

Abstract
The EFSA Scientific Committee was asked to provide guidance on the most appropriate in vivo tests to follow up on positive in vitro results for aneugenicity, and on the approach to risk assessment for substances that are aneugenic but not clastogenic nor causing gene mutations. The Scientific Committee confirmed that the preferred approach is to perform an in vivo mammalian erythrocyte micronucleus test with a relevant route of administration. If this is positive, it demonstrates that the substance is aneugenic in vivo. A negative result with evidence that the bone marrow is exposed to the test substance supports a conclusion that aneugenic activity is not expressed in vivo. If there is no evidence of exposure to the bone marrow, a negative result is viewed as inconclusive and further studies are required. The liver micronucleus assay, even though not yet fully validated, can provide supporting information for substances that are aneugenic following metabolic activation. The gastrointestinal micronucleus test, conversely, to be further developed, may help to assess aneugenic potential at the initial site of contact for substances that are aneugenic in vitro without metabolic activation. Based on the evidence in relation to mechanisms of aneugenicity, the Scientific Committee concluded that, in principle, health‐based guidance values can be established for substances that are aneugenic but not clastogenic nor causing gene mutations, provided that a comprehensive toxicological database is available. For situations in which the toxicological database is not sufficient to establish health‐based guidance values, some approaches to risk assessment are proposed. The Scientific Committee recommends further development of the gastrointestinal micronucleus test, and research to improve the understanding of aneugenicity to support risk assessment.
Keywords: aneugenicity, micronucleus test, genotoxicity in vivo and in vitro
Short abstract
This publication is linked to the following EFSA Supporting Publications article: http://onlinelibrary.wiley.com/doi/10.2903/sp.efsa.2021.EN-6814/full
1. Background and Terms of Reference as provided by EFSA
The genotoxicity testing strategy1 indicated in the EFSA Scientific Committee opinion is designed to investigate the genotoxic potential of substances through the detection of three genotoxic endpoints: gene mutations, structural chromosomal aberrations (i.e. clastogenicity) and numerical chromosomal aberrations (i.e. aneuploidy).
The testing strategy is developed as a step‐wise approach, beginning with a basic battery of in vitro tests, comprising:
a bacterial reverse mutation assay [Organisation for Economic Co‐operation and Development (OECD) TG 471, end‐point: gene mutations]; and
an in vitro mammalian cell micronucleus (MN) test (OECD TG 487, endpoints: clastogenicity and aneugenicity).
The ability of the compound to induce clastogenicity and/or aneugenicity can be discriminated in vitro through the in vitro MN test with centromere labelling [i.e. fluorescence in situ hybridisation (FISH) technique or CREST analysis]. Clarification of the mechanism of action would make it possible to identify the most appropriate follow‐up in vivo study, but this is not a mandatory requirement of the current testing strategy.
If positive results are observed in vitro, the substance should be tested in an appropriate in vivo test depending on the relevant end‐point to be followed up.
The currently recommended in vivo tests are:
the transgenic rodent assay (OECD TG 488, end‐point: gene mutations);
the in vivo mammalian alkaline comet assay (OECD TG 489, endpoints: DNA strand breaks as follow‐up of compounds inducing gene mutations and/or clastogenicity);
the in vivo mammalian erythrocyte micronucleus (MN) test (OECD TG 474, endpoints: clastogenicity and/or aneugenicity).
Clastogenic substances induce structural chromosomal aberrations through breaks in DNA. Aneugenic substances induce numerical chromosomal aberrations through interactions with cellular targets other than DNA, such as proteins involved in the segregation of chromosomes during mitosis or meiosis. This difference in molecular targets results in features that are typical of aneugenic substances:
a critical number of target sites must be affected before the aneugenic effect occurs;
the onset of numerical chromosomal aberrations generally occurs with steep dose–response curves in a narrow range of concentrations/doses;
aneuploidy‐inducing agents exhibit non‐linear dose–response curves and a threshold is usually estimated.
According to the EFSA Scientific Committee (SC) Opinion on genotoxicity testing strategies (EFSA Scientific Committee, 2011) (Scenario IIa) if the available data show an aneugenic effect in vitro, an in vivo mammalian erythrocyte micronucleus MN test (in bone marrow or peripheral blood) would typically be considered appropriate. If an adequately conducted in vivo MN test (with evidence for significant exposure of the target tissue) is negative, it will be possible to conclude that the test substance is not aneugenic in vivo. However, if the in vivo MN test is negative and there is no demonstration that the target tissue was exposed, it cannot be concluded that there is no concern with respect to the aneugenic potential. Lack of demonstration of target tissue exposure could be due to several reasons including:
the tested substance could be poorly absorbed or metabolised and eliminated before reaching the bone marrow;
a clear demonstration that the aneugenic substance reaches the bone marrow could be difficult.
At present there are no internationally validated methods to investigate the potential aneugenic effect in tissues other than bone marrow, including the site of first contact. For food chemicals, the gastrointestinal tract (GIT) may represent a particularly relevant target because of the potential higher exposure, especially for substances that are poorly absorbed and scarcely bioavailable to systemic circulation.
Terms of Reference
Considering the current limitations in the evaluation of aneugenic substances, the SC is requested to develop guidance in relation to:
what is the most appropriate in vivo follow‐up for substances that are aneugenic in vitro;
how should risk to human health be assessed for a substance exhibiting aneugenicity.
1.1. Audience and degree of obligation
This guidance is unconditional (i.e. required, see EFSA Scientific Committee, 2015) for the EFSA panels and units evaluating the genotoxicity of chemical substances in the food and feed safety area. It should be supported by sectoral guidance, if available. It also provides guidance to applicants submitting dossiers to EFSA.
2. Introduction
Mutational events can be subdivided into three categories: gene mutations, structural chromosomal aberrations (clastogenicity) and numerical chromosomal aberrations (aneugenicity and polyploidy). Aneugenicity designates chemically induced aneuploidy, which means a change in the chromosome number from the normal diploid or haploid number of chromosomes that is not a multiple of the haploid number. By contrast, polyploidy means a multiple of the haploid number of chromosomes. The two main processes leading to aneuploidy are non‐disjunction at anaphase or lagging of chromosomes during cell division, both during mitosis and meiosis, resulting in loss or gain of individual chromosomes in the daughter cells, while polyploidy may result from mitotic failure, endoreduplication or cell fusion (Kirsch‐Volders et al., 2019).
Chemicals inducing aneuploidy are called aneugens. They may interact with a number of different targets in the cell, including centromeres and telomeres, kinetochores and chromatid glue proteins involved in chromatid attachment and separation, tubulin, microtubule‐associated proteins (MAPs), centrioles and other components of the spindle apparatus, the anaphase promoting complex, proteins involved in cell cycle control such as cyclins, cyclin‐dependent kinases (CDKs) and p53, or targets indirectly involved in the cell cycle such as calmodulin, and the cellular or nuclear membrane (Kirsch‐Volders et al., 2002). Interaction with one or more of these molecular targets may represent the molecular initiating event triggering an adverse outcome pathway (AOP), leading through subsequent key events to aneuploidy as the final outcome. Examples of AOP leading to aneugenicity, triggered by tubulin binding or aurora kinase inhibition, have been proposed (Lynch et al., 2019). The AOP framework represents a potentially useful approach also in hazard identification, linking subcellular mechanistic data with aneuploidy, but its application for regulatory purpose appears still premature.
The consequence of aneuploidy is a difference in the number of copies of genes that are located on the missing or additional chromosome, and so a difference in the relative dosage of these gene products (Kirsch‐Volders et al., 2019). If the genes concerned are transcriptional regulators, the dosage of other genes, located on the normally segregated chromosomes, may also be affected. Aneuploidy is recognised as a significant cause of infertility and pregnancy loss when impacting germ cells (Pacchierotti et al., 2019), and a hallmark of genomic instability and consequently cancer when occurring in somatic cells (Naylor and van Deursen, 2016).
The impact of aneuploidy during gametogenesis and embryogenesis is a significant cause of reproduction failure. Heritable aneugenic hazard has been characterised for a few chemicals in experimental models. Limited evidence suggests that male human germ cells might be more sensitive than mouse spermatocytes to chemically induced aneuploidy (Baumgartner et al., 2001; Adler et al., 2002). Although aneuploidy in germ cells is a significant cause of infertility and pregnancy loss in humans, there is currently limited evidence that aneugens induce hereditary diseases in human populations because the great majority of aneuploid conceptuses die in utero (Pacchierotti et al., 2019).
The role of somatic cell aneuploidy in carcinogenesis is less firmly established. Aneuploidy was observed at many stages of cancer progression in humans, although the evidence that it is a primary driver of carcinogenesis is limited. Human constitutive aneuploidies show increased risks of cancer early in life. As an example, individuals with Down's syndrome have an increased risk of acute megakaryoblastic leukaemia (AMKL) in childhood and constitutive trisomy 21 is viewed as an early event in carcinogenesis. The available rodent carcinogenicity studies of known chemical aneugens have produced mixed results, and in cases of a positive outcome, it is difficult to characterise the exact role of chemically induced aneuploidy in tumour induction and/or progression, as carcinogenic aneugens often show, in addition, other properties including gene mutation, structural chromosomal damage, toxicity, immunosuppression, epigenetic and hormonal effects (Tweats et al., 2019). However, growing scientific evidence indicates that aneuploidy in somatic cells may be involved in the development of cancer. Chromosome loss (included in micronuclei) can trigger chromothripsis (massive chromosome fragmentation), followed by a random recombination of the fragments in any position on chromosomes, leading to increased chromosomal instability (Guo et al., 2019). In addition, recognition of cytosolic self‐DNA contained in micronuclei (MN) by cyclic guanosine monophosphate–adenosine monophosphate (cyclic GMP–AMP) or cGAMP synthase (cGAS) may activate a proinflammatory pathway that promotes the metastatic phenotype (Bakhoum et al., 2018).
In its Opinion on genotoxicity testing strategies for risk assessment of substances in food and feed, the SC of the European Food Safety Authority (EFSA) recommended an in vitro test battery consisting of a bacterial reverse mutation test (OECD TG 471), and an in vitro mammalian cell micronucleus (MN) test (OECD TG 487) to cover all three genotoxic endpoints (EFSA Scientific Committee, 2011). The bacterial reverse mutation assay covers gene mutations and the in vitro mammalian cell MN test covers both structural and numerical chromosome aberrations. Aneugens can be distinguished from clastogens in the mammalian cell MN test if a staining technique (FISH or CREST, see Annex A) is used to distinguish whole chromosomes from acentric chromosome fragments.
For a positive result in vitro, an in vivo follow‐up test should cover the same end‐point that was found to be positive in vitro (EFSA Scientific Committee, 2011). The only validated in vivo MN test for which an OECD test guideline is available is carried out in mammalian erythrocytes, sampled either from the bone marrow or peripheral blood cells of rodents (OECD TG 474). However, the usefulness of this test in the follow‐up of in vitro aneugens may be limited when there is not sufficient evidence that the test substance reaches the bone marrow and, therefore, effects at other potentially relevant targets, such as the site of first contact, need to be evaluated (EFSA, 2017b). This is particularly the case when the test substance itself, rather than a metabolite, is the aneugenic substance, i.e. positive results are seen in the absence of metabolic activation. Because of these limitations, and the lack of an internationally agreed strategy for the in vivo assessment of aneugenic hazards, in this document the SC provides guidance on the in vivo follow‐up of in vitro aneugens, and on the risk characterisation of substances evaluated as aneugenic in vivo.
The present guidance addresses aneugenic substances for which a concern about clastogenicity has been ruled out. The SC notes that there are currently no predictive models that can distinguish between aneugenic and clastogenic substances.
3. In vivo follow‐up of in vitro aneugens
For substances aneugenic in vitro, it is necessary to establish if the aneugenic activity is also expressed in vivo. In that case the substance is considered to be of potential safety concern and a specific risk assessment is required.
The mammalian erythrocyte MN Test (OECD TG 474), covering the endpoints of structural and numerical chromosomal aberrations of erythroblasts, is the only validated assay for the in vivo follow‐up of in vitro aneugenic compounds. The experimental protocol was standardised to evaluate MN formation in young erythrocytes (polychromatic) sampled in bone marrow and/or reticulocytes in peripheral blood cells of rodents. Newly formed micronucleated erythrocytes are identified and quantitated by staining followed by visual scoring using a microscope. In the case of positive results, the application of kinetochore staining or FISH with a pan‐centromeric probe is not mandatory to confirm in vivo the aneugenic mode of action. Automated analysis of micronuclei on cell suspensions using flow cytometry allows scoring of a large number of cells, reducing the scorer subjectivity and increasing the statistical power.
The mammalian erythrocyte MN test is one of the most widely used in vivo genotoxicity tests and it has been shown to be very sensitive in detecting aneugenic compounds that are systemically available. However, organ‐specific compounds and unstable compounds or metabolites might not be detected with this assay. Negative results obtained in immature erythrocytes need to be supported by a proof of bone marrow exposure. The EFSA opinion (EFSA, 2017b) specifically addressed the lines of evidence to demonstrate bone marrow exposure and to decide on the validity of the assay. Toxicity at the bone marrow level, detected as a reduction in the percent of immature erythrocytes, in total erythrocytes was considered as direct evidence of bone marrow exposure. However, evidence of systemic bioavailability can also be obtained from plasma level or from independent toxicokinetic and/or toxicity studies using the same route and same species.
Based on these considerations, the interpretation of the outcome from the in vivo erythrocyte MN assay needs to take into account the results from the in vitro studies (metabolic activation, condition of exposure) and all the available data on the substance, such as the chemical reactivity (which might predispose to site of contact effects), bioavailability, metabolism, toxicokinetics and any target organ specificity.
The scheme in Figure 1 describes the recommended follow‐up with substances that are aneugenic in vitro. Positive results in the mammalian erythrocyte MN test may be detected for systemically available compounds. These results should be viewed as reliable and risk assessment conducted as described in Section 5. On the other hand, negative results obtained together with evidence of bone marrow exposure allow the conclusion that the substance is not aneugenic in vivo, and therefore, aneugenicity does not raise a safety concern.
Figure 1.
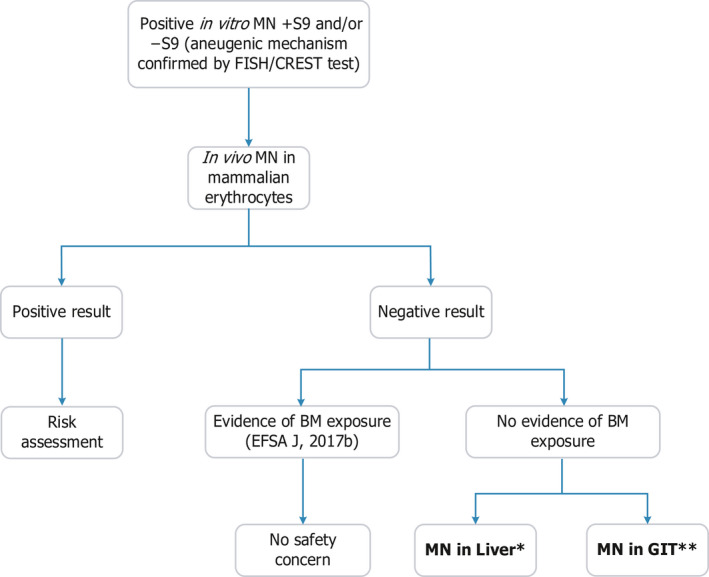
- BM: bone marrow; GIT: gastrointestinal tract; MN: micronucleus test. *: In the process of being considered for the development of an OECD TG (Kirkland et al., 2019). **: For a positive in vitro MN test in the absence of S9, and after negative results in an in vivo MN with no evidence of BM exposure, a GIT MN assay would be appropriate, but more work is required for an OECD guideline (see text).
Negative results observed in the mammalian erythrocyte MN test without evidence of bone marrow exposure may be unreliable for hazard assessment, and further analyses are needed to rule out the concern for in vivo aneugenic effects. In this case, the MN assay could be applied in different target organs, i.e. liver and gastrointestinal tract (GIT) for chemicals acting with and without metabolic activation.
The state of the standardisation and validation of the MN assays in tissues other than bone marrow and peripheral blood were discussed at different International Workshops on Genotoxicity Testing (IWGT) during the meetings of the International Association of Environmental Mutagen Societies (IAEMS) (Hayashi et al., 2000, 2007; Uno, 2013; Uno et al., 2015a,b). The IWGT Working Group in 2017 concluded that the liver MN assay is sufficiently validated for the development of an OECD guideline. The GIT MN assay appeared to be useful for the site‐specific analysis of micronuclei induction and some evaluation of the sensitivity and specificity was possible, based on a very limited number of substances. However, more substances would need to be tested to draw firm conclusions and the IWGT Working Group concluded that more work is required for an OECD guideline (Kirkland et al., 2019).
Aneugenic compounds detected in vitro exclusively or predominantly in the presence of liver S9 fraction, suggesting the involvement of liver‐specific metabolites, can be evaluated in the liver MN assay.
Aneugenic substances inducing increased in vitro micronuclei frequency in the absence of S9 fraction, can be evaluated with the MN assay in the GIT. The SC noted that at present further development and validation of the MN test in the GIT is required before it can be recommended as routine test in the follow‐up of substances that are aneugenic in vitro. However, data generated by applying the MN test in the GIT (stomach and colon) will be considered by EFSA and evaluated case by case as supporting evidence when obtained using appropriate dosing period and dose levels (Kirkland et al., 2019) in the comprehensive evaluation of all data to assess the in vivo aneugenic hazard.
Combined analysis of micronuclei in erythrocytes and in liver or GIT of the same animals could be considered to reduce the animal usage, cost and time required for consecutive testing.
MN analysis has the potential to be integrated into repeat‐dose toxicity studies (Hayashi, 2016; Kirkland et al., 2019), providing that the highest dose applied be consistent with the requirements of the OECD TG for the mammalian erythrocyte MN assay (OECD TG 474) and/or ICH criteria. The major advantages of incorporating the MN analysis into conventional repeat‐dose toxicity studies are the reduction in the number of animals used, saving time and the possibility to use general toxicology observations for the interpretation of genotoxicity results. Short‐term studies (14 or 28 days) should preferentially be considered for integration, as the lower doses used in longer studies may reduce the ability to detect aneugens in the MN assay. Blood samples could be obtained from the animals used in toxicity assays to assess micronuclei in reticulocytes at an early time, and in bone marrow and GIT at the end of treatment.
Since proliferative activity of hepatocytes is required, which reduces with age, the MN assay in liver was validated in juvenile rodents (6 weeks old). Therefore, its integration in routine repeat dose toxicity studies, in which the animals are older at termination, is not recommended (Kirkland et al., 2019).
Following the testing scheme described above, the results obtained in the mammalian erythrocytes MN test, and eventually in the liver or GIT MN assays, provide evidence of whether the aneugenic activity observed in vitro is also expressed in somatic cells in vivo. Substances which are evaluated as aneugenic in vivo are also considered aneugenic in germ cells, unless there is evidence that the chemical or its metabolites will not reach germ cells or gonadal tissue (Martus et al., 2015).
4. Evidence for thresholds in aneugenicity
4.1. Theoretical considerations
The analysis of the dose–effect relationship (hazard characterisation) is a key component in chemical risk assessment, in which usually a reference point (also known as a point of departure) is identified. This is commonly the lower confidence limit of the benchmark dose (BMDL) associated with a specific benchmark response (BMR), or the no observed adverse effect level (NOAEL), identified from the dose–effect relationship for the critical effect (i.e. the relevant effect occurring at the lowest doses). Both approaches have limitations; however, the BMD approach aims at estimating the dose that corresponds to a low, but measurable change in an adverse response. Health‐based guidance values (HBGVs) are derived from the reference point. As a convention, this approach is currently not applied to substances that induce gene mutation or clastogenicity, for which a linear dose–effect relationship is widely assumed for the reaction with DNA. The hypothesis of linearity was first proposed for ionising radiations, and subsequently extended to DNA reactive chemicals, in consideration of the prevailing mechanisms of mutagenicity that rely on single molecular interactions between the agent and its target (DNA).
Aneugens have non‐DNA targets and induce abnormal chromosome segregation, interacting with a variety of molecular and structural targets of the mitotic/meiotic machinery within the cell. The induction of chromosome mis‐segregation, leading to aneuploidy, requires multiple molecular interactions due to the multiplicity of copies of critical targets present in the cell, and the need to affect a critical number of these to elicit a functional effect. As a consequence, the shape of the dose–effect relationship will not be linear but will have a variable slope, depending on the number of involved events.
Aneugenicity is typically observed in a narrow dose range. As the dose of aneugen increases, the cell division apparatus becomes more compromised resulting in cells with chromosome imbalances, and the potential for the effects of aneuploidy to be detected. Doses causing moderate cell cycle alterations may allow cells with imbalanced chromosome number to survive mitosis and aneuploidy, while higher doses inducing strong cell cycle alterations may not allow cell survival and/or completion of mitosis (Lynch et al., 2019). It is therefore widely accepted that genotoxicants with an aneugenic mode of action exhibit non‐linear dose–response relationships with a dose below which no effects are observed and defined as the ‘threshold dose’.
A threshold dose was defined by the World Health Organization (WHO, 2009, EHC 240) as ‘The dose at which an effect just begins to occur ‐ that is, at a dose immediately below the threshold dose, the effect will not occur, and immediately above the threshold dose, the effect will occur. For a given chemical, there can be multiple threshold doses, in essence one for each definable effect. For a given effect, there may be different threshold doses in different individuals. Furthermore, the same individual may vary from time to time as to his or her threshold dose for any effect.’
Therefore, a threshold cannot be observed experimentally. However, the general existence of an underlying threshold‐based mechanism can be assumed on the basis of various experiments focusing on different aneugenic endpoints (Elhajouji et al., 2011).
4.2. Threshold in aneuploidy – empirical evidence
Based on mechanistic considerations, some aneugens can be assumed to have well‐characterised threshold responses in vitro and in vivo, using the induction of micronuclei in a specific cell population or tissue as a surrogate end‐point to measure chromosome loss, or the induction of non‐disjunction using chromosome‐specific probes (Elhajouji et al., 2011).
Classical spindle poisons, such as colchicine, carbendazim, mebendazole and nocodazole, were chosen as model molecules to evaluate the possible threshold, because their mechanism of action is well known, consisting of direct interaction with the spindle fibres by inhibiting the polymerisation of tubulin. No increase in the frequencies of centromere‐positive micronuclei, using FISH with a pancentromeric probe, in human peripheral lymphocytes was observed over a range of low concentrations, while a steep increase to highly statistically significant values of centromere‐positive micronuclei was observed over a range of higher concentrations (Elhajouji et al., 1995). Similarly, shaped dose–response curves have been obtained from another study using similar in vitro experimental conditions for two spindle inhibitors, benomyl and carbendazim (Bentley et al., 2000). The shape of this dose–response relationship is strongly indicative of a threshold.
Studies (Elhajouji et al., 1995; Marshall et al., 1996; Bentley et al., 2000; Tweats et al., 2016) on benomyl and carbendazim, frequently used as model aneugenic compounds, indicate that threshold values may differ depending on the end‐point and scoring method applied, e.g. polyploidy or aneuploidy in metaphase preparations, non‐disjunction or frequency of chromosome loss with the MN assay.
The possibility of an aneuploidy threshold for these compounds was also investigated based on non‐disjunction using chromosome‐specific probes on binucleated cells in the cytokinesis‐blocked MN assay. The results indicated that non‐disjunction occurred at lower concentrations than chromosome loss (Zijno et al., 1996; Elhajouji et al., 1997). Results described in the published literature on the sensitivity of the two main endpoints (the chromosome loss and non‐disjunction) and analysed to evaluate an aneugenicity threshold, varied among the aneugens and the cell systems used, the chromosome chosen and the interpretation of results for non‐disjunction and the method applied to analyse chromosome loss (Elloway et al., 2017).
Evidence for the existence of thresholds for aneuploidy induced by spindle poisons was also obtained in vivo using the flow cytometry peripheral blood MN test in mice. Dose–response curves observed for vinblastine, vincristine and colchicine, based on chromosome loss, were not linear and the results were consistent with in vitro findings (Cammerer et al., 2010).
5. Risk assessment of substances that are aneugenic but not clastogenic nor causing gene mutations
5.1. Identifying reference points for aneugenicity
There is a general consensus that genotoxic agents acting via non‐DNA‐reactive mechanisms exhibit a thresholded, non‐linear dose–response. Therefore, the SC considers that reference points may be identified for establishing a HBGV, or be used in a margin of exposure approach.
There are currently no internationally accepted guidelines for the quantitative analyses of genetic toxicity. Protocols to evaluate the dose–response curves for aneugenic compounds have been reported using the MN assay in vitro and in vivo as described below (Elhajouji et al., 1995; Cammerer et al., 2010; Tweats et al., 2016).
Dose–response analysis of data from the in vitro mammalian MN test using flow cytometry sorting and FISH with pancentromeric probe in human peripheral lymphocytes treated with colchicine, carbendazim, mebendazole and nocodazole was carried out by Elhajouji et al. (1995) applying a piecewise linear regression analysis. The piecewise linear regression analysis (or breakpoint regression) is a discontinuous regression model that reflects a ‘jump’ in the regression line. The equation used is the sum of a constant function (if the concentration is lower than the breakpoint) and a linear function (if the concentration is higher than the breakpoint). Determination of the breakpoint was carried out based on the results of the chi‐squared test p‐values. The first concentration that induced a statistically significant increase in the MN‐centromere‐positive frequency compared with the control was chosen as the breakpoint. During the calculations of regressions, the programme calculates the expected values (values of the regression curve at the given concentrations) and the correlation coefficients based on the data and the fixed breakpoints. Analysis comparing the observed and predicted values showed a high correlation for the tested substances (Elhajouji et al., 1995).
Evaluation of the threshold dose has been carried out in vivo using the flow cytometry peripheral blood MN test in mice (Cammerer et al., 2010). Vinblastine, vincristine and colchicine were tested. A specific study design was applied based on small size groups of animals, high range of doses (10–12) and the automated scoring of a high number of cells (around 20,000) to increase the statistical power. A specific statistical analysis of the dose–response curves was performed to determine the breakpoint dose. The intersection point of piecewise regression lines was determined as the breakpoint of the dose–response curve (Cammerer et al., 2010).
An example of application of the Benchmark Dose (BMD) approach to evaluate in vitro and in vivo dose–responses and to derive a reference point was reported for the aneugenic anti‐parasitic benzimidazole flubendazole, which has been used for many years to treat intestinal infections in humans and animals. The compound was shown to be aneugenic in vitro with the MN test in cultured human peripheral lymphocytes testing nine concentrations with the standardised protocol. A first in vivo experiment using the mouse bone marrow MN assay was negative without demonstration of bone marrow exposure. In a further experiment using a new oral formulation with improved bioavailability of flubendazole, micronuclei induction was observed. Analysis of the plasma from the treated animals showed that there was exposure to flubendazole. Analysis of the in vivo data allowed establishment of a reference point for aneugenicity, which could be compared with therapeutic exposures of flubendazole (Tweats et al., 2016). The BMR of one standard deviation above the spontaneous value was used in calculating the BMDL as the reference point for the in vivo MN dose–response data, following the recommendations of the International Workgroup on Genotoxicity Testing (MacGregor et al., 2015). The EPA's Benchmark Dose Software was applied and the polynomial model was selected as the most suitable for these data.
The SC considers that reference points for aneugenicity can be identified using dose–response analysis. The preferred approach is BMD modelling following the EFSA guidance (EFSA, 2017a) and selection of the BMR needs to be justified case by case. If BMD modelling is not possible, a breakpoint may be identified and used as the reference point based on examples provided by relevant scientific literature (e.g. Cammerer et al., 2010) and using expert judgement.
5.2. Hazard characterisation for substances that are aneugenic but not clastogenic nor causing gene mutations
As described above, a thresholded mode of action is plausible for aneugenic substances and, therefore, in principle, a health‐based guidance value (HBGV) can be established based on the most sensitive end‐point. So, the first step is to look at the entire toxicological database. Several scenarios can be envisaged, depending on the completeness of the database for the substance under consideration and its toxicological properties. Figure 2 summarises the different scenarios for the hazard and risk characterisation of aneugenic substances based on the toxicological data available.
Figure 2.
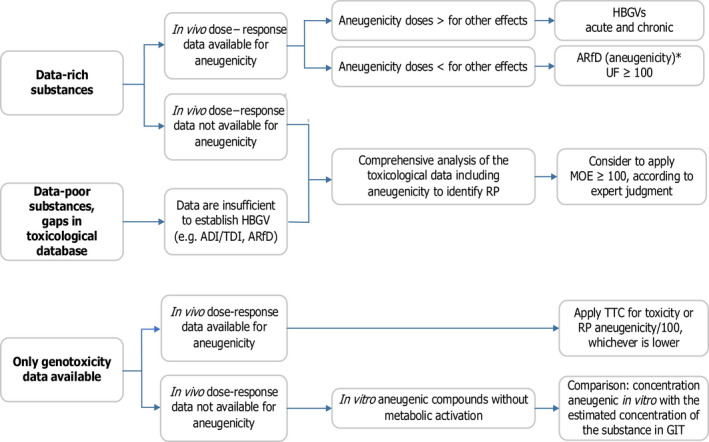
- *: May also be applied as a chronic HBGV if required.
5.2.1. Data‐rich substances, in vivo dose–response data are available for aneugenicity
If it has been possible to identify a reference point for aneugenicity, generally from an in vivo mammalian erythrocyte MN test using an appropriate study protocol, then this can be compared with the reference points for other effects. The reference point would preferably be identified applying the benchmark dose approach. However, even if it has not been possible to calculate a reliable BMDL, due to insufficient dose–response information, then comparison of the breakpoint (see Section 5.1) for in vivo induction of micronuclei with the reference points for other toxicity endpoints can be informative about whether aneugenicity should be viewed as the most sensitive effect, i.e. that occurring at lowest dose levels.
If the reference point for aneugenicity in vivo is higher than that for another effect, HBGVs (acute and chronic) can be set up using the well‐established principles (EHC 240), as is seen in the examples in Table B.1 (Annex B). Whether an acute and a chronic HBGV (e.g. ARfD and an ADI) is needed will depend on the nature of the critical effect.
Table B.1.
Examples of active substances (possibly) causing aneugenic effects that have previously been evaluated for the maximum residue limit (MRL) classification for use in food producing animals. Note that these are published examples and have not been re‐evaluated by EFSA for this guidance document
| Active substance | Genotoxicity | Experi‐mental evidence of aneu‐genicity | Carcinogenicity | Risk assessment | CR (EU) 37/2010 entry* | Reference** |
|---|---|---|---|---|---|---|
| Colchicine | Positive results in vivo and in vitro for gene and chromosome mutation. Aneugenic potential in vivo (1 mg/kg) and in vitro (0.006 μg/ml) | Yes | No data available | No ADI could be set up based on genotoxicity, teratogenicity, effects on fertility, no data on absorption after local treatment of cattle and horses) | Table 2 | EMEA/MRL/044/95‐FINAL |
| Benzimidazole derivatives | ||||||
| Albendazole | In vivo aneugens | Yes | No evidence in either rats or mice in suitable carcinogenicity bioassays | ADI of 0–0.005 mg/kg bw per day based on NOEL for teratogenicity of 5 mg/kg per day and a safety factor of 1,000 | Table 1 | MRL Summary Report (reference here) and EMEA/MRL/865/03‐FINAL |
| Albendazole oxide | Yes | No studies with albendazole oxide | ADI of 0–0.005 mg/kg bw per day based on albendazole data | Table 1 | EMEA/MRL/555/99‐FINAL February 1999 | |
| Netobimin | Yes | No studies with netobimin | ADI of 0–0.005 mg/kg bw per day based on albendazole data | Table 1 | EMEA/MRL/556/99‐FINAL April 1999 | |
| Mebendazole | Not a direct acting mutagen or clastogenic. Aneugenic in mammalian somatic cells. Not possible to identify an in vivo NOEL for aneugenicity, but a no‐effect concentration of 85 ng/ml was identified from in vitro FISH studies | Yes | No evidence of carcinogenicity in rats and mice but studies were considered inadequate | ADI of 0.0125 mg/kg bw per day based on NOEL of 2.5 mg/kg bw per day in a 13‐week study in dogs and in developmental toxicity studies in rats and mice using a safety factor of 200 | Table 1 | EMEA/MRL/625/99‐FINAL July 1999 and EMEA/MRL/781/01‐FINAL March 2001 |
| Thiabendazole | No gene mutation or structural chromosomal damage. Consistent evidence of aneugenicity in vitro. Negative in validated oral in vivo mutagenicity assays. Some reports of aneuploidy in bone marrow cells in vivo following intraperitoneal administration | Yes, but conclusion that negative in vivo with the oral route | No excess incidence of any type of tumour in mice. In rats, no increases in tumours except thyroid follicular adenomas, which was considered to be due to by a nongenotoxic mechanism related to liver enlargement | ADI of 0.1 mg/kg bw based on an overall toxicological NOEL of 10 mg/kg bw/day for a range of toxicological endpoints including effects on the liver, thyroid and bone marrow, spleen, effects on reproductive performance, teratogenicity in mice and fetotoxicity in rats. A safety factor of 100 was applied | Table 1 | EMEA/MRL/868/03‐FINAL June 2004 |
| Oxfendazole | Benzimidazole compound, which are known to be mitotic spindle poisons. The mutagenicity data available for febantel, fenbendazole and oxfendazole show no clear evidence of genotoxicity and although no specific tests for aneugenicity have been conducted, the clastogenicity studies that have been conducted are generally reassuring’ | No | No evidence of carcinogenicity in rats or mice | ADI of 7 μg/kg bw per day based on the NOEL of 0.65 mg/kg bw per day for hepatic vacuolation in a carcinogenicity study in rats with a safety factor of 100 to febantel, fenbendazole and oxfendazole share the same metabolism with oxfendazole being most toxic | Table 1 | EMEA/MRL/888/03‐FINAL June 2004 |
| Fenbendazole | Benzimidazole compound, which are known to be mitotic spindle poisons. The mutagenicity data available for febantel, fenbendazole and oxfendazole show no clear evidence of genotoxicity and although no specific tests for aneugenicity have been conducted, the clastogenicity studies that have been conducted are generally reassuring’ | No | No evidence of carcinogenicity in rats or mice | ADI of 7 μg/kg bw per day, based on toxicity data for oxfendazole | Table 1 | EMA/CVMP/914694/2011 |
| Febantel | Benzimidazole compound, which is known to be a mitotic spindle poison. The mutagenicity data available for febantel, fenbendazole and oxfendazole show no clear evidence of genotoxicity and although no specific tests for aneugenicity have been conducted, the clastogenicity studies that have been conducted are generally reassuring | No | No evidence of carcinogenicity in rats and mice | ADI of 7 μg/kg bw per day, based on toxicity data for oxfendazole | Table 1 | EMEA/MRL/192/97‐FINAL June 1997 |
| Benzimidazole derivatives without indication of aneugenicity in the MRL summary reports | ||||||
| Flubendazole | Negative for gene mutation, DNA damage, a sex‐linked recessive lethal assay in Drosophila melanogaster, a dominant lethal assay in mice and in vivo micronucleus tests in rats and mice | No | No evidence of carcinogenicity in rats and mice although both studies were marred by poor survival | ADI of 0–12 μg/kg bw per day based on the NOEL of 2.5 mg/kg bw per day in a 3‐month study in dogs, with a safety factor of 200 | Table 1 | EMEA/CVMP/33128/2006‐FINAL |
| Triclabendazole | Negative in a range of genotoxicity studies, including MN in hamster bone marrow | No | No evidence of carcinogenicity in rats and mice | ADI of 0.0015 mg/kg bw, based on a NOEL of 0.15 mg/kg bw/day for increased post‐partum mortality of the F2 generation in a two‐generation rat reproduction study and an UF of 100 | Table 1 | No reference number of CVMP's MRL Summary Report (1) available*** |
CR (EU) 37/2010: Use of active substances in veterinary medicinal products for food producing animals; Table 1 substances with numerical MRLs or ‘No MRL required’ classification, made it possible to be used in veterinary medicines; Table 2 – prohibited substances.
If the reference point for aneugenicity in vivo is lower than or close to those for other effects, an acute HBGV (i.e. an ARfD) can be established from the reference point by applying the 100‐fold default uncertainty factor (UF, i.e. 10 for interspecies and 10 for intraspecies differences; EFSA, 2012) or higher, using an expert judgement. As an example, an additional UF may be considered to allow for the possible higher sensitivity to aneuploidy induction of germ cells, and especially oocytes, compared to somatic cells, as suggested by limited experimental data (Kirkland et al., 2019). Establishing an acute reference dose is appropriate because aneugenicity is an acute effect and exhibits a steep dose–response relationship at doses higher than the breakpoint. In this situation, unless there is a legal requirement, establishing a chronic HBGV (e.g. ADI) is not considered necessary as the reference point for the acute HBGV would be the lowest from all the relevant reference points in the database and so any chronic HBGV based on another effect would be higher.
5.2.2. Data‐rich substances, in vivo data are not sufficient to set up a dose–response relationship for aneugenicity
If it has not been possible to identify doses associated with aneugenicity in vivo, for example if the mammalian erythrocyte MN assay is negative with insufficient evidence of exposure to the bone marrow, then the first approach is to seek additional data to follow up on the in vitro data. If that is not an option, or in vivo dose–response data for aneugenicity cannot be obtained, then a HBGV should not be established and a margin of exposure approach (MOE) for risk characterisation should be considered, as described in Section 5.3.
5.2.3. Data‐poor substances, i.e. gaps in the toxicological database
In vivo aneugenicity data might or might not be available but overall, the data are insufficient for establishment of an HBGV, one of the other approaches described in Section 5.3 might be applied.
5.3. Risk characterisation for substances that are aneugenic but not clastogenic nor causing gene mutations
If an HBGV (e.g. ADI/TDI, ARfD) has been established following the approaches described in Section 5.2, then the ADI or ARfD is compared with the estimated chronic or acute dietary exposure, in line with the commonly accepted risk assessment paradigm.
If the data are insufficient to establish an HBGV due to the absence of sufficient dose‐response data for aneugenicity and/or the absence of other key toxicological data, the MOE approach should be used to assess human risk. Similarly to establishing an HBGV, the entire toxicological database and its limitations should be taken into account. The MOE should be calculated based on the lowest reference point from the available in vivo toxicological studies and interpreted taking into account the overall uncertainties in the toxicological database. For data‐poor chemical substances, the reference point might be from a subchronic study. The concentration or dose range for which aneugenicity was observed needs to be considered and the overall uncertainties should be assessed in determining the size of MOE that is not expected to be a safety concern, using expert judgement. This would normally be larger than the default uncertainty factor (UF) of 100 applied to allow for interspecies and intraspecies differences because of the additional uncertainties related to gaps in the database, such as absence of NOAEL or key toxicological studies. As described in the EFSA guidance on default values (EFSA, 2012), the preferred option is to request additional data but, if this is not feasible, then ‘the use of an additional UF to take account of the deficiency of a database should be considered on a case‐by-case basis and justified. It is not possible to propose a default value for this UF, as it will be directly dependent on the dataset available’.
In the event that there is no concern for gene mutations and clastogenicity and aneugenicity is observed but no other toxicological data are available, in principle, application of the threshold of toxicological concern (TTC) could be considered. If a reference point for aneugenicity can be identified, then the TTC approach can be applied, provided that the substance does not belong to one of the exclusion categories (EFSA, 2019b). The relevant TTC value can be applied unless the reference point for aneugenicity divided by 100 is lower than the relevant TTC value. The SC stressed that the TTC approach should not be used when toxicological data are available, or for regulated products requiring toxicological data.
For aneugens for which a reference point cannot be identified, the SC notes that the TTC approach cannot be currently recommended. This is because there is insufficient information on chemical structures leading to aneugenicity and whether aneugens are adequately represented in the TTC databases. Therefore, further investigations are needed and no conclusions on the risk to human health can be made.
A further possibility for substances that are aneugenic without metabolic activation (i.e. most likely would have effects at the site of contact) is to compare the concentrations resulting in aneugenicity in vitro with the estimated concentration of the substance in the GIT following ingestion of food or beverage.
The dilution in the upper parts of the GIT can be considered to be small and the concentration to be in the same order of magnitude to the concentrations in food and beverages (ICRP Publication 89, 2002).
Factors which would lower the concentration of the substance in the oesophagus and stomach and which should be taken into consideration are (1) possible dilution by the presence of other foods/beverages at the time of consumption and (2) whether the substance is likely to be altered by gastric juices.
If there is no presence of other foods/beverages and if the substance is stable, it is assumed that the concentration in the oesophagus and stomach is likely to be of the same order of magnitude as the concentrations in food and beverages. If the concentration in the oesophagus/stomach is estimated to be of the same order of magnitude as the concentrations shown to be aneugenic in vitro, there is concern for aneugenicity in vivo.
The SC noted that comparison with in vitro data is suggested as a pragmatic approach to obtain information on the possibility of effects at the first site of contact when the approaches proposed above are not feasible.
5.4. Risk assessment for combined exposure to multiple substances that are aneugenic but not clastogenic nor causing gene mutations
For simultaneous exposure to multiple aneugenic agents, the default assumption of dose additivity (EFSA, 2019a) should be used. The possibility of combination effects of aneugenic compounds with the same mechanism of action has been demonstrated in vitro by analysis of micronuclei induced by a mixture of seven benzimidazoles acting by binding to the colchicine‐binding site on tubulin monomers and disrupting microtubule polymerisation. Additive effects were observed with the mixture of seven compounds at their estimated threshold concentrations as predicted according to the principles of concentration addition (Ermler et al., 2013). Studies on combined exposure to aneugens acting by different modes of action have not been found. The combined effects should be addressed case by case following the EFSA guidance (EFSA, 2019a).
6. Conclusions
The SC emphasises that this guidance applies specifically to substances that are aneugenic, but do not induce clastogenicity or gene mutations. Based on the information on mechanisms and testing methods on aneugenicity, the current limitations of in vivo genotoxicity assays (EFSA, 2017b) and recent developments in genotoxicity risk characterisation, the SC considered that the questions presented in the Terms of Reference (ToR) of this opinion can be addressed as follows:
6.1. Question 1: What is the most appropriate in vivo follow‐up for substances that are aneugenic in vitro?
The preferred approach commences with an in vivo mammalian erythrocyte MN test, conducted with a relevant route of administration and in accordance with OECD TG 474 (OECD). Evidence that the bone marrow is exposed to the test substance should be obtained following the EFSA opinion (EFSA, 2017b). There are three possible outcomes to this test:
A positive result, which confirms that the substance is aneugenic in vivo and risk assessment is required.
A negative result in a study testing an acceptable maximum dose with evidence of exposure to the bone marrow. This supports the conclusion that the aneugenic activity observed in vitro is not expressed in vivo, and aneugenicity does not raise a safety concern (see Section 3).
A negative result in a study testing an acceptable maximum dose without evidence of exposure to the bone marrow. This is viewed as an inconclusive result and it is considered prudent to regard the substance as potentially active also under in vivo conditions, unless further follow‐up studies are performed.
Aneugenic substances are frequently active without requiring metabolic activation and therefore are likely to act at the first site of contact. There is no validated in vivo test that detects aneugenicity at the first site of contact. Progress has been made in the development of the MN assay in the GIT. However, further validation is required before the development of an OECD guideline.
The SC noted that at present the MN test in the GIT cannot be recommended as a routine test in the follow‐up of substances that are aneugenic in vitro. However, data generated by applying the MN test in the GIT (stomach and colon) will be considered by EFSA case‐by‐case in the comprehensive evaluation of all data to assess the in vivo hazard of substances aneugenic in vitro without metabolic activation.
For aneugenic substances that produce in vitro positive results in the presence of metabolic activation appropriate follow‐up could include a liver MN assay. The SC notes that there is as yet no OECD TG for the liver MN assay, but that it is sufficiently validated for development of a guideline (see also Section 3).
The SC suggests that consideration be given to combining analysis micronuclei in erythrocytes and in liver or GIT of the same animals to minimise use of experimental animals, cost and time. The MN analysis in erythrocytes has also the potential to be integrated into repeat‐dose toxicity studies, providing that the highest dose applied is consistent with the requirements of the OECD TG for MN test (OECD TG 474) and/or ICH criteria.
For a positive aneugenic response in vivo, or if a conclusion on in vivo aneugenic potential cannot be drawn based on the available data, the evaluation should continue with risk characterisation (Question 2).
6.2. Question 2: How should risk to human health be assessed for a substance exhibiting aneugenicity?
There is a general consensus that genotoxic agents acting by non‐DNA‐reactive mechanisms exhibit a thresholded, non‐linear dose–response (Elhajouji et al., 2011). Therefore, the first step in risk assessment for an aneugenic substance is to identify a suitable reference point, to be used in risk characterisation.
Reference points for aneugenicity should be identified by analysis of dose–response data for induction of micronuclei from in vivo studies, if available. The preferred approach is BMD modelling following the EFSA guidance (EFSA, 2017a) and selection of the BMR needs to be justified. If BMD modelling is not possible, a breakpoint may be identified as the reference point using expert judgement.
The subsequent steps of risk assessment should be determined by the completeness of the dataset for the substance under consideration.
For substances with a comprehensive toxicological database (data‐rich substances), for which dose‐response data for aneugenicity are available, this should be compared with the reference points for other toxicity endpoints, to identify the most sensitive effect. If the lowest dose resulting in aneugenicity in vivo is higher than that for other toxic effects, acute and chronic HBGVs can be established using the commonly accepted principles (EHC 240).
If the reference point for aneugenicity in vivo is lower than or close to those for other effects, an acute HBGV (i.e. an ARfD) can be established from the reference point by applying the 100‐fold default uncertainty factor (UF, i.e. 10 for interspecies and 10 for intraspecies differences) or higher, using an expert judgment. Establishing an acute reference dose is appropriate because aneugenicity is an acute effect and exhibits a steep dose–response relationship at doses higher than the breakpoint. In this situation, unless there is a legal requirement, establishing a chronic HBGV (e.g. ADI) is not considered necessary as the reference point for the acute HBGV would be the lowest from all the relevant reference points in the database and so any chronic HBGV based on another effect would be higher. In this case, the acute HBGV may also be applied as a chronic HBGV, such as an ADI, as any chronic HBGV based on another effect would be higher than the ARfD based on aneugenicity reference point. Risk characterisation follows the commonly accepted paradigm of comparing the estimated acute and chronic exposure to the ARfD and ADI, respectively (this applies to data‐rich substances with dose‐response data for aneugenicity, see Section 5.2.1).
If a reference point for aneugenicity can be identified, but no toxicological data other than genotoxicity are available, the TTC approach could be considered, as described in Section 5.3. If a reference point for aneugenicity cannot be identified, the TTC approach cannot be currently recommended and the safety assessment cannot be completed without further investigations.
If it has not been possible to identify doses associated with aneugenicity in vivo, then the first approach is to seek additional data. If that is not an option, or in vivo dose–response data for aneugenicity cannot be obtained, then it is not possible to establish an HBGV, and an alternative approach may be considered (and justified) based on the available data and expert judgement. Possible approaches include:
A margin of exposure (MOE) approach applied to the reference point for another toxicological end‐point, taking into account the uncertainties in the entire toxicological database. The size of MOE that is not expected to be a safety concern, should be assessed using expert judgement and would normally be larger than the default UF of 100 applied to allow for interspecies and intraspecies differences in setting HBGVs, because of the additional uncertainties related to gaps in the database.
Comparison of the concentrations resulting in aneugenicity in vitro, with the concentration of substance estimated to be present in the GIT following ingestion of a food or beverage might provide additional information for site of contact exposure (see Section 5.3)
7. Recommendations
The SC recommends further development and validation of the GIT and liver MN test, including establishing OECD TG.
In addition, research to improve understanding and evaluation of aneugenicity is recommended, including:
Investigation of potential structure–activity relationships for aneugens;
investigation of the applicability of the TTC approach to aneugens;
quantitative comparison of aneugenic activity of model compounds in immortalised cell lines and human primary cells;
development and validation of in vitro 3D models for GIT;
development and validation of AOPs to be used in risk characterisation;
investigation of the relative sensitivity of rodent and human somatic and germ cells to aneuploidy induced by model compounds;
comparison of plasma or tissue levels associated with lowest effective doses in vivo with lowest effective concentrations in vitro (for the calibration of in vitro versus in vivo data).
Annex A – Methods to detect aneuploidy
1.
Aneuploidy is the result of malsegregation events, namely chromosome loss and chromosome non‐disjunction, which give rise to changes in chromosome number. For chromosome loss, the chromosome lost during cell division may lead to the formation of an MN or may be re‐incorporated in one of the two daughter nuclei randomly forming monosomic, trisomic or diploid cells. Chromosome non‐disjunction results in one of the two daughter nuclei becoming trisomic and the other one monosomic. Methods are available to detect these effects in mitotic and interphase cells by microscopy analysis.
A.1. Detection of aneuploidy in mitotic cells
In mitotic cells, condensed chromosomes can be easily visualised and counted to determine whether increase (hyperploidy) or reduction (hypoploidy) of their number are induced. To allow the expression of the aneugenic effect of a treatment, the analysis needs to be restricted to metaphase cells that have divided twice in culture (Parry et al., 1995). In addition, only hyperploid cells should be recorded to avoid the inclusion of false aneuploid cells resulting from the mechanical loss of chromosomes during the preparation of the slides.
Besides chromosome counting methods, also the study of abnormalities in cell division through the investigation of mitotic spindle structure and chromosome segregation can be indicative of a potential aneugenic activity. The analysis of the spindle apparatus can be achieved by use of specific staining procedures involving a fixation technique to maintain the integrity of the spindle apparatus and differential staining to visualise proteins of the spindle and chromosomes. In particular, the application of this visualisation technique is suitable for the quantification of the so‐called C‐metaphases, namely cells lacking a mitotic spindle, typically resulting from the action of inhibitors of the spindle apparatus, such as colchicine (Parry and Parry, 1987). Conversely, analysis of chromosomes at anaphase makes it possible to identify lagging chromosomes. Single lagging chromosomes or chromatids can be detected in bipolar ana‐telophases, while more severe damage can be detected in cells at anaphase, i.e. C‐anaphases (Minissi et al., 1999).
A.2. Detection of aneuploidy in interphase cells
A.2.1. Analysis of binucleated cells
Acentric chromosome fragments or whole chromosomes not included in the daughter nuclei at the end of mitosis may give rise to micronuclei when the cells enter subsequent interphase. The analysis of micronuclei in interphase cells allows faster and easier measurement of chromosome damage compared with the count of chromosomes particularly when the cytokinesis‐block assay is applied. This is a method that improves the accuracy and sensitivity of the MN test, focusing the analysis on cells that have divided only once after treatment (Fenech, 2007). These cells can be detected as binucleated cells using the inhibitor of cytokinesis cytochalasin B (CytB), which prevents the separation of the cytoplasm into two daughter cells after mitosis and leads to the formation of cells with two nuclei within a cytoplasm (binucleated cells).
As the origin of micronuclei is associated with the induction of both structural and numerical aberrations, it is necessary to characterise the content of micronuclei to clarify the mechanism of their induction; here, micronuclei containing whole chromosomes are considered the result of an aneugenic event, while micronuclei containing acentric chromosome fragments are related to clastogenic activity. Several methods have been applied to discriminate aneugens from clastogens, such as the size‐classified counting of micronuclei based on the assumption that increased frequency of large‐sized micronuclei could be an alerting index for aneugenic effects; in addition, C‐banding was applied as well as measurement of DNA content in micronuclei (Parry et al., 2002). However, the use of molecular cytogenetic methods allows a more precise and reliable classification of micronuclei.
Here, the approaches most widely used to distinguish between clastogenic and aneugenic effects are based on the detection of the centromere into the MN by immunochemical labelling of kinetochores using anti‐kinetochore antibodies obtained from the serum of patients with the autoimmune disease scleroderma, formerly called CREST (calcinosis, Raynaud's phenomenon, oesophageal dysmotility, sclerodactyly and telangiectasia) syndrome, (CREST analysis) and FISH with DNA probes specific for centromeric regions (FISH analysis). Occasionally CREST staining could falsely identify micronuclei with whole chromosomes as micronuclei containing acentric chromosome fragments when the test compound: (i) causes the detachment of the kinetochore; (ii) produces an alteration of the kinetochore epitope targeted by the CREST‐antibody; and (iii) binds to the kinetochore epitope, interfering with antibody recognition. The interaction of substances such as caffeine and mitomycin C with the kinetochore and consequences for reliability of the CREST analysis has been known for decades (Miller et al., 1991). However, while the specific characteristics of the two techniques should be taken into account in the evaluation of the data, both CREST and FISH can be considered reliable tools to obtain information on the origin of micronuclei and the mechanisms of genotoxicity, as also indicated in OECD TG 487 (OECD, 2016a).
A.2.2. Simultaneous detection of chromosome loss and non‐disjunction
The relative contribution of chromosome loss and non‐disjunction to aneuploidy induction can be investigated following the segregation of single chromosomes in the two daughter nuclei at the completion of mitosis; this can be achieved applying the FISH technique with centromeric DNA probes for a specific chromosome in association with the cytokinesis‐block MN assay (Zijno et al., 1996). By this approach it is possible to recognise the correct segregation of an autosomic chromosome when two fluorescent spots per nucleus are observed in the binucleated cells (two diploid cells), while malsegregation events are identified by an unbalanced distribution of the specific chromosome. For non‐disjunction, one nucleus contains three spots (hyperploid cell) and the other nucleus one spot (hypoploid cell). Conversely, chromosome loss is identified when a nucleus contains two spots (diploid), the other nucleus one fluorescent spot (hypoploid) and the MN one spot, corresponding to the lost chromosome. With this method, technical artefacts can be excluded from the total counting as only binucleated cells with the expected number of labelled chromosomes are included in the analysis independently of the distribution of the spots in the two daughter nuclei.
A.2.3. Detection of monosomy and trisomy in mononucleated cells
The in situ hybridisation technique, DNA probes complementary to centromeric sequences of specific chromosomes have also been applied to easily estimate the frequency of monosomic and trisomic cells (Eastmond and Pinkel, 1990). This procedure allows the measurement of aneuploidy induction by counting the number of labelled signals, representing the chromosome of interest. Simultaneous hybridisation with adjacent double‐labelled pericentromeric probes (Eastmond et al., 1993) has been proposed to partially overcome any technical limitations that may reduce the reliability of this approach. Indeed, elevated frequency of apparently trisomic cells has been reported and associated with non‐specific hybridisation or breakage within the hybridisation region (Eastmond and Pinkel, 1990). In addition, the original method could be relatively insensitive for detection of hypoploidy, like most of the classical cytogenetic techniques, because overlap of two spots may occur resulting in visualisation of a single signal. Another limitation is related to the application of this approach to cell culture in the absence of cytB. The inability to distinguish subpopulations of cells that have divided once from the others, in fact, may introduce a bias in the assessment of aneugenic effects.
A.2.4. Battery of tests for the identification of aneugens
An additional approach to identify an aneugenic effect could rely on the comparison, in the same experimental system, of results obtained in the MN test and those obtained in assays that measure only DNA strand breaks (chromosome aberrations and comet assays). Positive results obtained in both the MN and DNA strand breaks assays would be indicative of clastogenic activity. In contrast, negative results in the DNA strand break assays associated with a positive MN test are indicative that MN formation is due to the loss of chromosomes. The subsequent characterisation of the content of MN by FISH analysis will confirm the induction of aneuploidy and elucidate the mechanism of malsegregation underneath.
A.2.5. Detection of aneuploidy in mononucleated cells
It is generally recommended to restrict the analysis of micronuclei to binucleate cells, but some studies have reported that aneugens increase the frequency of micronuclei in mononucleate and binucleate cells, while clastogens induce the formation of micronuclei only in binucleated cells (Elhajouji et al., 1995; Kirsch‐Volders and Fenech, 2001; Dutta et al., 2007; Kirkland, 2010). The unknown origin of micronuclei in mononucleated cells, however, represents a strong limitation for the reliability of this approach.
A.3. Micronucleus assay techniques in tissues other than bone marrow and peripheral blood
The mammalian erythrocyte MN test (OECD 474, 2016b), which is performed on bone marrow or peripheral blood may not detect substances or their metabolites that do not reach the target cells or which are too short lived to reach the target cells. Therefore, several attempts have been made in the past to perform the MN assay in liver and other tissues.
Amendments to the MN assay have already been developed several years ago (see e.g. Barbason et al., 1975; Tates et al., 1980; Braithwaite and Ashby, 1988; Das and Roy, 1988; Cliet et al., 1989; Uryvaeva and Delone, 1995; Parton and Garriott, 1997; Igarashi and Shimada, 1997; etc.). Most studies were carried out using MN assays in liver (reviewed by Morita et al., 2011; Uno et al., 2015a) but there are also studies on several other organs including spleen (Krishna et al., 1990; Benning et al., 1992; Benning et al., 1994), oesophagus (Mehta et al., 1987), stomach and colon (reviewed by Uno et al., 2015b).
As proliferating cells are required for the MN assay and hepatocyte turnover is slow, several methods have been developed for stimulation of hepatocyte proliferation (Uno, 2013; Uno et al., 2015a). Partial hepatectomy was initially used to stimulate hepatocyte proliferation, e.g. by Barbason et al. (1975) and Tates et al. (1980). Chemical mitogens were later used by Braithwaite and Ashby (1988) and Uryvaeva and Delone (1995). However, this method seems to be no longer in use, as the mitogens might interfere with the genotoxic response (Uno, 2013; Martus et al., 2015; Uno et al., 2015a). Parton and Garriott (1997) developed a MN assay using juvenile rats without partial hepatectomy or chemical mitogens considering that the percentage of hepatocytes in S‐phase is about 5% in 4‐week‐old rats, while it is much less in adult rats. More recently, a MN assay using young (6‐week‐old) adult rats and repeated‐dose administration (for 14 or 28 days) have been developed based on the observation that micronuclei could accumulate in the liver of adult rats (Narumi et al., 2012, 2013; Takasawa et al., 2013).
A Collaborative Study Group of the Micronucleus Test (CSGMT), a subgroup of the Mammalian Mutagenicity Study Group (MMS), of the Japanese Environmental Mutagen Society (JEMS), organised several collaborative studies to evaluate MN methods detecting chromosomal aberrations in the liver and the GIT (Ohyama et al., 2002; Suzuki et al., 2005; Suzuki et al., 2009; Takasawa et al., 2010a,b; Hamada et al., 2015).
Micronucleus assays in tissues other than bone marrow and peripheral blood were discussed at the 2nd, 4th, 6th and 7th International Workshop on Genotoxicity Test Procedures (IWGTP) under the umbrella of the IAEMS in 1999, 2005, 2013 and 2017, respectively (Hayashi et al., 2000, 2007; Uno, 2013; Uno et al., 2015a,b; Kirkland et al., 2019). The strengths and limitations of these methods and the need for further data were described in the report of the 6th IWGTP (Uno et al., 2015a,b). Since then, further studies have been performed on specific aspects of these methods addressing the issues identified at the 6th IWGTP (e.g. Itoh et al., 2015; Shimada et al., 2015; Shigano et al., 2016; Avlasevich et al., 2018; Hori et al., 2019; Itoh and Hattori, 2019).
In 2013, the IWGT working group recommended protocols for the liver MN test (LMNT), agreed on the most likely situations in which this method would be applicable and ‘reached consensus that the LMNT is a useful and promising assay for regulatory use’ (Uno et al., 2015a). At that time, the Working Group, however, also pointed out that further actions for development will be needed for the liver MN assay (Uno et al., 2015a). Based on additional data, the IWGT Working Group concluded in 2017 that the liver MN assay is sufficiently validated for the development of an OECD guideline and that for the GIT MN assay, some evaluation of the sensitivity and specificity is possible. However, the Working Group also noted that more work is required for an OECD guideline for the GIT MN test (Kirkland et al., 2019).
Overall, among the MN assays in tissues other than bone marrow and peripheral blood, the repeated‐dose liver MN assay in young (6‐week‐old) adult rats reached the highest level of development and validation. It belongs to the assays for which internationally agreed protocols have already been set up and it can be combined with repeated‐dose general toxicity studies and with other repeated‐dose genotoxicity assays such as the bone marrow/peripheral blood MN test (OECD, 2016b). In addition, there is some evidence that it can also be combined with a gene mutation assay (Hori et al., 2019). The available data suggest that the GIT MN test could be integrated into general toxicity studies. However, additional data are required for the evaluation of its sensitivity and specificity.
A.4. Other in vitro assays
MultiFlow DNA Damage Assay and ToxTracker
Cell‐based assays, such as MultiFlow DNA Damage Assay and ToxTracker, considering additional endpoints to the induction of micronuclei and the characterisation of their content have been proposed. In particular, the in vitro MultiFlow DNA Damage assay integrates multiple biomarkers, such as gamma H2AX, p53, phospho‐histone H3 and polyploidisation, into a single flow cytometric analysis to distinguish between aneugenic and clastogenic mode of action (Bryce et al., 2016). It has also been described a tiered bioassay and data analysis scheme allowing to investigate some of the molecular targets possibly affected by aneugenic compounds, i.e. tubulin destabilisation, tubulin stabilisation and inhibition of Aurora kinase B (Derek et al., 2019).
ToxTracker is a mouse stem cell‐based reporter assay that is reported to identify genotoxic compounds by the activation of specific cellular signalling responses allowing to discriminate between the induction of DNA damage, oxidative stress and protein damage in a single test (Hendriks et al., 2016). Genotoxicity is indicated by the activation of two reporter genes responding to the formation of bulky DNA adducts or DNA double‐strand breaks. The differential activation of these two genotoxicity reporters has been applied in ToxTracker to further differentiate between a clastogenic and an aneugenic mode of action in association with cell cycle and polyploidy analysis by flow cytometry (Brandsma et al., 2020).
The SC considers that these assays are not yet sufficiently developed for regulatory purposes.
References
Avlasevich S, Labash C, Torous D, Bemis J, MacGregor J and Dertinger S, 2018. In vivo pig‐a and micronucleus study of the prototypical aneugen vinblastine sulfate. Environmental Molecular Mutagenesis, 59, 30–37.
Barbason H, Fridman‐Manduzio A and Betz EH, 1975. Long term effects of a single dose of dimethylnitrosamine on the rat liver. [Z Krebsforsch Klin Onkol] Journal of Cancer Research and Clinical Oncology, 84, 135–142.
Benning V, Depasse F, Melcion C and Cordier A, 1992. Detection of micronuclei after exposure to mitomycin C, cyclophosphamide and diethylnitrosamine by the in vivo micronucleus test in mouse splenocytes. Mutation Research, 280, 137–142.
Benning V, Brault D, Duvinage C, Thybaud V and Melcion C, 1994. Validation of the in vivo CD1 mouse splenocyte micronucleus test. Mutagenesis, 9, 199–204.
Braithwaite I and Ashby J, 1988. A non‐invasive micronucleus assay in the rat liver. Mutation Research, 203, 23–32.
Brandsma I, Moelijker N, Derr R and Hendriks G, 2020. Aneugen versus clastogen evaluation and oxidative stress‐related mode‐of‐ action assessment of genotoxic compounds using the ToxTracker reporter assay.
Bryce SM, Bernacki DT, Bemis JC and Dertinger SD, 2016. Genotoxic mode of action predictions from a multi‐ plexed flow cytometric assay and a machine learning ap‐ proach. Environmental Molecular Mutagenesis, 57, 171–189.
Cliet I, Fournier E, Melcion C and Cordier A, 1989. In vivo micronucleus test using mouse hepatocytes. Mutation Research, 216, 321–326.
Das RK and Roy B, 1988. A simplified method for micronucleus preparation from hepatic cells. Stain Technology, 63, 71–74.
Bernacki DT, Bryce SM, Bemis JC and Dertinger SD, 2019. Aneugen molecular mechanism assay: proof‐of‐concept with 27 reference chemicals. Toxicological Sciences, 170, 382–393.
Dutta D, Sundaram SK and Teeguarden JG, 2007. Adsorbed proteins influence the biological activity and molecular targeting of nanomaterials. Toxicological Sciences, 100, 303–315.
Eastmond DA and Pinkel D, 1990. Detection of aneuploidy and aneuploidy‐inducing agents in human lymphocytes using fluorescence in situ hybridization with chromosome‐specific dna probes. Mutation Research, 234, 303–318.
Eastmond DA, Rupa DS, Chen H and Hasegawa LS, 1993. Multicolor fluorescence in situ hybridization with centromeric DNA probes as a new approach to distinguish chromosome breakage from aneuploidy in interphase cells and micronuclei. In: Vig BK (eds.), Chromosome Segregation and Aneuploidy. NATO ASI Series (Series H: Cell Biology), 72, 377–390. Springer, Berlin, Heidelberg.
Elhajouji A, Van Hummelen P and Kirsch‐Volders M, 1995. Indications for a threshold of chemically induced aneuploidy in vitro in human lymphocytes. Environmental and Molecular Mutagenesis, 26, 292–304.
Fenech M, 2007. Cytokinesis‐block micronucleus cytome assay. Nature Protocols, 2, 1084–1104.
Hamada S, Ohyama W, Takashima R, Shimada K, Matsumoto K, Kawakami S, Uno F, Sui H, Shimada Y, Imamura T, Matsumura S, Sanada H, Inoue K, Muto S, Ogawa I, Hayashi A, Takayanagi T, Ogiwara Y, Maeda A, Okada E, Terashima Y, Takasawa H, Narumi K, Wako Y, Kawasako K, Sano M, Ohashi N, Morita T, Kojima H, Honma M and Hayashi M, 2015. Evaluation of the repeated‐dose liver and gastrointestinal tract micronucleus assays with 22 chemicals using young adult rats: summary of the collaborative study by the Collaborative Study Group for the Micronucleus Test (CSGMT)/The Japanese Environmental Mutagen Society (JEMS) – Mammalian Mutagenicity Study Group (MMS). Mutation Research Genetic Toxicology and Environmental Mutagenesis, 780–781, 2–17.
Hayashi M, MacGregor JT, Gatehouse DG, Adler ID, Blakey DH, Dertinger SD, Krishna G, Morita T, Russo A and Sutou S, 2000. In vivo rodent erythrocyte micronucleus assay. II. Some aspects of protocol design including repeated treatments, integration with toxicity testing, and automated scoring. Environmental and Molecular Mutagenesis, 35, 234–252.
Hayashi M, MacGregor JT, Gatehouse DG, Blakey DH, Dertinger SD, Abramsson‐Zetterberg L, Krishna G, Morita T, Russo A, Asano N, Suzuki H, Ohyama W, Gibson D; and the In Vivo Micronucleus Assay Working Group, IWGT, 2007. In vivo erythrocyte micronucleus assay III. Validation and regulatory acceptance of automated scoring and the use of rat peripheral blood reticulocytes, with discussion of non‐hematopoietic target cells and a single dose‐level limit test. Mutation Research, 627, 10–30.
Hendriks G, Derr RS, Misovic B, Morolli B, Calléja FMGR and Vrieling H, 2016. The extended toxtracker assay discriminates between induction of dna damage, oxidative stress, and protein misfolding. Toxicological Sciences, 150, 190–203.
Hori H, Shimoyoshi S, Tanaka Y, Momonami A, Masumura K, Yamada M, Fujii W and Kitagawa Y, 2019. Integration of Micronucleus Tests with a Gene Mutation Assay in F344 Gpt Delta Transgenic Rats Using Benzo[a]pyrene. Mutation Research, 837, 1–7.
Igarashi M and Shimada H, 1997. An improved method for the mouse liver micronucleus test. Mutation Research, 391, 49–55.
Itoh S and Hattori C, 2019. In vivo genotoxicity of 1,4‐dioxane evaluated by liver and bone marrow micronucleus tests and Pig‐a assay in rats. Mutation Research, 837, 8–14.
Itoh S, Igarashi M, Nagata M and Hattori C, 2015. Assessment of a twice dosing regimen both before and after partial hepatectomy in the rat liver micronucleus test. Mutation Research Genetic Toxicology and Environmental Mutagenesis, 782, 18–23.
Kirkland D, 2010. Evaluation of different cytotoxic and cytostatic measures for the in vitro micronucleus test (MNVit): summary of results in the collaborative trial. Mutation Research, 702, 139–147.
Kirkland D, Uno Y, Luijten M, Beevers C, van Benthem J, Burlinson B, Dertinger S, Douglas GR, Hamada S, Horibata K, Lovell DP, Manjanatha M, Martus HJ, Mei N, Morita T, Ohyama W and Williams A, 2019. In vivo genotoxicity testing strategies: report from the 7th International workshop on genotoxicity testing (IWGT). Mutation Research, 847, 403035.
Kirsch‐Volders M and Fenech M, 2001. Inclusion of micronuclei in nondivided mononuclear lymphocytes and necrosis/apoptosis may provide a more comprehensive cytokinesis block micronucleus assay for biomonitoring purposes. Mutagenesis, 16, 51–58.
Krishna G, Kropko ML and Theiss JC, 1990. Dimethylnitrosamine‐induced micronucleus formation in mouse bone marrow and spleen. Mutation Research, 242, 345–351.
Martus HJ, Hayashi M, Honma M, Kasper P, Gollapudi B, Mueller L, Schoeny R, Uno Y and Kirkland DJ, 2015. Summary of major conclusions from the 6th International Workshop on Genotoxicity Testing (IWGT), Foz do Iguaçu, Brazil. Mutation Research Genetic Toxicology and Environmental Mutagenesis, 783, 1–5.
Mehta R, Silinskas KC, Zucker PF, Ronen A, Heddle JA and Archer MC, 1987. Micronucleus formation induced in rat liver and esophagus by nitrosamines. Cancer Letters, 35, 313–320.
Miller BM, Zitzelsberger HF, Weier H‐UG and Adler I‐D, 1991. Classification of micronuclei in murine erythrocytes: immunofluorescent staining using CREST antibodies compared to in situ hybridization with biotinylated gamma satellite DNA. Mutagenesis, 6, 297–302.
Minissi S, Gustavino B, Degrassi F, Tanzarella C and Rizzoni M, 1999. Effect of cytochalasin B on the induction of chromosome mis‐segregation by colchicine at low concentrations in human lymphocytes. Mutagenesis, 14, 43–49.
Morita T, MacGregor JT and Hayashi M, 2011. Micronucleus assays in rodent tissues other than bone marrow. Mutagenesis, 26, 223–230.
Narumi K, Ashizawa K, Takashima R, Takasawa H, Katayama S, Tsuzuki Y, Tatemoto H, Morita T, Hayashi M and Hamada S, 2012. Development of a repeated‐dose liver micronucleus assay using adult rats: an investigation of diethylnitrosamine and 2,4‐diaminotoluene. Mutation Research, 747, 234–239.
Narumi K, Ashizawa K, Fujiishi Y, Tochinai R, Okada E, Tsuzuki Y, Tatemoto H, Hamada S, Kaneko K and Ohyama W, 2013. Persistence and accumulation of micronucleated hepatocytes in liver of rats after repeated administration of diethylnitrosamine. Mutation Research, 755, 100–107.
OECD (Organisation for Economic Co‐operation and Development), 2016a. Test No. 487: In Vitro Mammalian Cell Micronucleus Test, OECD Guidelines for the Testing of Chemicals, Section 4, OECD Publishing. Available online: https://doi.org/10.1787/9789264224438-en
OECD (Organisation for Economic Co‐operation and Development), 2016b. Test No. 474: Mammalian Erythrocyte Micronucleus Test, OECD Guidelines for the Testing of Chemicals, Section 4, OECD Publishing. Available online: https://doi.org/10.1787/9789264264762-en
Ohyama W, Gonda M, Miyajima H, Kondo K, Noguchi T, Yoshida J, Hatakeyama S, Watabe E, Ueno Y, Hayashi M and Tokumitsu T, 2002. Collaborative validation study of the in vivo micronucleus test using mouse colonic epithelial cells. Mutation Research, 518, 39–45.
Parry EM, Henderson L, Mackay JM, 1995. Procedures for the detection of chemically induced aneuploidy: recommendations of a UK environmental mutagen society working group. Mutagenesis, 10, 1–14.
Parry EM, Parry JM, Corso C, Doherty A, Haddad F, Hermine TF, Johnson G, Kayani M, Quick E, Warr T and Williamson J, 2002. Detection and characterization of mechanisms of action of aneugenic chemicals. Mutagenesis, 17, 509–521.
Parry JM and Parry EM, 1987. Comparisons of Tests for Aneuploidy. Mutation Research, 181, 267–287.
Parton JW and Garriott ML, 1997. An evaluation of micronucleus induction in bone marrow and in hepatocytes isolated from collagenase perfused liver or from formalin‐fixed liver using four‐week‐old rats treated with known clastogens. Environmental and Molecular Mutagenesis, 29, 379–385.
Shigano M, Takashima R, Takasawa H and Hamada S, 2016. Optimization of specimen preparation from formalin‐fixed liver tissues for liver micronucleus assays: Hepatocyte staining with fluorescent dyes. Mutation Research Genetic Toxicology and Environmental Mutagenesis, 800–801, 35–39.
Shimada K, Yamamoto M, Takashima M, Seki J, Miyamae Y and Wakata A, 2015. Prolonged rest period enables the detection of micronucleated hepatocytes in the liver of young adult rats after a single dose of diethylnitrosamine or mitomycin C. Mutation Research Genetic Toxicology and Environmental Mutagenesis, 791, 38–41.
Suzuki H, Ikeda N, Kobayashi K, Terashima Y, Shimada Y, Suzuki T, Hagiwara T, Hatakeyama S, Nagaoka K, Yoshida J, Saito Y, Tanaka J and Hayashi M, 2005. Evaluation of liver and peripheral blood micronucleus assays with 9 chemicals using young rats. A study by the Collaborative Study Group for the Micronucleus Test (CSGMT)/Japanese Environmental Mutagen Society (JEMS) – Mammalian Mutagenicity Study Group (MMS). Mutation Research, 583, 133–145.
Suzuki H, Takasawa H, Kobayashi K, Terashima Y, Shimada Y, Ogawa I, Tanaka J, Imamura T, Miyazaki A and Hayashi M, 2009. Evaluation of a liver micronucleus assay with 12 chemicals using young rats (II): a study by the Collaborative Study Group for the Micronucleus Test/Japanese Environmental Mutagen Society‐Mammalian Mutagenicity Study Group. Mutagenesis, 24, 9–16.
Takasawa H, Suzuki H, Ogawa I, Shimada Y, Kobayashi K, Terashima Y, Matsumoto H, Aruga C, Oshida K, Ohta R, Imamura T, Miyazaki A, Kawabata M, Minowa S and Hayashi M, 2010a. Evaluation of a liver micronucleus assay in young rats (III): a study using nine hepatotoxicants by the Collaborative Study Group for the Micronucleus Test (CSGMT)/Japanese Environmental Mutagen Society (JEMS)‐Mammalian Mutagenicity Study Group (MMS). Mutation Research, 698, 30–37.
Takasawa H, Suzuki H, Ogawa I, Shimada Y, Kobayashi K, Terashima Y, Matsumoto H, Oshida K, Ohta R, Imamura T, Miyazaki A, Kawabata M, Minowa S, Maeda A and Hayashi M, 2010b. Evaluation of a liver micronucleus assay in young rats (IV): a study using a double‐dosing/single‐sampling method by the Collaborative Study Group for the Micronucleus Test (CSGMT)/Japanese Environmental Mutagen Society (JEMS)‐Mammalian Mutagenicity Study Group (MMS). Mutation Research, 698, 24–29.
Takasawa H, Takashima R, Hattori A, Narumi K, Kawasako K, Morita T, Hayashi M and Hamada S, 2013. Development of a repeated‐dose liver micronucleus assay using adult rats (II): further investigation of 1,2‐dimethylhydrazine and 2,6‐diaminotoluene. Mutation Research, 751, 12–18.
Tates AD, Neuteboom I and Hofker M, 1980. A micronucleus technique for detecting clastogenic effects of mutagens/carcinogens (DEN, DMN) in hepatocytes of rat liver in vivo. Mutation Research, 74, 11–20.
Uno Y, 2013. Liver Micronucleus Test (LMNT) Subgroup, 6th International Workshop on Genotoxicity Testing, Iguassu, Brazil. Available online: http://www.iaemgs.org/IWGT2013_Liver_MNT_summary.pdf
Uno Y, Morita T, Luijten M, Beevers C, Hamada S, Itoh S, Ohyama W and Takasawa H, 2015a. Recommended protocols for the liver micronucleus test: Report of the IWGT working group. Mutation Research Genetic Toxicology and Environmental Mutagenesis, 783, 13–18.
Uno Y, Morita T, Luijten M, Beevers C, Hamada S, Itoh S, Ohyama W and Takasawa H, 2015b. Micronucleus test in rodent tissues other than liver or erythrocytes: Report of the IWGT working group. Mutation Research Genetic Toxicology and Environmental Mutagenesis, 783, 19–22. (review)
Uryvaeva IV and Delone GV, 1995. An improved method of mouse liver micronucleus analysis: an application to age‐related genetic alteration and polyploidy study. Mutation Research, 334, 71–80.
Annex B – Examples of active substances (possibly) causing aneugenic effects
1.
Annex C – List of Abbreviations
1.
| ADI | Acceptable Daily Intake |
| ADME | Absorption, Distribution, Metabolism, Excretion |
| AMKL | Acute Megakaryoblastic Leukaemia |
| ARfD | Acute Reference Dose |
| BMD | Benchmark Dose |
| BMDL | Lower confidence Limit of the Benchmark Dose |
| BMR | Benchmark Response |
| CDKs | Cyclin‐dependent kinases |
| cGAMP or cyclic GMP–AMP | Cyclic guanosine monophosphate–adenosine monophosphate |
| cGAS | Cyclic guanosine monophosphate–adenosine monophosphate synthase |
| CREST | Calcinosis, Raynaud's phenomenon, oesophageal dysmotility, sclerodactyly and telangiectasia |
| CSGMT | Collaborative Study Group of the Micronucleus Test |
| CytB | Cytochalasin B |
| FISH | Fluorescence in situ hybridisation |
| GIT | Gastrointestinal Tract |
| GIT MN assay | Gastrointestinal Tract Micronucleus assay |
| HBGV | Health‐Based Guidance Value |
| IWGT | International Workshop on Genotoxicity Test |
| IWGTP | International Workshop on Genotoxicity Test Procedures |
| JEMS | Japanese Environmental Mutagen Society |
| MMS | Mammalian Mutagenicity Study |
| MN | Micronucleus/micronuclei |
| MOE | Margin of Exposure |
| NOAEL | No Observed Adverse Effect Level |
| NOEL | No Observed Effect Level |
| OECD | Organisation for Economic Co‐operation and Development |
| OECD TG 471 | Bacteria reverse mutation text guideline (AMES test) |
| OECD TG 474 | The mammalian erythrocyte micronucleus test guideline |
| OECD TG 487 | In vitro mammalian cell micronucleus test guideline |
| OECD TG 488 | Transgenic rodent assay |
| OECD TG 489 | In vivo mammalian alkaline comet assay |
| SC | Scientific Committee |
| TDI | Tolerable Daily Intake |
| ToR | Terms of Reference |
| TTC | Threshold of Toxicological Concern |
| UF | Uncertainty Factor |
| WHO | World Health Organization |
Suggested citation: EFSA Scientific Committee , More SJ, Bampidis V, Bragard C, Halldorsson TI, Hernández‐Jerez AF, Hougaard Bennekou S, Koutsoumanis K, Lambré C, Machera K, Naegeli H, Nielsen SS, Schlatter J, Schrenk D, Turck D, Younes M, Aquilina G, Bignami M, Bolognesi C, Crebelli R, Gürtler R, Marcon F, Nielsen E, Vleminckx C, Carfì M, Martino C, Maurici D, Parra Morte J, Rossi A and Benford D, 2021. Scientific Opinion on the guidance on aneugenicity assessment. EFSA Journal 2021;19(8):6770, 27 pp. 10.2903/j.efsa.2021.6770
Requestor: European Food Safety Authority
Question number: EFSA‐Q‐2019‐00262
Panel members: Simon John More, Diane Benford, Vasileios Bampidis, Claude Bragard, Thorhallur Ingi Halldorsson, Antonio F Hernández‐Jerez, Susanne Hougaard Bennekou, Kostas Koutsoumanis, Claude Lambré, Kyriaki Machera, Hanspeter Naegeli, Søren Saxmose Nielsen, Josef Schlatter, Dieter Schrenk, Dominique Turck and Maged Younes.
Declarations of interest: The declarations of interest of all scientific experts active in EFSA's work are available at https://ess.efsa.europa.eu/doi/doiweb/doisearch.
Acknowledgements: The EFSA Scientific Committee wishes to acknowledge the following hearing experts for the views provided to this scientific output: Roland Froetschl, Bodo Haas, Uta Herbst, David Kirkland, Christina Tlustos; the observer Frank Le Curieux (European Chemicals Agency ‐ ECHA); the experts Maria Dusinska and Ursula Gunder‐Remy for the critical review of the manuscript; the EFSA staff Sara De Berardis, Christina Kyrkou and Esraa Elewa.
Adopted: 1 July 2021
This publication is linked to the following EFSA Supporting Publications article: http://onlinelibrary.wiley.com/doi/10.2903/sp.efsa.2021.EN-6814/full
Note
EFSA Scientific Committee, 2011. Scientific Opinion on Genotoxicity Testing Strategies Applicable to Food and Feed Safety Assessment. EFSA Journal 2011;9(9):2379, 69 pp. https://doi.org/10.2903/j.efsa.2011.2379
References
- Adler ID, Schmid TE and Baumgartner A, 2002. Induction of aneuploidy in male mouse germ cells detected using the sperm‐FISH assay: a review of the present data base. Mutation Research, 504, 173–182. [DOI] [PubMed] [Google Scholar]
- Bakhoum SF, Ngo B, Laughney AM, Cavallo JA, Murphy CJ, Ly P, Shah P, Sriram RK, Watkins TBK, Taunk NK, Duran M, Pauli C, Shaw C, Chadalavada K, Rajasekhar VK, Genovese G, Venkatesan S, Birkbak NJ, McGranahan N, Lundquist M, LaPlant Q, Healey JH, Elemento O, Chung CH, Lee NY, Imielenski M, Nanjangud G, Pe'er D, Cleveland DW, Powell SN, Lammerding J, Swanton C and Cantley LC, 2018. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature, 553, 467–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgartner A, Schmid TE, Schuetz CG and Adler ID, 2001. Detection of aneuploidy in rodent and human sperm using multicolor FISH after chronic exposure to diazepam. Mutation Research, 490, 11–19. [DOI] [PubMed] [Google Scholar]
- Bentley KS, Kirkland D, Murphy M and Marshall R, 2000. Evaluation of thresholds for benomyl‐ and carbendazim‐induced aneuploidy in cultured human lymphocytes using fluorescence in situ hybridization. Mutation Research, 464, 41–51. [DOI] [PubMed] [Google Scholar]
- Cammerer Z, Schumacher MM, Kirsch‐Volders M, Suter W and Elhajouji A, 2010. Flow cytometry peripheral blood micronucleus test in vivo: determination of potential thresholds for aneuploidy induced by spindle poisons. Environmental and Molecular Mutagenesis, 51, 278–284. [DOI] [PubMed] [Google Scholar]
- EFSA (European Food Safety Authority), 2012. Guidance on selected default values to be used by the EFSA Scientific Committee, Scientific Panels and Units in the absence of actual measured data. EFSA Journal 2012;10(3):2579, 32 pp. 10.2903/j.efsa.2012.2579 Available online: https://www.efsa.europa.eu/en/efsajournal/pub/2579 [DOI] [Google Scholar]
- EFSA (European Food Safety Authority), 2017a. Update: use of the benchmark dose approach in risk assessment. EFSA Journal 2017;15(1):4658, 55 pp. 10.2903/j.efsa.2017.4658 Available online: https://www.efsa.europa.eu/it/efsajournal/pub/4658 [DOI] [Google Scholar]
- EFSA (European Food Safety Authority), 2017b. Clarification of some aspects related to genotoxicity assessments. EFSA Journal 2017;15(12):5113, 25 pp. 10.2903/j.efsa.5113 Available online: https://efsa.onlinelibrary.wiley.com/doi/full/10.2903/j.efsa.2017.5113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA (European Food Safety Authority), 2019a. Guidance on harmonised methodologies for human health, animal health and ecological risk assessment of combined exposure to multiple chemicals. EFSA Journal 2019;17(3):5634, 77 pp. 10.2903/j.efsa.2019.5634 Available online: https://www.efsa.europa.eu/it/efsajournal/pub/5634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA (European Food Safety Authority), 2019b. Guidance on the use of the threshold of toxicological concern approach in food safety assessment. EFSA Journal 2019;17(6):5708, 17 pp. 10.2903/j.efsa.2019.5708 Available online: https://www.efsa.europa.eu/en/efsajournal/pub/5708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA Scientific Committee , 2011. Scientific Opinion on genotoxicity testing strategies applicable to food and feed safety assessment. EFSA Journal 2011;9(9):2379, 69 pp. 10.2903/j.efsa.2011.2379 Available online: https://www.efsa.europa.eu/en/efsajournal/pub/2379 [DOI] [Google Scholar]
- EFSA Scientific Committee , 2015. Scientific Opinion: guidance on the review, revision and development of EFSA's Cross‐cutting Guidance Documents. EFSA Journal 2015;13(4):4080, 11 pp. 10.2903/j.efsa.2015.4080 [DOI] [Google Scholar]
- Elhajouji A, Van Hummelen P and Kirsch‐Volders M, 1995. Indications for a threshold of chemically‐induced aneuploidy in vitro in human lymphocytes. Environmental and Molecular Mutagenesis, 26, 292–304. [DOI] [PubMed] [Google Scholar]
- Elhajouji A, Tibaldi F and Kirsch‐Volders M, 1997. Indication for thresholds of chromosome non‐disjunction versus chromosome lagging induced by spindle inhibitors in vitro in human lymphocytes. Mutagenesis, 12, 133–140. [DOI] [PubMed] [Google Scholar]
- Elhajouji A, Lukamowicz M, Cammerer Z and Kirsch‐Volders M, 2011. Potential thresholds for genotoxic effects by micronucleus scoring. Mutagenesis, 26, 199–204. [DOI] [PubMed] [Google Scholar]
- Elloway JM, Davies AK, Hayes JE and Doherty AT, 2017. From the cover: does the assessment of nondisjunction provide a more sensitive assay for the detection of aneugens? Toxicological Sciences, 157, 20–29. [DOI] [PubMed] [Google Scholar]
- Ermler S, Scholze M and Kortenkamp A, 2013. Seven benzimidazole pesticides combined at sub‐threshold levels induce micronuclei in vitro . Mutagenesis, 28, 417–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Ni J, Liang Z, Xue J, Fenech MF and Wang X, 2019. The molecular origins and pathophysiological consequences of micronuclei: new insights into an age‐old problem. Mutation Research, 779, 1–35. [DOI] [PubMed] [Google Scholar]
- Hayashi M, 2016. The micronucleus test‐most widely used in vivo genotoxicity test. Genes and Environment: The Official Journal of the Japanese Environmental Mutagen Society, 38, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi M, MacGregor JT, Gatehouse DG, Adler ID, Blakey DH, Dertinger SD, Krishna G, Morita T, Russo A and Sutou S, 2000. In vivo rodent erythrocyte micronucleus assay. II. Some aspects of protocol design including repeated treatments, integration with toxicity testing, and automated scoring. Environmental and Molecular Mutagenesis, 35, 234–252. [PubMed] [Google Scholar]
- Hayashi M, MacGregor JT, Gatehouse DG, Blakey DH, Dertinger SD, Abramsson‐Zetterberg L, Krishna G, Morita T, Russo A, Asano N, Suzuki H, Ohyama W and Gibson D; and the In Vivo Micronucleus Assay Working Group, IWGT, 2007. In vivo erythrocyte micronucleus assay III. Validation and regulatory acceptance of automated scoring and the use of rat peripheral blood reticulocytes, with discussion of non‐hematopoietic target cells and a single dose‐level limit test. Mutation Research, 627, 10–30. [DOI] [PubMed] [Google Scholar]
- ICRP (International Commission on Radiological Protection), 2002. Basic Anatomical and Physiological Data for Use in Radiological Protection Reference Values. ICRP Publication 89. Ann. ICRP 32 (3‐4). [PubMed]
- Kirkland D, Uno Y, Luijten M, Beevers C, van Benthem J, Burlinson B, Dertinger S, Douglas GR, Hamada S, Horibata K, Lovell DP, Manjanatha M, Martus HJ, Mei N, Morita T, Ohyama W and Williams A, 2019. In vivo genotoxicity testing strategies: report from the 7th International workshop on genotoxicity testing (IWGT). Mutation Research, 847. [DOI] [PubMed] [Google Scholar]
- Kirsch‐Volders M, Vanhauwaert A, De Boeck M and Decordier I, 2002. Importance of detecting numerical versus structural chromosome aberrations. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis, 504, 137–148. [DOI] [PubMed] [Google Scholar]
- Kirsch‐Volders M, Pacchierotti F, Parry EM, Russo A, Eichenlaub‐Ritter U and Adler ID, 2019. Risks of aneuploidy induction from chemical exposure: twenty years of collaborative research in Europe from basic science to regulatory implications. Mutation Research, 779, 126–147. [DOI] [PubMed] [Google Scholar]
- Lynch AM, Eastmond D, Elhajouji A, Froetschl R, Kirsch‐Volders M, Marchetti F, Masumura K, Pacchierotti F, Schuler M and Tweats D, 2019. Targets and mechanisms of chemically induced aneuploidy. Part 1 of the Report of the 2017 IWGT workgroup on assessing the risk of aneugens for carcinogenesis and hereditary diseases. Mutation Research, 847, 403025. [DOI] [PubMed] [Google Scholar]
- MacGregor JT, Frötschl R, White PA, Crump KS, Eastmond DA, Fukushima S, Guérard M, Hayashi M, Soeteman‐Hernández LG, Kasamatsu T, Levy DD, Morita T, Müller L, Schoeny R, Schuler MJ, Thybaud V and Johnson GE, 2015. IWGT report on quantitative approaches to genotoxicity risk assessment I. Methods and metrics for defining exposure–response relationships and points of departure (PoDs). Mutation Research Genetic Toxicology and Environmental Mutagenesis, 783, 55–65. [DOI] [PubMed] [Google Scholar]
- Marshall RR, Murphy M, Kirkland DJ and Bentley KS, 1996. Fluorescence in situ hybridisation with chromosome‐specific centromeric probes: a sensitive method to detect aneuploidy. Mutation Research, 372, 233–245. [DOI] [PubMed] [Google Scholar]
- Martus HJ, Hayashi M, Honma M, Kasper P, Gollapudi B, Mueller L, Schoeny R, Uno Y and Kirkland DJ, 2015. Summary of major conclusions from the 6th International Workshop on Genotoxicity Testing (IWGT), Foz do Iguaçu, Brazil. Mutation Research Genetic Toxicology and Environmental Mutagenesis, 783, 1–5. [DOI] [PubMed] [Google Scholar]
- Naylor RM and van Deursen JM, 2016. Aneuploidy in cancer and aging. Annual Review of Genetics, 50, 45–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OECD (Organisation for Economic Co‐operation and Development), 2016. Test No. 474: Mammalian Erythrocyte Micronucleus Test, OECD Guidelines for the Testing of Chemicals, Section 4, OECD Publishing. Available at: 10.1787/9789264264762-en [DOI] [Google Scholar]
- Pacchierotti F, Masumura K, Eastmond DA, Elhajouji A, Froetschl R, Kirsch‐Volders M, Lynch A, Schuler M, Tweats D and Marchetti F, 2019. Chemically induced aneuploidy in germ cells. Part II of the report of the 2017 IWGT workgroup on assessing the risk of aneugens for carcinogenesis and hereditary diseases. Mutation Research, 848, 403023. [DOI] [PubMed] [Google Scholar]
- Tweats DJ, Johnson GE, Scandale I, Whitwell J and Evans DB, 2016. Genotoxicity of flubendazole and its metabolites in vitro and the impact of a new formulation on in vivo aneugenicity. Mutagenesis, 31, 309–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tweats D, Eastmond DA, Lynch AM, Elhajouji A, Froetschl R, Kirsch‐Volders M, Marchetti F, Masumura K, Pacchierotti F and Schuler M, 2019. Role of aneuploidy in the carcinogenic process: part 3 of the report of the 2017 IWGT workgroup on assessing the risk of aneugens for carcinogenesis and hereditary diseases. Mutation Reserach, 847. [DOI] [PubMed] [Google Scholar]
- Uno Y, 2013. Liver Micronucleus Test (LMNT) Subgroup, 6th International Workshop on Genotoxicity Testing. Iguassu, Brazil. Available online: http://www.iaemgs.org/IWGT2013_Liver_MNT_summary.pdf [Google Scholar]
- Uno Y, Morita T, Luijten M, Beevers C, Hamada S, Itoh S, Ohyama W and Takasawa H, 2015a. Recommended protocols for the liver micronucleus test: report of the IWGT working group. Mutation Research Genetic Toxicology and Environmental Mutagenesis, 783, 13–18. [DOI] [PubMed] [Google Scholar]
- Uno Y, Morita T, Luijten M, Beevers C, Hamada S, Itoh S, Ohyama W and Takasawa H, 2015b. Micronucleus test in rodent tissues other than liver or erythrocytes: report of the IWGT working group. Mutation Research Genetic Toxicology and Environmental Mutagenesis, 783, 19–22. [DOI] [PubMed] [Google Scholar]
- WHO (World Health Organization), 2009. Principles and methods for the risk assessment of chemicals in food. Environmental health criteria, 240. ISBN: 9789241572408. [Google Scholar]
- Zijno A, Marcon F, Leopardi P and Crebelli R, 1996. Analysis of chromosome segregation in cytokinesis‐blocked human lymphocytes: non‐disjunction is the prevalent damage resulting from low‐dose exposure to spindle poisons. Mutagenesis, 11, 335–340. [DOI] [PubMed] [Google Scholar]