

Abstract
Protein–protein interactions are vital to biological processes, but the shape and size of their interfaces makes them hard to target using small molecules. Cyclic peptides have shown promise as protein–protein interaction modulators, as they can bind protein surfaces with high affinity and specificity. Dozens of cyclic peptides are already FDA-approved, and many more are in various stages of development as immunosuppressants, antibiotics, antivirals, or anticancer drugs. However, most cyclic peptide drugs so far have been natural products or derivatives thereof, with de novo design having proven challenging. A key obstacle is structural characterization: cyclic peptides frequently adopt multiple conformations in solution, which are difficult to resolve using techniques like NMR spectroscopy. The lack of solution structural information prevents a thorough understanding of cyclic peptides’ sequence–structure–function relationship. Here we review recent development and application of molecular dynamics simulations with enhanced sampling to studying the solution structures of cyclic peptides. We describe novel computational methods capable of sampling cyclic peptides’ conformational space and provide examples of computational studies that relate peptides’ sequence and structure to biological activity. We demonstrate that molecular dynamics simulations have grown from an explanatory technique to a full-fledged tool for systematic studies at the forefront of cyclic peptide therapeutic design.
Graphical Abstract
1. Introduction
1.1. Cyclic peptides
In the study of amino acid polymers, linear peptides and proteins could be described as the “default”. The ubiquity of linear peptides and protein chains in biological systems, as well as their high level of involvement in biological processes, have led to these systems being immensely well-studied. However, peptides certainly come in other “flavors” – and the biological and medicinal utility of cyclic peptides, in which the peptide chain is closed (e.g. head-to-tail, or via a disulfide bond), is becoming increasingly well-recognized.1 Many cyclic peptide hormones participate in important signaling pathways – somatostatin,2 vasopressin,3,4 and oxytocin,4,5 to name a few. Cyclic peptide natural products such as cyclosporin A (an immunosuppressant),6–8 gramicidin S (an antibiotic),9,10 and vancomycin (an antibiotic),11 have also found important medicinal use. The biosynthesis of cyclic peptides occurs along diverse pathways, being either non-ribosomal or following a series of post-translational modifications on precursor proteins.12–15
The unique and interesting properties of cyclic peptides have captured many scientists’ and pharmaceutical companies’ attention.1 With sizes situated in between those of small molecules and antibodies, peptides can selectively and effectively target receptors and modulate protein–protein interactions. However, owing to their structural flexibility, linear peptides typically have relatively poor affinity, selectivity, and bioavailability. In contrast, cyclic peptides are more rigid and exhibit desirable druglike properties, including high affinity for protein surfaces, increased specificity, and improved enzyme stability. Over the last several decades, more than 40 cyclic peptide therapeutics have been approved for clinical use by the FDA and the EMA, with dozens more in clinical trials or early stages of design.16–18 However, an overwhelming majority of cyclic peptide therapeutics currently on the market or undergoing testing are natural products or their derivatives.16,19 This group includes some of the best-known cyclic peptide drugs, such as cyclosporin A, isolated from Tolypocladium inflatum, or vancomycin, biosynthesized by Amycolatopsis orientalis. It is important to note that relatively few well-characterized natural cyclic peptides remain available for research, and fewer still can be easily adapted to new functions. On the other hand, many protein–protein interactions have been identified as potential therapeutic targets and the ability to use cyclic peptides to modulate these protein–protein interactions will provide a transformative means to control cellular functions for both fundamental research and therapeutic intervention. De novo design of cyclic peptides, however, has proven substantially more difficult, especially in comparison to the design of small molecules and antibodies.1 One of the primary challenges encountered, in both cyclic peptide drug design and the study of naturally occurring cyclic peptides, is an inability to establish the cyclic peptides’ sequence–structure relationships.
1.2. The elusive solution structures of cyclic peptides
When designing inhibitors for protein–protein interactions, occasionally the structures of the target interactions are available. In some cases, X-ray structures of cyclic peptides bound to their target proteins may exist.20 This information on the “end-point” or desired conformations should, in principle, enable structure-based rational design of excellent cyclic peptide binders and inhibitors. However, such a strategy requires that we also know the solution structures adopted by cyclic peptides to understand how different modifications change the peptide structure and their binding affinity for the target. While X-ray structures may be obtained for some cyclic peptides,21–86 they can be different from the solution structures, meaning that the X-ray structures of cyclic peptides cannot be used as surrogates of their solution structures.81,87–90 While solution NMR spectroscopy is the standard method to derive a structural model for a molecule, cyclic peptides typically exhibit few relevant NOE cross-peaks due to their low core-to-surface ratio, and in the case of N-methylated residues, which are commonly observed in cyclic peptide natural products and a popular means to improve membrane permeability, important NH–Hα couplings are absent altogether.91 Even more importantly, cyclic peptides tend to adopt multiple conformations in solution, making the development of a structural model using NMR spectroscopy extremely difficult, if not impossible, as most protocols assume the existence of a single conformation and collect time- and ensemble-averaged data.92–107 This issue isn’t specific to synthetic peptides either. The study of cyclic peptide hormones and natural products can also be complicated by limited utility of NMR in structure determination of disordered molecules.108–112 The inability to experimentally observe and distinguish the relevant conformations of a cyclic peptide in solution prevents scientists from both understanding and optimizing natural cyclic peptides and establishing a clear sequence–structure–activity relationship for de novo cyclic peptide designs. In this review, we focus on exactly this fundamental obstacle to the rational design of cyclic peptides: their elusive solution structures.
1.3. Simulations of cyclic peptides
Because of the therapeutic potential of cyclic peptides, many computational design platforms have been adopted to treat cyclic peptides. Tools like I-TASSER,113 PEP-FOLD,114–116 Peplook,117 PEPstrMOD,118 OMEGA,119,120 MacroModel,121 Rosetta,122–124 BRIKARD,125 or EGSCyP,126 to name a few, can be used to generate many structures for docking. However, the behavior of a peptide in a biological system can only be adequately predicted if we know its solution structural ensemble, necessitating solvated simulations, particularly using explicit solvent. Experiments and simulations both show that solvent plays a critical role in cyclic peptide structures – at times, even water molecules bridged or caged within a cyclic peptide have been observed.88,127–131 Because of their small size, closed topology, and the abundance of solvent-exposed H-bond donors and acceptors, accurate modeling of cyclic peptides is difficult using implicit-solvent models, which do not account for these consequential and direct interactions with solvent molecules.132
Molecular dynamics (MD) simulations are a powerful tool to understand the properties of peptides and proteins.133–163 In MD simulation, water molecules can be explicitly incorporated to accurately describe the solution behaviors. However, because explicit-water MD simulations are computationally expensive, they are rarely used in peptide design, which requires screening of many variants. Furthermore, when it comes to cyclic peptides in particular, their ring strain slows down the dynamics, making it difficult to sample cyclic peptides’ free-energy landscape effectively using MD simulation.
To address the sampling issues, a number of methods have been applied to or developed specifically for cyclic peptides to enhance their conformational sampling. As enhanced sampling in MD simulations is a topic of considerable breadth, in this review we describe in detail only those methods used in simulation work featured in this review. Following this theoretical overview, we review the latest studies focused on developing or using MD simulation with enhanced sampling as a powerful strategy to study the solution structures of cyclic peptides. Herein we define cyclic peptides as molecules that are solely or mostly composed of peptide moieties, linked via head-to-tail or other cyclization, and display no regular secondary structures of an α helix or a β sheet. In Section 2 we first discuss several enhanced sampling techniques used for sampling cyclic peptide conformations. In Section 3 we focus on the development of simulation methods, evaluation of force fields, and general approaches to modeling the solution structures of cyclic peptides. In Section 4 we review recent applications of MD simulations with enhanced sampling methods to understand or design cyclic peptides for specific targets. A summary focused on the computational performance of the cyclic peptide simulations discussed here is provided in Table 1 at the end of the review as well.
Table 1.
Summary of performance of enhanced sampling methods on cyclic peptides.
| Sec | System | Method1 | Solvent | #Replicas | Length per Rep | Total Length | Convergence Criteria | Converged? | Ref |
|---|---|---|---|---|---|---|---|---|---|
| 3.2 | Cyclo-(PSIDV) | cMD | Explicit | 1 | 1 μs | 1 μs | N/A | No | 201 |
| aMD | Explicit | 1 | 1 μs | 1 μs | N/A | N/A | |||
| Cyclo-(RRWWRF) | aMD | Explicit | 1 | 1 μs | 1 μs | N/A | N/A | ||
| Cyclo-(RGDfV) | aMD | Explicit | 20 | 50 ns | 1 μs | N/A | N/A | ||
| 3.3 | Cyclosporin A | cMD | Explicit | 10 | 2 ns | 20 ns | Ensemble diversity | 2224 confs | 196 |
| aMD | Explicit | 10 | 2 ns | 20 ns | 5912confs | ||||
| CoCo-MD | Explicit | 10 | 2 ns | 20 ns | 9822 confs | ||||
| cMD | Explicit | 100 | 100 ns | 10 μs | N/A | N/A | 214 | ||
| 3.4 | 20 cyclic peptides (Fig. 9) | REMD | Explicit | 24–32 | 100–200 ns | ≥2.4 μs | Block analysis2 | Yes | 221 |
| 3.5 | Cyclo-(YNPFEEGG) | REMD | Explicit | 51–59 | 300 ns | ≥15.3 μs | Two independent trajs3 | Yes and No4 | 171 |
| BE-META | Explicit | 18 | 300 ns | 5.4 μs | Yes | ||||
| cMD | Explicit | 1 | 500 ns | 500 ns | No | ||||
| 3.6 | Cyclo-(aAAAAA), cyclo-(aAAAAA) | BE-META | Explicit | 17 | 100–250 ns | ≥1.7 μs | Two independent trajs3 | Yes | 239 |
| 3.7 | Cyclo-(GGGGG), cyclo-(X1X2AAA)5 | BE-META | Explicit | 15 | 100–300 ns | ≥1.5 μs | Two independent trajs3 | Yes | 242 |
| 3.8 | Cyclo-(GnA6−n), cyclo-(GnV6−n) | BE-META | Explicit | 17 | 100 ns | 1.7 μs | Two independent trajs3 | Yes | 243 |
| 3.9 | Cyclo-(sarcosine8) | REMD | Implicit | ~24 | 500 ns | ~12 μs | N/A | N/A | 251 |
| Cyclo-(Nspe)9 | REMD | Implicit | 15 | 1 μS | 15 μs | N/A | N/A | 128 | |
| 3.10 | 8 cyclic hexapeptides (Fig. 15) | McMD | Explicit | 336 | 20 ns | 6.72 μs | Reached desired flat potential energy landscape | Yes | 190 |
| 3.11 | Disulfide-bonded tetrapeptides (Fig. 16) | REMD | Explicit | 23 | 60–120 ns | ≥1.38 μs | N/A | N/A | 131, 272, 274 |
| 3.12 | Cyclo-(GHGAYG), cyclo-(GRCTKSIPPICFPD) | PTWTE | Explicit | 7 | >300 ns | >2.1 μs | Following the cumulative average of the radius of gyration | Yes | 281 |
| 4.1 | α-Fetoprotein-derived cyclic peptides | REMD | Implicit | 8 | 20 ns | 160 ns | N/A | N/A | 283 |
| 4.2 | Apelin-derived cyclic peptides | REMD | Explicit | 16 | 25–35 ns | ≥400 ns | N/A | N/A | 303 |
| 4.3 | LapD-derived cyclic peptides | REMD | Implicit | 24 | 1–2 μs | ≥24 μs | N/A | N/A | 305 |
| 4.4 | Lens epithelium-derived growth factor-derived cyclic peptides | WT-META | Explicit | 1 | 100 ns | 100 ns | N/A | N/A | 312 |
| 4.5 | Vasopressin | cMD | Explicit | 1 | 11 μs | 11 μs | Author assertion | No | 323 |
| WT-META | Explicit | 4 walkers | 200 ns | 800 ns | Author assertion | Yes | 324 | ||
| Oxytocin/vasopressin systems (Fig. 20) | R-REMD | Explicit | 24 | 50 ns | 1.2 μs | Two independent trajs6 | Yes | 325 | |
| Urotensin II | cMD | Explicit | 1 | 35 μs | 35 μs | Author assertion | No | 340 | |
| REMD | Explicit | 64 | 500 ns | 32 μs | Three independent trajs7 | Yes | |||
| 4.6 | Photoswitch-embedded cyclic peptides (Fig. 22) | REMD | United-atom DMSO | 28 | 10 ns | 280 ns | Monitoring changes in Ramachandran plots8 | Yes | 354 |
| 4.7 | 18 RGD-related cyclic pentapeptides (Fig. 23) | REMD | Implicit | 8 | 2.4 μs | 19.2 μs | Author assertion | Yes | 370 |
| 5 RGD-related cyclic hexapeptides (Fig. 24) | BE-META | Explicit | 6 | 320 ns | 1.92 μs | Block analysis9 | Yes | 236 | |
| 4 RGD or isoDGR-related disulfide-bondec cyclic peptides (Fig. 25) | REMD | Explicit | 16 | 2 ns | 32 ns | N/A | N/A | 378 | |
| Cilengitide and two isoDGR-related disulfide-bonded cyclic peptides (Fig. 26) | WT-META | Explicit | 1 | 10 ns | 10 ns | Comparing to experiments | Yes | 170 | |
| Cyclo-(CGisoDGRG) | BE-META | Explicit | 13 | 30 ns | 390 ns | Block analysis9 | Yes | 381 | |
| 5 isoDGR-related cyclic peptides (Fig. 27) | BE-META | Explicit | 13–15 | 30–60 ns | ≥390 ns | Block analysis9 | Yes | 383 |
WT-META: well-tempered metadynamics; R-REMD: reservoir REMD.
Each trajectory (with the first 10% discarded) was divided into three equal-time-length sections. Convergence was achieved when all sections gave similar distributions of root-mean-square deviations to the corresponding crystal structures.
Simulations starting from the two different structures provided similar conformation density profiles.
The simulations using OPLS-AA/L force field converged after 50 ns; the simulations using AMBER-99SB-ILDN force field did not converge even after 300 ns.
X1, X2 were one of the eight amino acids: A, D, F, G, N, R, S and V.
The convergence was determined by monitoring the difference between the distributions of radius of gyration of two independent 45 ns production runs.
Three REMD simulations starting from different structures gave similar results.
The convergence was determined by comparing the conformational distributions of the longer simulations to 5 ns simulations and observing minor changes.
After an equilibration time, 1-D free-energy profiles obtained from two halves of the simulation were compared; convergence was considered to be obtained if the free-energy profiles were consistent within 2kBT.
2. Molecular dynamics methods
2.1. Replica exchange molecular dynamics (REMD)
First implemented by Sugita and Okamoto164, the replica exchange molecular dynamics (REMD) method simulates multiple copies, i.e. replicas, of the same system simultaneously at a series of temperatures. The exchanges between replicas of neighboring temperatures enable the higher-temperature replica to enhance the sampling of the lower-temperature replica.165 In the simulation process of REMD, two different types of steps are performed as shown in Figure 1: First, all replicas are run independently and simultaneously at their specified temperatures for a certain number of MD steps; then, a pair of replicas at neighboring temperatures are exchanged according to the Metropolis criterion (Eq.1) to ensure that the detailed balance condition is satisfied:
| Eq. 1 |
where A is the exchange probability, kB is the Boltzmann constant; (Ti, Ei) and (Tj, Ej) are the temperatures and potential energies of the two replicas i and j, respectively. The temperature range and the number of replicas to use need to be chosen carefully. The highest temperature must be high enough to ensure that MD simulation at this temperature is not trapped in local energy minima; the lowest temperature is usually near the temperature of interest.166 A variant of REMD named reservoir REMD also includes a reservoir of structures generated beforehand using MD simulations at a high temperature; by allowing replicas at the highest temperature to exchange with this reservoir, the sampling efficiency can be further improved.167
Figure 1.
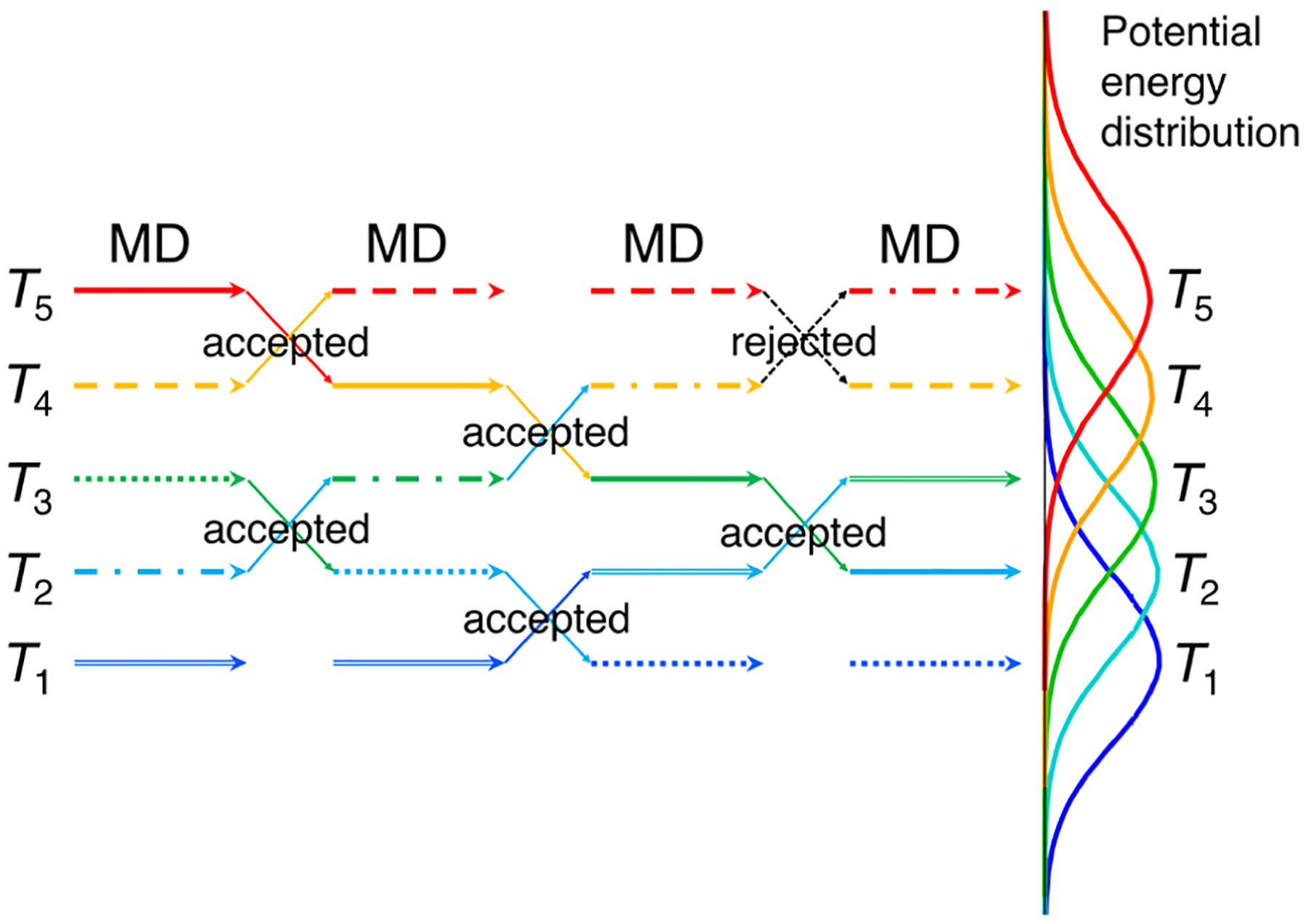
Schematic view of the REMD algorithm.164 An exchange between replicas at different temperatures is attempted every set number of steps of MD simulation. The right side shows the potential energy distributions for a model system at five different temperatures. The potential energy distributions of replicas at adjacent temperatures must have sufficient overlap to ensure reasonable exchange rates.
While REMD is likely one of the most popular enhanced sampling methods, if not the most popular enhanced sampling method, in an REMD run the number of replicas should be chosen carefully so that the potential energy distributions at neighboring temperatures overlap sufficiently to ensure a large enough exchange rate, as is shown in Figure 1.168 As the average potential energy of a system varies as N (the number of particles in the system), while the width of the potential energy distribution is proportional to , the number of replicas one needs for sufficient potential energy overlap increases as . Therefore, a large number of replicas are needed for biological systems that have many particles, which can make running REMD prohibitive. When it comes to using REMD simulations to sample conformations of cyclic peptides, the ring strain of cyclic peptides, in particular those of small sizes, can impose large energy barriers for conformation changes. These large barriers can potentially require very high temperatures in REMD to effectively enhance conformation sampling of cyclic peptides.
2.2. Metadynamics and bias-exchange metadynamics
2.2.1. Metadynamics
Metadynamics is a technique used to enhance sampling in MD simulations. Developed by Laio et al.,169 metadynamics assumes that a few coordinates (typically termed “collective variables”) could provide an essential description of the simulated system. Collective variables used to describe the conformational space of cyclic peptides are usually a number of selected degrees of freedom, such as backbone dihedral angles,169–172 which are assumed to direct the conformational changes of interest and progress along the free-energy landscape. During a 1-D-metadynamics simulation along a given collective variable (s), 1-D Gaussian potentials of defined width (σ) and height (w) centered at s(t) are added at a certain deposition rate (τG) to discourage the system from revisiting the same spot (Figure 2). As the simulation proceeds, more Gaussian potentials are deposited, filling the minima in the free-energy landscape. This process thus creates a history-dependent biasing potential, VG, i.e. the sum of all the previously deposited Gaussians. The full free-energy landscape can be explored and reconstructed as the opposite of VG, once the simulation reaches convergence:173
| Eq. 2 |
Here, x corresponds to the Cartesian coordinates of the atoms in the system; s is the defined collective variable as a function of x; VG is the summation of the Gaussians; τG, w, and σ are the deposition rate, height, and width of the Gaussians, respectively. If more than one collective variable is needed to describe the free-energy landscape, multidimensional metadynamics with n-D Gaussian potentials can be performed.
Figure 2.
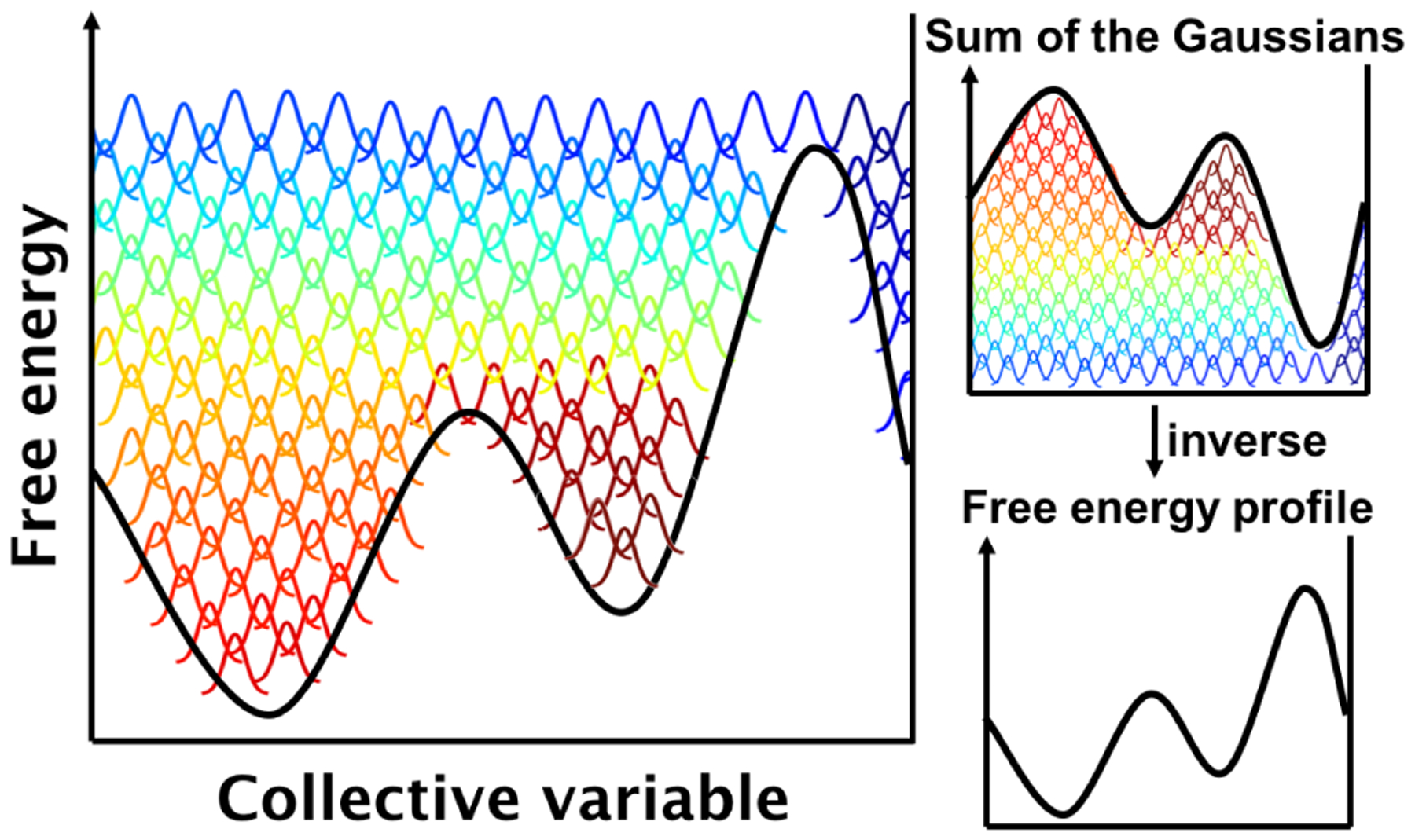
Schematic view of metadynamics.169 Gaussian potentials along a collective coordinate are deposited over time (bell-shaped curves changing from red to green to blue with time) to discourage the system from revisiting the same spot. The free-energy profile can be inferred by adding up the deposited Gaussian hills and inverting the result.
Although metadynamics simulations can be powerful sampling methods, their performance highly depends on the proper choice of collective variables, which is critical, but not always evident a priori. The selection of collective variables is usually based on the researcher’s chemical intuition for the system. Development of algorithms that can help design optimal collective variables is an active research area and it is important to note that finding simple and effective collective variables is nontrivial and can impact the efficiency of metadynamics simulations.174–178
2.2.2. Well-tempered metadynamics
Ordinary metadynamics bears potential challenges: It is often challenging to identify when the simulation has converged, and running the metadynamics simulation excessively long can push the system into an unphysical configurational space.179 In addition, free energy never truly converges to one value; instead, it fluctuates around the correct value, introducing an error proportional to the square root of the bias deposition rate. Lowering the error thus requires decreasing the bias deposition rate and inevitably increases the time it takes for the free-energy surface to be filled.179
To overcome these issues, well-tempered metadynamics was developed.179 With well-tempered metadynamics, the height of the Gaussians decreases with simulation time, allowing a smooth convergence of VG. It is important to note, however, that well-tempered metadynamics still relies on a history-dependent potential bias. Consequently, the presence of slow degrees of freedom outside the chosen collective variables could introduce hidden barriers and statistical errors associated therewith.180 Moreover, under the well-tempered metadynamics scheme, the rate of change of VG(s,t) decreases as 1/t. The idea of well-tempering is that the rate of change decreases fast enough for the bias to converge, but slowly enough that the resulting bias is independent of the initial condition VG(s,0). The proportionality to 1/t makes for mathematically simple scaling, but is not necessarily optimal.181
The ensemble generated by metadynamics or well-tempered metadynamics, i.e. well-tempered ensemble (WTE), can be subjected to further enhanced sampling using parallel tempering (PT).182,183 In parallel tempering, the simulation is run simultaneously using a number of replicas at different temperatures, which exchange similarly to REMD, but with the Metropolis criterion adjusted for the different bias potentials experienced by the replicas:
| Eq. 3 |
where x stands for the coordinates, E is the potential energy, and VG is the bias potential defined on collective variable s. The combined approach of well-tempered metadynamics and parallel tempering is referred to as PTWTE (parallel-tempering well-tempered ensemble).183
2.2.3. Bias-exchange metadynamics (BE-META)
Bias-exchange metadynamics (BE-META) is another variant of metadynamics.173 It is similar to REMD insofar as it also uses several replicas of the system in parallel, as is shown in Figure 3. However, in contrast to REMD, the replicas are simulated at the same temperature, but each replica is biased along different collective variables by time-dependent potentials according to the metadynamics scheme. The conformations of the replicas are exchanged periodically according to the Metropolis criterion (Eq. 4):184
| Eq. 4 |
where kB is the Boltzmann constant; T is the temperature; and are the summation of Gaussians for replica i and j, respectively; and xi and xj are the Cartesian coordinates of the system in replicas i and j before exchanges, respectively. A large number of collective variables can thus be simultaneously biased in this way, which enables efficient exploration of a high-dimensional free-energy space. One or multiple neutral replicas that are not biased by any time-dependent potential can also be added and allowed to exchange with the other replicas. When the ratio of the Gaussian height and the rate of deposition (w/τG) is small, the neutral replicas can be used to approximate the canonical distribution of states and thus can be analyzed to compute any equilibrium properties directly.173
Figure 3.
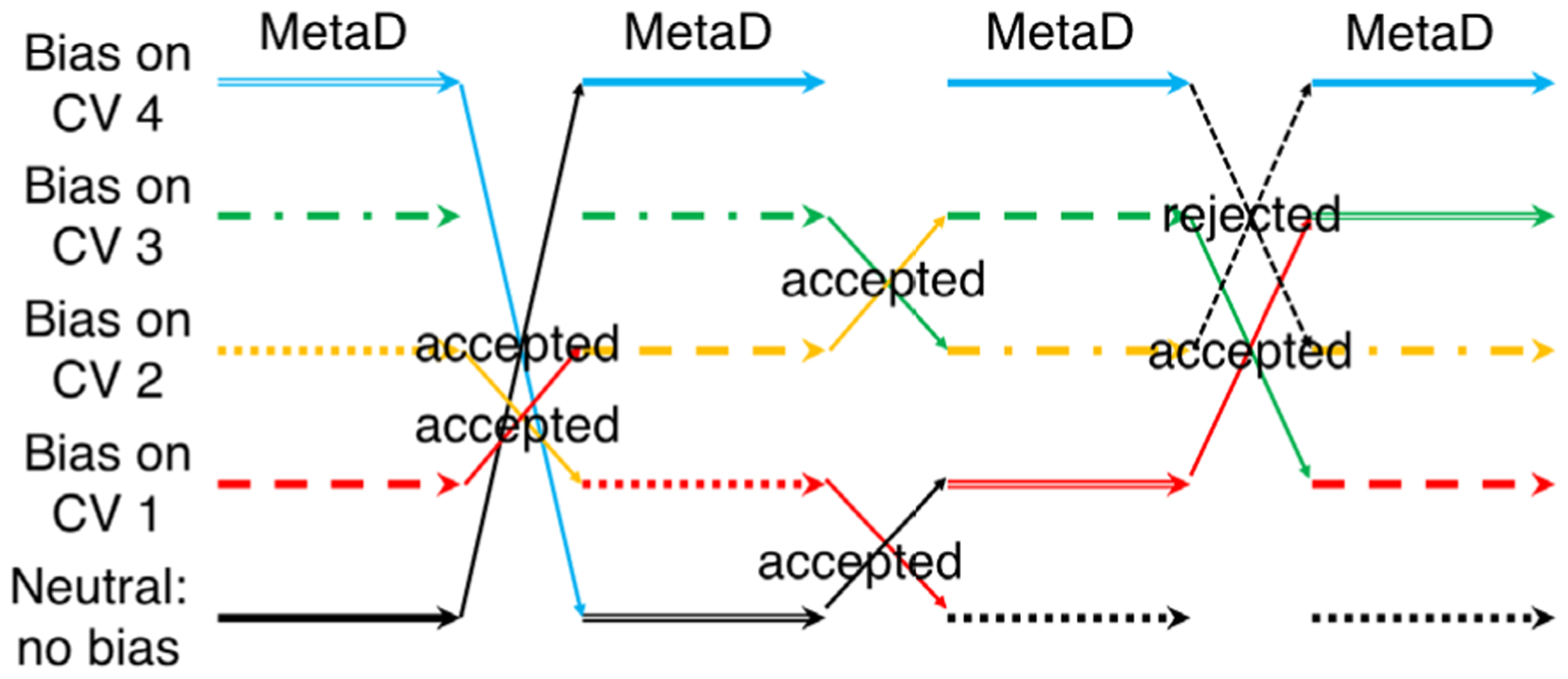
Schematic diagram of the BE-META algorithm.173 Each of the replicas in BE-META (shown in different colors) is a metadynamics (MetaD) simulation biased along a different collective variable (CV). Unlike in REMD, exchanges in BE-META can happen between any two replicas. and are not limited to just the neighboring replicas.
2.3. Multicanonical molecular dynamics (McMD)
In a canonical MD simulation, the conformations of a system are sampled under an NVT ensemble and the partition of all the possible states in thermal equilibrium follows the canonical distribution:
| Eq. 5 |
where E and n(E) are the potential energy and density of states, respectively, and Zc is the canonical partition function: . At a specific temperature T, Pc(E, T) displays a Gaussian-like distribution centered around 〈E〉. At low temperature, the probability of sampling a high-energy state is low, and thus it is difficult for the system to overcome an energy barrier (Figure 4A). At high temperature, Pc(E, T) shifts to higher energy, indicating that the system now samples high-energy states better and has a higher chance of crossing energy barriers (Figure 4A). However, at high temperature the sampling of low-energy states, which are the states most simulators are interested in, is poor. Inspired by the multicanonical Monte Carlo method developed by Berg and Neuhaus,185 Nakajima et al. developed multicanonical MD (McMD) to solve this conundrum, with the goal of enhancing the conformational sampling of peptides.186 McMD aims to flatten the energy landscape by modifying the potential energy of the system to Emc = E + kBTln[Pc(E, T)]. With this modified potential energy, the distribution of states in McMD is
| Eq. 6 |
where . McMD typically starts with a preliminary canonical simulation at a high temperature T0 to obtain Pc(E, T0). If a range of E wider than that sampled by Pc(E, T0) is desired, the modification to the potential energy can be refined to reach a flat sampling of states by iterating over several McMD runs at T0. The canonical distribution Pc(E, T) at any temperature can be obtained by reweighting Pmc(E, T0) (Figure 4B),
| Eq. 7 |
Figure 4.

Schematic view of multicanonical MD.186,190 (A) In a conventional canonical MD, a simulation at low temperature can be trapped in a local minimum while a simulation at high temperature might not sample low-energy conformations that are of interest. (B) In a multicanonical MD simulation, the potential energy of the system is modified to sample conformations with a wide range of potential energy evenly. The canonical distribution at a specific temperature is then obtained by reweighting.
Variant methods of McMD have been developed to further improve sampling. A McMD simulation can be coupled with a virtual system, in a protocol termed V-McMD.187,188 In V-McMD, a few virtual states are initialized with a modified potential energy. The virtual systems are held constant while the real system is in time evolution, and vice versa. Neighboring states’ energy distributions overlap and transition between the states occurs per the Metropolis criterion, similar to REMD. However, the probability of transition can be arbitrarily set by modulating the energy distribution function of the virtual system(s). McMD simulations can also undergo trivial trajectory parallelization (TTP), in a method named TTP-McMD.189 In TTP-McMD, multiple McMD runs starting from different coordinates are conducted independently and the resulting trajectories are concatenated in any order. The main goal of TTP-McMD is to increase computational efficiency by parallelizing trajectory calculations. V-McMD simulations can be parallelized in similar fashion (TTP-V-McMD).190 Even with these improvements, the large barriers imposed by ring strain in small cyclic peptides likely challenge McMD and its variant methods in much the same way they challenge REMD.
2.4. Accelerated molecular dynamics (aMD)
Accelerated molecular dynamics (aMD) simulations present another approach to overcoming the issue of systems being trapped in local free-energy minima. This method aims to increase the frequency of transitions across energy barriers by introducing a bias potential such that the potential surfaces near minima are raised, while those surfaces near the barriers or saddle points are left unmodified or less-modified, thus decreasing the barrier heights (Figure 5).191 During an aMD simulation, an energy boost is introduced when the system’s potential energy falls below a chosen threshold. The added bias potential, ΔV, depends on the system’s potential energy V:
| Eq. 8 |
where Vthreshold is the threshold boost energy, V(r) is the original potential, and α is a tuning parameter that determines the depth of the modified potential well. This expression for ΔV ensures that the bias potential mimics the shape of the underlying potential minima. The original free-energy landscape can be reconstructed using reweighting algorithms.192
Figure 5.
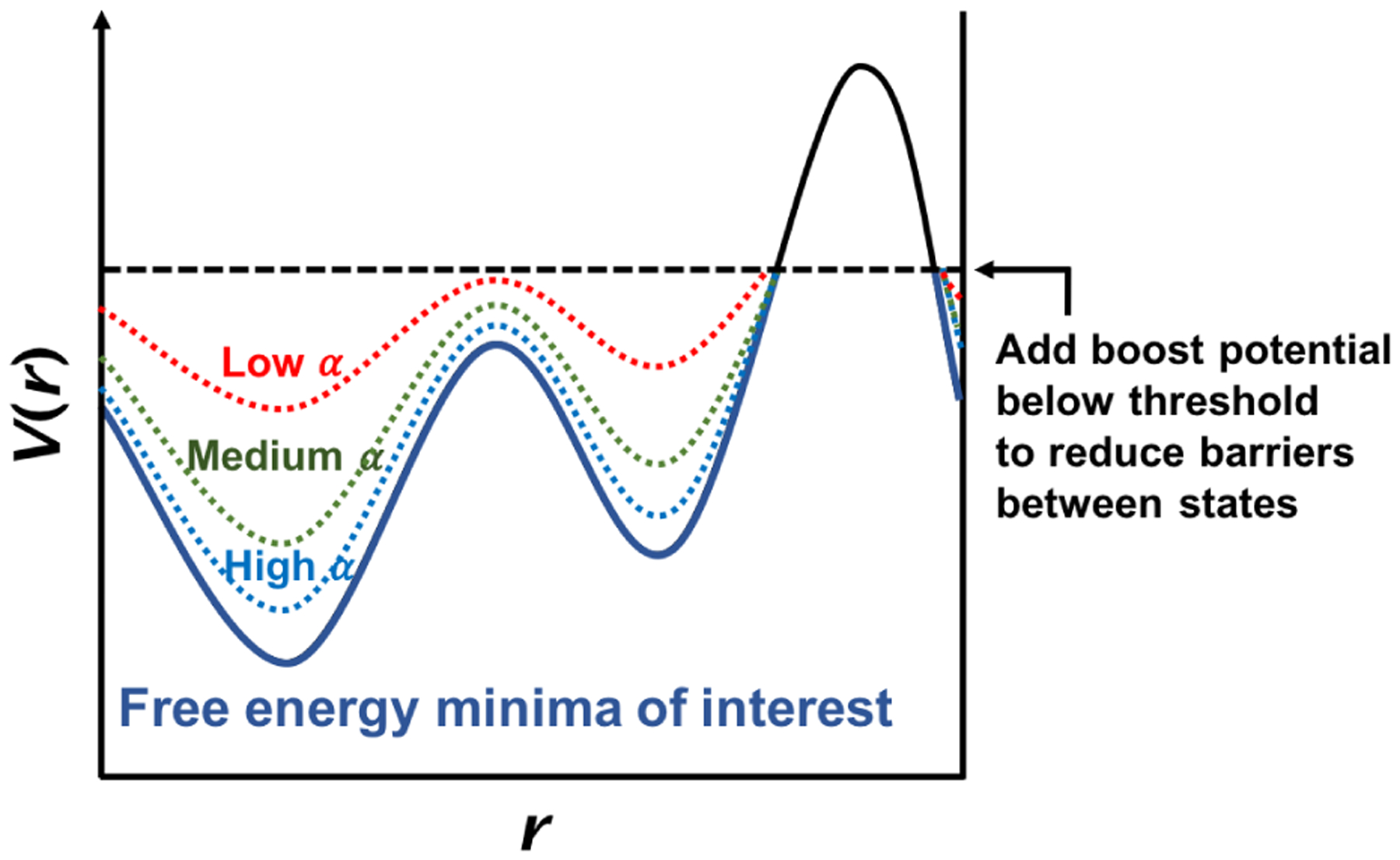
Schematic view of accelerated MD.191 The true potential is shown as a solid line. Boost potentials are added when the potential energy is below the threshold (dashed line). The modified potentials with various values of the tuning parameter α are shown in dotted lines; the smaller the α, the more significant the added biasing potential.
One noteworthy caveat is that aMD is not a fully standalone method – the boosting parameters α and Vthreshold are system-specific and have to be tuned, which is typically done based on the number of atoms or residues, as well as the average energies from prior conventional MD runs.193 One must also be careful when choosing the reweighting algorithm, as exponential-average reweighting (Boltzmann reweighting) is known to introduce high energetic fluctuations caused by a small number of high-boost-potential frames dominating the reweighting factors.192 This problem can be avoided by using a Maclaurin expansion,193 though this expansion does not always give the correct energy minimum positions.192 Cumulant expansion tends to avoid these issues and commonly proves to be the best choice for a wide array of biomolecules.192,194,195
2.5. Complementary-coordinates molecular dynamics (CoCo-MD)
Complementary-coordinates molecular dynamics (CoCo-MD) is a conformational sampling method developed by Shkurti et al. by combining the CoCo (“complementary coordinates”) method with MD simulations.196 CoCo is an ensemble enhancement method based on principal-component analysis (PCA) and initially built to increase the conformational variability of NMR-derived structures.197
The workflow of CoCo-MD consists of a number of iterative cycles (Figure 6).196 Starting with N short independent MD simulations, in each cycle, the CoCo method is used to generate N new starting structures for subsequent MD simulations. Specifically, Cartesian PCA is performed on the original MD trajectories and the structural ensemble is projected onto the first few principal components (PCs). The boundaries are then chosen in the PC space to include all the data points (Figure 6A). The data points are then binned into a multidimensional histogram, where N unoccupied bins are chosen iteratively such that the bin chosen in each step is the one most distant from all occupied or prior chosen bins (chosen bins are shown as crosses in Figure 6). The PCs of the centers of the chosen bins are then converted back into Cartesian coordinates to produce N new conformations. Because these new conformations might not be physically realistic, N short restrained MD simulations need to be performed, starting from the well-equilibrated initial structure with these new conformations as target structures, to generate conformations that can be used as a new starting point. N short MD simulations are then run, the trajectory data are saved as sampled conformations (Figure 6B), and the cycle (PCA→new start points→short MDs) repeats.
Figure 6.

Schematic view of the CoCo-MD algorithm.196 (A) In this example, starting from the initial configurations (black dot), 4 short, independent simulations are run (red curves). Principal-component analysis is performed using the Cartesian coordinates of the trajectories and boundaries in the principal-component space are chosen to include all the data points (dashed lines). The selected principal-component space is then binned and 4 unoccupied bins are chosen iteratively to be the most distant from all occupied or prior chosen bins (green ×’s). (B) The 4 points in the principal-component space are mapped back to the Cartesian space to generate 4 new structures. After equilibration, 4 short, independent simulations starting from these 4 new structures are run. A similar process is repeated to generate 4 new starting points (blue ×’s) for the next iteration.
Notably, the structures sampled by CoCo-MD do not follow the Boltzmann distribution, and thus must be unbiased to recover the equilibrium structural ensemble. Two methods were proposed by Shkurti et al. to obtain such an ensemble using conformations sampled by CoCo-MD.196 The first method is to use the structures sampled by CoCo-MD as the highest temperature reservoir in reservoir REMD.167,198 The other method is to reweight the structures sampled by CoCo-MD to match the potential energy distribution of conventional MD. The first method is computationally more intensive; on the other hand, the latter method relies on a short conventional MD run for its energy distribution, and does not necessarily offer rigorous free-energy estimates.196
3. Using MD simulations to elucidate the solution structures of cyclic peptides: Development of methods, evaluation of force fields, and general approaches to modeling cyclic peptides
3.1. Coupled two-dihedral motions for efficient metadynamics simulations of cyclic peptides [BE-META]
A key challenge in using metadynamics simulations to enhance conformation sampling lies in the selection of biasing coordinates. Ideally, the selected coordinates represent the slow degrees of freedom and describe the transition of interest. In recent published work, McHugh et al. investigated how cyclo-(GGGGGG) (cyclo-G6) and several other cyclic peptides switch conformations.172 It was found that the conformational switches of the cyclic peptides consistently require simultaneous changes of two dihedrals, either (ϕi, ψi) or (ψi, ϕi+1). The latter type of changes, involving the movement of ψi and ϕi+1 in opposite directions, is consistent with a rotation of the peptide plane around the axis between two consecutive Cα atoms, and is commonly referred to as a crankshaft flip.199,200 By targeting (ϕi, ψi) and (ψi, ϕi+1) using 2-D collective variables with bias exchanges, the time required to converge was significantly reduced (by 2–4×) for model cyclic peptides, cyclo-(AAAAAA) and cyclo-(YNPFEEGG) compared to standard BE-META targeting individual ϕ and ψ dihedrals.
3.2. Test of accelerated MD for cyclic peptide sampling [aMD]
To test whether accelerated MD (aMD) simulations with an explicit-solvent model could sample the conformational space of constrained systems such as cyclic peptides, Kamenik et al. used three small cyclic peptides as benchmarks: cyclo-(PSlDV), cyclo-(RGDfV), and cyclo-(RRWWRF) (Figure 7; in the peptide sequences, lowercase letters denote D-amino acids and underline denotes N-methylation).201 All three cyclic peptides have potential therapeutic applications: cyclo-(PSlDV) is an integrin binder,106 cyclo-(RGDfV), also called cilengitide, is a former anticancer drug,90,202–205 and cyclo-(RRWWRF) is an antimicrobial peptide.107 Moreover, solution NMR studies had been reported for all three cyclic peptides.90,106,107 NMR analysis of cyclo-(PSlDV) found up to five conformers in slow exchange. The two most abundant components were found to have a cis- and trans- amide bond between Pro1 and Val5, respectively, with all other bonds being trans. Their relative abundance was found to be 16% trans to 66% cis.106 No cis peptide bonds were observed in the NMR studies of the other two peptides.90,107 Cyclo-(RRWWRF) was determined to be highly flexible in aqueous solution, only assuming a more rigid structure when bound to a micelle.107
Figure 7.

Cyclo-(PSlDV), cyclo-(RGDfV), and cyclo-(RRWWRF) were simulated using accelerated MD simulations.201 Accelerated MD showed superior sampling compared to conventional MD and the simulation results were able to reproduce some NMR observations. However, the energetic noise introduced in the reweighting step of accelerated MD might prevent resolving subtle differences in dihedral distributions.
Accelerated MD simulations were carried out using the AMBER-14SB force field206 with the TIP3P water model.207 In the case of cilengitide, additional parameters for N-methylated amino acids were taken from Forcefield_NCAA.208 One-microsecond trajectories of both conventional MD and aMD were collected; for cilengitide, the authors performed twenty 50 ns simulations, combining them into a 1 μs trajectory. All aMD simulations used the dual-boost algorithm implemented in the Amber16209 software package – in other words, a bias was applied to the total potential, along with an additional bias component on the dihedral term.191,210 The global structural ensemble was analyzed using dihedral principal-component analysis211 and cluster analysis, following a Boltzmann reweighting process192 to recover the unbiased results. In order to make data analysis faster and more robust, the cluster analysis was performed on the 2,000 snapshots with the highest boosting potential, i.e. the lowest potential energy (0.4% of all trajectory snapshots).201
Kamenik et al. first showed that while a 1 μs conventional MD simulation exhibited limited sampling of cyclo-(PSlDV), aMD was able to explore both the cis and trans isomers for the amide bond between Pro1 and Val5 (ω15).201 The cis:trans distribution of ω15 in the reweighted aMD results was 25:75, which is consistent with the ratio reported in the solution NMR study.106 All average interproton distance restraints for the trans structures were satisfied; for the cis structures, 17 out of 18 NOE restraints were met, with one distance found to be 0.2 Å too small in the simulation. The cluster analysis showed cyclo-(PSlDV) adopted three distinct conformations, which was consistent with NMR findings. For cilengitide, all NOE restraints but one were met, with a small violation of 0.24 Å. The conformational landscape of cyclo-(RRWWRF) exhibited a large number of minima, which is, again, consistent with the NMR results, which had suggested that this cyclic peptide was highly flexible in aqueous solution.107 All NOE restraints were met for average interproton distances; however, individual structures show large violations, further supporting the idea that the experimental NOE distances resulted from averaging. While aMD simulations seemed to reproduce the experimental NOE results, the 3J(HN, Hα) coupling constants for cyclo-(PSlDV) determined from aMD were not found to be in good agreement with NMR-derived ones. The authors noted that this discrepancy suggests that as a result of energetic noise introduced in the reweighting step, aMD does not provide a high resolution when it comes to subtle differences between dihedral distributions, potentially.201
3.3. CoCo-MD: Development and benchmark on cyclosporin A [CoCo-MD]
Shkurti et al. tested CoCo-MD on several molecules, including cyclosporin A, which is an N-methylated cyclic peptide used as an immunosuppressant drug, well known for its good bioavailability (Figure 8).196 Witek et al. had previously performed 100×100 ns (10 μs in total; GROMOS-54a7 force field212 with the SPC water model213) conventional MD (cMD) simulations beginning with 100 diverse structures to investigate the membrane-permeating mechanism of cyclosporin A.214 Shkurti et al. also performed cMD, together with CoCo-MD and accelerated MD (aMD), and compared results to those obtained from Witek et al.’s 10 μs simulation. Parameters from the General Amber Force Field215 and the work of Khoury et al.208 were used, along with the TIP3P water model;207 the total lengths of MD simulations for CoCo-MD, cMD, and aMD were all 20 ns. Within this time scale, it appears that cMD and aMD failed to sample the conformations produced in Witek et al.’s 10 μs cMD simulation, while CoCo-MD sampled a wider conformational space than that sampled by Witek et al. The conformational space sampled by CoCo-MD included a number of rarely observed, but thermally accessible states.196 When conformational states were labeled by values of dihedrals ϕ (g+, t, or g−), ψ (g+, t, or g−), and ω (cis or trans), it was found that CoCo-MD identified 9,822 conformational states, while cMD identified 2,224 states, and aMD found 5,912 states.196
Figure 8.

Cyclosporin A was studied using conventional MD, accelerated MD, and CoCo-MD.196 N-methylated residues are underlined; Bmt: butenyl-methyl-threonine; Abu: aminobutyric acid. With the same total simulation lengths (20 ns), CoCo-MD identified 9,822 conformational states, while conventional MD identified 2,224 states, and accelerated MD found 5,912 states.
3.4. Evaluation of residue-specific force fields for cyclic peptides [REMD]
The development of robust and reliable computational methods for cyclic peptide structure prediction has faced a number of challenges. For one, cyclic peptides require high accuracy in free-energy determination, as they can assume distinct conformations with small free-energy differences.92–105 Moreover, sufficient conformational sampling is often necessary to gain insight into their biological activity. To that end, Geng et al. used REMD simulations to evaluate the performance of four force fields: OPLS-AA/L216+TIP4P-Ew217 and AMBER-99SB-ILDN218+TIP3P,207 as well as two residue-specific force fields, RSFF1219 and RSFF2,220 which were based on OPLS-AA/L and AMBER-99SB, respectively. The residue-specific modifications were added in order to reproduce the rotamer-dependent Ramachandran plots for each amino acid type. The benchmarks consisted of 20 cyclic peptides selected from the Cambridge Structure Database. These cyclic peptides were 5–12 residues in length, all-trans, head-to-tail cyclized, and without non-natural amino acids (Figure 9). REMD simulations were performed with 24–32 replicas spanning temperatures from 300 K to 600 K (300–500 K for four cyclic peptides susceptible to cis/trans isomerization under RSFF2). Simulations were found to have reached convergence within 100–200 ns.221 For each cyclic peptide, the 300 K trajectory was analyzed by first performing a cluster analysis of each residue’s backbone dihedrals (ϕ, ψ) using a density-based algorithm.222 Discrete conformations were then defined by a string of the cluster indices of all residues in the sequence, and their populations computed.
Figure 9.
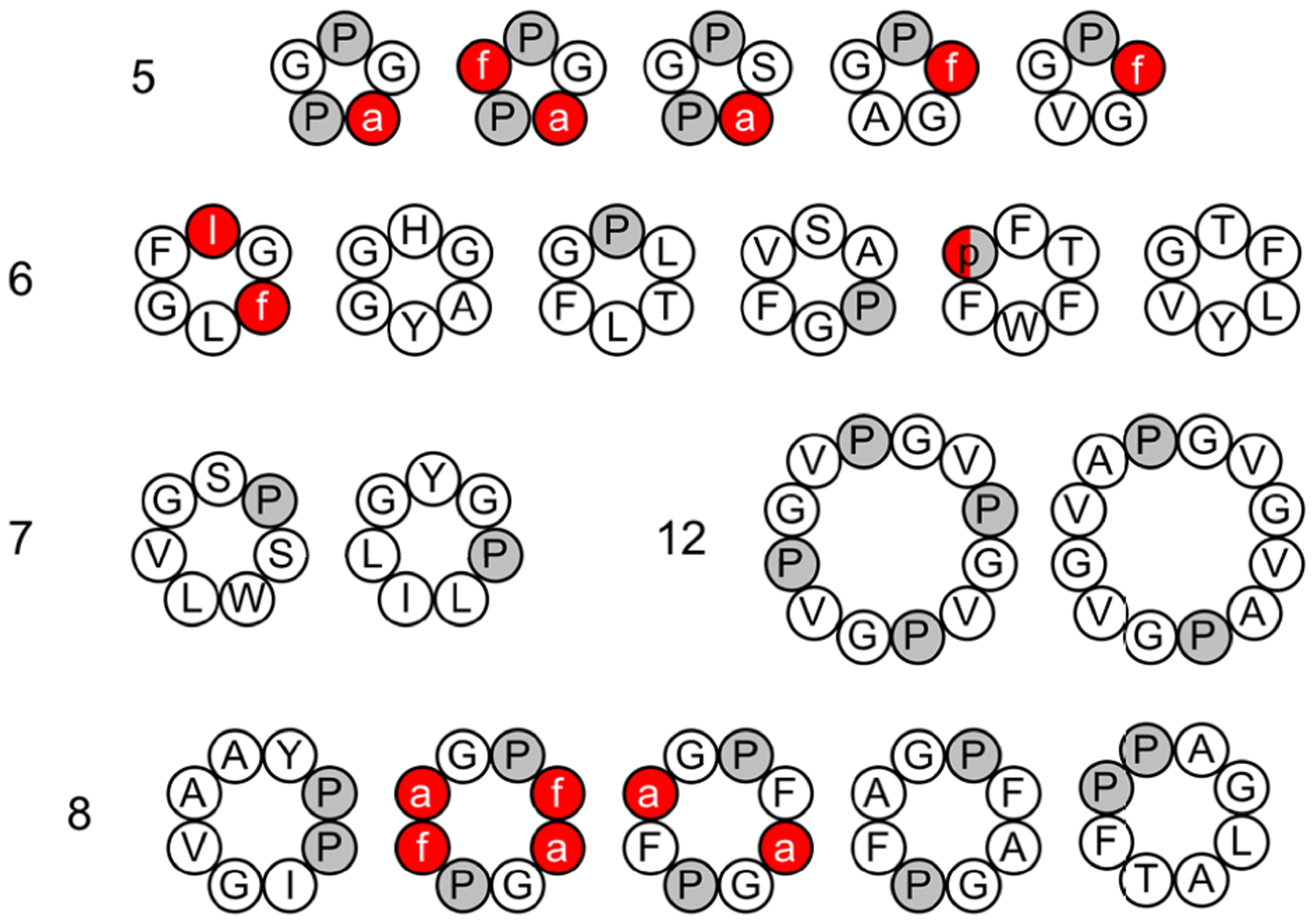
Twenty cyclic peptides were simulated using REMD simulation and four force fields. Residue-specific force field 2 was the best at recapitulating the X-ray structure of the cyclic peptides. D-amino acids are listed in lowercase and shown in red; Pro is colored in gray.221
In real-life scenarios, without knowing the experimental structure, the most populated cluster is generally taken as the basis for a structure prediction. The authors found that the most populated clusters obtained from simulations using RSFF2 match the crystal structures with backbone+Cβ RMSD < 1.0 Å for 15 cyclic peptides, compared to about 10 for the other force fields.221 The authors also reported the RMSD of the most crystal-like conformations observed in the REMD simulations. Crystal-like conformations with RMSD < 1.0 Å were observed for 17 cyclic peptides in simulations using OPLS-AA/L, whereas the other three force fields met this condition for 19 cyclic peptides.221 The most crystal-like conformations correspond to the global energy minima for 12 and 13 cyclic peptides under RSFF1 and RSFF2, respectively; this is only the case for 7 cyclic peptides under the other two force fields. Geng et al. also investigated the effect of water model, using RSFF2 with TIP4P-Ew. One fewer cyclic peptide met the RMSD cutoff under these conditions, suggesting that the water model used does influence simulations, but not as much as force field choice.
Geng et al. also compared the sampling of backbone dihedrals in cyclic peptides to that in linear peptides and globular proteins, in order to determine whether cyclic peptides are more likely to sample less-favored values of ϕ and ψ and whether residues in cyclic peptides are more conformationally rigid. The overall findings, based on local conformational free energies and backbone entropies, suggested that the backbone sampling of residues in cyclic peptides was more similar to that in globular proteins than that in linear peptides.221
3.5. Force field evaluation using cyclo-(YNPFEEGG) [REMD, BE-META]
The performance of popular force fields in simulating linear peptide and protein systems is quite well-documented.154,223,224 However, their ability to model cyclic peptides has yet to be thoroughly examined. The performance of six force fields (AMBER-96225+TIP3P,207 AMBER-99SB-ILDN218+TIP3P, AMBER-03226+TIP3P, OPLS-AA/L216+TIP4P, GROMOS-53a6227+SPC,213 and RSFF1219+TIP4P-Ew217) was recently tested on a benchmark cyclic peptide, cyclo-(YNPFEEGG) (Figure 10A).171 This cyclic octapeptide was designed to bind the EH domain of EHD1, and a structural model in water had been determined via NMR spectroscopy (Figure 10B).228 Yu et al. first found that REMD simulations were unable to consistently provide converged results even after 300 ns.
Figure 10.

(A) Cyclo-(YNPFEEGG) was simulated using AMBER-96, AMBER-99SB-ILDN, AMBER-03, OPLS-AA/L, GROMOS-53a6 and RSFF1.171 (B) Previous NMR results suggested the cyclic peptide adopted one dominant structure in water.228 Simulations showed that the cyclic peptide formed multiple conformations that do not recapitulate the NMR-derived structural model well. The results might suggest further re-parameterization of the force fields is needed, assuming the NMR structure is accurate.
To more efficiently explore the conformational landscape of the cyclic peptide, Yu et al. instead used BE-META to enhance sampling.173,229,230 Eighteen collective variables were used, targeting ϕ/ψ/χ1 of Tyr-1, Asn-2 and Phe-4, ϕ/ψ/χ1/χ2 of Glu-5 and Glu-6, and ψ of Pro-3; the simulations were performed for 300 ns. To obtain an equilibrium structural ensemble for further analysis, the biased trajectories were “unbiased” using Boltzmann reweighting, where the k-th frame of the i-th replica is either kept or discarded based on the Boltzmann probability criterion:
| Eq. 9 |
where ΔGi is the free-energy profile along collective variable i, and ski is the value of collective variable i in the k-th frame. Dihedral principal-component analysis211 and modified density-based cluster analysis222 were subsequently applied to characterize the structural ensemble. To verify whether convergence was reached, two sets of simulations starting from two significantly different initial structures were performed, and convergence was assumed to be achieved upon the two simulations yielding similar conformational density profiles after the principal-component analysis. The authors found that, instead of a single highly populated structure, as observed in NMR, results from all six force fields showed that cyclo-(YNPFEEGG) adopted multiple conformations with significant populations. In addition, none of the identified conformations matched the NMR-derived structure well.171 At first glance, these results suggest that further reparameterization of force fields is needed to robustly predict structures of cyclic peptides in solutions. It is not out of the question, however, that a single-conformation NMR model does not provide as accurate of a picture as the simulation conformational ensemble. A more detailed comparison, looking at, for instance, NOE-derived interproton distance restraints, may shed more light on the cause of the observed discrepancies.
3.6. Force field evaluation for cyclic peptides containing N-methylated residues [BE-META]
N-methylation has proven a popular and useful tool for structural design of cyclic peptides and optimizing their pharmacological properties. N-methylation has been found to increase membrane permeability,231 achieve better bioavailability,232,233 and improve the affinity and selectivity of receptor binding.234 Replacing the amide hydrogen with a methyl group increases the likelihood of observing a cis isomer for the amide bond preceding the N-methylated residue.235 The elimination of the amide proton on the N-methylated backbone also incurs a nonlocal effect by disenabling it from forming transannular hydrogen bonds.235 Therefore, with N-methylation of appropriate residues, cyclic peptide backbones can be rigidified to predominantly adopt bioactive conformations by allowing the cyclic peptides to only form certain hydrogen bonds.231
Computational methods have been used to predict the effects of N-methylation on various aspects of cyclic peptide structures and function – and membrane permeability in particular.231,236–238 These studies demonstrated the applications of established computational framework, such as BE-META sampling with cluster analysis,236 PLOP sampling with solvation-free-energy calculation,231 or extensive MD simulations with Markov state models,237,238 in studying N-methylated cyclic peptides. An issue that presents itself, however, is that N-methylation can lead to formation of cis peptide bonds, and thus, a well-designed computational model will need to include a reasonable prediction of the cis/trans isomer ratio. With that in mind, a major challenge is to accurately reproduce the experimentally determined structures with the correct cis/trans preferences, given only the sequence information.
In a recent study, Slough et al. examined the accuracy of two force fields, RSFF1219 with TIP4P-Ew water217 and RSFF2220 with TIP3P water,207 in recapitulating the structures of two N-methylated cyclic peptides, with an emphasis on the ability of the force fields to predict the isomerization states of N-methylated amide bonds.239 The two benchmark cyclic peptides were cyclo-(aAAAAA) and cyclo-(aAAAAA), where “a” in the sequences stands for D-alanine, and the underlined residues are N-methylated (Figure 11). The solution structures of these two cyclic peptides had been previously determined using NMR.104 In cyclo-(aAAAAA), residues 1 and 6 were N-methylated and the two N-methylated amide bonds both adopted a trans configuration (denoted as t1t6). This cyclic peptide formed two type-II β turns at residues 6–1 and at residues 3–4 with two transannular hydrogen bonds. In contrast, in cyclo-(aAAAAA), residues 1 and 5 were N-methylated and the N-methylated amide bonds adopted a trans and cis configuration, respectively (denoted as t1c5). This cyclic peptide formed a type-VIa1 β turn at residues 4–5.
Figure 11.
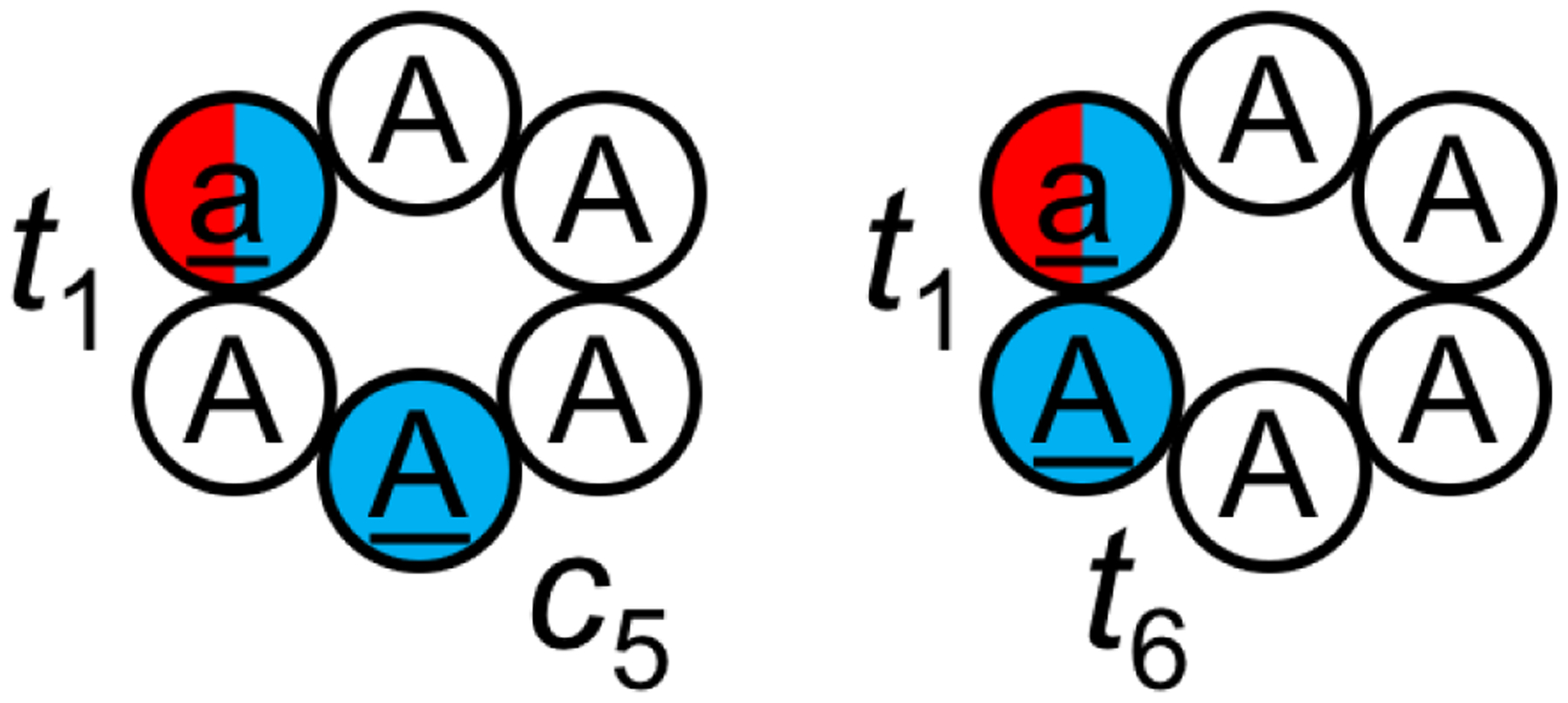
Cyclo-(aAAAAA) and cyclo-(aAAAAA) were simulated using BE-META.239 D-amino acids are listed in lowercase and shown in red; N-methylated amino acids are underlined and shown in blue. In cyclo-(aAAAAA), residues 1 and 5 were N-methylated and the N-methylated amide bonds adopted a trans and cis configuration, respectively (denoted as t1c5). In cyclo-(aAAAAA), residues 1 and 6 were N-methylated and the two N-methylated amide bonds both adopted a trans configuration (denoted as t1t6). It was found that if the correct isomer states were given, the RSFF2 force field was able to reproduce the solution NMR structures for both cyclic peptides; however, when the correct isomer states were not given, the RSFF2 force field was unable to predict the t1c5 isomer of cyclo-(aAAAAA).
To enable simulations of these two cyclic peptides, the authors first developed parameters that were compatible with the RSFF1 and RSFF2 force fields for the N-methylated alanine.239 BE-META was used to sample the structural ensembles of these two benchmark peptides. In the first set of simulations, the authors tested whether the two force fields were able to reproduce the NMR structures when the correct isomer states were given. In the initial structures, cyclo-(aAAAAA) was prepared in the t1t6 state while cyclo-(aAAAAA) was prepared in the t1c5 state. 2-D biases on all pairs of (ϕi', ψi') and (ψi', ϕi+1') were used as collective variables to enhance conformational sampling. Here, ϕi' is the dihedral Hi/CNi–Ni–Cαi–Ci (Hi for nonmethylated amino acids and CNi for N-methylated ones) and ψi' is the dihedral Ni–Cαi–Ci–O. Instead of (ϕi, ψi) and (ψi, ϕi+1),172 (ϕi', ψi') and (ψi', ϕi+1') were used as collective variables here to minimize artificial cis/trans isomerization due to the added biasing potentials in BE-META simulations. In the second set of simulations, 1-D biases on ω angles involving the N-methylated amino acids were also applied as part of the collective variables to evaluate whether the force fields could accurately predict the correct isomer states as well. In both sets of simulations, five neutral replicas with no bias were added to enable analysis of the equilibrium structural ensembles. The simulations were run for 100–250 ns, until convergence was achieved.
When the correct isomer states were provided and maintained through improper-dihedral restraints (without biasing the ω angle), RSFF2 was able to reproduce the structures observed in NMR for both cyclic peptides, while RSFF1 was only successful in the case of cyclo-(aAAAAA).239 It is worth noting, however, that unintended cis/trans isomerization was still observed in these simulations without directly biasing the ω angle. The (ϕi', ψi') and (ψi', ϕi+1') bias potentials added to the backbone atoms during BE-META may have compromised the improper dihedrals’ ability to maintain amide bond planarity.239
When the simulations needed to also predict the isomer states, it was found that both RSFF1 and RSFF2 were able to accurately predict the isomer state and the structure of cyclo-(aAAAAA). Notably, the most populated structure in RSFF2 simulations (>50% population) resembles the NMR structure, with the correct t1t6 configuration for the N-methylated residues. However, both force fields were unable to reproduce the NMR structure of cyclo-(aAAAAA). Unlike the t1c5 configuration observed in the experiments, the top clusters in the simulations adopted a t1t5 configuration. For the RSFF2 simulations, only 3.1% of the population had the correct t1c5 configuration. Although the top cluster of those conformations with t1c5 did resemble the NMR structure, its population was only 1.0%. Switching to other solvent models such as RSFF2 with SPC/E240 (1.5%), TIP4P/2005241 (3.3%), TIP4P-Ew217 (1.5%) did not improve matters, although the total population of the correct t1c5 isomer increased to 10.5% with DMSO in comparison to 3.1% in TIP3P.239 These results suggest that while the recently developed residue specific force fields are promising at predicting conformations of N-methylated cyclic peptide when the correct isomer states are given, further development is needed for complete de novo structure predictions.
3.7. Systematic study of cyclic pentapeptides [BE-META]
The BE-META sampling protocol targeting the (ϕi, ψi) and (ψi, ϕi+1) essential transitional motions of cyclic peptides172 enables rapid convergence of simulation results, which allowed Slough et al. to simulate >70 cyclic pentapeptides to study how sequences control their structures.132 First, simulation of cyclo-(GGGGG) was performed and the results showed that cyclic pentapeptides typically adopt a β turn (βI, βI’, βII, or βII’) and a tight turn (αR, αL, γ, or γ’) at the position opposite the β turn. Then, to understand how a single amino acid affects the cyclic peptide structural ensemble, simulations of cyclo-(X1AAAA) were performed, where X1 was one of the 20 standard amino acids. The most favorable turn combination among these CPs was found to be βII’+αR (a type-II’ β turn with a tight αR turn opposing it). The top cluster of these cyclic peptides typically had a population <30%, except for cyclo-(AAAAA), whose top cluster adopts the βII’+αR conformation with a population of ~53%. The structural ensembles of cyclo-(X1AAAA) allowed the authors to analyze the structural preference of each of the 20 standard amino acids. Based on the results, they predicted a sequence, cyclo-(GFSEV), to be most likely well-structured with a βII’+αR conformation. BE-META simulations showed that cyclo-(GFSEV) indeed adopts the βII’+αR as the most populated conformation, but the population (~55%) appeared to be only marginally improved compared to the ~53% for cyclo-(AAAAA) already present in the cyclo-(X1AAAA) database. This observation implies that the preferences of single amino acids are non-additive and neighboring residues likely play a part in determining the structures cyclic peptides adopt. To capture the influence of neighboring residues, the authors simulated and analyzed the structural ensembles of cyclo-(X1X2AAA), where X1, X2 were each one of the eight amino acids A, D, F, G, N, R, S and V. These amino acids were chosen as representatives of the different types among the 20 standard amino acids to reduce the number of sequences involved. With these data, the authors developed a scoring function that estimates cyclic peptides’ structural preference.242 Using this scoring function to enumerate all possible cyclic peptide sequences, cyclo-(GNSRV) had the highest score for the βII’+αR conformation (Figure 12). BE-META simulations showed that cyclo-(GNSRV) was indeed well structured in this conformation, with a population of 67%. This result was further supported by NMR experiments.242
Figure 12.
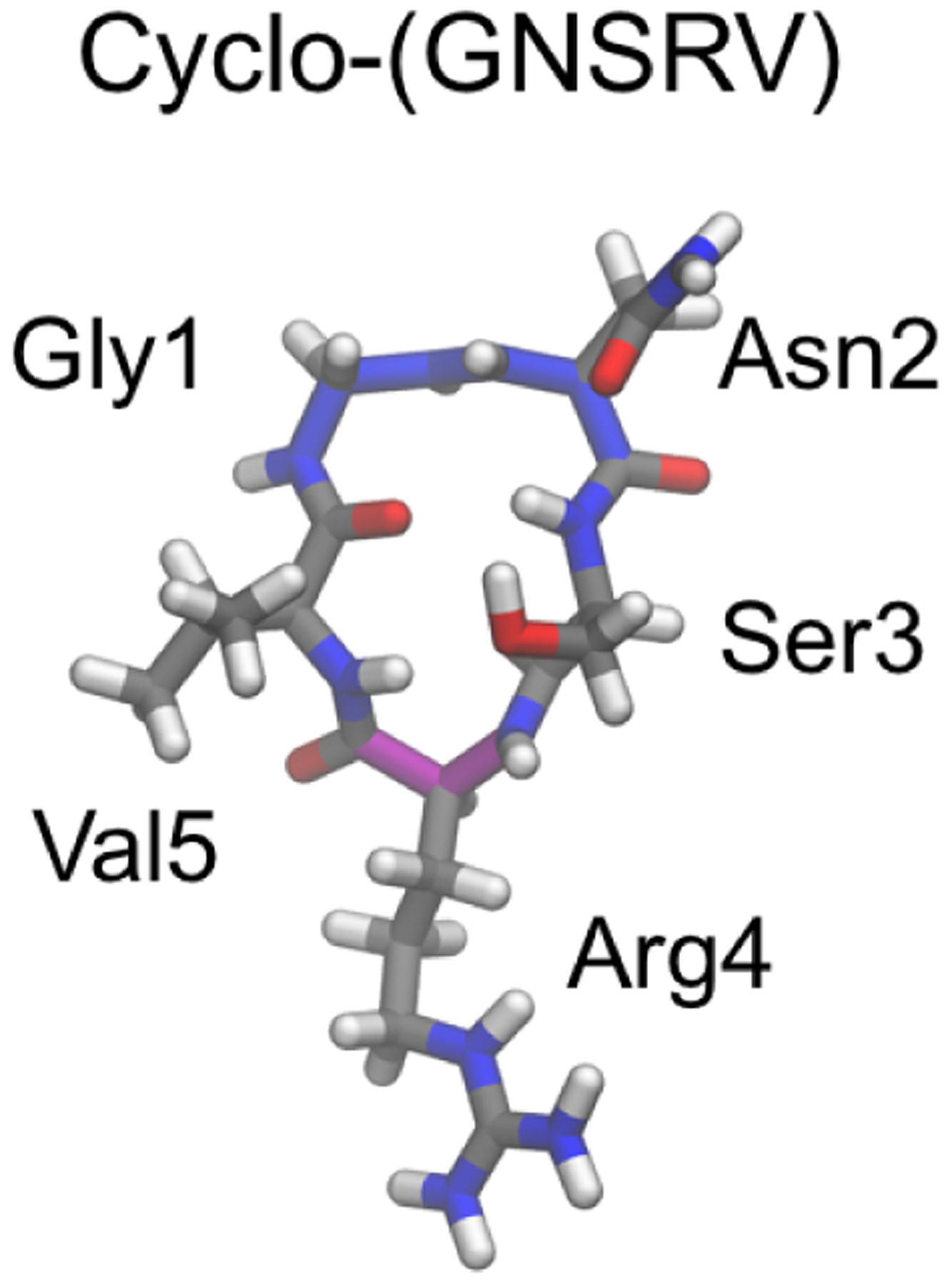
By combining 57 BE-META simulation results of cyclo-(X1X2AAA), where X1, X2 were one of the eight amino acids A, D, F, G, N, R, S and V, a scoring function that can be used to predict the likelihood of a cyclic pentapeptide adopting a specific structure was developed.242 Cyclo-(GNSRV) was predicted to be well-structured with a type-II’ β turn at GN (backbone highlighted in blue) and an αR tight turn at R (backbone highlighted in magenta). This prediction was supported by experimental NMR results.
3.8. Systematic study of cyclo-(GnA6–n) and cyclo-(GnV6–n) [BE-META]
Using an optimized BE-META simulation protocol they had previously developed, McHugh et al. systematically explored the global structure ensembles for two families of cyclic peptides: cyclo-(GnA6-n) and cyclo-(GnV6-n) to gain further insight into the sequence–structure relationship of cyclic hexapeptides.243 Using (ϕi, ψi) and (ψi, ϕi+1) as 2-D collective variables in BE-META, the simulations were able to achieve superior sampling and reach convergence sooner than targeting ϕ and ψ dihedrals individually. In all these BE-META simulations, five neutral replicas with no bias were added to obtain unbiased equilibrium structural ensembles.
These two families of cyclo-(GnA6-n) and cyclo-(GnV6-n) contain a total of 27 cyclic peptides, with sequence permutations and symmetry taken into account. Most of the cyclic peptides tested adopted multiple conformations in solution. However, one of the cyclic peptides, cyclo-(VVGGVG), exhibited a single highly populated conformation: ~80% of the population adopts two βII turns at residues 2–3 and residues 5–6 (Figure 13).243 This sequence is unique among the 27 cyclic peptides, as all the others (including, e.g., cyclo-(VVGVGG), which has the same amino acid composition but different ordering of residues) adopted multiple conformations with small populations in solution.
Figure 13.
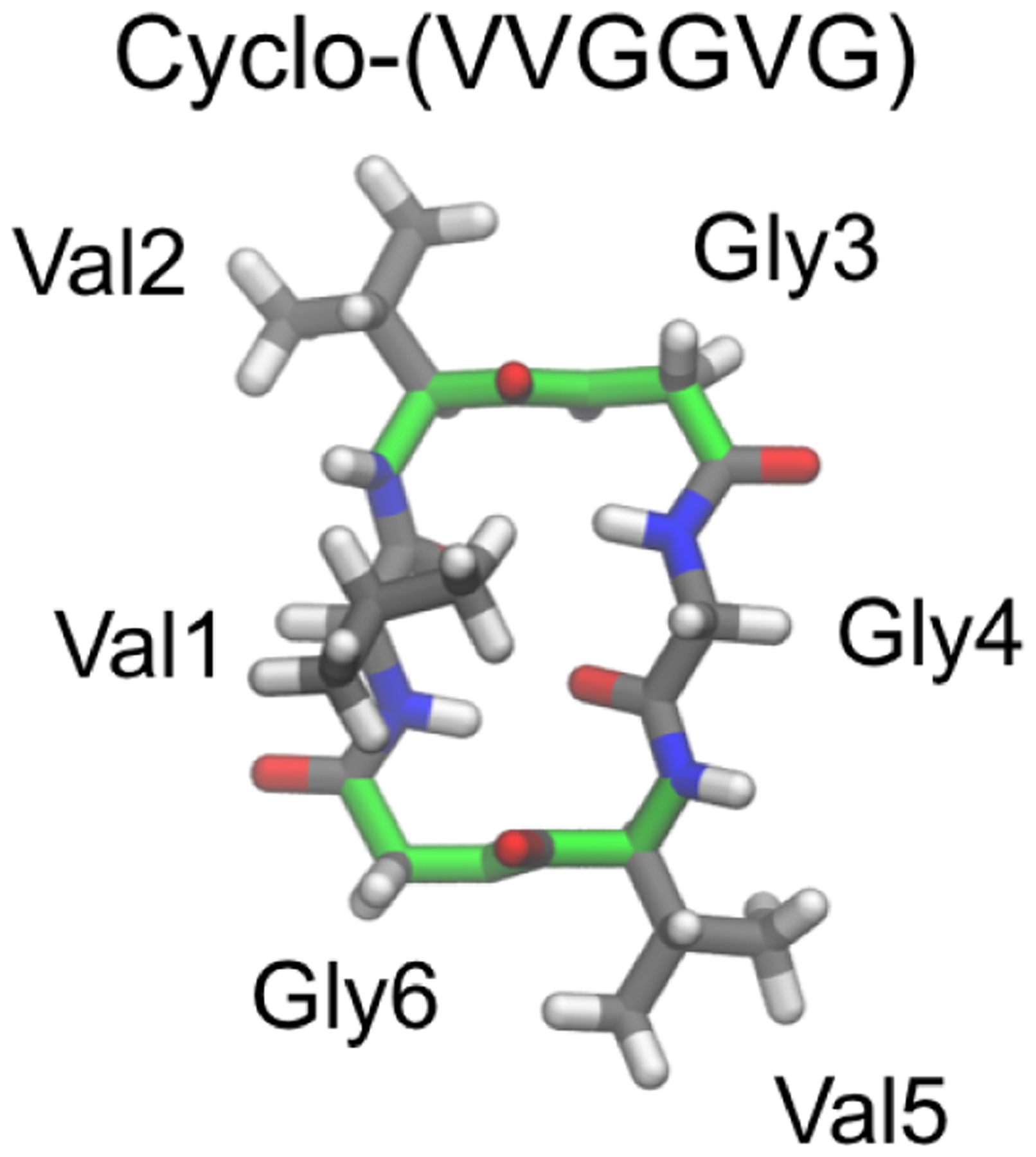
Among all the 27 cyclo-(GnA6-n) and cyclo-(GnV6-n) cyclic hexapeptides simulated using BE-META, cyclo-(VVGGVG) was the only cyclic peptide that showed a dominant structure in solution with population >50%.243 It was predicted that ~80% of the population of cyclo-(VVGGVG) adopts two βII turns (backbone highlighted in green) at residues 2–3 and residues 5–6, which was later supported by NMR experiments.244
In a recent study, Cummings et al. synthesized cyclo-(VVGGVG) and cyclo-(VVGVGG), and experimentally verified the respective structure predictions using NMR spectroscopy.244 Cyclo-(VVGGVG) showed a wide spread in the amide region of 1H NMR, indicating a well-defined structure; unique NOE patterns were observed and in agreement with the prediction of two βII turns at residues 2–3 and residues 5–6. On the other hand, cyclo-(VVGVGG) had a much narrower span in its amide 1H chemical shifts and exhibited multiple weak–strong NOEs, suggesting that the cyclic peptide adopts multiple conformations in solution.
To understand the importance of each Val residue at stabilizing the βII+βII structural motif adopted by cyclo-(VVGGVG), simulations were performed where each of the three Val residues was replaced with Ala. It was found that Val at position 1 was the most important at stabilizing this structure. Additional simulations of V1I, V1L, V1T, and V1S mutants suggested a critical role of β branching at position 1. These simulation predictions were also supported by experimental data from NMR spectroscopy.244
3.9. Cyclic peptoids [REMD]
Peptoids (poly-N-substituted glycines) are peptidomimetic oligomers in which side chains are appended to the backbone nitrogen, rather than connected to the Cα atoms, as they are in peptides. It is relatively easy to use various amines as monomers to incorporate a diverse range of side chains during peptoid synthesis, and even though peptoids do not have any backbone NH groups, they can fold into specific structures.245–249 Furthermore, peptoids are resistant to proteolysis,250 which makes them highly appealing as potential peptidomimetic therapeutics. While the trans isomer is generally preferred in most amide bonds in peptides (except those preceding a proline), cis amide bonds are frequently observed in peptoids, owing to the backbone N-substitution.128 Currently, the conformational landscape of peptoids is not as well understood as that of peptides, and the applicability of physical modeling to predicting the structures of peptoids has not been widely demonstrated. The work of Voelz et al.251 and Butterfoss et al.128 exemplifies the merit of REMD simulations in exploring the energy landscape of linear and cyclic peptoids. In these REMD simulations, to attain sufficient sampling across the cis/trans isomerization barrier, 800 K was chosen as the highest temperature; the number of replicas ranged from 8 to 24; each replica was run for 500 ns. It was shown that the General Amber Force Field (GAFF)215 and a Generalized Born/Surface Area (GBSA-OBC) implicit-solvent model252 were in general able to reproduce the experimental dihedral preferences of peptoids and the lowest-energy QM structures for small linear peptoids.251,253 However, acetyl-(S)-N-(1-phenylethyl)glycine-dimethylamine (Ace-Nspe-NMe2) showed the opposite ϕ angle preference compared to the QM results, likely due to the inability of GAFF/GBSA-OBC to describe the n→π* interaction well.251 Furthermore, REMD simulations were performed on three peptoids whose experimental structures were known, including one cyclic peptoid, cyclo-(sarcosine8) (i.e. cyclo-octa-NMeGly) (Figure 14). The REMD simulation of cyclo-(sarcosine8) showed a heterogeneous free-energy landscape with close to 30 cis/trans isomers present within 5 kcal/mol of the lowest free energy. The most populated cis/trans isomer state in the REMD simulation was cttttttt (c: cis; t: trans), while ccttcctt, the cis/trans isomer state seen in the crystal structure of cyclo-(sarcosine8)254 was the third lowest free-energy state with a heavy-atom RMSD of 0.64 Å from the experimental crystal structure. This computational prediction was consistent with the ambiguous isomeric states observed in NMR.254 Later on, Butterfoss et al.128 used REMD combined with QM refinement to predict the structure of three peptoids, including a cyclic peptoid cyclo-(Nspe)9, an (S)-N(1-phenylethyl) glycine nonamer. The predicted structure after QM refinement had the same cis/trans pattern of cccctccct and distorted planar amide bonds as the crystal structure, with a backbone RMSD of 1.0 Å. However, some discrepancy was observed between the experimental and predicted ϕ angles, likely due to crystal packing and the presence of a bound solvent molecule observed in the crystal structure.128
Figure 14.
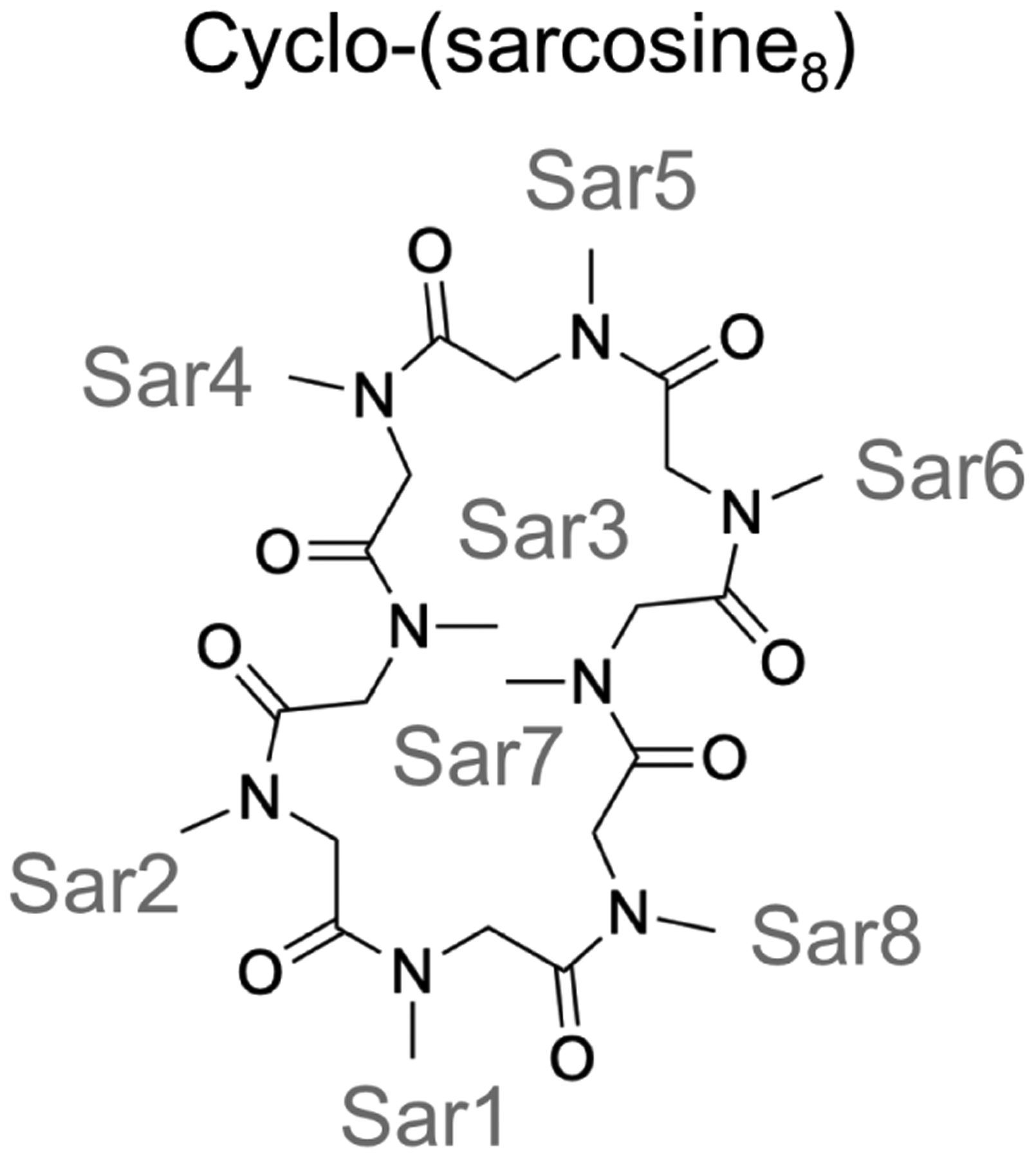
Cyclo-(sarcosine8) adopted a heterogeneous structural ensemble in the REMD simulations with about 30 cis/trans isomers within 5 kcal/mol, consistent with the ambiguous isomeric states observed in NMR.251,254
3.10. Water solubility and membrane permeability of cyclic peptidomimetics [McMD]
To investigate the relationship between the structural ensembles of cyclic peptides and their cell membrane permeability, Ono et al. simulated eight cyclic hexapeptides in explicit water, chloroform, and cyclohexane.190 For all the model cyclic peptides, residues 1, 2, and 6 were D-Pro, D-Leu, and L-Tyr, respectively; on the other hand, residues 3, 4, 5 could be D- or L-Leu, yielding a total of 8 diastereomers (Figure 15). Even though these cyclic peptides had the same molecular weight, their cell permeability varied by more than two orders of magnitude.190 To understand the origin of such differences in cell permeability, the structural ensembles of these cyclic peptides were simulated in water, chloroform, and cyclohexane using McMD186 coupled with virtual systems187,188 and trivial trajectory parallelization (TTP-V-McMD).255 Eight virtual states were employed and a flat potential-energy distribution between 280 K to 1525 K was obtained after 5–6 iterations. It was noted that although McMD simulations were usually performed to reach a flat potential energy distribution between 280 K to 700 K,256–261 1525 K was used as the highest temperature in this study to enhance the sampling of cyclic hexapeptides, which were expected to have higher energy barriers than linear peptides, as well as to sample the cis/trans isomerization of Pro. The AMBER-03 force field was used for the peptides;226 for the solvents, the TIP3P model was used for water,207 parameters from AmberTools 17262 were used for chloroform, and parameters derived from the Lipid14 force field263 and RESP264 charges were used for cyclohexane. The total length of the simulations was 6.72 μs for each system. The authors assessed the correlation between the cell permeability determined in a low-efflux MDCK cell line265 and the calculated structural properties of the cyclic peptides: intramolecular hydrogen bonds, solvent-accessible surface area (SASA), free-energy landscapes represented by molecular shapes (rod, sphere, and disk), and principal components from principal-component analysis of backbone RMSD. It was found that the average SASA in cyclohexane correlated well with the experimental cell permeability (R2=0.872). It was noted that, although chloroform was commonly used as a membrane mimetic, the correlation between the average SASA in chloroform and cell permeability was much weaker (R2=0.390). This observation is consistent with a previous finding that cyclohexane can serve as a good membrane-mimetic solvent.266 In addition, solubility in water was found to correlate with the average SASAs in explicit water (R2=0.755).
Figure 15.

Eight cyclic hexapeptide diastereomers were simulated using McMD simulation to characterize their structural ensembles in water and in organic solvents to explain their different water solubility and membrane permeability.190
Very recently, the same group studied the role of chameleonic properties in the membrane permeability of cyclic peptide-peptoid hybrids.267 It was previously proposed that macrocycles can adopt more polar conformations in water while adopting more nonpolar conformations in low-dielectric environment, and such chameleonic properties underpin their high water solubility and membrane permeability.268–270 By studying two libraries of cyclic peptide-peptoid hybrids, Furukawa et al. found that it is possible for both chameleonic and nonchameleonic compounds to achieve high membrane permeability and good water solubility.267 McMD was used to study the structural ensembles of two cyclic peptide-peptoid hybrids that both showed high membrane permeability. The simulation results confirmed that one compound was indeed nonchameleonic, adopting similar, rigid conformations in chloroform, acetonitrile, DMSO, and water, while the other compound was chameleonic, adopting different conformations in different solvents and also becoming more heterogeneous as the solvent polarity increased.267
3.11. Disulfide-bonded Boc-Cys-Pro-Xaa-Cys-OMe tetrapeptides [REMD]
Disulfide-bonded cyclic tetrapeptides with the sequence of Boc-Cys-Pro-Xaa-Cys-OMe have been shown to typically adopt a β turn at the Cys-Pro-Xaa-Cys motif (Figure 16), where Boc is a tert-butyloxycarbonyl protecting group, OMe a methoxy group, and Xaa a natural or artificial amino acid.271–273 The small size of these model peptides facilitates the use of spectroscopy analysis, including NMR, IR, CD, and VCD, as well as simulation methods, to characterize their structures. Merten et al. and Li et al. conducted a series of studies to characterize the conformational landscape of disulfide-bonded Boc-Cys-Pro-Xaa-Cys-OMe, with Xaa being glycine (peptide 1), L-leucine (peptide L-2) or D-leucine (peptide D-2) (Figure 16).131,272,274 The authors utilized REMD with quantum mechanics/molecular mechanics for conformation sampling and energy evaluation, together with a number of experimental techniques to investigate how solvent and the change of a single stereocenter (from L-Leu to D-Leu) affect the structures of these model peptides.131,272,274 For REMD simulations, the OPLSAA/L force field216 and a polar solvent, acetonitrile (CH3CN), were used; 23 replicas were spread in the temperature range of 290–400 K; the replica lengths ranged from 60 to 120 ns for different systems.131,274 The simulations showed that at 300 K, peptide 1 in Figure 16 predominantly adopts a type-II β turn (βII turn) at Pro2–Gly3, with a small population (7%) adopting a type-I β turn (βI turn) at the same location.131 On the other hand, peptide L-2 was found to adopt both the βI and βII conformations near 300 K, but the population of the βII structure decreased to 84%. A single stereocenter change of substituting Leu2 with a D-Leu, however, led to a well-structured peptide, which exclusively adopted the βII conformation at 300 K.274 These results were consistent with the NMR, IR and CD observations.272,274 VCD results suggested that peptide 1 notably adopted both βI- and βII-turn structures; L-2 predominantly adopted the βI-turn structure and D-2 predominantly adopted the βII-turn structure.131 Compared to the VCD results, REMD overestimated the amount of the βII-turn structure for peptides 1 and L-2, but recapitulated the relative trend that the likelihood of adopting the βII-turn structure was D-2>1>L-2.131,274 When it comes to the effects of solvent, MD simulations on peptide 1 in the gas phase, in acetonitrile, and in water revealed that the conformer group with a longer Cys1…Cys4 hydrogen bond distance (3.0 to 4.0 Å) was preferred (66.2%) in the gas phase. However, in solvent, the most preferred conformer group had a Cys1…Cys4 hydrogen bond length between 2.2 and 3.0 Å, showing the effect of peptide-solvent interactions. Differences between solvents were observed—in water, the population of the most preferred conformer group is lower than in acetonitrile (73.4% vs. 78.7%). The authors interpreted this to be a result of strong cooperative effects between the water molecules, which can form more hydrogen bonds, weakening the peptide–solvent interaction.272
Figure 16.
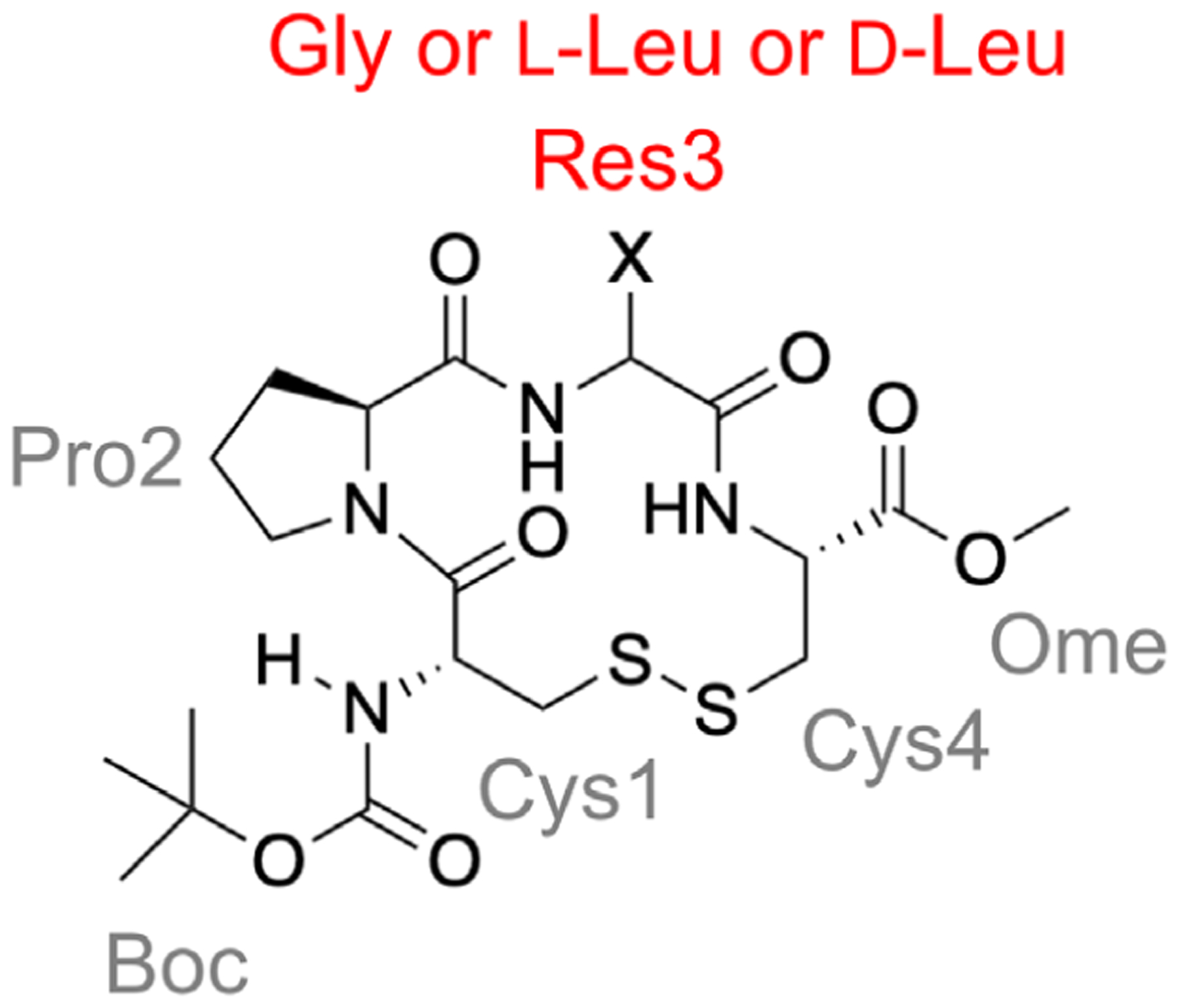
Structure of the disulfide-bonded model cyclic tetrapeptide used to model a β turn.131 Residue 3 is Gly for peptide 1 and L-Leu and D-Leu for peptides L-2 and D-2, respectively. Consistent with the experiment,272,274 in REMD simulations, the propensity of adopting a type-II β turn at Pro–Gly is D-2>1>L-2.
3.12. Sequence reversal effect on cyclic peptide structure [PTWTE]
The asymmetric structure of the amide bond (CONH) means that peptides are nonpalindromic; in other words, reversing the sequence of a peptide yields a different compound from the original peptide. While this observation may be trivial, it raises the question of whether there is a relationship between a peptide’s structural preferences, and those of its retroisomer. Retroisomerization is commonly applied in conjunction with chirality change, as part of retro-inverso drug design to achieve the same sidechain orientation as the original peptide but with improved enzyme stability.275–277 It is thus important to understand the impact sequence reversal has on peptide structure. An early hypothesis that retroisomers would assume structures that are mirror images of one another278 has since been disproven by CD, NMR, and X-ray crystallography experiments279,280.
Recently, Zerze et al. investigated the effect of sequence reversal on the conformational preferences of a number of peptide systems, including two cyclic peptides: cyclo-(GHGAYG) and cyclo-(GRCTKSIPPICFPD), the latter of which also contains a disulfide bridge between the cysteine residues.281 The authors performed PTWTE simulations using the AMBER-03w force field282 and the TIP4P/2005 water model.241 Seven replicas in the 300–450 K range were used for each system, with a bias factor of 10 for cyclo-(GHGAYG), and 16 for cyclo-(GRCTKSIPPICFPD); the replicas were run for at least 300 ns for each system. The same simulation parameters were used for their respective retroisomers.
The authors assigned structural features to the peptides’ residues based on the values of the backbone dihedral angles in the most populated cluster. The hexapeptide cyclo-(GHGAYG) and its retroisomer cyclo-(GYAGHG) displayed different conformational preferences between the two cyclic peptides when comparing residues 1–6 of cyclo-(GHGAYG) to residues 6–1 of cyclo-(GYAGHG). Both cyclic peptides were found to form type-I β turns at their respective residues 3–6, further suggesting that retroisomerization didn’t lead to a symmetric transformation of structural preferences. For cyclo-(GRCTKSIPPICFPD), Zerze et al. found that the retroisomer showed local α-region dihedral preferences centered at Lys-10, as well as polyproline-II-region dihedral angles at residues Ile-5 to Pro-7. Neither of these features were observed in cyclo-(GRCTKSIPPICFPD) itself, which predominantly assumed β-strand-like dihedral angle values. These findings suggest that, in retro-inverso drug design, it is important to consider structural changes brought about by retroisomerization, in addition to the impact of chirality change.
4. Using MD simulations to elucidate the solution structures of cyclic peptides: Target-focused studies
4.1. α-Fetoprotein-derived cyclic peptides [REMD]
REMD has been applied to enhance the conformational sampling of cyclic peptides and provide structural insights to help understand and design antiestrogenic peptides.283,284 These peptides are derived from α-fetoprotein, which displays antiestrogenic activity and can inhibit estrogen-dependent breast cancer.285,286 Further studies suggested that an octapeptide EMTPVNPG, residues 472–479 of the human α-fetoprotein, was the minimal sequence that afforded the antiestrogenic activity.287–289 Unfortunately, rational design of more lead compounds is challenging because little structural information is available on how α-fetoprotein interacts with its receptors. Nonetheless, it was reported that EMTPVNPG, EMTPTNPG, cyclo-(EMTPVNPGQ), cyclo-(EKTPVNPGQ), and cyclo-(EKTPVNPGN) all displayed antiestrogenic activity and were able to inhibit breast cancer to an extent comparable to the full α-fetoprotein (residues the same as those in the original octapeptide sequence are underlined).290 To identify common structural features these five active peptides share and help design more active compounds, Shields and coworkers used REMD simulations to characterize the solution structural ensembles of these peptides.283,291,292 The REMD simulations were carried out using the AMBER-99SB force field and an implicit water model,293,294 with 8 replicas spanning temperatures from 265 K to 700 K for 20 ns. Shields and coworkers found that all five of these active peptides adopted a type-I β turn at residues TPXN (Figure 17).283 To further test the importance of this β-turn motif, REMD simulations of EMTPVNP, TPVNP, TPVN, and PVNP were performed and their ability to inhibit estrogen-stimulated growth of immature mouse uterus was measured. Shields and coworkers found that the inhibition activity correlated with the extent to which the peptide forms the β-turn conformation at TPVN.
Figure 17.
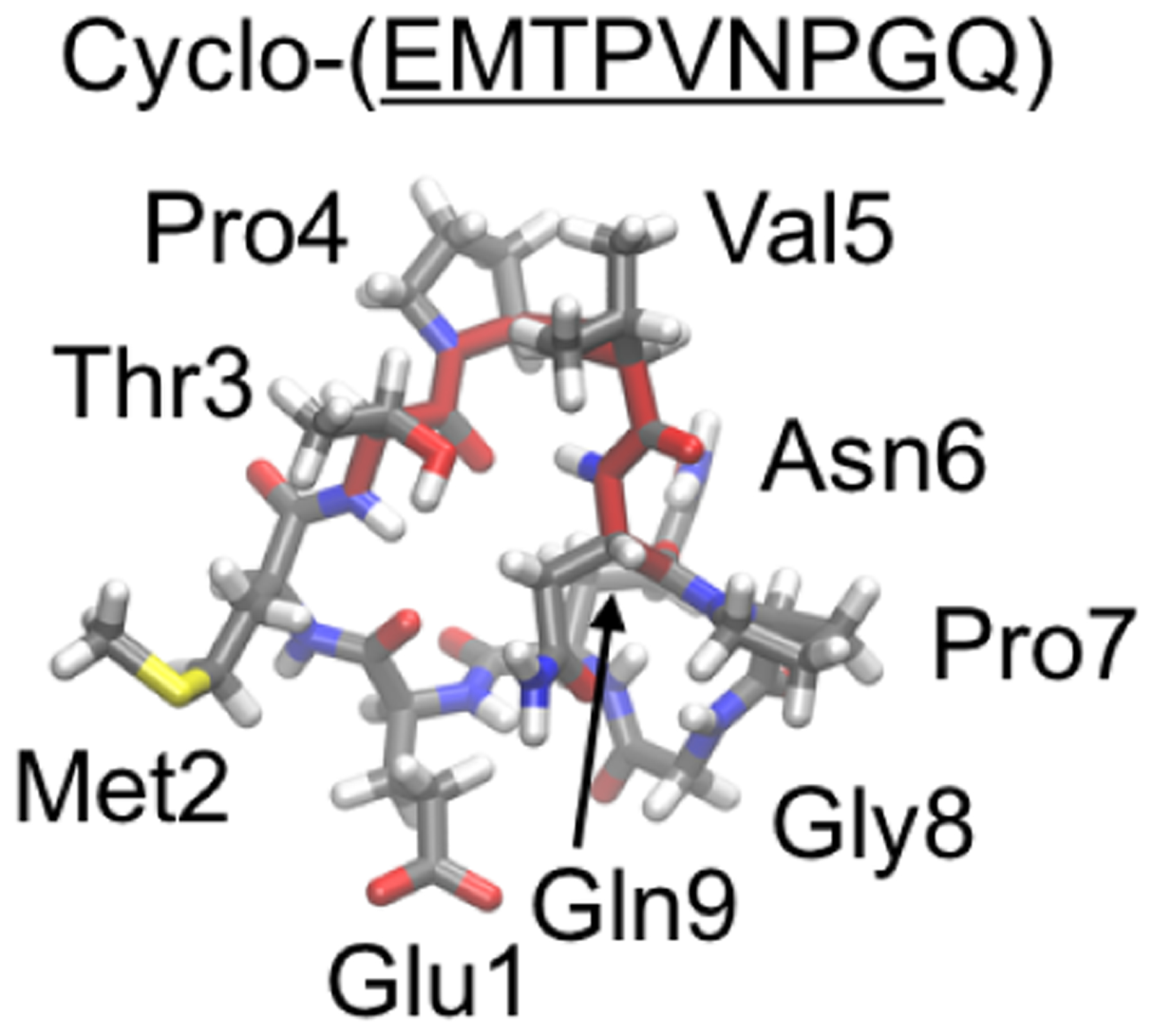
Representative structure of cyclo-(EMTPVNPGQ) in REMD simulation.283 The sequence (EMTPVNPG) derived from α-fetoprotein is underlined; type-I β turn at TPVN is highlighted in red.
4.2. Apelin-derived cyclic peptides [REMD]
The apelin receptor (also known as the APJ receptor) is a G protein-coupled receptor that is involved in a number of physiological functions, including the control of blood pressure and cardiac contractility.295 Apelin peptides are endogenous peptide ligands for the apelin receptor and exist in several isoforms, corresponding to C-terminal segments of varying lengths of the initial 55-residue proprotein. One of these peptides is apelin-13, a 13-residue peptide with the sequence QRPRLSHKGPMPF.296 Alanine-scanning studies on this peptide have shown that residues 2RPRLS6 and 8KGPM11 are the most important with regard to binding to the apelin receptor.297,298 NMR experiments on apelin-17, a 17-residue isoform of apelin-13, suggest that the 2RPRL5 residues adopt a type-IV β turn in solution and a type-I β turn in the presence of micelles.299,300 To understand the role of the RPRL motif in how the peptide binds to the apelin receptor, Macaluso and Glen designed 4 head-to-tail cyclic peptides, cyclo-(1QRPRLS6), cyclo-(1QRPRLSH7), cyclo-(1QRPRLSHK8), and cyclo-(1QRPRLSHKG9).301 REMD simulations were used to characterize the structural ensembles adopted by these cyclic peptides. The REMD simulations were carried out using the OPLS-AA force field and the TIP3P water model,207,302 with 16 replicas spanning temperatures from 298.0 to 352.8 K for 25–35 ns. Dihedral analysis showed that cyclo-(1QRPRLS6) and cyclo-(1QRPRLSH7) predominantly adopted β turns at residues RPRL (Figure 18), with 90% of cyclo-(1QRPRLS6) forming a type-II β turn and 51% and 10% of cyclo-(1QRPRLSH7) forming a type-II and type-I β turn, respectively, at residues RPRL during the simulations. In contrast, cyclo-(1QRPRLSHK8) and cyclo-(1QRPRLSHKG9) tended to adopt a β turn at residues RLSH instead, with 49% and 21% cyclo-(1QRPRLSHK8) forming a type-II and type-I β turn, respectively, and 21% of cyclo-(1QRPRLSHKG9) forming a type-I β turn at residues RLSH during the simulations (Figure 18). In vitro experiments showed that cyclo-(1QRPRLS6) and cyclo-(1QRPRLSH7) displayed much stronger inhibition than cyclo-(1QRPRLSHK8) and cyclo-(1QRPRLSHKG9), suggesting that a β turn at residues RPRL is indeed vital to binding to the apelin receptor. It was also noted that, although the larger cyclic peptides had more residues derived from apelin-13, the head-to-tail cyclization stabilized a β turn at residues RLSH via a side chain–backbone hydrogen bond. The subsequent absence of a β turn at residues RPRL led to a decrease in binding affinity for the apelin receptor.
Figure 18.
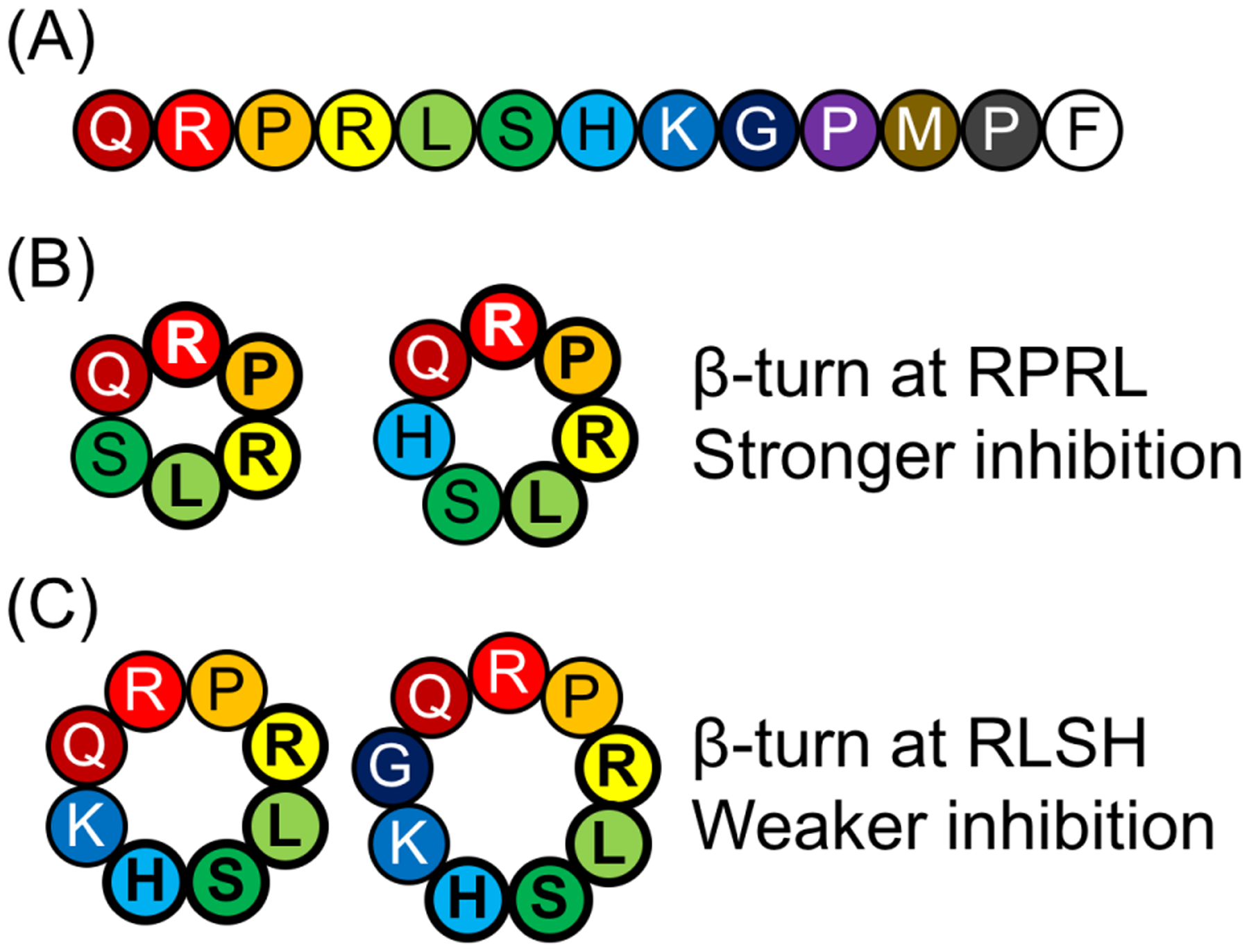
Cyclic peptides derived from apelin-13.301 (A) Sequence of apelin-13. (B) In REMD simulations, cyclo-(QRPRLS) and cyclo-(QRPRLSH) adopted a β turn at residues RPRL and showed stronger inhibition in experiment. (C) In contrast, cyclo-(QRPRLSHK) and cyclo-(QRPRLSHKG) adopted a β turn at residues RLSH and showed weaker inhibition in experiment.
Later on, to facilitate the design of bivalent ligands for the apelin receptor, REMD simulation was used to verify that disulfide-bonded CRPRLC also adopted a β turn at residues RPRL.303 Two disulfide-bonded CRPRLC motifs were then connected with various linkers, generating [CRPRLC]-A-[CRPRLC], [CRPRLC]-AA-[CRPRLC], [CRPRLC]-GG-[CRPRLC], [CRPRLC]-HK-[CRPRLC], and [CRPRLC]-KH-[CRPRLC]. REMD simulations showed that all these bivalent designs formed multiple structural clusters in solution but tended to adopt a β turn at residues RPRL. Experimental competition and affinity assays found that [CRPRLC]-KH-[CRPRLC] to be a promising competitive antagonist for the apelin receptor.303 In a later study, [CRPRLC]-KH-[CRPRLC] was shown to suppress glioblastoma growth, by preventing apelin-regulated expansion of glioblastoma stemlike cells.304
4.3. LapD-derived cyclic peptides [REMD]
Simulating the behavior of proposed candidate molecules can help streamline the process of designing and screening peptides or peptidomimetics that primarily adopt desired conformations. To demonstrate the potential usefulness of MD simulations in cyclic peptide design, Razavi et al. used REMD simulations to efficiently sample the conformational space and computationally screen 20 cyclic peptidomimetics designed to mimic a β-hairpin structure of the bacterial protein LapD.305 This β hairpin of LapD is critically involved in the interaction between LapD and LapG, a key protein–protein interaction for bacterial biofilm formation.306,307 Inspired by the hairpin sequence VSRGWEQAA of LapD from P. fluorescens, two scaffolds were designed to preserve its hydrogen bonding pattern and the salt bridge between the Arg and Glu residues. The first scaffold is a doubly cross-linked 9-mer bearing the same 9-amino-acid sequence 1VSRGWEQAA9, with one central linker connecting the side chains of Ser2 and Gln7, and a peripheral linker connecting the backbones of Val1 and Ala9 (Figure 19). For this scaffold, four stereochemical and substitutional effects were investigated: L- or D-valine at residue 1, E or Z double bond in the central linker, E or Z double bond in the peripheral linker, and H or CH3 as the R group of the peripheral linker, resulting in a total of 16 designs, herein called L-Val-E-E-H, L-Val-E-E-CH3, etc (Figure 19). The second scaffold contained only the middle seven residues, SRGWEQA, with a single peripheral linker connecting the backbones of Ser and Ala. For this scaffold, the double bond in the peripheral linker could be in the E or Z form and the R group of the peripheral linker was chosen to be either H or CH3, resulting in a total of 4 designs.
Figure 19.
Sixteen cyclic peptide designs derived from LapD. REMD simulation results show that L-Val-E-Z-H, L-Val-E-Z-CH3, D-Val-E-Z-CH3, and D-Val-E-Z-H are the best at mimicking the target β-hairpin structure.305
All 20 of these cyclic peptide designs were first simulated using REMD with implicit solvent. The AMBER-96 force field225 was used for the standard amino acids and the GAFF force field215 for other moieties of the cyclic peptide molecules, along with the GBSA-OBC implicit solvent;252 24 replicas spanning in the temperature range of 300–1000 K were used and each replica was run for 1–2 μs. Because the high temperatures used in the REMD simulations caused trans/cis isomerization of the backbone amide bonds, only all-trans configurations were selected for analysis.
To screen for cyclic peptide designs that best mimic the target β-hairpin structure, the free-energy profile of each system was generated with respect to the RMSD from the X-ray structure, calculated using the backbone and Cβ atoms of the six residues in the hairpin, SRGWEQ. It was found that while none of the 7-mer designs mimicked the target β-hairpin structure well, the free-energy profiles of the 9-mer designs did show a basin near an RMSD of 1.5 Å (called “near-native”) and a basin with RMSD <1 Å (defined as “nativelike”). Among all the sixteen 9-mer designs, L-Val-E-Z-H, L-Val-E-Z-CH3, D-Val-E-Z-H, and D-Val-E-Z-CH3 sampled the highest percentage of the nativelike structures at 300 K. It was also noticed that while changing the R group from H to CH3 as in the L-Val-E-Z-H and L-Val-E-Z-CH3 designs resulted in a destabilization of the nativelike conformation relative to the near-native conformation by ~0.5 kcal/mol, changing the R group from H to CH3 as in the D-Val-E-Z-H and D-Val-E-Z-CH3 designs resulted in a stabilization of the nativelike conformation relative to the near-native conformation by ~1 kcal/mol.
The four best designs (L-Val-E-Z-H, L-Val-E-Z-CH3, D-Val-E-Z-H, and D-Val-E-Z-CH3) were then subjected to large-scale parallel explicit-solvent MD simulations using Folding@home.308 The AMBER-99SB-ILDN force field218 with the TIP3P water model207 was used and a total of over a millisecond of simulation trajectory data were collected. Markov state models built from these simulations verified that these designs indeed adopted native or nativelike conformation with significant populations. Although these designs and insight were purely computational, the study demonstrated how MD-based methods can be applied to characterize and understand the conformational space of cyclic peptides and aid their screening and rational design.305
4.4. Lens epithelium-derived growth factor-derived cyclic peptides [metadynamics]
The viral enzyme HIV integrase is considered an anti-HIV drug target due to its involvement in HIV replication.309,310 More specifically, the entry of the HIV pre-integration complex (PIC) and its localization to host DNA is mediated by the interaction between HIV integrase and the lens-epithelium-derived growth factor (LEDGF).309,311 LEDGF contains a hot loop in the form of a type-I β turn at residues 364–367 (sequence KIDN). Northfield et al. designed four cyclic peptides with the goal of mimicking this hot loop: disulfide-bonded Ace-CKIDNC-NH2, cyclo-(PKIDNp), cyclo-(PKZDNv), and cyclo-(PKIDNG), with lowercase letters denoting D-amino acids, and Z denoting norleucine.312
X-ray structures of the peptide–integrase complex showed that these peptides bound to the HIV integrase via conformations closely matching the shape of the LEDGF binding loop; however, the measured affinities were > 10 mM for cyclo-(PKIDNG), and in the low millimolar (3–6 mM) range for the other peptides.312
To explain the extremely low binding affinity of cyclo-(PKIDNG), Northfield et al. calculated its binding free energy by first performing umbrella sampling simulations to gradually pull the ligand from its binding site, leading to a projected Kd of 2.49 mM at 300 K. However, as umbrella-sampling simulations can overestimate free energies due to insufficient sampling and underestimation of the entropic cost of binding, the authors further conducted well-tempered metadynamics simulations to characterize the cyclic peptide’s conformational ensemble in solution. Three transpeptide distances were used as the collective variables, and the simulations were run under the GROMOS-54a7 force field with SPC/E water for 100 ns.212,240 The conformation that had the lowest energy in solution had a backbone RMSD of 1.63 Å from the bound pose. In solution, the bound pose was 6.77 kJ/mol higher in energy compared to the lowest-energy solution conformation. The calculated Kd of 30.1 mM agreed with the experimental value. These results show that it is critical to know the solution structural ensembles adopted by cyclic peptides to understand their binding affinity.
4.5. Neuropeptides: oxytocin, vasopressin, and urotensin II [REMD, metadynamics]
Oxytocin and vasopressin are two cyclic peptide hormones that are involved in social cognition and bonding, with vasopressin also acting as an antidiuretic hormone.4 While their primary biological roles are distinct, both oxytocin and vasopressin are nonapeptides with a six-membered tocin ring formed by a Cys1–Cys6 disulfide bond and a three-residue-long amidated tail. The sequences of oxytocin and vasopressin, CYI3QNCPL8G-NH2 and CYF3QNCPR8G-NH2, respectively, differ in only two residues at positions 3 and 8 (Figure 20). Oxytocin and vasopressin are secreted by the posterior pituitary gland in complex with their neurophysin (NP) carrier proteins (oxytocin with NP I and vasopressin with NP II),313,314 which shepherd the peptide hormones to their corresponding target receptors. The oxytocin receptor is a G-protein coupled receptor, notably expressed in the mammary glands and the uterus at the end of pregnancy. The oxytocin pathway is involved in inducing contractions during birth. The oxytocin receptor is also expressed in the central nervous system and regulates a variety of social behaviors – bonding, anxiety, sexual and aggressive behavior, among others. Vasopressin, on the other hand, binds to a number of receptors, classified into three main subtypes – V1 (or V1A), V2, and V3 (or V1B). The V2 receptor is expressed in the kidney tubule, and loss of its function is associated with nephrogenic diabetes insipidus.315–317
Figure 20.
Oxytocin and vasopressin. Well-tempered metadynamics simulations show vasopressin adopts two structures in solution with 70% and 30% populations.324 Reservoir REMD was used to simulate oxytocin and vasopressin, along with various variants to understand how mutations affect their structural ensembles and the different binding affinities.325
Owing to their sequence similarities, it is not surprising that oxytocin and vasopressin can also interact with each other’s receptors.318 Similar to other intrinsically disordered peptides, the spectroscopic characterizations of these two cyclic peptides in solution suggest averaging over a number of conformations,108,109 and crystal structures have only been resolved for deamino-oxytocin,319 for the cyclic moiety of vasopressin,320 or in complexes with their carrier proteins.313,314 In addition, a crystal structure has been obtained for a trypsin–vasopressin complex, suggesting vasopressin could be used to inhibit this enzyme.321 Therefore, to better understand the solution structures adopted by these two peptide hormones and the effects of mutations, various computational methods including molecular mechanics, Monte Carlo, MD, REMD, and metadynamics have been employed.322–325 Below we focus on discussing recent computational studies that used modern force fields and enhanced sampling methods to characterize the solution structural ensembles of oxytocin and vasopressin.323–325
To characterize the solution structural ensemble of vasopressin, Haensele et al. used microsecond MD simulations and well-tempered metadynamics.323,324 In a 23 μs long MD simulation with the AMBER-99SB force field and the TIP4P-Ew water model,217,294 four major conformations of the tocin ring, open, saddle, clinched-open, and twisted saddle, were identified, while the three-residue tail was found to switch between an extended and a folded conformation frequently. Using these conformations as a path variable, Haensele et al. performed well-tempered metadynamics simulations to calculate the relative free energies for these four conformations, which showed that the saddle and clinched-open structures predominate in solution with ~70% and ~30% of the total population, respectively.324 DFT calculations were then performed on the representative structures for each conformation to calculate the isotropic magnetic shielding for 1H and 13C, which was then converted to chemical shifts and weighted by the equilibrium population from the metadynamics simulations. The calculated 1H chemical shifts reproduced the experimental values with a standard error of < 0.24 ppm, whereas the error was < 2.2 ppm for 13C chemical shifts.
In addition to the oxytocin receptor, it is known that native oxytocin is capable of interacting with the V2 receptor for vasopressin.326 Studies aiming to develop more specific agonists for these receptors have shown that the single mutation Q4T increases oxytocin’s binding affinity and activity for the oxytocin receptor twofold, but reduces the activity for the V2 receptor for vasopressin to 10–25% of that of native oxytocin.327–329 On the other hand, the double mutation Q4T/P7G reduces the activity for the oxytocin receptor to < 1/3 of native, and the activity for the V2 receptor for vasopressin to essentially zero. Additionally, two rare vasopressin mutations have been implicated in different mechanisms of the pathogenesis of diabetes insipidus.330 The Y2H mutant likely does not bind to the carrier protein NP II correctly, which makes it difficult for it to leave the posterior pituitary. P7L can bind NP II, but has a lower receptor binding affinity.331
Hypothesizing that geometric features of the solution structure of intrinsically disordered peptides like oxytocin and vasopressin may influence their binding affinity and specificity for receptors, Yedvabny et al. performed reservoir REMD simulations on the native and mutant sequences of these two cyclic peptides, i.e. oxytocin, oxytocin Q4T, oxytocin Q4T/P7G, vasopressin, vasopressin Y2H, vasopressin Y2H+ (where the histidine was protonated), and vasopressin P7L.325 Twenty-four replicas ranging from 298.15 to 442.45 K, and a reservoir of structures pre-equilibrated at 450 K were used. The production run was 50 ns long for each replica, under the AMBER-99SB-ILDN force field, with ϕ and ω corrections, and TIP4P-Ew water.217,218,332,333
The simulation results showed that both oxytocin and vasopressin adopt two sub-ensembles, one with a compact C-terminal tail, and another where the tail is extended. There were indeed differences in the populations of different transannular hydrogen bonds within the tocin ring between vasopressin and oxytocin. However, overall, both peptides adopted a β turn and an α turn within the tocin ring, stabilized by backbone hydrogen bonds between residues 2 and 5 and between residues 2 and 6, explaining the cross-agonism oxytocin and vasopressin display with each other’s receptors.
As mentioned above, the Q4T mutation of oxytocin results in a higher binding affinity for the oxytocin receptor, but a lower binding affinity for the V2 receptor for vasopressin, while Q4T/P7G mutations severely reduce the affinity for both receptors, virtually eliminating it in the case of the V2 receptor for vasopressin.327,328 In the simulations, oxytocin Q4T was found to be more rigid than native oxytocin, adopting a higher percentage of the compact structure and forming 6–9 and 7–9 hydrogen bonds more frequently. The double mutant maintained the canonical ring structure; however, the 6–9 β-turn population decreased to 11% (compared to 50% and 71% for the native and single mutant forms, respectively), meaning that the tail adopts a variety of other interactions with the tocin ring. These observations suggest that rigidity of the tocin ring and the tail compactness are factors which influence oxytocin binding affinity.
Simulations of vasopressin mutants also yielded results consistent with hypotheses based on experimental results. The Y2H mutant was found to frequently (82%) form a hydrogen bond between the His side chain and the tocin backbone, making it unlikely for the side chain to be exposed and available for neurophysin binding. The P7L mutant showed no distinct compact or extended conformations, and instead formed an evenly distributed ensemble. It also failed to maintain the canonical tocin ring, much like the Y2H+ mutant. These two mutants were found to have a much higher population within a 2.5 Å RMSD to a trypsin-inhibiting vasopressin structure than the others, lending support to Christensen et al.’s hypothesis that the P7L mutant could prevent enzymes from excising the hormone from the proform.325,330
Yedvabny et al. also showed that the predicted structural ensembles strongly depend on the force fields used.325 For example, while the AMBER-99SB-ILDN force field with ϕ and ω corrections218,332,333 predicted that oxytocin would adopt two subensembles with Rg centered around 4.3 Å and 5.4 Å, the AMBER-99SB294 force field predicted an additional structure with a much more extended tail conformation. This new structure lacks the typical 2–5 and 2–6 hydrogen bonds seen in a tocin ring.
The 6-membered disulfide-bridged ring is also seen in other neuropeptides, notably in urotensin II — the strongest known vasoconstrictor, involved in the function of the cardiovascular, endocrine, and renal systems.334–337 In addition to the 6-membered ring, it also contains an N-terminal tail, the length of which varies among species.335,337 Replacing the tail with a single alanine residue yields urotensin-related peptide (Figure 21), which displays similar binding affinity for the urotensin II receptor.338,339
Figure 21.
Human urotensin II and urotensin-related peptide. REMD simulations showed that both peptides adopted multiple conformations in solution with the open:folded ring conformation of 72:28 for urotensin II and 86:14 for urotensin-related peptide.340
Haensele et al. performed conventional MD and REMD simulations of both human urotensin II and urotensin-related peptide in solution in order to determine their conformational equilibria, and elucidate the effect of the tail presence on conformational behavior of urotensin II.340 The conventional MD simulations on the 10 μs scale (35 μs for urotensin II and 22.8 μs for urotensin-related peptide) yielded few interconversions between states, suggesting enhanced sampling was needed to exhaustively explore the conformational space of these peptides. To that end, for each peptide, the authors performed three sets of 500 ns REMD simulations with different initial conformations. 64 replicas were used, spanning a temperature range from 298 K to 543 K, using the AMBER-99SB force field294 and the TIP3P water model.207 Proton chemical shifts were calculated based on the equilibrium populations, and compared to NMR experimental data.
Both peptides were found to assume multiple conformations in solution but unlike vasopressin, urotensin II and urotensin-related peptide preferred an open-ring conformation than a folded-ring conformation (open: with no or sparsely populated transannular hydrogen bonds; folded: with highly populated transannular hydrogen bonds). The ratio of open:folded ring conformation was 72:28, 71:29, 79:21 from the three sets of REMD simulation for urotensin II and 86:14, 94:6, 91:9 for urotensin-related peptide, suggesting that the folded conformation is more stable in urotensin compared to urotensin-related peptide. By calculating the chemical shifts from each of the three sets of REMD simulations and comparing the results to the experiments, the authors found that the REMD simulation with 72:28 open:folded for urotensin II and the REMD simulation with 86:14 open:folded for urotensin-related peptide were the most consistent with the NMR results. It was noted that there were multiple subtypes of conformations within the open and folded states and it was rather challenging to converge the population distribution of these subtypes.340
When it comes to tail conformations, principal-component analysis results implied covariance between ring (open and folded) and tail (folded and extended) conformations in most cases. This correlation could potentially explain the differences in bioactivity between urotensin II and urotensin-related peptide. This behavior also distinguishes urotensin II from vasopressin,323 likely due to urotensin II’s longer tail enabling ring–tail interactions such as hydrogen bonding.340
4.6. Photoswitch-embedded cyclic peptides [REMD]
As peptides become increasingly promising inhibitors of protein–protein interactions, further incorporating a photoswitch linker in the construct has been proposed to reversibly control the structures and functions of the molecules.341–347 For example, Moroder and coworkers used ACATCDGF, residues 134–141 in the active site of thioredoxin reductase to develop photoswitchable cyclic peptides.348–351 In thioredoxin reductase, these 8 residues are disulfide-bonded and form a 310 helix;352 the octapeptide fragment in its oxidized form retains such helicity.353 Because of its high helix propensity, the sequence ACATCDGF was chosen to study the effects of embedding an azobenzene photoswitch. The backbone of the octapeptide was cyclized with 4-(4-aminomethyl)phenylazobenzoic acid to create a bicyclic peptide (bcAMPB in Figure 22). NMR experiments showed that the cis- and trans-azo-isomers of this photoswitch-embedded cyclic peptide had different flexibility.350 Using the NMR restraints, 10 lowest-energy structures were derived. The 10 structures of the trans-azo-state formed a rather tight ensemble with a backbone RMSD of 0.19 Å. On the other hand, the cis-azo-state appeared frustrated and adopted multiple conformations (backbone RMSD 1.54 Å).
Figure 22.
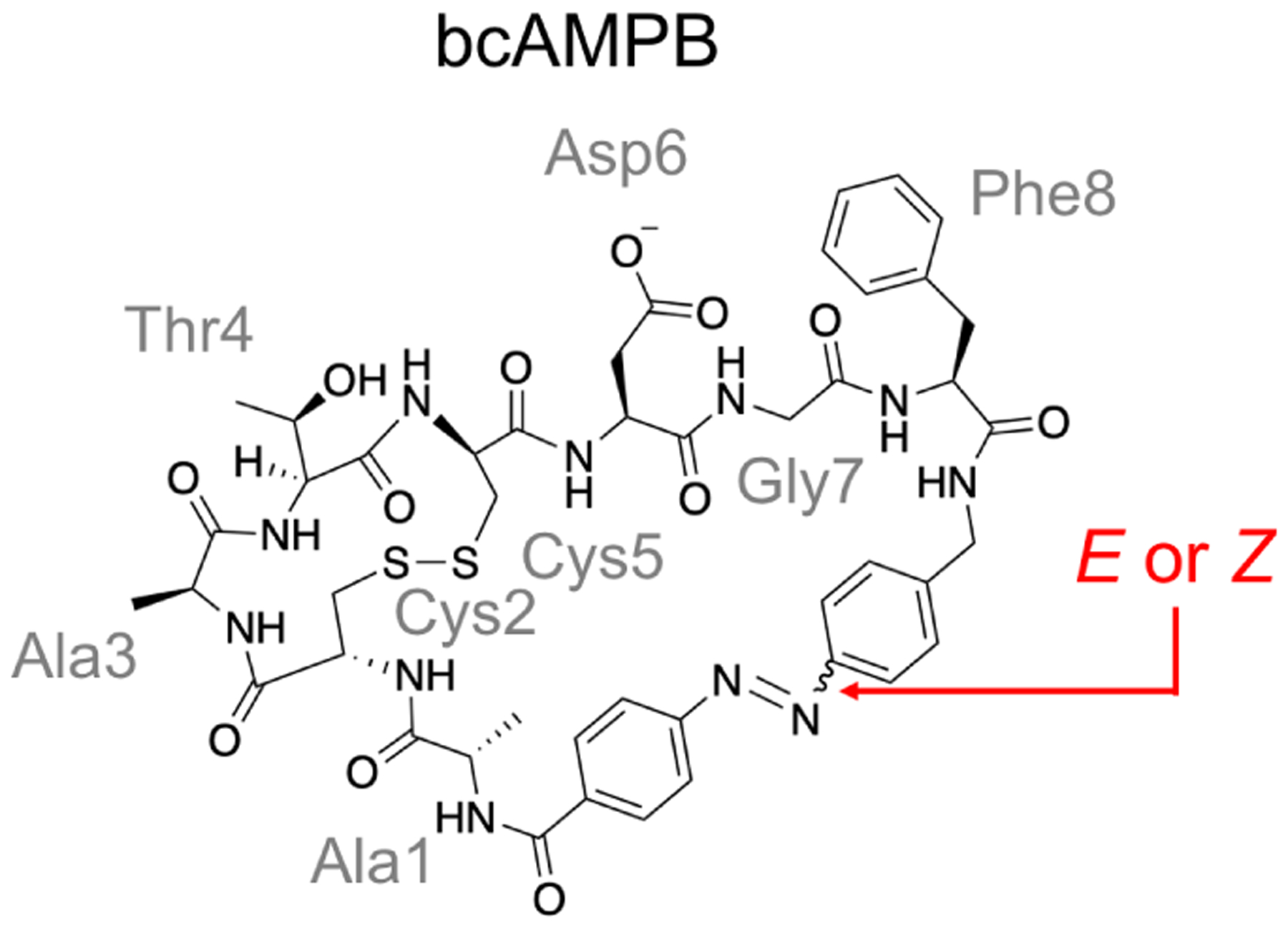
Structure of the bicyclic peptide bcAMPB (ACATCDGF with a disulfide bond and a cis/trans isomerizable photoswitch embedded). REMD simulations show that the trans-azo-isomer is well-structured, while at least four conformers are identified for the cis-azo-isomer, consistent with the NMR results.350,354
To test whether simulations can be used to predict the structures of such photoswitchable cyclic peptides, Nguyen et al. used REMD simulations to sample the structural ensembles of both the trans- and cis-azo-isomers and characterize their free-energy landscapes.354 Twenty eight replicas ranging from 295 K to 453 K were used and each replica was run for 10 ns. The GROMOS96–43a1 force field was used for the peptide,355 and united-atom DMSO by Liu et al. was used for the solvent.356 The parameters for the azobenzene unit were derived using Gaussian98.357 Two separate sets of REMD simulations were performed for the cis- and trans-azo-isomers, respectively. It was found that the REMD replica at 295 K agreed well with the experimentally derived NMR interproton distance restraints. The trans-azo-isomer was shown to be rather well-structured, adopting two states differing in the dihedral preference of residue Gly7. The simulated cis-azo peptide depicted a “frustrated” picture, in which at least four conformers were identified. Nguyen et al. further pointed out that REMD simulations were necessary to obtain the correct structural ensemble for bcAMPB. For example, the cis-azo-isomer was clearly trapped during the conventional MD simulation at 295 K; on the other hand, while the peptide sampled the free-energy landscape better during the conventional MD simulation at 453 K, the resulting conformational distribution did not resemble the results at room temperature.
4.7. RGD-related peptides [REMD, metadynamics, BE-META]
Integrins are transmembrane α/β heterodimer receptors and they interact with many proteins in the extracellular matrix (ECM). These integrin–protein interactions are critically involved in cell adhesion and migration, and their importance in cancer progression and metastasis makes integrin antagonists promising anticancer therapeutics.358 The tripeptide Arg-Gly-Asp (RGD) motif, first discovered in the FN-III10 repeat of fibronectin (FN),359 is found in a number of ECM proteins and is identified as an important motif that binds to several members of the integrin family, notably integrins αIIbβ3, αvβ3 and αvβ5.360–364 Various designs of RGD-containing cyclic peptides have since been developed to selectively target specific integrins.203,234,365,366 Among the cyclic peptides studied, cilengitide, cyclo-(RGDfV), where the lowercase f denotes D-Phe and the underlined V denotes N-methylated Val, had previously drawn special attention. Designed by Kessler et al.234,367, cilengitide had been shown to be a selective inhibitor for integrin αvβ3 in vitro234,367,368 and had demonstrated efficacious tumor regression in vivo, though clinical trials were eventually unsuccessful.204,205,369 The crystal structure of cilengitide in complex with a segment of integrin αvβ3 has also been resolved (PDB ID: 1L5G).202
The primary focus of a number of studies on RGD-containing peptides has been the extent to which the desired bioactive conformation is stabilized in solution. These efforts have resulted in a rich amount of information on the structural features of these peptides and provided ideal benchmarks for testing computational methods for structure prediction of cyclic peptides. For example, to verify whether simulation could be used for modeling cyclic peptides, Wakefield et al. performed REMD simulations of 18 RGD-containing cyclic pentapeptides (Figure 23).370 These cyclic peptides, including cilengitide, contained D-amino acids, N-methylated amino acids and/or other non-natural amino acids, and NMR data had been previously published for 13 of them.234,366,371,372 The REMD simulations were carried out using the AMBER-96 force field225 (with GAFF215 for non-standard amino acids) and the GBSA-OBC solvation model,252 with 8 replicas spanning temperatures from 300 K to 450 K and a simulation time of 2.4 μs for each replica. For the 13 cyclic peptides for which NMR results were available, the average violations against the experimental NOEs were all reasonably small (<0.4 Å) with R2 of 0.444–0.805. It was found that although cilengitide was not as well-structured as most of the other cyclic peptides, the most populated cluster (5.3%) of cilengitide indeed resembled the structure of cilengitide bound to integrin (PDB ID: 1L5G), consistent with its strong binding affinity. It was also observed that, although N-methylation has the potential to increase backbone flexibility by increasing the amide cis/trans isomer ratio, in the cyclic peptides tested N-methylation in general reduces the conformational entropy by about 0.5 kcal/mol.370
Figure 23.
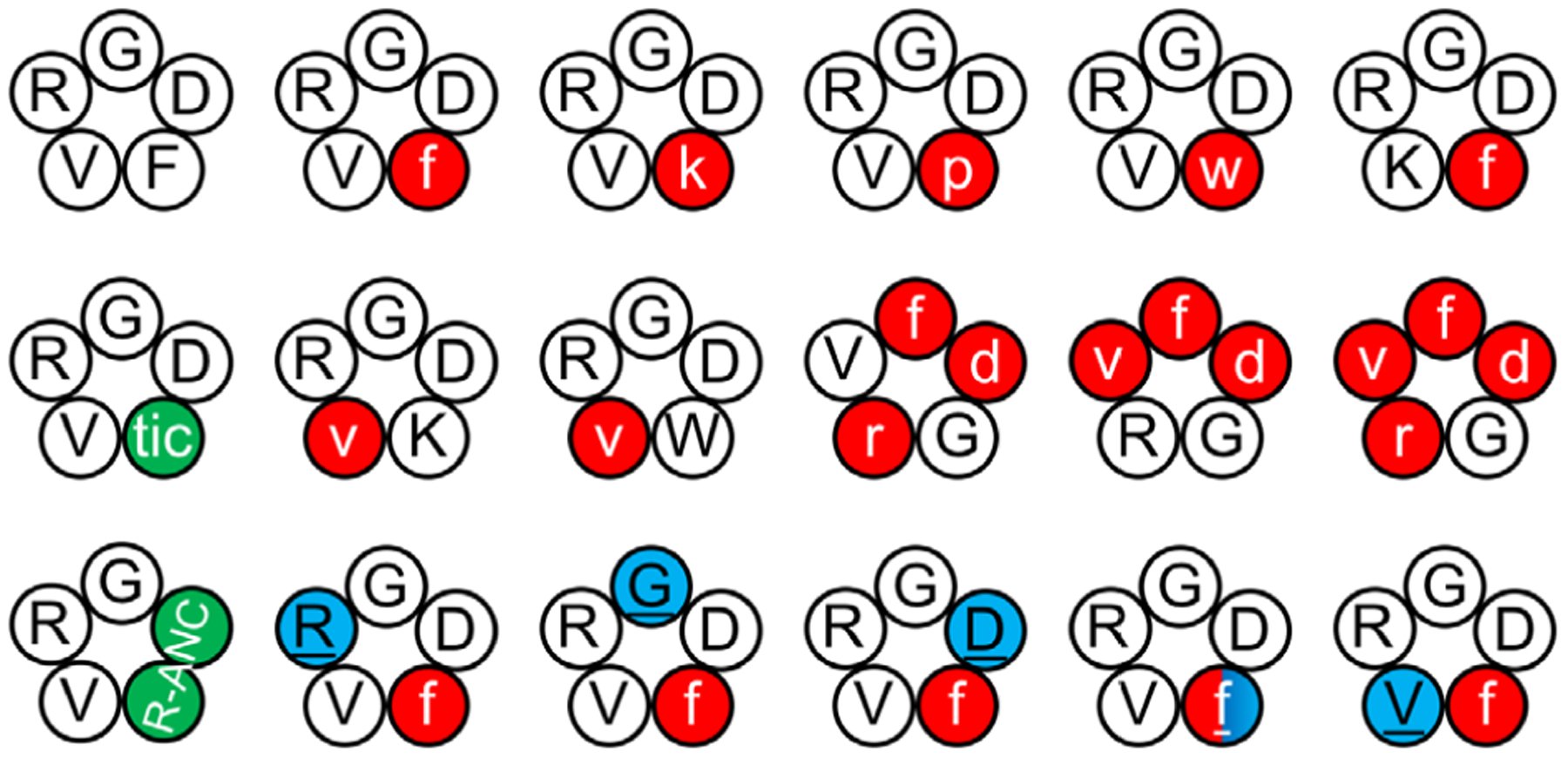
Eighteen RGD-related cyclic pentapeptides were simulated using REMD simulation.370 D-amino acids are listed in lowercase and shown in red; N-methylated amino acids are underlined and shown in blue; artificial amino acids are shown in green. All cyclic peptides formed multiple clusters in the simulations. The most populated cluster of cilengitide resembled the structure of cilengitide bound to integrin.
One of the greatest obstacles to effective therapeutic use of cyclic peptides is the difficulty associated with determining their structures, and additionally, the effects of popular modifications commonly used to fine-tune cyclic peptide properties, such as N-methylation. To examine the impact of N-methylation on cyclic peptide structure and binding affinity, Paissoni et al. simulated an RGD-containing cyclic hexapeptide, cyclo-(GRGDfL), along with four derivatives with various N-methylation patterns (Figure 24) using BE-META, docking, and Prime/MM-GBSA rescoring.236 More specifically, the sequences of the five cyclic peptides were: (1) cyclo-(GRGDfL), (2) cyclo-(GRGDfL), (3) cyclo-(GRGDfL), (4) cyclo-(GRGDfL), and (5) cyclo-(GRGDfL), where f denotes D-Phe, and N-methylated amino acids are underlined. The IC50 values for integrin αIIbβ3 of all five cyclic peptides had been previously published.373 Out of these five cyclic peptides, cyclic peptides 2 and 5 were the most potent binders for integrin αIIbβ3 with IC50 on the order of 10−8 M (Figure 24).373 To determine whether simulation could be used to characterize the structural ensembles of these cyclic peptides and rationalize their different binding affinity for integrin αIIbβ3, Paissoni et al. first used BE-META to exhaustively sample the conformational landscape of the cyclic peptides. Six replicas were used to bias the ϕ and ψ angles of non-glycine residues, and each replica was run for 320 ns. The all-atom OPLS force field216 and the TIP3P water model207 were used. Four major conformations (called minima A–D) were identified from the BE-META simulations and cluster analysis, and the populations of each minimum for the five cyclic peptides were calculated. It was found that cyclic peptides 2 and 5, the two cyclic peptides that have the lowest IC50 values, both favored minimum B (population of minimum B was 0%, 73%, 0%, 37%, and 82% for cyclic peptides 1–5, respectively).
Figure 24.
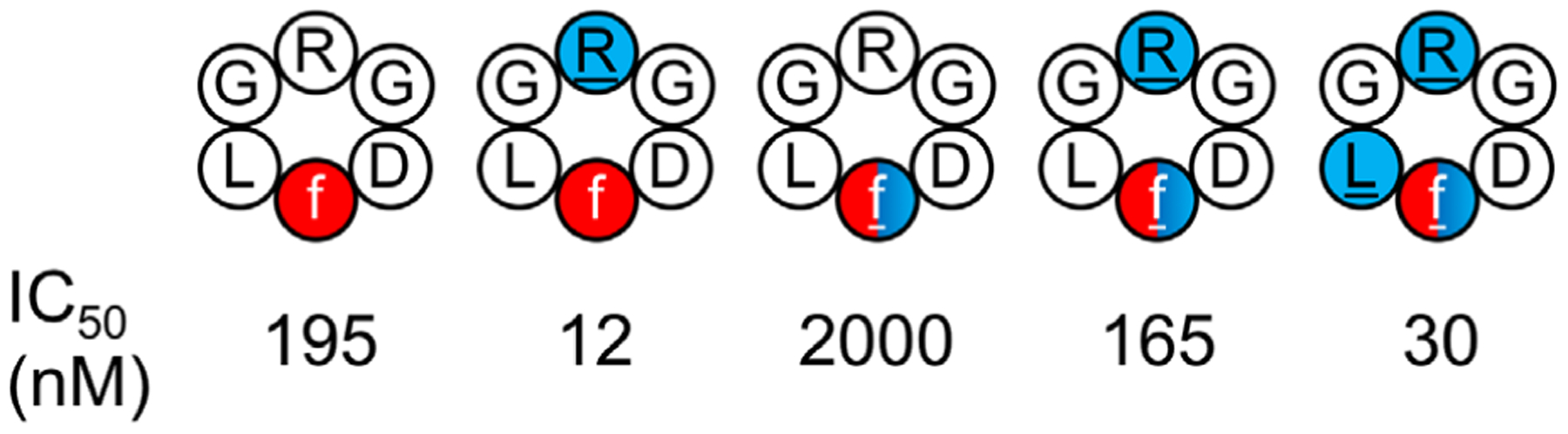
Five RGD-related cyclic hexapeptides were simulated using BE-META simulation.236 D-amino acids are listed in lowercase and shown in red; N-methylated amino acids are underlined and shown in blue. The IC50 values for integrin αIIbβ3 are listed. Four major conformations (called minima A–D) were identified. Cyclic peptides 2 and 5, the two cyclic peptides that have the lowest IC50 values, both favored minimum B (population of minimum B was 0%, 73%, 0%, 37%, and 82% for cyclic peptides 1–5, respectively).
To understand how these structural differences affect the cyclic peptides’ binding affinity for integrin αIIbβ3, the structures sampled from the four minima discovered in the BE-META simulations were then docked to the crystal structure of integrin αIIbβ3 (PDB ID: 2VDN374) using the Glide software tool to generate decoy poses.375 The decoy poses were then clustered based on RMSD and ranked using Prime/MM-GBSA, which had been shown to give more accurate binding affinity predictions for polypeptides than Glide.376 As a result of docking and Prime/MM-GBSA rescoring, the canonical interactions between Arg and Asp in the RGD motif and the integrin αIIbβ3 binding surface374 were reproduced in the three top-ranked clusters with the best cluster exhibiting additional stabilizing hydrophobic interactions compared to the other two. Importantly, all the conformations in the best cluster originated from minimum B. This finding is consistent with the observation that cyclic peptides 2 and 5, which strongly favored (>70%) conformation B, showed the highest affinities for integrin αIIbβ3 (Figure 24).
isoDGR.
Recently, another peptide sequence, Asn-Gly-Arg (NGR), located in the FN-I5 and FN-I7 repeats of fibronectin, has been found to also be able to bind to integrin αvβ3 upon deamidation of the asparagine residue into isoaspartate (isoD) (Figure 25A).377 The resulting isoD-Gly-Arg (isoDGR) motif binds to the RGD binding sites and can therefore inhibit endothelial cell adhesion.377 To gain structural insights that would help explain why the RGD and isoDGR motifs can bind to integrin αvβ3 while NGR and DGR cannot, Spitaleri et al. used REMD simulations to model four disulfide-bonded cyclic peptides: CRGDCGVRY (named RGD-2C), CisoDGRCGVRY (isoDGR-2C), CDGRCGVRY (DGR-2C), and CNGRCGVRY (NGR-2C) (Figure 25B).378 The REMD simulations were carried out using the OPLS force field302 and the SPC water model,213,379 with 16 replicas spanning temperatures from 293 K to 353 K, and a sampling time of 2 ns per replica. The initial structures for REMD simulations were the lowest-energy NMR-derived structures. It was found that, during the REMD simulations, RGD-2C and isoDGR-2C adopted a more extended conformation and placed the positive and negative groups at a distance of ~13–14 Å from each other, while this distance was shorter (~12–13 Å) in DGR-2C and NGR-2C. Furthermore, replacing Asp with Asn greatly reduced the magnitude of the negatively charged area in the calculated electrostatic surface potentials for the cyclic peptide. Docking simulations showed that for both RGD-2C and isoDGR-2C, the top cluster sampled in the REMD simulations was able to form highly favorable interactions with integrin αvβ3, and their binding poses captured the typical RGD–αvβ3 interactions. However, for DGR-2C and NGR-2C, no cluster sampled in the REMD simulations showed such favorable interactions.378
Figure 25.
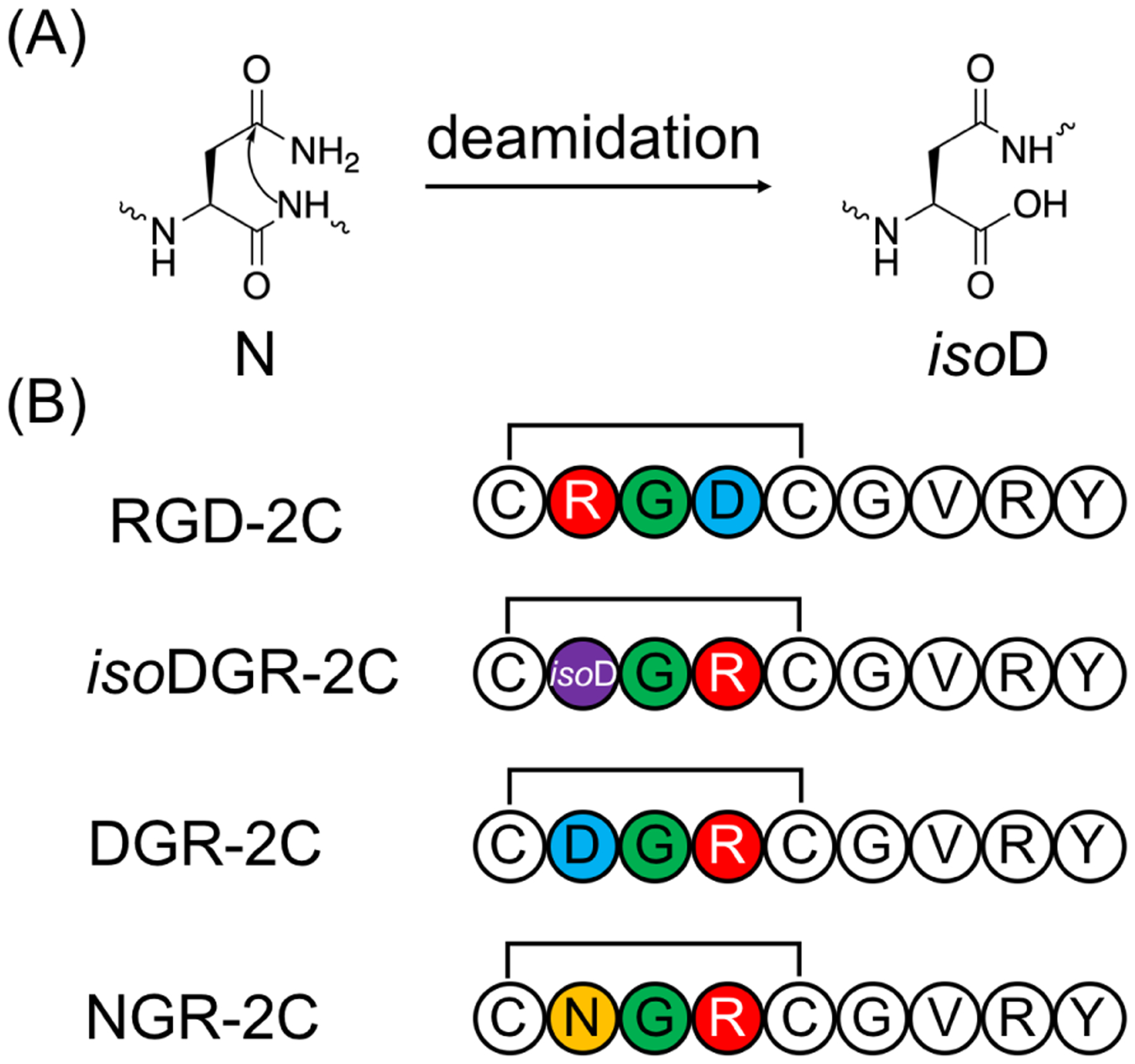
(A) Chemical structure of isoaspartate. (B) Four RGD and isoDGR-related disulfide-bonded cyclic peptides were simulated using REMD simulation. RGD-2C and isoDGR-2C adopted a more extended conformation and placed the positive and negative groups at a distance of ~13–14 Å from each other, while this distance was shorter (~12–13 Å) in DGR-2C and NGR-2C.378
Short REMD simulations, however, do not necessarily provide complete insight into conformational equilibria of cyclic peptides, due to the complex nature of their free-energy surfaces. Spitaleri et al. thus extended their previous work on isoDGR-related disulfide-bonded cyclic peptides,378 and used well-tempered metadynamics to explore the cyclic peptides’ rugged energy landscapes in a later study.170 They first simulated cilengitide, cyclo-(RGDfV), and two disulfide-bonded cyclic peptides, CisoDGRC and CDGRC, to compare their free-energy landscapes (Figure 26). The well-tempered metadynamics simulations were carried out for 10 ns using the OPLS-AA force field216 and the TIP3P water model,207 with ϕ and ψ angles of the central Gly used as a 2-D collective variable. By examining the calculated free-energy surfaces along this 2-D collective variable for cyclo-(RGDfV), CisoDGRC, and CDGRC, it was found that the central Gly in cyclo-(RGDfV) mainly populated the βL region (97.0%) in the Ramachandran plot, while in CDGRC the central Gly mainly occupied the αL region (90.0%), and in CisoDGRC it adopted the βL, βP, and α regions with 42.5%, 38.3%, and 18.4% population, respectively. Docking results showed that structures with the central Gly in the βL region were able to form favorable interactions with integrin αvβ3. The authors then showed in simulation that N-terminal acetylation of CisoDGRC led to an increase in population in the βL region (83.0%), thus suggesting that Ace-CisoDGRC would have an improved integrin αvβ3 binding affinity over CisoDGRC. Indeed, experimental results showed that cyclo-(RGDfV) had the highest binding affinity for integrin αvβ3, followed by Ace-CisoDGRC, CisoDGRC, and CDGRC.170
Figure 26.

Cyclo-(RGDfV) and disulfide-bonded CisoDGRC and CDGRC were simulated using well-tempered metadynamics simulations.170 The central Gly in cyclo-(RGDfV) mainly populated the βL region (97.0%); in CisoDGRC it adopted the βL, βP, and α regions with 42.5%, 38.3%, and 18.4% population, respectively; in CDGRC it mainly occupied the αL region (90.0%). Docking results showed that structures with the central Gly in the βL region were able to form favorable interactions with integrin αvβ3.
When it comes to drug development, isoDGR-containing ligands could be explored as a way to mitigate the severe side effects of RGD-containing peptides, believed to be due to conformational changes in integrin upon binding with the RGD motif.380 With this in mind, Nardelli et al. recently analyzed the behavior of several isoDGR-containing cyclic peptides, none of which were found to induce such conformational changes.381 Starting from the head-to-tail cyclized peptide cyclo-(CGisoDGRG), the authors previously found that the conjugation of this peptide with 4-(N-maleimidomethyl)cyclohexane-1-carboxamide (MCCA) at the cysteine thiol (resulting in a peptide labeled conjugate 2), yielded improved selectivity and binding affinity.382 This improvement is thought to result from the additional polar interactions brought about by the presence of the succinimide ring in MCCA. The authors used the AMBER-99SB force field and TIP3P water to carry out BE-META simulations of cyclo-(CGisoDGRG) and conjugate 2;207,294 GAFF was used for missing parameters.215 In the simulation of cyclo-(CGisoDGRG), all the backbone dihedrals with the exception of w, were used as collective variables. The results yielded 4 main minima in the conformational space. Because the NMR results suggested that the MCCA linker did not affect the conformation of the cyclic peptide system, in the simulations of conjugate 2, backbone dihedrals were restrained at each of the four detected minimum conformations of cyclo-(CGisoDGRG). Five dihedrals along the Cys side chain and the MCCA linker were used as collective variables to sample the linker conformations. Eight hundred conformations of conjugate 2 were selected from these BE-META trajectories for subsequent docking calculations, which further illustrated the stabilizing role played by the succinimide ring in MCCA.381
Since the force fields most commonly used for peptide simulations were not parameterized for β-amino acids such as isoD, their utility for in silico structure prediction of these compounds remains to be determined. In a recent work, Paissoni et al. evaluated the performance of eight force fields in simulating five cyclic peptides containing the isoDGR motif.383 The eight force fields tested were: AMBER-99SB,294 AMBER-99SB-ILDN,218 AMBER-99SB*,384 AMBER-14SB,206 OPLS-AA/L,216 OPLS-AA/LSTD,383 CHARMM-27,385 and GROMOS-54a7.212 The five benchmark cyclic peptides included two disulfide-bonded pentapeptides: CisoDGRC and Ace-CisoDGRC, and two head-to-tail cyclized hexapeptides: cyclo-(CGisoDGRG), cyclo-(GCisoDGRG), and cyclo-(CphgisoDGRG), where phg denotes phenylglycine (Figure 27). BE-META simulations were performed with 1-D biases of all backbone dihedrals except the ones associated with peptide bonds. The replica lengths varied between 30 and 60 ns for different systems. The authors found that, in terms of 3J(HN, Hα) prediction, the three AMBER force fields (99SB, 99SB-ILDN, and 99SB*) performed the best (χ2 < 1.6); AMBER-14SB, CHARMM27, and GROMOS-54a7 performed satisfactorily (χ2 < 3.0), while OPLS-AA/L and OPLS-AA/LSTD did not perform well (χ2 > 5.0). When it comes to the 3JisoD(Hα, Hβ) prediction, deviations > ±2 Hz from experimental values were observed for all eight force fields, suggesting that further force field improvement is necessary to accurately describe cyclic peptides containing β-amino acids.
Figure 27.
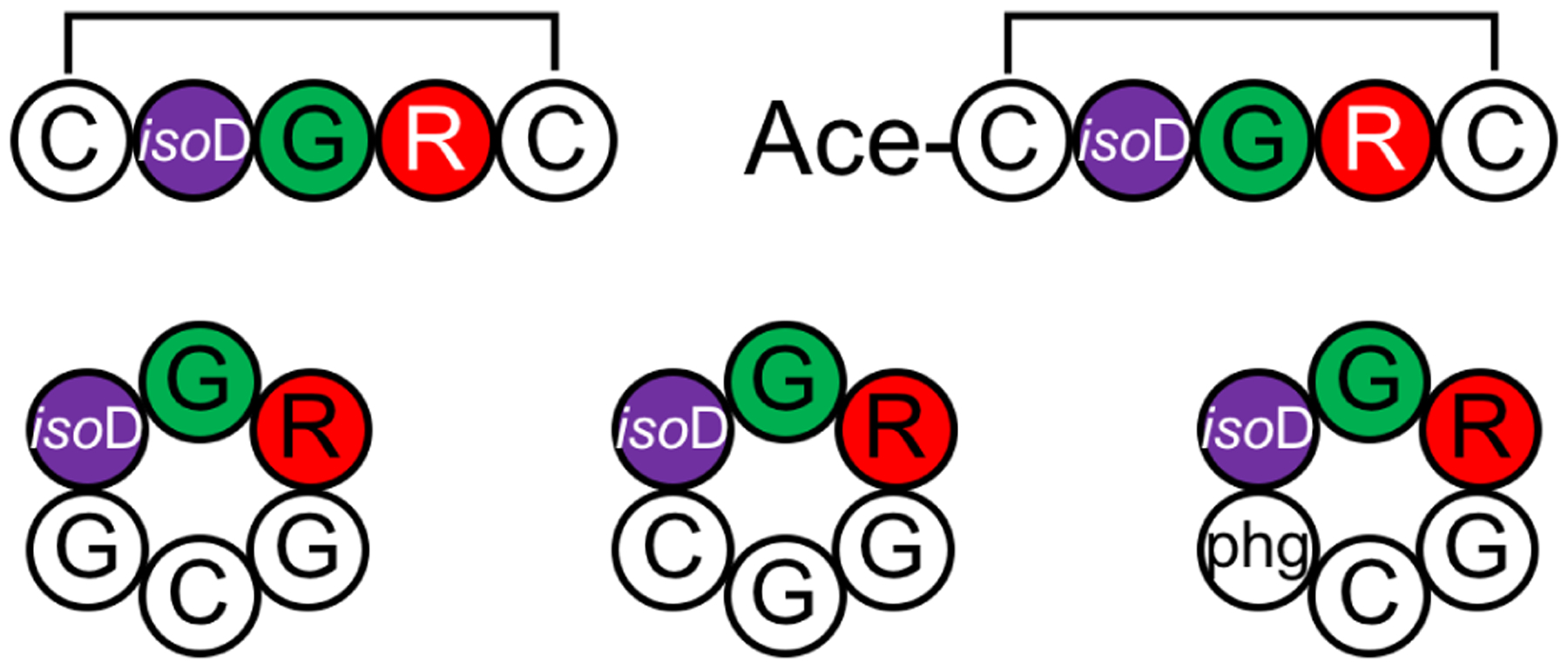
Five isoDGR-containing cyclic peptides were simulated using BE-META simulation to evaluate how well eight force fields could model isoD and recapitulate the 3J couplings measured in NMR.383
5. Conclusions and future prospects
The ability of cyclic peptides to target protein surfaces and protein–protein interaction interfaces makes them an attractive drug modality. Capturing the solution structure of a cyclic peptide is a crucial step toward any quantitative understanding of its properties. However, cyclic peptides tend to adopt multiple conformations in solution and such a feature makes it difficult to characterize their solution structures using traditional means such as NMR spectroscopy. This review shows that MD simulations with enhanced sampling techniques are capable of characterizing the solution structures of cyclic peptides and providing a rationale for their sequence–structure–activity relationships.
As simulation performance is an important aspect when applying enhanced sampling methods to studying cyclic peptides, Table 1 provides a summary focused on the computational performance of the cyclic peptide simulations discussed in this review. These enhanced sampling methods often take advantage of using multiple replicas, and some cyclic peptide systems could require ≤100 ns per replica to achieve convergence. We hope to encourage computational chemists to take on simulating this challenging class of molecules and also to consistently apply measures to verify simulation convergence. As computational tools tailored to simulations of cyclic peptides become more readily available, the utility of MD simulations in solving this problem continues to increase. Already, targeting cyclic peptides’ essential transitional motions using BE-META has enabled studies of dozens of sequences at a reasonable timescale.172,242 Further improvements on this front will be critical in enabling MD simulation to be used for systematic studies of large cyclic peptides to establish sequence–structure relationships, and for computational screening for therapeutic designs.
Cyclic peptides typically have fewer than 20 residues and a small modification in sequence oftentimes leads to a big change in structure and activity. The small sizes of cyclic peptides require high force field accuracy for each amino acid and residue-specific force fields provide more flexibility to achieve this goal.220,221 Further improvement of force fields to simulate cyclic peptides with N-methylated amino acids and artificial amino acids, along with the ability to predict cis/trans isomers, will enable MD simulation to model cyclic peptides with even more therapeutic relevance.
In addition to being used to explain known experimental results, we have recently seen MD simulation take a leading role in making predictions followed by experimental verification.242,244 Providing easy-to-follow, streamlined protocols will further enable researchers other than computational chemists to apply MD simulation with enhanced sampling as a routine method for studying cyclic peptides. MD simulation with enhanced sampling has proven to be a powerful tool to elucidate the elusive solution structures of cyclic peptides and we expect that its use will soon become a trend in cyclic peptide designs.
ACKNOWLEDGMENTS
We thank the support of the Tufts start-up fund, the Knez Family Faculty Investment Fund, and the National Institute of General Medical Sciences of the National Institutes of Health under award number R01GM124160 (PI: Y.-S. L). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We thank Professor George Shields, Dr. Karl Kirschner, Dr. David Chalmers, and Dr. Billy Williams-Noonan for providing the coordinate files of the cyclic peptides in their studies.
ABBREVIATIONS
- MD
molecular dynamics
- REMD
replica exchange molecular dynamics
- META
metadynamics
- PTWTE
parallel-tempering well-tempered ensemble
- BE
bias-exchange
- McMD
multicanonical molecular dynamics
- aMD
accelerated MD
- CoCo-MD
complementary-coordinates molecular dynamics
- RMSD
root-mean-squared deviation
- PCA
principal-component analysis
Biographies
Jovan Damjanovic received his B.A. degree in Chemistry and Mathematics from Amherst College (Amherst, MA) in 2016. At Amherst, he worked on continuous-time numerical simulations of hydrogen exchange mass spectrometry experiments in the group of Prof. Sheila Jaswal. Since 2018, he has been a graduate student at Tufts University (Medford, MA), in the group of Prof. Yu-Shan Lin, where he has worked on computational screening of cyclic peptide hot loop mimics, and methods for processing molecular dynamics trajectories. His academic interests include massively parallel computing, large-scale data analysis, and chemical applications of machine learning.
Jiayuan Miao received his B.S. in Physics from Shandong University (Jinan, China) in 2008. Then he received his M.S. in Physics from Shandong University in 2011, working on improving the performance of Thin Gap Chamber detectors for ATLAS experiment on LHC in Prof. Chengguang Zhu’s group. He received his Ph.D. in Physics from Case Western Reserve University (Cleveland, OH) in 2017, studying the responses of polymers to electric fields, stress, irradiation, and diffusive solvents employing theoretical and simulation tools, under the supervision of Prof. Philip Taylor and Prof. Mesfin Tsige. He joined Prof. Yu-Shan Lin’s group at Tufts University in 2017 as a postdoctoral researcher, working on understanding the structures of cyclic peptides using molecular dynamics simulations and enhanced sampling methods.
He Huang received her B.S. degree in chemistry from Lanzhou University (Gansu, China) in 2012 where she first learned molecular modeling in the group of Prof. Xiaojun Yao. She obtained her Ph.D. degree from Stony Brook University (Stony Brook, NY) in 2018, working with Prof. Carlos Simmerling on AMBER software development and application at the Laufer Center for Physical and Quantitative Biology. From 2018 to 2020, she was a postdoctoral scholar at Tufts University in the group of Prof. Yu-Shan Lin, where she employed molecular dynamics method to investigate the thermodynamic and dynamic properties of cyclic peptides. She then joined OpenEye Scientific Software (Boston, MA) where she is currently an application scientist. Her current research interests range from computational methods for early drug discovery and design, particularly large-scale virtual screening, free-energy calculations, machine learning, to new chemical modalities including macrocycles.
Yu-Shan Lin received her B.S. in Chemistry from National Taiwan University in 2004. During her Ph.D. study at University of Wisconsin–Madison, under the guidance of Prof. James Skinner she worked on computing and using theoretical vibrational spectroscopy to help interpret experimental results and understand water and peptide structure and dynamics. She then moved to Stanford University in 2009 as a Bio-X postdoctoral fellow in Prof. Vijay Pande’s group, using Folding@home to study how length and mutations impact the structural properties of amyloid beta peptides. She joined the Department of Chemistry at Tufts University in 2012 and received tenure in 2018. Her current research interests include understanding and designing cyclic peptides and automated analysis of molecular dynamics simulation data.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Morrison C Constrained peptides’ time to shine? Nat. Rev. Drug Discov 2018, 17, 531–533. [DOI] [PubMed] [Google Scholar]
- (2).Reichlin S Somatostatin. N. Engl. J. Med 1983, 309, 1495–1501. [DOI] [PubMed] [Google Scholar]
- (3).Treschan TA; Peters J The vasopressin system: Physiology and clinical strategies. Anesthesiology 2006, 105, 599–612. [DOI] [PubMed] [Google Scholar]
- (4).Donaldson ZR; Young LJ Oxytocin, vasopressin, and the neurogenetics of sociality. Science 2008, 322. [DOI] [PubMed] [Google Scholar]
- (5).Lee H-J; Macbeth AH; Pagani J; Young WS Oxytocin: The great facilitator of life. Prog. Neurobiol 2009, DOI:10.1016/j.pneurobio.2009.04.00110.1016/j.pneurobio.2009.04.00110.1016/j.pneurobio.2009.04.00110.1016/j.pneurobio.2009.04.001 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Dreyfuss M; Härri E; Hofmann H; Kobel H; Pache W; Tscherter H Cyclosporin a and c. New metabolites from trichodermapolysporum (link ex pers.) rifai. Ind. Microbiol 1976, 3, 125–133. [Google Scholar]
- (7).Borel JF; Feurer C; Gubler HU; Stähelin H Biological effects of cyclosporin a: A new antilymphocytic agent. Agents Actions 1976, 6, 468–475. [DOI] [PubMed] [Google Scholar]
- (8).Faulds D; Goa KL; Benfield P Cyclosporin. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic use in immunoregulatory disorders. Drugs 1993, 45, 953–1040. [DOI] [PubMed] [Google Scholar]
- (9).Gause GF; Brazhnikova MG Gramicidin S and its use in the treatment of infected wounds. Nature 1944, 154, 703. [Google Scholar]
- (10).Kondejewski LH; Farmer SW; Wishart DS; Hancock RE; Hodges RS Gramicidin S is active against both gram-positive and gram-negative bacteria. Int. J. Pept. Protein Res 1996, 47, 460–466. [DOI] [PubMed] [Google Scholar]
- (11).Levine DP Vancomycin: A history. Clin. Infect. Dis 2006, 42, S5–12. [DOI] [PubMed] [Google Scholar]
- (12).Sieber SA; Marahiel MA Learning from nature’s drug factories: Nonribosomal synthesis of macrocyclic peptides. J. Bacteriol 2003, 185, 7036–7043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Felnagle EA; Jackson EE; Chan YA; Podevels AM; Berti AD; McMahon MD; Thomas MG Nonribosomal peptide synthetases involved in the production of medically relevant natural products. Molecular Pharmaceutics 2008, 5, 191–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Nolan EM; Walsh CT How nature morphs peptide scaffolds into antibiotics. ChemBioChem 2009, 10, 34–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Mull RW; Harrington A; Sanchez LA; Tal-Gan Y Cyclic peptides that govern signal transduction pathways: From prokaryotes to multi-cellular organisms. Current Topics in Medicinal Chemistry 2018, 18, 625–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Zorzi A; Deyle K; Heinis C Cyclic peptide therapeutics: Past, present and future. Curr. Opin. Chem. Biol 2017, 38, 24–29. [DOI] [PubMed] [Google Scholar]
- (17).Wishart DS; Feunang YD; Guo AC; Lo EJ; Marcu A; Grant JR; Sajed T; Johnson D; Li C; Sayeeda Z et al. Drugbank 5.0: A major update to the drugbank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Jing X; Jin K A gold mine for drug discovery: Strategies to develop cyclic peptides into therapies. Med. Res. Rev 2020, 40, 753–810. [DOI] [PubMed] [Google Scholar]
- (19).Abdalla M; McGaw L Natural cyclic peptides as an attractive modality for therapeutics: A mini review. Molecules 2018, 23, 2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Malde AK; Hill TA; Iyer A; Fairlie DP Crystal structures of protein-bound cyclic peptides. Chem. Rev 2019, 119, 9861–9914. [DOI] [PubMed] [Google Scholar]
- (21).Karle IL; Karle J An application of a new phase determination procedure to the structure of cyclo(hexaglycyl)demihydrate. Acta Cryst. 1963, 16, 969–975. [Google Scholar]
- (22).Groth P Crystal structure of cyclotetrasarcosyl. Acta Chem. Scand 1970, 24, 780–790. [Google Scholar]
- (23).Karle IL; Gibson JW; Karle J Conformation and crystal structure of the cyclic polypeptide [Gly-Gly-D-Ala-D-Ala-Gly-Gly]·3H2O. J. Am. Chem. Soc 1970, 92, 3755–3760. [DOI] [PubMed] [Google Scholar]
- (24).Kartha G; Ambady G; Shankar PV Structure and conformation of a cyclic tripeptide. Nature 1974, 247, 204–205. [DOI] [PubMed] [Google Scholar]
- (25).Kartha G; Ambady GK The crystal structure and molecular conformation of cyclo-L-prolyl-L-prolyl-L-hydroxyproline, a cyclic tripeptide. Acta Cryst. 1975, B31, 2035–2039. [Google Scholar]
- (26).Brown JN; Teller RG Crystal structure and molecular conformation of the hydrated cyclic hexapeptide cyclo(L-Ala-L-Pro-D-Phe)2. J. Am. Chem. Soc 1976, 98, 7565–7569. [DOI] [PubMed] [Google Scholar]
- (27).Druyan ME; Coulter CL; Walter R; Kartha G; Ambady GK Structure and conformation of cyclo(tri-L-prolyl) in the crystalline state. J. Am. Chem. Soc 1976, 98, 5496–5502. [DOI] [PubMed] [Google Scholar]
- (28).Flippen JL; Karle IL Conformation of the cyclic tetrapeptide dihydrochlamydocin. L-abu-L-Phe-D-Pro-L-X, and experimental values for 3→1 intramolecular hydrogen bonds by X-ray diffraction. Biopolymers 1976, 15, 1081–1092. [DOI] [PubMed] [Google Scholar]
- (29).Hossain MB; Van der Helm D Conformation and crystal structures of two cycloisomeric hexapeptides: Cyclo-(L-Alanyl-L-alanylglycylglycyl-L-alanylglycyl) monohydrate (I) and cyclo-(L-Alanyl-L-alanylglycyl-L-alanylglycylglycyl) dihydrate (II). J. Am. Chem. Soc 1978, 100, 5191–5198. [Google Scholar]
- (30).Karle IL Crystal structure and conformation of cyclo-(glycylprolylglycyl-D-alanylprolyl) containing 4→1 and 3→1 intramolecular hydrogen bonds. J. Am. Chem. Soc 1978, 100, 1286–1289. [Google Scholar]
- (31).Brown JN; Yang CH Crystal and molecular structure of the cyclic hexapeptide cyclo-(Gly-Pro-D-Phe)2. J. Am. Chem. Soc 1979, 101, 445–449. [Google Scholar]
- (32).Hossain MB; Van der Helm D Conformation of cyclo(-L-Leu-L-Phe-Gly-D-Leu-D-Phe-Gly-) dihydrate. Acta Cryst. 1979, B35, 2781–2784. [DOI] [PubMed] [Google Scholar]
- (33).Karle IL Conformation of the cyclic pentapeptide Gly-L-Pro-L-Ser-D-Ala-L-Pro in the crystalline state and an example of rotational “isomerism” between analogs. J. Am. Chem. Soc 1979, 101, 181–184. [Google Scholar]
- (34).Kostansek EC; Lipscomb WN; Thiessen WE Crystal structure and conformation of the cyclic hexapeptide cyclo-(Gly-L-Pro-D-Ala)2. J. Am. Chem. Soc 1979, 101, 834–837. [Google Scholar]
- (35).Kostansek EC; Thiessen WE; Schomburg D; Lipscomb WN Crystal structure and molecular conformation of the cyclic hexapeptide cyclo-(Gly-L-Pro-Gly)2. J. Am. Chem. Soc 1979, 101, 5811–5815. [Google Scholar]
- (36).Swepston PN; Cordes AW; Kuyper LF; Meyer WL Dihyrotentoxin: A cyclic tetrapeptide. Acta Cryst. 1981, B37, 1139–1141. [Google Scholar]
- (37).Varughese KI; Kartha G; Kopple KD Crystal structure and conformation of cyclo-(glycyl-D-leucyl-L-leucyl)2. J. Am. Chem. Soc 1981, 103, 3310–3313. [Google Scholar]
- (38).Yang C-H; Brown JN; Kopple KD Crystal structure and solution studies of the molecular conformation of the cyclic hexapeptide cyclo-(Gly-L-His-Gly-L-Ala-L-Tyr-Gly). J. Am. Chem. Soc 1981, 103, 1715–1719. [Google Scholar]
- (39).Karle IL In Perspectives in peptide chemistry. Dedicated to robert schwyzer; Eberle AN;Wieland T;Geiger R, Eds.; Karger: Basel, 1981. [Google Scholar]
- (40).Barnes CL; Van der Helm D Conformation and structures of two cycloisomeric hexapeptides: Cyclo(-L-Phe-D-Leu-Gly-L-Phe-L-Leu-Gly-) tetrahydrate and cyclo(-L-Phe-D-Leu-Gly-D-Phe-L-Leu-Gly-) dihydrate. Acta Cryst. 1982, B38, 2589–2595. [DOI] [PubMed] [Google Scholar]
- (41).Chiang CC; Karle IL Crystal structure of the 1:1 mixture of cyclic (L-Ala-L-Pro-L-Phe-L-Pro) and cyclic (L-Ala-L-Pro-D-Phe-L-Pro). Int. J. Pept. Protein Res 1982, 20, 133–138. [DOI] [PubMed] [Google Scholar]
- (42).Czugler M; Sasvari K; Hollosi M Crystal structure of cyclo-(Gly-L-Pro-L-Pro-Gly-L-Pro-L-Pro) trihydrate. Unusual conformational characteristics of a cyclic hexapeptide. J. Am. Chem. Soc 1982, 104, 4465–4469. [Google Scholar]
- (43).Chiang CC; Karle IL; Wieland T Unusual intramolecular hydrogen bonding in cycloamanide a, cyclic (LPro-LVal-LPhe-LPhe-LAla-Gly). Int. J. Peptide Protein Res 1982, 20, 414–429. [DOI] [PubMed] [Google Scholar]
- (44).Steyn PS; Tuinman AA; van Heerden FR; van Rooyen PH; Wessels PL; Rabie CJ The isolation, structure, and absolute configuration of the mycotoxin, rhizonin a, a novel cyclic heptapeptide containing n-methyl-3-(3-furyl)alanine, produced by rhizopus microsporus. J. Chem. Soc., Chem. Commun 1983, 1983, 47–79. [Google Scholar]
- (45).Ueno K; Shimizu T Crystal structure and conformation of a cyclic tetrapeptide cyclo(L-Pro-sar)2 containing all-cis peptide units. Biopolymers 1983, 22, 633–641. [Google Scholar]
- (46).Kartha G; Bhandary KK; Kopple KD; Go A; Zhu P-P Synthesis and conformational studies by X-ray crystallography and nuclear magnetic resonance of cyclo(L-Phe-L-Pro-D-Ala)2. J. Am. Chem. Soc 1984, 106, 6844–6850. [Google Scholar]
- (47).Nakashima T; Yamane T; Tanaka I; Ashida T Structure of cyclo(-L-Pro-L-Pro-Gly-L-Pro-L-Leu-Gly-) methanol solvate monohydrate, c25h38n6o6·ch3oh·H2O. Acta Cryst. 1984, C40, 171–174. [Google Scholar]
- (48).Ueda I; Ueda T; Sada I; Kato T; Mikuriya M; Kida S; Izumiya N Structure of cyclo(-L-Pro-L-Val-L-Pro-L-Val-) dimethyl sulfoxide solvate, c20h32n4o4·c2h6os. Acta Cryst. 1984, C40, 111–113. [Google Scholar]
- (49).Gierasch LM; Karle IL; Rockwell AL; Yenal K Crystal and solution structures of cyclo(Ala-Pro-Gly-D-Phe-Pro): A new type of cyclic pentapeptide which undergoes cis-trans isomerization of the Ala-Pro bond. J. Am. Chem. Soc 1985, 107, 3321–3327. [Google Scholar]
- (50).Cook WJ; Trapane TL; Prasad KU Crystal structure and conformation of the cyclic tetramer of a repeat tripeptide of elastin, cyclo(l-valyl-l-prolylglycyl)4. Chem. Biol. Drug Des 1985, 25, 481–486. [DOI] [PubMed] [Google Scholar]
- (51).Kopple KD; Kartha G; Bhandary KK; Romanowska K Conformations of cyclic octapeptides. 2. Crystal structure of cyclo(D-Ala-Gly-Pro-D-Phe)2. Solvent exposure of backbone protons in crystal and solution conformations. J. Am. Chem. Soc 1985, 107, 4893–4897. [Google Scholar]
- (52).Karle IL Variability in the backbone conformation of cyclic pentapeptides. Int. J. Pept. Protein Res 1986, 28, 420–427. [DOI] [PubMed] [Google Scholar]
- (53).Kartha G; Aimoto S; Varughese KI Crystal and molecular structure of cyclo(L-prolyl-glycyl)3. Chem. Biol. Drug Des 1986, 27, 112–117. [DOI] [PubMed] [Google Scholar]
- (54).Kessler H; Klein M; Muller A; Wagner K; Bats JW; Ziegler K; Frimmer M Conformational prerequisites for the in vitro inhibition of cholate uptake in hepatocytes by cyclic analogues of antamanide and somatostatin. Angew. Chem 1986, 25, 997–999. [Google Scholar]
- (55).Kopple KD; Bhandary KK; Kartha G; Wang YS; Parameswaran KN Conformations of cyclic octapeptides. 3. Cyclo-(D-Ala-Gly-Pro-Phe)2. Conformations in crystals and a t1p examination of internal mobility in solution. J. Am. Chem. Soc 1986, 108, 4637–4642. [Google Scholar]
- (56).Stroup AN; Rheingold AL; Rockwell AL; Gierasch LM Crystal structure of cyclo(Gly-L-Pro-D-Phe-Gly-Val): An example of a new type of three-residue turn. J. Am. Chem. Soc 1987, 109, 7146–7150. [Google Scholar]
- (57).Stroup AN; Rockwell AL; Rheingold AL; Gierasch LM Crystal structure of cyclo(Gly1-L-Pro2-D-Phe3-L-Ala4-L-Pro5): A cyclic pentapeptide with a Gly-L-Pro delta turn. J. Am. Chem. Soc 1988, 110, 5157–5161. [Google Scholar]
- (58).Kessler H; Bats JW; Griesinger C; Koll S; Will M; Wagner K Peptide conformations. 46. Conformational analysis of a superpotent cytoprotective cyclic somatostatin analogue. J. Am. Chem. Soc 1988, 110, 1033–1049. [Google Scholar]
- (59).Pavone V; Benedetti E; Di Blasio B; Lombardi A; Pedone C; Tomasich L; Lorenzi GP Regularly alternating L,D-peptides. III. Hexacyclic peptides from valine or phenylalanine. Biopolymers 1989, 28, 215–223. [DOI] [PubMed] [Google Scholar]
- (60).Shoham G; Burley SK; Lipscomb WN Structure of cyclo(-L-prolylglycyl-)2 trihydrate. Acta Cryst. 1989, C45, 1944–1948. [DOI] [PubMed] [Google Scholar]
- (61).Barnes CL; Hassain MB; Fidelis K; Van der Helm D Structure and conformations of two cycloisomeric hexapeptides: Cyclo(L-Leu-L-Phe-Gly-D-Phe-L-Leu-Gly-) trihydrate and cyclo(L-Phe-L-Leu-Gly-D-Leu-L-Phe-Gly-) trihydrate. Acta Cryst. 1990, B46, 238–246. [DOI] [PubMed] [Google Scholar]
- (62).Declercq JP; Tinant B; Bashwira S; Hootele C Structure of the cyclohexapeptide cleromyrine II trihydrate. Acta Cryst. 1990, C46, 1259–1262. [DOI] [PubMed] [Google Scholar]
- (63).Bhandary KK; Kopple KD Conformation of cyclo-bis(-L-valyl-L-prolyl-D-Alanyl-), a synthetic cyclic hexapeptide. Acta Cryst. 1991, C47, 1280–1283. [DOI] [PubMed] [Google Scholar]
- (64).Bhandary KK; Kopple KD Conformation of cyclic octapeptides. VI. Structure of cyclo-bis-(-L-Alanylglycyl-L-prolyl-L-phenylalanyl-) tetrahydrate. Acta Cryst. 1991, C47, 1483–1487. [DOI] [PubMed] [Google Scholar]
- (65).Eggleston DS; Baures PW; Peishoff CE; Kopple KD Conformations of cyclic heptapeptides: Crystal structure and computational studies of evolidine. J. Am. Chem. Soc 1991, 113, 4410–4416. [Google Scholar]
- (66).Kessler H; Matter H; Gemmecker G; Kottenhahn M; Bats JW Structure and dynamics of synthetic O-glycosylated cyclopeptide in solution determined by NMR spectroscopy and MD calculations. J. Am. Chem. Soc 1992, 114, 4805–4818. [Google Scholar]
- (67).Pettit GR; Srirangam JK; Herald DL; Erickson KL; Doubek DL; Schmidt JM; Tackett LP; Bakus GJ Antineoplastic agents. 251. Isolation and structure of stylostatin 1 from the papua new guinea marine sponge stylotella aurantium. J. Org. Chem 1992, 57, 7217–7220. [Google Scholar]
- (68).Morita H; Kayashita T; Takeya K; Itokawa H; Shiro M Crystal and solution forms of a cyclic heptapeptide, pseudostellarin D. Tetrahedron 1995, 51, 12539–12548. [Google Scholar]
- (69).Morita H; Yun YS; Takeya K; Itokawa H; Shiro M Conformational analysis of a cyclic hexapeptide, segetalin a from vaccaria segetalis. Tetrahedron 1995, 51, 5987–6002. [Google Scholar]
- (70).Pettit GR; Srirangam JK; Herald DL; Xu J-P; Boyd MR; Cichacz Z; Kamano Y; Schmidt JM; Erickson KL Isolation and crystal structure of stylopeptide 1, a new marine porifera cycloheptapeptide. J. Org. Chem 1995, 60, 8257–8261. [Google Scholar]
- (71).Morita H; Kayashita T; Shishido A; Takeya K; Itokawa H; Shiro M Dichotomins A–E, new cyclic peptides from stellaria dichotoma L. Var. Lanceolata Bge. Tetrahedron 1996, 52, 1165–1176. [Google Scholar]
- (72).Morita H; Kayashita T; Takeya K; Itokawa H; Shiro M Conformation of cyclic heptapeptides: Solid and solution state conformation of yunnanin a. Tetrahedron 1997, 53, 1607–1616. [Google Scholar]
- (73).Zanotti G; Saviano M; Saviano G; Tancredi T; Rossi F; Pedone C; Benedetti E Structure of cyclic peptides: The crystal and solution conformation of cyclo(Phe-Phe-aib-Leu-Pro). J. Pept. Res 1998, 51, 460–466. [DOI] [PubMed] [Google Scholar]
- (74).Dittrich B; Koritsanszky T; Grosche M; Scherer W; Flaig R; Wagner A; Krane HG; Kessler H; Riemer C; Schreurs AMM et al. Reproducibility and transferability of topological properties; experimental charge density of the hexapeptide cyclo-(D,L-Pro)2-(L-Ala)4 monohydrate. Acta Cryst. 2002, B58, 721–727. [DOI] [PubMed] [Google Scholar]
- (75).Wang C; Zhang L-L; Lu Y; Zheng Q-T; Cheng Y-X; Zhou J; Tan N-H Study on the spatial structure of brachystemin c, a new cyclic peptide from brachystemma calycinum. J. Mol. Struct 2004, 688, 67–71. [Google Scholar]
- (76).Wiiliams DE; Patrick BO; Behrisch HW; Van Soest R; Roberge M; Andersen RJ Dominicin, a cyclic octapeptide, and laughine, a bromopyrrole alkaloid, isolated from the caribbean marine sponge eurypon laughlini. J. Nat. Prod 2005, 68, 327–330. [DOI] [PubMed] [Google Scholar]
- (77).Karle IL; Urry DW Crystal structure of cyclic (apgvgv)2, an analog of elastin, and a suggested mechanism for elongation/contraction of the molecule. Biopolymers 2005, 77, 198–204. [DOI] [PubMed] [Google Scholar]
- (78).Wu L; Lu Y; Zheng Q-T; Tan N-H; Li C-M; Zhou J Study on the spatial structure of annomuricatin a, a cyclohexapeptide from the seeds of Annona muricata. J. Mol. Struct 2007, 827, 145–148. [Google Scholar]
- (79).Dolle RE; Michaut M; Martinez-Teipel B; Seida PR; Ajello CW; Muller AL; DeHaven RN; Carroll PJ Nascent structure-activity relationship study of a diastereomeric series of kappa opioid receptor antagonists derived from cj-15,208. Bioorg. Med. Chem. Lett 2009, 19, 3647–3650. [DOI] [PubMed] [Google Scholar]
- (80).Vicente J; Vera B; Rodriguez AD; Rodriguez-Escudero I; Raptis RG Euryjanicin a: A new cycloheptapeptide from the caribbean marine sponge prosuberites laughlini. Tetrahedron Lett. 2009, 50, 4571–4574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Wélé A; Mayer C; Quentin D; Zhang Y; Blond A; Bodo B 3D-structure of cycloreticulin c and glabrin a, cyclopeptides from the seeds of annona reticulata. Tetrahedron 2009, 65, 275–281. [Google Scholar]
- (82).Zhao J; Zhou L-L; Li X; Xiao H-B; Hou F-F; Cheng Y-X Bioactive compounds from the aerial parts ofbrachystemma calycinumand structural revision of an octacyclopeptide. J. Nat. Prod 2011, 74, 1392–1400. [DOI] [PubMed] [Google Scholar]
- (83).Tian J-M; Ou-Yang S-S; Zhang X; Di Y-T; Jiang H-L; Li H-L; Dai W-X; Chen K-Y; Liu M-L; Hao X-J et al. Experimental and computational insights into the conformations of tunicyclin e, a new cycloheptapeptide from psammosilene tunicoides. RSC Adv. 2012, 2, 1126–1135. [Google Scholar]
- (84).Tong Y; Luo JG; Wang R; Wang XB; Kong LY New cyclic peptides with osteoblastic proliferative activity from dianthus superbus. Bioorg. Med. Chem. Lett 2012, 22, 1908–1911. [DOI] [PubMed] [Google Scholar]
- (85).Chakraborty S; Tyagi P; Tai D-F; Lee G-H; Peng S-M A lead(II) 3D coordination polymer based on a marine cyclic peptide motif. Molecules 2013, 18, 4972–4985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Barman AK; Gour N; Verma S Morphological transition triggered by mannose conjugation to a cyclic hexapeptide. Arkivoc 2013, 2013, 82–99. [Google Scholar]
- (87).Kostansek EC; Thiessen WE; Schomburg D; Lipscomb WN Crystal structure and molecular conformation of the cyclic hexapeptide cyclo-(Gly-L-Pro-Gly)2. J. Am. Chem. Soc 1979, 101, 5811–5815. [Google Scholar]
- (88).Morita H; Yun YS; Takeya K; Itokawa H; Shiro M Conformational analysis of a cyclic hexapeptide, segetalin a from vaccaria segetalis. Tetrahedron 1995, 51, 5987–6002. [Google Scholar]
- (89).Gierasch LM; Karle IL; Rockwell AL; Yenal K Crystal and solution structures of cyclo(Ala-Pro-Gly-D-Phe-Pro): A new type of cyclic pentapeptide which undergoes cis-trans isomerization of the Ala-Pro bond. J. Am. Chem. Soc 1985, 107, 3321–3327. [Google Scholar]
- (90).Marelli UK; Frank AO; Wahl B; La Pietra V; Novellino E; Marinelli L; Herdtweck E; Groll M; Kessler H Receptor-bound conformation of cilengitide better represented by its solution-state structure than the solid-state structure. Chemistry 2014, 20, 14201–14206. [DOI] [PubMed] [Google Scholar]
- (91).Nguyen QNN; Schwochert J; Tantillo DJ; Lokey RS Using 1H and 13C NMR chemical shifts to determine cyclic peptide conformations: A combined molecular dynamics and quantum mechanics approach. Phys. Chem. Chem. Phys 2018, 20, 14003–14012. [DOI] [PubMed] [Google Scholar]
- (92).Kopple KD; Go A; Logan RJ Jr.; Savrda J Conformations of cyclic peptides. VI. Factors influencing mono-, 1,4-di-, and 1,2,4-trisubstituted cyclic hexapeptide backbones. J. Am. Chem. Soc 1972, 94, 973–981. [DOI] [PubMed] [Google Scholar]
- (93).Tonelli AE; Brewster AI The conformational characteristics in solution of the cyclic hexapeptide Gly-Gly-D-Ala-D-Ala-Gly-Gly. J. Am. Chem. Soc 1972, 94, 2851–2854. [DOI] [PubMed] [Google Scholar]
- (94).Kopple KD; Go A; Schamper TJ Conformation of cyclic peptides. 10. Conformational averaging in peptides with the sequence cyclo-(Gly-D-Xxx-L-Yyy)2. J. Am. Chem. Soc 1978, 100, 4289–4295. [Google Scholar]
- (95).Blout ER Cyclic peptides: Past, present, and future. Biopolymers 1981, 20, 1901–1912. [Google Scholar]
- (96).Yang C-H; Brown JN; Kopple KD Crystal structure and solution studies of the molecular conformation of the cyclic hexapeptide cyclo-(Gly-L-His-Gly-L-Ala-L-Tyr-Gly). J. Am. Chem. Soc 1981, 103, 1715–1719. [Google Scholar]
- (97).Varughese KI; Kartha G; Kopple KD Crystal structure and conformation of cyclo-(glycyl-D-leucyl-L-leucyl)2. J. Am. Chem. Soc 1981, 103, 3310–3313. [Google Scholar]
- (98).Kopple KD; Wang Y-S; Cheng AG; Bhandary KK Conformations of cyclic octapeptides. 5. Crystal structure of cyclo(Cys-Gly-Pro-Phe)2 and rotating frame relaxation (t1ρ NMR studies of internal mobility in cyclic octapeptides. J. Am. Chem. Soc 1988, 110, 4168–4176. [Google Scholar]
- (99).Stradley SJ; Rizo J; Bruch MD; Stroup AN; Gierasch LM Cyclic pentapeptides as models for reverse turns: Determination of the equilibrium distribution between type I and type II conformations of Pro-Asn and Pro-Ala β-turns. Biopolymers 1990, 29, 263–287. [DOI] [PubMed] [Google Scholar]
- (100).Kopple KD; Bean JW; Bhandary KK; Briand J; D’Ambrosio CA; Peishoff CE Conformational mobility in cyclic oligopeptides. Biopolymers 1993, 33, 1093–1099. [DOI] [PubMed] [Google Scholar]
- (101).Alberg DG; Schreiber SL Structure-based design of a cyclophilin-calcineurin bridging ligand. Science 1993, 262, 248–250. [DOI] [PubMed] [Google Scholar]
- (102).Marshall GR; Beusen DD; Nikiforovich GV In Peptides: Synthesis, structures, and applications; Gutte B, Ed., 1995.
- (103).Porter CJ; Wilce JA NMR analysis of g7-18nate, a nonphosphorylated cyclic peptide inhibitor of the grb7 adapter protein. Biopolymers 2007, 88, 174–181. [DOI] [PubMed] [Google Scholar]
- (104).Beck JG; Chatterjee J; Laufer B; Kiran MU; Frank AO; Neubauer S; Ovadia O; Greenberg S; Gilon C; Hoffman A et al. Intestinal permeability of cyclic peptides: Common key backbone motifs identified. J. Am. Chem. Soc 2012, 134, 12125–12133. [DOI] [PubMed] [Google Scholar]
- (105).Cuniasse P; Raynal I; Yiotakis A; Dive V Accounting for conformational variability in NMR structure of cyclopeptides: Ensemble averaging of interproton distance and coupling constant restraints. J. Am. Chem. Soc 1997, 119, 5239–5248. [Google Scholar]
- (106).Viles JH; Mitchell JBO; Gough SL; Doyle PM; Harris CJ; Sadler PJ; Thornton JM Multiple solution conformations of the integrin-binding cyclic pentapeptide cyclo(-Ser-D-Leu-Asp-Val-Pro-) analysis of the (phi, psi) space available to cyclic pentapeptides. Eur. J. Biochem 1996, 242, 352–362. [DOI] [PubMed] [Google Scholar]
- (107).Appelt C; Wessolowski A; Soderhall JA; Dathe M; Schmieder P Structure of the antimicrobial, cationic hexapeptide cyclo(RRWWRF) and its analogues in solution and bound to detergent micelles. ChemBioChem 2005, 6, 1654–1662. [DOI] [PubMed] [Google Scholar]
- (108).Feeney J; Roberts GC; Rockey JH; Burgen AS Conformational studies of oxytocin and lysine vasopressin in aqueous solution using high resolution NMR spectroscopy. Nat. New Biol 1971, 232, 108–110. [DOI] [PubMed] [Google Scholar]
- (109).Brewster AIR; Hruby VJ 300-MHz nuclear magnetic resonance study of oxytocin in aqueous solution: Conformational implications. Proc. Natl. Acad. Sci. U.S.A 1973, 70, 3806–3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Sterner O; Etzel W; Mayer A; Anke H Omphalotin, a new cyclic peptide with potent nematicidal activity fromomphalotus oleariusii. Isolation and structure determination. Nat. Prod. Lett 1997, 10, 33–38. [Google Scholar]
- (111).Korsinczky MLJ; Schirra HJ; Rosengren KJ; West J; Condie BA; Otvos L; Anderson MA; Craik DJ Solution structures by 1H NMR of the novel cyclic trypsin inhibitor sfti-1 from sunflower seeds and an acyclic permutant. J. Mol. Biol 2001, 311, 579–591. [DOI] [PubMed] [Google Scholar]
- (112).Sikorska E; Rodziewicz-Motowidlo S Conformational studies of vasopressin and mesotocin using NMR spectroscopy and molecular modelling methods. Part I: Studies in water. J. Pept. Sci 2008, 14, 76–84. [DOI] [PubMed] [Google Scholar]
- (113).Zhang Y I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 2008, 9, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (114).Thevenet P; Shen Y; Maupetit J; Guyon F; Derreumaux P; Tuffery P PEP-FOLD: An updated de novo structure prediction server for both linear and disulfide bonded cyclic peptides. Nucleic Acids Res. 2012, 40, W288–W293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (115).Shen Y; Maupetit J; Derreumaux P; Tufféry P Improved PEP-FOLD approach for peptide and miniprotein structure prediction. J. Chem. Theory Comp 2014, 10, 4745–4758. [DOI] [PubMed] [Google Scholar]
- (116).Lamiable A; Thevenet P; Rey J; Vavrusa M; Derreumaux P; Tuffery P PEP-FOLD3: Faster de novo structure prediction for linear peptides in solution and in complex. Nucleic Acids Res. 2016, 44, W449–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).Beaufays J; Lins L; Thomas A; Brasseur R In silico predictions of 3D structures of linear and cyclic peptides with natural and non-proteinogenic residues. J. Pept. Sci 2012, 18, 17–24. [DOI] [PubMed] [Google Scholar]
- (118).Singh S; Singh H; Tuknait A; Chaudhary K; Singh B; Kumaran S; Raghava GP PEPstrMOD: Structure prediction of peptides containing natural, non-natural and modified residues. Biol. Direct 2015, 10, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (119).Hawkins PCD; Skillman AG; Warren GL; Ellingson BA; Stahl MT Conformer generation with OMEGA: Algorithm and validation using high quality structures from the protein databank and cambridge structural database. J. Chem. Inf. Model 2010, 50, 572–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (120).Poongavanam V; Danelius E; Peintner S; Alcaraz L; Caron G; Cummings MD; Wlodek S; Erdelyi M; Hawkins PCD; Ermondi G et al. Conformational sampling of macrocyclic drugs in different environments: Can we find the relevant conformations? ACS Omega 2018, 3, 11742–11757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (121).Watts KS; Dalal P; Tebben AJ; Cheney DL; Shelley JC Macrocycle conformational sampling with macromodel. J. Chem. Inf. Model 2014, 54, 2680–2696. [DOI] [PubMed] [Google Scholar]
- (122).Mandell DJ; Coutsias EA; Kortemme T Sub-angstrom accuracy in protein loop reconstruction by robotics-inspired conformational sampling. Nat. Methods 2009, 6, 551–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (123).Bhardwaj G; Mulligan VK; Bahl CD; Gilmore JM; Harvey PJ; Cheneval O; Buchko GW; Pulavarti SV; Kaas Q; Eletsky A et al. Accurate de novo design of hyperstable constrained peptides. Nature 2016, 538, 329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (124).Hosseinzadeh P; Bhardwaj G; Mulligan VK; Shortridge MD; Craven TW; Pardo-Avila F; Rettie SA; Kim DE; Silva D-A; Ibrahim YM et al. Comprehensive computational design of ordered peptide macrocycles. Science 2017, 358, 1461–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (125).Coutsias EA; Lexa KW; Wester MJ; Pollock SN; Jacobson MP Exhaustive conformational sampling of complex fused ring macrocycles using inverse kinematics. J. Chem. Theory Comput 2016, 12, 4674–4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (126).Jusot M; Stratmann D; Vaisset M; Chomilier J; Cortes J Exhaustive exploration of the conformational landscape of small cyclic peptides using a robotics approach. J. Chem. Inf. Model 2018, 58, 2355–2368. [DOI] [PubMed] [Google Scholar]
- (127).Tayar NE; Mark AE; Vallat P; Brunne RM; Testa B; van Gunsteren WF Solvent-dependent conformation and hydrogen-bonding capacity of cyclosporin A: Evidence from partition coefficients and molecular dynamics simulations. J. Med. Chem 1993, 36, 3757–3764. [DOI] [PubMed] [Google Scholar]
- (128).Butterfoss GL; Yoo B; Jaworski JN; Chorny I; Dill KA; Zuckermann RN; Bonneau R; Kirshenbaum K; Voelz VA De novo structure prediction and experimental characterization of folded peptoid oligomers. Proc. Natl. Acad. Sci. U.S.A 2012, 109, 14320–14325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (129).Chen Y; Deng K; Qiu X; Wang C Visualizing cyclic peptide hydration at the single-molecule level. Sci. Rep 2013, 3, 2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (130).Quartararo JS; Eshelman MR; Peraro L; Yu H; Baleja JD; Lin Y-S; Kritzer JA A bicyclic peptide scaffold promotes phosphotyrosine mimicry and cellular uptake. Bioorg. Med. Chem 2014, 22, 6387–6391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (131).Merten C; Li F; Bravo-Rodriguez K; Sanchez-Garcia E; Xu Y; Sander W Solvent-induced conformational changes in cyclic peptides: A vibrational circular dichroism study. Phys. Chem. Chem. Phys 2014, 16, 5627–5633. [DOI] [PubMed] [Google Scholar]
- (132).Slough DP; McHugh SM; Lin Y-S Understanding and designing head-to-tail cyclic peptides. Biopolymers 2018, 109, e23113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (133).Karplus M; Petsko GA Molecular dynamics simulations in biology. Nature 1990, 347, 631–639. [DOI] [PubMed] [Google Scholar]
- (134).Ichiye T; Karplus M Collective motions in proteins: A covariance analysis of atomic fluctuations in molecular dynamics and normal mode simulations. Proteins 1991, 11, 205–217. [DOI] [PubMed] [Google Scholar]
- (135).Brooks CL 3rd; Case DA Simulations of peptide conformational dynamics and thermodynamics. Chem. Rev 1993, 93, 2487–2502. [Google Scholar]
- (136).Cheatham TEI; Miller JL; Fox T; Darden TA; Kollman PA Molecular dynamics simulations on solvated biomolecular systems: The particle mesh ewald method leads to stable trajectories of DNA, rna, and proteins. J. Am. Chem. Soc 1995, 117, 4193–4194. [Google Scholar]
- (137).Hunenberger PH; Mark AE; Gunsteren W. F. v. Fluctuation and cross-correlation analysis of protein motions observed in nanosecond molecular dynamics simulations. J. Mol. Biol 1995, 252, 492–503. [DOI] [PubMed] [Google Scholar]
- (138).Daura X; Jaun B; Seebach D; Gunsteren W. F. v.; Mark AE Reversible peptide folding in solution by molecular dynamics simulation. J. Mol. Biol 1998, 280, 925–932. [DOI] [PubMed] [Google Scholar]
- (139).Schaefer M; Bartels C; Karplus M Solution conformations and thermodynamics of structured peptides: Molecular dynamics simulation with an implicit solvation model. J. Mol. Biol 1998, 284, 835–848. [DOI] [PubMed] [Google Scholar]
- (140).Buck M; Karplus M Internal and overall peptide group motion in proteins: Molecular dynamics simulations for lysozyme compared with results from X-ray and NMR spectroscopy. J. Am. Chem. Soc 1999, 121, 9645–9658. [Google Scholar]
- (141).Pande VS; Rokhsar DS Molecular dynamics simulations of unfolding and refolding of a beta-hairpin fragment of protein g. Proc. Natl. Acad. Sci. U.S.A 1999, 96, 9062–9067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (142).Karplus M; McCammon JA Molecular dynamics simulations of biomolecules. Nat. Struct. Biol 2002, 9, 646–652. [DOI] [PubMed] [Google Scholar]
- (143).Snow CD; Nguyen N; Pande VS; Gruebele M Absolute comparison of simulated and experimental protein-folding dynamics. Nature 2002, 420, 102–106. [DOI] [PubMed] [Google Scholar]
- (144).Ponder JW; Case DA Force fields for protein simulations. Adv. Prot. Chem 2003, 66, 27–85. [DOI] [PubMed] [Google Scholar]
- (145).Snow CD; Qiu LL; Du DG; Gai F; Hagen SJ; Pande VS Trp zipper folding kinetics by molecular dynamics and temperature-jump spectroscopy. Proc. Natl. Acad. Sci. U.S.A 2004, 101, 4077–4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (146).Nguyen HD; Hall CK Molecular dynamics simulations of spontaneous fibril formation by random-coil peptides. Proc. Natl. Acad. Sci. U.S.A 2004, 101, 16180–16185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (147).Chong LT; Snow CD; Rhee YM; Pande VS Dimerization of the p53 oligomerization domain: Identification of a folding nucleus by molecular dynamics simulations. J. Mol. Biol 2005, 345, 869–878. [DOI] [PubMed] [Google Scholar]
- (148).Karplus M; Kuriyan J Molecular dynamics and protein function. Proc. Natl. Acad. Sci. U.S.A 2005, 102, 6679–6685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (149).Bond PJ; Holyoake J; Ivetac A; Khalid S; Sansom MSP Coarse-grained molecular dynamics simulations of membrane proteins and peptides. J. Struct. Biol 2007, 157, 593–605. [DOI] [PubMed] [Google Scholar]
- (150).Lindahl E; Sansom M Membrane proteins: Molecular dynamics simulations. Curr. Opin. Struct. Biol 2008, 18, 425–431. [DOI] [PubMed] [Google Scholar]
- (151).Klepeis JL; Lindorff-Larsen K; Dror RO; Shaw DE Long-timescale molecular dynamics simulations of protein structure and function. Curr. Opin. Struct. Biol 2009, 19, 120–127. [DOI] [PubMed] [Google Scholar]
- (152).Shaw DE; Maragakis P; Lindorff-Larsen K; Piana S; Dror RO; Eastwood MP; Bank JA; Jumper JM; Salmon JK; Shan Y et al. Atomic-level characterization of the structural dynamics of proteins. Science 2010, 330, 341–346. [DOI] [PubMed] [Google Scholar]
- (153).Lindorff-Larsen K; Piana S; Dror RO; Shaw DE How fast-folding proteins fold. Science 2011, 334, 517–520. [DOI] [PubMed] [Google Scholar]
- (154).Piana S; Klepeis JL; Shaw DE Assessing the accuracy of physical models used in protein-folding simulations: Quantitative evidence from long molecular dynamics simulations. Curr. Opin. Struct. Biol 2014, 24, 98–105. [DOI] [PubMed] [Google Scholar]
- (155).Shukla D; Hernandez CX; Weber JK; Pande VS Markov state models provide insights into dynamic modulation of protein function. Acc. Chem. Res 2015, 48, 414–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (156).Robertson MJ; Tirado-Rives J; Jorgensen WL Improved peptide and protein torsional energetics with the OPLS-AA force field. J. Chem. Theory Comp 2015, 11, 3499–3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (157).Brown Anne M.; Bevan David R. Molecular dynamics simulations of amyloid β-peptide (1–42): Tetramer formation and membrane interactions. Biophys. J 2016, 111, 937–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (158).Chavent M; Duncan AL; Sansom MSP Molecular dynamics simulations of membrane proteins and their interactions: From nanoscale to mesoscale. Curr. Opin. Struct. Biol 2016, 40, 8–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (159).Kuroda Y; Suenaga A; Sato Y; Kosuda S; Taiji M All-atom molecular dynamics analysis of multi-peptide systems reproduces peptide solubility in line with experimental observations. Sci. Rep 2016, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (160).Mori T; Miyashita N; Im W; Feig M; Sugita Y Molecular dynamics simulations of biological membranes and membrane proteins using enhanced conformational sampling algorithms. Biochim. Biophys. Acta Biomembr 2016, 1858, 1635–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (161).Childers MC; Daggett V Insights from molecular dynamics simulations for computational protein design. Mol. Syst. Des. Eng 2017, 2, 9–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (162).Robustelli P; Piana S; Shaw DE Developing a molecular dynamics force field for both folded and disordered protein states. Proc. Natl. Acad. Sci. U.S.A 2018, 115, E4758–E4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (163).Shrestha UR; Juneja P; Zhang Q; Gurumoorthy V; Borreguero JM; Urban V; Cheng X; Pingali SV; Smith JC; O’Neill HM et al. Generation of the configurational ensemble of an intrinsically disordered protein from unbiased molecular dynamics simulation. Proc. Natl. Acad. Sci. U.S.A 2019, 116, 20446–20452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (164).Sugita Y; Okamoto Y Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett 1999, 314, 141–151. [Google Scholar]
- (165).Nymeyer H How efficient is replica exchange molecular dynamics? An analytic approach. J. Chem. Theory Comput 2008, 4, 626–636. [DOI] [PubMed] [Google Scholar]
- (166).Sindhikara DJ; Emerson DJ; Roitberg A Exchange often and properly in replica exchange molecular dynamics. J. Chem. Theory Comput 2010, 6, 2804–2808. [DOI] [PubMed] [Google Scholar]
- (167).Okur A; Roe DR; Cui G; Hornak V; Simmerling C Improving convergence of replica-exchange simulations through coupling to a high-temperature structure reservoir. J. Chem. Theory Comput 2007, 3, 557–568. [DOI] [PubMed] [Google Scholar]
- (168).Patriksson A; van der Spoel D A temperature predictor for parallel tempering simulations. Phys. Chem. Chem. Phys 2008, 10, 2073–2077. [DOI] [PubMed] [Google Scholar]
- (169).Laio A; Parrinello M Escaping free-energy minima. Proc. Natl. Acad. Sci. U.S.A 2002, 99, 12562–12566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (170).Spitaleri A; Ghitti M; Mari S; Alberici L; Traversari C; Rizzardi GP; Musco G Use of metadynamics in the design of isoDGR-based αvβ3 antagonists to fine-tune the conformational ensemble. Angew. Chem. Int. Ed 2011, 50, 1832–1836. [DOI] [PubMed] [Google Scholar]
- (171).Yu H; Lin Y-S Toward structure prediction of cyclic peptides. Phys. Chem. Chem. Phys 2015, 17, 4210–4219. [DOI] [PubMed] [Google Scholar]
- (172).McHugh SM; Rogers JR; Yu H; Lin Y-S Insights into how cyclic peptides switch conformations. J. Chem. Theory Comp 2016, 12, 2480–2488. [DOI] [PubMed] [Google Scholar]
- (173).Piana S; Laio A A bias-exchange approach to protein folding. J. Phys. Chem. B 2007, 111, 4553–4559. [DOI] [PubMed] [Google Scholar]
- (174).Branduardi D; Gervasio FL; Parrinello M From a to b in free energy space. J. Chem. Phys 2007, 126, 054103. [DOI] [PubMed] [Google Scholar]
- (175).Spiwok V; Lipovova P; Kralova B Metadynamics in essential coordinates: Free energy simulation of conformational changes. J. Phys. Chem. B 2007, 111, 3073–3076. [DOI] [PubMed] [Google Scholar]
- (176).Sultan MM; Pande VS Tica-metadynamics: Accelerating metadynamics by using kinetically selected collective variables. J. Chem. Theory Comput 2017, 13, 2440–2447. [DOI] [PubMed] [Google Scholar]
- (177).Trapl D; Horvacanin I; Mareska V; Ozcelik F; Unal G; Spiwok V Anncolvar: Approximation of complex collective variables by artificial neural networks for analysis and biasing of molecular simulations. Frontiers in Molecular Biosciences 2019, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (178).Bonati L; Rizzi V; Parrinello M Data-driven collective variables for enhanced sampling. J. Phys. Chem. Lett 2020, 11, 2998–3004. [DOI] [PubMed] [Google Scholar]
- (179).Barducci A; Bussi G; Parrinello M Well-tempered metadynamics: A smoothly converging and tunable free-energy method. Phys. Rev. Lett 2008, 100. [DOI] [PubMed] [Google Scholar]
- (180).Paz SA; Abrams CF Free energy and hidden barriers of the beta-sheet structure of prion protein. J. Chem. Theory Comput 2015, 11, 5024–5034. [DOI] [PubMed] [Google Scholar]
- (181).Poulain P; Calvo F; Antoine R; Broyer M; Dugourd P Performances of wang-landau algorithms for continuous systems. Phys. Rev. E Stat. Nonlin. Soft Matter Phys 2006, 73, 056704. [DOI] [PubMed] [Google Scholar]
- (182).Bussi G; Gervasio FL; Laio A; Parrinello M Free-energy landscape for beta hairpin folding from combined parallel tempering and metadynamics. J. Am. Chem. Soc 2006, 128, 13435–13441. [DOI] [PubMed] [Google Scholar]
- (183).Bonomi M; Parrinello M Enhanced sampling in the well-tempered ensemble. Phys. Rev. Lett 2010, 104. [DOI] [PubMed] [Google Scholar]
- (184).Sugita Y; Kitao A; Okamoto Y Multidimensional replica-exchange method for free-energy calculations. J. Chem. Phys 2000, 113, 6042. [Google Scholar]
- (185).Berg B; Neuhaus T Multicanonical ensemble: A new approach to simulate first-order phase transitions. Phys. Rev. Lett 1992, 68, 9–12. [DOI] [PubMed] [Google Scholar]
- (186).Nakajima N; Nakamura H; Kidera A Multicanonical ensemble generated by molecular dynamics simulation for enhanced conformational sampling of peptides. J. Phys. Chem. B 1997, 101, 817–824. [Google Scholar]
- (187).Higo J; Nakamura H Virtual states introduced for overcoming entropic barriers in conformational space. Biophysics (Nagoya-shi) 2012, 8, 139–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (188).Higo J; Umezawa K; Nakamura H A virtual-system coupled multicanonical molecular dynamics simulation: Principles and applications to free-energy landscape of protein–protein interaction with an all-atom model in explicit solvent. J. Chem. Phys 2013, 138, 184106. [DOI] [PubMed] [Google Scholar]
- (189).Ikebe J; Umezawa K; Kamiya N; Sugihara T; Yonezawa Y; Takano Y; Nakamura H; Higo J Theory for trivial trajectory parallelization of multicanonical molecular dynamics and application to a polypeptide in water. J. Comput. Chem 2011, 32, 1286–1297. [DOI] [PubMed] [Google Scholar]
- (190).Ono S; Naylor MR; Townsend CE; Okumura C; Okada O; Lokey RS Conformation and permeability: Cyclic hexapeptide diastereomers. J. Chem. Inf. Model 2019, 59, 2952–2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (191).Hamelberg D; Mongan J; McCammon JA Accelerated molecular dynamics: A promising and efficient simulation method for biomolecules. J. Chem. Phys 2004, 120, 11919–11929. [DOI] [PubMed] [Google Scholar]
- (192).Miao Y; Sinko W; Pierce L; Bucher D; Walker RC; McCammon JA Improved reweighting of accelerated molecular dynamics simulations for free energy calculation. J. Chem. Theory Comput 2014, 10, 2677–2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (193).Pierce LC; Salomon-Ferrer R; Augusto F. d. O. C.; McCammon JA; Walker RC Routine access to millisecond time scale events with accelerated molecular dynamics. J. Chem. Theory Comput 2012, 8, 2997–3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (194).Jarzynski C Nonequilibrium equality for free energy differences. Phys. Rev. Lett 1997, 78, 2690–2693. [Google Scholar]
- (195).Park S; Khalili-Araghi F; Takjkhorshid E; Schulten K Free energy calculation from steered molecular dynamics simulations using jarzynski’s equality. J. Chem. Phys 2003, 119, 3559–3566. [Google Scholar]
- (196).Shkurti A; Styliari ID; Balasubramanian V; Bethune I; Pedebos C; Jha S; Laughton CA Coco-MD: A simple and effective method for the enhanced sampling of conformational space. J. Chem. Theory Comput 2019, 15, 2587–2596. [DOI] [PubMed] [Google Scholar]
- (197).Laughton CA; Orozco M; Vranken W Coco: A simple tool to enrich the representation of conformational variability in NMR structures. Proteins 2009, 75, 206–216. [DOI] [PubMed] [Google Scholar]
- (198).Roitberg AE; Okur A; Simmerling C Coupling of replica exchange simulations to a non-Boltzmann structure reservoir. J. Phys. Chem. B 2007, 111, 2415–2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (199).Helfand E Theory of the kinetics of conformational transitions in polymers. J. Chem. Phys 1971, 54, 4651. [Google Scholar]
- (200).Fadel AR; D. JR; Montelione GT; Levy RM Crankshaft motions of the polypeptide backbone in molecular dynamics simulations of human type-a transforming growth factor. J. Biomol. NMR 1995, 6, 221–226. [DOI] [PubMed] [Google Scholar]
- (201).Kamenik AS; Lessel U; Fuchs JE; Fox T; Liedl KR Peptidic macrocycles - conformational sampling and thermodynamic characterization. J. Chem. Inf. Model 2018, 58, 982–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (202).Xiong J-P; Stehle T; Zhang R; Joachimiak A; Frech M; Goodman SL; Arnaout MA Crystal structure of the extracellular segment of integrin αvβ3 in complex with an Arg-Gly-Asp ligand. Science 2002, 296, 151–155. [DOI] [PubMed] [Google Scholar]
- (203).Mas-Moruno C; Rechenmacher F; Kessler H Cilengitide: The first anti-angiogenic small molecule drug candidate. Design, synthesis and clinical evaluation. Anti-Cancer Agents Med. Chem 2010, 10, 753–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (204).Burke PA; DeNardo SJ; Miers LA; Lamborn KR; Matzku S; DeNardo GL Cilengitide targeting of alphavbeta3 integrin receptor synergizes with radioimmunotherapy to increase efficacy and apoptosis in breast cancer xenografts. Cancer Res. 2002, 62, 4263–4272. [PubMed] [Google Scholar]
- (205).Eisele G; Wick A; Eisele AC; Clement PM; Tonn J; Tabatabai G; Ochsenbein A; Schlegel U; Neyns B; Krex D et al. Cilengitide treatment of newly diagnosed glioblastoma patients does not alter patterns of progression. J. Neurooncol 2014, 117, 141–145. [DOI] [PubMed] [Google Scholar]
- (206).Maier JA; Martinez C; Kasavajhala K; Wickstrom L; Hauser K; Simmerling C ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comp 2015, 11, 3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (207).Jorgensen WL; Chandrasekhar J; Madura JD; Impey RW; Klein ML Comparison of simple potential functions for simulating liquid water. J. Chem. Phys 1983, 79, 926–935. [Google Scholar]
- (208).Khoury GA; Smadbeck J; Tamamis P; Vandris AC; Kieslich CA; Floudas CA Forcefield_ncaa:Ab initiocharge parameters to aid in the discovery and design of therapeutic proteins and peptides with unnatural amino acids and their application to complement inhibitors of the compstatin family. ACS Synth. Biol 2014, 3, 855–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (209).Case DA; Betz RM; Botello-Smith W; Cerutti DS; Cheatham I,TE;; Darden TA; Duke RE; Giese TJ; Gohlke H; Goetz AW et al. University of California: San Francisco, CA, 2016.
- (210).Hamelberg D; de Oliveira CA; McCammon JA Sampling of slow diffusive conformational transitions with accelerated molecular dynamics. J. Chem. Phys 2007, 127, 155102. [DOI] [PubMed] [Google Scholar]
- (211).Sittel F; Jain A; Stock G Principal component analysis of molecular dynamics: On the use of Cartesian vs. Internal coordinates. J. Chem. Phys 2014, 141, 014111. [DOI] [PubMed] [Google Scholar]
- (212).Schmid N; Eichenberger AP; Choutko A; Riniker S; Winger M; Mark AE; van Gunsteren WF Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur. Biophys. J 2011, 40, 843–856. [DOI] [PubMed] [Google Scholar]
- (213).Berendsen HJC; Postma JPM; van Gunsteren WF; Hermans J In Intermolecular forces; Pullman B, Ed., 1981.
- (214).Witek J; Keller BG; Blatter M; Meissner A; Wagner T; Riniker S Kinetic models of cyclosporin a in polar and apolar environments reveal multiple congruent conformational states. J. Chem. Inf. Model 2016, 56, 1547–1562. [DOI] [PubMed] [Google Scholar]
- (215).Wang J; Wolf RM; Caldwell JW; Kollman PA; Case DA Development and testing of a general Amber force field. J. Comput. Chem 2004, 25, 1157–1174. [DOI] [PubMed] [Google Scholar]
- (216).Kaminski GA; Friesner RA Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J. Phys. Chem. B 2001, 105, 6474–6487. [Google Scholar]
- (217).Horn HW; Swope WC; Pitera JW; Madura JD; Dick TJ; Hura GL; Head-Gordon T Development of an improved four-site water model for biomolecular simulations: TIP4P-ew. J. Chem. Phys 2004, 120, 9665–9678. [DOI] [PubMed] [Google Scholar]
- (218).Lindorff-Larsen K; Piana S; Palmo K; Maragakis P; Klepeis JL; Dror RO; Shaw DE Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 78, 1950–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (219).Jiang F; Zhou CY; Wu YD Residue-specific force field based on the protein coil library. RSFF1: Modification of OPLS-AA/L. J. Phys. Chem. B 2014, 118, 6983–6998. [DOI] [PubMed] [Google Scholar]
- (220).Zhou CY; Jiang F; Wu YD Residue-specific force field based on protein coil library. RSFF2: Modification of AMBER ff99SB. J. Phys. Chem. B 2015, 119, 1035–1048. [DOI] [PubMed] [Google Scholar]
- (221).Geng H; Jiang F; Wu YD Accurate structure prediction and conformational analysis of cyclic peptides with residue-specific force fields. J. Phys. Chem. Lett 2016, 7, 1805–1810. [DOI] [PubMed] [Google Scholar]
- (222).Rodriguez A; Laio A Clustering by fast search and find of density peaks. Science 2014, 344, 1492–1496. [DOI] [PubMed] [Google Scholar]
- (223).Piana S; Lindorff-Larsen K; Shaw DE How robust are protein folding simulations with respect to force field parameterization? Biophys. J 2011, 100, L47–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (224).Beauchamp KA; Lin Y-S; Das R; Pande VS Are protein force fields getting better? A systematic benchmark on 524 diverse NMR measurements. J. Chem. Theory Comput 2012, 8, 1409–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (225).Kollman PA Advances and continuing challenges in achieving realistic and predictive simulations of the properties of organic and biological molecules. Acc. Chem. Res 1996, 29, 461–469. [Google Scholar]
- (226).Duan Y; Wu C; Chowdhury S; Lee MC; Xiong G; Zhang W; Yang R; Cieplak P; Luo R; Lee T et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem 2003, 24, 1999–2012. [DOI] [PubMed] [Google Scholar]
- (227).Oostenbrink C; Villa A; Mark AE; van Gunsteren WF A biomolecular force field based on the free enthalpy of hydration and solvation: The GROMOS force-field parameter sets 53A5 and 53A6. J. Comput. Chem 2004, 25, 1656–1676. [DOI] [PubMed] [Google Scholar]
- (228).Kamens AJ; Eisert RJ; Corlin T; Baleja JD; Kritzer JA Structured cyclic peptides that bind the EH domain of EHD1. Biochemistry 2014, 53, 4758–4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (229).Todorova N; Marinelli F; Piana S; Yarovsky I Exploring the folding free energy landscape of insulin using bias exchange metadynamics. J. Phys. Chem. B 2009, 113, 3556–3564. [DOI] [PubMed] [Google Scholar]
- (230).Baftizadeh F; Cossio P; Pietrucci F; Laio A Protein folding and ligand-enzyme binding from bias-exchange meta-dynamics simulations Curr. Phys. Chem 2012, 2, 79–91. [Google Scholar]
- (231).White TR; Renzelman CM; Rand AC; Rezai T; McEwen CM; Gelev VM; Turner RA; Linington RG; Leung SSF; Kalgutkar AS et al. On-resin N-methylation of cyclic peptides for discovery of orally bioavailable scaffolds. Nat. Chem. Biol 2011, 7, 810–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (232).Biron E; Chatterjee J; Ovadia O; Langenegger D; Brueggen J; Hoyer D; Schmid HA; Jelinek R; Gilon C; Hoffman A et al. Improving oral bioavailability of peptides by multiple N-methylation: Somatostatin analogues. Angew. Chem. Int. Ed 2008, 47, 2595–2599. [DOI] [PubMed] [Google Scholar]
- (233).Rader AFB; Reichart F; Weinmuller M; Kessler H Improving oral bioavailability of cyclic peptides by N-methylation. Bioorg. Med. Chem 2018, 26, 2766–2773. [DOI] [PubMed] [Google Scholar]
- (234).Dechantsreiter M; Planker E; Matha B; Lohof E; Holzemann G; Jonczyk A; Goodman SL; Kessler H N-methylated cyclic RGD peptides as highly active and selective αvβ3 integrin antagonists. J. Med. Chem 1999, 42, 3033–3040. [DOI] [PubMed] [Google Scholar]
- (235).Chatterjee J; Rechenmacher F; Kessler H N-methylation of peptides and proteins: An important element for modulating biological functions. Angew. Chem. Int. Ed 2013, 52, 254–269. [DOI] [PubMed] [Google Scholar]
- (236).Paissoni C; Ghitti M; Belvisi L; Spitaleri A; Musco G Metadynamics simulations rationalise the conformational effects induced by N-methylation of RGD cyclic hexapeptides. Chem. Eur. J 2015, 21, 14165–14670. [DOI] [PubMed] [Google Scholar]
- (237).Witek J; Muhlbauer M; Keller BG; Blatter M; Meissner A; Wagner T; Riniker S Interconversion rates between conformational states as rationale for the membrane permeability of cyclosporines. ChemPhysChem 2017, 18, 3309–3314. [DOI] [PubMed] [Google Scholar]
- (238).Witek J; Wang S; Schroeder B; Lingwood R; Dounas A; Roth HJ; Fouche M; Blatter M; Lemke O; Keller B et al. Rationalization of the membrane permeability differences in a series of analogue cyclic decapeptides. J. Chem. Inf. Model 2019, 59, 294–308. [DOI] [PubMed] [Google Scholar]
- (239).Slough DP; Yu H; McHugh SM; Lin YS Toward accurately modeling N-methylated cyclic peptides. Phys. Chem. Chem. Phys 2017, 19, 5377–5388. [DOI] [PubMed] [Google Scholar]
- (240).Berendsen HJC; Grigera JR; Straatsma TP The missing term in effective pair potentials. J. Phys. Chem 1987, 91, 6269–6271. [Google Scholar]
- (241).Abascal JL; Vega C A general purpose model for the condensed phases of water: TIP4P/2005. J. Chem. Phys 2005, 123, 234505. [DOI] [PubMed] [Google Scholar]
- (242).Slough DP; McHugh SM; Cummings AE; Dai P; Pentelute BL; Kritzer JA; Lin YS Designing well-structured cyclic pentapeptides based on sequence-structure relationships. J. Phys. Chem. B 2018, 122, 3908–3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (243).McHugh SM; Yu H; Slough DP; Lin YS Mapping the sequence-structure relationships of simple cyclic hexapeptides. Phys. Chem. Chem. Phys 2017, 19, 3315–3324. [DOI] [PubMed] [Google Scholar]
- (244).Cummings AE; Miao J; Slough DP; McHugh SM; Kritzer JA; Lin YS β-branched amino acids stabilize specific conformations of cyclic hexapeptides. Biophys. J 2019, 116, 433–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (245).Kirshenbaum K; Barron AE; Goldsmith RA; Armand P; Bradley EK; Truong KTV; Dill KA; Cohen FE; Zuckermann RN Sequence-specific polypeptoids: A diverse family of heteropolymers with stable secondary structure. Proc. Natl. Acad. Sci. U.S.A 1998, 95, 4303–4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (246).Wu CW; Sanborn TJ; Huang K; Zuckermann RN; Barron AE Peptoid oligomers with r-chiral, aromatic side chains: Sequence requirements for the formation of stable peptoid helices. J. Am. Chem. Soc 2001, 123, 6778–6784. [DOI] [PubMed] [Google Scholar]
- (247).Lee B-C; Zuckermann RN; Dil KA Folding a nonbiological polymer into a compact multihelical structure. J. Am. Chem. Soc 2005, 127, 10999–11009. [DOI] [PubMed] [Google Scholar]
- (248).Lee B-C; Chu TK; Dill KA; Zuckermann RN Biomimetic nanostructures: Creating a high-affinity zinc-binding site in a folded nonbiological polymer. J. Am. Chem. Soc 2008, 130, 8847–8855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (249).Stringer JR; Crapster JA; Guzei IA; Blackwell HE Extraordinarily robust polyproline type I peptoid helices generatedviathe incorporation of α-chiral aromaticn-1-naphthylethyl side chains. J. Am. Chem. Soc 2011, 133, 15559–15567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (250).Miller SM; Simon RJ; Ng S; Zuckermann RN; Kerr JM; Moos WH Comparison of the proteolytic susceptibilities of homologous L-amino acid, D-amino acid, and n-substituted clycine peptide and peptoid oligomers. Drug Development Research 1995, 35, 20–32. [Google Scholar]
- (251).Voelz VA; Dill KA; Chorny I Peptoid conformational free energy landscapes from implicit-solvent molecular simulations in AMBER. Biopolymers 2011, 96, 639–650. [DOI] [PubMed] [Google Scholar]
- (252).Onufriev A; Bashford D; Case DA Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins 2004, 55, 383–394. [DOI] [PubMed] [Google Scholar]
- (253).Butterfoss GL; Renfrew PD; Kuhlman B; Krishenbaum K; Bonneau R A preliminary survey of the peptoid folding landscape. J. Am. Chem. Soc 2009, 131, 16798–16807. [DOI] [PubMed] [Google Scholar]
- (254).Titlestad K; Groth P; Dale J; Ali MY Unique conformation of the cyclic octapeptide of sarcosine and a related depsipeptide. J. C. S. Chem. Comm 1973, 346–347. [Google Scholar]
- (255).Higo J; Kamiya N; Sugihara T; Yonezawa Y; Nakamura H Verifying trivial parallelization of multicanonical molecular dynamics for conformational sampling of a polypeptide in explicit water. Chem. Phys. Lett 2009, 473, 326–329. [Google Scholar]
- (256).Nakajima N; Higo J; Kidera A; Nakamura H Free energy landscapes of peptides by enhanced conformational sampling. J. Mol. Biol 2000, 296, 197–216. [DOI] [PubMed] [Google Scholar]
- (257).Ono S; Nakajima N; Higo J; Nakamura H Peptide free-energy profile is strongly dependent on the force field: Comparison of C96 and AMBER95. J. Comput. Chem 2000, 21, 748–762. [Google Scholar]
- (258).Higo J; Galzitskaya OV; Ono S; Nakamura H Energy landscape of a ~-hairpin peptide in explicit water studied by multicanonical molecular dynamics. Chem. Phys. Lett 2001, 337, 169–175. [Google Scholar]
- (259).Higo J; Nishimura Y; Nakamura H A free-energy landscape for coupled folding and binding of an intrinsically disordered protein in explicit solvent from detailed all-atom computations. J. Am. Chem. Soc 2011, 133, 10448–10458. [DOI] [PubMed] [Google Scholar]
- (260).Nishigami H; Kamiya N; Nakamura H Revisiting antibody modeling assessment for cdr-h3 loop. Protein Engineering Design and Selection 2016, 29, 477–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (261).Bekker G-J; Kamiya N; Araki M; Fukuda I; Okuno Y; Nakamura H Accurate prediction of complex structure and affinity for a flexible protein receptor and its inhibitor. J. Chem. Theory Comp 2017, 13, 2389–2399. [DOI] [PubMed] [Google Scholar]
- (262).Case DA; Cerutti DS; Cheatham TE III; Darden TA; Duke RE; Giese TJ; Gohlke H; Goetz AW; Greene D; Homeyer N et al. University of California: San Francisco, CA, 2017. [Google Scholar]
- (263).Dickson CJ; Madej BD; Skjevik ÅA; Betz RM; Teigen K; Gould IR; Walker RC Lipid14: The Amber lipid force field. J. Chem. Theory Comp 2014, 10, 865–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (264).Dupradeau F-Y; Pigache A; Zaffran T; Savineau C; Lelong R; Grivel N; Lelong D; Rosanski W; Cieplak P The R.E.D. Tools: Advances in RESP and esp charge derivation and force field library building. Phys. Chem. Chem. Phys 2010, 12, 7821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (265).Di L; Whitney-Pickett C; Umland JP; Zhang H; Zhang X; Gebhard DF; Lai Y; Federico JJ; Davidson RE; Smith R et al. Development of a new permeability assay using low-efflux MDCKII cells. J. Pharm. Sci 2011, 100, 4974–4985. [DOI] [PubMed] [Google Scholar]
- (266).Naylor MR; Ly AM; Handford MJ; Ramos DP; Pye CR; Furukawa A; Klein V; Noland RP; Edmondson Q; Turmon AC et al. Lipophilic permeability efficiency (LPE) reconciles the opposing roles of lipophilicity in membrane permeability and aqueous solubility. J. Med. Chem 2018, 61, 11169–11182. [DOI] [PubMed] [Google Scholar]
- (267).Furukawa A; Schwochert J; Pye CR; Asano D; Edmondson QD; Turmon AC; Klein VG; Ono S; Okada O; Lokey RS Drug-like properties in macrocycles above mw 1000: Backbone rigidity versus side-chain lipophilicity. Angew. Chem. Int. Ed 2020, Online Version of Record before inclusion in an issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (268).Whitty A; Zhong M; Viarengo L; Beglov D; Hall DR; Vajda S Quantifying the chameleonic properties of macrocycles and other high-molecular-weight drugs. Drug Discov. Today 2016, 21, 712–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (269).Rossi Sebastiano M; Doak BC; Backlund M; Poongavanam V; Over B; Ermondi G; Caron G; Matsson P; Kihlberg J Impact of dynamically exposed polarity on permeability and solubility of chameleonic drugs beyond the rule of 5. J. Med. Chem 2018, 61, 4189–4202. [DOI] [PubMed] [Google Scholar]
- (270).Danelius E; Poongavanam V; Peintner S; Wieske LHE; Erdélyi M; Kihlberg J Solution conformations explain the chameleonic behaviour of macrocyclic drugs. Chem. Eur. J 2020, 26, 5231–5244. [DOI] [PubMed] [Google Scholar]
- (271).Kolano C; Helbing J; Kozinski M; Sander W; Hamm P Watching hydrogen-bond dynamics in a beta-turn by transient two-dimensional infrared spectroscopy. Nature 2006, 444, 469–472. [DOI] [PubMed] [Google Scholar]
- (272).Li F; Bravo-Rodriguez K; Phillips C; Seidel RW; Wieberneit F; Stoll R; Doltsinis NL; Sanchez-Garcia E; Sander W Conformation and dynamics of a cyclic disulfide-bridged peptide: Effects of temperature and solvent. J. Phys. Chem. B 2013, 117, 3560–3570. [DOI] [PubMed] [Google Scholar]
- (273).Kolano C; Gomann K; Sander W Small cyclic disulfide peptides: Synthesis in preparative amounts and characterization by means of NMR and ft-IR spectroscopy. European Journal of Organic Chemistry 2004, 2004, 4167–4176. [Google Scholar]
- (274).Li F; Bravo-Rodriguez K; Fernandez-Oliva M; Ramirez-Anguita JM; Merz K; Winter M; Lehmann CW; Sander W; Sanchez-Garcia E Stereochemistry rules: A single stereocenter changes the conformation of a cyclic tetrapeptide. J. Phys. Chem. B 2013, 117, 10785–10791. [DOI] [PubMed] [Google Scholar]
- (275).Cheley S; Braha O; Lu X; Conlan S; Bayley H A functional protein pore with a “retro” transmembrane domain. Prot. Sci 1999, 8, 1256–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (276).Ben-Yedidia T; Beignon A-S; Partidos CD; Muller S; Arnon R A retro-inverso peptide analogue of influenza virus hemagglutinin b-cell epitope 91–108 induces a strong mucosal and systemic immune response and confers protection in mice after intranasal immunization. Mol. Immunol 2002, 39, 323–331. [DOI] [PubMed] [Google Scholar]
- (277).Li C; Zhan C; Zhao L; Chen X; Lu W-Y; Lu W Functional consequences of retro-inverso isomerization of a miniature protein inhibitor of the p53–mdm2 interaction. Bioorg. Med. Chem 2013, 21, 4045–4050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (278).Guptasarma P Reversal of peptide backbone direction may result in the mirroring of protein structure. FEBS Lett. 1992, 310, 205–210. [DOI] [PubMed] [Google Scholar]
- (279).Lacroix E; Viguera AR; Serrano L Reading protein sequences backwards. Fold. Des 1998, 3, 79–85. [DOI] [PubMed] [Google Scholar]
- (280).Mittl PRE; Deillon C; Sargent D; Liu N; Klauser S; Thomas RM; Gutte B; Grutter MG The retro-GCN4 leucine zipper sequence forms a stable three-dimensional structure. Proc. Natl. Acad. Sci. U.S.A 2000, 97, 2562–2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (281).Zerze GH; Stillinger FH; Debenedetti PG Computational investigation of retro-isomer equilibrium structures: Intrinsically disordered, foldable, and cyclic peptides. FEBS Letters 2019, 594, 104–113. [DOI] [PubMed] [Google Scholar]
- (282).Best RB; Mittal J Protein simulations with an optimized water model: Cooperative helix formation and temperature-induced unfolded state collapse. J. Phys. Chem. B 2010, 114, 14916–14923. [DOI] [PubMed] [Google Scholar]
- (283).Kirschner KN; Lexa KW; Salisburg AM; Alser KA; Joseph L; Andersen TT; Bennett JA; Jacobson HI; Shields GC Computational design and experimental discovery of an antiestrogenic peptide derived from alpha-fetoprotein. J. Am. Chem. Soc 2007, 129, 6263–6268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (284).Lexa KW; Alser KA; Salisburg AM; Ellens DJ; Hernandez L; Bono SJ; Michael HC; Derby JR; Skiba JG; Feldgus S et al. The search for low energy conformational families of small peptides: Searching for active conformations of small peptides in the absence of a known receptor. Int. J. Quantum Chem 2007, 107, 3001–3012. [Google Scholar]
- (285).Jacobson HI; Bennett JA; Mizejewski GJ Inhibition of estrogen-dependent breast cancer growth by a reaction product of α-fetoprotein and estradiol. Cancer Res. 1990, 50, 415–420. [PubMed] [Google Scholar]
- (286).Bennett JA; Zhu S; Pagano-Mirarchi A; Kellom TA; Jacobson HI Alpha-fetoprotein derived from a human hepatoma prevents growth of estrogen-dependent human breast cancer xenografts. Clin. Cancer Res 1998, 4, 2877–2884. [PubMed] [Google Scholar]
- (287).Mesfin FB; Bennett JA; Jacobson HI; Zhu S; Andersen TT Alpha-fetoprotein-derived antiestrotrophic octapeptide. Biochim. Biophys. Acta 2000, 1501, 33–43. [DOI] [PubMed] [Google Scholar]
- (288).Bennett JA; Mesfin FB; Andersen TT; Gierthy JF; Jacobson HI A peptide derived from alpha-fetoprotein prevents the growth of estrogen-dependent human breast cancers sensitive and resistant to tamoxifen. Proc. Natl. Acad. Sci. U.S.A 2002, 99, 2211–2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (289).Parikh RR Prevention of n-methyl-n-nitrosourea-induced breast cancer by alpha-fetoprotein (afp)-derived peptide, a peptide derived from the active site of afp. Clin. Cancer Res 2005, 11, 8512–8520. [DOI] [PubMed] [Google Scholar]
- (290).DeFreest LA; Mesfin FB; Joseph L; Stallmer DJMA; Reddy S; Balulad SS; Jacobson HI; Andersen TT; Bennett JA Synthetic peptide derived from a-fetoprotein inhibits growth of human breast cancer: Investigation of the pharmacophore and synthesis optimization. J. Peptide Res 2004, 63, 409–419. [DOI] [PubMed] [Google Scholar]
- (291).Shields GC Computational approaches for the design of peptides with anti-breast cancer properties. Future Med. Chem 2009, 1, 201–212. [DOI] [PubMed] [Google Scholar]
- (292).Temelso B; Alser KA; Gauthier A; Palmer AK; Shields GC Structural analysis of α-fetoprotein (afp)-like peptides with anti-breast-cancer properties. J. Phys. Chem. B 2014, 118, 4514–4526. [DOI] [PubMed] [Google Scholar]
- (293).Cramer CJ Essentials of computational chemistry: Theories and models; 2 ed.; Wiley & Sons Ltd: Chichester, U.K., 2004. [Google Scholar]
- (294).Hornak V; Abel R; Okur A; Strockbine B; Roitberg A; Simmerling C Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 2006, 65, 712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (295).Chapman NA; Dupré DJ; Rainey JK The apelin receptor: Physiology, pathology, cell signalling, and ligand modulation of a peptide-activated class a GPCR. Biochem. Cell Biol 2014, 92, 431–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (296).Tatemoto K; Hosoya M; Habata Y; Fujii R; Kakegawa T; Zou M-X; Kawamata Y; Fukusumi S; Hinuma S; Kitada C et al. Isolation and characterization of a novel endogenous peptide ligand for the human APJ receptor. Biochem. Biophys. Res. Commun 1998, 251, 471–476. [DOI] [PubMed] [Google Scholar]
- (297).Medhurst AD; Jennings CA; Robbins MJ; Davis RP; Ellis C; Winborn KY; Lawrie KWM; Hervieu G; Riley G; Bolaky JE et al. Pharmacological and immunohistochemical characterization of the APJ receptor and its endogenous ligand apelin. J. Neurochem 2003, 84, 1162–1172. [DOI] [PubMed] [Google Scholar]
- (298).Fan X; Zhou N; Zhang X; Mukhtar M; Lu Z; Fang J; DuBois GC; Pomerantz RJ Structural and functional study of the apelin-13 peptide, an endogenous ligand of the HIV-1 coreceptor, APJ. Biochemistry 2003, 42, 10163–10168. [DOI] [PubMed] [Google Scholar]
- (299).Langelaan DN; Bebbington EM; Reddy T; Rainey JK Structural insight into g-protein coupled receptor binding by apelin. Biochemistry 2009, 48, 537–548. [DOI] [PubMed] [Google Scholar]
- (300).Langelaan DN; Rainey JK Headgroup-dependent membrane catalysis of apelin-receptor interactions is likely. J. Phys. Chem. B 2009, 113, 10465–10471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (301).Macaluso NJ; Glen RC Exploring the ‘rprl’ motif of apelin-13 through molecular simulation and biological evaluation of cyclic peptide analogues. ChemMedChem 2010, 5, 1247–1253. [DOI] [PubMed] [Google Scholar]
- (302).Jorgensen WL; Maxwell DS; Tirado-Rives J Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc 1996, 118, 11225–11236. [Google Scholar]
- (303).Macaluso NJ; Pitkin SL; Maguire JJ; Davenport AP; Glen RC Discovery of a competitive apelin receptor (APJ) antagonist. ChemMedChem 2011, 6, 1017–1023. [DOI] [PubMed] [Google Scholar]
- (304).Harford-Wright E; Andre-Gregoire G; Jacobs KA; Treps L; Le Gonidec S; Leclair HM; Gonzalez-Diest S; Roux Q; Guillonneau F; Loussouarn D et al. Pharmacological targeting of apelin impairs glioblastoma growth. Brain 2017, 140, 2939–2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (305).Razavi AM; Wuest WM; Voelz VA Computational screening and selection of cyclic peptide hairpin mimetics by molecular simulation and kinetic network models. J. Chem. Inf. Model 2014, 54, 1425–1432. [DOI] [PubMed] [Google Scholar]
- (306).Newell PD; Boyd CD; Sondermann H; O’Toole GA A c-di-gmp effector system controls cell adhesion by inside-out signaling and surface protein cleavage. PLoS Biol. 2011, 9, e1000587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (307).Navarro MV; Newell PD; Krasteva PV; Chatterjee D; Madden DR; O’Toole GA; Sondermann H Structural basis for c-di-gmp-mediated inside-out signaling controlling periplasmic proteolysis. PLoS Biol. 2011, 9, e1000588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (308).Shirts M; Pande VS Screen savers of the world unite! Science 2000, 290, 1903–1904. [DOI] [PubMed] [Google Scholar]
- (309).Krishnan L; Engelman A Retroviral integrase proteins and HIV-1 DNA integration. J. Biol. Chem 2012, 287, 40858–40866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (310).Jurado KA; Wang H; Slaughter A; Feng L; Kessl JJ; Koh Y; Wang W; Ballandras-Colas A; Patel PA; Fuchs JR et al. Allosteric integrase inhibitor potency is determined through the inhibition of HIV-1 particle maturation. Proc. Natl. Acad. Sci. U.S.A 2013, 110, 8690–8695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (311).Engelman A; Cherepanov P The lentiviral integrase binding protein LEDGF/p75 and HIV-1 replication. PLoS Pathogens 2008, 4, e1000046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (312).Northfield SE; Wielens J; Headey SJ; Williams-Noonan BJ; Mulcair M; Scanlon MJ; Parker MW; Thompson PE; Chalmers DK Cyclic hexapeptide mimics of the LEDGF integrase recognition loop in complex with HIV-1 integrase. ChemMedChem 2018, 13, 1–12. [DOI] [PubMed] [Google Scholar]
- (313).Rose JP; Wu C-K; Hsiao C-D; Breslow E; Wang B-C Crystal structure of the neurophysin-oxytocin complex. Nat. Struct. Biol 1996, 3, 163–169. [DOI] [PubMed] [Google Scholar]
- (314).Wu CK; Hu B; Rose JP; Liu Z-J; Nguyen TL; Zheng C; Breslow E; Wang B-C Structures of an unliganded neurophysin and its vasopressin complex: Implications for binding and allosteric mechanisms. Protein Sci. 2001, 10, 1869–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (315).Bichet DG; Arthus MF; Lonergan M; Hendy GN; Paradis AJ; Fujiwara TM; Morgan K; Gregory MC; Rosenthal W; Didwania A X-linked nephrogenic diabetes insipidus mutations in north america and the hopewell hypothesis. J. Clin. Invest 1993, 92, 1262–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (316).Birnbaumer M; Gilbert S; Rosenthal W An extracellular congenital nephrogenic diabetes insipidus mutation of the vasopressin receptor reduces cell surface expression, affinity for ligand, and coupling to the gjadenylyl cyclase system. Mol. Endocrinol 1994, 8, 886–894. [DOI] [PubMed] [Google Scholar]
- (317).Baribeau DA; Anagnostou E Oxytocin and vasopressin: Linking pituitary neuropeptides and their receptors to social neurocircuits. Front. Neurosci 2015, 9, 335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (318).Baribeau DA; Anagnostou E Oxytocin and vasopressin: Linking pituitary neuropeptides and their receptors to social neurocircuits. Front Neurosci-Switz 2015, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (319).Wood SP; Tickle IJ; Treharne AM; Pitts JE; Mascarenhas Y; Li JY; Husain J; Cooper S; Blundell TL; Hruby VJ et al. Crystal structure analysis of deamino-oxytocin: Conformational flexibility and receptor binding. Science 1986, 232, 633–636. [DOI] [PubMed] [Google Scholar]
- (320).Langs DA; Smith GD; Stezowski JJ; Hughes RE Structure of pressinoic acid: The cyclic moiety of vasopressin. Science 1986, 232, 1240–1242. [DOI] [PubMed] [Google Scholar]
- (321).Syed Ibrahim B; Pattabhi V Trypsin inhibition by a peptide hormone: Crystal structure of trypsin–vasopressin complex. J. Mol. Biol 2005, 348, 1191–1198. [DOI] [PubMed] [Google Scholar]
- (322).Liwo A; Tempczyk A; Oldziej S; Shenderovich MD; Hruby VJ; Talluri S; Ciarkowski J; Kasprzykowski F; Lankiewicz L; Grzonka Z Explorationof the conformational space of oxytocin and arginine-vasopressin using the electrostatically driven Monte Carlo and molecular dynamics methods. Biopolymers 1996, 38, 157–175. [DOI] [PubMed] [Google Scholar]
- (323).Haensele E; Banting L; Whitley DC; Clark T Conformation and dynamics of 8-Arg-vasopressin in solution. J. Mol. Model 2014, 20, 2485. [DOI] [PubMed] [Google Scholar]
- (324).Haensele E; Saleh N; Read CM; Banting L; Whitley DC; Clark T Can simulations and modeling decipher NMR data for conformational equilibria? Arginine-vasopressin. J. Chem. Inf. Model 2016, 56, 1798–1807. [DOI] [PubMed] [Google Scholar]
- (325).Yedvabny E; Nerenberg PS; So C; Head-Gordon T Disordered structural ensembles of vasopressin and oxytocin and their mutants. J. Phys. Chem. B 2015, 119, 896–905. [DOI] [PubMed] [Google Scholar]
- (326).Chou CL; DiGiovanni SR; Luther A; Lolait SJ; Knepper MA Oxytocin as an antidiuretic hormone II. Role of v2 vasopressin receptor. Am. J. Physiol 1995, 269, F78–F85. [DOI] [PubMed] [Google Scholar]
- (327).Lowbridge J; Manning M; Haidar J. a.; Sawyer WH Synthesis and some pharmacological properties of [4-threonine,7-glycine]oxytocin, [l-(L-2-hydroxy-3-mercaptopropanoic acid),4-threonine,7-glycine]oxytocin (hydroxy[thr4,gly7]oxytocin), and [7-glycine]oxytocin, peptides with high oxytocic-antidiuretic selectivity. J. Med. Chem 1977, 20. [DOI] [PubMed] [Google Scholar]
- (328).Hibert M; Hoflack J; Trumpp-Kallmeyer S; Mouillac B; Chini B; Mahé E; Cotte N; Jard S; Manning M; Barberis C Functional architecture of vasopressin/oxytocin receptors. J. Recept. Signal Transduct. Res 1999, 19, 589–596. [DOI] [PubMed] [Google Scholar]
- (329).Manning M; Stoev S; Chini B; Durroux T; Mouillac B; Guillon G Peptide and non-peptide agonists and antagonists for the vasopressin and oxytocin v1a, v1b, v2 and ot receptors: Research tools and potential therapeutic agents. 2008, 170, 473–512. [DOI] [PubMed] [Google Scholar]
- (330).Christensen JH; Siggaard C; Corydon TJ; Robertson GL; Gregersen N; Bolund L; Rittig S Differential cellular handling of defective arginine vasopressin (AVP) prohormones in cells expressing mutations of the AVP gene associated with autosomal dominant and recessive familial neurohypophyseal diabetes insipidus. J. Clin. Endocrinol. Metab 2004, 89, 4521–4531. [DOI] [PubMed] [Google Scholar]
- (331).Willcutts MD; Felner E; White PC Autosomal recessive familial neurohypophyseal diabetes insipidus with continued secretion of mutant weakly active vasopressin. Hum. Mol. Genet 1999, 8, 1303–1307. [DOI] [PubMed] [Google Scholar]
- (332).Nerenberg PS; Head-Gordon T Optimizing protein−solvent force fields to reproduce intrinsic conformational preferences of model peptides. J. Chem. Theory Comput 2011, 7, 1220–1230. [DOI] [PubMed] [Google Scholar]
- (333).Doshi U; Hamelberg D Reoptimization of the AMBER force field parameters for peptide bond (omega) torsions using accelerated molecular dynamics. J. Phys. Chem. B 2009, 113, 16590–16595. [DOI] [PubMed] [Google Scholar]
- (334).Clozel M; Binkert C; Birker-Robaczewska M; Boukhadra C; Ding S-S; Fischli W; Hess P; Mathys B; Morrison K; Müller C et al. Pharmacology of the urotensin-II receptor antagonist palosuran (act-058362; 1-[2-(4-benzyl-4-hydroxy-piperidin-1-yl)-ethyl]-3-(2-methyl-quinolin-4-yl)-urea sulfate salt): First demonstration of a pathophysiological role of the urotensin system. J. Pharmacol. Exp. Ther 2004, 311, 204–212. [DOI] [PubMed] [Google Scholar]
- (335).Vaudry H; Do Rego J-C; Le Mevel J-C; Chatenet D; Tostivint H; Fournier A; Tonon M-C; Pelletier G; Michael Conlon J; Leprince J Urotensin II, from fish to human. Ann. N. Y. Acad. Sci 2010, 1200, 53–66. [DOI] [PubMed] [Google Scholar]
- (336).Tostivint H; Ocampo Daza D; Bergqvist CA; Quan FB; Bougerol M; Lihrmann I; Larhammar D Molecular evolution of gpcrs: Somatostatin/urotensin II receptors. J. Mol. Endocrinol 2014, 52, T61–T86. [DOI] [PubMed] [Google Scholar]
- (337).Vaudry H; Leprince J; Chatenet D; Fournier A; Lambert DG; Le Mével J-C; Ohlstein EH; Schwertani A; Tostivint H; Vaudry D et al. International union of basic and clinical pharmacology. Xcii. Urotensin II, urotensin II–related peptide, and their receptor: From structure to function. Pharmacol. Rev 2015, 67, 214–258. [DOI] [PubMed] [Google Scholar]
- (338).Sugo T; Murakami Y; Shimomura Y; Harada M; Abe M; Ishibashi Y; Kitada C; Miyajima N; Suzuki N; Mori M et al. Identification of urotensin II-related peptide as the urotensin II-immunoreactive molecule in the rat brain. Biochem. Biophys. Res. Commun 2003, 310, 860–868. [DOI] [PubMed] [Google Scholar]
- (339).Chatenet D; Dubessy C; Leprince J; Boularan C; Carlier L; Ségalas-Milazzo I; Guilhaudis L; Oulyadi H; Davoust D; Scalbert E et al. Structure–activity relationships and structural conformation of a novel urotensin II-related peptide. Peptides 2004, 25, 1819–1830. [DOI] [PubMed] [Google Scholar]
- (340).Haensele E; Mele N; Miljak M; Read CM; Whitley DC; Banting L; Delépée C; Sopkova-de Oliveira Santos J; Lepailleur A; Bureau R et al. Conformation and dynamics of human urotensin II and urotensin related peptide in aqueous solution. J. Chem. Inf. Model 2017, 57, 298–310. [DOI] [PubMed] [Google Scholar]
- (341).Ulysse L; Cubillos J; Chmielewsk J Photoregulation of cyclic peptide conformation. J. Am. Chem. Soc 1995, 117, 8466–8467. [Google Scholar]
- (342).Renner C; Moroder L Azobenzene as conformational switch in model peptides. ChemBioChem 2006, 7, 868–878. [DOI] [PubMed] [Google Scholar]
- (343).Beharry AA; Woolley GA Azobenzene photoswitches for biomolecules. Chem. Soc. Rev 2011, 40, 4422–4437. [DOI] [PubMed] [Google Scholar]
- (344).Babii O; Afonin S; Berditsch M; Reiβer S; Mykhailiuk PK; Kubyshkin VS; Steinbrecher T; Ulrich AS; Komarov IV Controlling biological activity with light: Diarylethene-containing cyclic peptidomimetics. Angew. Chem. Int. Ed 2014, 53, 3392–3395. [DOI] [PubMed] [Google Scholar]
- (345).Yeoh YQ; Yu J; Polyak SW; Horsley JR; Abell AD Photopharmacological control of cyclic antimicrobial peptides. ChemBioChem 2018, 19, 2591–2597. [DOI] [PubMed] [Google Scholar]
- (346).Albert L; Vazquez O Photoswitchable peptides for spatiotemporal control of biological functions. Chem. Commun. (Camb.) 2019, 55, 10192–10213. [DOI] [PubMed] [Google Scholar]
- (347).Afonin S; Babii O; Reuter A; Middel V; Takamiya M; Strähle U; Komarov IV; Ulrich AS Light-controllable dithienylethene-modified cyclic peptides: Photoswitching the in vivo toxicity in zebrafish embryos. Beilstein Journal of Organic Chemistry 2020, 16, 39–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (348).Behrendt R; Schenk M; Musiol HJ; Moroder L Photomodulation of conformational states. Synthesis of cyclic peptides with backbone-azobenzene moieties. J. Pep. Sci 1999, 5, 519–529. [DOI] [PubMed] [Google Scholar]
- (349).Renner C; Behrendt R; Sporlein S; Wachtveitl J; Moroder L Photomodulation of conformational states. I. Mono- and bicyclic peptides with (4-amino)phenylazobenzoic acid as backbone constituent. Biopolymers 2000, 54, 489–500. [DOI] [PubMed] [Google Scholar]
- (350).Renner C; Cramer J; Behrendt R; Moroder L Photomodulation of conformational states. II. Mono- and bicyclic peptides with (4-aminomethyl)-phenylazobenzoic acid as backbone constituent. Biopolymers 2000, 54, 501–514. [DOI] [PubMed] [Google Scholar]
- (351).Carstens H; Renner C; Milbradt AG; Moroder L; Tavan P Multiple loop conformations of peptides predicted by molecular dynamics simulations are compatible with nuclear magnetic resonance. Biochemistry 2005, 44, 4829–4840. [DOI] [PubMed] [Google Scholar]
- (352).Kuriyan J; Krishna TSR; Wong L; Guenther B; Pahler A; Jr CHW; Model P Convergent evolution of similar function in two structurally divergent enzymes. Nature 1991, 352, 172–174. [DOI] [PubMed] [Google Scholar]
- (353).Moroder L; Besse D; Musiol HJ; Rudolph-Böhner S; Siedler F Oxidative folding of cystine-rich peptides vs regioselective cysteine pairing strategies. Biopolymers 1996, 40, 207–234. [DOI] [PubMed] [Google Scholar]
- (354).Nguyen PH; Mu Y; Stock G Structure and energy landscape of a photoswitchable peptide: A replica exchange molecular dynamics study. Proteins 2005, 60, 485–494. [DOI] [PubMed] [Google Scholar]
- (355).Lindahl E; Hess B; Van Der Spoel D Gromacs 3.0: A package for molecular simulation and trajectory analysis. J. Mol. Model 2001, 7, 306–317. [Google Scholar]
- (356).Liu H; Miiller-Plathe F; Gunsteren WF v. A force field for liquid dimethyl sulfoxide and physical properties of liquid dimethyl sulfoxide calculated using molecular dynamics simulation. J. Am. Chem. Soc 1995, 117, 4363–4366. [Google Scholar]
- (357).Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Zakrzewski VG; J. A. M.; Stratmann RE; Burant JC et al. ; Gaussian, Inc.: Pittsburgh, PA, 1998.
- (358).Hynes RO Integrins: Bidirectional, allosteric signaling machines. Cell 2002, 110, 673–687. [DOI] [PubMed] [Google Scholar]
- (359).Pierschbacher MD; Ruoslahti E Cell attachment activity of fibronectin can be duplicated by small synthetic fragments of the molecule. Nature 1984, 309, 30–33. [DOI] [PubMed] [Google Scholar]
- (360).Pierschbacher MD; Ruoslahti E Variants of the cell recognition site of fibronectin that retain attachment-promotingactivity. Proc. Natl. Acad. Sci. U.S.A 1984, 81, 5985–5988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (361).Ruoslahti E; Pierschbacher MD Arg-Gly-Asp: A versatile cell recognition signal. Cell 1986, 44, 517–518. [DOI] [PubMed] [Google Scholar]
- (362).Ruoslahti E; Pierschbacher MD New perspectives in cell adhesion: RGD and integrins. Science 1987, 238, 491–497. [DOI] [PubMed] [Google Scholar]
- (363).Ruoslahti E Fibronectin and its receptors. Annu. Rev. Biochem 1988, 57, 375–413. [DOI] [PubMed] [Google Scholar]
- (364).Plow EF; Haas TA; Zhang L; Loftus J; Smith JW Ligand binding to integrins. J. Biol. Chem 2000, 275, 21785–21788. [DOI] [PubMed] [Google Scholar]
- (365).Aumailley M; Gurrath M; Muller G; Calvete J; Timpi R; Kessler H Arg-Gly-Asp constrained within cyclic pentapoptides strong and selective inhibitors of cell adhesion to vitronectin and laminin fragment p1. FEBS Lett. 1991, 291, 50–54. [DOI] [PubMed] [Google Scholar]
- (366).Haubner R; Gratias R; Diefenbach B; Goodman SL; Jonczyk A; Kessler H Structural and functional aspects of RGD-containing cyclic pentapeptides as highly potent and selective integrin αvβ3 antagonists. J. Am. Chem. Soc 1996, 118, 7461–7472. [Google Scholar]
- (367).Kessler H; Diefenbach B; Finsinger D; Geyer A; Gurrath M; Goodman SL; Holzemann G; Haubner R; Jonczyk A; Muller G et al. Design of superactive and selective integrin receptor antagonists containing the RGD sequence. Lett. Peptide Sci 1995, 2, 155–160. [Google Scholar]
- (368).Goodman SL; Holzemann G; Sulyok AG; Kessler H Nanomolar small molecule inhibitors for alphavbeta6, alphavbeta5, and alphavbeta3 integrins. J. Med. Chem 2002, 45, 1045–1051. [DOI] [PubMed] [Google Scholar]
- (369).Yamada S; Bu XY; Khankaldyyan V; Gonzales-Gomez I; McComb JG; Laug WE Effect of the angiogenesis inhibitor cilengitide (EMD 121974) on glioblastoma growth in nude mice. Neurosurgery 2006, 59, 1304–1312; discussion 1312. [DOI] [PubMed] [Google Scholar]
- (370).Wakefield AE; Wuest WM; Voelz VA Molecular simulation of conformational pre-organization in cyclic RGD peptides. J. Chem. Inf. Model 2015, 55, 806–813. [DOI] [PubMed] [Google Scholar]
- (371).Haubner R; Schmitt W; Holzemann G; Goodman SL; Jonczyk A; Kessler H Cyclic RGD peptides containing beta-turn mimetics. J. Am. Chem. Soc 1996, 118, 7881–7891. [Google Scholar]
- (372).Wermuth J; Goodman SL; Jonczyk A; Kessler H Stereoisomerism and biological activity of the selective and superactive αvβ3 integrin inhibitor cyclo(-RGDfV-) and its retro-inverso peptide. J. Am. Chem. Soc 1997, 119, 1328–1335. [Google Scholar]
- (373).Chatterjee J; Ovadia O; Zahn G; Marinelli L; Hoffman A; Gilon C; Kessler H Multiple N-Methylation by a designed approach enhances receptor selectivity. J. Med. Chem 2007, 50, 5878–5881. [DOI] [PubMed] [Google Scholar]
- (374).Springer TA; Zhu J; Xiao T Structural basis for distinctive recognition of fibrinogen gammac peptide by the platelet integrin alphaiibbeta3. J. Cell Biol 2008, 182, 791–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (375).Friesner RA; Murphy RB; Repasky MP; Frye LL; Greenwood JR; Halgren TA; Sanschagrin PC; Mainz DT Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem 2006, 49, 6177–6196. [DOI] [PubMed] [Google Scholar]
- (376).Tubert-Brohman I; Sherman W; Repasky M; Beuming T Improved docking of polypeptides with glide. J. Chem. Inf. Model 2013, 53, 1689–1699. [DOI] [PubMed] [Google Scholar]
- (377).Curnis F; Longhi R; Crippa L; Cattaneo A; Dondossola E; Bachi A; Corti A Spontaneous formation of L-isoaspartate and gain of function in fibronectin. J. Biol. Chem 2006, 281, 36466–36476. [DOI] [PubMed] [Google Scholar]
- (378).Spitaleri A; Mari S; Curnis F; Traversari C; Longhi R; Bordignon C; Corti A; Rizzardi GP; Musco G Structural basis for the interaction of isoDGR with the RGD-binding site of αvβ3 integrin. J. Biol. Chem 2008, 283, 19757–19768. [DOI] [PubMed] [Google Scholar]
- (379).Hermans J; Berendsen HJC; Gunsteren WFV; Postma JPM A consistent empirical potential for water–protein interactions. Biopolymers 1984, 23, 1513–1518. [Google Scholar]
- (380).Takagi J; Petre BM; Walz T; Springer TA Global conformational rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell 2002, 110, 599–611. [DOI] [PubMed] [Google Scholar]
- (381).Nardelli F; Paissoni C; Quilici G; Gori A; Traversari C; Valentinis B; Sacchi A; Corti A; Curnis F; Ghitti M et al. Succinimide-based conjugates improve isodgr cyclopeptide affinity to alphavbeta3 without promoting integrin allosteric activation. J. Med. Chem 2018, 61, 7474–7485. [DOI] [PubMed] [Google Scholar]
- (382).Curnis F; Sacchi A; Longhi R; Colombo B; Gasparri A; Corti A Isodgr-tagged albumin: A new α v β 3 selective carrier for nanodrug delivery to tumors. Small 2013, 9, 673–678. [DOI] [PubMed] [Google Scholar]
- (383).Paissoni C; Nardelli F; Zanella S; Curnis F; Belvisi L; Musco G; Ghitti M A critical assessment of force field accuracy against NMR data for cyclic peptides containing beta-amino acids. Phys. Chem. Chem. Phys 2018, 20, 15807–15816. [DOI] [PubMed] [Google Scholar]
- (384).Best RB; Hummer G Optimized molecular dynamics force fields applied to the helix-coil transition of polypeptides. J. Phys. Chem. B 2009, 113, 9004–9015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (385).Mackerell AD Jr.; Feig M; Brooks CL III. Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem 2004, 25, 1400–1415. [DOI] [PubMed] [Google Scholar]