

Abstract
Malignant transformation may occur in the background of post-translational modification, such as ADP-ribosylation, phosphorylation and acetylation. Recent genomic analysis of ADP-ribosylation lead to the discovery of more than twenty ADP-ribosyltransferases (ARTs), which catalyze either mono- or poly- ADP-ribosylation. ARTs catalyze the attachment of ADP-ribose to acceptor molecules. The ADP-ribose-acceptor bond can then be cleaved by a family of hydrolases in a substrate-specific manner, which is dependent on the acceptor and its functional group, e.g., arginine (guanidino), serine (hydroxyl), aspartate (carboxyl). These hydrolases vary in structure and function, and include poly-ADP-ribose glycohydrolase (PARG), MacroD1, MacroD2, terminal ADP-ribose protein glycohydrolase 1 (TARG1) and ADP-ribosyl-acceptor hydrolases (ARHs). In murine models, PARG deficiency increased susceptibility to alkylating agents-induced carcinogenesis. Similarly, by cleaving mono-ADP-ribosylated arginine on target proteins, ARH1 appears to inhibit tumor formation, suggesting that ARH1 is a tumor-suppressor gene. Although ARH3 is similar to ARH1 in amino acid sequence and crystal structure, ARH3 does not cleave ADP-ribose-arginine, rather it degrades in an exocidic manner, the PAR polymer and cleaves O-acetyl-ADP-ribose (OAADPr) and the ADP-ribose-serine linkage in acceptor proteins. Under conditions of oxidative stress, ARH3-deficient cells showed increased cytosolic PAR accumulation and PARP-1 mediated cell death. These findings expand our understanding of ADP-ribosylation and provide new therapeutic targets for cancer treatment. In the present review, research on ARH1-regulated tumorigenesis and cell death pathways that are enhanced by ARH3 deficiency are discussed.
Keywords: ADP-ribosylation, tumorigenesis, ADP-ribosyl-acceptor hydrolase 1, poly-ADP-ribose glycohydrolase, ARH
Graphical Abstract
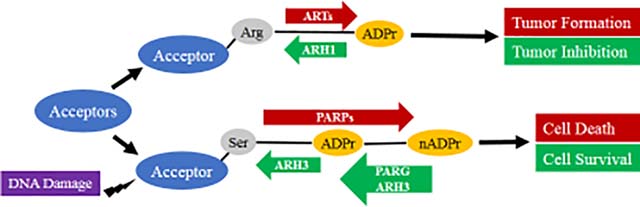
1. Transferases and hydrolases that regulate the extent of ADP-ribosylation
ADP-ribosylation, a post-translational modification, refers to the covalent attachment of one or more ADP-ribose (ADPr) units to proteins or small molecules [1]. Mono-ADP-ribosylation involves the transfer of a single ADP-ribose moiety to an acceptor, whereas poly-ADP-ribosylation involves synthesis of long linear or branched poly-ADP-ribose-(PAR) chains attached to an acceptor [2]. Mammalian transferases involved in ADP-ribosylation are generally classified as members of ADP-ribosyltransferases (ARTs) and poly-ADP-ribose polymerases (PARPs) genomic families, based on the similarity of their transferase folds respectively to cholera toxin or diphtheria toxin [3, 4]. Using NAD as a cofactor, these transferases add ADP-ribose moiety to specific amino acid residues of various protein targets or ADP-ribose itself in the elongation process, releasing nicotinamide (NAM) [3]. The ART family consists of five members (ART1–5), four (ART1–4) are anchored to the cell membrane where they can exert their effects on extracellular targets, whereas ART5 is secreted [5–8]. ART1, ART2 and ART5 specifically transfer an ADP-ribose to the arginine of protein acceptors, while no amino acid substrates have been validated for ART3 and ART4 [2]. ART2 is not found in humans. For example, in vitro ART1 modified the arginine of cell surface T-cell co-receptors, and inhibited proliferation and cytolytic activity of T-lymphocytes [9, 10]. In vivo human neutrophil peptide 1 (HNP-1), isolated from airways of patients with idiopathic pulmonary fibrosis and asthma, was ADP-ribosylated on specific arginines. The mono-ADP-ribosylated HNP-1 exhibited decreased antimicrobial and cytotoxic activities. The data supported the conclusion that HNP-1 was ADP-ribosylated by ART1 [11]. In contrast to ARTs, PARPs have a broad spectrum of substrates, including lysine, serine, glutamate, and aspartate [12]. Seventeen members belonging to the PARP superfamily have been identified according to sequence and structural homology: PARP1–4, tankyrase1/2, and PARP6–16 [4]. PARP1 and 2 can synthesize polymers of ADP-ribose units, through O-glycosidic bonds into linear or branched chains [1, 13]. Most PARPs tend to catalyze the addition of a single ADP-ribose to target proteins [1, 14]. PARP9, in a complex with Deltex E3 Ubiquitin Ligase 3L (Dtx3L), can mediate NAD+-dependent mono-ADP-ribosylation of ubiquitin [15]. PARP13, missing a PARP consensus sequence motif, lacks ADP-ribosylation activity [16]. PARP1 is activated by DNA strand breaks and is responsible for the majority of nuclear protein poly-ADP-ribosylation, catalyzing auto-ADP-ribosylation as well as poly-ADP-ribosylation of other proteins [17, 18]. PAR-modified PARP1 serves as a scaffold to recruit a protein complex that may include XRCC1 (X-ray repair cross-complementing 1), DNA ligase III and the Ku70 subunit of DNA-dependent protein kinase, to facilitate DNA base-excision repair [19–21]. Clinically, PARP1 inhibitors (i.e. olaparib, rucaparib, niraparib) have been approved for anti-cancer treatment and others are being evaluated in clinical trials of various solid tumors [22].
ADP-ribosylation is reversible and is catalyzed by substrate-specific hydrolases. PARG degrades ADP-ribose polymers, catalyzing both endo- and exo- glycosidic cleavage [1, 12]. Mono-ADP-ribosylated proteins can be de-modified by several amino acid-specific ADP-ribose-acceptor hydrolases [12]. MacroD1, MacroD2 and terminal ADP-ribose protein glycohydrolase 1 (TARG1) cleave mono-ADP-ribose directly linked to glutamate/aspartate [23, 24]. ARHs, a mammalian family of around 39 kD proteins with significant amino acid sequence and structural similarities, also possess the ability to cleave ADP-ribosylated proteins or PAR chains [2, 25].
Another family of proteins, containing the NUDIX (nucleoside diphosphates linked to other moieties X) domain, hydrolyzes ADP-ribose to adenosine mono-phosphate (AMP) and D-ribose 5-phosphate [26, 27]. These enzymes cleave the PAR chain by targeting the phosphodiester bond in the protein-proximal ADP-ribose unit, leaving a phosphoribose moiety on the acceptor amino acid [28, 29]. The NUDIX hydrolases can also cleave OAADPr, an NAD+-dependent sirtuin reaction product, producing AMP and O-acetylated ribose-5’-phosphate [30]. Thus, these enzymes appear to regulate intracellular ADP-ribose levels and are crucial in cellular bioenergetic metabolism [28].
Interestingly, hydrolases are localized to different subcellular components: mitochondria (MacroD1), nucleus (TARG1 and PARG), and cytosol (NUDT9) [12, 28]. Turnover of the ADP-ribose modification is crucial for many cellular processes and related to human diseases [31]. Depletion of TARG1 protein in cells leads to DNA repair defects and reduced proliferation [31]. Patients with truncated TARG1 (a dysfunctional mutant) due to a homozygous mutation of the TARG1 gene, presented with severe progressive neurodegeneration and a seizure disorder, showing the importance of this protein in supporting normal neural cell function [31].
2. ARH family: structure, activity and distribution
Three members, ARH1–3, are found in the ARH family. ADP-ribose acceptor hydrolysis assays demonstrated that ARH1 and ARH3 have different substrate specificities [25]. ARH2 plays a direct role in modifying Z-disc and actin dynamics as heart chambers develop; thus far, an enzymatic activity has not been identified [32].
ARH1 is a cytosolic protein that is ubiquitously expressed in mouse tissues and cells [25]; it has a robust hydrolytic activity on ADP-ribosylated arginine [33]. As a mono-ADP-ribosyl-arginine hydrolase, ARH1 specially recognizes and cleaves the α-anomer at the C-1” position of ADP-ribose attached to the arginine (N-glycosidic bond) [34, 35]. In addition, ARH1 has a weak hydrolytic activity towards PAR and OAADPr when compared to ARH3 [2]. ARH1 activity is highly dependent on two conserved aspartates at positions 60 and 61 [2, 36]. Replacement of Asp-60 or Asp-61 with Ala, Gln, or Asn, significantly reduced enzyme activity, while the Glu-60 and Glu-61 double mutant was completely inactive [36]. Human ARH1 crystal structure has been solved in a complex with ADP and K+; it exhibits α -helical protein folds composed of 4 helical bundle subdomains [37, 38]. Mg2+ is essential for ARH1 activity. In some species, thiol dependence activation was also noted, which was dependent on a specific cysteine [2] (Table 1).
Table 1.
Summary of ARH1 and ARH3 enzymatic characteristics.
| N-glycosidic bond (guanidino) | O-glycosidic bond | ADP-ribosyl-arginine | ADP-ribosyl-serine | PAR | OAADPr | Mg2+ requirement | |
|---|---|---|---|---|---|---|---|
| ARH1 | +++ | + | +++ | – | + | + | Yes |
| ARH3 | – | +++ | – | +++ | +++ | +++ | Yes |
| PARG | – | +++ | – | – | +++ | Unknown | Yes |
ARH3 cleaves PAR and OAADPr [39], and is the main hydrolase responsible for cleaving ADP-ribosylated serine [40, 41]. ARH3 hydrolyzes the O-glycosidic bond in PAR and serine-ADP-ribose, generating free ADP-ribose and removing ADP-ribose attached to serine residues in proteins respectively [25, 41]. Likewise, OAADPr is hydrolyzed by ARH3, attacking the O-glycosidic bond between ADP-ribose and the acetyl group of OAADPr (Table 1)[42]. The acetyl group of OAADPr is reversibly interconverted among the hydroxyl groups attached to C-1”, 2”, and 3” of ribose in OAADPr in a pH-dependent equilibrium. However, ARH3 only hydrolyzes the acetyl group attached to a carbon at the C-1” position of ribose in OAADPr [42]. Crystal structure of ARH3 has also been solved in presence of ADP-ribose and Mg2+ [43]. Interestingly, structural analysis revealed that a conformational change of the Glu41-containing flap motif in ARH3, enabled the specific substrate recognition and cleavage [43]. ARH3 hydrolysis activity relied on the di-Mg2+-containing catalytic center as did ARH1 [25]. Aspartates at positions of 77 and 78, are essential for ARH3 hydrolase activity in the presence of Mg2+ [25]. Replacement of these two aspartates with alanine, glutamine or asparagine dramatically reduced the enzyme activity although the mutated ARH3 retains its ability to bind ADP-ribose [35, 44]. Moreover, G115, S148, Y149, H182, D314, and T317 ARH3 mutants could no longer hydrolyze ADP-ribosylated substrates [40]. ARH1 and ARH3 activities were inhibited by ADP-ribose [2, 25]. In addition, ARH3 was inhibited by adenosine diphosphate-(hydroxymethyl) pyrrolidine-2′,3′-diol (ADP-HPD), an analog of the oxocarbenium ion intermediate [43]. Most ARH3 was expressed in cytosol. However, ARH3 also contains a mitochondrial-targeting sequence, which suggests ARH3 may be responsible for PAR degradation in mitochondria [45, 46]. Indeed, in cells with artificially induced mitochondrial PAR accumulation, ARH3 dramatically lowered PAR content [45]. ARH3 in the nucleus, together with PARG, regulates PAR content in response to DNA damage caused by oxidative stress [47] (Table 1).
3. Importance of reversible ADP-ribosylation in cancer pathogenesis
ADP-ribosylation has been implicated in various biological processes including DNA repair, transcription, aging, chromatin remodeling and cell replication. In some cases, transferases and hydrolases participate in an ADP-ribosylation cycle to maintain biological homeostasis [48]. During nitrogen fixation in the photosynthetic bacterium Rhodospirillum, dinitrogenase reductase activity is regulated via arginine ADP-ribosylation by coupling of the ADP-ribosyltransferase (DraT)/ hydrolase (DraG) system [49]. In mammalian cells, ART1 and ARH1 are similar enzymes that catalyze, respectively, addition and removal of ADP-ribose from (arginine) protein. Many of the targets for the specific ARTs and ARHs have been definitively established. Using an ARH1-deficient mouse, we first described the anti-bacterial toxin effects of endogenous ARH1 [33]. In a murine intestinal loop model, cholera toxin ADP-ribosylated arginine on the α subunit of the stimulatory guanine nucleotide-binding protein (Gsα) of the adenylyl cyclase system [33]. ADP-ribosylation of Gsα inhibited its GTPase activity, resulting in increased adenylyl cyclase activity and intracellular cAMP, resulting in the toxin’s effects on water and electrolyte transport [33]. Intestinal loops of ARH1−/− mice, exposed to cholera toxin, showed significantly enhanced fluid accumulation compared to those of wild-type (WT) mice [33]. In agreement, ARH1−/− mouse embryonic fibroblasts (MEFs) exhibited enhanced sensitivity to cholera toxin.
ARH1-deficient MEFs injected into nude mice developed tumors. Moreover, the ARH1-deficient mice developed multiple tumors (e.g., lymphoma, carcinoma, sarcoma) and increased metastasis; tumor formation, both in vitro and in vivo, may be due to an increase in ADP-ribosyl-arginine proteins [50] (Figure 1). Similarly, ART1 expression was upregulated in colorectal cancer (CRC) and showed a positive correlation with the expression of vascular endothelial growth factor (VEGF) [51]. ART1 overexpression in CT26 cells could promote the change of cells from apical-basal polarized epithelioid cells to mesenchymal-like cells (the epithelial-mesenchymal transition, EMT) [52], while ART1 gene silencing inhibited the growth and development of transplanted CT26 tumor cells in vivo [51]. Hyperglycemia in patients with type 2 diabeters mellitus (T2DM), is a risk factor of colorectal cancer. ART1 overexpression increased glycolysis and energy metabolism of CT26 CRC cells, which could be partially reversed by β-caryophyllene treatment, a compound exerting anticancer and hypoglycemic effects [53]. Moreover, ART1 upregulation in glioma patients is associated with poor prognosis and significantly decreases the susceptibility to vincristine treatment [54]. Together, ART1 overexpression was involved in multiple aspects of cancer progression and therefore may be a promising target for therapy.
Figure 1. Substrate specificity and biological processes regulated by ARH1, ARH3 and PARG.
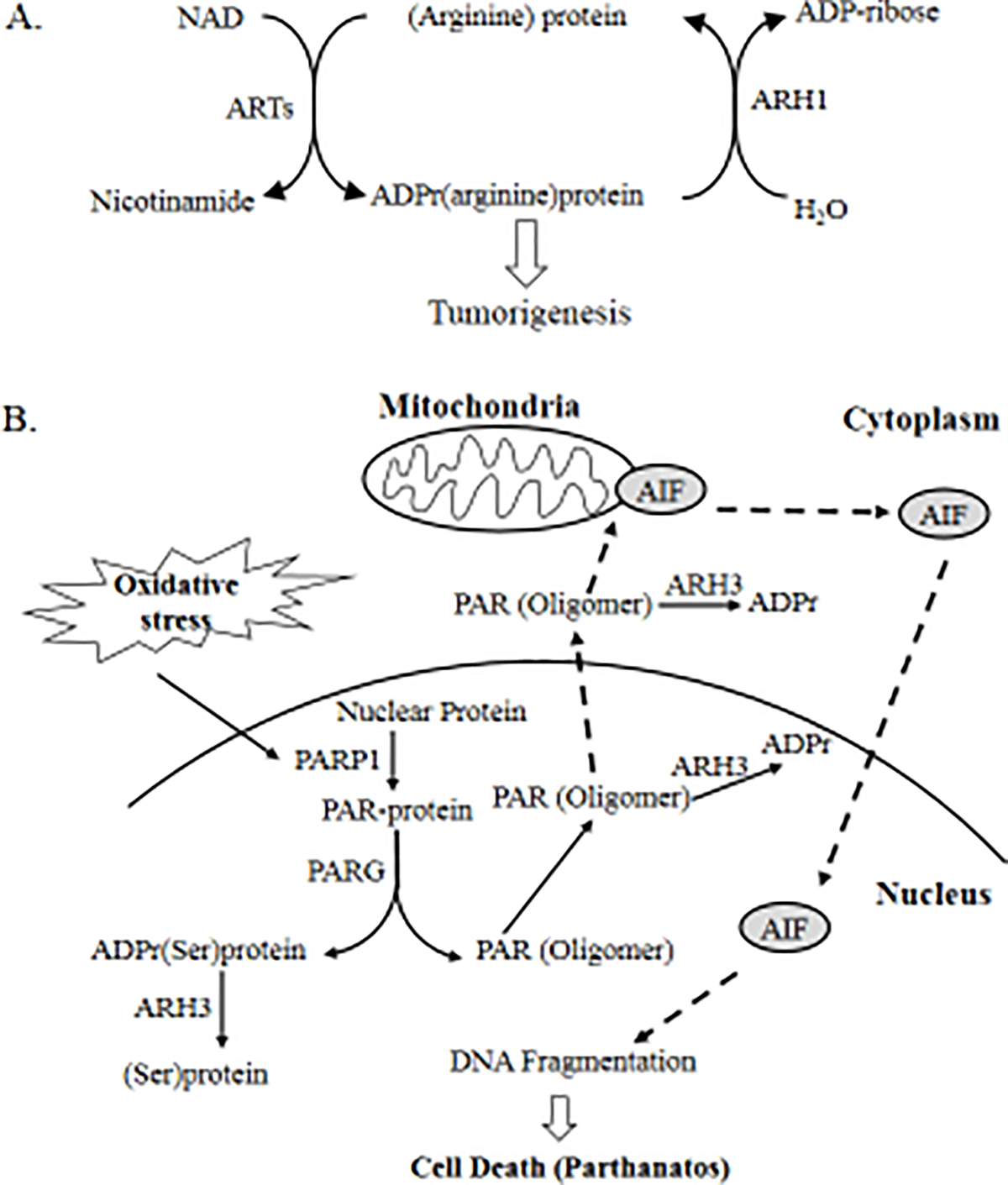
A, Using NAD as substrate, ARTs transfer the ADP-ribose moiety to arginine of acceptor proteins with release of nicotinamide. The mono-ADP-ribosylation is reversed by ARH1, restoring unmodified proteins. ARH1 appears to be a tumor-suppressor gene as ARH1 deficiency leads to tumor development. B, Under oxidative stress, PARP1 was activated and poly-ADP-ribosylated nuclear acceptor proteins. PARG degrades PAR polymers, producing ADP-ribose monomers and small PAR fragments. ARH3 hydrolyzes the ADP-ribose polymer or ADP-ribose linked to the serine of acceptors, generating free ADP-ribose (solid arrow). By participation in PAR degradation, ARH3 prevents the PAR translocation and its binding to mitochondrial apoptosis inducing factors (AIF), blocking the initiation of parthanatos (dotted arrow). Therefore, ARH3 is protective against PARP-1 mediated cell death.
3.1. Tumor-suppressor function of ARH1
3.1.1. Increased tumorigenesis in ARH1-deficient mice
ARH1-deficient mice showed severed phenotypic changes including tumor formation and infections [50]. ARH1−/− and ARH1+/− mice were prone to spontaneous tumor formation, first observed at 3 months. After a two-year observation of ARH1 littermates, over 20% ARH1−/− and 10% ARH1+/− mice developed spontaneous tumors, a frequency that was greater than that seen in wild-type mice [50]. Malignancies included lymphoma, adeno-/hepatocellular- carcinoma, and hemangio-/rhabdomyo- sarcoma and were found in lung, liver, lymph nodes, abdomen, gastrointestinal tract, uterus, and mammary glands. Moreover, more frequent metastasis and occurrence of multiple tumors were seen in ARH1−/− mice [50]. Analysis of ARH1 gene/protein in tumors found in ARH1 heterozygous mice demonstrated the loss of heterozygosity and absence or mutation ARH1 protein [50]. Data support the conclusion that ARH1 deficiency is strongly associated with tumor formation and progression.
3.1.2. Malignant properties of ARH1-deficient MEFs
Consistent with the findings in animals, MEFs generated from ARH1 knockout and heterozygous mice developed tumors in nude mice and showed increased proliferation relative to wild-type MEFs [50]. Further, ARH1 knockout and heterozygous MEFs formed colonies in soft agar [50]. Overexpression of functional ARH1 protein in ARH1−/− MEFs partially reversed the malignant potential [50].
Estrogen seems to promote xenograft growth of ARH1−/− MEFs in vivo. Tumors from ARH1−/− MEFs enlarged more rapidly in female than in male nude mice. This effect was further validated to be estrogen dependent. Tumors in ovariectomized female nude mice exhibited reduced growth rate compared to tumors in female, and ovariectomized female nude mice supplemented with estrogen. Further, estrogen enhanced the survival of ARH1−/− MEFs in the murine circulation and promoted tumor metastasis in the lung [2, 55].
3.1.3. ARH1 mutations affect tumorigenesis
ARH1 heterozygous mice or MEFs demonstrated tumors in nude mice similar to their respective ARH1-deficient mice due to loss of heterozygosity (LOH) of the remaining ARH1 allele or loss of ARH1 gene activity due to mutagenesis. Several mutations were identified in tumors of ARH1+/− mice and in tumors in nude mice injected with ARH1+/− MEFs [44, 50]. Some ARH1 gene mutations exhibited low catalytic activity ranging from 4% to 55% of WT ARH1, when transformed into ARH1−/− MEFs [44, 50]. Mutations tended to be in exons that comprised the catalytic site. These transformed MEFs proliferated much faster than ARH1−/− MEFs that stably expressed WT ARH1, as well as displayed anchorage-independent growth [44]. More importantly, transformed ARH1−/− MEFs with various ARH1 mutations showed reduced tumor growth compared to ARH1−/− MEFs after being implanted into nude mice [44]. Altogether, these findings demonstrated the correlation between ARH1 enzyme activity and tumorigenesis. ARH1 mutations were observed in human cancers according to a human somatic tumor mutation database and were also located in the region of the protein that comprises the catalytic site in mouse tumors [44], implying the application of our finding with murine tumors to human cancers.
3.1.4. ARH1 and CD38
Since all PARPs and ARTs use NAD+ as a substrate, major NAD consumers (CD38 and sirtuins) should also be considered as potential regulators of ADP-ribosylation [1]. CD38 is a multi-functional enzyme; it is a primary NAD glycohydrolase regulating NAD+ homeostasis, and a cyclic ADPR synthase and hydrolase contributing to intracellular calcium signaling [56]. CD38 knockout led to elevated tissue NAD+ levels in mice [57]. In a combined CD38 and ARH1 double knockout, we evaluated the potential synergistic effects on NAD metabolism and tumor development. NAD+ levels were significantly higher in double and CD38-single knockout mice but were not changed in ARH1 knockouts and WT mice [58]. CD38 deficiency significantly reduced the tumorigenic effects of ARH1 deficiency in mice, perhaps via indirectly regulating the NAD pool [58]. The underlying mechanism is unclear. Fewer tumors were seen in CD38 knockout than in WT mice, implying that CD38 deficiency may directly inhibit tumor development [58]. Most malignant plasma cells in all stages of multiple myeloma (MM) strongly express CD38 [59]. Development and application of anti-CD38 antibody against MM, represent an important advance in targeting NAD for cancer therapy [60]. In a human cancer database, CD38 mRNA levels were seen frequently to be significantly upregulated in human lung carcinoma compared to normal lung tissues, simultaneously with down-regulation of ARH1 mRNA levels [58]. CD38 protein levels were also elevated in some lung cancers and cell lines [58]. Given the overexpression of CD38 in tumors, we propose that CD38 may be involved in regulating the growth of human lung cancers. Consistent with this model, CD38 knockout in A549 cells suppressed invasiveness, and reduced clonogenicity and tumor growth in nude mice [58]. Based on our data and the success of anti-CD38 antibodies in MM, targeting CD38 by antibodies or specific inhibitors might improve the prognosis of lung cancer in patients whose tumors overexpress CD38.
3.2. PARG function in carcinogenesis
A single parg gene gives rise to several PARG isoforms including the major nuclear 110-kD isoform and the cytosolic 60-kD isoform [61]. PARG exhibits both exo- and endo- glycosidic hydrolytic activities on PAR polymers, and prefers long PAR chains as substrates [13]. PARP1 and PARG, acting in concert, efficiently modulate PAR chains, which have half-lives probably less than 1 min [13]. PARG has a crucial role in preventing DNA insult-induced cell death. In a HeLa cell model, depletion of PARG led to a delay in DNA strand repair as well as mitotic aberrations after X-irradiation [62]. Consequently, PARG-depleted cells are radiosensitive and die by mitotic catastrophe [62]. The absence of PARG in embryonic trophoblast stem cells led to enhanced DNA damage in response to genotoxic stress, resulting in increased apoptosis-inducing factor (AIF) translocation to the nucleus independent of caspase-generated apoptotic signals [63]. PARG is essential for embryogenesis [64]. Drosophila mutant lacking PARG activity showed lethality at the larval stage at normal development temperatures [64]. At high temperatures, PARG mutants that survived and progressed to the adult stage showed progressive neurodegeneration, with reduced locomotor activity and a short lifespan [64]. Similarly, complete knockout of the parg gene resulted in a lethal phenotype in a mouse embryo [64]. Most findings arise from studies of mice lacking the nuclear 110-kD isoform, which were viable and fertile [65, 66]. PARG110−/− MEFs and embryonic stem cells were hypersensitive to treatment with alkylating agents or exposure to γ-irradiation [65, 67]. Loss of PARG110 led to a high rate of sister chromatid exchange (SCE), more micronuclei and a high degree of chromosomal aberration in PARG110−/− MEFs. These MEFs showed more severe DNA repair defects than wild-type counterparts when subjected to H2O2, possibly due to an impaired ATM (ataxia-telangiectasia, mutated)-dependent repair pathway [66]. Although PARG110−/− mice do not develop spontaneous tumors, diethylnitrosamine (DEN) treatment greatly increased the number of hepatocellular carcinomas (HCC) in PARG110−/− mice relative to their WT counterparts [66]. All these observations indicate that PARG deletion increased the risk of carcinogenesis following external or internal stress.
4. Cellular function of ARH3 related to cell death
ARH3 has more than a 40% sequence similarity and 20% identity with ARH1, as well as a ubiquitous tissue distribution [39]. ARH3 enzymatic activities differ from ARH1. ARH3 degrades long PAR polymers, cleaves the ADP-ribose-serine linkage, and hydrolyzes OAADPr [25, 40]. The best-defined cellular function of ARH3 is its participation in oxidative stress-induced, PARP1-mediated cell death (parthanatos) [47] (Figure 1). ARH3−/− MEFs were more sensitive to hydrogen peroxide-induced cell death than WT MEFs, with features of programmed cell death that were mediated by PARP1 and independent of caspase activation; following exposure to H2O2, ARH3−/− MEFs showed rapid PAR elevation in nuclei; PARG preferentially cleaved long PAR chains consisting of more than 20 units into short PAR oligomers which passed through the nuclear membrane and were exported to cytosol [13]; cytoplasmic PAR triggered AIF release from mitochondria and nuclear translocation, which caused large-scale DNA fragmentation and chromatin condensation, leading to cell death. Overexpression of ARH3 protein in ARH3−/− MEFs suppressed cytosolic PAR accumulation and AIF release, thus protecting the cell from death due to oxidative stress [47]. Knockdown of PARP1 or PARG in ARH3−/− MEFs, reduced the cell cytotoxicity of H2O2, respectively by preventing PAR accumulation or reducing free PAR generation and cytosolic translocation [47]. ARH3 may be also responsible for hydrolysis of PAR in the mitochondrial matrix [68]. Using a model expressing mitochondrial PARP1 catalytic activity, ARH3 but not the mitochondrial PARG isoform effectively suppressed PAR accumulation in the mitochondrial matrix [68]. Given the importance of ARH3 in PAR metabolism, ARH3 gene aberration appears to affect fundamental physiological processes. Indeed, a family-based genomic analysis revealed that affected individuals displaying age-dependent, recessive, epilepsy-ataxia syndrome, had mutated ARH3 alleles [69]. We have made a similar observation in a different family (Mashimo et al., unpublished data).
Histone PARylation factor 1 (HPF1) is a PARP1-regulating factor involved in cellular response to DNA damage [70]. A newly discovered form of ADP-ribosylation that specifically occurs in PARP1/2-HPF1 complexes, results in the ADP-ribosylation of serine residues in target proteins (e.g. histones, PARP1) [70, 71]. Further, DNA strand break-induced serine-ADP-ribose modification of histones was found to be reversible in human cells, with ARH3 being the main hydrolase capable of efficiently cleaving serine-ADP-ribose proteins [40, 41] (Figure 1). Surprisingly, serine appeared to be the major amino acid ADP-ribosylated following hydrogen peroxide-induced DNA damage, which relied on the PARP1, ARH3 and HPF1 network [72]. Further, proteomic analysis revealed a list of poly-ADP-ribosylated protein substrates of ARH3 including PARP1, PARP14 and HMGB1 [40]. Because of its ubiquitous expression in mitochondria, cytosol and nucleus, these findings placed significant importance on ARH3 in protection and resolution of DNA damage-associated cellular responses. The biological consequences of removal of ADP-ribose from its linkage to serine under physiological or pathological conditions are largely unknown. The widespread identification of serine modification suggests that ARH3 may participate in many cellular processes beyond DNA damage [40, 70]. For example, HMGB1 performs a pivotal function in various human diseases, involving autoimmunity, neurodegeneration and cancer [73].
5. Concluding remarks
Accumulating evidence, largely from genetic knockout mice, shows that hydrolases are important in regulating the response to stress (e.g., genotoxic insults, oxidative stress) [50, 64, 67, 74]. For example, PARG deficiency alone does not result in tumor formation, but it did increase the tumorigenic sensitivity to genotoxic agents [1, 13, 66]. Our findings from ARH1-deficient mice and cells suggests that ARH1 act as a tumor suppressor gene and is a promising target for cancer therapy [50] (Figure 2). To date, no reports have appeared regarding the role of ARH3 in carcinogenesis. In contrast, ARH3 controls cell death under oxidative stress [47] (Figure 2). Due to lack of specific inhibitors for ARHs, the therapeutic potential of targeting these enzymes in human diseases is still unclear. Fortunately, advances in structural studies may provide useful basis for structure-guided design of ARH-specific inhibitors [43, 46]. Moreover, there is still a huge gap in our knowledge about how cells translate the ADP-ribosylation signal into tumor formation. Mass spectrometry-based approaches are being developed to decipher the cellular ADP-ribosylome under various conditions [40, 75]. Therefore, determination of the protein substrates as well as their ADP-ribosylation sites, is necessary to explain the biological consequences of ADP-ribosylation, and potentially lead to the development of new therapeutic options.
Figure 2. Association of ARHs/PARG deficiency and diseases.
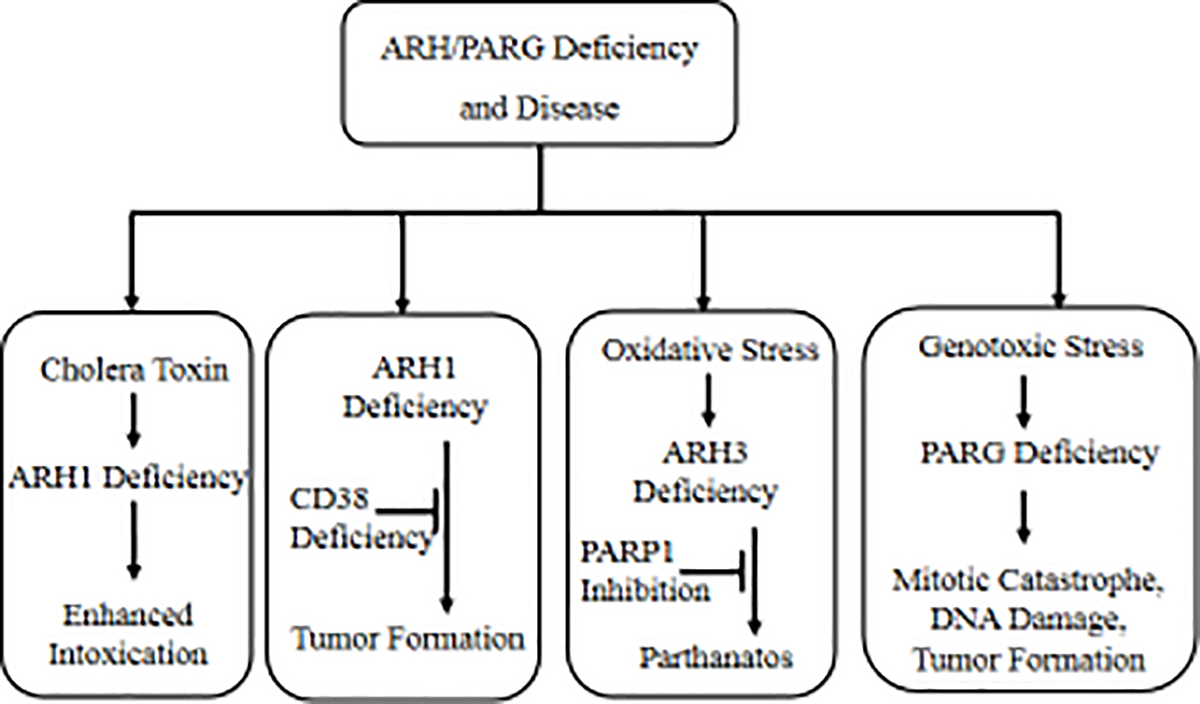
ARHs are key enzymes in maintaining intracellular ADP-ribosylation homeostasis. ARH deficiencies lead to severe consequences under certain pathological conditions. ARH1 plays an important role in the intoxication process caused by toxin ADP-ribosyltransferases. Effects of intoxication induced by cholera toxin on ARH1-deficient mouse cells and intestinal loops were greater than those on wild-type counterparts. ARH1 deficiency causes tumor formation in murine models, which could be partially overcome by iCD38 deficiency. Upon oxidative stress, parthanatos, a mechanism of PARP1/PAR/AIF-mediated cell death, was induced in ARH3-deficient cells. Therefore, PARP1 inhibition is a promising option for treatment. Similarly, PARG-deficient cells displayed lethal DNA repair defects and mitotic abnormality when exposed to genotoxic stress (radiation or hydrogen peroxide). Also, alkylation agents induced more tumor formation in PARG-deficient mice relative to their WT counterparts [66].
Acknowledgments
This study was supported by the Intramural Research Program, National Institutes of Health, National Heart, Lung, and Blood Institute.
Abbreviations:
- ARH
ADP-ribose-acceptor hydrolase
- PAR
poly-ADP-ribose
- OAADPr
O-acetyl-ADP-ribose
- PARG
poly-ADP-ribose glycohydrolase
- PARP
poly-ADP-ribose polymerase
- NUDIX
nucleoside diphosphate-linked to other moieties (“X”)
- AIF
apoptosis-inducing factor
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- [1].Gupte R, Liu Z, Kraus WL, PARPs and ADP-ribosylation: recent advances linking molecular functions to biological outcomes, Genes Dev 31(2) (2017) 101–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Mashimo M, Kato J, Moss J, Structure and function of the ARH family of ADP-ribosyl-acceptor hydrolases, DNA Repair (Amst) 23 (2014) 88–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Cohen MS, Chang P, Insights into the biogenesis, function, and regulation of ADP-ribosylation, Nat Chem Biol 14(3) (2018) 236–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Hottiger MO, Hassa PO, Luscher B, Schuler H, Koch-Nolte F, Toward a unified nomenclature for mammalian ADP-ribosyltransferases, Trends Biochem Sci 35(4) (2010) 208–19. [DOI] [PubMed] [Google Scholar]
- [5].Scarpa ES, Fabrizio G, Di Girolamo M, A role of intracellular mono-ADP-ribosylation in cancer biology, FEBS J 280(15) (2013) 3551–62. [DOI] [PubMed] [Google Scholar]
- [6].Yadollahi-Farsani M, Kefalas P, Saxty BA, MacDermot J, Polymorphic forms of human ADP-ribosyltransferase-1 differences in their catalytic activities revealed by labeling of membrane-associated substrates, Eur J Biochem 262(2) (1999) 342–8. [DOI] [PubMed] [Google Scholar]
- [7].Weng B, Thompson WC, Kim HJ, Levine RL, Moss J, Modification of the ADP-ribosyltransferase and NAD glycohydrolase activities of a mammalian transferase (ADP-ribosyltransferase 5) by auto-ADPribosylation, J Biol Chem 274(45) (1999) 31797–803. [DOI] [PubMed] [Google Scholar]
- [8].Bourgeois C, Okazaki I, Cavanaugh E, Nightingale M, Moss J, Identification of regulatory domains in ADP-ribosyltransferase-1 that determine transferase and NAD glycohydrolase activities, J Biol Chem 278(29) (2003) 26351–5. [DOI] [PubMed] [Google Scholar]
- [9].Wang J, Nemoto E, Dennert G, Regulation of CTL by ecto-nictinamide adenine dinucleotide (NAD) involves ADP-ribosylation of a p56lck-associated protein, J Immunol 156(8) (1996) 2819–27. [PubMed] [Google Scholar]
- [10].Liu ZX, Yu Y, Dennert G, A cell surface ADP-ribosyltransferase modulates T cell receptor association and signaling, J Biol Chem 274(25) (1999) 17399–401. [DOI] [PubMed] [Google Scholar]
- [11].Paone G, Wada A, Stevens LA, Matin A, Hirayama T, Levine RL, Moss J, ADP ribosylation of human neutrophil peptide-1 regulates its biological properties, Proc Natl Acad Sci U S A 99(12) (2002) 8231–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Crawford K, Bonfiglio JJ, Mikoc A, Matic I, Ahel I, Specificity of reversible ADP-ribosylation and regulation of cellular processes, Crit Rev Biochem Mol Biol 53(1) (2018) 64–82. [DOI] [PubMed] [Google Scholar]
- [13].Tanuma S, Sato A, Oyama T, Yoshimori A, Abe H, Uchiumi F, New Insights into the Roles of NAD+−Poly(ADP-ribose) Metabolism and Poly(ADP-ribose) Glycohydrolase, Curr Protein Pept Sci 17(7) (2016) 668–682. [DOI] [PubMed] [Google Scholar]
- [14].Vyas S, Matic I, Uchima L, Rood J, Zaja R, Hay RT, Ahel I, Chang P, Family-wide analysis of poly(ADP-ribose) polymerase activity, Nat Commun 5 (2014) 4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Yang CS, Jividen K, Spencer A, Dworak N, Ni L, Oostdyk LT, Chatterjee M, Kusmider B, Reon B, Parlak M, Gorbunova V, Abbas T, Jeffery E, Sherman NE, Paschal BM, Ubiquitin Modification by the E3 Ligase/ADP-Ribosyltransferase Dtx3L/Parp9, Mol Cell 66(4) (2017) 503–516 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Karlberg T, Klepsch M, Thorsell AG, Andersson CD, Linusson A, Schuler H, Structural basis for lack of ADP-ribosyltransferase activity in poly(ADP-ribose) polymerase-13/zinc finger antiviral protein, J Biol Chem 290(12) (2015) 7336–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Langelier MF, Planck JL, Roy S, Pascal JM, Structural basis for DNA damage-dependent poly(ADP-ribosyl)ation by human PARP-1, Science 336(6082) (2012) 728–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Altmeyer M, Messner S, Hassa PO, Fey M, Hottiger MO, Molecular mechanism of poly(ADP-ribosyl)ation by PARP1 and identification of lysine residues as ADP-ribose acceptor sites, Nucleic Acids Res 37(11) (2009) 3723–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ahel D, Horejsi Z, Wiechens N, Polo SE, Garcia-Wilson E, Ahel I, Flynn H, Skehel M, West SC, Jackson SP, Owen-Hughes T, Boulton SJ, Poly(ADP-ribose)-dependent regulation of DNA repair by the chromatin remodeling enzyme ALC1, Science 325(5945) (2009) 1240–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].El-Khamisy SF, Masutani M, Suzuki H, Caldecott KW, A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage, Nucleic Acids Res 31(19) (2003) 5526–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG, PARP inhibition: PARP1 and beyond, Nat Rev Cancer 10(4) (2010) 293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ohmoto A, Yachida S, Current status of poly(ADP-ribose) polymerase inhibitors and future directions, Onco Targets Ther 10 (2017) 5195–5208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Rosenthal F, Feijs KL, Frugier E, Bonalli M, Forst AH, Imhof R, Winkler HC, Fischer D, Caflisch A, Hassa PO, Luscher B, Hottiger MO, Macrodomain-containing proteins are new mono-ADP-ribosylhydrolases, Nat Struct Mol Biol 20(4) (2013) 502–7. [DOI] [PubMed] [Google Scholar]
- [24].Feijs KL, Forst AH, Verheugd P, Luscher B, Macrodomain-containing proteins: regulating new intracellular functions of mono(ADP-ribosyl)ation, Nat Rev Mol Cell Biol 14(7) (2013) 443–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Oka S, Kato J, Moss J, Identification and characterization of a mammalian 39-kDa poly(ADP-ribose) glycohydrolase, J Biol Chem 281(2) (2006) 705–13. [DOI] [PubMed] [Google Scholar]
- [26].Ribeiro JM, Cameselle JC, Fernandez A, Canales J, Pinto RM, Costas MJ, Inhibition and ADP-ribose pyrophosphatase-I by nitric-oxide-generating systems: a mechanism linking nitric oxide to processes dependent on free ADP-ribose, Biochem Biophys Res Commun 213(3) (1995) 1075–81. [DOI] [PubMed] [Google Scholar]
- [27].Fernandez A, Ribeiro JM, Costas MJ, Pinto RM, Canales J, Cameselle JC, Specific ADP-ribose pyrophosphatase from Artemia cysts and rat liver: effects of nitroprusside, fluoride and ionic strength, Biochim Biophys Acta 1290(1) (1996) 121–7. [PubMed] [Google Scholar]
- [28].Long A, Klimova N, Kristian T, Mitochondrial NUDIX hydrolases: A metabolic link between NAD catabolism, GTP and mitochondrial dynamics, Neurochem Int 109 (2017) 193–201. [DOI] [PubMed] [Google Scholar]
- [29].Palazzo L, Thomas B, Jemth AS, Colby T, Leidecker O, Feijs KL, Zaja R, Loseva O, Puigvert JC, Matic I, Helleday T, Ahel I, Processing of protein ADP-ribosylation by Nudix hydrolases, Biochem J 468(2) (2015) 293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Tong L, Denu JM, Function and metabolism of sirtuin metabolite O-acetyl-ADP-ribose, Biochim Biophys Acta 1804(8) (2010) 1617–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Sharifi R, Morra R, Appel CD, Tallis M, Chioza B, Jankevicius G, Simpson MA, Matic I, Ozkan E, Golia B, Schellenberg MJ, Weston R, Williams JG, Rossi MN, Galehdari H, Krahn J, Wan A, Trembath RC, Crosby AH, Ahel D, Hay R, Ladurner AG, Timinszky G, Williams RS, Ahel I, Deficiency of terminal ADP-ribose protein glycohydrolase TARG1/C6orf130 in neurodegenerative disease, EMBO J 32(9) (2013) 1225–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Smith SJ, Towers N, Saldanha JW, Shang CA, Mahmood SR, Taylor WR, Mohun TJ, The cardiac-restricted protein ADP-ribosylhydrolase-like 1 is essential for heart chamber outgrowth and acts on muscle actin filament assembly, Dev Biol 416(2) (2016) 373–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Kato J, Zhu J, Liu C, Moss J, Enhanced sensitivity to cholera toxin in ADP-ribosylarginine hydrolasedeficient mice, Mol Cell Biol 27(15) (2007) 5534–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Laing S, Unger M, Koch-Nolte F, Haag F, ADP-ribosylation of arginine, Amino Acids 41(2) (2011) 257–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Moss J, Tsai SC, Adamik R, Chen HC, Stanley SJ, Purification and characterization of ADP-ribosylarginine hydrolase from turkey erythrocytes, Biochemistry 27(15) (1988) 5819–23. [DOI] [PubMed] [Google Scholar]
- [36].Konczalik P, Moss J, Identification of critical, conserved vicinal aspartate residues in mammalian and bacterial ADP-ribosylarginine hydrolases, J Biol Chem 274(24) (1999) 16736–40. [DOI] [PubMed] [Google Scholar]
- [37].Karlberg T, Langelier MF, Pascal JM, Schuler H, Structural biology of the writers, readers, and erasers in mono- and poly(ADP-ribose) mediated signaling, Mol Aspects Med 34(6) (2013) 1088–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kernstock S, Koch-Nolte F, Mueller-Dieckmann J, Weiss MS, Mueller-Dieckmann C, Cloning, expression, purification and crystallization as well as X-ray fluorescence and preliminary X-ray diffraction analyses of human ADP-ribosylhydrolase 1, Acta Crystallogr Sect F Struct Biol Cryst Commun 65(Pt 5) (2009) 529–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Mashimo M, Moss J, Functional Role of ADP-Ribosyl-Acceptor Hydrolase 3 in poly(ADP-Ribose) Polymerase-1 Response to Oxidative Stress, Curr Protein Pept Sci 17(7) (2016) 633–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Abplanalp J, Leutert M, Frugier E, Nowak K, Feurer R, Kato J, Kistemaker HVA, Filippov DV, Moss J, Caflisch A, Hottiger MO, Proteomic analyses identify ARH3 as a serine mono-ADP-ribosylhydrolase, Nat Commun 8(1) (2017) 2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Fontana P, Bonfiglio JJ, Palazzo L, Bartlett E, Matic I, Ahel I, Serine ADP-ribosylation reversal by the hydrolase ARH3, Elife 6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Kasamatsu A, Nakao M, Smith BC, Comstock LR, Ono T, Kato J, Denu JM, Moss J, Hydrolysis of O-acetyl-ADP-ribose isomers by ADP-ribosylhydrolase 3, J Biol Chem 286(24) (2011) 21110–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Pourfarjam Y, Ventura J, Kurinov I, Cho A, Moss J, Kim IK, Structure of human ADP-ribosylacceptor hydrolase 3 bound to ADP-ribose reveals a conformational switch that enables specific substrate recognition, J Biol Chem (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Kato J, Vekhter D, Heath J, Zhu J, Barbieri JT, Moss J, Mutations of the functional ARH1 allele in tumors from ARH1 heterozygous mice and cells affect ARH1 catalytic activity, cell proliferation and tumorigenesis, Oncogenesis 4 (2015) e151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Niere M, Kernstock S, Koch-Nolte F, Ziegler M, Functional localization of two poly(ADP-ribose)-degrading enzymes to the mitochondrial matrix, Mol Cell Biol 28(2) (2008) 814–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Mueller-Dieckmann C, Kernstock S, Lisurek M, von Kries JP, Haag F, Weiss MS, Koch-Nolte F, The structure of human ADP-ribosylhydrolase 3 (ARH3) provides insights into the reversibility of protein ADP-ribosylation, Proc Natl Acad Sci U S A 103(41) (2006) 15026–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Mashimo M, Kato J, Moss J, ADP-ribosyl-acceptor hydrolase 3 regulates poly (ADP-ribose) degradation and cell death during oxidative stress, Proc Natl Acad Sci U S A 110(47) (2013) 18964–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Moss J, Zolkiewska A, Okazaki I, ADP-ribosylarginine hydrolases and ADP-ribosyltransferases. Partners in ADP-ribosylation cycles, Adv Exp Med Biol 419 (1997) 25–33. [DOI] [PubMed] [Google Scholar]
- [49].Fu H, Burris RH, Roberts GP, Reversible ADP-ribosylation is demonstrated to be a regulatory mechanism in prokaryotes by heterologous expression, Proc Natl Acad Sci U S A 87(5) (1990) 1720–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Kato J, Zhu J, Liu C, Stylianou M, Hoffmann V, Lizak MJ, Glasgow CG, Moss J, ADP-ribosylarginine hydrolase regulates cell proliferation and tumorigenesis, Cancer Res 71(15) (2011) 5327–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Xu JX, Xiong W, Zeng Z, Tang Y, Wang YL, Xiao M, Li M, Li QS, Song GL, Kuang J, Effect of ART1 on the proliferation and migration of mouse colon carcinoma CT26 cells in vivo, Mol Med Rep 15(3) (2017) 1222–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Song GL, Jin CC, Zhao W, Tang Y, Wang YL, Li M, Xiao M, Li X, Li QS, Lin X, Chen WW, Kuang J, Regulation of the RhoA/ROCK/AKT/beta-catenin pathway by arginine-specific ADP-ribosytransferases 1 promotes migration and epithelial-mesenchymal transition in colon carcinoma, Int J Oncol 49(2) (2016) 646–56. [DOI] [PubMed] [Google Scholar]
- [53].Zhou L, Zhan ML, Tang Y, Xiao M, Li M, Li QS, Yang L, Li X, Chen WW, Wang YL, Effects of betacaryophyllene on arginine ADP-ribosyltransferase 1-mediated regulation of glycolysis in colorectal cancer under high-glucose conditions, Int J Oncol 53(4) (2018) 1613–1624. [DOI] [PubMed] [Google Scholar]
- [54].Li Z, Yan X, Sun Y, Yang X, Expression of ADP-ribosyltransferase 1 Is Associated with Poor Prognosis of Glioma Patients, Tohoku J Exp Med 239(4) (2016) 269–78. [DOI] [PubMed] [Google Scholar]
- [55].Shim B, Pacheco-Rodriguez G, Kato J, Darling TN, Vaughan M, Moss J, Sex-specific lung diseases: effect of oestrogen on cultured cells and in animal models, Eur Respir Rev 22(129) (2013) 302–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Quarona V, Zaccarello G, Chillemi A, Brunetti E, Singh VK, Ferrero E, Funaro A, Horenstein AL, Malavasi F, CD38 and CD157: a long journey from activation markers to multifunctional molecules, Cytometry B Clin Cytom 84(4) (2013) 207–17. [DOI] [PubMed] [Google Scholar]
- [57].Young GS, Choleris E, Lund FE, Kirkland JB, Decreased cADPR and increased NAD+ in the Cd38−/− mouse, Biochem Biophys Res Commun 346(1) (2006) 188–92. [DOI] [PubMed] [Google Scholar]
- [58].Bu X, Kato J, Hong JA, Merino MJ, Schrump DS, Lund FE, Moss J, CD38 knockout suppresses tumorigenesis in mice and clonogenic growth of human lung cancer cells, Carcinogenesis 39(2) (2018) 242–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Lin P, Owens R, Tricot G, Wilson CS, Flow cytometric immunophenotypic analysis of 306 cases of multiple myeloma, Am J Clin Pathol 121(4) (2004) 482–8. [DOI] [PubMed] [Google Scholar]
- [60].Chini EN, Chini CCS, Espindola Netto JM, de Oliveira GC, van Schooten W, The Pharmacology of CD38/NADase: An Emerging Target in Cancer and Diseases of Aging, Trends Pharmacol Sci 39(4) (2018) 424–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Meyer-Ficca ML, Meyer RG, Coyle DL, Jacobson EL, Jacobson MK, Human poly(ADP-ribose) glycohydrolase is expressed in alternative splice variants yielding isoforms that localize to different cell compartments, Exp Cell Res 297(2) (2004) 521–32. [DOI] [PubMed] [Google Scholar]
- [62].Ame JC, Fouquerel E, Gauthier LR, Biard D, Boussin FD, Dantzer F, de Murcia G, Schreiber V, Radiation-induced mitotic catastrophe in PARG-deficient cells, J Cell Sci 122(12) (2009) 1990–2002. [DOI] [PubMed] [Google Scholar]
- [63].Zhou Y, Feng X, Koh DW, Activation of cell death mediated by apoptosis-inducing factor due to the absence of poly(ADP-ribose) glycohydrolase, Biochemistry 50(14) (2011) 2850–9. [DOI] [PubMed] [Google Scholar]
- [64].Hanai S, Kanai M, Ohashi S, Okamoto K, Yamada M, Takahashi H, Miwa M, Loss of poly(ADP-ribose) glycohydrolase causes progressive neurodegeneration in Drosophila melanogaster, Proc Natl Acad Sci U S A 101(1) (2004) 82–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Koh DW, Lawler AM, Poitras MF, Sasaki M, Wattler S, Nehls MC, Stoger T, Poirier GG, Dawson VL, Dawson TM, Failure to degrade poly(ADP-ribose) causes increased sensitivity to cytotoxicity and early embryonic lethality, Proc Natl Acad Sci U S A 101(51) (2004) 17699–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Min W, Cortes U, Herceg Z, Tong WM, Wang ZQ, Deletion of the nuclear isoform of poly(ADP-ribose) glycohydrolase (PARG) reveals its function in DNA repair, genomic stability and tumorigenesis, Carcinogenesis 31(12) (2010) 2058–65. [DOI] [PubMed] [Google Scholar]
- [67].Fujihara H, Ogino H, Maeda D, Shirai H, Nozaki T, Kamada N, Jishage K, Tanuma S, Takato T, Ochiya T, Sugimura T, Masutani M, Poly(ADP-ribose) Glycohydrolase deficiency sensitizes mouse ES cells to DNA damaging agents, Curr Cancer Drug Targets 9(8) (2009) 953–62. [DOI] [PubMed] [Google Scholar]
- [68].Niere M, Mashimo M, Agledal L, Dolle C, Kasamatsu A, Kato J, Moss J, Ziegler M, ADP-ribosylhydrolase 3 (ARH3), not poly(ADP-ribose) glycohydrolase (PARG) isoforms, is responsible for degradation of mitochondrial matrix-associated poly(ADP-ribose), J Biol Chem 287(20) (2012) 16088–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Ghosh SG, Becker K, Huang H, Salazar TD, Chai G, Salpietro V, Al-Gazali L, Waisfisz Q, Wang H, Vaux KK, Stanley V, Manole A, Akpulat U, Weiss MM, Efthymiou S, Hanna MG, Minetti C, Striano P, Pisciotta L, De Grandis E, Altmuller J, Nurnberg P, Thiele H, Yis U, Okur TD, Polat AI, Amiri N, Doosti M, Karimani EG, Toosi MB, Haddad G, Karakaya M, Wirth B, van Hagen JM, Wolf NI, Maroofian R, Houlden H, Cirak S, Gleeson JG, Biallelic Mutations in ADPRHL2, Encoding ADP-Ribosylhydrolase 3, Lead to a Degenerative Pediatric Stress-Induced Epileptic Ataxia Syndrome, Am J Hum Genet 103(3) (2018) 431–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Bonfiglio JJ, Fontana P, Zhang Q, Colby T, Gibbs-Seymour I, Atanassov I, Bartlett E, Zaja R, Ahel I, Matic I, Serine ADP-Ribosylation Depends on HPF1, Mol Cell 65(5) (2017) 932–940 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Leidecker O, Bonfiglio JJ, Colby T, Zhang Q, Atanassov I, Zaja R, Palazzo L, Stockum A, Ahel I, Matic I, Serine is a new target residue for endogenous ADP-ribosylation on histones, Nat Chem Biol 12(12) (2016) 998–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Palazzo L, Leidecker O, Prokhorova E, Dauben H, Matic I, Ahel I, Serine is the major residue for ADP-ribosylation upon DNA damage, Elife 7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Wu L, Yang L, The function and mechanism of HMGB1 in lung cancer and its potential therapeutic implications, Oncol Lett 15(5) (2018) 6799–6805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Masutani M, Fujimori H, Poly(ADP-ribosyl)ation in carcinogenesis, Mol Aspects Med 34(6) (2013) 1202–16. [DOI] [PubMed] [Google Scholar]
- [75].Bilan V, Selevsek N, Kistemaker HAV, Abplanalp J, Feurer R, Filippov DV, Hottiger MO, New Quantitative Mass Spectrometry Approaches Reveal Different ADP-ribosylation Phases Dependent On the Levels of Oxidative Stress, Mol Cell Proteomics 16(5) (2017) 949–958. [DOI] [PMC free article] [PubMed] [Google Scholar]