

Abstract
The discovery of the FOXP2 transcription factor, and its implication in a rare severe human speech and language disorder, has led to two decades of empirical studies focused on uncovering its roles in the brain using a range of in vitro and in vivo methods. Here, we discuss what we have learned about the regulation of FOXP2, its downstream effectors, and its modes of action as a transcription factor in brain development and function, providing an integrated overview of what is currently known about the critical molecular networks.
Keywords: FOXP2, molecular network, neurodevelopment, speech disorder, transcription factor
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; Neuroscience
FOXP2 was discovered 20 years ago to be required for human speech and language development. This review discusses molecular networks of FOXP2 regulation, downstream effectors, and how it acts as a transcription factor in brain development and function.
Glossary
- ADHD
attention‐deficit/hyperactivity disorder
- BCL11B
B‐cell lymphoma/leukemia 11B
- BRET
bioluminescence resonance energy transfer
- CASK
calcium/calmodulin‐dependent serine protein kinase 3
- CHD
chromodomain‐helicase‐DNA‐binding protein
- ChIP
chromatin immunoprecipitation
- CNTNAP2/CASPR2
contactin‐associated protein‐like 2
- CTBP
C‐terminal‐binding protein
- DISC1
disrupted in schizophrenia 1
- FOXP
forkhead box/winged‐helix protein
- GATAD2B
GATA zinc finger domain‐containing 2B
- GRIN2A
glutamate ionotropic receptor NMDA type subunit 2A
- GSK3β
glycogen‐synthase kinase 3 beta
- HDAC
histone deacetylase
- Int.
protein interactors
- LZ
leucine zipper
- MET
MET proto‐oncogene, receptor tyrosine kinase
- MRI
magnetic resonance imaging
- MTA
metastasis‐associated protein
- NEDD9
neural precursor cell expressed developmentally downregulated protein 9
- NEUROD
neurogenic differentiation 1
- NFI
nuclear factor 1
- NGN2
neurogenin 2
- NR2F
nuclear receptor subfamily 2, group F
- NuRD
nucleosome remodeling and histone deacetylase
- PAX6
paired box protein 6
- pcw
post‐conception week
- PIAS
protein inhibitor of activated STAT
- POU3F2
POU class 3 homeobox 2
- PTM
post‐translational modification
- RAR
retinoic acid receptor
- RELN
reelin
- ROR
RAR‐related orphan receptor
- SATB
special AT‐rich binding protein
- SNP
single nucleotide polymorphism
- SOX5
SRY (sex determining region Y)‐box 5
- SRPX2
sushi repeat‐containing protein X‐linked 2
- SUMO
small ubiquitin‐like modifier
- TBR
T‐box, brain
- TCF/LEF
T‐cell factor/lymphoid enhancer‐binding factor
- TF
transcription factor
- VLDLR
very‐low‐density lipoprotein receptor
- WNT
wingless‐related MMTV integration site 1
- WNT3
wnt family member 3
- YY1
yin yang 1
- ZBTB20
zinc finger and BTB domain‐containing 20
- ZF
zinc finger
- ZMYM2
zinc finger MYM‐type protein 2
Introduction
FOXP2 was the first gene to be clearly linked to speech and language development. The initial finding was made through studies of a large multi‐generational family (the KE family) with a severe dominantly inherited developmental speech and language disorder (MIM #602081) (Lai et al, 2001). All fifteen affected family members carried a heterozygous missense mutation (p.R553H) disrupting FOXP2. In the two decades since then, additional cases of FOXP2‐related speech and language disorders have been discovered, both inherited and de novo (MacDermot et al, 2005; Feuk et al, 2006; Reuter et al, 2017), with childhood apraxia of speech (also called developmental verbal dyspraxia) as a core phenotypic feature, characterized by difficulties in coordinating sequences of articulatory movements underlying proficient speech. In a subset of individuals, broader phenotypes are observed including oral motor deficits, global developmental delays, and/or autism spectrum disorder (Morgan et al, 2016). Beyond the well‐documented consequences of rare highly penetrant genetic disruptions, studies have investigated contributions of common variation in FOXP2 to genetically complex traits. For example, some studies of small samples proposed that single nucleotide polymorphisms (SNPs) in the FOXP2 gene are associated with schizophrenia risk (Spaniel et al, 2011; Li et al, 2013; Rao et al, 2017), but there is little evidence of replication (Yin et al, 2018). Large‐scale systematic genome‐wide association studies have identified significant associations of intronic FOXP2 SNPs with several traits, including attention‐deficit/hyperactivity disorder (ADHD) (Demontis et al, 2019) and risk‐taking behaviors (Clifton et al, 2018). Although rare disruptions in FOXP2 have been associated with changes in brain activity (Liégeois et al, 2003) and structure (Watkins et al, 2002; Liégeois et al, 2016; Argyropoulos et al, 2019), common variation could not be linked to task‐based neural activations on language tasks (Uddén et al, 2019) or neuroanatomical differences between individuals (Hoogman et al, 2014).
FOXP2 belongs to the forkhead box/winged‐helix (FOX) family of proteins, a large group of transcription factors that share a highly conserved DNA‐binding domain of ˜ 80–100 amino acids, called the forkhead box (Weigel & Jackle, 1990; Hannenhalli & Kaestner, 2009) (following nomenclature guidelines, we use FOXP2 for humans, Foxp2 for mice, and FoxP2 for other species). There are 19 subclasses of FOX proteins, from FOXA to FOXS (Kaestner et al, 2000; Hannenhalli & Kaestner, 2009), with important roles in various biological processes, including cell differentiation, proliferation, and development (Hannenhalli & Kaestner, 2009; Zhang et al, 2017). Although they all share a characteristic DNA‐binding domain, different FOX proteins have distinct expression patterns and are involved in diverse mechanisms (Benayoun et al, 2011).
The FOXP subclass comprises four members, FOXP1–4 (Shu et al, 2001; Li et al, 2004). As well as the DNA‐binding domain, FOXP proteins share a zinc finger and leucine zipper motif (Fig 1A) (Wang et al, 2003; Li et al, 2004). Moreover, FOXP1, FOXP2, and FOXP4 contain long N‐terminal glutamine‐rich regions of unknown function (Wang et al, 2003; Li et al, 2004). A unique feature of the FOXP subclass is that they form homo‐ and heterodimers via the conserved leucine zipper, which appears essential for DNA binding and transcription regulation (Li et al, 2004). They may even form oligomer complexes, as detected for FoxP1, FoxP2, and FoxP4 in studies of zebra finch brain (Mendoza & Scharff, 2017). Formation of FOXP homo‐ and heterodimers in any particular tissue/cell type is likely mediated by expression and availability of the different FOXP proteins, providing potential for more complex regulation of downstream pathways.
Figure 1. FOXP expression in the brain.

(A) Schematic representation of the FOXP family of proteins. The polyglutamine‐rich region is shaded in light gray (Q‐rich), the zinc finger domain in light blue (ZF), the leucine zipper in regular gray (LZ), and the forkhead domain in dark gray (FOX). (B) Expression patterns of FOXP1, FOXP2, and FOXP4 in the brain, based on the developmental human RNA sequencing dataset of BrainSpan (http://www.brainspan.org/). (C) Expression patterns of FOXP2 in a selection of cortical regions. These regions were selected based on structural MRI studies with KE family members carrying a FOXP2 mutation (Vargha‐Khadem et al, 1998; Watkins et al, 2002; Belton et al, 2003): Gray matter differences were found in the cortical motor‐related areas, the inferior frontal gyrus and the superior temporal gyrus, among other regions. While the expression in the primary motor cortex (M1C) and the primary sensory cortex (S1C) peaks during development, the expression of FOXP2 in the superior temporal cortex (STC) and the ventromedial prefrontal cortex (VFC) seems to be maintained during adulthood. (B, C) Each individual dot represents a brain sample, and the lines are loess curves fitted through the data points. The dashed vertical line represents time of birth. Abbreviations for the analyzed brain regions are A1C, primary auditory cortex; CB, cerebellum; CBC, cerebellar cortex; DFC, dorsolateral prefrontal cortex; DTH, dorsal thalamic nucleus; HIP, hippocampus; IPC, inferior parietal cortex; ITC, inferior temporal cortex; M1C, primary motor cortex; MD, mediodorsal thalamic nucleus; MFC, medial frontal cortex; OFC, orbitofrontal; S1C, primary sensory cortex; STR, striatum; TC, superior temporal cortex; V1C, primary visual cortex; VFC, ventromedial prefrontal cortex. Other abbreviations are mos, months; pcw, post‐conception week.
While FOXP3 expression and function is largely limited to the immune system (Fontenot et al, 2003), FOXP1, FOXP2, and FOXP4 are expressed in various tissues throughout the body, including the brain, where they show distinctive, yet partially overlapping, expression patterns (human fetal and post‐natal expression of FOXP1, FOXP2, and FOXP4 based on BrainSpan expression data: Fig 1B and C. For a detailed review on the expression patterns of FOXP genes in the brain, see (Co et al, 2020)). FOXP1 expression is enriched in layers III‐IV of the cerebral cortex (Ferland et al, 2003; Hisaoka et al, 2010), as well as the thalamus, striatum, and CA1 subregion of the hippocampus (Ferland et al, 2003). Main sites of FOXP2 expression include layers IV‐VI of the cerebral cortex (Ferland et al, 2003; Lai et al, 2003; Campbell et al, 2009; Hisaoka et al, 2010), the striatum (Ferland et al, 2003; Lai et al, 2003; Campbell et al, 2009; Garcia‐Calero et al, 2016), the posterior and lateral thalamic nuclei (Ferland et al, 2003; Lai et al, 2003; Campbell et al, 2009), the Purkinje cells in the cerebellum (Lai et al, 2003; Campbell et al, 2009), and the inferior olive (Ferland et al, 2003; Lai et al, 2003; Campbell et al, 2009). FOXP4 has been less well studied than the other FOXP proteins, but is expressed in the subventricular zone, throughout the cortical plate and in the striatum during embryonic development (Takahashi et al, 2008), and in Purkinje cells (Tam et al, 2011).
The roles of FOXP2 have been investigated by studying its orthologues in an array of animal models. Mice that lack both alleles of Foxp2 have severe motor impairments, developmental delays, and typically die by post‐natal day 21 (Shu et al, 2005), while heterozygous animals show no obvious differences compared to wild‐type littermates, but display some altered vocal behaviors (Castellucci et al, 2016). Mice that are heterozygous for the mutation originally identified in the KE family display reduced motor‐skill learning (Groszer et al, 2008) and produce shorter sequences of ultrasonic vocalizations with less complex syntax (Chabout et al, 2016), as compared to wild‐type littermates. Foxp2 expression in the mouse cortex, striatum, and cerebellum modulates different aspects of motor function, as demonstrated by conditional homozygous knockouts targeting these structures (French et al, 2019). However, selective deletion of the gene in each of these brain regions does not significantly alter production of ultrasonic vocalizations (Urbanus et al, 2020). Interestingly, while selective deletion of Foxp2 in the mouse cortex does not appear to impact development of cortical structures during embryogenesis (Co et al, 2019; Kast et al, 2019), cortical‐specific knockouts are reported to nonetheless show altered social behaviors (Co et al, 2019; Medvedeva et al, 2019). When mouse Foxp2 is constitutively replaced by a partially humanized version, medium spiny neurons in the striatum show increases in dendrite length and synaptic plasticity (Enard et al, 2009), consistent with multiple studies implicating the gene in development and function of corticostriatal circuitry (Vernes et al, 2011; French et al, 2012; Chen et al, 2016; Hachigian et al, 2017; van Rhijn et al, 2018; French et al, 2019). Moreover, knockdown and overexpression studies in the brains of zebra finches suggest that avian FoxP2 is important not only in auditory‐guided vocal learning during development, but also for maintenance of vocal behaviors in adulthood (Haesler et al, 2004; Heston & White, 2015; Day et al, 2019; Norton et al, 2019; Xiao et al, 2021).
Notably, in humans, heterozygous disruptions of FOXP1 and FOXP4 have also been linked to neurodevelopmental disorders: an intellectual disability syndrome, frequently accompanied with autistic features and language impairment (MIM #613670) (Hamdan et al, 2010; O'Roak et al, 2011; Srivastava et al, 2014; Lozano et al, 2015; Sollis et al, 2016), and a milder developmental disorder with speech/language delays and congenital abnormalities (Snijders Blok et al, 2021), respectively. Some of the etiological variants affect equivalent residues in the DNA‐binding domain of these genes (Sollis et al, 2017; Snijders Blok et al, 2021). While differences in the associated phenotypes are likely explained by the distinct expression patterns of the FOXP proteins, there are also regions of overlap where they can potentially form heterodimers. More thorough phenotypic comparison studies between these distinct neurodevelopmental disorders and functional follow‐up would be required to uncover whether equivalent variants in FOXP1 and FOXP4 directly impact speech and language or whether they have an indirect effect on the function of FOXP2.
In‐depth studies of the functions of FOXP2 and its orthologues in brain development have involved not only mice and zebra finches (as noted above), but also other models such as zebrafish and cell‐based systems. These investigations have uncovered upstream regulators of its expression, downstream targets that it regulates, and protein interactions that modulate its functions. Here, we give an up‐to‐date overview of the molecular networks of FOXP2 in the brain, highlighting how this information promises to deliver novel insights into roles of the gene in cognition and behavior.
Regulation of FOXP2 expression
Although the specific spatiotemporal expression patterns of FOXP2 in the brain imply tight regulation, little is known about the upstream mechanisms involved. Only a few transcription factors have been shown to bind to the genomic locus and/or to directly regulate its expression.
Tbr1 activates Foxp2 expression in the developing cortex
TBR1 is a neural transcription factor with high expression in deep layers of the cortex, where it promotes a layer‐VI identity, largely via repression of layer‐V genes (Han et al, 2011; McKenna et al, 2011). In adult mice, almost 70% of FOXP2‐positive cells in layer VI express TBR1 (Medvedeva et al, 2019), and cell‐based assays have demonstrated that TBR1, in complex with its co‐regulator CASK, can activate FOXP2 expression (Fig 2A) (Becker et al, 2018; Fazel Darbandi et al, 2018). Conditional deletion of Tbr1 in layer‐VI neurons of mice leads to reduced Foxp2 expression in these neurons, which shift to a layer‐V‐like identity (Fazel Darbandi et al, 2018). Although the role of FOXP2 in cortical lamination is limited, based on studies with cortical‐specific knockout mice (Kast et al, 2019), the gene may be part of the regulatory program involved in formation, maintenance, and connectivity of corticothalamic neurons in layer VI (Druart et al, 2020), under control of TBR1. People with heterozygous FOXP2 disruptions have been reported to show subtle differences in gray matter density in several parts of the cortex (Watkins et al, 2002), based on voxel‐based morphometry of MRI scans, although it is not known whether this involves altered connectivity and/or function of layer‐VI neurons in those regions. Recurrent de novo mutations of TBR1 have been linked to a neurodevelopmental syndrome involving intellectual disability and/or autism spectrum disorder, and sometimes language impairments (MIM #606053), suggesting some phenotypic overlaps with FOXP2‐related disorder (Deriziotis et al, 2014).
Figure 2. FOXP2 molecular networks.
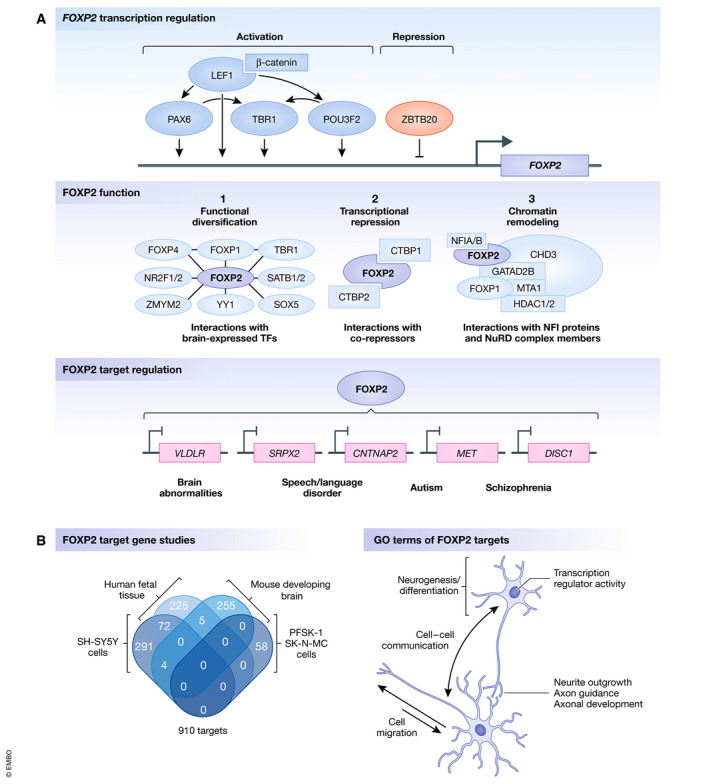
(A) An overview of FOXP2 molecular networks in the brain, at the level of transcription regulation, function, and target regulation. This overview represents results from a selection of separate studies using different types of model systems. TFs: transcription factors. (B) Left, a Venn diagram showing the overlap between FOXP2 target genes identified in four FOXP2 ChIP‐chip/seq studies. SH‐SY5Y and SK‐N‐MC are human neuroblastoma cell lines, and PFSK‐1 is a neuroectodermal tumor cell line. Right, a schematic with a selection of gene ontology (GO) terms that are associated with the identified FOXP2 target genes.
Regulation of FOXP2 by the canonical WNT/β‐catenin signaling pathway
The genomic region upstream of the FOXP2 locus contains six highly conserved binding regions for TCF/LEF transcription factors (Hallikas et al, 2006; Bonkowsky et al, 2008), regulatory proteins that are activated by canonical WNT/β‐catenin signaling, and involved in proliferation and direction of cell fate (Bonkowsky et al, 2008). Binding of WNT to its receptor, Frizzled, leads to inhibition of GSK3β and accumulation of β‐catenin, which translocates to the nucleus and activates transcription via TCF/LEF transcription factors (Ciani & Salinas, 2005). One such TCF/LEF transcription factor is LEF1. FoxP2 and Lef1 are co‐expressed in the developing zebrafish brain, where knockdown of Lef1 expression yields loss of FoxP2 expression (Bonkowsky et al, 2008). Chromatin immunoprecipitation (ChIP) against Lef1 showed enrichment of the predicted Tcf/Lef binding regions upstream of FoxP2, suggesting that Lef1 directly binds to these enhancers to activate FoxP2 expression (Bonkowsky et al, 2008).
The FOXP2 locus also includes multiple highly conserved binding sites for PAX6, a key regulator of central nervous system development (Coutinho et al, 2011). Knockdown of Pax6 in developing zebrafish embryos disrupts FoxP2 expression, while for knockout mice lacking Pax6, expression of Foxp2 is absent in the dorsolateral telencephalon (Coutinho et al, 2011). ChIP against Pax6 in zebrafish embryos showed enrichment of binding sites in the FoxP2 locus, confirming it as a direct target (Coutinho et al, 2011). In the developing neocortex, PAX6 is expressed in neural progenitor cells in the ventricular zone, regulating the cell cycle and differentiation (Gotz et al, 1998), while FOXP2 is expressed at low levels in progenitor cells (Tsui et al, 2013; Garcia‐Calero et al, 2016) but at higher levels in neurons in the cortical plate (Lai et al, 2003; Garcia‐Calero et al, 2016) and (as noted above) later in deep cortical layers (Hisaoka et al, 2010). Under control of WNT3, secreted by thalamic axons that grow into the developing neocortex, FOXP2 mRNA has been shown to be actively translated, driving differentiation of early neurons into deep layer neurons (Kraushar et al, 2015). Activation of FOXP2 by PAX6 might therefore be one of the steps that lead to differentiation of neural progenitor cells into neurons, fine‐tuning their activity and connectivity.
The middle of the FOXP2 locus contains an intronic regulatory element with a binding site for POU3F2, a well‐known neural transcription factor (Maricic et al, 2013). This element drew the attention of molecular anthropologists studying the evolution of FOXP2, because the POU3F2‐binding site contains a DNA variant that arose specifically on the human lineage after splitting from our common ancestor with Neanderthals/Denisovans. However, the site is not fixed in modern human populations; analysis of next‐generation sequencing data from around the world shows that it remains polymorphic in southern Africa, casting doubt on the significance of this variant for human evolution (Atkinson et al, 2018) (see (Fisher, 2019) for a recent account of how views of the relevance of FOXP2 for human evolution have shifted with the availability of comprehensive genome‐wide sequencing datasets and enhanced methods for assessing signals of selection). Based on reporter gene assays with the intronic enhancer, it has been suggested that binding of POU3F2 to this site may lead to increased FOXP2 expression (Maricic et al, 2013), although this finding has not been confirmed in a more physiologically relevant model and it is possible that the element instead regulates the expression of a different gene in the vicinity. Pou3f2 plays important roles in the formation and radial migration of upper‐layer cortical neurons (McEvilly et al, 2002; Sugitani et al, 2002) and is known to drive expression of Ngn2, Tbr2, and Tbr1, facilitating the differentiation of glutamatergic neurons (Dominguez et al, 2013).
PAX6 and POU3F2 are, like FOXP2, direct downstream targets of LEF1 (Goodall et al, 2004; Gan et al, 2014; Belinson et al, 2016). The LEF1‐β‐catenin/PAX6 signaling pathway is involved in self‐renewal of neural progenitors and neurogenesis during neocortical development, initiating the PAX6/NGN2/TBR2/NEUROD/TBR1 cascade (Gan et al, 2014). LEF1‐β‐catenin/POU3F2 signaling has been found to contribute to expansion of cortical neural progenitors and neurogenesis via the POU3F2/TBR2 and POU3F2/TBR1 cascades (Dominguez et al, 2013; Belinson et al, 2016). We speculate that FOXP2 and its transcriptional regulators LEF1, PAX6, and POU3F2 may all be downstream effectors of WNT/β‐catenin signaling (Fig 2A), a suggestion that could be tested in future with targeted experiments. Intriguingly, ectopic activation of Wnt signaling in the chicken optic cup has been shown to lead to upregulation of FoxP2 expression (Trimarchi et al, 2009).
FOXP2 has been reported to regulate the transcription of WNT pathway genes and to directly interact with β‐catenin (Richter et al, 2021). Moreover, the FOXP2‐regulator TBR1 promotes maturation of layer‐VI cortical neurons by enhancing WNT signaling (Fazel Darbandi et al, 2020). As both an upstream and downstream player of this pathway, FOXP2 may potentially fulfill a central role in WNT/β‐catenin signaling in the brain, a hypothesis that would be interesting to explore with future studies.
Zbtb20 represses Foxp2 expression in the hippocampus
To our knowledge, the only well‐characterized repressor of FOXP2 identified through animal models is ZBTB20 (Fig 2A) (Nielsen et al, 2014), a transcription factor expressed in hippocampal projection neurons, cerebellar granular cells, and gliogenic progenitors (Mitchelmore et al, 2002). Zbtb20 was found to bind to and repress cortical layer marker genes, including Foxp2, in the developing mouse hippocampus, thereby directing a hippocampal fate while repressing other neuronal identities (Nielsen et al, 2014). Consistently, transgenic expression of Zbtb20 in mice results in reduced Foxp2 expression (Nielsen et al, 2014). Mouse Zbtb20 and human ZBTB20 proteins are highly conserved, with similar neural expression patterns (Nielsen et al, 2014), suggesting that the human orthologue may be important for FOXP2 repression in the human hippocampus.
Downstream effectors of FOXP2
Multiple studies have sought downstream neural targets of FOXP2, yielding insights into pathways that it regulates in the context of brain development, function, and disease.
FOXP2 targets are important for neurite outgrowth and cell migration
In early work on identifying targets of FOXP2, three studies performed ChIP‐chip experiments on human fetal tissue (Spiteri et al, 2007), human neuroblastoma cells (Vernes et al, 2007), and embryonic mouse brain tissue (Vernes et al, 2011). Although no identified targets were common to all three studies, they are enriched for genes associated with similar gene ontology categories, namely cell communication/migration and nervous system development including neurogenesis, neurite development and axon guidance (Spiteri et al, 2007; Vernes et al, 2007; Vernes et al, 2011) (Fig 2B). A ChIP‐sequencing study of FOXP2 in neuroectodermal tumor cells and neuroblastoma cells identified 58 targets near high‐confidence ChIP peaks from a merged dataset, that were mostly enriched for genes linked to transcriptional (regulatory) activity (Nelson et al, 2013).
Follow‐up experiments confirmed that Foxp2 promotes neurite outgrowth in both mouse neuroblastoma cells and mouse striatal primary neurons (Vernes et al, 2011). Indeed, genetic manipulations of Foxp2 in an array of mouse models have been found to have effects on dendrite length. Specifically, introducing a partially humanized version of Foxp2 into mice results in increased dendrite length of medium spiny neurons (Enard et al, 2009), while a loss‐of‐function mutation of the gene is reported to lead to decreased dendrite length of layer‐VI excitatory neurons in the cortex (Druart et al, 2020). The roles of Foxp2 in neuronal migration are less clear‐cut; although in vitro studies support effects of the gene on cell migration phenotypes (Devanna et al, 2014), in vivo data from different mouse models are somewhat inconsistent with each other. For example, studies in which Foxp2 expression was knocked down during embryonic development identified changes in cortical neurogenesis (Tsui et al, 2013) and in migration of neural progenitors out of the subventricular zone (Garcia‐Calero et al, 2016), but selective deletion of the gene was not found to have such effects (Kast et al, 2019).
Although large ChIP‐chip/sequencing datasets do not provide detailed directional insights into regulatory mechanisms, these data are valuable for further targeted investigations of relevant molecular pathways. In one such study, multiple targets from prior ChIP‐chip studies (Spiteri et al, 2007; Vernes et al, 2007; Vernes et al, 2011) were found to be differentially expressed in human neuroblastoma cells stably transfected with FOXP2, including retinoic acid signaling genes, such as the retinoic acid receptor (RAR)‐β, RAR‐related orphan receptor (ROR)‐α, ROR‐β, ROR‐γ, and NEDD9 (Devanna et al, 2014). Retinoic acid signaling is involved in forebrain and hindbrain development and directs the differentiation of embryonic stem cells into neural progenitors (Rhinn & Dolle, 2012). Retinoic acid treatment of human neuroblastoma cells induces neurite outgrowth and reduces cell migration, effects that are enhanced by concurrent FOXP2 overexpression (Devanna et al, 2014), suggesting that the gene may modulate retinoic acid signaling in the developing brain.
FOXP2 target genes are implicated in neurodevelopmental disorders
Out of the hundreds of putative targets of FOXP2, a small subset have received special attention through validation and follow‐up in animal or cell‐based models. One of the first targets to be studied in this way was CNTNAP2, which encodes CASPR2, a neurexin transmembrane protein expressed widely in the brain, with roles in nerve conduction, neuronal migration, neurite outgrowth, and connectivity (Rodenas‐Cuadrado et al, 2014). FOXP2 directly binds to regulatory regions of the CNTNAP2 locus to repress expression (Vernes et al, 2008; Mendoza & Scharff, 2017). This is consistent with complementary expression patterns reported for the two genes in human fetal cerebral cortex (Vernes et al, 2008) and increased Cntnap2 expression in the cerebellum of a Foxp2‐R552H mouse model (based on the human KE family mutation) (Fujita et al, 2012). However, CNTNAP2 expression changes temporally (Gordon et al, 2016) and expression patterns of these genes could potentially show different relationships at distinct stages of development and/or in different brain regions. Interestingly, a cluster of SNPs in CNTNAP2 has been associated with reduced performance on a nonsense‐word repetition task in a cohort of children with developmental language disorders (Vernes et al, 2008) and with a measure of early communicative behavior in a general population sample (Whitehouse et al, 2011). Furthermore, homozygous and compound heterozygous loss‐of‐function variants cause a severe neurodevelopmental disorder with epilepsy and intellectual disability (MIM #610042) (Strauss et al, 2006; Zweier et al, 2009; Smogavec et al, 2016). Although in prior work both common and rare CNTNAP2 variation has been linked to a range of brain‐related phenotypes (Fig 2A), including autism (MIM #612100) (Alarcon et al, 2008; Arking et al, 2008) and schizophrenia (Friedman et al, 2008; Ji et al, 2013), data from a recent large‐scale study argue that the contributions of this gene to risk of these psychiatric disorders have been overstated (Toma et al, 2018).
Other genes that are repressed by FOXP2, and where links have been investigated in follow‐up studies, include SRPX2 (Roll et al, 2010), MET (Mukamel et al, 2011), and DISC1 (Spiteri et al, 2007; Walker et al, 2012; Nelson et al, 2013). FOXP2 overexpression in cell‐based assays lowers the expression of SRPX2 (Roll et al, 2010), MET (Mukamel et al, 2011), and DISC1 (Walker et al, 2012), and FOXP2 directly binds to regulatory sequences in MET and SRPX2 (Roll et al, 2010; Mukamel et al, 2011). Cell‐based assays additionally suggest that the FOXP2‐R553H mutation disrupts regulation of SRPX2 and DISC1 (Roll et al, 2010; Walker et al, 2012). SRPX2 variants have been identified in people with epilepsy of the rolandic speech area, speech apraxia, polymicrogyria, and intellectual disability (MIM #300643) (Roll et al, 2006; Roll et al, 2010; Chen et al, 2017), although their etiological relevance is uncertain given subsequent discovery of GRIN2A disruptions in the affected individuals (Lesca et al, 2013). Common variation in MET has been associated with autism spectrum disorder (MIM %611015) (Campbell et al, 2006; Thanseem et al, 2010) and schizophrenia (Burdick et al, 2010), and post‐mortem brain studies have shown altered MET expression in individuals with autism (Campbell et al, 2007). The DISC1 gene has been linked to schizophrenia (MIM #604906) (Hennah et al, 2003; Hodgkinson et al, 2004; Schumacher et al, 2009).
Beyond its effects as a transcriptional repressor, noted above, FOXP2 has been reported to be a direct activator of VLDLR expression (Spiteri et al, 2007; Vernes et al, 2007; Adam et al, 2016; Mendoza & Scharff, 2017). VLDLR is a receptor for RELN, expressed in the apical processes of migrating neurons in the developing cortex, with roles in neuronal migration, dendrite and spine development, and synaptic function (Lee & D'Arcangelo, 2016). Studies of zebra finch brain have found that FoxP2 protein directly binds to regulatory sequences of the Vldlr locus and that knockdown of the former reduces expression of the latter (Adam et al, 2016). Homozygous disruptions of the human VLDLR gene have been discovered in patients with cerebellar hypoplasia, mild cerebral gyral simplification, and intellectual disability (MIM #224050) (Boycott et al, 2005; Ozcelik et al, 2008; Dixon‐Salazar et al, 2012).
Based on data thus far collected on downstream pathways, FOXP2 and its targets belong to molecular networks that are crucial for aspects of brain function and that are implicated in a range of neurodevelopmental disorders with partially overlapping phenotypes, raising the possibility that etiological variants of these genes affect shared mechanisms (Fig 2A).
FOXP2 transcriptional regulation
Although studies of FOXP2 have probed its expression patterns, regulation, and transcriptional targets, the molecular mechanisms by which this regulatory protein acts as a transcription factor have been much less explored.
FOXP2 interacts with the CTBP transcriptional co‐repressors
The first proteins to be identified as putative binding partners of FOXP2 were CTBP1 and CTBP2 (Li et al, 2004), transcriptional co‐repressors that also interact with FOXP1 via a consensus binding site, which is lacking in FOXP4 (Li et al, 2004; Estruch et al, 2016a). Drosophila CtBP enhances repression by directly blocking the transcription initiation complex or inhibiting adjacent transcriptional activators (Nibu et al, 2003). Moreover, CTBP1 and CTBP2 were identified in a core protein complex that contained DNA‐binding proteins, histone‐modifying enzymes, histone methyltransferases, and chromodomain‐containing proteins (Shi et al, 2003), and may thereby aid FOXP2 in its transcriptional repressive functions (Fig 2A). Indeed, in cell‐based assays, CTBP1 is able to increase FOXP1 and FOXP2 repression of reporter constructs (Li et al, 2004). The FOXP2‐R553H protein, which harbors an etiological substitution disrupting the DNA‐binding domain (Vernes et al, 2006), retains its ability to bind to CTBP1 and CTBP2, suggesting that DNA binding of FOXP2 is not essential for the CTBP‐FOXP2 interaction (Estruch et al, 2016a). Since CTBP proteins depend on their interaction partners to be recruited to DNA, and FOXP2‐R553H is unable to bind to DNA, it is unlikely that this complex represses target genes.
SUMOylation of FOXP2 modulates its function
Post‐translational modifications are another way to dynamically regulate transcription factor activity. One such modification is SUMOylation, the reversible coupling of small ubiquitin‐like modifiers (SUMOs), which are ubiquitously expressed polypeptides, to specific sites in proteins. FOXP2 has a SUMOylation site at position K674, which is SUMOylated by SUMO1/2/3 via interaction with PIAS1/3 (Estruch et al, 2016b; Usui et al, 2017). K674 SUMOylation is not critical for FOXP2 protein stability, dimerization, and subcellular localization in human cell lines (Estruch et al, 2016b; Meredith et al, 2016), but may alter its transcriptional activity (Meredith et al, 2016). Although one study did not detect changes in transcriptional repression of a non‐SUMOylated FOXP2 K674R mutant (Estruch et al, 2016b), another found this mutant to be less effective in repressing target promoters compared to wild‐type protein (Meredith et al, 2016). Disrupting the equivalent SUMOylation site in FOXP1 (K670) abolishes FOXP1 repression, while K670 SUMOylation in wild‐type FOXP1 enhances binding to the CTBP1 co‐repressor (Rocca et al, 2017). Studies of mice suggest that FOXP2 SUMOylation in the cerebellum is important for Purkinje cell development and motor functions (Usui et al, 2017). In cell‐based studies, ubiquitination, another form of post‐translational modification, has been found for an alternatively spliced short isoform of unknown significance (FOXP2.10+), while the canonical isoform was not ubiquitinated (Vernes et al, 2006). Whether other post‐translational modifications beyond SUMOylation and ubiquitination, such as phosphorylation and acetylation, significantly contribute to regulation of FOXP2 functions has yet to be elucidated.
FOXP2 interacts with other brain‐expressed transcription factors
A mass spectrometry study to characterize the FOXP2 interactome identified multiple transcription factors binding to FOXP2 in HEK293 cells, including NR2F1, NR2F2, SATB1, SATB2, SOX5, YY1, and ZMYM2 (Estruch et al, 2018). Foxp2 is co‐expressed with Sox5, Satb1, Satb2, and Nr2f1 in a subset of neurons in the mouse cerebral cortex and with Nr2f2 in Purkinje cells (Estruch et al, 2018). The interactions were validated in cell lines using bioluminescence resonance energy transfer (BRET) assays (Estruch et al, 2018). Additionally, the cortical transcription factor TBR1 was identified as a putative FOXP2 interactor in a yeast‐two‐hybrid assay (Sakai et al, 2011) and confirmed with BRET (Deriziotis et al, 2014). The etiological FOXP2 p.R553H mutation disrupts the interactions with these brain‐expressed transcription factors (Deriziotis et al, 2014; Estruch et al, 2018). The functional importance of these interactions for in vivo brain development has not yet been studied, but may contribute to diversification of FOXP2 activity, guiding the protein to specific transcriptional complexes, changing its affinity for certain targets, and/or helping to recruit transcriptional co‐factors (Fig 2A).
FOXP2 regulatory activity may be mediated via the NuRD chromatin remodeling complex
FOXP1, FOXP2, and FOXP4 all interact with the nucleosome remodeling and histone deacetylase (NuRD) complex (Chokas et al, 2010), a multiprotein complex that couples two independent chromatin‐regulatory functions, (i) ATP‐dependent histone remodeling and (ii) histone deacetylation (Tong et al, 1998; Xue et al, 1998). The complex, involved in both activation and repression of genes (Basta & Rauchman, 2015), is the most abundant form of deacetylase in mammals (Torchy et al, 2015) and is linked to fundamental biological processes, including cell cycle progression, genomic integrity (Lai & Wade, 2011), and differentiation of embryonic stem cells (Basta & Rauchman, 2015; Torchy et al, 2015). FOXP1 interacts with NuRD complex members HDAC1/2, GATAD2B, and MTA1 (Chokas et al, 2010), FOXP4 with HDAC1/2 and GATAD2B (Chokas et al, 2010), and FOXP2 with GATAD2B (Chokas et al, 2010) and CHD3 (Estruch et al, 2016b). For FOXP1 and FOXP4, these interactions further reduce target gene expression in cell‐based reporter assays, suggesting that these NuRD complex interaction partners act as co‐repressors. For the FOXP2‐GATAD2B interaction however, assays found no evidence of synergistic repression (Chokas et al, 2010).
Interestingly, the NuRD complex plays an important role in the developing brain, apparent from the links of multiple of the core NuRD complex members with neurodevelopmental disorders that are characterized by features that partly overlap with the FOXP2‐associated phenotypes. Mutations in the CHD4 gene result in an intellectual disability syndrome that includes global developmental delay and in some cases macrocephaly (MIM #617159) (Sifrim et al, 2016; Weiss et al, 2016). A mutation in CHD3 was first discovered in a child with childhood apraxia of speech (Eising et al, 2019), whereafter additional etiological variants were found in a number of patients that displayed intellectual disability, accompanied by speech/language problems and brain abnormalities including both macrocephaly and microcephaly (MIM #618205) (Snijders Blok et al, 2018). Furthermore, GATAD2B disruptions have been identified in patients with intellectual disability and limited speech (MIM #615074) (de Ligt et al, 2012; Willemsen et al, 2013; Shieh et al, 2020).
In addition to the direct interactions of FOXPs with NuRD complex members, there are multiple indirect links. FOXP2 and the HDAC1/2 proteins share at least three common interaction partners, the cortical transcription factors YY1 (Yang et al, 1996; Yao et al, 2001; Estruch et al, 2018), SATB1 (Yasui et al, 2002; Estruch et al, 2018), and SATB2 (Gyorgy et al, 2008; Estruch et al, 2018). In layer‐IV neurons of the cortex, Satb2 has been shown to assemble the NuRD complex upstream of Bcl11b, resulting in Bcl11b repression, via the Satb2‐Hdac1 interaction (Britanova et al, 2008). Repression of BCL11B in SATB2‐positive neurons is an essential mechanism in cortical lamination, resulting in upper‐layer neuron specification (Britanova et al, 2008). In humans, YY1 (MIM #617557), SATB1 (MIM # 619228 and #619229), and SATB2 (MIM #612313) are all implicated in neurodevelopmental disorders (Bengani et al, 2017; den Hoed et al, 2021; Gabriele et al, 2017). Notably, SATB2 mutations cause severe language impairments (Zarate & Fish, 2017). Furthermore, CTBP2, a direct FOXP2 interactor and co‐repressor (Estruch et al, 2016a), interacts with several NuRD complex members, namely HDAC2, MTA2, GATAD2B, and CHD4 (Zhao et al, 2014). Whether these FOXP2 interactors interact with FOXP2 and the NuRD complex simultaneously has not been studied.
Most FOXP‐NuRD complex interactions have only been characterized in cell lines or in the context of lung function (another tissue where FOXP proteins are expressed) (Chokas et al, 2010), and the importance of such interactions for brain development remains to be uncovered. The NuRD complex plays major roles in the proliferation, migration, and differentiation of neurons (Nitarska et al, 2016), and interactions with cortical transcription factors, such as SATB2, seem to recruit it to specific targets (Britanova et al, 2008). Hence, the FOXP proteins (as homo/heterodimers or together with other co‐factors) may guide the NuRD complex to the DNA, to repress or activate target sequences via chromatin remodeling (Fig 2A). FOXP2 mutations may disrupt this mechanism by abolishing either DNA binding or interaction with NuRD complex members, resulting in abnormal regulation of downstream targets. Mutations in NuRD complex members may result in similar transcriptional regulatory defects, contributing to partial overlaps in the neurodevelopmental phenotypes that are associated with FOXP2, GATAD2B, and SATB2 mutations.
In addition to potential chromatin remodeling functions via interactions with the NuRD complex, FOXP2 has been reported to mediate chromatin accessibility by interacting with transcriptional co‐factors NFIA and NFIB in neuronal cell‐based models (Hickey et al, 2019). Direct interactions of FOXP2 with DNA were found to yield repression of proliferation‐promoting genes, while FOXP2‐NFI complexes activated expression of genes driving neuronal differentiation via chromatin alterations (Hickey et al, 2019). Although FOXP2‐R553H in complex with NFIA was still able to open chromatin, it did not activate gene expression. Thus, these data suggest the existence of distinct FOXP2 regulatory modes that together mediate target gene expression.
Future perspectives
Two decades of molecular studies on the functions of FOXP2 have shown that it belongs to an extensive molecular network with brain‐expressed transcription factors and co‐regulators, mediating neuronal differentiation, neurite outgrowth and cell migration in human cell‐based assays, and shaping the development, plasticity and maturation of corticostriatal and corticocerebellar circuits important for behavioral phenotypes in animal models. Despite the attention FOXP2 has received over the years, much remains to be learned regarding its regulatory capabilities, position in molecular pathways, roles in cellular functions, and ultimately its effects on brain development and human speech and language capacities (Fig 3).
Figure 3. Open questions on the molecular aspects of FOXP2 in the brain.
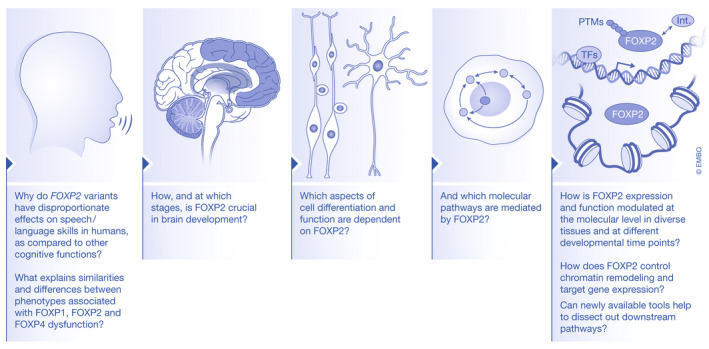
Schematic with different levels of FOXP2 functioning. For each level, questions are included that have remained largely unanswered and should be focus of future studies. The shaded brain areas in the schematic in the second left panel represent regions of expression of FOXP2 that have been main focus in current literature. Int., protein interactors; PTMs, post‐translational modifications; TFs, transcription factors.
New and more sophisticated models may hold special promise for furthering our understanding of FOXP2 functions, particularly in light of links to speech and language. Human brain organoids grown from stem cells can model early stages of development of various parts of the nervous system (Kelava & Lancaster, 2016; Marton & Pașca, 2020) and overcome species‐specific developmental programs (Kanton et al, 2019), providing the opportunity to study the human transcriptome during brain development. Long‐term (Gordon et al, 2021) and slice cultures (Giandomenico et al, 2019; Qian et al, 2020) of these brain organoids result in maturation up to late fetal and early post‐natal stages, while merging of region‐specific organoids make it possible to model early establishment of brain circuitries (Andersen et al, 2020; Miura et al, 2020). Genetic manipulation of FOXP2 in such model systems could reveal human‐specific functions that have been unable to be studied in traditional in vitro settings so far.
For studying FOXP2 functions in vivo, more relevant and non‐traditional animal models are also being explored (Lattenkamp & Vernes, 2018). In addition to zebra finches, other species of birds display auditory‐guided vocal learning (Pfenning et al, 2014), as well as bats (Knörnschild, 2014; Vernes, 2017) and ocean mammals (Ravignani et al, 2016). The latter two are evolutionarily closer to us, with brain structures and circuitries more similar to human brains. Indeed, analyses of FoxP expression patterns in the brains of bat species are already proving informative (Rodenas‐Cuadrado et al, 2018). Although the genetic tools in such species are not yet as well established as in the traditional animal models, optimization and validation of these in the coming years will open up exciting new avenues for investigations of FOXP2 and its orthologues, placing the critical molecular networks in their broader evolutionary context.
Author contributions
All authors contributed to writing and revising of the manuscript and approved the final version.
Conflict of interest
The authors declare that they have no conflict of interest.
Acknowledgements
We would like to thank Dr. Cleo J.L.M. Smeets (L'Institut du Cerveau et de la Moelle Épinière, Paris Brain Institute) for her suggestions and feedback and Dr. Else Eising (Max Planck Institute for Psycholinguistics) for help with analyzing BrainSpan data. This work was supported by the Max Planck Society.
EMBO reports (2021) 22: e52803.
See the Glossary for abbreviations used in this article
References
- Adam I, Mendoza E, Kobalz U, Wohlgemuth S, Scharff C (2016) FoxP2 directly regulates the reelin receptor VLDLR developmentally and by singing. Mol Cell Neurosci 74: 96–105 [DOI] [PubMed] [Google Scholar]
- Alarcón M, Abrahams BS, Stone JL, Duvall JA, Perederiy JV, Bomar JM, Sebat J, Wigler M, Martin CL, Ledbetter DH et al (2008) Linkage, association, and gene‐expression analyses identify CNTNAP2 as an autism‐susceptibility gene. Am J Hum Genet 82: 150–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen J, Revah O, Miura Y, Thom N, Amin ND, Kelley KW, Singh M, Chen X, Thete MV, Walczak EM et al (2020) Generation of functional human 3D cortico‐motor assembloids. Cell 183: 1913–1929.e1926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argyropoulos GPD, Watkins KE, Belton‐Pagnamenta E, Liégeois F, Saleem KS, Mishkin M, Vargha‐Khadem F (2019) Neocerebellar crus I abnormalities associated with a speech and language disorder due to a mutation in FOXP2. Cerebellum 18: 309–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arking DE, Cutler DJ, Brune CW, Teslovich TM, West K, Ikeda M, Rea A, Guy M, Lin S, Cook EH et al (2008) A common genetic variant in the neurexin superfamily member CNTNAP2 increases familial risk of autism. Am J Hum Genet 82: 160–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson EG, Audesse AJ, Palacios JA, Bobo DM, Webb AE, Ramachandran S, Henn BM (2018) No evidence for recent selection at FOXP2 among diverse human populations. Cell 174: 1424–1435.e1415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basta J, Rauchman M (2015) The nucleosome remodeling and deacetylase complex in development and disease. Transl Res 165: 36–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker M, Devanna P, Fisher SE, Vernes SC (2018) Mapping of human FOXP2 enhancers reveals complex regulation. Front Mol Neurosci 11: 47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belinson H, Nakatani J, Babineau BA, Birnbaum RY, Ellegood J, Bershteyn M, McEvilly RJ, Long JM, Willert K, Klein OD et al (2016) Prenatal β‐catenin/Brn2/Tbr2 transcriptional cascade regulates adult social and stereotypic behaviors. Mol Psychiatry 21: 1417–1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belton E, Salmond CH, Watkins KE, Vargha‐Khadem F, Gadian DG (2003) Bilateral brain abnormalities associated with dominantly inherited verbal and orofacial dyspraxia. Hum Brain Mapp 18: 194–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benayoun BA, Caburet S, Veitia RA (2011) Forkhead transcription factors: key players in health and disease. Trends Genet 27: 224–232 [DOI] [PubMed] [Google Scholar]
- Bengani H, Handley M, Alvi M, Ibitoye R, Lees M, Lynch SA, Lam W, Fannemel M, Nordgren A, Malmgren H et al (2017) Clinical and molecular consequences of disease‐associated de novo mutations in SATB2. Genet Med 19: 900–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonkowsky JL, Wang X, Fujimoto E, Lee JE, Chien CB, Dorsky RI (2008) Domain‐specific regulation of foxP2 CNS expression by lef1. BMC Dev Biol 8: 103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boycott KM, Flavelle S, Bureau A, Glass HC, Fujiwara TM, Wirrell E, Davey K, Chudley AE, Scott JN, McLeod DR et al (2005) Homozygous deletion of the very low density lipoprotein receptor gene causes autosomal recessive cerebellar hypoplasia with cerebral Gyral simplification. Am J Hum Genet 77: 477–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britanova O, de Juan Romero C , Cheung A, Kwan KY, Schwark M, Gyorgy A, Vogel T, Akopov S, Mitkovski M, Agoston D et al (2008) Satb2 is a postmitotic determinant for upper‐layer neuron specification in the neocortex. Neuron 57: 378–392 [DOI] [PubMed] [Google Scholar]
- Burdick KE, DeRosse P, Kane JM, Lencz T, Malhotra AK (2010) Association of genetic variation in the MET proto‐oncogene with schizophrenia and general cognitive ability. Am J Psychiatry 167: 436–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell DB, Sutcliffe JS, Ebert PJ, Militerni R, Bravaccio C, Trillo S, Elia M, Schneider C, Melmed R, Sacco R et al (2006) A genetic variant that disrupts MET transcription is associated with autism. Proc Natl Acad Sci USA 103: 16834–16839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell DB, D'Oronzio R, Garbett K, Ebert PJ, Mirnics K, Levitt P, Persico AM (2007) Disruption of cerebral cortex MET signaling in autism spectrum disorder. Ann Neurol 62: 243–250 [DOI] [PubMed] [Google Scholar]
- Campbell P, Reep RL, Stoll ML, Ophir AG, Phelps SM (2009) Conservation and diversity of Foxp2 expression in muroid rodents: functional implications. J Comp Neurol 512: 84–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellucci GA, McGinley MJ, McCormick DA (2016) Knockout of Foxp2 disrupts vocal development in mice. Sci Rep 6: 23305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabout J, Sarkar A, Patel SR, Radden T, Dunson DB, Fisher SE, Jarvis ED (2016) A Foxp2 mutation implicated in human speech deficits alters sequencing of ultrasonic vocalizations in adult male mice. Front Behav Neurosci 10: 197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y‐C, Kuo H‐Y, Bornschein U, Takahashi H, Chen S‐Y, Lu K‐M, Yang H‐Y, Chen G‐M, Lin J‐R, Lee Y‐H et al (2016) Foxp2 controls synaptic wiring of corticostriatal circuits and vocal communication by opposing Mef2c. Nat Neurosci 19: 1513–1522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XS, Reader RH, Hoischen A, Veltman JA, Simpson NH, Francks C, Newbury DF, Fisher SE (2017) Next‐generation DNA sequencing identifies novel gene variants and pathways involved in specific language impairment. Sci Rep 7: 46105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chokas AL, Trivedi CM, Lu MM, Tucker PW, Li S, Epstein JA, Morrisey EE (2010) Foxp1/2/4‐NuRD interactions regulate gene expression and epithelial injury response in the lung via regulation of interleukin‐6. J Biol Chem 285: 13304–13313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciani L, Salinas PC (2005) WNTS in the vertebrate nervous system: from patterning to neuronal connectivity. Nat Rev Neurosci 6: 351 [DOI] [PubMed] [Google Scholar]
- Clifton EAD, Perry JRB, Imamura F, Lotta LA, Brage S, Forouhi NG, Griffin SJ, Wareham NJ, Ong KK, Day FR (2018) Genome–wide association study for risk taking propensity indicates shared pathways with body mass index. Commun Biol 1: 36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Co M, Hickey SL, Kulkarni A, Harper M, Konopka G (2019) Cortical Foxp2 supports behavioral flexibility and developmental dopamine D1 receptor expression. Cereb Cortex 30: 1855–1870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Co M, Anderson AG, Konopka G (2020) FOXP transcription factors in vertebrate brain development, function, and disorders. WIREs Dev Biol 9: e375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutinho P, Pavlou S, Bhatia S, Chalmers KJ, Kleinjan DA, van Heyningen V (2011) Discovery and assessment of conserved Pax6 target genes and enhancers. Genome Res 21: 1349–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day NF, Hobbs TG, Heston JB, White SA (2019) Beyond critical period learning: striatal FoxP2 affects the active maintenance of learned vocalizations in adulthood. eneuro 6: ENEURO.0071‐19.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demontis D, Walters RK, Martin J, Mattheisen M, Als TD, Agerbo E, Baldursson G, Belliveau R, Bybjerg‐Grauholm J, Bækvad‐Hansen M et al (2019) Discovery of the first genome‐wide significant risk loci for attention deficit/hyperactivity disorder. Nat Genet 51: 63–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deriziotis P, O’Roak BJ, Graham SA, Estruch SB, Dimitropoulou D, Bernier RA, Gerdts J, Shendure J, Eichler EE, Fisher SE (2014) De novo TBR1 mutations in sporadic autism disrupt protein functions. Nat Commun 5: 4954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devanna P, Middelbeek J, Vernes SC (2014) FOXP2 drives neuronal differentiation by interacting with retinoic acid signaling pathways. Front Cell Neurosci 8: 305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon‐Salazar TJ, Silhavy JL, Udpa N, Schroth J, Bielas S, Schaffer AE, Olvera J, Bafna V, Zaki MS, Abdel‐Salam GH et al (2012) Exome sequencing can improve diagnosis and alter patient management. Sci Transl Med 4: 138ra178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez MH, Ayoub AE, Rakic P (2013) POU‐III transcription factors (Brn1, Brn2, and Oct6) influence neurogenesis, molecular identity, and migratory destination of upper‐layer cells of the cerebral cortex. Cerebral Cortex 23: 2632–2643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Druart M, Groszer M, Le Magueresse C (2020) An etiological Foxp2 mutation impairs neuronal gain in layer VI cortico‐thalamic cells through increased GABAB/GIRK signaling. J Neurosci 40: 8543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eising E, Carrion‐Castillo A, Vino A, Strand EA, Jakielski KJ, Scerri TS, Hildebrand MS, Webster R, Ma A, Mazoyer B et al (2019) A set of regulatory genes co‐expressed in embryonic human brain is implicated in disrupted speech development. Mol Psychiatry 24: 1065–1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enard W, Gehre S, Hammerschmidt K, Hölter SM, Blass T, Somel M, Brückner MK, Schreiweis C, Winter C, Sohr R et al (2009) A humanized version of Foxp2 affects cortico‐basal ganglia circuits in mice. Cell 137: 961–971 [DOI] [PubMed] [Google Scholar]
- Estruch SB, Graham SA, Chinnappa SM, Deriziotis P, Fisher SE (2016a) Functional characterization of rare FOXP2 variants in neurodevelopmental disorder. J Neurodev Disord 8: 44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estruch SB, Graham SA, Deriziotis P, Fisher SE (2016b) The language‐related transcription factor FOXP2 is post‐translationally modified with small ubiquitin‐like modifiers. Sci Rep 6: 20911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estruch SB, Graham SA, Quevedo M, Vino A, Dekkers DHW, Deriziotis P, Sollis E, Demmers J, Poot RA, Fisher SE (2018) Proteomic analysis of FOXP proteins reveals interactions between cortical transcription factors associated with neurodevelopmental disorders. Hum Mol Genet 27: 1212–1227 [DOI] [PubMed] [Google Scholar]
- Fazel Darbandi S, Robinson Schwartz SE, Qi Q, Catta‐Preta R, Pai E‐L, Mandell JD, Everitt A, Rubin A, Krasnoff RA, Katzman S et al (2018) Neonatal Tbr1 dosage controls cortical layer 6 connectivity. Neuron 100: 831–845.e837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazel Darbandi S, Robinson Schwartz SE, Pai EL‐L, Everitt A, Turner ML, Cheyette BNR, Willsey AJ, State MW, Sohal VS, Rubenstein JLR (2020) Enhancing WNT signaling restores cortical neuronal spine maturation and synaptogenesis in Tbr1 mutants. Cell Rep 31: 107495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferland RJ, Cherry TJ, Preware PO, Morrisey EE, Walsh CA (2003) Characterization of Foxp2 and Foxp1 mRNA and protein in the developing and mature brain. J Comp Neurol 460: 266–279 [DOI] [PubMed] [Google Scholar]
- Feuk L, Kalervo A, Lipsanen‐Nyman M, Skaug J, Nakabayashi K, Finucane B, Hartung D, Innes M, Kerem B, Nowaczyk MJ et al (2006) Absence of a paternally inherited FOXP2 gene in developmental verbal dyspraxia. Am J Hum Genet 79: 965–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher SE (2019) Human genetics: the evolving story of FOXP2. Curr Biol 29: R65–R67 [DOI] [PubMed] [Google Scholar]
- Fontenot JD, Gavin MA, Rudensky AY (2003) Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol 4: 330–336 [DOI] [PubMed] [Google Scholar]
- French CA, Jin X, Campbell TG, Gerfen E, Groszer M, Fisher SE, Costa RM (2012) An aetiological Foxp2 mutation causes aberrant striatal activity and alters plasticity during skill learning. Mol Psychiatry 17: 1077–1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- French CA, Vinueza Veloz MF, Zhou K, Peter S, Fisher SE, Costa RM, De Zeeuw CI (2019) Differential effects of Foxp2 disruption in distinct motor circuits. Mol Psychiatry 24: 447–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman JI, Vrijenhoek T, Markx S, Janssen IM, van der Vliet WA , Faas B, Knoers NV, Cahn W, Kahn RS, Edelmann L et al (2008) CNTNAP2 gene dosage variation is associated with schizophrenia and epilepsy. Mol Psychiatry 13: 261–266 [DOI] [PubMed] [Google Scholar]
- Fujita E, Tanabe Y, Momoi MY, Momoi T (2012) Cntnap2 expression in the cerebellum of Foxp 2(R552H) mice, with a mutation related to speech‐language disorder. Neurosci Lett 506: 277–280 [DOI] [PubMed] [Google Scholar]
- Gabriele M, Vulto‐van Silfhout AT, Germain P‐L, Vitriolo A, Kumar R, Douglas E, Haan E, Kosaki K, Takenouchi T, Rauch A et al (2017) YY1 Haploinsufficiency causes an intellectual disability syndrome featuring transcriptional and chromatin dysfunction. Am J Hum Genet 100: 907–925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan Q, Lee A, Suzuki R, Yamagami T, Stokes A, Nguyen BC, Pleasure D, Wang J, Chen HW, Zhou CJ (2014) Pax6 mediates β‐catenin signaling for self‐renewal and neurogenesis by neocortical radial glial stem cells. Stem Cells 32: 45–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Calero E, Botella‐Lopez A, Bahamonde O, Perez‐Balaguer A, Martinez S (2016) FoxP2 protein levels regulate cell morphology changes and migration patterns in the vertebrate developing telencephalon. Brain Struct Funct 221: 2905–2917 [DOI] [PubMed] [Google Scholar]
- Giandomenico SL, Mierau SB, Gibbons GM, Wenger LMD, Masullo L, Sit T, Sutcliffe M, Boulanger J, Tripodi M, Derivery E et al (2019) Cerebral organoids at the air‐liquid interface generate diverse nerve tracts with functional output. Nat Neurosci 22: 669–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodall J, Martinozzi S, Dexter TJ, Champeval D, Carreira S, Larue L, Goding CR (2004) Brn‐2 expression controls melanoma proliferation and is directly regulated by β‐catenin. Mol Cell Biol 24: 2915–2922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon A, Salomon D, Barak N, Pen Y, Tsoory M, Kimchi T, Peles E (2016) Expression of Cntnap2 (Caspr2) in multiple levels of sensory systems. Mol Cell Neurosci 70: 42–53 [DOI] [PubMed] [Google Scholar]
- Gordon A, Yoon S‐J, Tran SS, Makinson CD, Park JY, Andersen J, Valencia AM, Horvath S, Xiao X, Huguenard JR et al (2021) Long‐term maturation of human cortical organoids matches key early postnatal transitions. Nat Neurosci 24: 331–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotz M, Stoykova A, Gruss P (1998) Pax6 controls radial glia differentiation in the cerebral cortex. Neuron 21: 1031–1044 [DOI] [PubMed] [Google Scholar]
- Groszer M, Keays DA, Deacon RMJ, de Bono JP , Prasad‐Mulcare S, Gaub S, Baum MG, French CA, Nicod J, Coventry JA et al (2008) Impaired synaptic plasticity and motor learning in mice with a point mutation implicated in human speech deficits. Curr Biol 18: 354–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyorgy AB, Szemes M, De Juan RC, Tarabykin V, Agoston DV (2008) SATB2 interacts with chromatin‐remodeling molecules in differentiating cortical neurons. Eur J Neurosci 27: 865–873 [DOI] [PubMed] [Google Scholar]
- Hachigian LJ, Carmona V, Fenster RJ, Kulicke R, Heilbut A, Sittler A, Pereira de Almeida L, Mesirov JP, Gao F, Kolaczyk ED et al (2017) Control of Huntington's disease‐associated phenotypes by the striatum‐enriched transcription factor Foxp2. Cell Rep 21: 2688–2695 [DOI] [PubMed] [Google Scholar]
- Haesler S, Wada K, Nshdejan A, Morrisey EE, Lints T, Jarvis ED, Scharff C (2004) FoxP2 expression in avian vocal learners and non‐learners. J Neurosci 24: 3164–3175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallikas O, Palin K, Sinjushina N, Rautiainen R, Partanen J, Ukkonen E, Taipale J (2006) Genome‐wide prediction of mammalian enhancers based on analysis of transcription‐factor binding affinity. Cell 124: 47–59 [DOI] [PubMed] [Google Scholar]
- Hamdan FF, Daoud H, Rochefort D, Piton A, Gauthier J, Langlois M, Foomani G, Dobrzeniecka S, Krebs M‐O, Joober R et al (2010) De novo mutations in FOXP1 in cases with intellectual disability, autism, and language impairment. Am J Hum Genet 87: 671–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han W, Kwan KY, Shim S, Lam MM, Shin Y, Xu X, Zhu Y, Li M, Sestan N (2011) TBR1 directly represses Fezf2 to control the laminar origin and development of the corticospinal tract. Proc Natl Acad Sci USA 108: 3041–3046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannenhalli S, Kaestner KH (2009) The evolution of Fox genes and their role in development and disease. Nat Rev Genet 10: 233–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennah W, Varilo T, Kestila M, Paunio T, Arajarvi R, Haukka J, Parker A, Martin R, Levitzky S, Partonen T et al (2003) Haplotype transmission analysis provides evidence of association for DISC1 to schizophrenia and suggests sex‐dependent effects. Hum Mol Genet 12: 3151–3159 [DOI] [PubMed] [Google Scholar]
- Heston JB, White SA (2015) Behavior‐linked FoxP2 regulation enables zebra finch vocal learning. J Neurosci 35: 2885–2894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickey SL, Berto S, Konopka G (2019) Chromatin decondensation by FOXP2 promotes human neuron maturation and expression of neurodevelopmental disease genes. Cell Rep 27: 1699–1711.e1699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hisaoka T, Nakamura Y, Senba E, Morikawa Y (2010) The forkhead transcription factors, Foxp1 and Foxp2, identify different subpopulations of projection neurons in the mouse cerebral cortex. Neuroscience 166: 551–563 [DOI] [PubMed] [Google Scholar]
- Hodgkinson CA, Goldman D, Jaeger J, Persaud S, Kane JM, Lipsky RH, Malhotra AK (2004) Disrupted in schizophrenia 1 (DISC1): association with schizophrenia, schizoaffective disorder, and bipolar disorder. Am J Hum Genet 75: 862–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Hoed J , de Boer E , Voisin N, Dingemans AJM, Guex N, Wiel L, Nellaker C, Amudhavalli SM, Banka S, Bena FS et al (2021) Mutation‐specific pathophysiological mechanisms define different neurodevelopmental disorders associated with SATB1 dysfunction. Am J Hum Genet 108: 346–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogman M, Guadalupe T, Zwiers MP, Klarenbeek P, Francks C, Fisher SE (2014) Assessing the effects of common variation in the FOXP2 gene on human brain structure. Front Hum Neurosci 8: 473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji W, Li T, Pan Y, Tao H, Ju K, Wen Z, Fu Y, An Z, Zhao Q, Wang T et al (2013) CNTNAP2 is significantly associated with schizophrenia and major depression in the Han Chinese population. Psychiatry Res 207: 225–228 [DOI] [PubMed] [Google Scholar]
- Kaestner KH, Knochel W, Martinez DE (2000) Unified nomenclature for the winged helix/forkhead transcription factors. Genes Dev 14: 142–146 [PubMed] [Google Scholar]
- Kanton S, Boyle MJ, He Z, Santel M, Weigert A, Sanchís‐Calleja F, Guijarro P, Sidow L, Fleck JS, Han D et al (2019) Organoid single‐cell genomic atlas uncovers human‐specific features of brain development. Nature 574: 418–422 [DOI] [PubMed] [Google Scholar]
- Kast RJ, Lanjewar AL, Smith CD, Levitt P (2019) FOXP2 exhibits projection neuron class specific expression, but is not required for multiple aspects of cortical histogenesis. eLife 8: e42012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelava I, Lancaster MA (2016) Dishing out mini‐brains: current progress and future prospects in brain organoid research. Dev Biol 420: 199–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knörnschild M (2014) Vocal production learning in bats. Curr Opin Neurobiol 28: 80–85 [DOI] [PubMed] [Google Scholar]
- Kraushar ML, Viljetic B, Wijeratne H, Thompson K, Jiao X, Pike JW, Medvedeva V, Groszer M, Kiledjian M, Hart RP et al (2015) Thalamic WNT3 Secretion spatiotemporally regulates the neocortical ribosome signature and mRNA translation to specify neocortical cell subtypes. J Neurosci 35: 10911–10926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai CS, Fisher SE, Hurst JA, Vargha‐Khadem F, Monaco AP (2001) A forkhead‐domain gene is mutated in a severe speech and language disorder. Nature 413: 519–523 [DOI] [PubMed] [Google Scholar]
- Lai CS, Gerrelli D, Monaco AP, Fisher SE, Copp AJ (2003) FOXP2 expression during brain development coincides with adult sites of pathology in a severe speech and language disorder. Brain 126: 2455–2462 [DOI] [PubMed] [Google Scholar]
- Lai AY, Wade PA (2011) Cancer biology and NuRD: a multifaceted chromatin remodelling complex. Nat Rev Cancer 11: 588–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lattenkamp EZ, Vernes SC (2018) Vocal learning: a language‐relevant trait in need of a broad cross‐species approach. Curr Opin Behav Sci 21: 209–215 [Google Scholar]
- Lee GH, D'Arcangelo G (2016) New insights into reelin‐mediated signaling pathways. Front Cell Neurosci 10: 122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesca G, Rudolf G, Bruneau N, Lozovaya N, Labalme A, Boutry‐Kryza N, Salmi M, Tsintsadze T, Addis L, Motte J et al (2013) GRIN2A mutations in acquired epileptic aphasia and related childhood focal epilepsies and encephalopathies with speech and language dysfunction. Nat Genet 45: 1061–1066 [DOI] [PubMed] [Google Scholar]
- Li S, Weidenfeld J, Morrisey EE (2004) Transcriptional and DNA binding activity of the Foxp1/2/4 family is modulated by heterotypic and homotypic protein interactions. Mol Cell Biol 24: 809–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Zeng Z, Zhao Q, Wang T, Huang K, Li J, Li Y, Liu J, Wei Z, Wang Y et al (2013) FoxP2 is significantly associated with schizophrenia and major depression in the Chinese Han population. World J Biol Psychiatry 14: 146–150 [DOI] [PubMed] [Google Scholar]
- Liégeois F, Baldeweg T, Connelly A, Gadian DG, Mishkin M, Vargha‐Khadem F (2003) Language fMRI abnormalities associated with FOXP2 gene mutation. Nat Neurosci 6: 1230–1237 [DOI] [PubMed] [Google Scholar]
- Liégeois FJ, Hildebrand MS, Bonthrone A, Turner SJ, Scheffer IE, Bahlo M, Connelly A, Morgan AT (2016) Early neuroimaging markers of FOXP2 intragenic deletion. Sci Rep 6: 35192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Ligt J , Willemsen MH, van Bon BWM , Kleefstra T, Yntema HG, Kroes T, Vulto‐van Silfhout AT, Koolen DA, de Vries P , Gilissen C et al (2012) Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med 367: 1921–1929 [DOI] [PubMed] [Google Scholar]
- Lozano R, Vino A, Lozano C, Fisher SE, Deriziotis P (2015) A de novo FOXP1 variant in a patient with autism, intellectual disability and severe speech and language impairment. Eur J Hum Genet 23: 1702–1707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDermot KD, Bonora E, Sykes N, Coupe A‐M, Lai CSL, Vernes SC, Vargha‐Khadem F, McKenzie F, Smith RL, Monaco AP et al (2005) Identification of FOXP2 truncation as a novel cause of developmental speech and language deficits. Am J Hum Genet 76: 1074–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maricic T, Günther V, Georgiev O, Gehre S, Ćurlin M, Schreiweis C, Naumann R, Burbano HA, Meyer M, Lalueza‐Fox C et al (2013) A recent evolutionary change affects a regulatory element in the human FOXP2 gene. Mol Biol Evol 30: 844–852 [DOI] [PubMed] [Google Scholar]
- Marton RM, Pașca SP (2020) Organoid and assembloid technologies for investigating cellular crosstalk in human brain development and disease. Trends Cell Biol 30: 133–143 [DOI] [PubMed] [Google Scholar]
- McEvilly RJ, de Diaz MO , Schonemann MD, Hooshmand F, Rosenfeld MG (2002) Transcriptional regulation of cortical neuron migration by POU domain factors. Science 295: 1528–1532 [DOI] [PubMed] [Google Scholar]
- McKenna WL, Betancourt J, Larkin KA, Abrams B, Guo C, Rubenstein JL, Chen B (2011) Tbr1 and Fezf2 regulate alternate corticofugal neuronal identities during neocortical development. J Neurosci 31: 549–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvedeva VP, Rieger MA, Vieth B, Mombereau C, Ziegenhain C, Ghosh T, Cressant A, Enard W, Granon S, Dougherty JD et al (2019) Altered social behavior in mice carrying a cortical Foxp2 deletion. Hum Mol Genet 28: 701–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendoza E, Scharff C (2017) Protein‐protein interaction among the FoxP family members and their regulation of two target genes, VLDLR and CNTNAP2 in the zebra finch song system. Front Mol Neurosci 10: 112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meredith LJ, Wang CM, Nascimento L, Liu R, Wang L, Yang WH (2016) The Key Regulator for Language and Speech Development, FOXP2, is a Novel Substrate for SUMOylation. J Cell Biochem 117: 426–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchelmore C, Kjaerulff KM, Pedersen HC, Nielsen JV, Rasmussen TE, Fisker MF, Finsen B, Pedersen KM, Jensen NA (2002) Characterization of two novel nuclear BTB/POZ domain zinc finger isoforms. Association with differentiation of hippocampal neurons, cerebellar granule cells, and macroglia. J Biol Chem 277: 7598–7609 [DOI] [PubMed] [Google Scholar]
- Miura Y, Li M‐Y, Birey F, Ikeda K, Revah O, Thete MV, Park J‐Y, Puno A, Lee SH, Porteus MH et al (2020) Generation of human striatal organoids and cortico‐striatal assembloids from human pluripotent stem cells. Nat Biotechnol 38: 1421–1430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan A, Fisher S, Scheffer I, Hildebrand M (2016) FOXP2‐related speech and language disorders. In Gene Reviews(R), Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mirzaa G, Amemiya A (eds). Seattle, WA: University of Washington; [updated 2017] [Google Scholar]
- Mukamel Z, Konopka G, Wexler E, Osborn GE, Dong H, Bergman MY, Levitt P, Geschwind DH (2011) Regulation of MET by FOXP2, genes implicated in higher cognitive dysfunction and autism risk. J Neurosci 31: 11437–11442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson CS, Fuller CK, Fordyce PM, Greninger AL, Li H, DeRisi JL (2013) Microfluidic affinity and ChIP‐seq analyses converge on a conserved FOXP2‐binding motif in chimp and human, which enables the detection of evolutionarily novel targets. Nucleic Acids Res 41: 5991–6004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nibu Y, Senger K, Levine M (2003) CtBP‐independent repression in the Drosophila embryo. Mol Cell Biol 23: 3990–3999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen JV, Thomassen M, Mollgard K, Noraberg J, Jensen NA (2014) Zbtb20 defines a hippocampal neuronal identity through direct repression of genes that control projection neuron development in the isocortex. Cerebral Cortex 24: 1216–1229 [DOI] [PubMed] [Google Scholar]
- Nitarska J, Smith JG, Sherlock WT, Hillege MM, Nott A, Barshop WD, Vashisht AA, Wohlschlegel JA, Mitter R, Riccio A (2016) A functional switch of NuRD chromatin remodeling complex subunits regulates mouse cortical development. Cell Rep 17: 1683–1698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norton P, Barschke P, Scharff C, Mendoza E (2019) Differential song deficits after lentivirus‐mediated knockdown of FoxP1, FoxP2, or FoxP4 in Area X of juvenile zebra finches. J Neurosci 39: 9782–9796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Roak BJ, Deriziotis P, Lee C, Vives L, Schwartz JJ, Girirajan S, Karakoc E, Mackenzie AP, Ng SB, Baker C et al (2011) Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet 43: 585–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozcelik T, Akarsu N, Uz E, Caglayan S, Gulsuner S, Onat OE, Tan M, Tan U (2008) Mutations in the very low‐density lipoprotein receptor VLDLR cause cerebellar hypoplasia and quadrupedal locomotion in humans. Proc Natl Acad Sci USA 105: 4232–4236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfenning AR, Hara E, Whitney O, Rivas MV, Wang R, Roulhac PL, Howard JT, Wirthlin M, Lovell PV, Ganapathy G et al (2014) Convergent transcriptional specializations in the brains of humans and song‐learning birds. Science 346: 1256846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian X, Su Y, Adam CD, Deutschmann AU, Pather SR, Goldberg EM, Su K, Li S, Lu Lu, Jacob F et al (2020) Sliced human cortical organoids for modeling distinct cortical layer formation. Cell Stem Cell 26: 766–781.e769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao W, Du X, Zhang Y, Yu Q, Hui L, Yu Y, Kou C, Yin G, Zhu X, Man L et al (2017) Association between forkhead‐box P2 gene polymorphism and clinical symptoms in chronic schizophrenia in a Chinese population. J Neural Transm 124: 891–897 [DOI] [PubMed] [Google Scholar]
- Ravignani A, Fitch WT, Hanke FD, Heinrich T, Hurgitsch B, Kotz SA, Scharff C, Stoeger AS, de Boer B (2016) What pinnipeds have to say about human speech, music, and the evolution of rhythm. Front Neurosci 10: 274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter MS, Riess A, Moog U, Briggs TA, Chandler KE, Rauch A, Stampfer M, Steindl K, Glaser D, Joset P et al (2017) FOXP2 variants in 14 individuals with developmental speech and language disorders broaden the mutational and clinical spectrum. J Med Genet 54: 64–72 [DOI] [PubMed] [Google Scholar]
- van Rhijn J‐R , Fisher SE, Vernes SC, Nadif Kasri N (2018) Foxp2 loss of function increases striatal direct pathway inhibition via increased GABA release. Brain Structure & Function 223: 4211–4226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhinn M, Dolle P (2012) Retinoic acid signalling during development. Development 139: 843–858 [DOI] [PubMed] [Google Scholar]
- Richter G, Gui T, Bourgeois B, Koyani CN, Ulz P, Heitzer E, von Lewinski D, Burgering BMT, Malle E, Madl T (2021) β‐catenin regulates FOXP2 transcriptional activity via multiple binding sites. The FEBS J 288: 3261–3284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocca DL, Wilkinson KA, Henley JM (2017) SUMOylation of FOXP1 regulates transcriptional repression via CtBP1 to drive dendritic morphogenesis. Sci Rep 7: 877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodenas‐Cuadrado P, Ho J, Vernes SC (2014) Shining a light on CNTNAP2: complex functions to complex disorders. Eur J Hum Genet 22: 171–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodenas‐Cuadrado PM, Mengede J, Baas L, Devanna P, Schmid TA, Yartsev M, Firzlaff U, Vernes SC (2018) Mapping the distribution of language related genes FoxP1, FoxP2, and CntnaP2 in the brains of vocal learning bat species. J Comp Neurol 526: 1235–1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roll P, Rudolf G, Pereira S, Royer B, Scheffer IE, Massacrier A, Valenti M‐P, Roeckel‐Trevisiol N, Jamali S, Beclin C et al (2006) SRPX2 mutations in disorders of language cortex and cognition. Hum Mol Genet 15: 1195–1207 [DOI] [PubMed] [Google Scholar]
- Roll P, Vernes SC, Bruneau N, Cillario J, Ponsole‐Lenfant M, Massacrier A, Rudolf G, Khalife M, Hirsch E, Fisher SE et al (2010) Molecular networks implicated in speech‐related disorders: FOXP2 regulates the SRPX2/uPAR complex. Hum Mol Genet 19: 4848–4860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai Y, Shaw CA, Dawson BC, Dugas DV, Al‐Mohtaseb Z, Hill DE, Zoghbi HY (2011) Protein interactome reveals converging molecular pathways among autism disorders. Sci Transl Med 3: 86ra49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher J, Laje G, Jamra RA, Becker T, Mühleisen TW, Vasilescu C, Mattheisen M, Herms S, Hoffmann P, Hillmer AM et al (2009) The DISC locus and schizophrenia: evidence from an association study in a central European sample and from a meta‐analysis across different European populations. Hum Mol Genet 18: 2719–2727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Sawada J‐I, Sui G, Affar EB, Whetstine JR, Lan F, Ogawa H, Po‐Shan Luke M, Nakatani Y, Shi Y (2003) Coordinated histone modifications mediated by a CtBP co‐repressor complex. Nature 422: 735 [DOI] [PubMed] [Google Scholar]
- Shieh C, Jones N, Vanle B, Au M, Huang AY, Silva APG, Lee H, Douine ED, Otero MG, Choi A et al (2020) GATAD2B‐associatedneurodevelopmental disorder (GAND): clinical and molecular insights into a NuRD‐related disorder. Genet Med 22: 878–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu W, Yang H, Zhang L, Lu MM, Morrisey EE (2001) Characterization of a new subfamily of winged‐helix/forkhead (Fox) genes that are expressed in the lung and act as transcriptional repressors. J Biol Chem 276: 27488–27497 [DOI] [PubMed] [Google Scholar]
- Shu W, Cho JY, Jiang Y, Zhang M, Weisz D, Elder GA, Schmeidler J, De Gasperi R, Sosa M, Rabidou D et al (2005) Altered ultrasonic vocalization in mice with a disruption in the Foxp2 gene. Proc Natl Acad Sci USA 102: 9643–9648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sifrim A, Hitz M‐P, Wilsdon A, Breckpot J, Turki SHA, Thienpont B, McRae J, Fitzgerald TW, Singh T, Swaminathan GJ et al (2016) Distinct genetic architectures for syndromic and nonsyndromic congenital heart defects identified by exome sequencing. Nat Genet 48: 1060–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smogavec M, Cleall A, Hoyer J, Lederer D, Nassogne MC, Palmer EE, Deprez M, Benoit V, Maystadt I, Noakes C et al (2016) Eight further individuals with intellectual disability and epilepsy carrying bi‐allelic CNTNAP2 aberrations allow delineation of the mutational and phenotypic spectrum. J Med Genet 53: 820–827 [DOI] [PubMed] [Google Scholar]
- Snijders Blok L, Rousseau J, Twist J, Ehresmann S, Takaku M, Venselaar H, Rodan LH, Nowak CB, Douglas J, Swoboda KJ et al (2018) CHD3 helicase domain mutations cause a neurodevelopmental syndrome with macrocephaly and impaired speech and language. Nat Commun 9: 4619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijders Blok L, Vino A, den Hoed J, Underhill HR, Monteil D, Li H, Reynoso Santos FJ, Chung WK, Amaral MD, Schnur RE et al (2021) Heterozygous variants that disturb the transcriptional repressor activity of FOXP4 cause a developmental disorder with speech/language delays and multiple congenital abnormalities. Genet Med 23: 534–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sollis E, Graham SA, Vino A, Froehlich H, Vreeburg M, Dimitropoulou D, Gilissen C, Pfundt R, Rappold GA, Brunner HG et al (2016) Identification and functional characterization of de novo FOXP1 variants provides novel insights into the etiology of neurodevelopmental disorder. Hum Mol Genet 25: 546–557 [DOI] [PubMed] [Google Scholar]
- Sollis E, Deriziotis P, Saitsu H, Miyake N, Matsumoto N, Hoffer MJV, Ruivenkamp CAL, Alders M, Okamoto N, Bijlsma EK et al (2017) Equivalent missense variant in the FOXP2 and FOXP1 transcription factors causes distinct neurodevelopmental disorders. Hum Mutat 38: 1542–1554 [DOI] [PubMed] [Google Scholar]
- Spaniel F, Horacek J, Tintera J, Ibrahim I, Novak T, Cermak J, Klirova M, Hoschl C (2011) Genetic variation in FOXP2 alters grey matter concentrations in schizophrenia patients. Neurosci Lett 493: 131–135 [DOI] [PubMed] [Google Scholar]
- Spiteri E, Konopka G, Coppola G, Bomar J, Oldham M, Ou J, Vernes SC, Fisher SE, Ren B, Geschwind DH (2007) Identification of the transcriptional targets of FOXP2, a gene linked to speech and language, in developing human brain. Am J Hum Genet 81: 1144–1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S, Cohen JS, Vernon H, Baranano K, McClellan R, Jamal L, Naidu S, Fatemi A (2014) Clinical whole exome sequencing in child neurology practice. Ann Neurol 76: 473–483 [DOI] [PubMed] [Google Scholar]
- Strauss KA, Puffenberger EG, Huentelman MJ, Gottlieb S, Dobrin SE, Parod JM, Stephan DA, Morton DH (2006) Recessive symptomatic focal epilepsy and mutant contactin‐associated protein‐like 2. New Engl J Med 354: 1370–1377 [DOI] [PubMed] [Google Scholar]
- Sugitani Y, Nakai S, Minowa O, Nishi M, Jishage K, Kawano H, Mori K, Ogawa M, Noda T (2002) Brn‐1 and Brn‐2 share crucial roles in the production and positioning of mouse neocortical neurons. Genes Dev 16: 1760–1765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Liu FC, Hirokawa K, Takahashi H (2008) Expression of Foxp4 in the developing and adult rat forebrain. J Neurosci Res 86: 3106–3116 [DOI] [PubMed] [Google Scholar]
- Tam WY, Leung CK, Tong KK, Kwan KM (2011) Foxp4 is essential in maintenance of Purkinje cell dendritic arborization in the mouse cerebellum. Neuroscience 172: 562–571 [DOI] [PubMed] [Google Scholar]
- Thanseem I, Nakamura K, Miyachi T, Toyota T, Yamada S, Tsujii M, Tsuchiya KJ, Anitha A, Iwayama Y, Yamada K et al (2010) Further evidence for the role of MET in autism susceptibility. Neurosci Res 68: 137–141 [DOI] [PubMed] [Google Scholar]
- Toma C, Pierce KD, Shaw AD, Heath A, Mitchell PB, Schofield PR, Fullerton JM (2018) Comprehensive cross‐disorder analyses of CNTNAP2 suggest it is unlikely to be a primary risk gene for psychiatric disorders. PLoS Genet 14: e1007535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong JK, Hassig CA, Schnitzler GR, Kingston RE, Schreiber SL (1998) Chromatin deacetylation by an ATP‐dependent nucleosome remodelling complex. Nature 395: 917–921 [DOI] [PubMed] [Google Scholar]
- Torchy MP, Hamiche A, Klaholz BP (2015) Structure and function insights into the NuRD chromatin remodeling complex. Cell Mol Life Sci 72: 2491–2507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimarchi JM, Cho S‐H, Cepko CL (2009) Identification of genes expressed preferentially in the developing peripheral margin of the optic cup. Dev Dyn 238: 2327–2329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsui D, Vessey JP, Tomita H, Kaplan DR, Miller FD (2013) FoxP2 regulates neurogenesis during embryonic cortical development. J Neurosci 33: 244–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uddén J, Hultén A, Bendtz K, Mineroff Z, Kucera KS, Vino A, Fedorenko E, Hagoort P, Fisher SE (2019) Toward robust functional neuroimaging genetics of cognition. J Neurosci 39: 8778–8787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbanus BHA, Peter S, Fisher SE, De Zeeuw CI (2020) Region‐specific Foxp2 deletions in cortex, striatum or cerebellum cannot explain vocalization deficits observed in spontaneous global knockouts. Sci Rep 10: 21631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usui N, Co M, Harper M, Rieger MA, Dougherty JD, Konopka G (2017) Sumoylation of FOXP2 regulates motor function and vocal communication through purkinje cell development. Biol Psychiat 81: 220–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargha‐Khadem F, Watkins KE, Price CJ, Ashburner J, Alcock KJ, Connelly A, Frackowiak R, Friston KJ, Pembrey ME, Mishkin M et al (1998) Neural basis of an inherited speech and language disorder. Proc Natl Acad Sci USA 95: 12695–12700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernes SC, Nicod J, Elahi FM, Coventry JA, Kenny N, Coupe AM, Bird LE, Davies KE, Fisher SE (2006) Functional genetic analysis of mutations implicated in a human speech and language disorder. Hum Mol Genet 15: 3154–3167 [DOI] [PubMed] [Google Scholar]
- Vernes SC, Spiteri E, Nicod J, Groszer M, Taylor JM, Davies KE, Geschwind DH, Fisher SE (2007) High‐throughput analysis of promoter occupancy reveals direct neural targets of FOXP2, a gene mutated in speech and language disorders. Am J Hum Genet 81: 1232–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernes SC, Newbury DF, Abrahams BS, Winchester L, Nicod J, Groszer M, Alarcón M, Oliver PL, Davies KE, Geschwind DH et al (2008) A functional genetic link between distinct developmental language disorders. N Engl J Med 359: 2337–2345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernes SC, Oliver PL, Spiteri E, Lockstone HE, Puliyadi R, Taylor JM, Ho J, Mombereau C, Brewer A, Lowy E et al (2011) Foxp2 regulates gene networks implicated in neurite outgrowth in the developing brain. PLoS Genet 7: e1002145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernes SC (2017) What bats have to say about speech and language. Psychon Bull Rev 24: 111–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker RM, Hill AE, Newman AC, Hamilton G, Torrance HS, Anderson SM, Ogawa F, Derizioti P, Nicod J, Vernes SC et al (2012) The DISC1 promoter: characterization and regulation by FOXP2. Hum Mol Genet 21: 2862–2872 [DOI] [PubMed] [Google Scholar]
- Wang B, Lin D, Li C, Tucker P (2003) Multiple domains define the expression and regulatory properties of Foxp1 forkhead transcriptional repressors. J Biol Chem 278: 24259–24268 [DOI] [PubMed] [Google Scholar]
- Watkins KE, Vargha‐Khadem F, Ashburner J, Passingham RE, Connelly A, Friston KJ, Frackowiak RS, Mishkin M, Gadian DG (2002) MRI analysis of an inherited speech and language disorder: structural brain abnormalities. Brain 125: 465–478 [DOI] [PubMed] [Google Scholar]
- Weigel D, Jackle H (1990) The fork head domain: a novel DNA binding motif of eukaryotic transcription factors? Cell 63: 455–456 [DOI] [PubMed] [Google Scholar]
- Weiss K, Terhal PA, Cohen L, Bruccoleri M, Irving M, Martinez AF, Rosenfeld JA, Machol K, Yang Y, Liu P et al (2016) De novo mutations in CHD4, an ATP‐dependent chromatin remodeler gene, cause an intellectual disability syndrome with distinctive dysmorphisms. Am J Hum Genet 99: 934–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehouse AJO, Bishop DVM, Ang QW, Pennell CE, Fisher SE (2011) CNTNAP2 variants affect early language development in the general population. Genes Brain Behav 10: 451–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willemsen MH, Nijhof B, Fenckova M, Nillesen WM, Bongers EM, Castells‐Nobau A, Asztalos L, Viragh E, van Bon BW , Tezel E et al (2013) GATAD2B loss‐of‐function mutations cause a recognisable syndrome with intellectual disability and are associated with learning deficits and synaptic undergrowth in Drosophila . J Med Genet 50: 507–514 [DOI] [PubMed] [Google Scholar]
- Xiao L, Merullo DP, Koch TMI, Cao M, Co M, Kulkarni A, Konopka G, Roberts TF (2021) Expression of FoxP2 in the basal ganglia regulates vocal motor sequences in the adult songbird. Nat Commun 12: 2617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Wong J, Moreno GT, Young MK, Cote J, Wang W (1998) NURD, a novel complex with both ATP‐dependent chromatin‐remodeling and histone deacetylase activities. Mol Cell 2: 851–861 [DOI] [PubMed] [Google Scholar]
- Yang W‐M, Inouye C, Zeng Y, Bearss D, Seto E (1996) Transcriptional repression by YY1 is mediated by interaction with a mammalian homolog of the yeast global regulator RPD3. Proc Natl Acad Sci USA 93: 12845–12850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao YL, Yang WM, Seto E (2001) Regulation of transcription factor YY1 by acetylation and deacetylation. Mol Cell Biol 21: 5979–5991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasui D, Miyano M, Cai S, Varga‐Weisz P, Kohwi‐Shigematsu T (2002) SATB1 targets chromatin remodelling to regulate genes over long distances. Nature 419: 641–645 [DOI] [PubMed] [Google Scholar]
- Yin J, Jia N, Liu Y, Jin C, Zhang F, Yu S, Wang J, Yuan J (2018) No association between FOXP2 rs10447760 and schizophrenia in a replication study of the Chinese Han population. Psychiatr Genet 28: 19–23 [DOI] [PubMed] [Google Scholar]
- Zarate YA, Fish JL (2017) SATB2‐associated syndrome: mechanisms, phenotype, and practical recommendations. Am J Med Genet A 173: 327–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Duan N, Song T, Li Z, Zhang C, Chen X (2017) The emerging roles of forkhead box (FOX) proteins in osteosarcoma. J Cancer 8: 1619–1628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L‐J, Subramanian T, Vijayalingam S, Chinnadurai G (2014) CtBP2 proteome: role of CtBP in E2F7‐mediated repression and cell proliferation. Genes Cancer 5: 31–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweier C, de Jong EK , Zweier M, Orrico A, Ousager LB, Collins AL, Bijlsma EK, Oortveld MAW, Ekici AB, Reis A et al (2009) CNTNAP2 and NRXN1 are mutated in autosomal‐recessive Pitt‐Hopkins‐like mental retardation and determine the level of a common synaptic protein in Drosophila . Am J Hum Genet 85: 655–666 [DOI] [PMC free article] [PubMed] [Google Scholar]