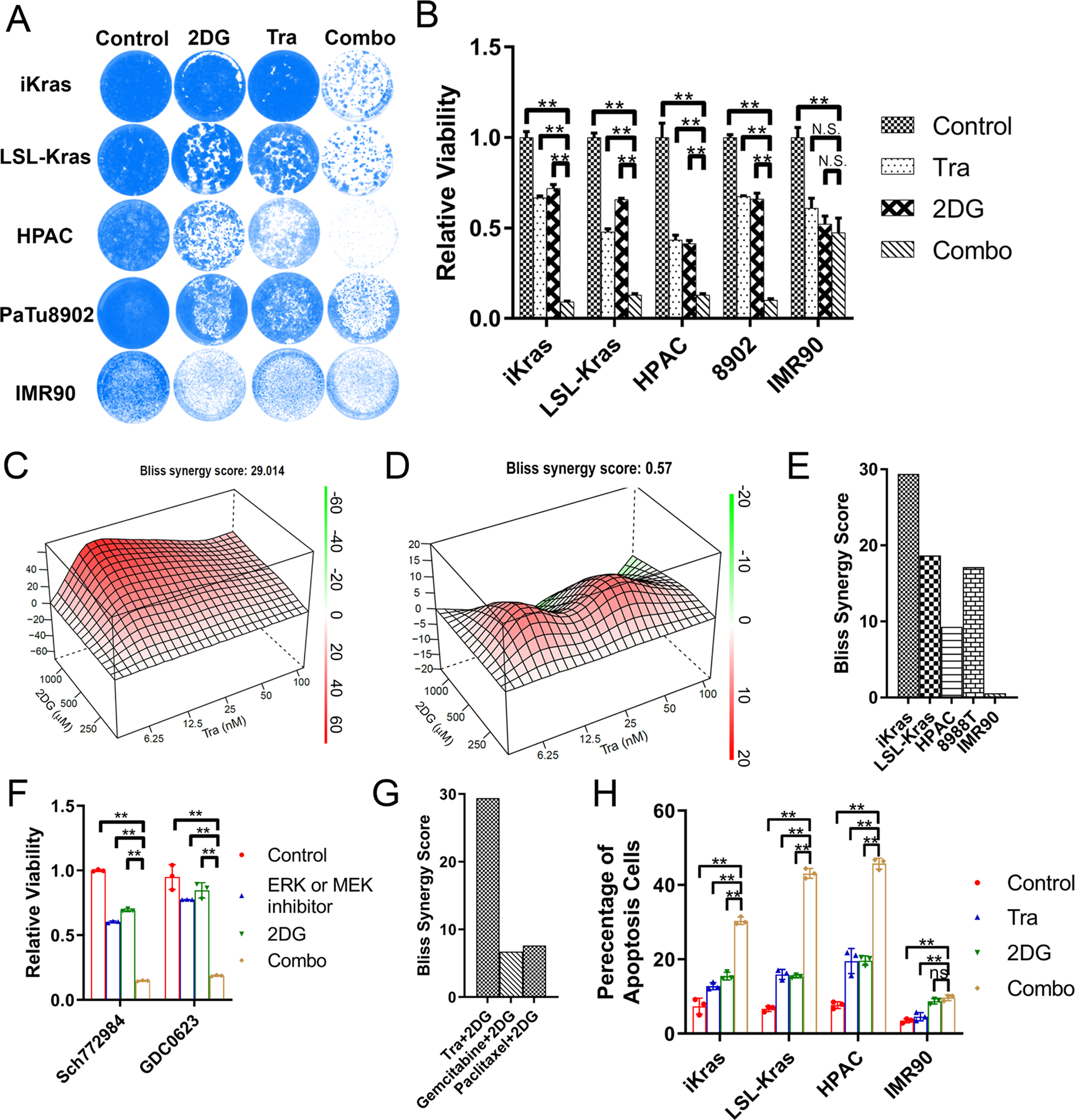
(A) Crystal violet staining of the cells treated with 25nM Tra, 1mM 2DG or combination for 4 days. (B) Quantification of crystal violet staining in B (n=3, Mean ± SD). (C-D) Representative Bliss score for combination of Tra and 2DG in iKras or IMR90 cell. (E) The synergistic effect of Tra and 2DG combination was analyzed using Bliss score in mouse (iKras and LSL-Kras), human PDAC line (HPAC and 8988T) and IMR90 cell line. (F) Quantification of crystal violet staining for the cell treated with SCH772984 (200nM)/GDC063 (100nM), 2DG (1mM) or combination (n=3, Mean ± SD). (G) The Bliss synergy score was calculated for the combination of Tra/2DG, gemcitabine/2DG and paclitaxel/2DG in iKras cell. (H) Apoptosis of the cell treated with 25nM Tra, 1mM 2DG or combination by annexin V/7-AAD staining (n=3, Mean ± SD).
2DG and MEK inhibition synergistically induces apoptosis through ER stress
To gain molecular insight into the mechanisms underlying the synergy between 2DG and MAPK inhibition, transcriptomic analysis was conducted in the iKras/p53 tumor cells treated with Trametinib, 2DG or in combination. By using a cut-off of fold change>2 and p<0.01, a total of 850 up-regulated and 310 down-regulated genes were identified from three treatment groups compared to vehicle control group (Fig. S5A–B). IPA analysis of the 430 differentially expressed genes (308 up and 112 down) in the Trametinib group indicated the enrichment of signaling pathways such as the sirtuin signaling pathway (Fig. 5A). On the other hand, the 209 differentially expressed genes (177 up and 32 down) from the 2DG-treated group were enriched in metabolism pathways such as N-acetylglucosamine degradation (Fig. 5B). The largest number of differentially expressed genes were identified in the combination treatment group, including 526 up- and 210 down-regulated genes. The top two enriched pathways are autophagosome maturation and unfold protein response (UPR), which are not among the top enriched pathways in the single treatment groups (Fig. 5C).
Figure 5. 2DG and Trametinib has a synergy to induce ER stress in PDAC cell lines.
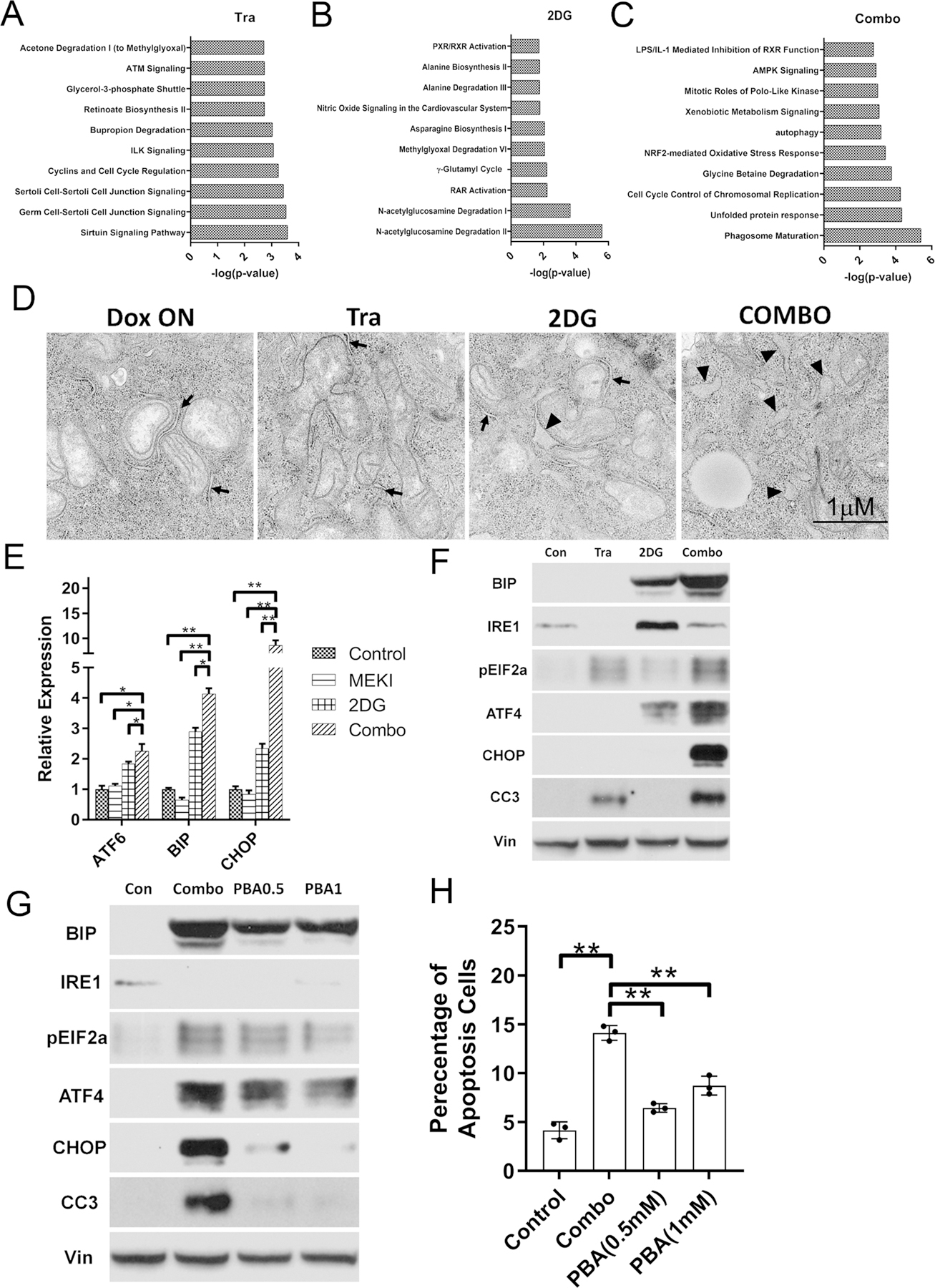
(A-C) IPA analysis of differentially expressed genes in iKras lines treated with 2DG and Tra combination. (D) Representative TEM images of iKras cell treated with Tra, 2DG or Combination for 48h. The normal ER were indicated by arrow and swollen ER were indicates by arrowheads. (E-F) Expression of the ER stress markers in iKras treated with 25nM Tra, 1mM 2DG or combination for 48h detected by qPCR (E, n=3, Mean ± SD) or WB (F). (G) Expression of the ER stress markers in iKras treated with Tra/2DG or Tra/2DG/PBA by WB. (H) Apoptosis analysis of iKras cell treated with Tra/2DG or Tra/2DG/PBA by Annexin-V staining (n=3, Mean ± SD). Tra: 25nM; 2DG: 1mM and PBA: 0.5mM or 1mM.
Transmission Electron Microscope (TEM) revealed swollen ER in 2DG/Trametinib-treated iKras/p53 tumor cells (Fig. 5D), a morphology change indicating the induction of UPR. The activation of UPR was further supported by the drastic upregulation of ER stress markers, including ATF4, ATF6 and BIP, following 2DG/Trametinib treatment compared with vehicle control or single treatment groups (Fig. 5E–F). Moreover, CHOP, a well-known apoptosis activator downstream of UPR, along with cleaved caspase 3, was also specifically upregulated by the combination treatment in iKras/p53 tumor cells, but not IMR90 cells (Fig. 5E–F, Fig. S5C), indicating 2DG and MEK inhibition results in lethal ER Stress in PDAC cells. To further validate if UPR is responsible for the induction of apoptosis by the combination treatment, cells were treated with the chemical chaperon PBA to decrease the UPR. As expected, PBA inhibited the induction of multiple ER-stress markers, such as BIP, ATF4, and phosphor-EIF2 α. Importantly, PBA treatment dramatically decreased the expression of CHOP, prevented caspase-3 cleavage and suppressed apoptosis induced by 2DG and Trametinib treatment (Fig. 5G–H), indicating the cell death induced by the combination treatment is mediated by hyper-activation of UPR. However, PBA has minimal effect to rescue the cell proliferation both in the iKras or LSL-Kras cell lines (Fig. S5D), indicating additional mechanisms are likely involved to mediate the cooperation between 2DG and MEK inhibition.
Trametinib in combination with 2DG exhibits antitumor activity in vivo
Next, we sought to evaluate the therapeutic potential of the combination in vivo. Compared with single treatment, combine 2DG and Trametinib treatment in subcutaneous xenograft tumors of mouse iKras/p53 PDAC cells or PDX-derived human PDAC cells resulted in significant decrease of tumor size (Fig. 6A–B). Moreover, the 2DG and Trametinib combination also showed a synergistic effect to decrease the proliferation of iKras/p53 tumor cells derived from C57BL/6J background (Fig. S6A) and dramatically decreases the tumor growth compared with the single treatment in syngeneic xenograft model (Fig. S6B). Lastly, we further evaluated the effect of the combination in the GEMM (iKras/p53L/+). After 8 weeks’ induction of KRASG12D expression with doxycycline, a time point previously showed to induce invasive carcinoma (14), the mice were randomized into four groups, including Vehicle control, Trametinib, 2DG, and Combo groups. While single treatment failed to elicit therapeutic benefit, we observed a statistically significant prolong of overall survival in the combination group (Fig. 6C). Immunohistochemistry revealed that the percentage of Ki67 positive cells was significantly decreased in the combo-treated tumors compared to the single treatment groups, indicating inhibition of tumor cell proliferation (Fig. 6D–E). More importantly, BIP expression and percentage of cleaved Caspase-3 positive cells were significantly upregulated in the tumors treated with 2DG/Trametinib combination (Fig. 6D and 6F), supporting the induction of UPR-related apoptosis. Notably, no body weight loss was observed during the treatment, indicating limited toxicity of the combination in preclinical model (Fig. S6C–D).
Figure 6. 2DG and MEK inhibitor combination has a synergy to treat PDAC in vivo.
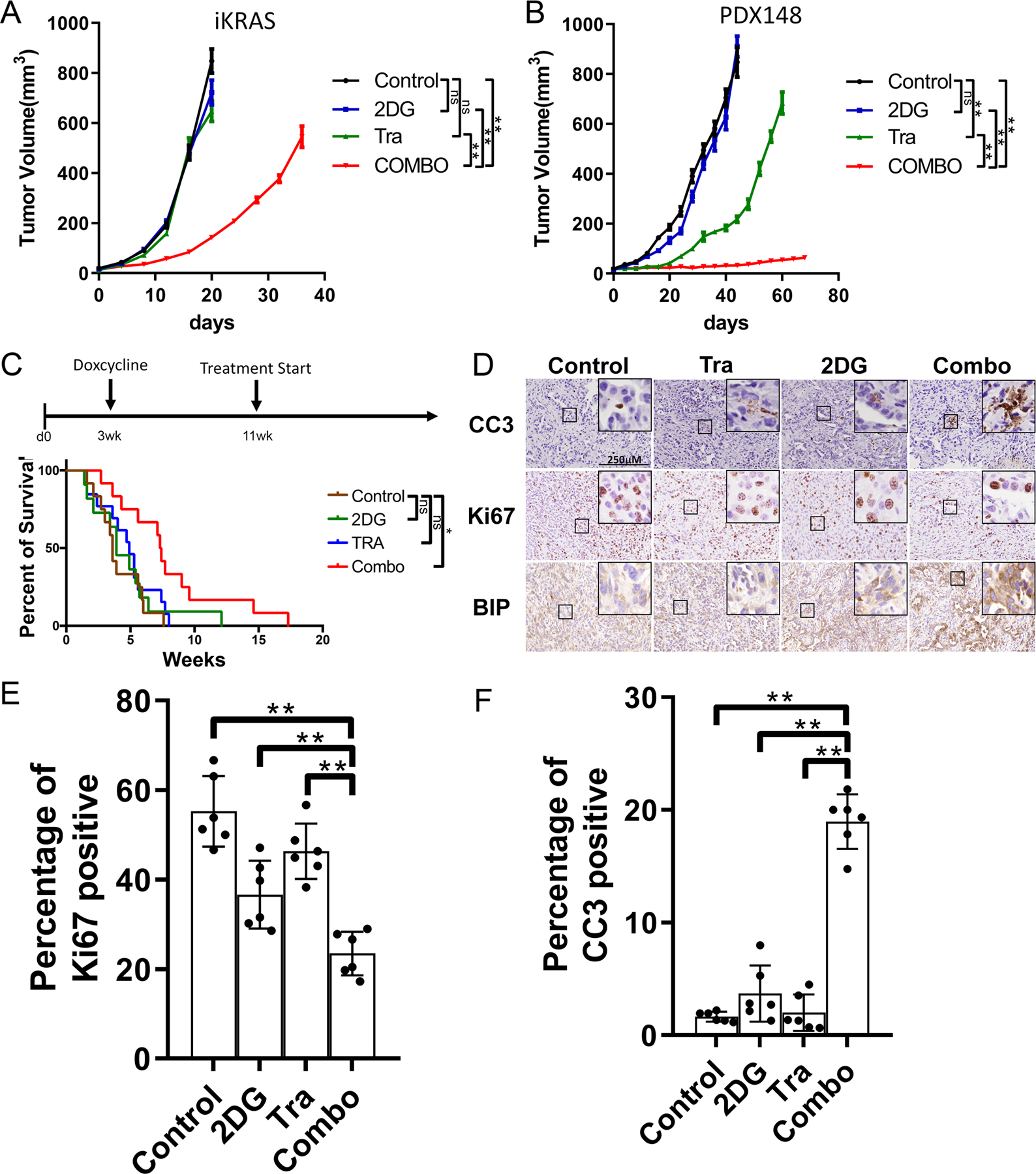
(A) Xenograft tumor volume of iKras cell in nude mice treated with Tra, 2DG or Combo (n=5, Mean ± SE). Tra: 1mg/kg/day; 2DG: 1000mg/kg/d. (B) Xenograft tumor volume of PDX-148 cell in nude mice treated with Tra, 2DG or Combo (n=5, Mean ± SE). Tra: 1mg/kg/day; 2DG: 1000mg/kg/d. (C) Kaplan–Meier survival analysis of iKras GEMM treated with vehicle, Tra, 2DG or Combo (n=12). The survival of the mice refers to weeks of treatment. Tra: 1mg/kg/day; 2DG: 1000mg/kg/d. (D) The iKras xenograft tumors were collected from the nude mice after 12-day treatment as indicated. Representative images of immunohistochemical staining of paraffin embedded xenograft tumors using antibodies for CC3, Ki67 or BIP. (E-F) Quantification of Ki67 or CC3 positive cell in D (n=6, Mean ± SD).
Discussion
As the major surrogates of KRAS signaling, the roles of MAPK and PI3K pathway in KRAS-driven tumors have been extensively studied. Mutations of BRAF, a dominant mediator for oncogenic KRAS signaling to activate MAPK signaling (32), were found to be mutually exclusive with the KRAS mutations in PDAC (5). Pancreatic-specific expression of oncogenic BrafV600E is sufficient to induce intraepithelial neoplasia (PanIN) lesions and invasive PDA in the autochthonous models (9). Moreover, CRAF has been shown to be essential for development of KRAS-driven non-small cell lung carcinoma (33,34), further supporting the central role of MAPK pathway in KRAS-driven tumorigenesis. On the other hand, PI3K and PDK1 have also been shown to be critical effectors downstream of oncogenic Kras in mediating cell plasticity and PDAC development (10). These data indicate that both MAPK and PI3K pathways are essential for KRAS-mediated tumor initiation. However, the requirement of these KRAS surrogates in advanced tumors has been less clear.
Recent study showed that ablation of CRAF expression leads to significant tumor regression in advanced tumors driven by KRASG12V/Trp53 mutations (11), underscoring the requirement of MAPK pathway for tumor maintenance. Here our data provide additional evidences that MAPK pathway is necessary and sufficient for PDAC maintenance whereas PI3K activation is less competent to sustain tumor growth by itself, supporting the need to target MAPK pathway in KRAS-driven tumors. However, it has been well documented that targeting MAPK alone failed to elicit therapeutic benefit in KRAS-driven tumors, likely due to the feedback activation of PI3K pathway (12,35). Although co-targeting MAPK and PI3K was able to induce tumor regression and prolong survival in PDAC GEMM (12,13), the combination is too toxic to be tolerant in human patients (36). Previous studies have identified reactivation of multiple RTKs as a prominent mechanism of adaptive resistance to MEK inhibition in KRAS-driven tumors (13,35,37). However, co-targeting multiple RTKs is difficult to achieve therapeutically, pointing to the need to identify additional strategies to target PI3K or its downstream effector pathways.
In this study, we identified the potential role of PI3K-mediated glycolysis in the adaptive resistance to MEK inhibition in KRAS-driven PDAC. In line with our findings, recent study in BRAF-driven melanoma showed that glycolysis inhibitors were able to potentiate the effects of Braf inhibitor (38). Although MAPK signaling has been shown to mediate the transcription of multiple glycolysis genes downstream of oncogenic KRAS (14,17), here we showed that feedback activation of PI3K pathway is sufficient to maintain glycolysis flux in KRAS-driven tumors following MAPK inhibition. PI3K has been shown to be a master regulator for the transcription of glucose transporters (39). PI3K can also activate glycolysis at post-translational level by controlling the cytoskeleton remodeling and thus relieving the sequestration of glycolysis enzymes (40). Whether such mechanisms are also involved in the feedback activation of glycolysis upon MEK inhibition in KRAS-driven tumor cells remains to be elucidated.
Our data indicates that the maintenance of glycolysis activity is essential for the survival of PDAC cells following the inhibition of MAPK signaling. Blocking glycolysis with 2DG in combination with MAPK inhibition leads to induction of apoptosis. 2DG, a derivative of glucose, could be phosphorylated to 2DG-6-phosphate in cell. The accumulated 2DG-6-phosphate inhibits hexokinase in a noncompetitive manner (41) and can lead to the inhibition of multiple anabolic processes branched from glycolysis pathway. It’s possible that the synergy between 2DG and MAPK inhibition is due to the blockade of multiple glucose-dependent metabolism pathways. Among them, 2DG has been shown to impair the pentose-phosphate pathway (PPP) and dramatically decreases R5P level (42). Interestingly, recent study has shown that the activation of PPP-mediated ribose metabolism is critical for the adaptation to the inhibition of KRAS signaling in PDAC cells (17). In addition, 2DG treatment has been shown to induce ER stress, likely due to its impact on HBP or mannose metabolism (43,44). While UPR is considered as a survival mechanism to maintain protein homeostasis, excessive ER stress will result in cell death (45,46). Here, we provide evidence that the combination of 2DG and MAPK inhibition dramatically amplified the ER stress in PDAC cells which results in apoptosis. This is supported by our finding that the cell death was partially rescued with chemical chaperon that mitigates the UPR. Therefore, our data indicates that the maintenance of protein homeostasis is critical for the survival of KRAS-driven PDAC cells upon the inhibition of MAPK signaling. It will be interesting to evaluate whether targeting additional regulators of protein homeostasis may also sensitize PDAC cells to MAPK inhibition.
Overall, our study provides evidence supporting the potential of co-targeting glycolysis and MAPK has an alternative approach to treat KRAS-driven PDAC.
Supplementary Material
Significance:
This study demonstrates the critical role of glucose metabolism in resistance to MAPK inhibition in KRAS-driven pancreatic cancer, uncovering a potential therapeutic approach for treating this aggressive disease.
Acknowledgment
We thank the Institute for Applied Cancer Science and Dr. Florian Muller for sharing reagents. We would like to thank Small Animal Imaging Facility, Histopathology Core, High Resolution Electron Microscopy Facility, the Flow Cytometry and Cellular Imaging Core at The University of Texas MD Anderson Cancer Center, and the Veterinary Medicine Department at MD Anderson (Cancer Center Support Grant CA016672). The research was supported by the National Cancer Institute (NCI) grant R01CA214793 and NCI P01 grant P01CA117969 to HY, and the 2016 Pancreatic Cancer Action Network -AACR Pathway to Leadership Grant, in memory of Carina Rogerson (16-70-25-YAO) to WY.
Footnotes
Conflict of interest: T.P. Heffernan reports personal fees from Cullgen Inc. and Silicon Therapeutics; other support from Taiho Pharmaceuticals and Boehringer Ingelheim outside the submitted work. No disclosures were reported by the other authors.
References
- 1.Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res 2014;74:2913–21 [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin 2020;70:7–30 [DOI] [PubMed] [Google Scholar]
- 3.Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: Mission possible? Nat Rev Drug Discov 2014;13:828–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019;575:217–23 [DOI] [PubMed] [Google Scholar]
- 5.Witkiewicz AK, McMillan EA, Balaji U, Baek G, Lin WC, Mansour J, et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun 2015;6:6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Downward J Targeting RAS signalling pathways in cancer therapy. Nature reviews Cancer 2003;3:11–22 [DOI] [PubMed] [Google Scholar]
- 7.Bryant KL, Mancias JD, Kimmelman AC, Der CJ. KRAS: feeding pancreatic cancer proliferation. Trends Biochem Sci 2014;39:91–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rodriguez-Viciana P, Warne PH, Khwaja A, Marte BM, Pappin D, Das P. et al. Role of phosphoinositide 3-OH kinase in cell transformation and control of the actin cytoskeleton by Ras. Cell 1997;89:457–67 [DOI] [PubMed] [Google Scholar]
- 9.Collisson EA, Trejo CL, Silva JM, Gu S, Korkola JE, Heiser LM. et al. A central role for RAF-->MEK-->ERK signaling in the genesis of pancreatic ductal adenocarcinoma. Cancer discovery 2012;2:685–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eser S, Reiff N, Messer M, Seidler B, Gottschalk K, Dobler M. et al. Selective requirement of PI3K/PDK1 signaling for Kras oncogene-driven pancreatic cell plasticity and cancer. Cancer Cell 2013;23:406–20 [DOI] [PubMed] [Google Scholar]
- 11.Sanclemente M, Francoz S, Esteban-Burgos L, Bousquet-Mur E, Djurec M, Lopez-Casas PP. et al. c-RAF Ablation Induces Regression of Advanced Kras/Trp53 Mutant Lung Adenocarcinomas by a Mechanism Independent of MAPK Signaling. Cancer Cell 2018;33:217–28 e4 [DOI] [PubMed] [Google Scholar]
- 12.Alagesan B, Contino G, Guimaraes AR, Corcoran RB, Deshpande V, Wojtkiewicz GR. et al. Combined MEK and PI3K inhibition in a mouse model of pancreatic cancer. Clin Cancer Res 2015;21:396–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pettazzoni P, Viale A, Shah P, Carugo A, Ying H, Wang H. et al. Genetic events that limit the efficacy of MEK and RTK inhibitor therapies in a mouse model of KRAS-driven pancreatic cancer. Cancer Res 2015;75:1091–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E. et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012;149:656–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bryant KL, Stalnecker CA, Zeitouni D, Klomp JE, Peng S, Tikunov AP. et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat Med 2019;25:628–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kinsey CG, Camolotto SA, Boespflug AM, Guillen KP, Foth M, Truong A. et al. Protective autophagy elicited by RAF-->MEK-->ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat Med 2019;25:620–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Santana-Codina N, Roeth AA, Zhang Y, Yang A, Mashadova O, Asara JM. et al. Oncogenic KRAS supports pancreatic cancer through regulation of nucleotide synthesis. Nat Commun 2018;9:4945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brown WS, McDonald PC, Nemirovsky O, Awrey S, Chafe SC, Schaeffer DF. et al. Overcoming Adaptive Resistance to KRAS and MEK Inhibitors by Co-targeting mTORC1/2 Complexes in Pancreatic Cancer. Cell Rep Med 2020;1:100131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chauhan SC, Ebeling MC, Maher DM, Koch MD, Watanabe A, Aburatani H. et al. MUC13 mucin augments pancreatic tumorigenesis. Molecular cancer therapeutics 2012;11:24–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Auciello FR, Bulusu V, Oon C, Tait-Mulder J, Berry M, Bhattacharyya S. et al. A Stromal Lysolipid-Autotaxin Signaling Axis Promotes Pancreatic Tumor Progression. Cancer discovery 2019 [DOI] [PMC free article] [PubMed]
- 21.Yao W, Rose JL, Wang W, Seth S, Jiang H, Taguchi A. et al. Syndecan 1 is a critical mediator of macropinocytosis in pancreatic cancer. Nature 2019;568:410–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carugo A, Genovese G, Seth S, Nezi L, Rose JL, Bossi D. et al. In Vivo Functional Platform Targeting Patient-Derived Xenografts Identifies WDR5-Myc Association as a Critical Determinant of Pancreatic Cancer. Cell Rep 2016;16:133–47 [DOI] [PubMed] [Google Scholar]
- 23.Aguirre AJ, Bardeesy N, Sinha M, Lopez L, Tuveson DA, Horner J. et al. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev 2003;17:3112–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.White MA, Nicolette C, Minden A, Polverino A, Van Aelst L, Karin M. et al. Multiple Ras functions can contribute to mammalian cell transformation. Cell 1995;80:533–41 [DOI] [PubMed] [Google Scholar]
- 25.Fiordalisi JJ, Holly SP, Johnson RL, 2nd, Parise LV, Cox AD. A distinct class of dominant negative Ras mutants: cytosolic GTP-bound Ras effector domain mutants that inhibit Ras signaling and transformation and enhance cell adhesion. J Biol Chem 2002;277:10813–23 [DOI] [PubMed] [Google Scholar]
- 26.Boehm JS, Zhao JJ, Yao J, Kim SY, Firestein R, Dunn IF. et al. Integrative genomic approaches identify IKBKE as a breast cancer oncogene. Cell 2007;129:1065–79 [DOI] [PubMed] [Google Scholar]
- 27.Infante JR, Somer BG, Park JO, Li CP, Scheulen ME, Kasubhai SM. et al. A randomised, double-blind, placebo-controlled trial of trametinib, an oral MEK inhibitor, in combination with gemcitabine for patients with untreated metastatic adenocarcinoma of the pancreas. Eur J Cancer 2014;50:2072–81 [DOI] [PubMed] [Google Scholar]
- 28.Van Cutsem E, Hidalgo M, Canon JL, Macarulla T, Bazin I, Poddubskaya E. et al. Phase I/II trial of pimasertib plus gemcitabine in patients with metastatic pancreatic cancer. Int J Cancer 2018;143:2053–64 [DOI] [PubMed] [Google Scholar]
- 29.Kasuga A, Nakagawa K, Nagashima F, Shimizu T, Naruge D, Nishina S. et al. A phase I/Ib study of trametinib (GSK1120212) alone and in combination with gemcitabine in Japanese patients with advanced solid tumors. Invest New Drugs 2015;33:1058–67 [DOI] [PubMed] [Google Scholar]
- 30.Kwong LN, Costello JC, Liu H, Jiang S, Helms TL, Langsdorf AE. et al. Oncogenic NRAS signaling differentially regulates survival and proliferation in melanoma. Nat Med 2012;18:1503–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lin YH, Satani N, Hammoudi N, Yan VC, Barekatain Y, Khadka S. et al. An enolase inhibitor for the targeted treatment of ENO1-deleted cancers. Nat Metab 2020;2:1413–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cseh B, Doma E, Baccarini M. “RAF” neighborhood: protein-protein interaction in the Raf/Mek/Erk pathway. FEBS letters 2014;588:2398–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karreth FA, Frese KK, DeNicola GM, Baccarini M, Tuveson DA. C-Raf is required for the initiation of lung cancer by K-Ras(G12D). Cancer Discov 2011;1:128–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Blasco RB, Francoz S, Santamaria D, Canamero M, Dubus P, Charron J. et al. c-Raf, but not B-Raf, is essential for development of K-Ras oncogene-driven non-small cell lung carcinoma. Cancer Cell 2011;19:652–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Manchado E, Weissmueller S, Morris JPt, Chen CC, Wullenkord R, Lujambio A. et al. A combinatorial strategy for treating KRAS-mutant lung cancer. Nature 2016;534:647–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Do K, Speranza G, Bishop R, Khin S, Rubinstein L, Kinders RJ. et al. Biomarker-driven phase 2 study of MK-2206 and selumetinib (AZD6244, ARRY-142886) in patients with colorectal cancer. Invest New Drugs 2015;33:720–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fedele C, Ran H, Diskin B, Wei W, Jen J, Geer MJ. et al. SHP2 Inhibition Prevents Adaptive Resistance to MEK Inhibitors in Multiple Cancer Models. Cancer Discov 2018;8:1237–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parmenter TJ, Kleinschmidt M, Kinross KM, Bond ST, Li J, Kaadige MR. et al. Response of BRAF-mutant melanoma to BRAF inhibition is mediated by a network of transcriptional regulators of glycolysis. Cancer Discov 2014;4:423–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hoxhaj G, Manning BD. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat Rev Cancer 2020;20:74–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hu H, Juvekar A, Lyssiotis CA, Lien EC, Albeck JG, Oh D. et al. Phosphoinositide 3-Kinase Regulates Glycolysis through Mobilization of Aldolase from the Actin Cytoskeleton. Cell 2016;164:433–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen W, Gueron M. The inhibition of bovine heart hexokinase by 2-deoxy-D-glucose-6-phosphate: characterization by 31P NMR and metabolic implications. Biochimie 1992;74:867–73 [DOI] [PubMed] [Google Scholar]
- 42.Ralser M, Wamelink MM, Struys EA, Joppich C, Krobitsch S, Jakobs C. et al. A catabolic block does not sufficiently explain how 2-deoxy-D-glucose inhibits cell growth. Proc Natl Acad Sci U S A 2008;105:17807–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xi H, Kurtoglu M, Liu H, Wangpaichitr M, You M, Liu X. et al. 2-Deoxy-D-glucose activates autophagy via endoplasmic reticulum stress rather than ATP depletion. Cancer Chemother Pharmacol 2011;67:899–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ishino K, Kudo M, Peng WX, Kure S, Kawahara K, Teduka K. et al. 2-Deoxy-d-glucose increases GFAT1 phosphorylation resulting in endoplasmic reticulum-related apoptosis via disruption of protein N-glycosylation in pancreatic cancer cells. Biochem Biophys Res Commun 2018;501:668–73 [DOI] [PubMed] [Google Scholar]
- 45.Sano R, Reed JC. ER stress-induced cell death mechanisms. Biochim Biophys Acta 2013;1833:3460–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep 2006;7:880–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.