

Abstract
Introduction
Diversity in cognition among apolipoprotein E (APOE) ε4 homozygotes can range from early‐onset Alzheimer's disease (AD) to a lifetime with no symptoms.
Methods
We evaluated a phenotypic extreme polygenic risk score (PRS) for AD between cognitively healthy APOE ε4 homozygotes aged ≥75 years (n = 213) and early‐onset APOE ε4 homozygote AD cases aged ≤65 years (n = 223) as an explanation for this diversity.
Results
The PRS for AD was significantly higher in APOE ε4 homozygote AD cases compared to older cognitively healthy APOE ε4/ε4 controls (odds ratio [OR] 8.39; confidence interval [CI] 2.0–35.2; P = .003). The difference in the same PRS between APOE ε3/ε3 extremes was not as significant (OR 3.13; CI 0.98–9.92; P = .053) despite similar numbers and power. There was no statistical difference in an educational attainment PRS between these age extreme case‐controls.
Discussion
A PRS for AD contributes to modified cognitive expression of the APOE ε4/ε4 genotype at phenotypic extremes of risk.
Keywords: Alzheimer's disease, Alzheimer's disease dementia, apolipoprotein E, dementia resilience, genetic modifiers, polygenic risk score
1. BACKGROUND
Alzheimer's disease (AD) has a strong underlying genetic component. 1 , 2 , 3 However, in the majority of individuals with non‐Mendelian AD, no single gene mutation can be identified as causative, with studies showing that AD is either an oligogenic or a polygenic disease. 4 , 5 , 6 The apolipoprotein E (APOE) ε4 allele has been identified as the single biggest risk factor. 7 The presence of APOE ε4 in the heterozygous form confers a 2‐ to 3‐fold increase in the odds of developing AD and in the homozygous form this confers up to a 14.9‐fold increase compared to the most common APOE genotype of ε3/ε3. 8 Moreover, the presence of APOE ε4 accelerates the age of onset (AOO) of AD, with the mean AOO being 84.3 years in non‐carriers as opposed to 68.4 years in those who are APOE ε4/ε4. 9
Despite the high risk for AD, it has been recognized that there is considerable phenotypic diversity among APOE ε4 homozygotes, ranging from early‐onset AD to a lifetime with no symptoms of cognitive impairment. 10 , 11 , 12 , 13 The reasons for this phenotypic diversity remain largely unexplained. Due to this variability in risk, APOE ε4 genotype, even in the homozygous state, has not demonstrated reliable utility for individual prediction of AD susceptibility or AOO of AD. 14 , 15 As there is a large polygenic component to AD, genetic factors beyond the APOE ε4 genotype may account for some of this modification in risk. Using data from large AD‐related genome‐wide association studies (GWAS), 16 , 17 , 18 polygenic risk scores (PRS) have been developed and used to predict risk for AD. 6 , 16 , 19 , 20 , 21 , 22 , 23 , 24 However, to our knowledge, no study has been designed specifically to examine the modification of risk by a PRS for AD between phenotypic extremes of the APOE ε4/ε4 risk spectrum. In this study, we investigate the role of an AD PRS, excluding the APOE region, as a potential modifier of risk between the two phenotypic extreme ends of the APOE ε4/ε4 AD risk spectrum, comparing the PRS between cognitively healthy older APOE ε4/ε4 controls without AD and APOE ε4/ε4 early‐onset clinically diagnosed AD cases.
2. METHODS
2.1. Participants
To compare a PRS for AD between the phenotypic extreme of the APOE ε4/ε4 risk spectrum, we obtained young onset AD cases and cognitively healthy older controls of European origin with APOE ε4/ε4 genotype from various cohorts. This included genotype data from APOE ε4/ε4 AD cases with AOO ≤ 65 years (n = 223) and cognitively healthy older APOE ε4/ε4 controls without a diagnosis of AD aged ≥ 75 years (n = 213).
Cases came from the Alzheimer's Disease Genetics Consortium (ADGC) (n = 200) and The Australian Imaging, Biomarkers & Lifestyle Flagship Study of Ageing (AIBL; n = 23). Diagnosis of probable AD in the cases was made using the Diagnostic and Statistical Manual of Mental Disorders (DSM‐IV) or the National Institute of Neurological and Communicative Disorders and Stroke– Alzheimer's Disease and Related Disorders Association (NINDS‐ADRDA) criteria or based on detailed clinical assessment in individual cohorts. Further details of these cohorts can be found in supporting information, the National Institute on Aging Genetics of Alzheimer's Disease Data Storage Site (NIAGADS; https://www.niagads.org/home), Kunkle et al., 18 and Ellis et al. 25
Cognitively healthy older APOE ε4/ε4 controls without a diagnosis of AD were from the Aspirin in Reducing Events in the Elderly (ASPREE) study 26 (n = 175), AIBL (n = 12), and ADGC (n = 26). ASPREE participants in this group had no clinical diagnosis of AD as determined by a multidisciplinary adjudicating committee and passed a test of global cognition (Modified Mini‐Mental State Examination [3MS] score of > 77) at enrolment. Control participants from AIBL had no clinical AD or mild cognitive impairment also determined by a multidisciplinary adjudicating committee. Controls with no reported clinical AD were also included from ADGC, in which individual cohorts used specifically designed cognitive screening criteria to determine “non demented” status (https://www.niagads.org/home). Any ADGC sample that was included in IGAP stage 1 or IGAP stage 2 in the GWAS by Lambert et al. 16 was excluded, to remove overlap with current PRS analysis. Demographic characteristics of cases and controls are shown in Table 1.
TABLE 1.
Demographic characteristics of APOE ε4/ε4 and APOE ε3/ε3 participants
| Characteristics | Young onset AD cases with APOE ε4/ε4 | Cognitively healthy older controls with APOE ε4/ε4 | Young onset cases with APOE ε3/ε3 | Cognitively healthy older controls with APOE ε3/ε3 |
|---|---|---|---|---|
| Total numbers | 223 | 213 | 223 | 213 |
| Numbers by cohort: | ||||
| ASPREE | 0 | 175 | 0 | 0 |
| AIBL | 23 | 12 | 0 | 0 |
| ADGC | 200 | 26 | 223 | 213 |
| Median AOO/AAA (range) in years | 62.5 (47–65) | 80.5 (75–91) | 57 (34–64) | 83 (76–97) |
| Female sex | 53.8% | 52.6% | 53.4% | 62.9% |
Abbreviations: AAA, age at assessment; AD, Alzheimer's disease; ADGC, Alzheimer's Disease Genetics Consortium; AIBL, The Australian Imaging, Biomarkers & Lifestyle Flagship Study of Ageing; AOO, age of onset; ASPREE, Aspirin in Reducing Events in the Elderly; APOE, apolipoprotein E.
Matched numbers of APOE ε3/ε3 AD cases (n = 223) with AOO ≤ 65 years and APOE ε3/ε3 cognitively healthy controls (n = 213) without AD aged ≥ 75 years were also included to compare the effect of the PRS in APOE ε3/ε3 extremes. Cases and controls for the APOE ε3/ε3 comparison were European ancestry participants, sourced from ADGC. As APOE ε3 is the most common genotype in the general population, this genotype was chosen for the comparison analysis. 27
This study was approved by the Royal Melbourne Hospital Ethics Committee (HREC/17/MH/444) for use of pre‐collected data. Ethical approval for the individual cohort participants was provided by their respective institutional ethics boards. All participants had provided DNA samples to the respective cohorts with consent for genotyping and data use. All patient data was anonymized prior to analysis. The reporting of this study follows the Strengthening the Reporting of Observational studies in Epidemiology (STROBE) guidelines for case‐control studies (https://www.strobe‐statement.org/index.php?id = strobe‐home).
2.2. Generating AD PRS in phenotypic extremes
A phenotypic extremes study design was used to select cases and controls for this study. 28 , 29 Study design is depicted in Figure 1.
FIGURE 1.
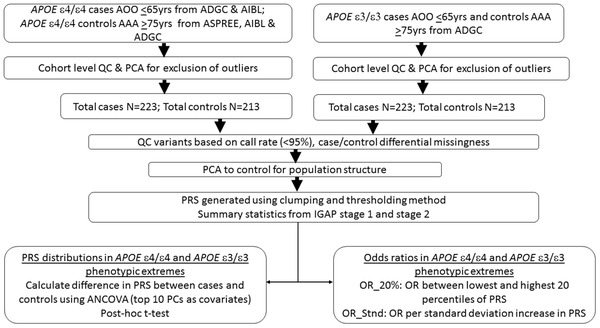
Flow‐chart detailing the study design. AAA, Age at assessment; ADGC, Alzheimer's Disease Genetics Consortium; AIBL, The Australian Imaging, Biomarkers & Lifestyle Flagship Study of Ageing; AOO, age of onset; ASPREE, Aspirin in Reducing Events in the Elderly study; IGAP, International Genomics of Alzheimer's Project; OR, odds ratio; PC, principal components; PRS, polygenic risk score; QC, quality control. Summary statistics for IGAP stage 1 and 2 samples derived from Lambert et al. 16 Principal component analysis (PCA) was done using first ten PCs based on the 1000 Genomes reference population
Detailed information on genotyping and quality control (QC) steps is included in supporting information. To mitigate the amount of technical variability introduced by combining samples from multiple cohorts, only samples that passed QC filters based on sex, relatedness, and European ancestry were included. Principal component analysis (PCA) on the top 10 principal components (PC) was done in each cohort to exclude outliers. Variants with call rates < 95% and those likely to have been improperly genotyped or imputed based on a test of Hardy‐Weinberg equilibrium were excluded. QC was repeated after merging the cohorts and PCA was again performed to control for population stratification.
RESEARCH IN CONTEXT
Systematic review: Despite the high risk for Alzheimer's disease (AD), there is considerable diversity in cognition among apolipoprotein E (APOE) ε4 homozygotes, ranging from early‐onset AD to a lifetime with no dementia. Literature review (PubMed) revealed that the reasons for this phenotypic diversity remain largely unexplained. In this study, we investigated the effect of a polygenic risk score (PRS) in this modification.
Interpretation: Using an extremes phenotype study model, we demonstrate that a PRS for AD contributes to modified cognitive expression of the APOE ε4/ε4 genotype.
Future directions: This study demonstrates an effective framework for investigation of risk modifiers in AD. A similar model can be used to investigate other AD risk modifiers. Conducting genome‐wide association studies using this framework, with larger participant numbers, may lead to discovery of novel risk‐modifying loci. Inclusion of AD risk modifiers along with APOE genotyping will aid in more accurate AD risk prediction.
PRS is calculated as a single score generated by aggregating the effects of genetic variants across the genome relevant for that particular trait. 30 As there is no published PRS available for a phenotypic and age extreme AD dataset of homozygous APOE genotypes, we undertook a clumping and thresholding method (described in supporting information) to generate a PRS in our age and phenotypic extreme APOE ε4/ε4 and APOE ε3/ε3 samples.
Clumping and thresholding is a common method used to compute PRS. Single nucleotide polymorphisms (SNPs) are first selected from GWAS summary statistics. The clumping step ensures that only variants that are weakly correlated with one another are retained in a pre‐specified window of the genome (in this case 1000 kilobase windows). Then the thresholding step is used to remove variants with a P‐value larger than a chosen level of significance (in this study, SNPs from the IGAP GWAS were threshold at r 2 > 0.1). Only the most significant P‐value threshold was used to select the SNPs that form the PRS.
The phenotypic extreme APOE ε4/ε4 as well as APOE ε3/ε3 participants (total cases n = 446; total controls n = 426) were combined to generate the PRS. To calculate PRS without APOE, variants within 750 kilobases of the start or end of the APOE gene (chr19:44659011‐ 46162650, hg19) were excluded. Effect sizes for the weighting of the SNPs used for the PRS generation was from the GWAS analysis by Lambert et al. 16 The more recent GWAS by Kunkle et al. was not used as the ADGC samples in this study overlap with their GWAS study. 18 The software PRSice‐2 was used to calculate and optimize PRS using clumping and thresholding. 31 , 32 The steps followed in the PRS generation are shown in Figure 1.
2.3. Statistical analysis
Using the PRS thus generated, the means of PRS distributions between the age extreme APOE ε4/ε4 cases and APOE ε4/ε4 controls were first analyzed using the statistical test analysis of covariance (with 10 PCs as covariates) and post hoc t test. This was done to check that there was significant difference in PRS between the cases and controls in each group before calculating odds ratios (OR). Subsequently, OR were calculated between the lowest and highest 20% of PRS (OR_20%) by performing logistic regressions between PRS quintile and AD status, in the APOE ε4/ε4 case‐controls, and also in the APOE ε3/ε3 case‐controls. OR were also calculated per standard deviation increase in PRS (OR_Stnd) by performing logistic regressions with Z‐standardized PRS as the predictor and AD status as the response. Level of significance was set at P < .05.
To verify if the variation in PRS was influenced by differences in other AD‐related risk factors in the APOE ε4/ε4 phenotypic extremes, we intended to check for differences in educational attainment between the cases and controls. Of the various modifiable risk factors, low level of education is the only trait to exhibit consistent association with AD. 33 Large GWAS studies have shown that genetically predicted education correlates with actual level of education and that high education attainment PRS is protective against AD. 34 As level of education was not available as a variable across the different cohorts included in our study, we calculated an education attainment PRS based on the GWAS study by Lee et al. 35 as a proxy for level of education in the APOE ε4/ε4 extremes. To generate the education attainment PRS, we applied clumping and thresholding to the Lee et al. GWAS using the same methods as described above for AD. The statistical package R (version 3.6.2) was used for statistical analysis and figures. 36
3. RESULTS
Out of a total of 5,295,512 SNPs that passed QC in the combined APOE ε4/ε4 and APOE ε3/ε3 age‐extreme samples, after excluding SNPs at a minor allele frequency (MAF) < 0.05 and clumping, 33,780 SNPs remained. These SNPs were then subjected to P‐value thresholding. Figure 2 shows the results of the clumping and thresholding process. The r 2 explained by PRS was calculated at IGAP GWAS P‐values from 5 × 10–08 to 1. There were 21 SNPs that fell in the most significant threshold, with the corresponding P‐value bracket being P < 5 × 10–08. This P‐value happened to correspond to the P‐value universally used to select the most significant SNPs in GWAS studies, that is, at genome‐wide significance level. Details of these SNPs are provided in Table S1 in supporting information.
FIGURE 2.
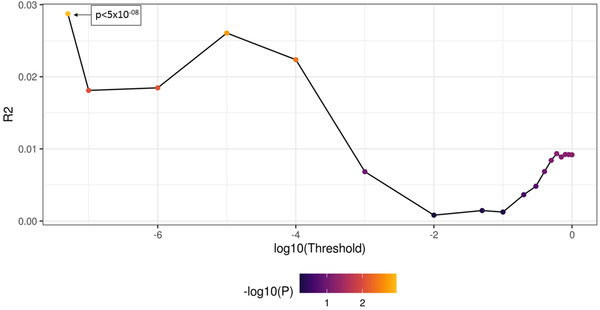
Line plot depicting the thresholding of single nucleotide polymorphisms. Each dot represents a different thresholding window. Best threshold in this case was at P < 5 × 10–08
The difference in means between the extreme cases and controls was significant in both the APOE ε4/ε4 case‐controls (P < .001) and the APOE ε3/ε3 case‐controls (P < .001) showing that the participants with AD have a significantly higher PRS compared to controls (Figure 3). The OR_20% in APOE ε4/ε4 extremes was 8.39 (confidence interval [CI] 2.0–35.2; P = .003), indicating a significant depletion of high risk PRS SNPs in the cognitively healthy older controls with APOE ε4/ε4. We also calculated the OR per standard deviation for the entire distribution of the APOE ε4/ε4 extreme cases and controls. OR_Stnd was 1.58 (CI 1.1–2.3; P = .013; Figure 4).
FIGURE 3.
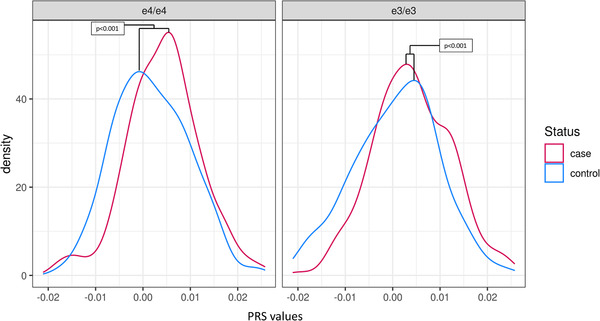
Density plot showing the difference in polygenic risk score distribution in cases and controls in the apolipoprotein E (APOE) ε4/ε4 extremes and APOE ε3/ε3 extremes
FIGURE 4.
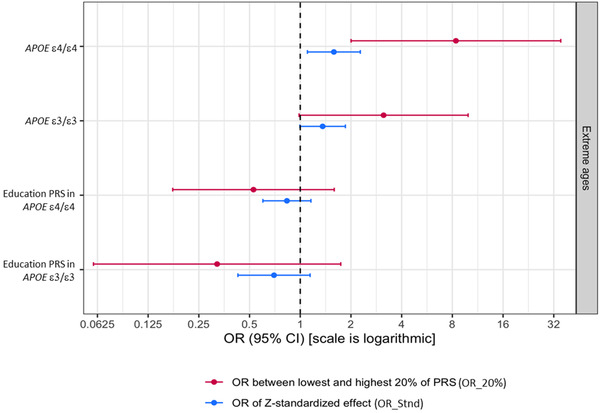
Odds ratio of risk‐modifying polygenic risk score (PRS) as well as education attainment PRS in apolipoprotein E (APOE) ε4/ε4 extremes and APOE ε3/ε3 extremes. CI, confidence interval; OR, odds ratio
As APOE ε3/ε3 is the most common genotype in the general population and considered the population reference, analysis in participants with this genotype was used as a comparison to determine whether there is a modifying effect of the PRS in APOE ε4‐negative phenotypic extremes. OR between the highest and lowest 20th percentile, that is, OR_20% was 3.13 (CI 0.98–9.92; P = .053), showing a relatively lower influence of this modifying PRS in the APOE ε3/ε3 phenotypic extremes, as opposed to the APOE ε4/ε4 phenotypic extremes at the two extreme quintile ends of the PRS distribution. OR_Stnd was 1.36 (CI 0.99–1.85; P = .054; Figure 4).
To clarify if the risk modification conferred by the PRS in APOE ε4/ε4s may have been influenced by a difference in education attainment, we checked for differences in genetically determined education attainment between the APOE ε4/ε4 cases and controls as well as the APOE ε3/ε3 cases and controls. The education attainment PRS was more polygenic with 24,502 SNPs falling under the most significant threshold of P = .3. The education attainment PRS was not significantly different between the APOE ε4/ε4 extreme cases and controls with OR_20% 0.52 (CI 0.17–1.60; P = .26) and OR_Stnd 0.83 (CI 0.6–1.16; P = .28), indicating that the influence of genetically determined education attainment was not confounding. Similarly, there was no statistically significant difference in the education attainment PRS between the APOE ε3/ε3 extreme cases and controls with OR_20% being 3.12 (CI 0.98–9.92; P = .05) and OR_Stnd being 0.70 (CI 0.42–1.14; P = .15; Figure 4).
4. DISCUSSION
In this study, we compared a PRS between phenotypic extremes of the APOE ε4/ε4 spectrum and demonstrated that the PRS was significantly higher in APOE ε4 homozygotes diagnosed with AD earlier in life, compared to APOE ε4 homozygotes who remained unaffected by AD to an advanced age. The PRS was also compared between a matched number of APOE ε3/ε3 young onset cases and unaffected controls, but was not as significant as the APOE ε4/ε4 case‐controls. Our findings illustrate how genetic risk modification in AD can be driven by common AD‐associated variants beyond the APOE locus, and that this risk modification may partially explain the phenotypic diversity among APOE ε4 homozygotes.
Our extreme phenotyping study design increased the ability to detect this PRS modifying effect. Extreme phenotyping is known to increase statistical power and variant effect sizes, enabling better identification of SNPs strongly associated with a trait. 28 For AD, an extreme phenotyping study translates to comparing risk factors between those at the highest risk, that is, AD cases with APOE ε4/ε4 and onset ≤ 65 years, with those who are most resilient to AD, being those with APOE ε4/ε4 genotype, aged ≥ 75 years and no AD. 37 Here, we have identified a PRS that modifies risk between the phenotypic extreme ends of the APOE ε4/ε4 spectrum. This finding has important implications for the potential risk stratification of this high‐risk genotype.
The loci that yielded the 21 SNPs forming risk‐modifying PRS in this study have all been previously described in AD GWAS with no new loci found in this study. This shows that the currently known non‐APOE loci still play an important role in risk modification. Thus far, up to 44 loci have been reported to be associated with AD in large GWAS. 5 , 16 , 17 , 18 , 38 , 39 However, it remains unresolved if AD is oligogenic, with risk determined by a smaller number of SNPs compared to other common diseases such as coronary artery disease and cancer; or is polygenic with similar genetic architecture to these diseases. 5 , 6 The present analysis has demonstrated that a detectable risk‐modifying effect in APOE ε4/ε4 extremes is driven by a relatively small number of SNPs in the common frequency range. Our study showed that a lower burden of some non‐APOE SNPs in APOE ε4 homozygotes could buffer disease risk and delay AD onset to ≥ 75 years.
An improved understanding of the modifying effect of PRS on the APOE ε4/ε4 genotype could assist in increasing the accuracy of risk prediction for AD. Similar risk modification by PRS has been shown recently in the context of autosomal dominant adult‐onset monogenic conditions, in which polygenic factors have been shown to modify the penetrance of clinically significant monogenic variants. 40 Such improved risk stratification is useful in identifying people at increased risk or at decreased risk despite their APOE ε4/ε4 genotype.
In the clinical setting, the APOE genotype has posed several challenges. The variability in AD phenotype despite the high risk has meant that testing for the APOE ε4 genotype has been discouraged by the American College of Medical Genetics and Genomics, especially in the predictive context in asymptomatic individuals. 41 Addition of PRS to the APOE ε4/ε4 genotyping increases the predictive value of such testing and may allow the incorporation of APOE ε4/ε4 testing in the clinic, where more accurate prediction of the chances of developing AD is considered useful. It will also become more relevant as effective therapies for AD are developed.
Although phenotypic variability may also be true for the heterozygous APOE ε4 genotype, given that the elevated OR of developing late‐onset AD in APOE ε4 homozygotes (up to 14.9) is markedly different from having one APOE ε4 allele (up to 4), the factors that modify the risk in APOE ε4 heterozygotes will be much broader, possibly with smaller effect sizes compared to those modifying APOE ε4/ε4 risk. 8 The predictive value for AD by inclusion of a modifying PRS in addition to APOE ε4/ε4 genotyping would be significantly higher than the predictive value of adding a modifying PRS to APOE ε4 heterozygotes. The APOE ε4/ε4 modifying PRS, especially in the extremes of phenotypes as described in this study, is therefore valuable in selecting appropriate participants for study of risk and resilience and will also contribute toward a better understanding of the genetic and non‐genetic factors underpinning AD and how they interact.
We were able to successfully incorporate an extreme phenotype design to identify the modifying PRS by using well‐phenotyped, resilient older controls in our study. Resilient controls are defined as those that do not develop a particular condition, despite being at a high risk for developing it. As the average age of onset of AD in APOE ε4 homozygotes is 68.4 years, 9 those who have the APOE ε4/ε4 genotype and are aged at least 75 years or older without major cognitive impairments, can be considered to be harboring factors that buffer the development of AD, despite their high risk. In some previous AD case‐control studies, participants too young be considered controls for AD have been used. 42 , 43 , 44 This confounds the ability to accurately determine risk‐modifying factors, as many of the controls may go on to develop AD when older. In the current study, we had access to well‐phenotyped, advanced aged elderly control cohorts of APOE ε4/ε4 participants, who fit the definition of resilience for AD. Using appropriately phenotyped extreme cases and controls strengthens the ability to find meaningful modifying factors.
Although many lifestyle and environmental factors can also play a part in the modification of risk of AD, no single environmental/lifestyle risk factor has been shown to be strongly associated with AD. 33 A recent study analyzing causal associations between various modifiable risk factors and the AD phenome, using PRS and Mendelian randomization, showed only genetically determined education attainment was causally associated with decreased risk of AD, delayed AOO, and increased cortical surface area and thickness. 45 Studies have shown that the effect of education is particularly prominent in early years and that the effect of education is difficult to separate out from overall cognitive ability. 46 , 47 In a large GWAS study, Lee et al. were able to show that the SNPs associated with education attainment explained a significant proportion of educational variance. 35 We were also able to show that the difference in the APOE ε4/ε4 modifying PRS was not influenced by the difference in education attainment PRS.
4.1. Limitations
The main constraint in following an approach of extreme phenotyping for AD is the reduced number of participants available for the study. Although our results are encouraging, the number of participants in our study was still relatively small. It is likely that the current PRS used in our study only captures a fraction of the genetic variation or divergence that may drive phenotypic expression between APOE ε4/ε4 extremes. The smaller size has limited the power of our study to identify novel SNPs from the existing GWAS data. A substantially larger number of participants would be required to perform an independent GWAS using the extreme phenotype approach or to investigate the role of rarer variants of strong effect. Larger independent studies are also needed to validate the results from our study. However, ascertaining individuals at either end of the APOE ε4/ε4 risk spectrum is particularly challenging given that the population genotype frequency of APOE ε4/ε4 in Europeans is only 2%. This necessitated combining samples from multiple cohorts in our analysis, which may have introduced some technical variation between cohorts and issues related to population stratification. We have tried to account for this by various QC checks and PCA, but acknowledge that despite this, there may be dissimilarities between the cohorts.
Moreover, risk prediction for AD remains complicated due to the complex genetic–environmental interactions and likely involvement of epigenetic mechanisms. We acknowledge that despite having small effect sizes individually, in combination, many lifestyle/environmental factors may play a larger part in modifying AD risk and this effect could not be accounted for in the present study. 48
There may also be other rare, high‐effect genetic variants influencing risk or resilience that have not been captured by our analysis. In addition, our analysis does not cover genomic structural variants such as deletions, duplications, and short tandem repeats that may contribute toward modification of AD risk. It is also to be noted that this PRS is not transferable to the non‐White population as our study population was predominantly of European White ethnicity.
5. CONCLUSIONS
In conclusion, our study demonstrates that a PRS for AD modifies the phenotypic expression of AD between extreme ends of the APOE ε4/ε4 risk spectrum. This suggests that common genetic variants beyond the APOE locus contribute to risk modification in AD, yet it is likely that far more genetic and non‐genetic factors contribute, beyond those captured by the PRS. Further studies are required to better understand the underlying biology of genetic risk modifiers in AD. Although not available in all our study cohorts, positron emission tomography or cerebrospinal fluid biomarkers of amyloid beta (Aβ) should be explored in the resilient controls in future studies to investigate if the non‐amyloidogenic loci represented by the 21 SNPs described here counter the effects of Aβ in the brain.
CONFLICTS OF INTEREST
AMG has consulted for Eisai, Biogen, Pfizer, AbbVie, Cognition Therapeutics, and GSK. She also served on the SAB at Denali Therapeutics from 2015–2018. RCS serves as a non‐compensated member of the Board of Directors of the Alzheimer's Association–Illinois Chapter. RCS's institution, Rush University Medical Center, receives research support for his role as a Site Principal Investigator or Site Sub‐Investigator for industry initiated clinical trials and research studies of Alzheimer's disease sponsored by Amylyx Pharmaceuticals, Inc.; Eli Lilly & Co., Inc.; Genentech, Inc.; Lundbeck, Inc.; Merck & Co., Inc.; Navidea Biopharmaceuticals; Novartis Pharmaceuticals, Inc.; Roche Holdings AG; and Takeda Development Center Americas, Inc. All other authors have no interests to declare.
Supporting information
Supplementary information
ACKNOWLEDGMENTS
ASPREE is supported by a Flagship cluster grant (including the Commonwealth Scientific and Industrial Research Organisation, Monash University, Menzies Research Institute, Australian National University, University of Melbourne); and grants (U01AG029824) from the National Institute on Aging and the National Cancer Institute at the National Institutes of Health, by grants (334047 and 1127060) from the National Health and Medical Research Council of Australia, and by Monash University and the Victorian Cancer Agency. Paul Lacaze is supported by a National Heart Foundation Future Leader Fellowship. Dr. Raj Shah serves as a non‐compensated member of the Board of Directors of the Alzheimer's Association–Illinois Chapter. Dr. Shah's institution, Rush University Medical Center, receives research support for his role as a Site Principal Investigator or Site Sub‐Investigator for industry‐initiated clinical trials and research studies of Alzheimer's disease sponsored by Amylyx Pharmaceuticals, Inc.; Eli Lilly & Co., Inc.; Genentech, Inc.; Lundbeck, Inc.; Merck & Co., Inc.; Navidea Biopharmaceuticals; Novartis Pharmaceuticals, Inc.; Roche Holdings AG; and Takeda Development Center Americas, Inc. The National Institutes of Health, National Institute on Aging (NIH‐NIA) supported this work through the following grants: ADGC, U01 AG032984, RC2 AG036528. Samples from the National Cell Repository for Alzheimer's Disease (NCRAD), which receives government support under a cooperative agreement grant (U24 AG21886) awarded by the National Institute on Aging (NIA), were used in this study. We thank contributors who collected samples used in this study, as well as patients and their families, whose help and participation made this work possible. The NACC database is funded by NIA/NIH Grant U01 AG016976. Data for this study were prepared, archived, and distributed by the National Institute on Aging Alzheimer's Disease Data Storage Site (NIAGADS) at the University of Pennsylvania (U24‐AG041689‐01). Dr. Aamira Huq is supported by the Commonwealth Research Training Program Scholarship, Australia (University of Melbourne reference number 96756) and the Yulgilbar Alzheimer Research Program. The authors would also like to acknowledge and thank all the participants in the various cohorts that took part in this study, and their families.
Huq AJ, Fulton‐Howard B, Riaz M, et al. Polygenic score modifies risk for Alzheimer's disease in APOE ε4 homozygotes at phenotypic extremes. Alzheimer's Dement. 2021;13:e12226. 10.1002/dad2.12226
Aamira J. Huq and Brian Fulton‐Howard are joint first authors.
REFERENCES
- 1. Gatz M, Mortimer JA, Fratiglioni L, et al. Potentially modifiable risk factors for dementia in identical twins. Alzheimers Dement. 2006;2:110‐117. [DOI] [PubMed] [Google Scholar]
- 2. Kukull WA, Higdon R, Bowen JD, et al. Dementia and Alzheimer disease incidence: a prospective cohort study. Arch Neurol. 2002;59:1737‐1746. [DOI] [PubMed] [Google Scholar]
- 3. Ridge PG, Hoyt KB, Boehme K, et al. Assessment of the genetic variance of late‐onset Alzheimer's disease. Neurobiol Aging. 2016;41:200 e13‐ e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Neuner SM, Tcw J, Goate AM. Genetic architecture of Alzheimer's disease. Neurobiol Dis. 2020;143:104976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhang Q, Sidorenko J, Couvy‐Duchesne B, et al. Risk prediction of late‐onset Alzheimer's disease implies an oligogenic architecture. Nat Commun. 2020;11:4799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Escott‐Price V, Sims R, Bannister C, et al. Common polygenic variation enhances risk prediction for Alzheimer's disease. Brain. 2015;138:3673‐3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Michaelson DM. APOE epsilon4: the most prevalent yet understudied risk factor for Alzheimer's disease. Alzheimers Dement. 2014;10:861‐868. [DOI] [PubMed] [Google Scholar]
- 8. Farrer LA, Cupples LA, Haines JL, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta‐analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA. 1997;278:1349‐1356. [PubMed] [Google Scholar]
- 9. Corder EH, Saunders AM, Strittmatter WJ, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993;261:921‐923. [DOI] [PubMed] [Google Scholar]
- 10. Wisdom NM, Callahan JL, Hawkins KA. The effects of apolipoprotein E on non‐impaired cognitive functioning: a meta‐analysis. Neurobiol Aging. 2011;32:63‐74. [DOI] [PubMed] [Google Scholar]
- 11. Davies G, Armstrong N, Bis JC, et al. Genetic contributions to variation in general cognitive function: a meta‐analysis of genome‐wide association studies in the CHARGE consortium (N = 53949). Mol Psychiatry. 2015;20:183‐192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Marioni RE, Campbell A, Scotland G, Hayward C, Porteous DJ, Deary IJ. Differential effects of the APOE e4 allele on different domains of cognitive ability across the life‐course. Eur J Hum Genet. 2016;24:919‐923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lyall DM, Ward J, Ritchie SJ, et al. Alzheimer disease genetic risk factor APOE e4 and cognitive abilities in 111,739 UK Biobank participants. Age Ageing. 2016;45:511‐517. [DOI] [PubMed] [Google Scholar]
- 14. Mayeux R, Saunders AM, Shea S, et al. Utility of the apolipoprotein E genotype in the diagnosis of Alzheimer's disease. Alzheimer's Disease Centers Consortium on Apolipoprotein E and Alzheimer's Disease. N Engl J Med. 1998;338:506‐511. [DOI] [PubMed] [Google Scholar]
- 15. Goldman JS, Hahn SE, Catania JW, et al. Genetic counseling and testing for Alzheimer disease: joint practice guidelines of the American College of Medical Genetics and the National Society of Genetic Counselors. Genet Med. 2011;13:597‐605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lambert JC, Ibrahim‐Verbaas CA, Harold D, et al. Meta‐analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet. 2013;45:1452‐1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jansen IE, Savage JE, Watanabe K, et al. Genome‐wide meta‐analysis identifies new loci and functional pathways influencing Alzheimer's disease risk. Nat Genet. 2019;51:404‐413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kunkle BW, Grenier‐Boley B, Sims R, et al. Genetic meta‐analysis of diagnosed Alzheimer's disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat Genet. 2019;51:414‐430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chouraki V, Reitz C, Maury F, et al. Evaluation of a genetic risk score to improve risk prediction for Alzheimer's disease. J Alzheimers Dis. 2016;53:921‐932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Escott‐Price V, Shoai M, Pither R, Williams J, Hardy J. Polygenic score prediction captures nearly all common genetic risk for Alzheimer's disease. Neurobiol Aging. 2017;49:214 e7‐ e11. [DOI] [PubMed] [Google Scholar]
- 21. Desikan RS, Fan CC, Wang Y, et al. Genetic assessment of age‐associated Alzheimer disease risk: development and validation of a polygenic hazard score. PLoS Med. 2017;14:e1002258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mormino EC, Sperling RA, Holmes AJ, et al. Polygenic risk of Alzheimer disease is associated with early‐ and late‐life processes. Neurology. 2016;87:481‐488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Martiskainen H, Helisalmi S, Viswanathan J. Effects of Alzheimer's disease‐associated risk loci on cerebrospinal fluid biomarkers and disease progression: a polygenic risk score approach. J Alzheimers Dis. 2015;43:565‐573. [DOI] [PubMed] [Google Scholar]
- 24. Lacour A, Espinosa A, Louwersheimer E, et al. Genome‐wide significant risk factors for Alzheimer's disease: role in progression to dementia due to Alzheimer's disease among subjects with mild cognitive impairment. Mol Psychiatry. 2017;22:153‐160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ellis KA, Bush AI, Darby D, et al. The Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging: methodology and baseline characteristics of 1112 individuals recruited for a longitudinal study of Alzheimer's disease. Int Psychogeriatr. 2009;21:672‐687. [DOI] [PubMed] [Google Scholar]
- 26. McNeil JJ, Woods RL, Nelson MR, et al. Baseline characteristics of participants in the ASPREE (ASPirin in Reducing Events in the Elderly) study. J Gerontol A Biol Sci Med Sci. 2017;72:1586‐1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Corbo RM, Scacchi R. Apolipoprotein E (APOE) allele distribution in the world. Is APOE*4 a ‘thrifty’ allele?. Ann Hum Genet. 1999;63:301‐310. [DOI] [PubMed] [Google Scholar]
- 28. Tesi N, van der Lee SJ, Hulsman M, et al. Centenarian controls increase variant effect sizes by an average twofold in an extreme case‐extreme control analysis of Alzheimer's disease. Eur J Hum Genet. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Barnett IJ, Lee S, Lin X. Detecting rare variant effects using extreme phenotype sampling in sequencing association studies. Genet Epidemiol. 2013;37:142‐151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lambert SA, Abraham G, Inouye M. Towards clinical utility of polygenic risk scores. Hum Mol Genet. 2019;28:R133‐R42. [DOI] [PubMed] [Google Scholar]
- 31. Choi SW, O'Reilly PF. PRSice‐2: polygenic Risk Score software for biobank‐scale data. Gigascience. 2019;8:giz082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Euesden J, Lewis CM, PRSice O'ReillyPF. Polygenic Risk Score software. Bioinformatics. 2015;31:1466‐1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Xu W, Tan L, Wang HF, et al. Meta‐analysis of modifiable risk factors for Alzheimer's disease. J Neurol Neurosurg Psychiatry. 2015;86:1299‐1306. [DOI] [PubMed] [Google Scholar]
- 34. Larsson SC, Traylor M, Malik R, et al. Modifiable pathways in Alzheimer's disease: mendelian randomisation analysis. BMJ. 2017;359:j5375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lee JJ, Wedow R, Okbay A, et al. Gene discovery and polygenic prediction from a genome‐wide association study of educational attainment in 1.1 million individuals. Nat Genet. 2018;50:1112‐1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chan BKC. Data analysis using R programming. Adv Exp Med Biol. 2018;1082:47‐122. [DOI] [PubMed] [Google Scholar]
- 37. Huq AJ, Fransquet P, Laws SM, et al. Genetic resilience to Alzheimer's disease in APOE epsilon4 homozygotes: a systematic review. Alzheimers Dement. 2019;15:1612‐1623. [DOI] [PubMed] [Google Scholar]
- 38. Khera AV, Chaffin M, Aragam KG, et al. Genome‐wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet. 2018;50:1219‐1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Andrews SJ, Fulton‐Howard B, Goate A. Interpretation of risk loci from genome‐wide association studies of Alzheimer's disease. Lancet Neurol. 2020;19:326‐335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Barnes DR, Rookus MA, McGuffog L, et al. Polygenic risk scores and breast and epithelial ovarian cancer risks for carriers of BRCA1 and BRCA2 pathogenic variants. Genet Med. 2020;22(10):1653‐1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Choosing_Wisely. American College of Medical Genetics and Genomics: Five Things Physicians and Patients Should Question. 2017.
- 42. Kamboh MI, Sanghera DK, Ferrell RE, DeKosky ST. APOE*4‐associated Alzheimer's disease risk is modified by alpha1‐ antichymotrypsin polymorphism. Nature Genetics. 1995;10:486‐488. [DOI] [PubMed] [Google Scholar]
- 43. DeKosky ST, Aston CE, Kamboh MI. Polygenic determinants of Alzheimer's disease: modulation of the risk by alpha‐1‐antichymotrypsin. Annals of the New York Academy of Sciences. 1996;802:27‐34. [DOI] [PubMed] [Google Scholar]
- 44. Harold D, Abraham R, Hollingworth P, et al. Genome‐wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat Genet. 2009;41:1088‐1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Andrews SJ, Fulton‐Howard B, O'Reilly P, Marcora E, Goate AM. collaborators of the Alzheimer's Disease Genetics C. Causal associations between modifiable risk factors and the Alzheimer's phenome. Ann Neurol. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Livingston G, Huntley J, Sommerlad A, et al. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet. 2020;396:413‐446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kremen WS, Beck A, Elman JA, et al. Influence of young adult cognitive ability and additional education on later‐life cognition. Proc Natl Acad Sci U S A. 2019;116:2021‐2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Livingston G, Sommerlad A, Orgeta V, et al. Dementia prevention, intervention, and care. Lancet. 2017;390:2673‐2734. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information