

Abstract
Aurora kinase A (AURKA) is a conserved kinase that plays crucial roles in numerous cellular processes. Although AURKA overexpression is frequent in human cancers, its pleiotropic functions and multifaceted regulation present challenges in its therapeutic targeting. Key to overcoming these challenges is to identify and characterize the full range of AURKA interactors, which are often weak and transient. Previous proteomic studies were limited in monitoring dynamic and non‐mitotic AURKA interactions. Here, we generate the proximity interactome of AURKA in asynchronous cells, which consists of 440 proteins involving multiple biological processes and cellular compartments. Importantly, AURKA has extensive proximate and physical interactions to centriolar satellites, key regulators of the primary cilium. Loss‐of‐function experiments identify satellites as negative regulators of AURKA activity, abundance, and localization in quiescent cells. Notably, loss of satellites activates AURKA at the basal body, decreases centrosomal IFT88 levels, and causes ciliogenesis defects. Collectively, our results provide a resource for dissecting spatiotemporal regulation of AURKA and uncover its proteostatic regulation by satellites as a new mechanism for its ciliary functions.
Keywords: Aurora Kinase A, BioID, centriolar satellites, centrosome, primary cilium
Subject Categories: Cell Adhesion, Polarity & Cytoskeleton; Cell Cycle
This study presents the in vivo proximity interactome of AURKA in asynchronous cells, which reveals a new regulatory and functional relationship between AURKA and centriolar satellites.

Introduction
Aurora kinase A (AURKA) is a member of the evolutionarily conserved Aurora serine/threonine kinase family and a pleiotropic regulator of cellular processes that are deregulated in cancer. Genetic amplification and overexpression of AURKA are prevalent in various solid and hematologic malignancies, and it correlates with aggressive cancer progression and poor survival (Nikonova et al, 2013). AURKA overexpression was shown to induce oncogenic phenotypes including aneuploidy, centrosome amplification, mitotic spindle defects, and resistance to apoptosis in cultured cell lines and mammary tumors in mice (Bischoff et al, 1998; Zhang et al, 2004; Wang et al, 2006; Zhang et al, 2008; Nikonova et al, 2013; Treekitkarnmongkol et al, 2016; Wang et al, 2019). Due to its causal link to cancer, AURKA has been extensively investigated as a drug target for anti‐cancer therapeutics. Although multiple small molecule inhibitors of AURKA activity such as alisertib (MLN8237) were shown to repress growth and progression of various cancers in vitro and in vivo, the inhibitors did not exhibit similar efficacies in cancer patients in clinical trials (Maris et al, 2010; Manfredi et al, 2011; Brockmann et al, 2013; Nikonova et al, 2013). An emerging therapeutic paradigm for overcoming the clinical limitations of monotherapies is combining AURKA inhibitors with drugs targeting cancer‐related AURKA functions and interactors (Chen & Lahav, 2016; Tang et al, 2017). Designing specific and effective combinatorial treatments requires identification of the full extent of AURKA interactors and activities in cells.
Since its discovery as a mitotic kinase, AURKA has been extensively studied for its mitotic functions and regulation (Glover et al, 1995; Barr & Gergely, 2007; Hegarat et al, 2011; Lioutas & Vernos, 2013; Nikonova et al, 2013; Courtheoux et al, 2018). AURKA interacts with and/or phosphorylates multiple proteins at the centrosomes and mitotic spindle such as PLK1, NDEL1, TACC, and CDC25B and is required for centrosome maturation, mitotic entry and exit, spindle assembly, and cytokinesis (Dutertre et al, 2004; Barr & Gergely, 2007; LeRoy et al, 2007; Mori et al, 2007; Joukov & De Nicolo, 2018). Spatiotemporal regulation of the pleiotropic functions of AURKA during mitosis is achieved by tight control of its activity, localization, and stability. More recently, non‐mitotic AURKA functions have been discovered, which include primary cilium biogenesis (Plotnikova et al, 2012; Golemis et al, 2018), stability and transcriptional activity of N‐Myc (Otto et al, 2009; Buchel et al, 2017), DNA damage repair and replication fork stability (Byrum et al, 2019), mitochondrial morphology and function (Kashatus et al, 2011; Bertolin et al, 2018), and neurite extension (Liem et al, 2009). Consistent with its diverse functions, AURKA has multiple cellular pools with differential kinase activity and substrate specificity (Willems et al, 2018). Little is known about how AURKA mediates its diverse non‐mitotic functions and the mechanisms by which it is regulated in these contexts. This is in part due to lack of systematic studies for identification of the spatially and temporally constrained interactions that govern the non‐mitotic regulation of AURKA. Addressing these questions is essential for designing combinatorial therapeutics targeting cancers linked to AURKA overexpression and to prevent the unanticipated adverse effects of AURKA inhibition (Gautschi et al, 2008; Perez de Castro et al, 2008; Vader & Lens, 2008; Karthigeyan et al, 2011; Tang et al, 2017).
The primary cilium functions as the signaling center of cells, and its biogenesis is tightly coordinated with cell cycle (Mirvis et al, 2018; Conkar & Firat‐Karalar, 2020). The relationship between AURKA and cilia was first described by studies that identified AURKA as a regulator of flagellar disassembly in Chlamydomonas reinhardtii and primary cilium disassembly in mammalian cells (Pan et al, 2004; DeVaul et al, 2017; Korobeynikov et al, 2017). Upon induction of cilium disassembly by growth factor stimulation, AURKA is activated at the basal body by HEF1/NEDD1 (Pugacheva et al, 2007). Active AURKA phosphorylates Histone Deacetylase 6 (HDAC6), which promotes deacetylation of axonemal microtubules and ciliary resorption. Additionally, Ca2+/calmodulin (CaM) (Plotnikova et al, 2012), trichoplein (TCHP) (Inoko et al, 2012), nuclear distribution element (NDE)‐like 1 (NDEL1) (Inaba et al, 2016), Pitchfork (Pifo) (Kinzel et al, 2010), and von Hippel‐Lindau (VHL) (Hasanov et al, 2017) were shown to regulate AURKA abundance and activation during cilium disassembly. In ciliated cells, AURKA is degraded and kept inactive to assemble and maintain primary cilium (Korobeynikov et al, 2017). Aberrant activation of AURKA in quiescent cells disrupts basal body biogenesis (i.e., recruitment of appendage proteins, IFT machinery) and upregulates the ciliary resorption pathway, which together identified AURKA as a negative regulator of cilium assembly (Kim et al, 2011; Inaba et al, 2016; Pejskova et al, 2020). The mechanisms by which AURKA localization, stability, and abundance are regulated in quiescent cells remain poorly understood.
Our current knowledge of the AURKA interactome is largely based on conventional proteomic and computational‐based identification of its mitotic substrates and regulators (Dephoure et al, 2008; Kettenbach et al, 2011; Koch et al, 2011; Santamaria et al, 2011; Deretic et al, 2019). Given the highly dynamic spatiotemporal regulation of AURKA interactome as well as its association with cytoskeletal structures (i.e., centrosomes, microtubules), these approaches were limited in probing the weak, transient, and insoluble AURKA interactors such as enzyme‐substrate interactions and interactions with cytoskeletal structures. Proximity‐based labeling combined with mass spectrometry has emerged as a powerful approach to circumvent these inherent challenges of traditional biochemical methods (Roux, 2013; Chen & Perrimon, 2017; Gentzel et al, 2019; Trinkle‐Mulcahy, 2019).
In this study, we applied the in vivo proximity‐dependent biotin identification (BioID) approach to map the proximity interactome of AURKA of asynchronous cells. BioID makes use of the promiscuous E. coli biotin ligase BirA(R118G) (hereafter BirA*), which enables covalent biotinylation of proteins within about 10–20 nm proximity of BirA*‐fusions of bait proteins (Roux et al, 2012; Roux et al, 2013; Roux, 2013; Lambert et al, 2014; Chen & Perrimon, 2017; Gentzel et al, 2019; Arslanhan et al, 2020). The resulting interactome provides a powerful resource for dissecting the dynamic AURKA sub‐complexes that enable its multifaceted regulation and diverse AURKA functions. Importantly, it revealed previously undescribed, extensive connections to centriolar satellites, which we verified by affinity pulldown and functional analysis. We showed that AURKA and PCM1 have antagonistic functions during basal body maturation and cilium assembly. Our results described proteostatic regulation of AURKA by centriolar satellites as a new regulatory mechanism for its functions during ciliogenesis.
Results
BirA*‐AURKA localizes and induces biotinylation at the centrosomes and spindle microtubules
To identify the AURKA proximity interactome, we applied BioID approach to human AURKA in human embryonic kidney (HEK293T) cells, which is a widely used cell line for proximity proteomics and thus makes benchmarking easier. As depicted in Fig 1A, our experimental workflow consists of generation of cells stably expressing V5‐BirA*‐AURKA or V5‐BirA* (control), induction of proximate biotinylation, pulldown of biotinylated proteins, and mass spectrometry analysis. Since BirA* covalently marks all proteins proximate to the bait, 18‐h biotin stimulation of asynchronous cells will reveal proximate relationships of AURKA during its entire lifetime (Roux, 2013). We generated stable cells that overexpress V5‐BirA*‐AURKA fusion to a level similar to the endogenous protein (Fig 1B). Because AURKA overexpression has dominant‐active cellular phenotypes such as centrosome amplification, spindle multipolarity, and multinucleation (Meraldi et al, 2002; Anand et al, 2003), we characterized the stable line for these phenotypes. To this end, we immunostained HEK293T::V5‐BirA*‐AURKA and control cells for markers of the centrosome, microtubules, and DNA and quantified their mitotic phenotypes. For both cell lines, there was no difference in the percentage of cells with centrosome amplification, multinucleation, and spindle multipolarity, and their mitotic index was similar (Fig EV1A–F). In agreement, flow cytometry analysis of asynchronous cells showed that both control and V5‐BirA*‐AURKA‐expressing cells had similar cell cycle profiles (Fig EV1G). Finally, immunoblotting for caspase‐3 showed that apoptosis was not activated in these cells (Fig EV1H).
Figure 1. Localization and activity of BirA*‐AURKA during cell cycle.

- Experimental workflow for the BioID experiment. HEK293T cells stably expressing V5‐BirA*‐AURKA and V5‐BirA* were treated with 50 μM biotin for 18 h, harvested, and lysed by denaturing lysis. Biotinylated proteins were captured with streptavidin affinity beads and analyzed by mass spectrometry.
- V5‐BirA*‐AURKA is expressed at near endogenous levels in the stable line. HEK293T cells and HEK293T::V5‐BirA*‐AURKA lysates were immunoblotted with antibodies against V5, AURKA, and vinculin (loading control).
- Biotinylation of the centrosome and microtubules by V5‐BirA*‐AURKA in different cell cycle stages. After 18‐h biotin incubation, HEK293T::V5‐BirA*‐AURKA were fixed and stained for the fusion protein with V5 antibody, biotinylated proteins with the fluorescent streptavidin, and the centrosome with gamma‐tubulin antibody. DNA was stained with DAPI. Scale bar, 5 μm.
- Control and biotin‐treated HEK293T cells stably expressing V5‐BirA* and V5‐BirA*‐AURKA were lysed, and biotinylated proteins were precipitated by streptavidin beads. The initial sample (initial) and captured biotinylated proteins (pulldown) were run on an SDS gel and immunoblotted with the fluorescent streptavidin and antibodies against V5 and vinculin (loading control).
Figure EV1. Validation and phenotypic characterization of HEK293T cells stably expressing V5‐BirA*‐AURKA.
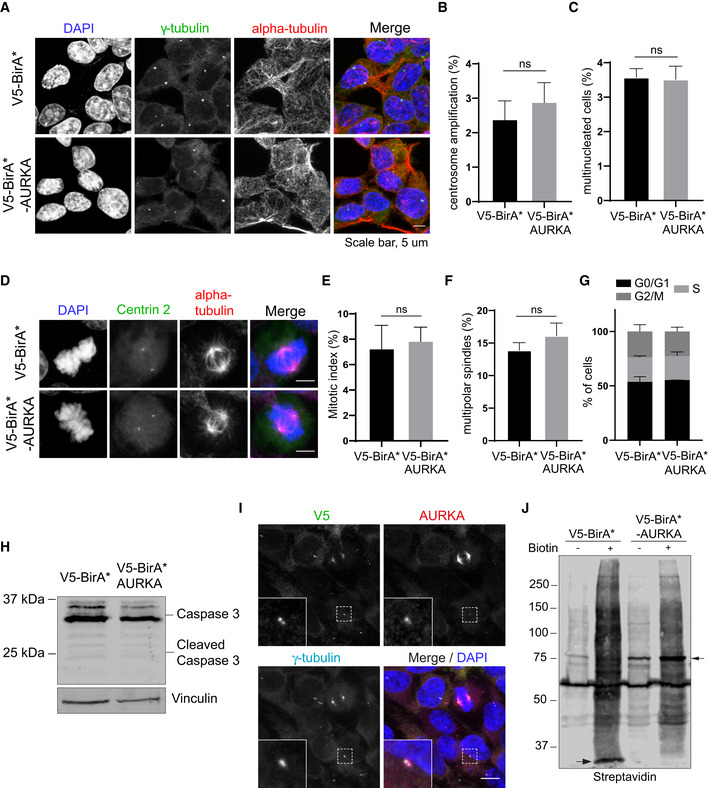
-
ARepresentative immunofluorescence images of HEK293T::V5‐BirA*‐AURKA fixed and stained for gamma‐tubulin, alpha‐tubulin, and DAPI. Centrosome number higher than 2 was quantified as “centrosome amplification”. Cells with more than one nucleus were quantified as “multinucleated”). Scale bar, 5 μm
-
B, CQuantification for (B) centrosome amplification and (C) multinucleation for A. Data represent mean value from two experiments per condition ± SEM (ns: non‐significant, unpaired Student's t‐test). n > 100 cells per experiment.
-
DRepresentative immunofluorescence images of mitotic HEK293T::V5‐BirA*‐AURKA fixed and stained for centrin 2 (centrioles), alpha‐tubulin, and DAPI. Scale bar, 5 μm.
-
E, FQuantification for (E) mitotic index (F) percentage of multipolar spindles for D. Data represent mean value from two experiments per condition ± SEM (ns: non‐significant, unpaired Student's t‐test). n > 100 cells per experiment.
-
GCell cycle analysis of asynchronous HEK293T::V5‐BirA*‐AURKA cells. Cells were fixed and stained with propidium iodide (PI) and in flow cytometer. Data represent mean value from two experiments per condition ± SEM (ns: non‐significant, unpaired Student's t‐test).
-
HRelative expression of Caspase3 in control cells andHEK293T::V5‐BirA*‐AURKA cells. Cells were lysed and immunoblotted with antibodies against Caspase3 and vinculin (loading control).
-
IRepresentative images of HEK293T::V5‐BirA*AURKA cells stained for V5, AURKA, and gamma‐tubulin. DNA was stained with DAPI. Scale bar is 10 μm.
-
JExpression and biotinylation of BirA* (control) and BirA*‐AURKA in the absence and the presence of biotin (50 μM biotin, 18 h). Cells were lysed, run on an SDS–PAGE gel, and immunoblotted with streptavidin. Arrows indicate the bait proteins V5‐BirA* and V5‐BirA*‐AURKA.
We then examined the expression of V5‐BirA*‐AURKA and biotinylation of its proximate interactors by staining and blotting for V5 to detect the fusion protein, streptavidin to detect biotinylated proteins, and/or gamma‐tubulin to mark the centrosome (Figs 1C and D, and EV1I and J). V5‐BirA*‐AURKA localized to the centrosome in interphase and to the spindle poles and microtubules in mitosis (Figs 1C and EV1I). Importantly, in cells incubated with 50 μm biotin for 18 h, V5‐BirA*‐AURKA induced localized biotinylation at these structures (Fig 1C). Immunoblotting of lysates from cells stably expressing V5‐BirA* and V5‐BirA*‐AURKA confirmed their expression as well as the successful streptavidin pulldown of the baits and the biotinylated proteins in their proximity (Figs 1D and EV1J). As expected, the levels of biotinylated proteins in lysates from V5‐BirA*‐AURKA and V5‐BirA* cells treated with biotin were substantially higher relative to no biotin control. (Fig EV1J). Together, these results show that V5‐BirA*‐AURKA is active and induces biotinylation of proteins at the centrosomes and microtubules.
Identification and network analysis of the AURKA proximity interactome
We performed label‐free quantitative proteomics to generate the AURKA proximity interactome. For large‐scale pulldowns, asynchronous cells stably expressing V5‐BirA*‐AURKA and V5‐BirA* were grown in 5x15 cm plates and incubated with 50 μM biotin for 18 h. Following denaturing lysis of cells, biotinylated proteins were captured by streptavidin beads and analyzed by mass spectrometry. Cells expressing V5‐BirA* were processed as controls. Two biological replicates for V5‐BirA*‐AURKA and four biological replicates for V5‐BirA* were analyzed by mass spectrometry (Dataset EV1).
We defined the high‐confidence proximity interactome of AURKA by performing significance analysis of interactome (SAINT) analysis, which assigns confidence scores for each protein interaction (Teo et al, 2014). We used a SAINT score cutoff of > 0.95, which yielded 440 proteins as high‐confidence AURKA proximity interactors. We next compared the AURKA proximity map with the AURKA interactors reported in the Biological General Repository for Interaction (BioGRID), which consisted of 393 unique proteins identified by low‐ and high–throughput experiments (Dataset EV2) (Stark et al, 2006). About 12% of the high‐confidence AURKA interactors (52 proteins) overlapped with BioGRID, identifying the remaining 388 proteins in the proximity map as previously undescribed AURKA interactors (Fig 2A, Dataset EV2). Shared interactors were enriched in functions such as centrosome maturation, microtubule dynamics, centriole duplication, mitotic progression, and cilium assembly. We also compared the AURKA proximity interactome to BioGRID interactors of AURKB, which identified 20% of AURKA interactors as overlapping interactions (Fig EV2A).
Figure 2. Identification, validation, and networking analysis of high‐confidence AURKA proximity interactors.
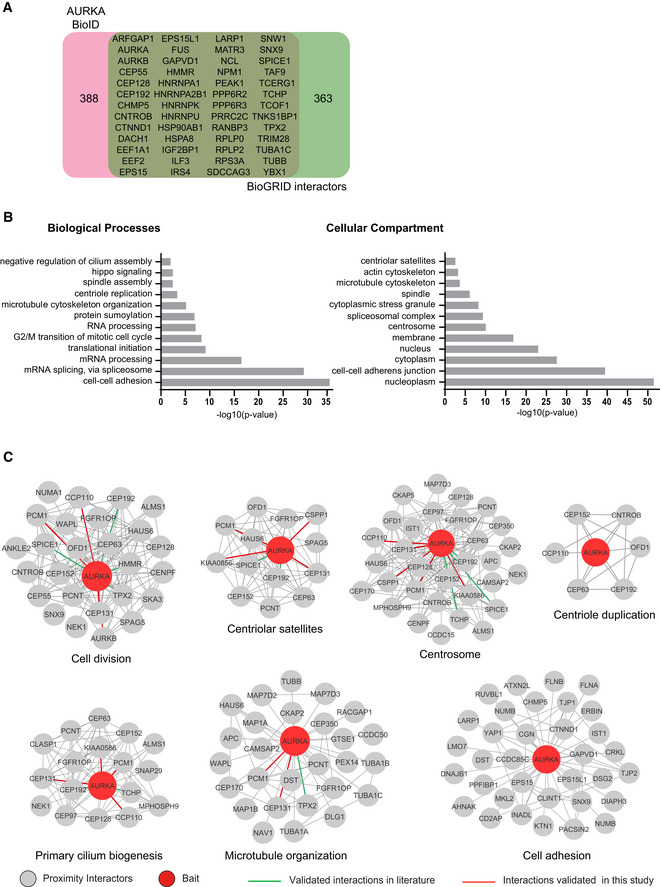
- AURKA proximity interactome consists of both overlapping and distinct interactors with the published AURKA interactors. AURKA proximity interactome was compared with published physical interactors/substrates and proteins curated from the BioGRID repository. Venn diagram shows the identity of the overlapping 52 proteins and the number of proteins specific to the AURKA proximity interactome and BioGRID dataset.
- GO‐enrichment analysis of the AURKA proximity interactors based on their biological process and cellular compartment. The x‐axis represents the log‐transformed p‐value (Fisher’s exact test) of GO terms.
- Sub‐interaction networks of AURKA based on biological process and cellular compartment. The AURKA proximity interactors were grouped using DAVID functional annotation tool and literature mining. The interaction networks visualized for the biological processes include cell cycle, centriole duplication, primary cilium biogenesis, microtubule organization, and cell adhesion. The interaction networks visualized for cellular compartments include the centrosome and the centriolar satellites. The interconnectedness among the proteins of each network was determined by the STRING database. Edges in green color indicate the published physical AURKA interactors/substrates, and edges in red color indicate the proximate interactors validated in this study.
Figure EV2. Networking analysis of the AURKA proximity interactome.
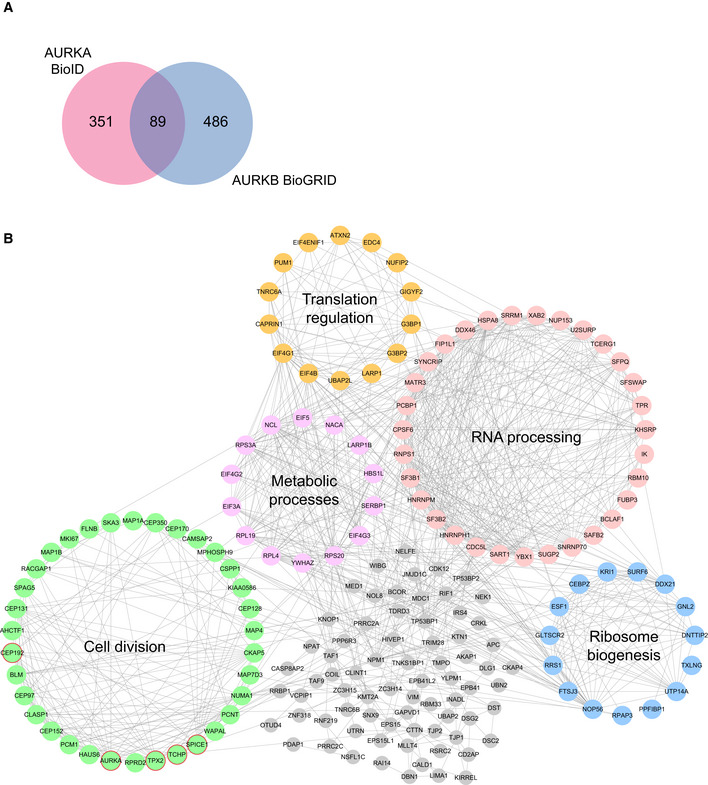
- Comparison of AURKA proximity interactome with Aurora Kinase B (AURKB) interactors derived from BioGRID. Pink circle indicates the AURKA proximity interactome, and blue circle indicates AURKB interactome.
- High‐confidence proximity interactors of AURKA were determined using SAINT analysis. AURKA proximity interactors were ranked by their fold change in the V5‐BirA*‐AURKA dataset relative to V5‐BirA*‐dataset. The first 200 proteins were visualized in CytoScape, and the functional clusters were determined by combining STRING analysis with the ClusterONE plug‐in. Five functional clusters were enriched in the interactome, which include cell cycle (green), RNA processing (pink), ribosome biogenesis (blue), translation regulation (yellow) metabolic processes (purple). Uncategorized interactors are shown in gray, and the published interactors are shown in red circles.
To define new AURKA functions and regulatory mechanisms, we performed Gene Ontology (GO) enrichment analysis of the high‐confidence AURKA interactors based on their “biological process” and “cellular compartment” (Fig 2B; Dataset EV3). Consistent with the previously described AURKA functions, GO term analysis of biological processes showed significant (P < 0.05) enrichment for cell division, microtubule‐based cellular processes such as spindle assembly, DNA damage response, and cilium assembly. There was also enrichment of biological processes including cell adhesion, mRNA processing, and centriole duplication, identifying them as putative new AURKA functions. Likewise, the enriched GO terms for cellular compartment were not limited to previously described ones such as “Centrosome” and “Spindle poles” but also included new ones such as “Centriolar Satellites” and “Cell‐cell adherens junctions” (Fig 2B; Dataset EV3). Additionally, we organized the top 200 AURKA proximity interactors ranked by their fold change into an interaction network by combining STRING database and ClusterONE plug‐in on Cytoscape. This analysis grouped proteins based on their interconnection and identified the functional clusters representing subnetworks and potential multiprotein complexes. The resulting network identified a diverse array of proteins from five major functional clusters (P < 0.005), which include cell division, translation regulation, RNA processing, ribosome biogenesis, and metabolic processes (Fig EV2B).
To generate testable hypothesis about AURKA function and regulation, we generated sub‐interaction interaction networks by grouping proteins based on enriched subcellular compartments (centriolar satellite and centrosome) and biological processes (cell division, centriole duplication, primary cilium biogenesis, microtubule organization, cell adhesion) (Fig 2C). In addition to suggesting sub‐complexes that function together and/or localize to the same compartment, this analysis revealed the proteins that are shared between different subcellular compartments and functions. Taken together, these results validated the AURKA proximity interactome and revealed a substantial number of new interactors.
AURKA has extensive proximity and physical interactions with centriolar satellites
Aurora kinase A proximity interactome shares a substantial number of interactors with the published proximity interactomes of the centrosome (38%) and the centriolar satellites (23%) and physical interactome of centriolar satellites (6%) (Fig 3A and B; Dataset EV2) (Gupta et al, 2015; Gheiratmand et al, 2019; Quarantotti et al, 2019). While localization and function of AURKA at the centrosome was previously described, its nature of relationship to centriolar satellites is not known. To validate the AURKA proximity interactome and to test whether AURKA is a component of centriolar satellites, we performed a set of different affinity purification experiments. First, we performed streptavidin pulldowns in FLAG‐BirA* (control) and FLAG‐BirA*‐AURKA‐expressing cells and immunoblotted for new AURKA interactors as predicted by the proximity map. Among these proteins were the satellite scaffolding protein PCM1 (Kubo et al, 1999); the ciliogenesis factors CEP131, CEP72, BBS4, KIAA0586/Talpid3, and CP110 (Breslow & Holland, 2019); the centriole duplication factors CEP63; and the microtubule‐associated protein CSPP1 (Shaheen et al, 2014; Frikstad et al, 2019). To test the specificity of AURKA‐satellite interactions, we also chose satellite proteins BBS4 and SSX2IP as proteins that were not identified in the AURKA proximity map (Nachury et al, 2007; Klinger et al, 2014; Hori et al, 2015). FLAG‐BirA*‐AURKA co‐pelleted with PCM1, CEP131, CEP72, CSPP1, Talpid3, CP110, but not with CEP63, BBS4, and SSX2IP (Fig 3C). Notably, the proximity interaction between PCM1 and AURKA was abolished upon inhibition of AURKA activity (Fig EV3A). These results suggest that AURKA interacts only with a subset of satellite proteins and that these interactions might underlie its functions at the primary cilium.
Figure 3. AURKA has extensive proximity interactions with centrosomes and centriolar satellites.
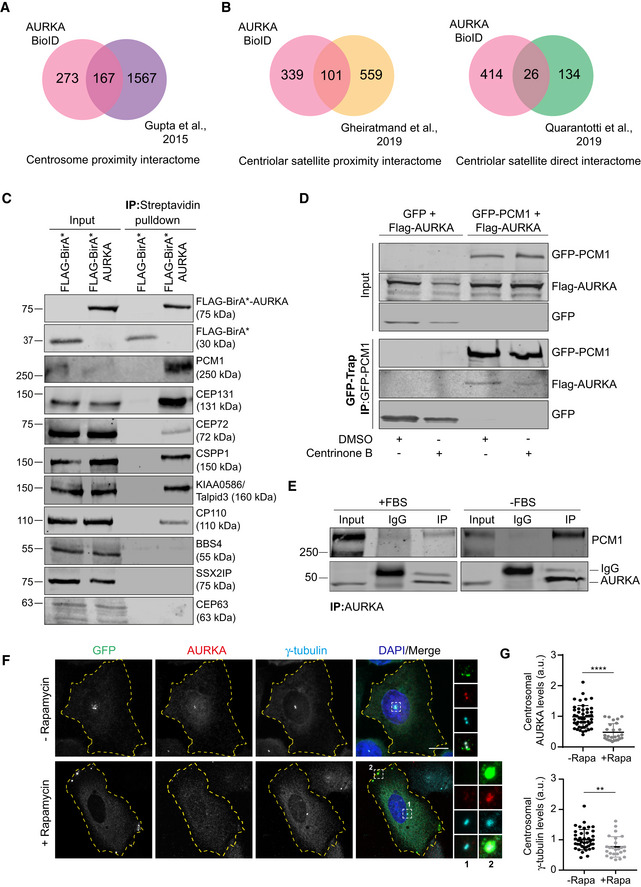
- A, B
-
CAURKA interacts with the centriolar satellite proteins PCM1, CEP131, CEP72, CSPP1, KIAA0586/Talpid3, and CP110. HEK293T cells were transiently transfected with FLAG‐BirA* or FLAG‐BirA*‐AURKA. Following 18‐h biotin incubation, cells were lysed, and biotinylated proteins were precipitated by streptavidin beads. The initial sample and immunoprecipitated biotinylated proteins were run on a gel and immunoblotted with fluorescent streptavidin and antibodies against FLAG, CEP72, CEP131, PCM1, CEP63, CSPP1, KIAA0586/Talpid3, CP110, BBS4, and SSX2IP.
-
DAURKA interacts with PCM1 in centriole‐less cells. GFP‐PCM1 or GFP‐PCM1 was co‐expressed with FLAG‐AURKA in HEK293T cells treated with DMSO (control) or 500 nM centrinone B for 7 days. Complexes were precipitated with GFP‐Trap beads. The initial sample and immunoprecipitated proteins were run on a gel and immunoblotted with antibodies against GFP and FLAG.
-
EEndogenous interaction of PCM1 and AURKA. Lysates from synchronous and serum‐starved HEK293T were immunoprecipitated with AURKA antibody and rabbit IgG control. The initial sample and immunoprecipitated proteins were run on a gel and immunoblotted with antibodies against PCM1 and AURKA.
-
FAURKA co‐localize with PCM1 at the peripheral satellite clusters upon chemical dimerization of satellites with Kif5b motor domain. U2OS cells were co‐transfected with GFP‐PCM1‐FKBP and HA‐Kif5b(1–269 a.a.)‐FRB and treated with 500 nM rapamycin for 1 h followed by fixation at 6 h. Cells that were not treated with rapamycin were processed in parallel as a control. Fixed cells were stained with antibodies against GFP, AURKA, and gamma‐tubulin. DNA was stained with DAPI. Cell edges are outlined. Scale bar, 10 μm.
-
GQuantification of pericentrosomal levels of AURKA and gamma‐tubulin for F. n > 25 cells per experiment. Data represent mean value from two experiments per condition ± SEM (****P < 0.0001; **P < 0.01; unpaired Student's t‐test.).
Figure EV3. AURKA interacts with centriolar satellites.
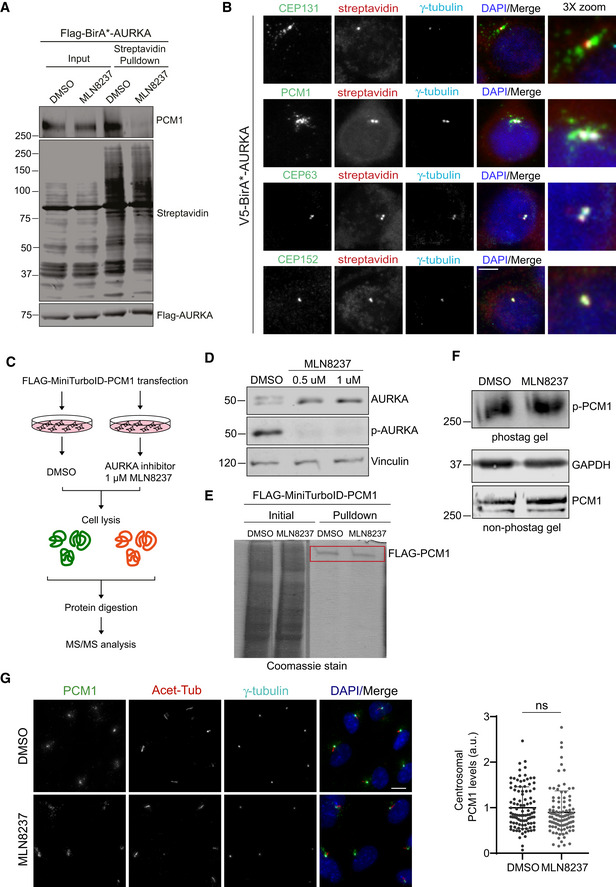
- AURKA activity is required for its interaction with PCM1. HEK293T cells were transfected with FLAG‐BirA*‐AURKA. 24 h post‐transfection, cells were treated with DMSO+biotin or MLN8237+biotin, lysed, and biotinylated proteins were precipitated by streptavidin beads. The initial sample and precipitated proteins were run on an SDS–PAGE gel and immunoblotted for streptavidin, FLAG, and PCM1.
- Localization of V5‐BirA*‐AURKA relative to markers of the centrosome and centriolar satellites. HEK293T::V5‐BirA*‐AURKA cells were incubated with 50 μM biotin for 18 h and immunostained with fluorescent streptavidin and antibodies against CEP63, CEP152, CEP131, and PCM1. DNA was stained with DAPI. Scale bar, 5 μm.
- Experimental workflow for quantitative phosphoproteomics. HEK293T cells were transfected with FLAG‐PCM1 (4 × 15 cm plates), treated either DMSO (vehicle control) or 1 μM MLN8237. Following cell lysis and FLAG pulldown, FLAG‐PCM1 was excised, digested with trypsin, and analyzed by mass spectrometry.
- Validation of AURKA inhibition upon MLN8237 treatment. Cells were treated with DMSO, 0.5 μM MLN8237 and 1 μM MLN8237, lysed, run on an SDS–PAGE, and immunoblotted with antibodies against AURKA, p‐AURKA, and vinculin (loading control).
- Coomassie gel image of FLAG‐PCM1 pulldown. The rectangle indicates the corresponding band excised for mass spectrometry.
- Phos‐tag and non‐phos‐tag immunoblots of cells were treated with DMSO (vehicle) or 1 μM MLN8237. Cells were lysed and immunoblotted for PCM1 and GAPDH.
- Representative images and quantification of pericentrosomal levels of PCM1. RPE1 cells were treated with 0.5 μM MLN8237, fixed, and immunostained with PCM1, acetylated tubulin, and gamma‐tubulin. Centrosomal AURKA and p‐AURKA fluorescence intensities were measured from maximum projections, and average means of the levels in control cells were normalized to 1. Data represent mean value from two experiments per condition ± SEM (ns: non‐significant, unpaired Student's t‐test). n > 100 cells per experiment Scale bar, 5 μm.
Next, we tested whether the proximity interaction between AURKA and satellites is reflected at the physical level by GFP pulldown experiments in cells expressing GFP‐PCM1 and FLAG‐BirA*‐AURKA. We found that FLAG‐BirA*‐AURKA co‐precipitated with GFP‐PCM1, but not GFP (Fig 3D). To test whether centrosomes are required for this interaction, we performed similar experiments in centriole‐less cells, which was generated by treatment of cells with the PLK4 inhibitor centrinone over 7 days (Wong et al, 2015; Gheiratmand et al, 2019). FLAG‐BirA*‐AURKA interacted with GFP‐PCM1 in centriole‐less cells, which further supports their interaction at the satellites (Fig 3D). Finally, immunoprecipitation experiments indicated that AURKA interacts with PCM1 at the endogenous level in both asynchronous and quiescent cells (Fig 3E). Collectively, these results confirm the physical interaction between AURKA and centriolar satellites.
Although pulldown experiments confirmed interactions between AURKA and satellites, we did not detect localization of AURKA to satellites. V5‐BirA*AURKA induced biotinylation at the centrosome but not at the satellites, as assessed by staining for centrosome proteins CEP63 and CEP152 and satellite proteins PCM1 and CEP131 (Fig EV3B). This might be due to the low abundance and transient nature of this interaction. To test this, we performed inducible satellite trafficking assay to distribute satellites to the cell periphery and asked whether endogenous AURKA, like other satellite residents, is sequestered at the peripheral satellites that localize away from the centrosome (Aydin et al, 2020). In cells expressing GFP‐PCM1‐FKBP and HA‐Kif5b motor domain, AURKA and gamma‐tubulin localized to the centrosome before rapamycin treatment (Fig 3F). 6 h after rapamycin treatment, both AURKA and gamma‐tubulin were recruited to the peripheral satellite clusters and their centrosomal levels were reduced (Fig 3F and G). These experiments indicate that AURKA specifically interacts with PCM1 at the peripheral satellites and that its centrosomal targeting might be regulated by sequestration at the satellites.
Aurora kinase A mediates its diverse functions in part through phosphorylating proteins implicated in these processes. To examine whether PCM1 is a substrate for AURKA, we applied quantitative phosphoproteomics combined with chemical inhibition of AURKA. To this end, we expressed Flag‐PCM1 in HEK293T cells and treated cells with DMSO (vehicle control) or AURKA inhibitor MLN8237 (Fig EV3C). Immunoblotting of extracts from cells treated with 0.5 and 1 μM MLN8237 for p‐AURKA confirmed inhibition of AURKA phosphorylation at Thr288 (Fig EV3D). Following FLAG pulldowns, in‐gel digestion of FLAG‐PCM1 band was performed and the resulting peptides were analyzed by mass spectrometry (Fig EV3E). Label‐free quantification of the results from three experimental replicates using MaxQuant did not identify any putative AURKA phosphosites on PCM1 (Dataset EV4). In a complementary approach, we ran lysates from cells treated with DMSO and MLN8237 on Phos‐tag SDS–PAGE (Fig EV3F) and found that the migratory behavior PCM1 in control and MLN8237‐treated quiescent cells were similar (Fig EV3F). Finally, we investigated whether AURKA activity is required for satellite integrity and distribution in cells. Pericentrosomal PCM1 levels did not change upon MLN8237 treatment as compared to control cells (Fig EV3G). Collectively, these results do not support phosphorylation of PCM1 and regulation of satellites by AURKA kinase activity.
Centriolar satellites regulate AURKA localization, abundance, and activity during cilium biogenesis
Centriolar satellites interact with a wide range of centrosome proteins and mediate their functions at the primary cilium in part by regulating cellular localization and abundance of their residents (Odabasi et al, 2020; Prosser & Pelletier, 2020). We hypothesized that satellites might regulate AURKA localization, stability, and/or kinase activity during cilium biogenesis. To test this, we investigated how these properties are altered upon loss of satellites in human retinal pigmented epithelial (RPE1) cells, which are widely used models to study cilium biogenesis. As previously described, we induced loss of satellites by RNAi‐mediated depletion of their scaffolding protein PCM1 (Dammermann & Merdes, 2002; Kim et al, 2008). We used immunoblotting and immunofluorescence to confirm the efficiency of PCM1 depletion and loss of satellites using two different PCM1 siRNAs (Fig EV4A and B).
Figure EV4. Validation of PCM1 depletion and regulation of AURKA abundance by satellites.

- Immunoblotting analysis of PCM1 depletion by RNAi. RPE1 cells were transfected with control or PCM1 siRNA #1 and #2 for 72 h. Cells were lysed and immunoblotted with PCM1 and Actin (loading control). Band intensities for immunoblots were quantified from two experimental replicates. Scale bar, 5 μm.
- Immunofluorescence analysis of PCM1 depletion and loss of satellites by RNAi. RPE1 cells were transfected with control or PCM1 siRNA #1 and #2 for 72 h. Cells were fixed and stained with PCM1, gamma‐tubulin, and DNA. DNA was stained with DAPI. Scale bar, 10 μm.
- Confirmation of CCDC66 depletion by RNAi and quantification of AURKA and p‐AURKA levels upon CCDC66 depletion. RPE1 cells were transfected with control or CCDC66 siRNA for 72 h. Cells were lysed and immunoblotted for AURKA, p‐AURKA, CCDC66, vinculin (loading control), and alpha‐tubulin (loading control).
- Depletion of PCM1 in RPE1 GFPCCDC66 line. RPE1 cells were transfected with control or PCM1 siRNA 72 h. Cells were fixed and immunostained with GFP, PCM1, and centrin3. DNA was stained with DAPI. Scale bar, 10 μm.
- Total AURKA in control and RPE1 PCM1 KO cells. RPE1 WT and RPE1 PCM1 KO cells were serum‐starved for 24 h, cell lysates were prepared and run on an SDS–PAGE gel. Proteins were detected by immunoblotting with antibodies against AURKA and vinculin (loading control). The band intensities were measured from two replicates (unpaired Student's t‐test).
- Representative images and quantification of basal body levels of AURKA and phospho‐AURKA (p‐AURKA) in control and RPE1 PCM1 KO cells. Following 24 h serum starvation, cells were fixed and stained with the indicated antibodies and DAPI. Scale bar, 10 μm.
- Quantification of AURKA and p‐AURKA levels in RPE1 WT and PCM1 KO cells for G. Centrosomal AURKA and p‐AURKA fluorescence intensities were measured from maximum projections, and average means of the levels in control cells were normalized to 1. n > 100 cells per experiment. Data represent mean value from two experiments per condition ± SEM (**P < 0.01, ns: non‐significant, unpaired Student's t‐test).
Data information: (B, D, F) Sizes of the scale bars are indicated in the figure legends.
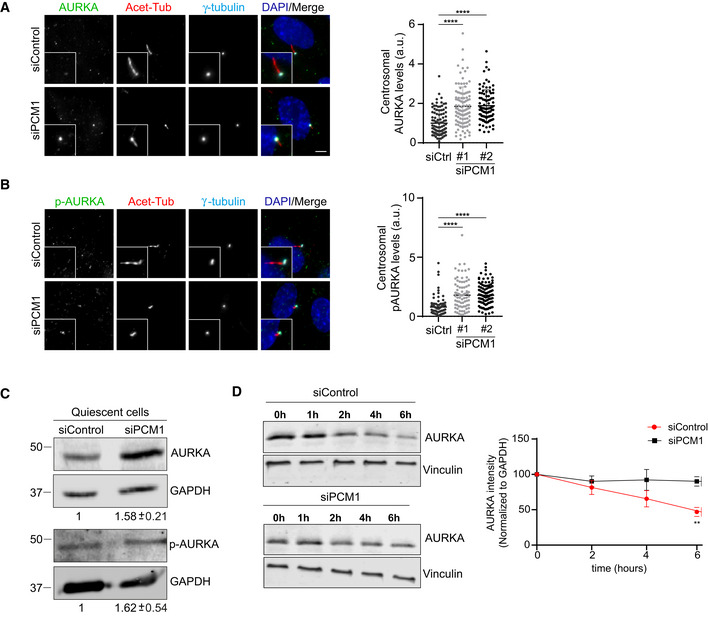
First, we first examined the levels of AURKA and Thr288‐phosphorylated AURKA (p‐AURKA) in control and PCM1‐depleted cells serum‐starved for 24 h. Phosphorylation of AURKA on Thr288 located in its activation T loop was shown to result in its activation and increase in its enzymatic activity (Walter et al, 2000). The basal body levels of AURKA and p‐AURKA increased significantly (P < 0.01) in PCM1‐depleted cells as compared to control cells, suggesting that satellites negatively regulate centrosomal abundance and activity of AURKA (Fig 4A and B). Next, we examined the cellular levels of AURKA by immunoblotting lysates from control and PCM1‐depleted quiescent cells for AURKA and p‐AURKA (Fig 4C). Cellular AURKA and p‐AURKA levels were significantly higher (P < 0.01) in PCM1‐depleted cells relative to control cells. To test whether satellites regulate AURKA stability, we assayed the degradation rate of AURKA by cycloheximide block‐chase experiments. While more than 53.05 ± 3.71% AURKA was degraded in control cells after 6 h cycloheximide treatment, only 10 ± 3.86% of PCM1 depletion was degraded (P < 0.01) (Fig 4D). Inhibition of AURKA degradation suggests that satellites destabilize AURKA. Finally, to investigate whether these phenotypes are specific to loss of satellites associated with PCM1 depletion, we performed similar experiments in cells depleted for another satellite protein CCDC66, which is required for pericentrosomal satellite clustering but not for satellite assembly (Fig EV4C and D; Conkar et al, 2017). In contrast to PCM1 depletion, control and CCDC66‐depleted cells had similar levels of AURKA and p‐AURKA (Fig EV4C). Taken together, these results identify PCM1 and thereby satellites as regulators of AURKA stability, activity, and localization in quiescent cells.
Figure 4. Centriolar satellites regulate AURKA localization, abundance, and activity in quiescent cells.
-
A, BRepresentative images and quantification of basal body levels of (A) AURKA and (B) phospho‐AURKA (p‐AURKA) in control and PCM1‐depleted cells. RPE1 cells were transfected with control or PCM1 siRNA #1 or siRNA #2 for 48 h. Following 24 h serum starvation, cells were fixed and stained with the indicated antibodies. Centrosomal AURKA and p‐AURKA fluorescence intensities were measured from maximum projections, and average means of the levels in control cells were normalized to 1. n > 500 cells per experiment. Data for (A) AURKA and (B) p‐AURKA represent mean value from three experiments per condition ± SEM (****P < 0.0001, unpaired Student's t‐test). Scale bar, 10 μm.
-
CTotal AURKA and p‐AURKA levels in control and PCM1‐depleted cells. RPE1 cells were transfected with control or PCM1 siRNA#1 for 48 h. Following 24 h serum starvation, cell lysates were prepared and run on an SDS–PAGE gel. Proteins were detected by immunoblotting with antibodies against AURKA, p‐AURKA, and GAPDH (loading control).
-
DCycloheximide chase experiment for quantification of AURKA half‐life. Cells were transfected with control or PCM1 siRNA#1 for 48 h and then treated with 200 nM cycloheximide along with serum starvation for indicated time points. AURKA intensities were quantified by immunoblotting for AURKA and vinculin (loading control) and AURKA levels were normalized to vinculin levels. Data represent mean value from three experiments per condition ± SEM (**P < 0.01, unpaired Student's t‐test).
To investigate AURKA regulation by satellites in an orthogonal system, we quantified cellular and centrosomal abundance and activation of AURKA in the satellite‐less RPE1::PCM1−/− cells (Wang et al, 2016; Odabasi et al, 2019). The cellular levels of AURKA were higher in RPE1::PCM1−/− cells relative to control cells (P < 0.05) (Fig EV4E). While the basal body levels of AURKA increased significantly in RPE1::PCM1−/− cells (P < 0.01), the levels of p‐AURKA did not change (P = 0.16) (Fig EV4F and G). The difference in AURKA activation at the basal body in PCM1‐depleted and RPE1::PCM1−/− might be due to differential effects of acute versus chronic depletion of PCM1, which was previously reported for other proteins such as CEP131 (Hall et al, 2013).
Centriolar satellites and AURKA have antagonistic functions in cilium assembly and disassembly
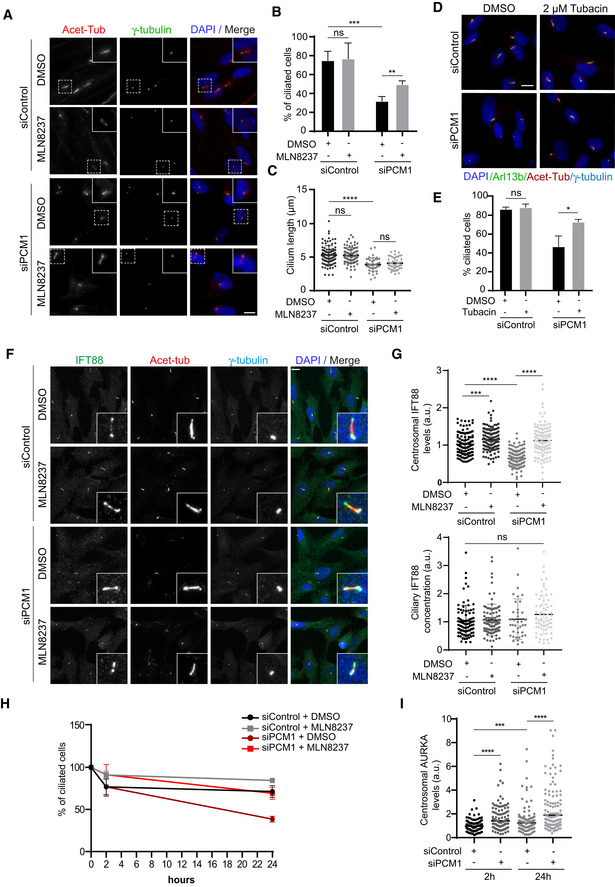
Having established the link between satellites and AURKA regulation in quiescent cells, we next investigated the functional consequences of this regulatory relationship. Recent studies showed that satellites function as trafficking and storage sites for key ciliogenesis factors and regulate primary cilium assembly, maintenance, and disassembly (Wang et al, 2016; Odabasi et al, 2019; Aydin et al, 2020). However, the proteins regulated by satellites during cilium biogenesis have not been fully characterized. We hypothesized that satellites might regulate cilium assembly and disassembly in part through AURKA and tested this by examining the effects of inhibition of AURKA activity in PCM1‐depleted cells. To this end, we treated control and PCM1‐depleted RPE1 cells with DMSO or MLN8237 and quantified the percentage of ciliated cells 24 h post‐serum starvation (Fig 5A and B). PCM1 depletion resulted in a significant decrease in the percentage of ciliated cells relative to control cells (P < 0.001) and the cilia that formed in these cells were significantly shorter (P < 0.0001) (Figs 5A–C and Fig EV5A). While DMSO‐ and MLN8237‐treated cells ciliated with similar efficiencies in control (P = 0.92), AURKA inhibition partially restored the ciliogenesis defect in PCM1‐depleted cells (P < 0.01) (Figs 5A and B, and EV5AA). Defective cilium length phenotype was not rescued by AURKA inhibition, suggesting that satellites regulate cilium length independent of AURKA activity (P = 0.32) (Figs 5C and EV5A). Of note, MLN8237 treatment did not restore ciliation in RPE1::PCM1−/− cells, further supporting the difference between ciliogenesis phenotypes of cells depleted or deleted for PCM1 (Fig EV5B). Finally, we examined whether the ciliogenesis phenotypes associated with PCM1 depletion and MLN8237 treatment were due to cell cycle defects. As assessed by staining cells for the proliferation marker Ki67, PCM1 depletion and AURKA inhibition did not change the percentage of quiescent cells serum‐starved for 24 h and the ciliogenesis phenotypes were similar when we quantified ciliated Ki67‐ cells (Fig EV5C). Together, these results identify regulation AURKA activity as a mechanism by which satellites function during cilium assembly.
Figure 5. Centriolar satellites and AURKA exhibit antagonistic effects during cilium assembly and disassembly.
-
ARepresentative immunofluorescence images of cilium assembly experiments in control and PCM1‐depleted RPE1 cells treated with DMSO or MLN8237. Cells were transfected with control or PCM1 siRNA#1 for 48 h and treated with DMSO (vehicle control) or 0.5 μM MLN8237 in serum starvation medium for 24 h. Cells were fixed and immunostained for the primary cilium with acetylated tubulin antibody (Acet‐tub) and the centrosome with gamma‐tubulin antibody. DNA was stained with DAPI. Scale bar, 10 μm.
-
BQuantification of ciliogenesis efficiency for A. n > 100 cells per experiment. Data represent mean value from three experiments per condition ± SEM (***P < 0.001; **P < 0.01; ns, non‐significant, unpaired Student's t‐test.).
-
CQuantification of cilium length for A. n > 100 cells per experiment. Data represent mean value from three experiments per condition ± SEM (****P < 0.0001; ns, non‐significant, unpaired Student's t‐test.).
-
DRepresentative immunofluorescence images of cilium assembly experiments in control and PCM1‐depleted RPE1 cells treated with DMSO or tubacin. Cells were transfected with control or PCM1 siRNA#1 for 48 h and treated with DMSO or 2 μM tubacin in serum starvation medium for 24 h. Cells were fixed and immunostained for the primary cilium with Arl13b and acetylated tubulin antibody (Acet‐tub) and the centrosome with gamma‐tubulin antibody. DNA was stained with DAPI. Scale bar, 10 μm.
-
EQuantification of ciliogenesis efficiency for D. n > 100 cells per experiment. Data represent mean value from three experiments per condition ± SEM (*P < 0.05; ns, non‐significant, unpaired Student's t‐test).
-
FRepresentative immunofluorescence images and quantification of IFT88 levels at the centrosome and primary cilium. RPE1 cells were transfected with control or PCM1 siRNA#1 for 48 h, serum‐starved for 24 h, and treated with DMSO or 0.5 μM MLN8237. Cells were fixed and immunostained with antibodies against IFT88, acetylated tubulin, and gamma‐tubulin. DNA was stained with DAPI. Scale bar, 10 μm.
-
GQuantification of IFT88 centrosomal intensity and ciliary concentration for F. IFT88 fluorescence intensities at the centrosome and axoneme were measured from maximum projections, and average means of the levels in control cells were normalized to 1. Ciliary concentration was calculated by dividing ciliary fluorescence intensity to the cilium length. n > 100 cells per experiment. Data represent mean value from three experiments per condition ± SEM (***P < 0.001; ****P < 0.0001, unpaired Student's t‐test).
-
H, IQuantification of cilium disassembly and centrosomal AURKA levels after serum stimulation. RPE1 cells were transfected with control or PCM1 siRNA#1 for 48 h, serum‐starved for 24 h, and treated with DMSO or 0.5 μM MLN8237 in serum stimulation medium for 2 and 24 h. Cells were fixed and immunostained with antibodies against acetylated tubulin and gamma‐tubulin, and percentage of ciliated cells were quantified. X‐axis indicates the hours after serum stimulation. (H) n > 100 cells per experiment. Data represent mean value from three experiments per condition ± SEM. (I) n > 95 cells per experiment. Data represent mean value from three experiments per condition ± SEM (***P < 0.001; ****P < 0.0001, unpaired Student's t‐test).
Figure EV5. Characterization of ciliogenesis and cell cycle phenotypes in cilium assembly and disassembly experiments.
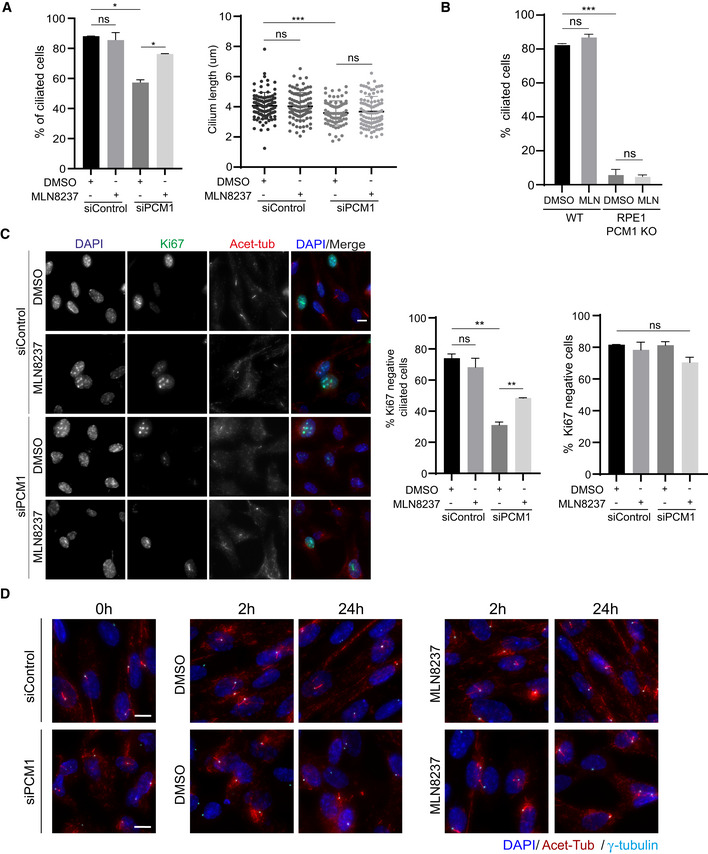
- Quantification of ciliogenesis efficiency and cilium length in control and PCM1‐depleted RPE1 cells treated with DMSO or MLN8237. Cells were transfected with control or PCM1 siRNA#2 for 48 h and treated with DMSO (vehicle control) or 0.5 μM MLN8237 in serum starvation medium for 24 h. Cells were fixed and immunostained for the primary cilium with acetylated tubulin antibody (Acet‐tub) and the centrosome with gamma‐tubulin antibody. n > 100 cells per experiment. Data represent mean value from two experiments per condition ± SEM (***P < 0.001; *P < 0.1; ns, non‐significant, unpaired Student's t‐test).
- Quantification of ciliogenesis percentage in RPE1 WT and PCM1 KO cells treated with DMSO (control) and 0.5 μM MLN8237. n > 100 cells per experiment. Data represent mean value from two experiments per condition ± SEM (***P < 0.001, ns: non‐significant, unpaired Student's t‐test).
- Representative images and quantification of proliferating cells upon serum starvation. RPE1 cells were transfected with control or PCM1 siRNA for 48 h, serum‐starved for 24 h, and treated with DMSO or 0.5 μM MLN8237. Cells were fixed and immunostained with Ki67 to mark proliferating cells and acetylated tubulin to mark the primary cilium. DNA was stained with DAPI. Scale bar, 10 μm. Ciliated Ki67‐ cells and Ki67‐ cells were quantified. Data represent mean value from two experiments per condition ± SEM (**P < 0.01, ns: non‐significant, unpaired Student's t‐test).
- Representative immunofluorescence images for cilium disassembly experiments. RPE1 cells were transfected with control or PCM1 siRNA for 48 h, serum‐starved for 24 h, and treated with DMSO (vehicle control) or 0.5 μM MLN8237 in serum stimulation medium for 2 and 24 h. Cells were fixed and immunostained with antibodies against acetylated tubulin antibody, gamma‐tubulin antibody to mark the centrosome. DNA was stained with DAPI. Scale bar, 10 μm.
Activated AURKA activates HDAC6 and induces cilium resorption in ciliated cells, suggesting that the ciliogenesis defect upon PCM1 depletion might be due to the induction of cilium disassembly (Pugacheva et al, 2007). To test this, we performed phenotypic rescue experiments with the HDAC6 inhibitor tubacin (Fig 5D and E). Like AURKA, inhibition of HDAC6 activity partially restored the cilium assembly defects of PCM1‐depleted cells (P < 0.05) (Fig 5D and E). Of note, there was no difference in the ciliation efficiency of DMSO and tubacin‐treated control cells (P = 0.59). These results are consistent with activation of AURKA/HDAC6 pathway and induction of cilium disassembly in PCM1‐depleted cells. Additionally, PCM1 and AURKA were both reported to mediate their functions during cilium assembly through regulating basal body maturation such as recruitment of the IFT‐B machinery (Odabasi et al, 2019; Pejskova et al, 2020). In agreement, we found that centrosomal levels of IFT88 were reduced upon PCM1 depletion, but its ciliary concentration remained unaltered (Fig 5F and G). Importantly, inhibition of AURKA activity rescued IFT88 centrosomal recruitment defect (Fig 5F and G). These results indicate that PCM1 and AURKA have antagonistic roles during ciliogenesis and basal body maturation.
Finally, we examined the functional significance of AURKA‐PCM1 interaction during cilium disassembly. First, we examined the consequences of loss of satellites during cilium disassembly upon PCM1 depletion. While control cells deciliated about 30% (28.7 ± 3.4%) after 24 h serum stimulation, PCM1‐depleted cells deciliated about 60% (61.4 ± 2%) (P < 0.01) (Figs 5H and EV5D). Consistent with enhanced cilium disassembly, PCM1 depletion resulted in a significant increase in the basal body levels of AURKA at 2 and 24 h post‐serum stimulation (P < 0.0001) (Fig 5I). Importantly, MLN8237 treatment rescued the enhanced cilium disassembly in control and PCM1‐depleted cells after 24 h serum stimulation (siPCM1+DMSO: 38.60 ± 2%, siPCM1+MLN8237: 69.19 ± 4.05%, P < 0.05) (Figs 5H and EV5D). While MLN8237 treatment inhibited cilium disassembly in control cells, it had a greater effect in PCM1‐depleted cells. This phenotypic difference was not due to cell cycle defects as the mitotic index of the confluent, ciliated RPE1 cells treated with DMSO or MLN8237 and stimulated with serum were similar (Fig EV5D). Together, these results suggest that satellites regulate the cilium disassembly through limiting the recruitment of AURKA to the basal body.
Discussion
In this study, we identified the first in vivo proximity interaction map of AURKA, which is composed of 440 proteins involving numerous biological processes and cellular compartments. The resulting interactome identified multiple well‐described AURKA interactors and revealed previously undescribed relationships, highlighting the diversity of AURKA functions and multifaceted spatiotemporal regulation. By investigating the extensive proximity relationship between AURKA and centriolar satellites, we showed that satellites negatively regulate the cellular abundance, localization, and activity of AURKA during cilium assembly and disassembly.
The AURKA proximity interactome partially overlapped with previously described AURKA interactors, which can be explained by two reasons. First, proximity mapping and conventional approaches monitor different types of interactions and they were shown to complement each other in comprehensive mapping of interactomes (Lambert et al, 2014; Liu et al, 2018). Second, previous proteomic studies for AURKA were designed to specifically identify its mitotic interactors. We note that a number of AURKA mitotic interactors such as TPX2 and CEP192 were relatively abundant among AURKA proximity interactors, confirming that information on spatially and temporally restricted interactions across different cell cycle stages are represented in its proximity map.
Importantly, the AURKA interactome was enriched for proteins implicated in non‐mitotic functions of AURKA such as cilium biogenesis and Hippo signaling, which provides insights into mechanisms by which AURKA regulates them (Bertolin & Tramier, 2020). Additionally, it revealed previously undescribed relationships with various biological processes and compartments such as centriolar satellites, mRNA processing, and translation regulations. Supporting these new relationships, recent work reported physical interactions of AURKA with splicing factors and defined its functions during alternative splicing (preprint: Damodaran et al, 2020). Future studies are required to investigate the importance and relevance of these new relationships to AURKA function and regulation and link to cancer.
Given the high representation of satellite proteins in the AURKA proximity map and emerging functions of satellites during cilium biogenesis, we elucidated the functional significance of the proximity between satellites and AURKA. Using pulldown experiments, we validated the proximity and physical interaction between AURKA and PCM1, which depends directly or indirectly on kinase activity of AURKA. Of note, we tested interactions of six more satellite proteins identified in the AURKA interactome and confirmed interaction for all except for CEP63, validating the confidence level of the AURKA proximity map as high. Although AURKA interacts with satellite proteins, we did not observe localization of endogenous or ectopically expressed AURKA to satellites. This is most likely due to the transient nature of this interaction, which would result in a small fraction of total AURKA to localize to satellites. In fact, this discrepancy between localization and interaction experiments were previously reported for other proteins identified in the satellite interactome such as CP110 (Spektor et al, 2007), CEP162 (Wang et al, 2013), MDM1 (Van de Mark et al, 2015), IFT74 (Bhogaraju et al, 2013). These results highlight the power of the BioID approach in accessing interactions that cannot be identified in traditional localization and interaction mapping studies.
AURKA localization, abundance, and activity are tightly regulated to ensure that it functions at the right time and place (Levinson, 2018). Importantly, our results identified satellites as a new mechanism by which AURKA is regulated at three different levels. First, satellites sequester AURKA and limit its recruitment to the basal body in ciliated cells. This result identifies AURKA as another protein regulated by satellites in a sequestration‐based mechanism and provides further support for satellite functions in centrosomal protein targeting (Odabasi et al, 2020; Prosser & Pelletier, 2020). Second, satellites negatively regulate AURKA stability, identifying them as active scaffolds for AURKA regulation. Given that a variety of enzymes including ubiquitin ligases, deubiquitinases, and kinases localize to satellites, it is likely that satellites spatially concentrate enzymes with their substrates to enhance biochemical reactions or modulate the activity of enzymes that reside at satellites (Gheiratmand et al, 2019; Quarantotti et al, 2019). A similar relationship was previously described for the ATG8 family member GABARAP and the ciliogenesis factor Talpid3 (Wang et al, 2016; Joachim et al, 2017). Third, AURKA activation at the basal body is regulated by satellites in ciliated cells for which we envision two possible mechanisms. It might be a consequence of increased AURKA levels at the basal body and self‐activation of AURKA by phosphorylation of Thr288. Alternatively, it might be indirect through regulation of AURKA activators by satellites, which is supported by the identification of NDEL1, TCHP1, and calmodulin in the satellite interactome (Inoko et al, 2012; Plotnikova et al, 2012; Gupta et al, 2015; Inaba et al, 2016; Gheiratmand et al, 2019).
Aurora kinase A overexpression was shown to suppress cilium assembly by interfering with basal body maturation as well as activation of the AURKA/HDAC6‐mediated cilium disassembly pathway (Inoko et al, 2012; Pejskova et al, 2020). Notably, AURKA inhibition restored the centrosomal IFT88 recruitment and cilium assembly defect in PCM1‐depleted cells, but not in control cells, which shows that these phenotypes associated with AURKA inhibition are synthetic. Importantly, our results define AURKA and PCM1 as antagonistic regulators of cilium biogenesis and basal body maturation. Intriguingly, key ciliogenesis factors such as CP110 and Cep97 were among abundant AURKA proximity interactors, identifying them as candidates by which AURKA regulates basal body maturation.
Although the relationship between AURKA and centriolar satellites might be two‐faceted, our results support regulation of AURKA by satellites. Specifically, phosphoproteomic and phos‐tag analysis of cells treated with MLN8237 did not support the possibility that PCM1 was a substrate for AURKA. Future experiments are required to test whether AURKA phosphorylates PCM1 in different contexts such as mitosis. Finally, dissection of the AURKA‐satellite relationship and identification of AURKA proximity maps in other contexts will lead to a comprehensive understanding of why AURKA localizes to satellites and have the potential to reveal a broader function for satellites in spatiotemporal regulation of AURKA function and regulation.
Materials and Methods
Cell culture and transfection
hTERT‐RPE1 cells were grown in DMEM/F12 50/50 (Pan Biotech, Cat. # P04‐41250) supplemented with 10% fetal bovine serum (FBS, Life Technologies, Ref. # 10270‐106, Lot # 42Q5283K)) and 1% penicillin‐streptomycin (P/S). HEK293T and U2OS cells were grown in DMEM (Pan Biotech, Cat. # P04‐03590) supplemented with 10% FBS and 1% P/S. All cells were cultured at 37°C and 5% CO2. All cell lines were tested for mycoplasma by MycoAlert Mycoplasma Detection Kit (Lonza) and authenticated. RPE1 cells were transfected with the plasmids using Lipofectamine for DNA transfections and Lipofectamine RNAiMax for siRNA transfections according to the manufacturer’s instructions (Thermo Fisher Scientific). The scrambled control siRNA and the siRNAs targeting Homo sapiens PCM1 and CCDC66 were previously described (Dammermann & Merdes, 2002; Conkar et al, 2019). HEK293T cells were transfected with the plasmids using 1 μg/μl polyethylenimine, MW 25 kDa (PEI, Sigma‐Aldrich, St. Louis, MO). Briefly, the plasmids were diluted in Opti‐MEM (Invitrogen) and incubated with PEI for 40 min at room temperature. The DNA/PEI complex was added to the cells, and the culture medium was replaced with fresh medium after 6 h of incubation with the transfection mix. Lentivirus expressing V5‐BirA*‐AURKA or V5‐BirA* was produced in HEK293T using transfer vector, packaging, and envelope vectors as previously described (Gurkaslar et al, 2020). HEK293T was infected with the lentivirus, and expression of fusion proteins in stable lines was confirmed by immunofluorescence and immunoblotting.
For cilium assembly experiments, hTERT‐RPE1 cells were washed twice with PBS and incubated with DMEM/F12 supplemented with 0.5% FBS for the indicated times. For cilium disassembly experiments, ciliated RPE1 cells were incubated with DMEM/F12 50/50 supplemented with 10% FBS for the indicated times. For inhibition of AURKA activity, cells were treated with 0.5 μM MLN8237 (Selleckchem, Cat#S1133, Germany) for AURKA inhibition in ciliogenesis experiments and 1 μM MLN823 for phosphoproteomics experiments. For HDAC6 inhibition, cells were treated with 2 μM tubacin (Cayman Chemicals, Cat #13691, Ann Arbor, MI). To inhibit protein translation, cells were treated with 200 nM cycloheximide (Sigma‐Aldrich) for the indicated times. To deplete centrioles, HEK293T cells were treated with 500 nM Centrinone B for 7 days (Wong et al, 2015).
For displacement of satellites to the cell periphery, U2OS cells were transfected with GFP‐PCM1‐FKBP and pCI‐neo‐HI‐Kif5b (1–807 bp)‐FRB. Twenty‐four hours post‐transfection, cells were treated with 500 nm rapamycin (Millipore, Cat #553210, Burlington, MA) for 1 h followed by 2× PBS washout. Cells were fixed 6 h after rapamycin induction.
Plasmids
Full‐length cDNA for Homo sapiens AURKA (GenBank accession no: BC002499.2) was obtained from DNASU Plasmid Repository (Seiler et al, 2014). pLVPT2‐V5‐BirA*‐AURKA, pDEST53‐AURKA, and pcDNA5.1‐FRT/TO‐FLAG‐AURKA were cloned by Gateway recombination between pDONR221‐AURKA and the indicated Gateway destination plasmids. Homo sapiens PCM1 (GenBank accession no: NM_006197) was cloned into pCDNA5.1‐FRT/TO‐Flag‐miniTurboID using AscI and NotI restriction sites in forward and reverse primers (gift from Bettencourt‐Dias laboratory, IGC Gulbenkian). pDEST53‐PCM1(1‐1200) was cloned by Gateway recombination between pDONR221(1‐1200) and pDEST53. Full‐length cDNA of Homo sapiens PCM1 was amplified from peGFP‐C1‐PCM1 and cloned into peGFP‐C1 (Clontech, Mountain View, CA) without stop codon using XhoI and KpnI sites. GFP‐PCM1‐FKBP and CI‐neoHI‐Kif5b (1–807)‐FRB plasmids were described previously (Aydin et al, 2020).
Biotin‐streptavidin affinity purification
For the BioID experiments, HEK293T stably expressing V5‐BirA*‐AURKA or V5‐BirA*‐AURKA were grown in 5 × 15 cm plates were grown in complete medium supplemented with 50 μM biotin for 18 h. Following biotin treatment, cells were lysed in lysis buffer (20 mM HEPES, pH 7.8, 5 mM K‐acetate, 0.5 mM MgCI2, 0.5 mM DTT, protease inhibitors) and sonicated. An equal volume of 4°C 50 mM Tris (pH 7.4) was added to the extracts, and insoluble material was pelleted. Soluble materials from whole cell lysates were incubated with Streptavidin agarose beads (Thermo Scientific). Beads were collected and washed twice in wash buffer 1 (2% SDS in dH2O), once with wash buffer 2 (0.2% deoxycholate, 1% Triton X‐100, 500 mM NaCI, 1 mM EDTA, and 50 mM Hepes, pH 7.5), once with wash buffer 3 (250 mM LiCI, 0.5% NP‐40, 0.5% deoxycholate, 1% Triton X‐100, 500 mM NaCI, 1 mM EDTA and 10 mM Tris, pH 8.1), and twice with wash buffer 4 (50 mM Tris, pH 7.4, and 50 mM NaCI). 10% of the sample was reserved for Western blot analysis, and 90% of the sample to be analyzed by mass spectrometry was washed twice in 50 mM NH4HCO3. Confirmation of proximity interactors of AURKA was carried out by immunoblotting for V5 or FLAG antibodies that detect fusion protein and streptavidin that detect biotinylated proteins.
Immunofluorescence, microscopy, and quantitation
For immunofluorescence experiments, cells were grown on coverslips and fixed in either methanol or 4% paraformaldehyde in PBS for indirect immunofluorescence. After rehydration in PBS, cells were blocked in 3% BSA (Sigma‐Aldrich) in PBS + 0.1% Triton X‐100. Coverslips were incubated in primary antibodies diluted in blocking solution. Anti‐PCM1 antibody was generated and used for immunofluorescence as previously described (Firat‐Karalar et al, 2014). Other antibodies used for immunofluorescence in this study were rabbit anti‐AURKA and anti‐p‐AURKA (Cell signaling, #3875T) at 1:1000, rabbit anti‐PCM1 (homemade) at 1:1,000, mouse anti‐γ‐tubulin (GTU‐88; Sigma‐Aldrich‐T6557) at 1:4,000, mouse anti‐acetylated tubulin (Santa Cruz Biotechnology, sc‐23950) at 1:5,000, mouse anti‐V5 (Invitrogen), mouse anti‐GFP (3e6; Invitrogen‐11120) T 1:1,000, mouse anti‐Ki67 (Santa Cruz Biotechnology, sc‐23900), and rabbit anti IFT88 (Proteintech‐12780‐1‐AP) 1:1,000. Following primary antibody incubation and 3× PBS wash, coverslips were incubated with Alexa Flour 488‐, 568‐, or 633‐coupled (Life Technologies), and they were used at 1:2,000. Biotinylated proteins were detected with streptavidin coupled to Alexa Fluor 488 or 594 (Thermo Fisher). DNA was stained with 4′,6‐diamidino‐2‐phenylindole (DAPI; 1 μg/ml). Samples were mounted using Mowiol mounting medium containing N‐propyl gallate (Sigma‐Aldrich). Images were acquired with a step size of 0.3 μm in 1024x1024 format with Leica DMi8 inverted fluorescent microscope or SP8 scanning confocal microscope using the HC PL APO CS2 63x 1.4 NA oil objective.
Quantitative immunofluorescence for AURKA was performed by acquiring a z stack of control and depleted cells using identical gain and exposure settings. The z‐stacks were used to assemble maximum‐intensity projections. The centrosome region for each cell was defined by staining for a centrosomal marker including gamma‐tubulin. The region of interest that encompassed the centrosome was defined as a circle 3 μm2 area centered on the centrosome in each cell. Total pixel intensity of fluorescence within the region of interest was measured using ImageJ (National Institutes of Health, Bethesda, MD) (Schneider et al, 2012). Background subtraction was performed by quantifying fluorescence intensity of a region of equal dimensions in the area proximate to the centrosome. Statistical analysis was done by normalizing these values to their mean. Primary cilium formation was assessed by counting the total number of cells, and the number of cells with primary cilia, as detected by DAPI and acetylated tubulin staining, respectively. Ciliary protein concentrations were determined by dividing fluorescence signal of the protein to the cilium length, which was quantified using Arl13b or acetylated tubulin staining. All data acquisition was done in a blinded manner.
Immunoprecipitation
HEK293T cells were co‐transfected with indicated plasmids. 48 post‐transfection, cells were washed and lysed with lysis buffer (10 mM Tris/Cl pH 7.5, 150 mM NaCl, 0.5 mM EDTA, 0.5% Nonidet™ P40 and protease inhibitor cocktail) for 30 min. Lysates were centrifuged at 13000 rpm for 10 min at +4°C, and supernatants were transferred to a tube. 100 μl from each sample was saved as input. The rest of the supernatant was immunoprecipitated with GFP‐Trap beads (ChromoTek) or anti‐FLAG M2 agarose beads (Sigma‐Aldrich) overnight at +4°C. After washing 3× with lysis buffer, samples were resuspended in SDS containing sample buffer and analyzed by immunoblotting. For FLAG‐PCM1 pulldowns processed for phosphoproteomics experiments, cells were lysed with lysis buffer freshly supplemented with protease inhibitors (10 µg/ml Leupeptin, Pepstatin, and Chymostatin, 1 mM PMSF) and phosphatase inhibitors (PhosStop, Sigma‐Aldrich). For endogenous immunoprecipitation, cells were lysed in lysis buffer (50 mM Tris–HCl pH 7.4, 266 mM NaCl, 2.27 mM KCl, 1.25 mM KH2PO4, 6.8 mM Na2HPO4‐7H2O, and 1% NP‐40, protease inhibitors) and lysates were incubated with anti‐AURKA antibody overnight. The protein complexes were precipitated with protein A agarose beads and immunoprecipitates were analyzed with Western blotting.
Cell lysis, immunoblotting, and Phos‐tag gels
Cells were lysed in 50 mM Tris (pH 7.6), 150 mM NaCI, 1% Triton X‐100, and protease inhibitors for 30 min at 4°C followed by centrifugation at 15,000 g for 15 min. The protein concentration of the resulting supernatants was determined with the Bradford solution (Bio‐Rad Laboratories, CA, USA). For immunoblotting, equal quantities of cell extracts were resolved on SDS–PAGE gels, transferred onto nitrocellulose membranes, blocked with TBST in 5% milk for 1 h at room temperature. Blots were incubated with primary antibodies diluted in 5% BSA in TBST overnight at 4°C, washed with TBST three times for 5 min, and blotted with secondary antibodies for 1 h at room temperature. After washing blots with TBST three times for 5 min, they were visualized with the LI‐COR Odyssey Infrared Imaging System and software at 169 mm (LI‐COR Biosciences).
For phos‐tag gel electrophoresis, the gels with Phos‐tag Acrylamide AAL‐107 were prepared and for the proteins bigger than 200 kDa, the acrylamide gel was mixed with agarose according to the manufacturer instructions (Wako Chemicals, Japan). Cells were lysed in lysis buffer (10 mM Tris–HCl, pH 8.0, 1 mM EDTA, 0.5 mM EGTA, 1% Triton X‐100, 0.1% Sodium Deoxycholate, 0.1% SDS, 140 mM NaCl) freshly supplemented with protease and phosphatase inhibitors. Samples were run in parallel on Phos‐tag gels, washed in transfer buffer with 1 mM EDTA twice, washed in transfer buffer without EDTA twice, and processed for immunoblotting.
Primary antibodies used for Western blotting were rabbit anti‐AURKA and anti‐p‐AURKA (Cell signaling, #3875T), rabbit anti‐PCM1 (Proteintech‐198561‐1‐AP) at 1:1,000, rabbit anti‐Flag (Cell signaling,#2368) at 1:1,000, mouse anti‐vinculin (Santa Cruz Biotechnology, sc‐55465), rabbit anti‐CEP131 (Bethyl Laboratories) at 1:1,000, rabbit anti‐CEP72 (Bethyl) at 1:1,000, Rabbit anti‐CEP63 (Thermo Fisher) at 1:1,000, rabbit anti‐GFP (homemade), rabbit anti‐CSPP1, rabbit anti‐Talpid3 (Proteintech‐24421‐1‐AP) at 1:500, rabbit anti‐CCP110 (Proteintech‐12780‐1‐AP) at 1:500, rabbit anti‐SSX2IP (Proteintech 13694‐1‐AP), rabbit anti‐BBS4 (Proteintech‐12766‐1‐AP), and rabbit anti‐GAPDH (Cell signaling #2118). Secondary antibodies used for Western blotting were IRDye 680‐ and IRDye 800‐coupled and were used 1:15,000 (LI‐COR Biosciences).
Flow cytometer
Cells were harvested and fixed in 70% ethanol at −20°C overnight, followed by washes in phosphate‐buffered saline (PBS) and staining with 40 μg/ml propidium iodide and 10 μg/ml RNaseA at 37°C for 2 h. DNA content was assessed by analyzing cells using flow cytometer (BD Biosciences).
Protein identification by Mass spectrometry
Proteins bound to beads were reduced by 20‐min incubation with 5 mM TCEP (tris(2‐carboxyethyl) phosphine) and alkylated in the dark by treatment with 10mM Iodoacetamide for 20 additional minutes. The proteins were subsequently digested by adding Sequencing Grade Modified Trypsin (Promega, Madison, WI, USA) and placing the reaction mixture in a Thermomixer (Eppendorf, Westbury, NY) and incubating overnight at 37°C at 750 rpm. The next day, the sample was acidified with formic acid to a final concentration of 5% and spun at 22,000 g for 30 min. The supernatant was carefully transferred to a separate microfuge tube so as not to disturb the bead pellet and pressure‐loaded into a biphasic trap column. MS analysis of the samples was performed using MudPIT technology [S7]. Capillary columns were prepared in‐house from particle slurries in methanol. An analytical column was generated by pulling a 100 μm ID/360 μm OD capillary (Polymicro Technologies, Inc., Phoenix, AZ) to 3 μm ID tip. The pulled column was packed with reverse‐phase particles (Aqua C18, 3 μm dia., 90 Å pores, Phenomenex, Torrance, CA) until 15 cm long. A biphasic trapping column was prepared by creating a Kasil frit at one end of an undeactivated 250 μm ID/360 μm OD capillary (Agilent Technologies, Inc., Santa Clara, CA), which was then successively packed with 2.5 cm strong cation exchange particles (Partisphere SCX, 5 μm dia., 100 Å pores, Phenomenex, Torrance, CA) and 2.5 cm reverse‐phase particles (Aqua C18, 5 μm dia., 90 Å pores, Phenomenex, Torrance, CA). The trapping column was equilibrated using buffer A (5% acetonitrile/0.1% formic acid) prior to sample loading. After sample loading and prior to MS analysis, the resin‐bound peptides were desalted with buffer A by letting it flow through the trap column. The trap and analytical columns were assembled using a zero‐dead volume union (Upchurch Scientific, Oak Harbor, WA). LC−MS/MS analysis was performed on LTQ Orbitrap or LTQ Orbitrap Velos (Thermo Scientific, San Jose, CA, USA) interfaced at the front end with a quaternary HP 1100 series HPLC pump (Agilent Technology, Santa Clara, CA, USA) using an in‐house built electrospray stage. Electrospray was performed directly from the analytical column by applying the ESI voltage at a tee (150 μm ID, Upchurch Scientific) directly downstream of a 1:1,000 split flow used to reduce the flow rate to 250 nl/min through the columns. A fully automated 6‐step MudPIT run was performed on each sample using a three mobile phase system consisting of buffer A (5% acetonitrile/0.1% formic acid), buffer B (80% acetonitrile/0.1% formic acid), and buffer C (500 mM ammonium acetate/5% acetonitrile/0.1% formic acid). The first step was 60 min reverse‐phase run, whereas five subsequent steps were of 120 min duration with different concentration of buffer C run for 4 min at the beginning of each of the gradient. In LTQ Orbitrap Velos, peptides were analyzed using a Top‐20 data‐dependent acquisition method in which fragmentation spectra are acquired for the top 20 peptide ions above a predetermined signal threshold. As peptides were eluted from the microcapillary column, they were electrosprayed directly into the mass spectrometer with the application of a distal 2.4 kV spray voltage. For each cycle, full−scan MS spectra (m/z range 300–1,600) were acquired in the Orbitrap with the resolution set to a value of 60,000 at m/z 400 and an automatic gain control (AGC) target of 1 × 106 ions and the maximal injection time of 250 ms. For MS/MS scans, the target value was 10,000 ions with injection time of 25 ms. Once analyzed, the selected peptide ions were dynamically excluded from further analysis for 120 s to allow for the selection of lower‐abundance ions for subsequent fragmentation and detection using the setting for repeat count = 1, repeat duration = 30 ms, and exclusion list size = 500. Charge state filtering, where ions with singly or unassigned charge states were rejected from fragmentation, was enabled. The minimum MS signal for triggering MS/MS was set to 500, and an activation time of 10 ms was used. All tandem mass spectra were collected using normalized collision energy of 35%, an isolation window of 2 Th. In LTQ Orbitrap, peptides were analyzed using a Top‐10 data‐dependent acquisition method. For protein identification, we used Integrated Proteomics Pipeline (IP2, San Diego, CA) software, a web‐based proteomics data analysis platform that supports both cloud and cluster computing, developed by Integrated Proteomics Applications, Inc. (http://www.integratedproteomics.com/). Tandem mass spectra were extracted from the Xcalibur data system format (.raw) into MS2 format using RawXtract1.9.9.2. The MS/MS spectra were searched with the ProLuCID algorithm against the EBI human IPI database (version 3.71, release date March 24, 2010) that was concatenated to a decoy database in which the sequence for each entry in the original database was reversed. The database also had sequence for two proteins, E. coli BirA‐R118G appended to it. The search parameters include 50 ppm peptide precursor mass tolerance and 0.6 Da for the fragment mass tolerance acquired in the ion trap. The initial wide precursor mass tolerance in the database search was subjected to post‐search filtering and eventually constrained to 20 ppm. Carbamidomethylating on cysteine was defined as fixed modification, and phosphorylation on STY was included as variable modification in the search criteria. The search space also included all fully and semi‐tryptic peptide candidates of length of at least six amino acids. Maximum number of internal miscleavages was kept unlimited, thereby allowing all cleavage points for consideration. ProLuCID outputs were assembled and filtered using the DTASelect2.0 program that groups related spectra by protein and removes those that do not pass basic data‐quality criteria [S8]. DTASelect2.0 combines XCorr and ΔCN measurements using a quadratic discriminant function to compute a confidence score to achieve a user‐specified false discovery rate (less than 1% in this analysis).
Mass Spectrometry data analysis for BioID experiments
Raw data of two biological replicates for V5‐BirA*‐AURKA and four biological replicates for V5‐BirA* were analyzed to identify proximity interactors of AURKA. The data were analyzed with SAINTExpress (Teo et al, 2014). High‐confidence interactors were defined as the ones with SAINT score > 0.95. Comparative analysis with BioID results and literature was carried out incorporating interactors in BioGRID. GO terms were determined by using Database for Annotation, Visualization, and Integrated Discovery (DAVID).
Network modeling and clustering analysis
Significance analysis of interactome analysis returned 440 high‐confidence interactors of AURKA. For Fig EV2, we performed the network analyses for the first 200 high‐confidence interactors ranked by their relative fold change in V5‐BirA*‐AURKA dataset versus V5‐BirA* dataset. The edges of the first 200 proteins in the proximity map are plotted with STRING database, and the map is visualized by CytoScape. The functional clusters and GO categories for these clusters are determined with the Clustering with Overlapping Neighborhood Expansion (ClusterONE) plug‐in of Cytoscape and BinGO plug‐ins (P < 0.05). The network output file was visualized using Cytoscape 3.7.2 (Nepusz et al, 2012).
Identification of phosphorylated peptides
Coomassie‐stained bands from two experimental replicates from FLAG‐PCM1 pulldown experiments were excised, chopped into small pieces, and transferred to 1.5 ml Eppendorf tubes. For all following steps, buffers were exchanged by two consecutive 15 min incubation steps of the gel pieces with 200 µl of acetonitrile (ACN) whereby the ACN was removed after each step. Proteins were reduced by the addition of 200 µl of a 10 mM DTT solution in 100 mM ammonium bicarbonate (AmBiC, Sigma‐Aldrich, A6141) and incubation at 56°C for 30 min. Proteins were alkylated by the addition of 200 µl of a 55 mM chloroacetamide (CAA) in 100 mM AmBiC and incubation for 20 min in the dark. A 0.1 µg/µl stock solution of trypsin (Promega, V511A) in trypsin resuspension buffer (Promega, V542A) was diluted with ice‐cold 50 mM AmBiC buffer to achieve a final concentration of 1 ng/µl. 50 µl thereof was added to gel pieces, which were incubated first for 30 min on ice and then over night at 37°C. Gel pieces were sonicated for 15 min, spun down, and the supernatant was transferred into a glass vial (VDS Optilab, 93908556). Remaining gel pieces were washed with 50 µl of an aqueous solution of 50% ACN and 1% formic acid and sonicated for 15 min. The combined supernatants were dried in a SpeedVac and reconstituted in 10 µl of an aqueous solution of 0.1% (v/v) formic acid.
Peptides were analyzed by LC‐MS/MS on an Orbitrap Fusion Lumos mass spectrometer (Thermo Scientific) as previously described (PMID:30858367). To this end, peptides were separated using an Ultimate 3000 nano RSLC system (Dionex) equipped with a trapping cartridge (Precolumn C18 PepMap100, 5 mm, 300 μm i.d., 5 μm, 100 Å) and an analytical column (Acclaim PepMap 100. 75 × 50 cm C18, 3 mm, 100 Å) connected to a nanospray‐Flex ion source. The peptides were loaded onto the trap column at 30 µl per min using solvent A (0.1% formic acid), and peptides were eluted using a gradient from 2 to 85% Solvent B (0.1% formic acid in acetonitrile) over 30 min at 0.3 µl per min (all solvents were of LC‐MS grade). For the detection of post‐translational modifications, peptides were eluted using a gradient from 2 to 80% solvent B over 60 min. The Orbitrap Fusion Lumos was operated in positive ion mode with a spray voltage of 2.2 kV and capillary temperature of 275°C. Full scan MS spectra with a mass range of 375–1200 m/z were acquired in profile mode using a resolution of 120,000 (maximum injections time of 50 ms, AGC target was set to 400%, and a max injection time of 86 ms). Precursors were isolated using the quadrupole with a window of 1.2 m/z, and fragmentation was triggered by HCD in fixed collision energy mode with fixed collision energy of 34%. MS2 spectra were acquired with the Orbitrap with a resolution of 30.000 and a max injection time of 86 ms.
The Orbitrap Fusion Lumos was operated in positive ion mode with a spray voltage of 2.2 kV and capillary temperature of 275°C. Full scan MS spectra with a mass range of 350–1500 m/z were acquired in profile mode using a resolution of 120,000 (maximum injections time of 100 ms, AGC target was set to standard, and a RF lens setting of 30%). Precursors were isolated using the quadrupole with a window of 1.2 m/z and Fragmentation was triggered by HCD in fixed collision energy mode with fixed collision energy of 30%. MS2 spectra were acquired in Ion Trap normal mode. The dynamic exclusion was set to 5 s. Acquired data were analyzed using IsobarQuant (PMID: 26379230) and Mascot V2.4 (Matrix Science) using a reversed Uniprot homo sapiens database (UP000005640) including common contaminants.
Raw data were analyzed as previously described (Cox & Mann, 2008). Briefly, it was searched against the Uniprot human reference proteome (75,093 entries, June 2016) including (please paste here name of PCM1 construct) using MaxQuant 1.6.17.0. The default parameters were used with the following adjustments: Carbamidomethyl (C) as fixed modification and phosphorylation of S, T, and Y was set as variable modification, as well as oxidation of M and acetylation of protein N‐termini. For digestion, Trypsin/P was enabled and the missed cleavages were set to 3. Label‐free quantification was enabled, with Fast LFQ disabled. IBAQ calculations were enabled. The minimum number of unique peptides was set to 1.
Statistical analysis
Statistical results, average, and standard deviation values were computed and plotted by using Prism (GraphPad, La Jolla, CA). Student’s t‐test and one‐way ANOVA test were applied to compare the statistical significance of the measurements. Following key is followed for asterisk placeholders for p‐values in the figures: *P < 0.05, **P < 0.01, ***P < 0.001, **** P < 0.0001.
Author contributions
Conceptualization, ENF‐K, MDA; Methodology, ENF‐K, MDA; Investigation (light microscopy, molecular biology, and biochemical analysis), MDA, NR; Investigation (proximity labeling and mass spectrometry), ENF‐K, NR; Investigation (network analysis), MDA; Resources, ENF‐K, MDA; Writing—Original Draft, ENF‐K, MDA; Writing—Review & Editing, ENF‐K, MDA, NR; Visualization, MDA; Supervision, ENF‐K, JRY; Project Administration, ENF‐K; Funding Acquisition, ENF‐K, JRY.
Conflict of interests
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Review Process File
Acknowledgements
We acknowledge the Firat‐Karalar laboratory members for insightful discussions regarding this work. We acknowledge Ahmet Aktas for cloning the pCDNA5.1‐FRT/TO‐FLAG‐miniTurboID‐PCM1 plasmid, Monica Bettencourt‐Dias (IGC Gulbenkian) for sharing the pCDNA5.1‐FRT/TO‐Flag‐miniTurbo plasmid and Sebastian Patzke (University of Oslo) for RPE1‐hTERT cells. We acknowledge the use of services of the European Molecular Biology Laboratory (Heidelberg) Proteomics Core Facility for phosphoproteomics experiments. We acknowledge the use of services and facilities of the Koc University Research Center for Translational Medicine (KUTTAM), funded by the Presidency of Turkey, Presidency of Strategy and Budget. This work was supported by European Research Council (ERC) grant 679140 to ENF, European Molecular Biology Organization (EMBO) Installation grant 3622 and an EMBO Young Investigator Award to ENF and the National Institutes of Health Grant to JRY.
EMBO reports (2021) 22: e51902.
Data availability
No data were deposited in a public database.
References
- Anand S, Penrhyn‐Lowe S, Venkitaraman AR (2003) AURORA‐A amplification overrides the mitotic spindle assembly checkpoint, inducing resistance to Taxol. Cancer Cell 3: 51–62 [DOI] [PubMed] [Google Scholar]
- Arslanhan MD, Gulensoy D, Firat‐Karalar EN (2020) A proximity mapping journey into the biology of the mammalian centrosome/cilium complex. Cells 9: 1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aydin OZ, Taflan SO, Gurkaslar C, Firat‐Karalar EN (2020) Acute inhibition of centriolar satellite function and positioning reveals their functions at the primary cilium. PLoS Biol 18: e3000679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr AR, Gergely F (2007) Aurora‐A: the maker and breaker of spindle poles. J Cell Sci 120: 2987–2996 [DOI] [PubMed] [Google Scholar]
- Bertolin G, Bulteau AL, Alves‐Guerra MC, Burel A, Lavault MT, Gavard O, Le Bras S, Gagne JP, Poirier GG, Le Borgne R et al (2018) Aurora kinase A localises to mitochondria to control organelle dynamics and energy production. eLife 7: e38111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertolin G, Tramier M (2020) Insights into the non‐mitotic functions of Aurora kinase A: more than just cell division. Cell Mol Life Sci 77: 1031–1047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhogaraju S, Cajanek L, Fort C, Blisnick T, Weber K, Taschner M, Mizuno N, Lamla S, Bastin P, Nigg EA et al (2013) Molecular basis of tubulin transport within the cilium by IFT74 and IFT81. Science 341: 1009–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischoff JR, Anderson L, Zhu Y, Mossie K, Ng L, Souza B, Schryver B, Flanagan P, Clairvoyant F, Ginther C et al (1998) A homologue of Drosophila aurora kinase is oncogenic and amplified in human colorectal cancers. EMBO J 17: 3052–3065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breslow DK, Holland AJ (2019) Mechanism and regulation of centriole and cilium biogenesis. Annu Rev Biochem 88: 691–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockmann M, Poon E, Berry T, Carstensen A, Deubzer H, Rycak L, Jamin Y, Thway K, Robinson S, Roels F et al (2013) Small molecule inhibitors of aurora‐a induce proteasomal degradation of N‐myc in childhood neuroblastoma. Cancer Cell 24: 75–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Büchel G, Carstensen A, Mak K‐Y, Roeschert I, Leen E, Sumara O, Hofstetter J, Herold S, Kalb J, Baluapuri A et al (2017) Association with aurora‐A controls N‐MYC‐dependent promoter escape and pause release of RNA polymerase II during the cell cycle. Cell Rep 21: 3483–3497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrum AK, Carvajal‐Maldonado D, Mudge MC, Valle‐Garcia D, Majid MC, Patel R, Sowa ME, Gygi SP, Harper JW, Shi Y et al (2019) Mitotic regulators TPX2 and Aurora A protect DNA forks during replication stress by counteracting 53BP1 function. J Cell Biol 218: 422–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CL, Perrimon N (2017) Proximity‐dependent labeling methods for proteomic profiling in living cells. Wiley Interdiscip Rev Dev Biol 6: e272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SH, Lahav G (2016) Two is better than one; toward a rational design of combinatorial therapy. Curr Opin Struct Biol 41: 145–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conkar D, Bayraktar H, Firat‐Karalar EN (2019) Centrosomal and ciliary targeting of CCDC66 requires cooperative action of centriolar satellites, microtubules and molecular motors. Sci Rep 9: 14250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conkar D, Culfa E, Odabasi E, Rauniyar N, Yates JR III, Firat‐Karalar EN (2017) Centriolar satellite protein CCDC66 interacts with CEP290 and functions in cilium formation and trafficking. J Cell Sci 130: 1450–1462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conkar D, Firat‐Karalar EN (2020) Microtubule‐associated proteins and emerging links to primary cilium structure, assembly, maintenance, and disassembly. FEBS J 288: 786–798 [DOI] [PubMed] [Google Scholar]
- Courtheoux T, Diallo A, Damodaran AP, Reboutier D, Watrin E, Prigent C (2018) Aurora A kinase activity is required to maintain an active spindle assembly checkpoint during prometaphase. J Cell Sci 131: jcs191353 [DOI] [PubMed] [Google Scholar]
- Cox J, Mann M (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.‐range mass accuracies and proteome‐wide protein quantification. Nat Biotechnol 26: 1367–1372 [DOI] [PubMed] [Google Scholar]
- Dammermann A, Merdes A (2002) Assembly of centrosomal proteins and microtubule organization depends on PCM‐1. J Cell Biol 159: 255–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damodaran AP, Gavard O, Gagne JP, Rogalska ME, Mancini E, Courtheoux C, Cailoce J, Poirier GG, Valcarcel J, Watrin E et al (2020) Aurora‐A phosphorylates splicing factors and regulates alternative splicing. bioRxiv 10.1101/2020.11.04.368498 [PREPRINT] [DOI] [Google Scholar]
- Dephoure N, Zhou C, Villen J, Beausoleil SA, Bakalarski CE, Elledge SJ, Gygi SP (2008) A quantitative atlas of mitotic phosphorylation. Proc Natl Acad Sci USA 105: 10762–10767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deretic J, Kerr A, Welburn JPI (2019) A rapid computational approach identifies SPICE1 as an Aurora kinase substrate. Mol Biol Cell 30: 312–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVaul N, Koloustroubis K, Wang R, Sperry AO (2017) A novel interaction between kinase activities in regulation of cilia formation. BMC Cell Biol 18: 33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutertre S, Cazales M, Quaranta M, Froment C, Trabut V, Dozier C, Mirey G, Bouché J‐P, Theis‐Febvre N, Schmitt E et al (2004) Phosphorylation of CDC25B by Aurora‐A at the centrosome contributes to the G2‐M transition. J Cell Sci 117: 2523–2531 [DOI] [PubMed] [Google Scholar]
- Firat‐Karalar EN, Rauniyar N, Yates JR III, Stearns T (2014) Proximity interactions among centrosome components identify regulators of centriole duplication. Curr Biol 24: 664–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frikstad K‐A, Molinari E, Thoresen M, Ramsbottom SA, Hughes F, Letteboer SJF, Gilani S, Schink KO, Stokke T, Geimer S et al (2019) A CEP104‐CSPP1 complex is required for formation of primary cilia competent in hedgehog signaling. Cell Rep 28: 1907–1922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautschi O, Heighway J, Mack PC, Purnell PR, Lara PN Jr, Gandara DR (2008) Aurora kinases as anticancer drug targets. Clin Cancer Res 14: 1639–1648 [DOI] [PubMed] [Google Scholar]
- Gentzel M, Pardo M, Subramaniam S, Stewart AF, Choudhary JS (2019) Proteomic navigation using proximity‐labeling. Methods 164–165: 67–72 [DOI] [PubMed] [Google Scholar]
- Gheiratmand L, Coyaud E, Gupta GD, Laurent EM, Hasegan M, Prosser SL, Goncalves J, Raught B, Pelletier L (2019) Spatial and proteomic profiling reveals centrosome‐independent features of centriolar satellites. EMBO J 38: e101109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover DM, Leibowitz MH, McLean DA, Parry H (1995) Mutations in aurora prevent centrosome separation leading to the formation of monopolar spindles. Cell 81: 95–105 [DOI] [PubMed] [Google Scholar]
- Golemis EA, Scheet P, Beck TN, Scolnick EM, Hunter DJ, Hawk E, Hopkins N (2018) Molecular mechanisms of the preventable causes of cancer in the United States. Genes Dev 32: 868–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta G, Coyaud É, Gonçalves J, Mojarad B, Liu Yi, Wu Q, Gheiratmand L, Comartin D, Tkach J, Cheung S et al (2015) A dynamic protein interaction landscape of the human centrosome‐cilium interface. Cell 163: 1484–1499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurkaslar HK, Culfa E, Arslanhan MD, Lince‐Faria M, Firat‐Karalar EN (2020) CCDC57 cooperates with microtubules and microcephaly protein CEP63 and regulates centriole duplication and mitotic progression. Cell Rep 31: 107630 [DOI] [PubMed] [Google Scholar]
- Hall EA, Keighren M, Ford MJ, Davey T, Jarman AP, Smith LB, Jackson IJ, Mill P (2013) Acute versus chronic loss of mammalian Azi1/Cep131 results in distinct ciliary phenotypes. PLoS Genet 9: e1003928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasanov E, Chen G, Chowdhury P, Weldon J, Ding Z, Jonasch E, Sen S, Walker CL, Dere R (2017) Ubiquitination and regulation of AURKA identifies a hypoxia‐independent E3 ligase activity of VHL. Oncogene 36: 3450–3463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegarat N, Smith E, Nayak G, Takeda S, Eyers PA, Hochegger H (2011) Aurora A and Aurora B jointly coordinate chromosome segregation and anaphase microtubule dynamics. J Cell Biol 195: 1103–1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori A, Peddie CJ, Collinson LM, Toda T (2015) Centriolar satellite‐ and hMsd1/SSX2IP‐dependent microtubule anchoring is critical for centriole assembly. Mol Biol Cell 26: 2005–2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inaba H, Goto H, Kasahara K, Kumamoto K, Yonemura S, Inoko A, Yamano S, Wanibuchi H, He D, Goshima N et al (2016) Ndel1 suppresses ciliogenesis in proliferating cells by regulating the trichoplein‐Aurora A pathway. J Cell Biol 212: 409–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoko A, Matsuyama M, Goto H, Ohmuro‐Matsuyama Y, Hayashi Y, Enomoto M, Ibi M, Urano T, Yonemura S, Kiyono T et al (2012) Trichoplein and Aurora A block aberrant primary cilia assembly in proliferating cells. J Cell Biol 197: 391–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joachim J, Razi M, Judith D, Wirth M, Calamita E, Encheva V, Dynlacht BD, Snijders AP, O’Reilly N, Jefferies HBJ et al (2017) Centriolar satellites control GABARAP ubiquitination and GABARAP‐mediated autophagy. Curr Biol 27: 2123–2136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joukov V, De Nicolo A (2018) Aurora‐PLK1 cascades as key signaling modules in the regulation of mitosis. Sci Signal 11: eaar4195 [DOI] [PubMed] [Google Scholar]
- Karthigeyan D, Prasad SB, Shandilya J, Agrawal S, Kundu TK (2011) Biology of Aurora A kinase: implications in cancer manifestation and therapy. Med Res Rev 31: 757–793 [DOI] [PubMed] [Google Scholar]
- Kashatus DF, Lim KH, Brady DC, Pershing NL, Cox AD, Counter CM (2011) RALA and RALBP1 regulate mitochondrial fission at mitosis. Nat Cell Biol 13: 1108–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettenbach AN, Schweppe DK, Faherty BK, Pechenick D, Pletnev AA, Gerber SA (2011) Quantitative phosphoproteomics identifies substrates and functional modules of Aurora and Polo‐like kinase activities in mitotic cells. Sci Signal 4: rs5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Krishnaswami SR, Gleeson JG (2008) CEP290 interacts with the centriolar satellite component PCM‐1 and is required for Rab8 localization to the primary cilium. Hum Mol Genet 17: 3796–3805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Zaghloul NA, Bubenshchikova E, Oh EC, Rankin S, Katsanis N, Obara T, Tsiokas L (2011) Nde1‐mediated inhibition of ciliogenesis affects cell cycle re‐entry. Nat Cell Biol 13: 351–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinzel D, Boldt K, Davis EE, Burtscher I, Trümbach D, Diplas B, Attié‐Bitach T, Wurst W, Katsanis N, Ueffing M et al (2010) Pitchfork regulates primary cilia disassembly and left‐right asymmetry. Dev Cell 19: 66–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinger M, Wang W, Kuhns S, Barenz F, Drager‐Meurer S, Pereira G, Gruss OJ (2014) The novel centriolar satellite protein SSX2IP targets Cep290 to the ciliary transition zone. Mol Biol Cell 25: 495–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch A, Krug K, Pengelley S, Macek B, Hauf S (2011) Mitotic substrates of the kinase aurora with roles in chromatin regulation identified through quantitative phosphoproteomics of fission yeast. Sci Signal 4: rs6 [DOI] [PubMed] [Google Scholar]
- Korobeynikov V, Deneka AY, Golemis EA (2017) Mechanisms for nonmitotic activation of Aurora‐A at cilia. Biochem Soc Trans 45: 37–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo A, Sasaki H, Yuba‐Kubo A, Tsukita S, Shiina N (1999) Centriolar satellites: molecular characterization, ATP‐dependent movement toward centrioles and possible involvement in ciliogenesis. J Cell Biol 147: 969–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert JP, Tucholska M, Go C, Knight JD, Gingras AC (2014) Proximity biotinylation and affinity purification are complementary approaches for the interactome mapping of chromatin‐associated protein complexes. J Proteomics 118: 81–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeRoy PJ, Hunter JJ, Hoar KM, Burke KE, Shinde V, Ruan J, Bowman D, Galvin K, Ecsedy JA (2007) Localization of human TACC3 to mitotic spindles is mediated by phosphorylation on Ser558 by Aurora A: a novel pharmacodynamic method for measuring Aurora A activity. Cancer Res 67: 5362–5370 [DOI] [PubMed] [Google Scholar]
- Levinson NM (2018) The multifaceted allosteric regulation of Aurora kinase A. Biochem J 475: 2025–2042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liem KF Jr, He M, Ocbina PJ, Anderson KV (2009) Mouse Kif7/Costal2 is a cilia‐associated protein that regulates Sonic hedgehog signaling. Proc Natl Acad Sci USA 106: 13377–13382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lioutas A, Vernos I (2013) Aurora A kinase and its substrate TACC3 are required for central spindle assembly. EMBO Rep 14: 829–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Salokas K, Tamene F, Jiu Y, Weldatsadik RG, Ohman T, Varjosalo M (2018) An AP‐MS‐ and BioID‐compatible MAC‐tag enables comprehensive mapping of protein interactions and subcellular localizations. Nat Commun 9: 1188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manfredi MG, Ecsedy JA, Chakravarty A, Silverman L, Zhang M, Hoar KM, Stroud SG, Chen W, Shinde V, Huck JJ et al (2011) Characterization of Alisertib (MLN8237), an investigational small‐molecule inhibitor of aurora A kinase using novel in vivo pharmacodynamic assays. Clin Cancer Res 17: 7614–7624 [DOI] [PubMed] [Google Scholar]
- Maris JM, Morton CL, Gorlick R, Kolb EA, Lock R, Carol H, Keir ST, Reynolds CP, Kang MH, Wu J et al (2010) Initial testing of the aurora kinase A inhibitor MLN8237 by the Pediatric Preclinical Testing Program (PPTP). Pediatr Blood Cancer 55: 26–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meraldi P, Honda R, Nigg EA (2002) Aurora‐A overexpression reveals tetraploidization as a major route to centrosome amplification in p53‐/‐ cells. EMBO J 21: 483–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirvis M, Stearns T, James Nelson W (2018) Cilium structure, assembly, and disassembly regulated by the cytoskeleton. Biochem J 475: 2329–2353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori D, Yano Y, Toyo‐oka K, Yoshida N, Yamada M, Muramatsu M, Zhang D, Saya H, Toyoshima YY, Kinoshita K et al (2007) NDEL1 phosphorylation by Aurora‐A kinase is essential for centrosomal maturation, separation, and TACC3 recruitment. Mol Cell Biol 27: 352–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachury MV, Loktev AV, Zhang Q, Westlake CJ, Peränen J, Merdes A, Slusarski DC, Scheller RH, Bazan JF, Sheffield VC et al (2007) A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell 129: 1201–1213 [DOI] [PubMed] [Google Scholar]
- Nepusz T, Yu H, Paccanaro A (2012) Detecting overlapping protein complexes in protein‐protein interaction networks. Nat Methods 9: 471–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikonova AS, Astsaturov I, Serebriiskii IG, Dunbrack RL Jr, Golemis EA (2013) Aurora A kinase (AURKA) in normal and pathological cell division. Cell Mol Life Sci 70: 661–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odabasi E, Batman U, Firat‐Karalar EN (2020) Unraveling the mysteries of centriolar satellites: time to rewrite the textbooks about the centrosome/cilium complex. Mol Biol Cell 31: 866–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odabasi E, Gul S, Kavakli IH, Firat‐Karalar EN (2019) Centriolar satellites are required for efficient ciliogenesis and ciliary content regulation. EMBO Rep 20: e47723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto T, Horn S, Brockmann M, Eilers U, Schüttrumpf L, Popov N, Kenney AM, Schulte JH, Beijersbergen R, Christiansen H et al (2009) Stabilization of N‐Myc is a critical function of Aurora A in human neuroblastoma. Cancer Cell 15: 67–78 [DOI] [PubMed] [Google Scholar]
- Pan J, Wang Q, Snell WJ (2004) An aurora kinase is essential for flagellar disassembly in Chlamydomonas. Dev Cell 6: 445–451 [DOI] [PubMed] [Google Scholar]
- Pejskova P, Reilly ML, Bino L, Bernatik O, Dolanska L, Ganji RS, Zdrahal Z, Benmerah A, Cajanek L (2020) KIF14 controls ciliogenesis via regulation of Aurora A and is important for Hedgehog signaling. J Cell Biol 219: e201904107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez de Castro I, de Carcer G , Montoya G, Malumbres M (2008) Emerging cancer therapeutic opportunities by inhibiting mitotic kinases. Curr Opin Pharmacol 8: 375–383 [DOI] [PubMed] [Google Scholar]
- Plotnikova OV, Nikonova AS, Loskutov YV, Kozyulina PY, Pugacheva EN, Golemis EA (2012) Calmodulin activation of Aurora‐A kinase (AURKA) is required during ciliary disassembly and in mitosis. Mol Biol Cell 23: 2658–2670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prosser SL, Pelletier L (2020) Centriolar satellite biogenesis and function in vertebrate cells. J Cell Sci 133: jcs239566 [DOI] [PubMed] [Google Scholar]
- Pugacheva EN, Jablonski SA, Hartman TR, Henske EP, Golemis EA (2007) HEF1‐dependent Aurora A activation induces disassembly of the primary cilium. Cell 129: 1351–1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quarantotti V, Chen JX, Tischer J, Gonzalez Tejedo C, Papachristou EK, D'Santos CS, Kilmartin JV, Miller ML, Gergely F (2019) Centriolar satellites are acentriolar assemblies of centrosomal proteins. EMBO J 38: e101082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux KJ (2013) Marked by association: techniques for proximity‐dependent labeling of proteins in eukaryotic cells. Cell Mol Life Sci 70: 3657–3664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux KJ, Kim DI, Raida M, Burke B (2012) A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J Cell Biol 196: 801–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux KJ, Kim DI, Burke B (2013) BioID: a screen for protein‐protein interactions. Curr Protoc Protein Sci 74: 19.23.1–19.23.14 [DOI] [PubMed] [Google Scholar]
- Santamaria A, Wang B, Elowe S, Malik R, Zhang F, Bauer M, Schmidt A, Sillje HH, Korner R, Nigg EA (2011) The Plk1‐dependent phosphoproteome of the early mitotic spindle. Mol Cell Proteomics 10: M110.004457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW (2012) NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9: 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiler CY, Park JG, Sharma A, Hunter P, Surapaneni P, Sedillo C, Field J, Algar R, Price A, Steel J et al (2014) DNASU plasmid and PSI:Biology‐Materials repositories: resources to accelerate biological research. Nucleic Acids Res 42: D1253–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaheen R, Shamseldin H, Loucks C, Seidahmed M, Ansari S, Ibrahim Khalil M, Al‐Yacoub N, Davis E, Mola N, Szymanska K et al (2014) Mutations in CSPP1, encoding a core centrosomal protein, cause a range of ciliopathy phenotypes in humans. Am J Hum Genet 94: 73–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spektor A, Tsang WY, Khoo D, Dynlacht BD (2007) Cep97 and CP110 suppress a cilia assembly program. Cell 130: 678–690 [DOI] [PubMed] [Google Scholar]
- Stark C, Breitkreutz BJ, Reguly T, Boucher L, Breitkreutz A, Tyers M (2006) BioGRID: a general repository for interaction datasets. Nucleic Acids Res 34: D535–D539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang A, Gao K, Chu L, Zhang R, Yang J, Zheng J (2017) Aurora kinases: novel therapy targets in cancers. Oncotarget 8: 23937–23954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teo G, Liu G, Zhang J, Nesvizhskii AI, Gingras AC, Choi H (2014) SAINTexpress: improvements and additional features in Significance Analysis of INTeractome software. J Proteomics 100: 37–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treekitkarnmongkol W, Katayama H, Kai K, Sasai K, Jones JC, Wang J, Shen Li, Sahin AA, Gagea M, Ueno NT et al (2016) Aurora kinase‐A overexpression in mouse mammary epithelium induces mammary adenocarcinomas harboring genetic alterations shared with human breast cancer. Carcinogenesis 37: 1180–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinkle‐Mulcahy L (2019) Recent advances in proximity‐based labeling methods for interactome mapping. F1000Research 8: 135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vader G, Lens SM (2008) The Aurora kinase family in cell division and cancer. Biochim Biophys Acta 1786: 60–72 [DOI] [PubMed] [Google Scholar]
- Van de Mark D, Kong D, Loncarek J, Stearns T (2015) MDM1 is a microtubule‐binding protein that negatively regulates centriole duplication. Mol Biol Cell 26: 3788–3802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter AO, Seghezzi W, Korver W, Sheung J, Lees E (2000) The mitotic serine/threonine kinase Aurora2/AIK is regulated by phosphorylation and degradation. Oncogene 19: 4906–4916 [DOI] [PubMed] [Google Scholar]
- Wang L, Lee K, Malonis R, Sanchez I, Dynlacht BD (2016) Tethering of an E3 ligase by PCM1 regulates the abundance of centrosomal KIAA0586/Talpid3 and promotes ciliogenesis. eLife 5: e12950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Gong Y, Guo T, Li M, Fang L, Yin S, Kamran M, Liu Y, Xu J, Xu L et al (2019) Activation of Aurora A kinase increases YAP stability via blockage of autophagy. Cell Death Dis 10: 432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WJ, Tay HG, Soni R, Perumal GS, Goll MG, Macaluso FP, Asara JM, Amack JD, Tsou MF (2013) CEP162 is an axoneme‐recognition protein promoting ciliary transition zone assembly at the cilia base. Nat Cell Biol 15: 591–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhou YX, Qiao W, Tominaga Y, Ouchi M, Ouchi T, Deng CX (2006) Overexpression of aurora kinase A in mouse mammary epithelium induces genetic instability preceding mammary tumor formation. Oncogene 25: 7148–7158 [DOI] [PubMed] [Google Scholar]
- Willems E, Dedobbeleer M, Digregorio M, Lombard A, Lumapat PN, Rogister B (2018) The functional diversity of Aurora kinases: a comprehensive review. Cell Div 13: 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong YL, Anzola JV, Davis RL, Yoon M, Motamedi A, Kroll A, Seo CP, Hsia JE, Kim SK, Mitchell JW et al (2015) Cell biology. Reversible centriole depletion with an inhibitor of Polo‐like kinase 4. Science 348: 1155–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Hirota T, Marumoto T, Shimizu M, Kunitoku N, Sasayama T, Arima Y, Feng L, Suzuki M, Takeya M et al (2004) Cre‐loxP‐controlled periodic Aurora‐A overexpression induces mitotic abnormalities and hyperplasia in mammary glands of mouse models. Oncogene 23: 8720–8730 [DOI] [PubMed] [Google Scholar]
- Zhang D, Shimizu T, Araki N, Hirota T, Yoshie M, Ogawa K, Nakagata N, Takeya M, Saya H (2008) Aurora A overexpression induces cellular senescence in mammary gland hyperplastic tumors developed in p53‐deficient mice. Oncogene 27: 4305–4314 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Review Process File
Data Availability Statement
No data were deposited in a public database.