

Abstract
Eukaryotes have evolved a variety of mRNA surveillance mechanisms to detect and degrade aberrant mRNAs with potential deleterious outcomes. Among them, nonsense-mediated mRNA decay (NMD) functions not only as a quality control mechanism targeting aberrant mRNAs containing a premature termination codon but also as a posttranscriptional gene regulation mechanism targeting numerous physiological mRNAs. Despite its well-characterized molecular basis, the regulatory scope and biological functions of NMD at an organismal level are incompletely understood. In humans, mutations in genes encoding core NMD factors cause specific developmental and neurological syndromes, suggesting a critical role of NMD in the central nervous system. Here, we review the accumulating biochemical and genetic evidence on the developmental regulation and physiological functions of NMD as well as an emerging role of NMD dysregulation in neurodegenerative diseases.
Keywords: RNA metabolism, nonsense-mediated mRNA decay, neurodegeneration, neurodevelopment, amyotrophic lateral sclerosis, frontal temporal dementia, mRNA translation
Introduction
A eukaryotic pre-mRNA undergoes multiple processing steps before the message is ready for export to the cytoplasm to fulfill its protein-coding function. Among these steps, intron recognition and excision by the spliceosome put the separate pieces of a gene (exons) back together into one consecutive mRNA sequence in a highly coordinated process known as splicing. Alternative usage of exons (i.e. alternative splicing) allows the production of multiple mRNA isoforms from a single gene, with each isoform having potentially distinct properties and functions. The resulting dramatic increase in the transcriptome complexity has been thought to contribute to organismal diversity (Keren et al., 2010). However, higher regulatory complexity also amplifies the chance of errors. While individual mRNA processing errors may be relatively inconsequential, misprocessed mRNAs from thousands of actively transcribed genes across the genome, each encoding a potentially defective protein, are likely detrimental to cellular and/or organismal fitness. In addition, a single mutation that disrupts mRNA processing may result in a substantial quantity of misprocessed mRNAs from that gene, also leading to the overproduction of defective polypeptides.
To protect cells from these potentially deleterious transcripts, multiple mechanisms, collectively known as mRNA surveillance, have evolved to detect misprocessed mRNAs and promote their rapid degradation. At least three highly conserved branches of mRNA surveillance have been characterized that target distinct classes of misprocessed mRNAs: nonsense-mediated decay (NMD), no-go decay (NGD), and nonstop decay (NSD). In the past, mRNA surveillance has been extensively studied in unicellular eukaryotes (e.g. the budding yeast) as well as in cultured mammalian cells. Recent biochemical and genetic studies are beginning to uncover unexpected roles of these ubiquitously present mechanisms in tissue-specific gene regulation, especially in the central nervous system (CNS). Here, we focus on NMD, perhaps the most well-studied mRNA surveillance mechanism. We review the current understanding of its molecular basis and the accumulating evidence for its roles in neural developmental and neurodegenerative disorders, while providing possible explanations for the CNS-specific vulnerability to defects in NMD.
Mechanisms of translation-dependent mRNA surveillance
Depending on the type of misprocessing, aberrant mRNAs can contain a wide range of errors. Large insertions and deletions may arise from intron retention or exon skipping, respectively, whereas genetic mutations may introduce small indels or substitutions. The aberrant mRNA per se is often structurally indistinguishable from a normal mRNA in that they are both 5ʹ-capped, 3ʹ-polyadenylated, spliced, and contain a linear RNA sequence. Unlike modern biologists, cells do not have access to a ‘reference transcriptome’ to which they can align and compare each RNA. The biological solution to the problem of detecting misprocessed RNAs lies in the aberrant events occurring during mRNA translation. As suggested by its name, an important role of NMD is to detect and degrade mRNAs carrying nonsense mutations, which generate premature termination codons (PTCs) and cause premature translation termination. In addition, it serves a gene regulatory function by lowering the abundance of a large subset of physiological mRNAs (Isken and Maquat, 2008; Nickless et al., 2017). In contrast, NSD and NGD facilitate the degradation of mRNAs lacking an in-frame stop codon and those with features causing prolonged stalling of ribosomes (Harigaya and Parker, 2010; Klauer and van Hoof, 2012; Shoemaker and Green, 2012), respectively.
In NMD, the molecular definition of PTCs differs in significant ways between yeast and metazoan cells. While PTCs are primarily defined by unusually long 3ʹ untranslated regions (3ʹ UTRs) in yeast, metazoan NMD is tightly linked to the nuclear processing history of an mRNA. Upon splicing of each intron, an exon junction complex (EJC) consisting of eukaryotic initiation factor 4A3 (eIF4A3), mago homolog (MAGOH), and RNA-binding motif protein 8A (RBM8A) is deposited near the exon‒exon junction and remains bound to the mRNA during nuclear export. In a normal mRNA, the stop codon typically resides in the last and often longest exon. As a result, all EJCs are upstream of the stop codon and therefore displaced by the ribosome during translation (Dostie and Dreyfuss, 2002). However, when translation terminates at a PTC at least 50‒55 nucleotides upstream from an EJC, that EJC would stay associated with the mRNA and trigger NMD (Thermann et al., 1998; Zhang et al., 1998). Based on this ‘50‒55 nucleotide rule’, PTCs within the last 50‒55 nucleotides of the second-to-last exon and those within the last exon would not trigger EJC-dependent NMD. The combination of translation termination and downstream EJCs results in a series of protein interactions between NMD factors, EJC components, and the eukaryotic release factors eRF1 and eRF3, ultimately initiating NMD (Chamieh et al., 2008; Lopez-Perrote et al., 2016; Neu-Yilik et al., 2017). Specifically, a protein complex known as SURF consisting of the ATP-dependent RNA helicase UPF1, the UPF1 kinase SMG1, and eRF1/3 plays an important role in triggering UPF1 phosphorylation and activating NMD (Kashima et al., 2006). Other NMD factors serve to bridge the SURF complex and the downstream EJC via additional protein‒protein interactions. For instance, UPF3 can recognize the release factors and facilitate their binding to EJCs (Neu-Yilik et al., 2017), while recruiting the adaptor protein UPF2 to further support the formation of NMD-inducing complexes (Chamieh et al., 2008; Lopez-Perrote et al., 2016). The initiation of NMD has been thought to be associated with less efficient translation termination, although this model has been recently challenged (Karousis et al., 2020).
NMD can also be induced in an EJC-independent manner. Particularly in the budding yeast, in which splicing is less prevalent than in metazoans, stop codons followed by long 3ʹ UTRs can be recognized as PTCs (Muhlrad and Parker, 1999; Amrani et al., 2004; Hogg and Goff, 2010) via either of two possible mechanisms. First, the key NMD factor UPF1 is highly abundant in the cytoplasm and can nonspecifically interact with RNAs (Hurt et al., 2013; Kurosaki and Maquat, 2016). During translation, the nonspecifically bound UPF1 is displaced by the ribosome, leaving the remaining UPF1 bound to 3ʹ UTR (Zund et al., 2013; Kurosaki and Maquat, 2016). Increased binding of UPF1 to an extended 3ʹ UTR may facilitate SURF complex formation. Second, a long distance between the PTC and additional factors that promote termination, such as PABP1, may increase the chance of the transcript being recognized as an NMD substrate (Amrani et al., 2004; Ivanov et al., 2008). Besides long 3ʹ UTRs, the presence of an upstream open reading frame (uORF) in the 5ʹ UTR can sometimes trigger NMD (Oliveira and McCarthy, 1995; Ruiz-Echevarría and Peltz, 2000), in part due to the uORF stop codon being recognized as a PTC. Alternatively, uORF translation represses the translation of the main ORF, which may increase the binding of UPF1.
Once phosphorylated by SMG1, UPF1 recruits additional factors including SMG5, SMG6, and SMG7 to promote RNA decay (Kashima et al., 2006). The endonuclease SMG6 cleaves the transcript near the PTC, which generates two RNA fragments further degraded by the 5ʹ-to-3ʹ exonuclease XRN1 and the 3ʹ-to-5ʹ exosome (Gatfield and Izaurralde, 2004; Glavan et al., 2006; Huntzinger et al., 2008; Eberle et al., 2009). RNA decay may also be promoted by the CCR4‒NOT deadenylase complex recruited by the SMG5‒SMG7 dimer. Additional NMD factors have recently been identified from NMD reporter-based genome-wide loss-of-function screens (Alexandrov et al., 2017; Zhu et al., 2020), although their exact functions in NMD are not fully understood.
With the exception of the universally required UPF1, the requirement of most NMD factors varies in a target-specific and perhaps cell type-specific manner, leading to the notion that metazoan NMD is a set of branched pathways with both shared and branch-specific components. In addition to the EJC-dependent and EJC-independent pathways, both UPF2-independent and UPF3B-independent NMD branches have been described (Gehring et al., 2005; Buhler et al., 2006; Chan et al., 2009). Consistent with their differential requirement for distinct subsets of NMD targets, loss-of-function mutations in each of them present overlapping yet distinct phenotypic outcomes, as we discuss further in the following sections.
The molecular mechanisms of other translation-coupled mRNA surveillance mechanisms such as NGD and NSD have been reviewed elsewhere (Harigaya and Parker, 2010; Klauer and van Hoof, 2012; Shoemaker and Green, 2012). Similar to NMD, these mechanisms also involve target recognition, decay complex assembly, ribosome rescue, and degradation of the faulty mRNAs. In addition, ribosome stalling-induced surveillance mechanisms such as NGD and NSD involve the degradation of nascent peptides by the ribosome-associated quality control pathways (Ito-Harashima et al., 2007; Wilson et al., 2007). Although these mRNA surveillance mechanisms appear to be distinct pathways, they are increasingly appreciated to be in many ways interconnected. For example, it had been initially thought that the position of ribosomal stalling (internal vs. 3ʹ end) determines which pathway is activated. However, recent studies have found that, on prematurely polyadenylated NSD targets, the highly charged poly-lysine nascent chain translated from the polyA tail would cause the ribosome to stall before it reaches the 3ʹ end (Ito-Harashima et al., 2007), thereby blurring the boundary between NSD and NGD. In addition, recent studies in invertebrates suggest that the post-endonucleolytic cleavage processing of NMD targets may involve NSD (Hashimoto et al., 2017; Arribere and Fire, 2018). Therefore, these seemingly distinct branches of mRNA surveillance mechanisms are often intertwined, enabling cells to recognize a large variety of aberrant mRNAs and mount a proper response.
Relationship between NMD and membraneless organelles
While NMD is widely recognized as an essential surveillance mechanism for both cellular RNA quality and quantity, the subcellular location in which NMD takes place has been under debate. In particular, the processing body (P body) has been proposed as the prime location in which NMD occurs either exclusively or preferentially. P bodies are membraneless cytoplasmic ribonucleoprotein (RNP) granules, which assemble and disassemble dynamically in response to the pool of translationally repressed mRNPs (Parker and Sheth, 2007). For example, P body size decreases when translating ribosomes are immobilized on mRNAs by elongation inhibitors such as cycloheximide (Sheth and Parker, 2003; Cougot et al., 2004; Teixeira et al., 2005). Conversely, a decrease in ribosome loading by inhibiting translation initiation can increase the size of P bodies (Kedersha et al., 2005; Teixeira et al., 2005; Koritzinsky et al., 2006). Most of the core proteins in P bodies are evolutionarily conserved across eukaryotes. In yeast, P body core proteins include the decapping enzymes Dcp1p and Dcp2p, the decapping enzyme activators (Dhh1p/RCK/p54, Pat1p, Scd6p/RAP55, Edc3p, and Lsm1p-7p), the 5′-to-3′ exonuclease Xrn1p, and the deadenylase complex CCR4/POP2/NOT (Parker and Sheth, 2007). Because these core proteins mediate the essential steps in mRNA degradation pathways, intuitively the degradation of normal and/or aberrant mRNAs may preferentially take place inside P bodies. Consistent with this hypothesis, the major NMD factors along with the mRNA decay intermediates have also been observed to accumulate in P bodies during mRNA decay in yeast and in mammalian cells (Unterholzner and Izaurralde, 2004; Sheth and Parker, 2006).
An important question is which step(s) of NMD might take place in P bodies. In yeast, the depletion of decapping enzymes Dcp1p/Dcp2p and 5′-to-3′ exonuclease Xrn1p results in the accumulation of UPF1‒UPF3 and NMD target mRNAs inside P bodies (Sheth and Parker, 2006). In mammalian cells treated with NMDI 1, a chemical inhibitor of UPF1 and SMG5 interaction, hyperphosphorylated UPF1, UPF3, and NMD target mRNAs also accumulate in P bodies (Durand et al., 2007). Interestingly, neither inhibiting NMD at an early step by depleting UPF2 nor at a late step by depleting XRN1 causes NMD factors to concentrate in P bodies, indicating a dynamic cycle of NMD factors associating with P bodies after target recognition and dissociating before processive mRNA decay (Durand et al., 2007). These results suggest that P bodies may contain mRNPs that have been marked for degradation, presumably in a translationally repressed state (Isken et al., 2008), while recruiting downstream NMD effectors and awaiting degradation.
However, the dynamic association of NMD factors and mRNA decay intermediates with P bodies does not prove a causal relationship between NMD and P bodies. In a seminal study, Eulalio et al. (2007) have shown that in Drosophila S2 cells, the disruption of P bodies by depleting their core components does not significantly impact RNA interference nor NMD. Instead, the evidence points to an opposite causal relationship, in which P body formation is a result of translationally repressed mRNAs and/or mRNAs undergoing decapping (Eulalio et al., 2007). P bodies are dispersed after polysomes are stabilized by cycloheximide, and also after decapping is inhibited by the depletion of NOT1, an essential component in the CAF1‒CCR4‒NOT deadenylase complex (Eulalio et al., 2007). Conversely, P body size is increased when Drosophila S2 cells are treated with puromycin, which induces premature termination and release of the elongating ribosomes (Eulalio et al., 2007). Similar results have been subsequently reported in mammalian cells, in which the depletion of Ge-1/EDC4, a P body core component, causes P body dispersion yet fails to stabilize endogenous NMD targets (Stalder and Muhlemann, 2009). It should be noted that in these experiments, P body loss-of-function was determined by the lack of microscopically visible P bodies with canonical molecular markers. The possibility remains that, under these conditions, P bodies without canonical markers or with sizes below the microscope detection limit may still exist. Using an orthogonal, live-cell RNA imaging approach, in which the 3ʹ fragments of mRNA degradation intermediates are stabilized and visualized by MS2 tagging, a study has found no enrichment of RNA decay intermediates in P bodies (Horvathova et al., 2017). Therefore, while P bodies appear to be dynamic hubs for storing translationally repressed mRNAs that are either tagged for degradation or released to cytoplasm for resumed translation (Parker and Sheth, 2007), the evidence supporting a functional role of P bodies in RNA decay including NMD is still lacking. Whether P bodies may be functionally required for RNA decay under specific conditions or in specific species, cell types, and/or genetic background remains an open question.
Another class of RNP granules that has been repeatedly shown to enrich for UPF1 and other NMD factors are stress granules (Markmiller et al., 2018). In contrast to P bodies, stress granules are typically detected in cells under stress conditions associated with eIF2α phosphorylation and, as a result, global translational repression. Consistent with previous studies (Markmiller et al., 2018), we have recently shown that in mammalian cells treated with either sodium arsenite or arginine-rich toxic peptides, UPF1 and, to a lesser extent, UPF3B are recruited to stress granules (Sun et al., 2020). The concentration of NMD factors in stress granules is accompanied by the global stabilization of NMD targets, leading us and others (Xu et al., 2019) to hypothesize that stress granules may be a negative regulator of NMD. However, in cells lacking G3BP1 and G3BP2, two essential components of stress granules, NMD target stabilization is largely unaffected even though stress granule formation is severely compromised (Sun et al., 2020). Therefore, the proposed role of stress granules in inhibiting NMD by sequestering NMD factors has also been challenged.
The examples of P bodies and stress granules have both shown us that, as tempting as it may be to assign functions to these visually compelling compartments, direct assessment of causality is necessary in which these granules are specifically disrupted by the depletion of their essential components. In addition, when interpreting the apparent requirement of an RNP granule for a regulatory process, one must keep in mind that even a ‘core’ granule protein may still have granule-independent functions. Nonetheless, the spatial organization and compartmentalization of NMD and other mRNA surveillance mechanisms, especially in morphologically complex cells like neurons, remains an intriguing topic certainly worth further investigation.
Developmental defects caused by mutations in NMD factors
The development of the nervous system requires precise control of gene expression of transcription factors and downstream effectors that direct neuronal differentiation, migration, dendritic and axonal growth, and synapse formation and pruning. Deviations from the proper developmental gene expression program can lead to a wide range of neurodevelopmental disorders. Remarkably, both point mutations and copy number variations in genes encoding NMD factors have been linked to intellectual disability (Nguyen et al., 2013; Sartor et al., 2015; Shaheen et al., 2016). While consistent with the significance of posttranscriptional gene regulation in neurons, the identification of disease-causing mutations in NMD factor genes also raises interesting questions regarding the tissue-specific functions and regulation of NMD in metazoans.
As expected from its critical roles in not only NMD but also several other RNA decay pathways (Kim and Maquat, 2019), the ATP-dependent RNA helicase UPF1 is essential for viability in most metazoans including Drosophila, zebrafish, and mice, with the exception of Caenorhabditis elegans (Table 1; Hodgkin et al., 1989; Pulak and Anderson, 1993; Medghalchi et al., 2001; Metzstein and Krasnow, 2006; Wittkopp et al., 2009). While the deeply conserved essentiality of UPF1 could be explained by the fact that nonsense mutation-containing and misprocessed mRNAs may be prevalent in all species, a recent study has challenged this notion (Nelson et al., 2016). Through a forward genetic screen searching for suppressors of the NMD mutant lethality, the authors found that the lethality observed in Drosophila embryos lacking either Upf1 or Upf2 can be partly rescued by depleting growth arrest and DNA damage-induced gene 45 (Gadd45) mRNA, which is one of many known physiological NMD targets (Table 1; Nelson et al., 2016). Consistent with these results, reducing GADD45 (mammals have three paralogs, GADD45A, GADD45B, and GADD45G) mRNA levels can also reduce cell death in UPF1-depleted mouse embryonic fibroblasts and human embryonic kidney cells (Nelson et al., 2016). Although in vivo rescue of UPF1 mutant lethality remains to be shown in species other than Drosophila, these results highlight the importance of NMD regulation of physiological mRNA targets rather than global nonsense mRNA surveillance.
Table 1.
Summary of phenotypes observed in NMD factor loss-of-function animal models.
| NMD factor | Functions | Species | Genotype | Phenotype | References |
|---|---|---|---|---|---|
| UPF1/SMG2 | ATP-dependent 5′−3′ RNA helicase; recruits UPF2 and UPF3 near the PTC; phosphorylated by SMG1 and activates NMD | C. elegans | Loss-of-function mutants | Viable, mild defects in tail, bursal, and vulval development | Hodgkin et al. (1989) |
| Drosophila | Missense and nonsense mutations | Lethality during larval development, rescued by loss of Gadd45 | Metzstein and Krasnow (2006); Avery et al. (2011); Nelson et al. (2016) | ||
| Zebrafish | Knockdown by morpholinos | Embryonic lethality, brain patterning and midbrain–hindbrain boundary defects, brain necrosis, somitogenesis impairment | Wittkopp et al. (2009) | ||
| Mouse | Global knockout | Embryonic lethality, apoptosis at blastocyst stage | Medghalchi et al. (2001) | ||
| UPF2/SMG3 | Branch-specific NMD factor; recruited to the EJC via UPF3B interactions; bridges UPF1 and the EJC | C. elegans | Loss-of-function mutants | Viable, mild defects in tail, bursal, and vulval development | Hodgkin et al. (1989) |
| Drosophila | Null mutant | Lethality during larval development, defects in NMJ synaptic transmission | Metzstein and Krasnow (2006); Long et al. (2010); Avery et al. (2011) | ||
| Drosophila | Brain-specific knockdown by RNAi | Impaired long-term memory | Johnson et al. (2019) | ||
| Zebrafish | Knockdown by morpholinos | Embryonic lethality, developmental defects similar to Upf1 knockdown | Wittkopp et al. (2009) | ||
| Mouse | Global knockout | Embryonic lethality between E3.5 and E7.5 | Weischenfeldt et al. (2008) | ||
| Mouse | Liver-specific knockout | Perinatal lethality, impaired liver development, activated DNA damage response | Thoren et al. (2010) | ||
| Mouse | Forebrain-specific knockout | Impaired hippocampal synaptic plasticity and long-term memory, deficits in social behaviors and behavioral inflexibility, neuroinflammation | Johnson et al. (2019) | ||
| UPF3/SMG4 | Branch-specific NMD factor; interacts with the EJC; recruits UPF2; in vertebrates, UPF3A is a partially redundant but less active paralog of UPF3B | C. elegans | Loss-of-function mutants | Viable, mild defects in tail, bursal, and vulval development | Hodgkin et al. (1989) |
| Drosophila | Null mutant | Viable, fertile, no severe developmental defects | Avery et al. (2011) | ||
| Zebrafish | Knockdown by morpholinos | Upf3a: weak brain patterning defect, viable; Upf3b: no apparent phenotype; double knockdown: mild phenotype similar to Upf1 mutant, few with brain necrosis, 19% lethality at 5 days post-fertilization | Wittkopp et al. (2009) | ||
| Mouse | Upf3a knockout | Global knockout: embryonic lethality between E4.5 and E8.5; male germ cell-specific knockout: spermatogenesis defects; olfactory epithelium-specific knockout: reduced NMD target abundance | Shum et al. (2016) | ||
| Mouse | Upf3b knockout | Impaired fear-conditioned learning and prepulse inhibition, dendritic spine maturation deficits | Huang et al. (2018) | ||
| SMG1 | PI3K-related kinase; phosphorylates UPF1 at S/TQ motifs and activates NMD | C. elegans | Loss-of-function mutants | Viable, mild defects in tail, bursal, and vulval development | Hodgkin et al. (1989) |
| Drosophila | Null mutant | Viable, fertile, impaired synaptic transmission, impaired synaptic vesicle recycling | Metzstein and Krasnow (2006); Long et al. (2010) | ||
| Zebrafish | Knockdown by morpholinos | No phenotype observed in embryos | Wittkopp et al. (2009) | ||
| Mouse | Global knockout | Embryonic lethality, developmental arrest at E8.5 | McIlwain et al. (2010) | ||
| SMG5 | NMD effector; recruited near the PTC by phosphorylated UPF1; promotes deadenylation and RNA decay; promotes dephosphorylation of UPF1 by recruiting PP2A | C. elegans | Loss-of-function mutants | Viable, mild defects in tail, bursal, and vulval development | Hodgkin et al. (1989) |
| Drosophila | Loss-of-function mutants | Developmental delay in larval stage and lethality during pupariation, synthetic lethality between Smg5 hypomorph and Smg1 null | Nelson et al. (2018) | ||
| Zebrafish | Knockdown by morpholinos | Embryonic lethality, developmental defects similar to Upf1 knockdown | Wittkopp et al. (2009) | ||
| SMG6 | Endonuclease; recruited by phosphorylated UPF1; cleaves mRNAs near the PTC | C. elegans | Loss-of-function mutants | Viable, mild defects in tail, bursal, and vulval development | Hodgkin et al. (1989) |
| Drosophila | Loss-of-function mutants | Partially functional NMD, moderate reduction in viability, growth disadvantage of mutant cells when competing with wild-type cells, impaired NMJ synaptic transmission | Long et al. (2010); Frizzell et al. (2012) | ||
| Zebrafish | Knockdown by morpholinos | Embryonic lethality, developmental defects similar to Upf1 knockdown | Wittkopp et al. (2009) | ||
| Mouse | Global knockout | Embryonic lethality at the blastocyst stage | Li et al. (2015) | ||
| SMG7 | NMD effector; promotes deadenylation and RNA decay; promotes dephosphorylation of UPF1 | C. elegans | Loss-of-function mutants | Viable, temperature-sensitive defect in NMD | Cali et al. (1999) |
| Zebrafish | Knockdown by morpholinos | Elongated hindbrain, altered midbrain to hindbrain boundary, stacked somites, bent tails | Wittkopp et al. (2009) |
Consistent with the presence of partially redundant NMD factors and branches of NMD, mutations in other NMD factor genes have been shown to cause more specific developmental defects. In particular, NMD factor gene mutations found in human diseases have allowed us to glimpse into the complexity of in vivo NMD functions. The first NMD factor gene that has been linked to neurodevelopmental disorders is UPF3B, of which loss-of-function mutations have been identified from families with X-linked mental retardation (Tarpey et al., 2007). Since this initial study, a variety of developmental phenotypes have been linked to UPF3B mutations, including facial abnormalities, autism spectrum disorder, schizophrenia, and attention deficit hyperactivity disorder (Addington et al., 2011; Szyszka et al., 2012). To investigate the role of UPF3B in mammalian development, Upf3b knockout (Upf3b-KO) mice have been generated (Table 1; Huang et al., 2018). Neural stem cells derived from these mice display prolonged proliferation and delayed differentiation into functional neurons in culture (Huang et al., 2018). These cellular phenotypes are reflected by the in vivo defect in the maturation of dendritic spines. This defect appears to be brain region-specific, primarily affecting pyramidal neurons in the prefrontal cortex while sparing those in the hippocampus. Upf3b-KO mice display behavioral phenotypes that recapitulate human UPF3B-associated neurodevelopmental disorders (Table 1). While Upf3b-KO mice display no apparent defects in working memory and spatial learning, they exhibit fear conditioning deficits and sleep alterations (Huang et al., 2018). That humans and mice can tolerate a complete loss of UPF3B, although with developmental defects, suggests two nonexclusive possibilities that either the loss of UPF3B can be at least partially compensated by other redundant factors such as UPF3A, or the UPF3B-dependent branch of NMD has tissue-specific functions in regulating developmental gene regulation, an important aspect of mammalian NMD functions that requires further investigation.
Subsequent to UPF3B, loss-of-function mutations in UPF2, SMG9, and RBM8A, as well as copy number variations in UPF2, UPF3A, SMG6, SMG9, eIF4A3, RBM8A, and RNPS1, have been identified to cause a variety of developmental disorders (Nguyen et al., 2013; Sartor et al., 2015; Shaheen et al., 2016; Johnson et al., 2019). For instance, individuals with frameshift mutations and copy number variations of UPF2 develop neurodevelopmental disorders with intellectual disability (Nguyen et al., 2013; Johnson et al., 2019). Transcriptomic changes due to either UPF2 or UPF3B mutations largely overlap. Neurological impairments observed in patients with mutations in either of these two genes are also similar, suggesting a primary role of NMD dysfunction in causing the developmental defects in brain regions critical for memory and learning (Nguyen et al., 2013; Johnson et al., 2019). Considering the embryonic lethality of complete Upf2 knockout in mice (Weischenfeldt et al., 2008), Johnson et al. (2019) have generated a conditional knockout mouse model that lacks UPF2 in the forebrain (fb-KO), resulting in a forebrain-specific increase in the abundance of UPF2-dependent NMD substrates (Table 1). Upf2 fb-KO mice exhibit decreased excitatory postsynaptic potentials, defects in long-term potentiation, and impairments in long-term memory, behavioral flexibility, and social behaviors (Table 1). Interestingly, the onset of behavioral deficits coincides with multiple signs of neuroinflammation, including increases in inflammation-related gene expression and in the numbers of immune cells in the brain (Table 1). Anti-inflammatory drugs reduce the overall numbers of immune cells in the brain and concomitantly ameliorate the synaptic and behavioral deficits in Upf2 fb-KO mice, suggesting a causal role of neuroinflammation in mediating these phenotypes. While it remains to be tested whether immune response mRNAs are direct NMD targets, these results uncover a novel anti-inflammatory role of UPF2-dependent NMD in the brain.
Both deletions and duplications of NMD factors have been identified in patients with intellectual disabilities and/or congenital anomalies, highlighting the importance of a proper balance of NMD factors (Nguyen et al., 2013). Studies in animal models have shown that the depletion of SMG1, SMG5, or SMG6 leads to impairments in embryogenesis and structural defects in the brain, similar to what has been observed in UPF1 and UPF2 mutants (Table 1). Zebrafish embryos depleted in either Upf2, Smg5, or Smg6 exhibit brain necrosis, aberrant eye development, and impairments in early brain patterning at the midbrain‒hindbrain boundary (Table 1; Wittkopp et al., 2009). In Drosophila, inhibition of NMD via either Smg1, Upf2, or Smg6 deletion leads to a wide array of synaptic deficits, including premature development of neuromuscular junction synapses, impaired synaptic vesicle recycling, and reduced neurotransmission (Table 1; Long et al., 2010). Complete knockout of SMG6 in mice causes embryonic lethality (Table 1; Li et al., 2015). Although the molecular mechanism remains unclear, loss of SMG6 inhibits stem cell differentiation partly due to the sustained expression of c-Myc, a key pluripotency factors that would be otherwise repressed by NMD during differentiation (Li et al., 2015). The wide spectrum of deleterious phenotypes observed in SMG mutants reiterate the critical role of NMD in maintaining the precision of gene expression required for normal development.
Lastly, genetic alterations in EJC components have been linked to microcephaly, a phenotypic hallmark of Eif4a3, Magoh, and Rbm8a haploinsufficient mouse models (Mao et al., 2016). A comparative analysis of the cortical transcriptomes derived from the Eif4a3, Magoh, and Rbm8a haploinsufficient mice shows increased abundance of intron-retaining and PTC-containing transcripts, suggesting aberrant alternative splicing and/or NMD inhibition (Mao et al., 2016). Among the EJC components, RBM8A has been extensively studied in the context of neurodevelopment due to its clinical implications. Patients with deletions within chromosome region 1q21.1 including the RBM8A gene develop a distinct set of neurological pathologies including microcephaly and developmental delays (Brunetti-Pierri et al., 2008). RBM8A appears to have tissue-specific functions, as reduced RBM8A expression in the dorsal telencephalon causes thinning of the neocortex in mice, accompanied by decreased numbers of radial glia and neuron-producing intermediate progenitor cells (Mao et al., 2015). In contrast, increased RBM8A expression in the mouse dentate gyrus leads to anxiety-like behaviors and abnormal social interactions (Alachkar et al., 2013). Although EJC components are critical players in EJC-dependent NMD, this multimeric complex also influences pre-mRNA processing, mature mRNA export, localization, and translation (Le Hir et al., 2001; Lykke-Andersen et al., 2001). Therefore, the extent to which EJC-associated developmental defects are NMD-dependent remains unclear.
For all the genetic diseases associated with NMD factors, the relative contributions of the loss of nonsense mRNA surveillance vs. the deregulation of physiological mRNA targets are not well understood. On one hand, compromised NMD functions would presumably lead to global changes in the transcriptome, in a manner similar to, for example, mutations in splicing factors such as Rbfox1, which also causes mental retardation, epilepsy, and autism spectrum disorder (Martin et al., 2007; Sebat et al., 2007; Zhao, 2013). On the other hand, aberrant derepression of specific physiological mRNA targets of NMD, such as GADD45 (Nelson et al., 2016) and immune response mRNAs (Johnson et al., 2019) in the absence of functional NMD may be more relevant for certain pathological phenotypes as well as the physiological functions of NMD in neurons, as we discuss next.
Neuron-specific regulation and functions of NMD
Aside from the important roles of NMD in neurodevelopment, the expression of NMD factors is highly regulated by microRNAs (miRNAs) during neural differentiation. miRNAs are ∼22-nucleotide noncoding RNAs that base-pair via their 5ʹ seed region to target mRNAs and repress their expression (Bartel, 2018). Because of their wide spectra of targets, miRNAs play critical roles in posttranscriptional gene regulation and control numerous biological processes. Several conserved miRNA families, including miR-9, miR-124, and miR-128, are abundantly and specifically expressed in neurons (Bruno et al., 2011). miR-128 is present in two isoforms, miR-128-1 and miR-128-2, both of which are predicted to repress the expression of critical NMD factors UPF1, MLN51/CASC3 (Bruno et al., 2011), as well as SMG1 (Agarwal et al., 2015). The expression of miR-128 is increased in the mouse brain around embryonic day 9.5 and continues throughout the embryonic and postnatal development (Bruno et al., 2011). Ectopic expression of miR-128 in primary mouse neural stem cells results in reduced NMD activity and the depression of neuronal mRNAs targeted by NMD, thereby promoting neural differentiation (Bruno et al., 2011). The conserved miRNA‒NMD regulatory circuit has been further expanded by the identification of additional miRNAs that regulate NMD, including miR-9, miR-124, and miR-125 (Wang et al., 2013; Lou et al., 2014). miR-125, a direct repressor of SMG1, has been shown to regulate dendritic spine morphology and synaptic plasticity in hippocampal neurons (Edbauer et al., 2010; Wang et al., 2013). That these conserved miRNAs downregulate NMD factor expression to promote neural differentiation and neuronal maturation indicates that the level of NMD itself must be strictly regulated. Since most of these neuron-specific miRNAs remain highly expressed in mature neurons (Ludwig et al., 2016), NMD may be kept at a minimal yet critical level in these cells throughout life. As a result, compared to other cell types with higher NMD factor expression, neurons may possess lower buffering capacities and become selectively vulnerable to perturbations of NMD (Figure 1).
Figure 1.
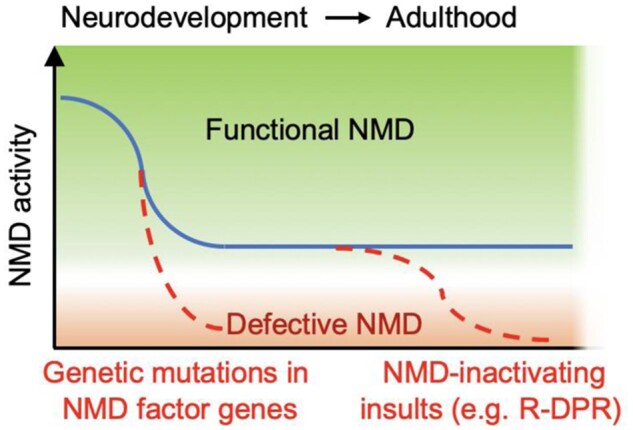
A hypothetical model of selective neuronal vulnerability to NMD perturbations. During neuronal differentiation, NMD activity is suppressed by miRNAs to a minimal level required for viability. Additional insults to NMD, either by genetic mutations in NMD factor genes (early onset, e.g. UPF3B) or by the accumulation of NMD-inactivating molecules (late onset, e.g. R-DPR), would further reduce the activity below the required threshold, leading to pathological outcomes.
The wiring of the developing CNS relies heavily on the intricate interplay between axon guidance cues and the growth cone, which allows an axon to find and form synaptic connections with its designated targets. Mechanisms of axon guidance and its requirement for precise spatiotemporal control of gene expression have been extensively studied in the context of the development of spinal cord circuits. The axons of commissural neurons are initially attracted to the ventral midline. Upon crossing, they become repelled from the midline. One of the better understood mechanisms that underlie this process is mediated by the Roundabout (Robo) immunoglobulin transmembrane receptor family. Robo3, a member of this family, encodes two alternatively spliced isoforms, Robo3.1 and Robo3.2. These two isoforms differ by the presence of a retained intron in the latter, which introduces a PTC and makes Robo3.2 mRNA an NMD substrate (Chen et al., 2008). The commissural axons express primarily Robo3.1 until they reach the midline, after which Robo3.2 is expressed (Chen et al., 2008). Robo3.2 mRNAs are localized and enriched in axons but translationally repressed early in development when the axons migrate toward the midline (Colak et al., 2013). After crossing, Robo3.2 mRNAs are translationally activated. This induction of Robo3.2 promotes the activity of Robo1 and Robo2, which along with Robo3.2 mediate the repulsion from the midline (Chen et al., 2008). Importantly, the translationally activated Robo3.2 mRNAs are quickly recognized and downregulated by the NMD factors that are also localized in the growth cone. Conditional knockout of Upf2 in mouse commissural neurons leads to an increase in the Robo3.2 protein levels and aberrant post-crossing behaviors (Colak et al., 2013), providing a compelling example of NMD ensuring the precision of gene expression in developing neurons at a subcellular level.
Fully differentiated mature neurons also employ posttranscriptional mechanisms to regulate gene expression in response to various stimuli, which plays important roles in neural plasticity and homeostasis. As previously mentioned, in vivo suppression of NMD causes synaptic dysfunctions and behavioral deficits in learning and memory (Table 1). One of the well-known NMD targets, activity-regulated cytoskeleton-associated protein (Arc), is an immediate-early mRNA encoding a cytoskeletal protein that accumulates at dendritic spines and regulates synaptic plasticity (Link et al., 1995; Lyford et al., 1995; Steward et al., 1998). The transcription of Arc is rapidly induced upon stimulation, and at least a fraction of the newly synthesized Arc mRNAs are transported to dendrites near activated synapses (Steward et al., 1998). The rapid increase of Arc mRNAs in depolarized neurons is transient, not only because its transcription quickly shuts off, but also because Arc mRNAs, with two conserved introns in the 3ʹ UTR, are targeted by EJC-dependent NMD. The depletion of either UPF1 or EIF4A3 results in an increased stability of Arc mRNAs (Giorgi et al., 2007). Consistent with the translation-dependence of NMD, Arc mRNAs are stabilized by translation inhibition (Farris et al., 2014). The depletion of EIF4A3, an EJC factor that physically binds to Arc mRNAs, increases AMPA receptors in dendritic spines and excitatory synaptic transmission (Giorgi et al., 2007). A computational analysis has identified 152 transcripts with conserved 3ʹ UTR introns similar to Arc mRNAs, including a variety of synaptogenesis factors such as cadherins, neurexins, and neuregulins (Giorgi et al., 2007). Collectively, these studies clearly demonstrate the integral and pleiotropic roles of NMD in governing neuronal development and functions by shaping the neuronal transcriptome in a spatiotemporally precise manner.
NMD dysregulation in neurodegenerative diseases
Considering the compelling genetic evidence supporting the essential role of NMD in neural development and homeostasis, it is perhaps unsurprising that NMD dysregulation may contribute to the pathogenesis of neurodegenerative diseases. Most studies on the role of NMD in neurodegeneration have focused on amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD, also known as frontotemporal lobal degeneration or FTLD). Although these diseases affect different neuronal types (upper and lower motor neurons in ALS, cortical neurons in FTD), they are now considered to form a continuous disease spectrum. Multiple lines of evidence indicate that RNA misprocessing plays an important role in the pathophysiology of ALS/FTD (Cook and Petrucelli, 2019; Abramzon et al., 2020). First, an overwhelming majority (>90%) of ALS cases exhibit the nuclear depletion and cytoplasmic aggregation of an RNA-binding protein (RBP) called transactive response DNA-binding protein 43 (TDP-43), with the exception of a small fraction of cases showing aggregates of superoxide dismutase 1 (SOD1) or fused in sarcoma (FUS). Because both TDP-43 and FUS are essential RBPs for nuclear RNA processing, their mislocalization and aggregation are expected to globally impair mRNA biogenesis, translation, and stability. Indeed, depletion of wild-type TDP-43 has been shown to activate widespread cryptic splicing events, which would generate aberrant mRNAs presumably targeted by NMD (Ling et al., 2015; Tan et al., 2016; Humphrey et al., 2017). In addition, global differences in RNA turnover have been identified between control and ALS patient-derived cells (Tank et al., 2018). Last but not least, mutations in genes encoding several RBPs, including TDP-43 and FUS, have been identified in familial ALS/FTD (Cook and Petrucelli, 2019; Abramzon et al., 2020), further establishing the causal role of RNA dysregulation in pathogenesis.
Although mutations in NMD factor genes have not yet been found to directly cause ALS or FTD, recent studies have shown that NMD may modify pathogenesis and/or disease progression in animal or cellular models. Early evidence suggesting a possible role of NMD in ALS has come from yeast genetic screens, in which both human UPF1 and a yeast homolog, ECM32, have been found to suppress the toxicity caused by overexpressed and mislocalized FUS (Ju et al., 2011). Later on, UPF1 overexpression has been shown to enhance the survival of mouse primary neurons expressing ALS-associated TDP-43 and FUS mutants but have little effect on polyglutamine- or mutant SOD1-associated toxicity (Barmada et al., 2015). The neuroprotective phenotype of UPF1 overexpression requires its helicase activity and can be phenocopied by UPF2 (Barmada et al., 2015), both suggesting that the phenotype is likely mediated by NMD. In a TDP-43-based rat paralysis model, UPF1 overexpression improves overall motor functions (Jackson et al., 2015), further suggesting a conserved and robust role of UPF1 in suppressing the neurotoxicity of mutant RBPs.
In a separate study, mutant FUS inclusions have been shown to enrich proteins involved in translation (eIF3, eIF4A, eIF4G, rpS6) and mRNA surveillance/decay (eIF4A3, UPF1, UPF3b, XRN1) (Kamelgarn et al., 2018). Consistent with the sequestration of translation factors by FUS inclusions, global translation is impaired in cells expressing FUS mutants. Paradoxically, a few tested NMD targets are less stable in these cells, which the authors attribute to the elevated expression levels of NMD factors including UPF1 and UPF3B (Kamelgarn et al., 2018). Whether NMD is indeed hyperactive on a transcriptome-wide scale, and if so, how NMD can proceed with NMD factors sequestered within FUS inclusions and with translation globally impaired remain open questions.
About 40% of familial ALS/FTD cases are caused by a (GGGGCC)N hexanucleotide repeat expansion within the first intron of the C9orf72 gene. The expanded repeats are bidirectionally transcribed into sense and antisense repeat-containing RNAs, both of which are translated in all three reading frames into six distinct dipeptide repeat (DPR) proteins: sense polyGA, sense polyGP, sense polyGR, antisense polyPA, antisense polyPG, and antisense polyPR, via a poorly understood process known as repeat-associated non-AUG (RAN) translation (Balendra and Isaacs, 2018). Among them, the arginine-rich DPRs (R-DPRs) are the most toxic species as they interfere with a wide range of cellular processes including nucleocytoplasmic transport (Zhang et al., 2015), mRNA translation (Zhang et al., 2018), and the dynamics of phase-separated organelles such as nucleoli and stress granules (Lee et al., 2016). One of the hallmarks of C9orf72 repeat expansion-associated ALS/FTD (C9ALS/FTD) is the high abundance of aberrant transcripts (Prudencio et al., 2015), which had previously been attributed to splicing changes caused by the repeat RNA sequestration of splicing factors (Conlon et al., 2016). Through bioinformatic analyses comparing the postmortem brain transcriptome profiles between control, C9-negative sporadic ALS, and C9ALS cases, we and others have found that NMD target mRNAs specifically accumulate in C9ALS brains (Xu et al., 2019; Sun et al., 2020). The accumulated NMD targets include intron-retaining mRNAs, physiological mRNA targets, as well as NMD factor mRNAs, suggesting global, rather than target-specific, dysregulation of NMD. Notably, changes in RNA decay in C9ALS are not limited to NMD but also affect histone mRNAs, a unique set of non-polyadenylated mRNAs that are stabilized by 3ʹ end stem-loop structures and degraded in a translation- and UPF1-dependent manner (Sun et al., 2020).
Using heterologous systems, both studies have found that R-DPRs are sufficient to recapitulate the NMD deficits observed in C9ALS cases (Xu et al., 2019; Sun et al., 2020), but distinct models have been proposed for the underlying mechanism. Xu et al. (2019) have shown that cells expressing R-DPRs had more stress granules and fewer P bodies. While we have observed similar effects of R-DPRs on stress granule formation and the recruitment of UPF1 to stress granules, multiple lines of evidence argue against stress granule formation being the cause of NMD deficits (Sun et al., 2020). Most importantly, disrupting stress granule formation either genetically by knocking out G3BP1 and G3BP2, two essential stress granule proteins, or pharmacologically by inhibiting the integrated stress response pathway has little effect on the NMD deficits induced by R-DPRs (Sun et al., 2020). Instead, by comparing the dose‒response relationships between R-DPR and cycloheximide, we have shown that the effect of R-DPRs on NMD can be largely explained by their negative impact on global translation (Sun et al., 2020).
Converging evidence supporting a pathophysiological effect of NMD dysregulation has come from another study focusing on protein mislocalization due to nucleocytoplasmic transport defects (Ortega et al., 2020), another pathological hallmark of C9ALS/FTD (Zhang et al., 2015). Using semiquantitative mass spectrometry, Ortega et al. (2020) have identified 126 proteins with altered nucleocytoplasmic ratios in cells ectopically expressing the GGGGCC repeat RNA. This list includes eRF1, which is partially mislocalized in the nuclear envelop invaginations in C9ALS motor neurons (Ortega et al., 2020). As mentioned previously, eRF1 forms part of the SURF complex at PTCs and plays a key role in triggering NMD. In C9ALS motor neurons, SMG1 is more highly expressed and UPF1 is more phosphorylated, leading to the interpretation that NMD may be hyperactive in these cells (Ortega et al., 2020). Although this interpretation may seem at odds with the above-mentioned C9ALS transcriptome analyses (Xu et al., 2019; Sun et al., 2020), UPF1 hyperphosphorylation may reflect the higher abundance of NMD targets in these cells. Hyperphosphorylation of UPF1 could also be a result of global translation inhibition by R-DPRs in C9ALS neurons (Sun et al., 2020), in a manner similar to cycloheximide-induced translation inhibition (Dang et al., 2009). Importantly, this study suggests that the GGGGCC repeat-containing C9orf72 RNA is an NMD target, as evidenced by an increase in cytoplasmic repeat RNA foci and the abundance of C9orf72 intron-retaining RNA upon eRF1 or UPF1 depletion (Ortega et al., 2020). This finding is significant, because it points to a vicious cycle in which NMD dysregulation allows C9orf72 intron-retaining RNAs to further accumulate in the cytoplasm, produce more R-DPRs, and further impair NMD (Figure 2). The accumulation of NMD targets in the cytoplasm may also in turn exacerbate the NMD deficits by overloading the already reduced NMD capacity in neurons.
Figure 2.

NMD inhibition by C9orf72 R-DPRs drives a positive feedback loop. In normal neurons, cytoplasmic intron-retaining mRNAs are efficiently detected and degraded by NMD. In neurons carrying the C9orf72 hexanucleotide repeat expansion, however, RAN translation of the repeat intron-retaining C9orf72 mRNAs produces R-DPRs, which inhibits global translation and NMD. This global NMD deficit allows more aberrant RNAs, presumably including the repeat-containing C9orf72 mRNA, to accumulate in cytoplasm. Excessive aberrant RNAs may also in turn overload the already reduced NMD capacity in C9ALS/FTD neurons.
Remarkably similar to TDP-43 and FUS models, a neuroprotective phenotype of NMD factor overexpression has been observed in multiple experimental C9ALS/FTD models. Xu et al. (2019) have shown that overexpression of UPF1 and, to a lesser extent, UPF2 can reduce the toxicity of R-DPRs in SH-SY5Y neuroblastoma cells and prolong the life span in Drosophila expressing R-DPRs. Ortega et al. (2020) have shown that overexpression of eRF1 and UPF1 can rescue the eye degeneration phenotype in Drosophila expressing the GGGGCC repeat RNA, which presumably produces some low level of R-DPRs via RAN translation. Lastly, we have found that overexpression of UPF1, but none of its NMD-deficient mutants, increases the survival of mouse primary cortical neurons treated with polyPR peptides (Sun et al., 2020).
While the neuroprotective effects of UPF1 overexpression have been repeatedly shown in a variety of ALS models, whether they all share a common mechanism remains unclear. On one hand, multiple studies have shown that the protective effect of UPF1 requires its helicase activity (Barmada et al., 2015; Sun et al., 2020), suggesting some mechanistic overlap between NMD and neuroprotection. On the other hand, most of these studies have not directly measured NMD activity in UPF1-overexpressing cells, leaving it unclear to what extent NMD function is restored by the overexpression of UPF1. Furthermore, non-NMD functions of UPF1, some of which also requires its helicase activity (Kim and Maquat, 2019), may also be involved. Future studies that comprehensively characterize and compare the transcriptome-wide impact of UPF1 overexpression in each model should provide important insight into the mechanism(s) underlying the observed neuroprotective phenotypes.
Concluding remark
Since the identification of NMD more than three decades ago, a tremendous amount of progress has been made in understanding the key molecular components and their interactions that collectively orchestrate this ancient and essential mRNA surveillance mechanism. Human genetics studies of diseases caused by variations in NMD factor genes, together with reverse genetics studies in animal models, have raised new and important questions on the in vivo functions and regulation of NMD. Going forward, it will be critical to systematically understand the scope of NMD substrates and their regulation in the native, organismal context and in a cell-type-specific manner. For both developmental and neurodegenerative diseases associated with NMD deficits, key questions remain whether the distinct pathological phenotypes may be due to the dysregulation of a small subset of physiological targets or global nonsense mRNA surveillance. Considering the convergent evidence of NMD factors being disease drivers and modifiers, pharmaceutical strategies to enhance NMD functions may hold promise in restoring homeostasis.
Acknowledgements
We thank members of the Guo Lab for discussions.
Funding
Our work is supported by an NIH New Innovator Award (DP2 GM132930) and the Muscular Dystrophy Association (MDA602934). J.U.G. is an NARSAD Young Investigator and a Klingenstein−Simons Fellow in Neuroscience.
Conflict of interest: none declared.
References
- Abramzon Y.A., Fratta P., Traynor B.J., et al. (2020). The overlapping genetics of amyotrophic lateral sclerosis and frontotemporal dementia. Front. Neurosci. 14, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Addington A.M., Gauthier J., Piton A., et al. (2011). A novel frameshift mutation in UPF3B identified in brothers affected with childhood onset schizophrenia and autism spectrum disorders. Mol. Psychiatry 16, 238–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal V., Bell G.W., Nam J.W., et al. (2015). Predicting effective microRNA target sites in mammalian mRNAs. eLife 4, e05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alachkar A., Jiang D., Harrison M., et al. (2013). An EJC factor RBM8a regulates anxiety behaviors. Curr. Mol. Med. 13, 887–899. [DOI] [PubMed] [Google Scholar]
- Alexandrov A., Shu M.D., Steitz J.A. (2017). Fluorescence amplification method for forward genetic discovery of factors in human mRNA degradation. Mol. Cell 65, 191–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amrani N., Ganesan R., Kervestin S., et al. (2004). A faux 3′-UTR promotes aberrant termination and triggers nonsense-mediated mRNA decay. Nature 432, 112–118. [DOI] [PubMed] [Google Scholar]
- Arribere J.A., Fire A.Z. (2018). Nonsense mRNA suppression via nonstop decay. eLife 7, e33292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avery P., Vicente-Crespo M., Francis D., et al. (2011). Drosophila Upf1 and Upf2 loss of function inhibits cell growth and causes animal death in a Upf3-independent manner. RNA 17, 624–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balendra R., Isaacs A.M. (2018). C9orf72-mediated ALS and FTD: multiple pathways to disease. Nat. Rev. Neurol. 14, 544–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barmada S.J., Ju S., Arjun A., et al. (2015). Amelioration of toxicity in neuronal models of amyotrophic lateral sclerosis by hUPF1. Proc. Natl Acad. Sci. USA 112, 7821–7826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel D.P. (2018). Metazoan microRNAs. Cell 173, 20–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunetti-Pierri N., Berg J.S., Scaglia F., et al. (2008). Recurrent reciprocal 1q21.1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities. Nat. Genet. 40, 1466–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno I.G., Karam R., Huang L., et al. (2011). Identification of a microRNA that activates gene expression by repressing nonsense-mediated RNA decay. Mol. Cell 42, 500–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhler M., Steiner S., Mohn F., et al. (2006). EJC-independent degradation of nonsense immunoglobulin-mu mRNA depends on 3′ UTR length. Nat. Struct. Mol. Biol. 13, 462–464. [DOI] [PubMed] [Google Scholar]
- Cali B.M., Kuchma S.L., Latham J., et al. (1999). smg-7 is required for mRNA surveillance in Caenorhabditis elegans. Genetics 151, 605–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamieh H., Ballut L., Bonneau F., et al. (2008). NMD factors UPF2 and UPF3 bridge UPF1 to the exon junction complex and stimulate its RNA helicase activity. Nat. Struct. Mol. Biol. 15, 85–93. [DOI] [PubMed] [Google Scholar]
- Chan W.K., Bhalla A.D., Le Hir H., et al. (2009). A UPF3-mediated regulatory switch that maintains RNA surveillance. Nat. Struct. Mol. Biol. 16, 747–753. [DOI] [PubMed] [Google Scholar]
- Chen Z., Gore B.B., Long H., et al. (2008). Alternative splicing of the Robo3 axon guidance receptor governs the midline switch from attraction to repulsion. Neuron 58, 325–332. [DOI] [PubMed] [Google Scholar]
- Colak D., Ji S.J., Porse B.T., et al. (2013). Regulation of axon guidance by compartmentalized nonsense-mediated mRNA decay. Cell 153, 1252–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conlon E.G., Lu L., Sharma A., et al. (2016). The C9ORF72 GGGGCC expansion forms RNA G-quadruplex inclusions and sequesters hnRNP H to disrupt splicing in ALS brains. eLife 5, e17820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook C., Petrucelli L. (2019). Genetic convergence brings clarity to the enigmatic red line in ALS. Neuron 101, 1057–1069. [DOI] [PubMed] [Google Scholar]
- Cougot N., Babajko S., Seraphin B. (2004). Cytoplasmic foci are sites of mRNA decay in human cells. J. Cell Biol. 165, 31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang Y., Low W.K., Xu J., et al. (2009). Inhibition of nonsense-mediated mRNA decay by the natural product pateamine A through eukaryotic initiation factor 4AIII. J. Biol. Chem. 284, 23613–23621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dostie J., Dreyfuss G. (2002). Translation is required to remove Y14 from mRNAs in the cytoplasm. Curr. Biol. 12, 1060–1067. [DOI] [PubMed] [Google Scholar]
- Durand S., Cougot N., Mahuteau-Betzer F., et al. (2007). Inhibition of nonsense-mediated mRNA decay (NMD) by a new chemical molecule reveals the dynamic of NMD factors in P-bodies. J. Cell Biol. 178, 1145–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberle A.B., Lykke-Andersen S., Muhlemann O., et al. (2009). SMG6 promotes endonucleolytic cleavage of nonsense mRNA in human cells. Nat. Struct. Mol. Biol. 16, 49–55. [DOI] [PubMed] [Google Scholar]
- Edbauer D., Neilson J.R., Foster K.A., et al. (2010). Regulation of synaptic structure and function by FMRP-associated microRNAs miR-125b and miR-132. Neuron 65, 373–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eulalio A., Behm-Ansmant I., Schweizer D., et al. (2007). P-body formation is a consequence, not the cause, of RNA-mediated gene silencing. Mol. Cell. Biol. 27, 3970–3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farris S., Lewandowski G., Cox C.D., et al. (2014). Selective localization of arc mRNA in dendrites involves activity- and translation-dependent mRNA degradation. J. Neurosci. 34, 4481–4493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frizzell K.A., Rynearson S.G., Metzstein M.M. (2012). Drosophila mutants show NMD pathway activity is reduced, but not eliminated, in the absence of Smg6. RNA 18, 1475–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatfield D., Izaurralde E. (2004). Nonsense-mediated messenger RNA decay is initiated by endonucleolytic cleavage in Drosophila. Nature 429, 575–578. [DOI] [PubMed] [Google Scholar]
- Gehring N.H., Kunz J.B., Neu-Yilik G., et al. (2005). Exon-junction complex components specify distinct routes of nonsense-mediated mRNA decay with differential cofactor requirements. Mol. Cell 20, 65–75. [DOI] [PubMed] [Google Scholar]
- Giorgi C., Yeo G.W., Stone M.E., et al. (2007). The EJC factor eIF4AIII modulates synaptic strength and neuronal protein expression. Cell 130, 179–191. [DOI] [PubMed] [Google Scholar]
- Glavan F., Behm-Ansmant I., Izaurralde E., et al. (2006). Structures of the PIN domains of SMG6 and SMG5 reveal a nuclease within the mRNA surveillance complex. EMBO J. 25, 5117–5125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harigaya Y., Parker R. (2010). No-go decay: a quality control mechanism for RNA in translation. Wiley Interdiscip. Rev. RNA 1, 132–141. [DOI] [PubMed] [Google Scholar]
- Hashimoto Y., Takahashi M., Sakota E., et al. (2017). Nonstop-mRNA decay machinery is involved in the clearance of mRNA 5′-fragments produced by RNAi and NMD in Drosophila melanogaster cells. Biochem. Biophys. Res. Commun. 484, 1–7. [DOI] [PubMed] [Google Scholar]
- Hodgkin J., Papp A., Pulak R., et al. (1989). A new kind of informational suppression in the nematode Caenorhabditis elegans. Genetics 123, 301–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogg J.R., Goff S.P. (2010). Upf1 senses 3′UTR length to potentiate mRNA decay. Cell 143, 379–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvathova I., Voigt F., Kotrys A.V., et al. (2017). The dynamics of mRNA turnover revealed by single-molecule imaging in single cells. Mol. Cell 68, 615–625.e9. [DOI] [PubMed] [Google Scholar]
- Huang L., Shum E.Y., Jones S.H., et al. (2018). A Upf3b-mutant mouse model with behavioral and neurogenesis defects. Mol. Psychiatry 23, 1773–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey J., Emmett W., Fratta P., et al. (2017). Quantitative analysis of cryptic splicing associated with TDP-43 depletion. BMC Med. Genomics 10, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntzinger E., Kashima I., Fauser M., et al. (2008). SMG6 is the catalytic endonuclease that cleaves mRNAs containing nonsense codons in metazoan. RNA 14, 2609–2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurt J.A., Robertson A.D., Burge C.B. (2013). Global analyses of UPF1 binding and function reveal expanded scope of nonsense-mediated mRNA decay. Genome Res. 23, 1636–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isken O., Kim Y.K., Hosoda N., et al. (2008). Upf1 phosphorylation triggers translational repression during nonsense-mediated mRNA decay. Cell 133, 314–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isken O., Maquat L.E. (2008). The multiple lives of NMD factors: balancing roles in gene and genome regulation. Nat. Rev. Genet. 9, 699–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito-Harashima S., Kuroha K., Tatematsu T., et al. (2007). Translation of the poly(A) tail plays crucial roles in nonstop mRNA surveillance via translation repression and protein destabilization by proteasome in yeast. Genes Dev. 21, 519–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov P.V., Gehring N.H., Kunz J.B., et al. (2008). Interactions between UPF1, eRFs, PABP and the exon junction complex suggest an integrated model for mammalian NMD pathways. EMBO J. 27, 736–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson K.L., Dayton R.D., Orchard E.A., et al. (2015). Preservation of forelimb function by UPF1 gene therapy in a rat model of TDP-43-induced motor paralysis. Gene Ther. 22, 20–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson J.L., Stoica L., Liu Y., et al. (2019). Inhibition of Upf2-dependent nonsense-mediated decay leads to behavioral and neurophysiological abnormalities by activating the immune response. Neuron 104, 665–679.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju S., Tardiff D.F., Han H., et al. (2011). A yeast model of FUS/TLS-dependent cytotoxicity. PLoS Biol. 9, e1001052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamelgarn M., Chen J., Kuang L., et al. (2018). ALS mutations of FUS suppress protein translation and disrupt the regulation of nonsense-mediated decay. Proc. Natl Acad. Sci. USA 115, E11904–E11913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karousis E.D., Gurzeler L.A., Annibaldis G., et al. (2020). Human NMD ensues independently of stable ribosome stalling. Nat. Commun. 11, 4134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashima I., Yamashita A., Izumi N., et al. (2006). Binding of a novel SMG-1‒Upf1‒eRF1‒eRF3 complex (SURF) to the exon junction complex triggers Upf1 phosphorylation and nonsense-mediated mRNA decay. Genes Dev. 20, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedersha N., Stoecklin G., Ayodele M., et al. (2005). Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. J. Cell Biol. 169, 871–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren H., Lev-Maor G., Ast G. (2010). Alternative splicing and evolution: diversification, exon definition and function. Nat. Rev. Genet. 11, 345–355. [DOI] [PubMed] [Google Scholar]
- Kim Y.K., Maquat L.E. (2019). UPFront and center in RNA decay: UPF1 in nonsense-mediated mRNA decay and beyond. RNA 25, 407–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klauer A.A., van Hoof A. (2012). Degradation of mRNAs that lack a stop codon: a decade of nonstop progress. Wiley Interdiscip. Rev. RNA 3, 649–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koritzinsky M., Magagnin M.G., van den Beucken T., et al. (2006). Gene expression during acute and prolonged hypoxia is regulated by distinct mechanisms of translational control. EMBO J. 25, 1114–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurosaki T., Maquat L.E. (2016). Nonsense-mediated mRNA decay in humans at a glance. J. Cell Sci. 129, 461–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Hir H., Gatfield D., Izaurralde E., et al. (2001). The exon–exon junction complex provides a binding platform for factors involved in mRNA export and nonsense-mediated mRNA decay. EMBO J. 20, 4987–4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K.H., Zhang P., Kim H.J., et al. (2016). C9orf72 dipeptide repeats impair the assembly, dynamics, and function of membrane-less organelles. Cell 167, 774–788.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T., Shi Y., Wang P., et al. (2015). Smg6/Est1 licenses embryonic stem cell differentiation via nonsense-mediated mRNA decay. EMBO J. 34, 1630–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling J.P., Pletnikova O., Troncoso J.C., et al. (2015). TDP-43 repression of nonconserved cryptic exons is compromised in ALS-FTD. Science 349, 650–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link W., Konietzko U., Kauselmann G., et al. (1995). Somatodendritic expression of an immediate early gene is regulated by synaptic activity. Proc. Natl Acad. Sci. USA 92, 5734–5738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long A.A., Mahapatra C.T., Woodruff E.A., 3rd, et al. (2010). The nonsense-mediated decay pathway maintains synapse architecture and synaptic vesicle cycle efficacy. J. Cell Sci. 123, 3303–3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Perrote A., Castano R., Melero R., et al. (2016). Human nonsense-mediated mRNA decay factor UPF2 interacts directly with eRF3 and the SURF complex. Nucleic Acids Res. 44, 1909–1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou C.H., Shao A., Shum E.Y., et al. (2014). Posttranscriptional control of the stem cell and neurogenic programs by the nonsense-mediated RNA decay pathway. Cell Rep. 6, 748–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig N., Leidinger P., Becker K., et al. (2016). Distribution of miRNA expression across human tissues. Nucleic Acids Res. 44, 3865–3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyford G.L., Yamagata K., Kaufmann W.E., et al. (1995). Arc, a growth factor and activity-regulated gene, encodes a novel cytoskeleton-associated protein that is enriched in neuronal dendrites. Neuron 14, 433–445. [DOI] [PubMed] [Google Scholar]
- Lykke-Andersen J., Shu M.D., Steitz J.A. (2001). Communication of the position of exon–exon junctions to the mRNA surveillance machinery by the protein RNPS1. Science 293, 1836–1839. [DOI] [PubMed] [Google Scholar]
- Mao H., McMahon J.J., Tsai Y.H., et al. (2016). Haploinsufficiency for core exon junction complex components disrupts embryonic neurogenesis and causes p53-mediated microcephaly. PLoS Genet. 12, e1006282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao H., Pilaz L.J., McMahon J.J., et al. (2015). Rbm8a haploinsufficiency disrupts embryonic cortical development resulting in microcephaly. J. Neurosci. 35, 7003–7018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markmiller S., Soltanieh S., Server K.L., et al. (2018). Context-dependent and disease-specific diversity in protein interactions within stress granules. Cell 172, 590–604.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin C.L., Duvall J.A., Ilkin Y., et al. (2007). Cytogenetic and molecular characterization of A2BP1/FOX1 as a candidate gene for autism. Am. J. Med. Genet. B Neuropsychiatr. Genet. 144B, 869–876. [DOI] [PubMed] [Google Scholar]
- McIlwain D.R., Pan Q., Reilly P.T., et al. (2010). Smg1 is required for embryogenesis and regulates diverse genes via alternative splicing coupled to nonsense-mediated mRNA decay. Proc. Natl Acad. Sci. USA 107, 12186–12191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medghalchi S.M., Frischmeyer P.A., Mendell J.T., et al. (2001). Rent1, a trans-effector of nonsense-mediated mRNA decay, is essential for mammalian embryonic viability. Hum. Mol. Genet. 10, 99–105. [DOI] [PubMed] [Google Scholar]
- Metzstein M.M., Krasnow M.A. (2006). Functions of the nonsense-mediated mRNA decay pathway in Drosophila development. PLoS Genet. 2, e180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhlrad D., Parker R. (1999). Aberrant mRNAs with extended 3′ UTRs are substrates for rapid degradation by mRNA surveillance. RNA 5, 1299–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson J.O., Forster D., Frizzell K.A., et al. (2018). Multiple nonsense-mediated mRNA processes require Smg5 in Drosophila. Genetics 209, 1073–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson J.O., Moore K.A., Chapin A., et al. (2016). Degradation of Gadd45 mRNA by nonsense-mediated decay is essential for viability. eLife 5, e12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neu-Yilik G., Raimondeau E., Eliseev B., et al. (2017). Dual function of UPF3B in early and late translation termination. EMBO J. 36, 2968–2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen L.S., Kim H.G., Rosenfeld J.A., et al. (2013). Contribution of copy number variants involving nonsense-mediated mRNA decay pathway genes to neuro-developmental disorders. Hum. Mol. Genet. 22, 1816–1825. [DOI] [PubMed] [Google Scholar]
- Nickless A., Bailis J.M., You Z. (2017). Control of gene expression through the nonsense-mediated RNA decay pathway. Cell Biosci. 7, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira C.C., McCarthy J.E. (1995). The relationship between eukaryotic translation and mRNA stability. A short upstream open reading frame strongly inhibits translational initiation and greatly accelerates mRNA degradation in the yeast Saccharomyces cerevisiae. J. Biol. Chem. 270, 8936–8943. [DOI] [PubMed] [Google Scholar]
- Ortega J.A., Daley E.L., Kour S., et al. (2020). Nucleocytoplasmic proteomic analysis uncovers eRF1 and nonsense-mediated decay as modifiers of ALS/FTD C9orf72 toxicity. Neuron 106, 90–107.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker R., Sheth U. (2007). P bodies and the control of mRNA translation and degradation. Mol. Cell 25, 635–646. [DOI] [PubMed] [Google Scholar]
- Prudencio M., Belzil V.V., Batra R., et al. (2015). Distinct brain transcriptome profiles in C9orf72-associated and sporadic ALS. Nat. Neurosci. 18, 1175–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulak R., Anderson P. (1993). mRNA surveillance by the Caenorhabditis elegans smg genes. Genes Dev. 7, 1885–1897. [DOI] [PubMed] [Google Scholar]
- Ruiz-Echevarría M.J., Peltz S.W. (2000). The RNA binding protein Pub1 modulates the stability of transcripts containing upstream open reading frames. Cell 101, 741–751. [DOI] [PubMed] [Google Scholar]
- Sartor F., Anderson J., McCaig C., et al. (2015). Mutation of genes controlling mRNA metabolism and protein synthesis predisposes to neurodevelopmental disorders. Biochem. Soc. Trans. 43, 1259–1265. [DOI] [PubMed] [Google Scholar]
- Sebat J., Lakshmi B., Malhotra D., et al. (2007). Strong association of de novo copy number mutations with autism. Science 316, 445–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaheen R., Anazi S., Ben-Omran T., et al. (2016). Mutations in SMG9, encoding an essential component of nonsense-mediated decay machinery, cause a multiple congenital anomaly syndrome in humans and mice. Am. J. Hum. Genet. 98, 643–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheth U., Parker R. (2003). Decapping and decay of messenger RNA occur in cytoplasmic processing bodies. Science 300, 805–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheth U., Parker R. (2006). Targeting of aberrant mRNAs to cytoplasmic processing bodies. Cell 125, 1095–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker C.J., Green R. (2012). Translation drives mRNA quality control. Nat. Struct. Mol. Biol. 19, 594–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shum E.Y., Jones S.H., Shao A., et al. (2016). The antagonistic gene paralogs Upf3a and Upf3b govern nonsense-mediated RNA decay. Cell 165, 382–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stalder L., Muhlemann O. (2009). Processing bodies are not required for mammalian nonsense-mediated mRNA decay. RNA 15, 1265–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steward O., Wallace C.S., Lyford G.L., et al. (1998). Synaptic activation causes the mRNA for the IEG Arc to localize selectively near activated postsynaptic sites on dendrites. Neuron 21, 741–751. [DOI] [PubMed] [Google Scholar]
- Sun Y., Eshov A., Zhou J., et al. (2020). C9orf72 arginine-rich dipeptide repeats inhibit UPF1-mediated RNA decay via translational repression. Nat. Commun. 11, 3354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szyszka P., Sharp S.I., Dedman A., et al. (2012). A nonconservative amino acid change in the UPF3B gene in a patient with schizophrenia. Psychiatr. Genet. 22, 150–151. [DOI] [PubMed] [Google Scholar]
- Tan Q., Yalamanchili H.K., Park J., et al. (2016). Extensive cryptic splicing upon loss of RBM17 and TDP43 in neurodegeneration models. Hum. Mol. Genet. 25, 5083–5093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tank E.M., Figueroa-Romero C., Hinder L.M., et al. (2018). Abnormal RNA stability in amyotrophic lateral sclerosis. Nat. Commun. 9, 2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarpey P.S., Raymond F.L., Nguyen L.S., et al. (2007). Mutations in UPF3B, a member of the nonsense-mediated mRNA decay complex, cause syndromic and nonsyndromic mental retardation. Nat. Genet. 39, 1127–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira D., Sheth U., Valencia-Sanchez M.A., et al. (2005). Processing bodies require RNA for assembly and contain nontranslating mRNAs. RNA 11, 371–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thermann R., Neu-Yilik G., Deters A., et al. (1998). Binary specification of nonsense codons by splicing and cytoplasmic translation. EMBO J. 17, 3484–3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoren L.A., Norgaard G.A., Weischenfeldt J., et al. (2010). UPF2 is a critical regulator of liver development, function and regeneration. PLoS One 5, e11650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unterholzner L., Izaurralde E. (2004). SMG7 acts as a molecular link between mRNA surveillance and mRNA decay. Mol. Cell 16, 587–596. [DOI] [PubMed] [Google Scholar]
- Wang G., Jiang B., Jia C., et al. (2013). MicroRNA 125 represses nonsense-mediated mRNA decay by regulating SMG1 expression. Biochem. Biophys. Res. Commun. 435, 16–20. [DOI] [PubMed] [Google Scholar]
- Weischenfeldt J., Damgaard I., Bryder D., et al. (2008). NMD is essential for hematopoietic stem and progenitor cells and for eliminating by-products of programmed DNA rearrangements. Genes Dev. 22, 1381–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson M.A., Meaux S., van Hoof A. (2007). A genomic screen in yeast reveals novel aspects of nonstop mRNA metabolism. Genetics 177, 773–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittkopp N., Huntzinger E., Weiler C., et al. (2009). Nonsense-mediated mRNA decay effectors are essential for zebrafish embryonic development and survival. Mol. Cell. Biol. 29, 3517–3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W., Bao P., Jiang X., et al. (2019). Reactivation of nonsense-mediated mRNA decay protects against C9orf72 dipeptide-repeat neurotoxicity. Brain 142, 1349–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Sun X., Qian Y., et al. (1998). Intron function in the nonsense-mediated decay of β-globin mRNA: indications that pre-mRNA splicing in the nucleus can influence mRNA translation in the cytoplasm. RNA 4, 801–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K., Donnelly C.J., Haeusler A.R., et al. (2015). The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 525, 56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.J., Gendron T.F., Ebbert M.T.W., et al. (2018). Poly(GR) impairs protein translation and stress granule dynamics in C9orf72-associated frontotemporal dementia and amyotrophic lateral sclerosis. Nat. Med. 24, 1136–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W.W. (2013). Intragenic deletion of RBFOX1 associated with neurodevelopmental/neuropsychiatric disorders and possibly other clinical presentations. Mol. Cytogenet. 6, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X., Zhang H., Mendell J.T. (2020). Ribosome recycling by ABCE1 links lysosomal function and iron homeostasis to 3′ UTR-directed regulation and nonsense-mediated decay. Cell Rep. 32, 107895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zund D., Gruber A.R., Zavolan M., et al. (2013). Translation-dependent displacement of UPF1 from coding sequences causes its enrichment in 3′UTRs. Nat. Struct. Mol. Biol. 20, 936–943. [DOI] [PubMed] [Google Scholar]