

Abstract
Recent experimental studies engaging isotopically substituted protein (heavy protein) have revealed that many, but not all, enzymatic systems exhibit altered chemical steps in response to an altered mass. The results have been interpreted as femtosecond protein dynamics at the active site being linked (or not) to transition-state barrier crossing. An altered enzyme mass can influence several kinetic parameters (kcat, Km, and kchem) in amounts of ≤30% relative to light enzymes. An early report on deuterium-labeled Escherichia coli alkaline phosphatase (AP) showed an unusually large enzyme kinetic isotope effect on kcat. We examined steady-state and chemical step properties of native AP, [2H]AP, and [2H,13C,15N]AP to characterize the role of heavy enzyme protein dynamics in reactions catalyzed by AP. Both [2H]- and [2H,13C,15N]APs showed unaltered steady-state and single-turnover rate constants. These findings characterize AP as one of the enzymes in which mass-dependent catalytic site dynamics is dominated by reactant-linked atomic motions. Two catalytic site zinc ions activate the oxygen nucleophiles in the catalytic site of AP. The mass of the zinc ions is unchanged in light and heavy APs. They are essentially linked to catalysis and provide a possible explanation for the loss of linkage between catalysis and protein mass in these enzymes.
Graphical Abstract
Enzymes display a hierarchy of motions on varying time scales, which range from seconds to femtoseconds. The role of slower time scale motions has been characterized by both experimental and computational methods, but local, fast catalytic site motions that influence the femtosecond lifetime of the transition states have been limited to computational regimes and require additional experimental investigation for a better understanding of enzyme catalysis.1–6 Experimental approaches that have been useful for unraveling the involvement of slower protein motions in catalysis and some aspects of transition-state properties include nuclear magnetic resonance spectroscopy (NMR),7,8 vibrational spectroscopy,9,10 and the temperature dependence of kinetic isotope effects (KIEs).11–14 A recently developed tool designed to experimentally perturb femtosecond to picosecond time scale fast protein motions involves the labeling of proteins with heavy isotopes (13C, 15N, and 2H), uniformly2,15 or by specific amino acids at the catalytic site.16–18 Covalent bond vibrations occur on the femtosecond time scale, and isotopic substitutions have immediate consequences on this time scale. However, heavy protein effects are certainly propagated throughout the protein, and effects on slower steps, including substrate binding, conformational changes, and product release, are also possible. Therefore, proper analysis of mass-altered enzymes requires definition of both steady-state and pre-steady-state analysis, where the rate of the chemical step can be observed.
An early study of heavy enzyme kinetic isotope effects was reported in 1969 by Rokop et al., using a deuterated version of Escherichia coli alkaline phosphatase (EcAP),19 substituting all non-exchangeable hydrogens with deuteriums. This work reported a decrease in the steady-state kinetic parameter, kcat, by a factor of 80% compared to that of native (natural abundance isotope) EcAP. More recent studies on isotopically labeled heavy enzymes have shown smaller (≤30%) changes, usually slowed chemical steps (kchem), or steady-state kinetic changes. In a few exceptional cases, faster chemical barrier crossing has been observed by favorable modulation of appropriate local catalytic site motions.16,18
The highly conserved EcAP is a homodimeric, two-metal ion enzyme and is among the most proficient natural catalysts.20 It is one of the most frequently referenced enzymes from the large number of studies that have elucidated its mechanistic and structural features and sought to understand its exceptional catalytic power (≤1027-fold rate acceleration).21,22 Alkaline phosphatase catalyzes the hydrolysis of diverse phosphate monoesters to yield inorganic phosphate and the alcohol products. It also exhibits transphosphorylation activity in the presence of phosphate acceptors.23 Following substrate binding, a covalent catalytic site phosphoserine intermediate (E-P) is formed by the nucleophilic attack of Ser102 (Scheme 1). The active site consists of two Zn2+ ions, which are favorably placed to ionize Ser102 to an anionic nucleophile and stabilize the negative charge on the alcohol leaving group.20 The E-P intermediate has been isolated, characterized, and observed in X-ray crystal structures.24 The subsequent hydrolysis of E-P by water forms a noncovalent enzyme phosphate (E·P) product complex followed by the dissociation of phosphate to regenerate the active enzyme. The rate-determining step of AP is pH-dependent, with limiting E·Pi product release at alkaline pH and E-P hydrolysis at acidic pH.25,26
Scheme 1.

Simplified Catalytic Mechanism of Alkaline Phosphatase
A review of the kinetic properties of isotopically heavy enzymes demonstrates small changes in steady-state kinetic constants, chemical steps, and, in some cases, no significant changes in any kinetic parameters.13 Human purine nucleoside phosphorylase (PNP) is characterized by a nearly fully dissociated ribocation-like transition state in the phosphorolysis of purine nucleosides and is one of the enzymes most thoroughly studied by protein isotope substitution. In a critical investigation of human PNP, mass-altered protein dynamics had no effect on the slower time scale steady-state kinetic parameters, suggesting that the binding equilibrium, transition-state structure, and product release were unaffected by the altered mass.2 In contrast, single-turnover kinetic experiments and forward commitment analysis demonstrated a reduced rate of chemical barrier crossing for isotopically labeled heavy PNP.
In the case of EcAP, under experimental conditions that reported large deuterium enzyme kinetic isotope effects on kcat, phosphate product release is expected to be the rate-limiting step, prompting us to reinvestigate the large reported enzyme KIE on steady-state kinetic parameters. Here, we explored the protein mass-modulated effects on steady-state and pre-steady-state kinetic parameters of EcAP by means of isotopically labeled enzymes.
We expressed and purified native AP and [2H]AP to reinvestigate the large reported heavy enzyme isotope effects of EcAP. We also expanded the heavy enzyme exploration by preparing [2H,13C,15N]EcAP and by measuring the rates of chemical steps.
Isotope substitution of EcAP alters the femtosecond bond vibration frequency of amino acids throughout the protein. Heavy atom effects were investigated by measuring steady-state kinetics for two different substrates (p-nitrophenyl phosphate and 4-methylumbelliferyl phosphate) and by measuring pre-steady-state kinetics under single-turnover conditions. The formation of the product p-nitrophenol from the most employed substrate of AP, p-nitrophenyl phosphate, is accompanied by the change in absorbance at 405 nm, whereas the hydrolysis of 4-methylumbelliieryl phosphate yields a highly fluorescent product, 4-methylumbelliferone. Steady-state kinetic parameters were unchanged for both substrates at pH 8.0 upon comparison of native EcAP, [2H]EcAP, and [2H,13C,15N]EcAP. The EcAP chemical step was measured in pre-steady-state kinetic measurements at 5 °C to provide experimentally accessible rates, and native EcAP, [2H]EcAP, and [2H,13C,15N]EcAP have statistically indistinguishable rates for the formation of the enzyme-bound product (E-P) from the enzyme-bound substrate (E·ROP). Thus, mass-dependent alteration of protein femtosecond bond vibrational states indicated the absence of fast dynamic involvement in the enzymatic phosphorylation step for EcAP. This finding differentiates EcAP from most other enzymes, where an altered protein mass alters the probability of locating the transition state.14 Heavy enzyme protein dynamics associated with EcAP transition-state formation are discussed in comparison to those of other enzyme systems.
MATERIALS AND METHODS
Deuterium oxide (99.9%), [1,2,3,4,5,6,6-2H7]glucose, [U-13C6,1,2,3,4,5,6,6-2H7]glucose, and [15N]ammonium chloride were purchased from Cambridge Isotope Laboratories, Inc. All other chemicals and reagents were obtained from commercially available sources and used without further purification.
Expression, Purification, and Characterization of Natural Abundance Isotope and Isotopically Labeled AP.
Native AP, [2H]AP, and [2H,13C,15N]AP were expressed in E. coli BL21(DE3) cells harboring a pMAL-p2X vector containing the codifying sequence for EcAP with an N-terminal signal peptide for periplasmic export, maltose binding protein (MBP) fusion, and a factor Xa cleavage site. Cells were grown in LB medium (for native AP) and M63 minimal medium prepared in D2O and supplemented with [1,2,3,4,5,6,6-2H7]glucose (for [2H]AP) or [U-13C6,1,2,3,4,5,6,6-2H7]glucose and [15N]ammonium chloride (for [2H,13C,15N]AP). The cells were grown in the presence of 100 mg/mL ampicillin at 37 °C until the OD600 reached to 0.8 and then induced by the addition of 1 mM isopropyl 1-thio-β-d-galactopyranoside (IPTG). Cells were grown for an additional 24 h, and APs were purified following a published protocol.22 The three constructs of AP were characterized using ESI-MS, and the mass increase confirmed the appropriate isotopic incorporation.
Preparation of the Phosphate-Free Enzyme.
Inorganic phosphate was copurified with EcAP and removed from the enzymes by following a published protocol with a higher concentration of ZnSO4.29 In brief, the APs were dialyzed at 4 °C against a buffer containing 10 mM nitrilotriacetate, 100 mM NaCl, and 10 mM Tris buffered at pH 7.4. After removal of phosphate, the APs were dialyzed against the buffer containing 1 mM ZnSO4, 100 mM NaCl, and 10 mM Tris at pH 7.4.
Steady-State Kinetics.
Steady-state kinetic parameters of native AP, [2H]AP, and [2H,13C,15N]AP were determined by measuring the initial reaction rates as a function of pNPP or 4-MUP concentration at 25 °C. The increase in absorbance at 405 nm (pNPP) or the increase in fluorescence with an excitation wavelength of 370 nm and an emission cutoff of 420 nm (4-MUP) was monitored using a Molecular Device SpectraMax M5 microplate reader with SoftMax pro 6 software. Reaction mixtures contained 50 mM Tris buffer (pH 8.0), varying concentrations of pNPP or 4-MUP (1–100 μM), and 10 nM AP. The relative fluorescence signals were normalized with a known concentration of product 4-methylumbelliferone by allowing the reaction to proceed to completion. Kinetic parameters were obtained by fitting the data to eq 1
| (1) |
where ν is the initial reaction rate, V is the maximal reaction rate, [S] is the substrate concentration, and Km is the Michaelis constant.
Single-Turnover Rate Constants.
Single-turnover rate constants for native and isotopically labeled APs were determined at 5 °C by monitoring the increase in fluorescence upon formation of enzyme-bound 4-methylumbelliferrone29 using an SX-20 stopped-flow instrument provided with temperature control (Applied Photophysics, dead time of ≤1.25 ms). The reaction chamber was excited at 370 nm with a 1 nm slit width, and the fluorescence signal above 420 nm was detected using a model WG420 Scott filter positioned between the photometer and the sample cells. The left syringe contained 100 mM Tris (pH 8.0), 500 mM NaCl, 100 μM ZnCl2, 1 mM MgCl2, and 10 μM 4-MUP, and the right syringe contained 100 mM Tris (pH 8.0), 500 mM NaCl, 100 μM ZnCl2, 1 mM MgCl2, and 50 μM native AP, [2H]AP, or [2H,13C,15N]AP. The final concentration of the 4-MUP was 5 μM, and the AP concentration was 25 μM for a single-turnover condition. The fluorescence spectra were monitored for 250 ms; 1000 data points were collected for each spectrum, and each experiment was performed five times. The data were fitted to eq 2 using GraphPad Prism 7, to obtain the observed rate constant.
| (2) |
RESULTS AND DISCUSSION
Expression, Purification, and Characterization of APs.
Native AP, [2H]AP, and [2H,13C,15N]AP were expressed and purified to homogeneity as explained in Materials and Methods. Electrospray ionization mass spectrometry (ESI-MS) determined masses of 47043, 49636, and 52117 Da for native AP, [2H]AP, and [2H,13C,15N]AP, respectively, confirming 5.5% and 10.8% mass increases for [2H]AP and [2H,13C,15N]AP, respectively (Figure S1). Enzymatic mass increases due to isotopic substitution are limited to non-exchangeable positions, as solvent-exchangeable 2H atoms are replaced by 1H atoms during enzyme purification and storage in buffer prepared in H2O.
Preparation of the Phosphate-Free Enzyme.
Purified alkaline phosphatase can contain ≤2 equiv of tightly bound phosphate per dimer.19,30 Co-purified phosphate can interfere with the pre-steady-state kinetics, and removal of this endogenous phosphate requires removal of the two catalytic site Zn2+ ions (Figure 1A). Phosphate-free enzymes were prepared as explained in Materials and Methods. The removal of phosphate from purified enzymes was quantitated by the malachite green phosphate assay,31 based on the complex formed by malachite green, molybdate, and free orthophosphate.
Figure 1.
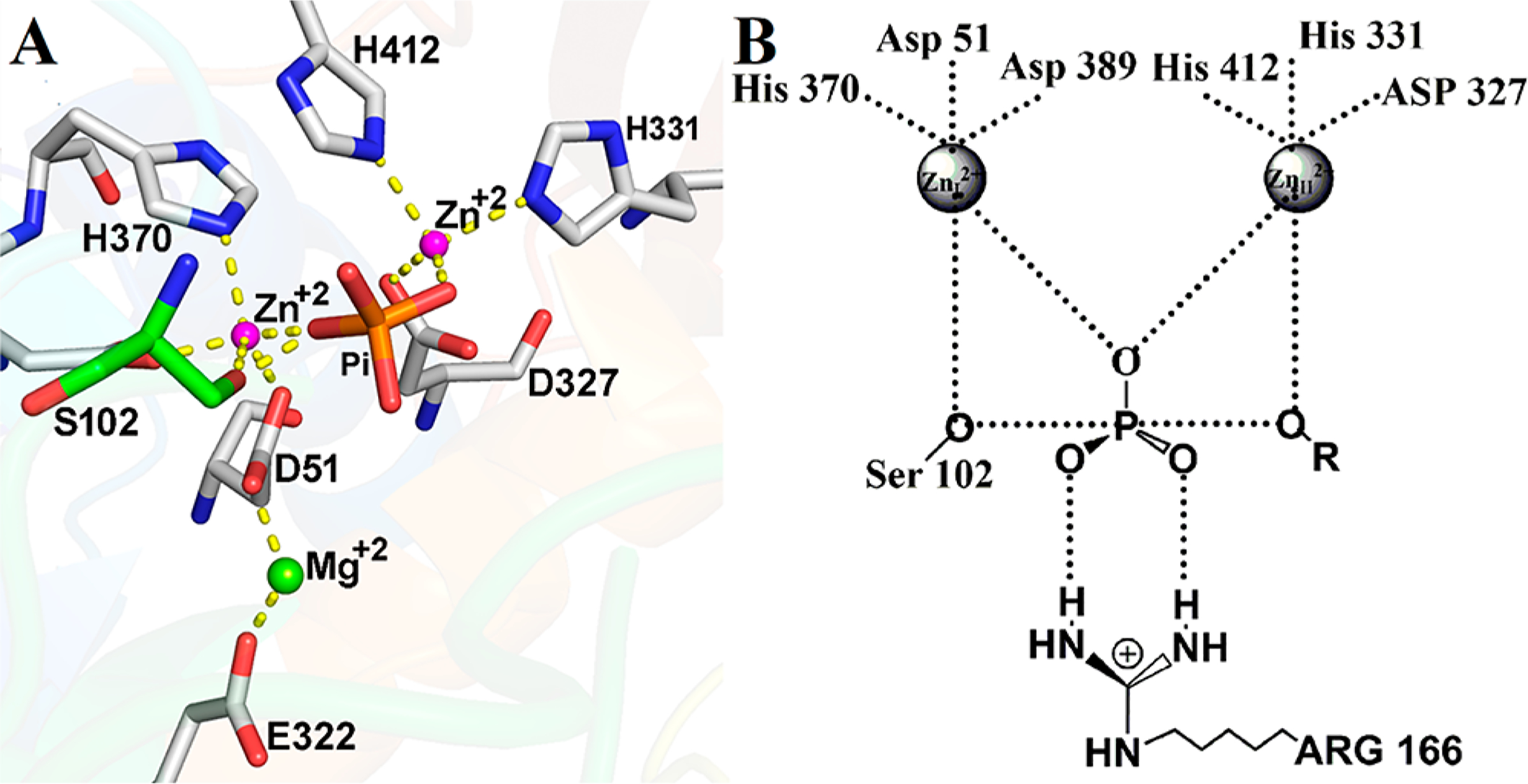
(A) Catalytic site configuration for EcAP based on the X-ray crystal structure of the enzyme with inorganic phosphate bound in the active site (Protein Data Bank entry 3TG0).27 The catalytic site serine is colored green, and the phosphate is colored orange and red. (B) Proposed transition-state structure for EcAP based upon a crystal structure of EcAP covalently bound to a pentavalent vanadate ester.28 The two Zn2+ ions are ~4 Å apart.
Steady-State Kinetics of Native AP, [2H]AP, and [2H,13C,15N]AP.
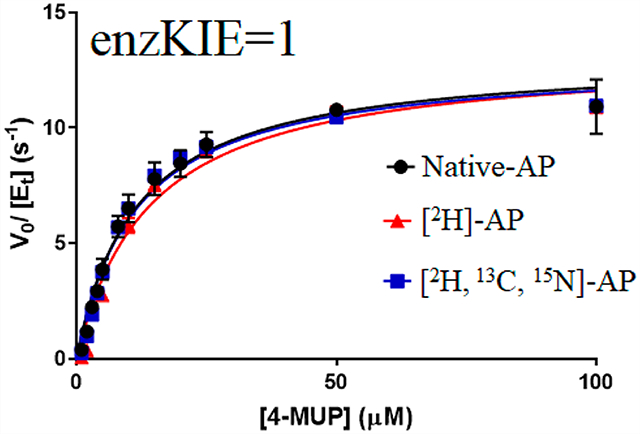
Steady-state kinetics of native AP, [2H]AP, and [2H,13C,15N]AP were measured for p-nitrophenyl phosphate (pNPP) and 4-methylumbelliferyl phosphate (4-MUP) substrates. The unpaired t-test for native and isotopically labeled APs showed no significant difference in kcat values for both pNPP and 4-MUP substrates (Figure 2, Table 1, and Figure S2). This result is a departure from earlier literature that indicated 2H-labeled EcAP was reported to exhibit an 80% decrease in kcat.18 Inorganic phosphate is a strong product inhibitor of EcAP, and its formation during the assay might account for the poor agreement in published rate constants among independent studies of EcAP.20,32–34 By comparing rate constants for two substrates with different assay sensitivities, we confirm the analysis of valid initial rates. Mass-independent steady-state kinetic rate constants have been reported for other enzymatic systems in which chemistry is not the rate-limiting step. Examples reported in the literature include studies of human purine nucleoside phosphorylase2 and the N23P/S148A mutant of EcDHFR (at pH 7.0).35 In these cases, steady-state kinetic parameters are not limited by the chemical step, rather by some other step(s) in the mechanism. For the steady-state hydrolysis of pNPP and 4-MUP, the chemical step for phosphorylation of the enzyme is also not rate-limiting.20,23 At alkaline pH, kcat is limited by the release of the product from the noncovalent enzyme–phosphate complex, whereas at acidic pH, the rate-limiting step is hydrolysis of the covalent enzyme–phosphate complex.20,25,26 Values of kcat/Km are limited by binding of substrates or an associated conformational change.36,37 The unchanged kinetic constants for native AP, [2H]AP, and [2H,13C,15N]AP at pH 8.0 indicate that the release of the product from noncovalent enzyme phosphate complex is not altered by isotopic substitution of EcAP.
Figure 2.
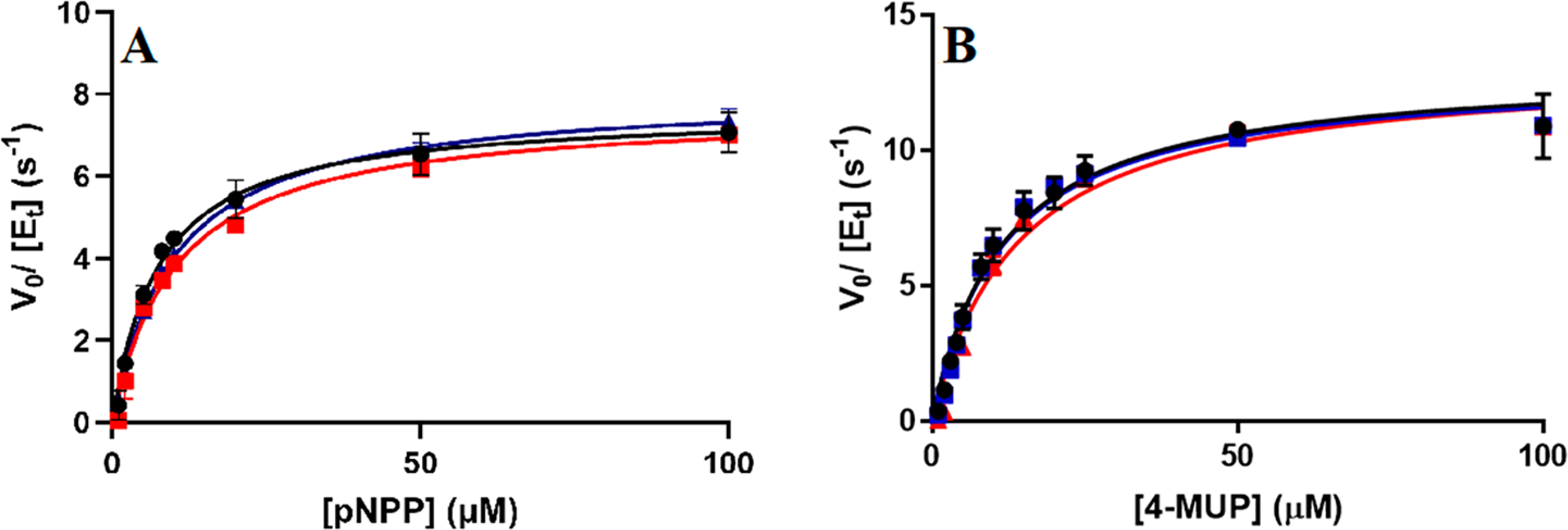
Steady-state substrate saturation curves for (A) pNPP and (B) 4-MUP substrates catalyzed by native EcAP (black), [2H]AP (red), and [2H,13C,15N]AP (blue). Lines are data fit to eq 1.
Table 1.
Steady-State Kinetic Constants for Native AP, [2H]AP, and [2H,13C,15N]AP for p-Nitrophenyl Phosphate (pNPP) and 4-Methylumbelliferyl Phosphate (4-MUP) Substrates
| native EcAP | [2H]EcAP | [2H,13C,15N]EcAP | |
|---|---|---|---|
| pNPP | |||
| kcat (s−1) | 7.6 ± 0.2 | 8.0 ± 0.2 | 7.6 ± 0.2 |
| Km (μM) | 7.4 ± 0.7 | 9.9 ± 0.9 | 10.3 ± 0.8 |
| kcat/Km (M−1 s−1) | (1.0 ± 0.1) × 106 | (0.80 ± 0.2) × 106 | (0.7 ± 0.1) × 106 |
| 4-MUP | |||
| kcat (s−1) | 13.1 ± 0.5 | 13.2 ± 0.6 | 12.9 ± 0.4 |
| Km (μM) | 11.4 ± 1.0 | 13.9 ± 1.8 | 11.4 ± 0.9 |
| kcat/Km (M−1 s−1) | (1.1 ± 0.2) × 106 | (0.9 ± 0.3) × 106 | (1.1 ± 0.2) × 106 |
| kobs (s−1) | 524 ± 3 | 515 ± 2 | 501 ± 2 |
Single-Turnover Rate Constants.
When femtosecond mass-influenced protein dynamics affect enzymatic catalysis, it is often on the chemical step, a consequence of linked fast motions at the catalytic site to formation of the transition state. Therefore, it is essential to be able to monitor the chemical step. As steady-state AP-catalyzed reactions of aryl phosphates are not limited by the chemical step for the phosphorylation of the enzyme,20,23 single-turnover rate constants were measured for native AP, [2H]AP, and [2H,13C,15N]AP for hydrolysis of 4-MUP. An experimental limitation of measuring on enzyme chemistry for AP is the very fast chemical step (>2000 s−1) in pre-steady-state kinetic measurements performed at room temperature, faster than conveniently measured in conventional rapid mixing instruments.38 We measured the rate for the phosphorylation of the enzyme at 5 °C for light and isotopically labeled APs using 4-MUP as a substrate. Hydrolysis of 4-methylumbelliferyl phosphate yields a highly fluorescent product 4-methylumbelliferone at the catalytic site, and this fluorescence signal was used to monitor the rate of on-enzyme chemistry (Figure 3, excitation at 370 nm and detection at >420 nm). The formation of 4-methylumbelliferone at the catalytic site is accompanied by phosphorylation of the enzyme (E-P), and the fluorescence signals provide a sensitive approach for measuring single-turnover chemistry for E-P formation using the stopped-flow instrument.
Figure 3.
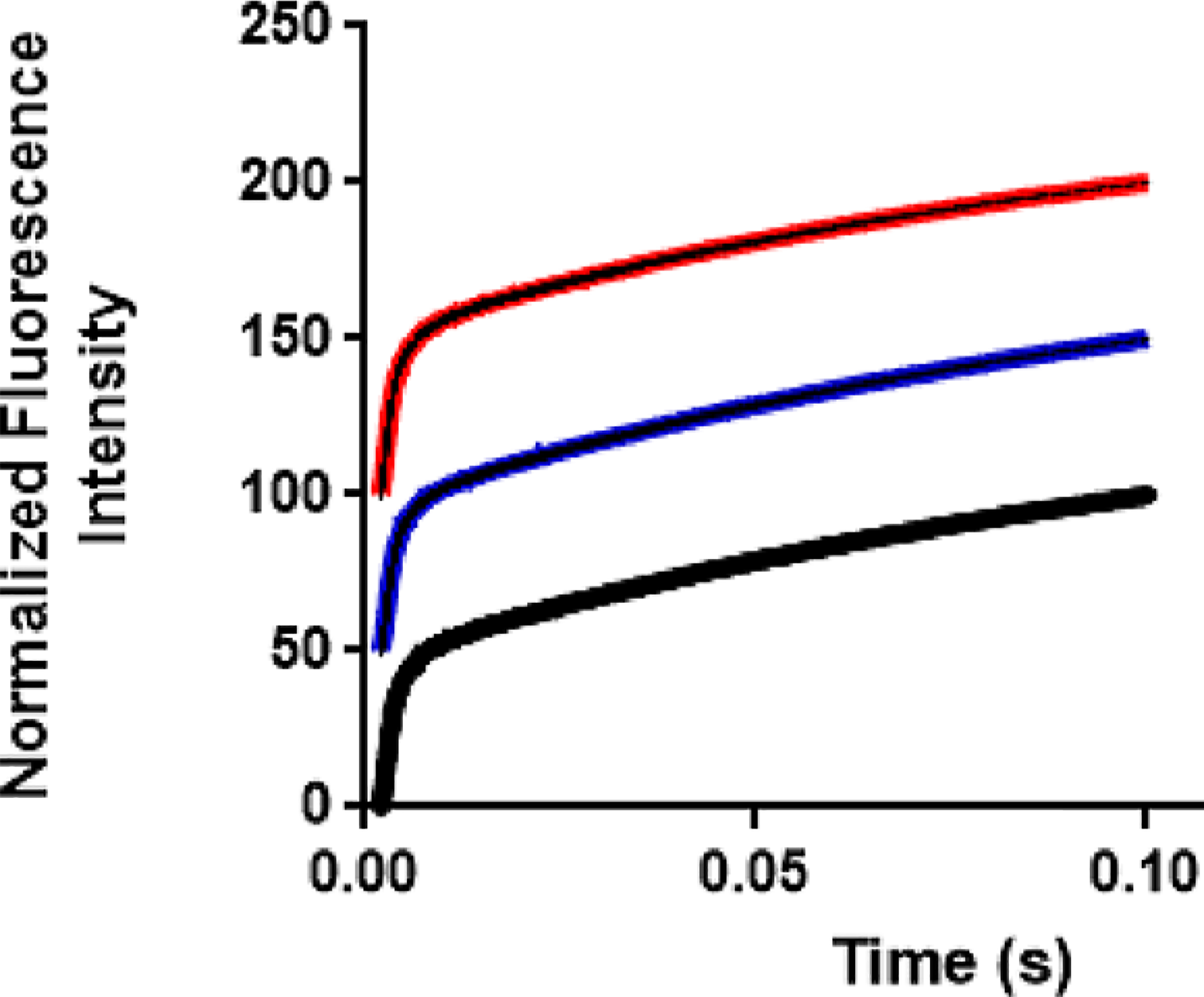
Representative stopped-flow traces for single-turnover experiments of 4-methylumbelliferyl phosphate. The fluorescence change resulting from reaction with APs is observed after rapid mixing. The traces were fitted to a single-exponential equation followed by a linear phase to determine the rate of phosphorylation of native AP (black), [2H]AP (red), and [2H,13C,15N]AP (blue). Traces are offset for the purpose of presentation.
The AP-catalyzed hydrolysis of 4-methylumbelliferyl phosphate can be described by the reaction in Scheme 2. When the measurement is performed under conditions for a single turnover ([E] ≫ [ROP]), the regeneration of E is not involved in the phosphoryl transfer of ROP and the reaction scheme reduces to
Under this condition of single turnover, the rate of formation of the product ROH at the active site (accompanied by the formation of E-P) is described by eq 329
| (3) |
Hence, the observed rate constant (kobs) is a function of the equilibrium constant (K1) for the formation of E·ROP and the chemical step (k2). The equilibrium constant K1 = k−1/k1, where k1 is the rate constant for the formation of E·ROP (“on rate”) and k−1 is the dissociation of E·ROP to the enzyme and substrate (“off rate”). Under the assumption of rapid equilibrium in substrate binding, the pseudo-first-order observed rate constant (kobs) represents the chemical step and becomes identical to the chemical step under saturating conditions. Studies using temperature jump techniques on the binding equilibrium between the E. coli alkaline phosphatase and an active site inhibitor 2-hydroxy-5-nitrobenzyl phosphonate under an identical reaction condition determined the equilibrium constant, K1, to be 2.5 × 10−4 M.39 Because the diffusion control rate constant for binding of a small molecule (on rate) with macromolecules is on the order of ~108 M−1 s−1, the k−1 can be estimated to be on the order of ~104 s−1.29 These rate constants are much greater than the subsequent chemical step and justified the rapid equilibrium treatment of the substrate binding to the enzyme. Under this rapid equilibrium assumption and identical reaction conditions, K1 is indistinguishable for light and heavy APs and any change in the chemical step should be apparent in the observed rate constant (kobs). Fitting the experimental data to a single exponential followed by a linear phase (eq 2) produces the rate of the chemical step (active site product fluorescence, exponential phase) and the release of the ROH, where it has an altered quantum yield (linear phase). The results show statistically indistinguishable single-turnover rate constants [kobs (Table 1)] for light and heavy APs, indicating that mass-altered protein dynamics do not affect the rate of the enzyme phosphorylation step or ROH release.
Scheme 2.
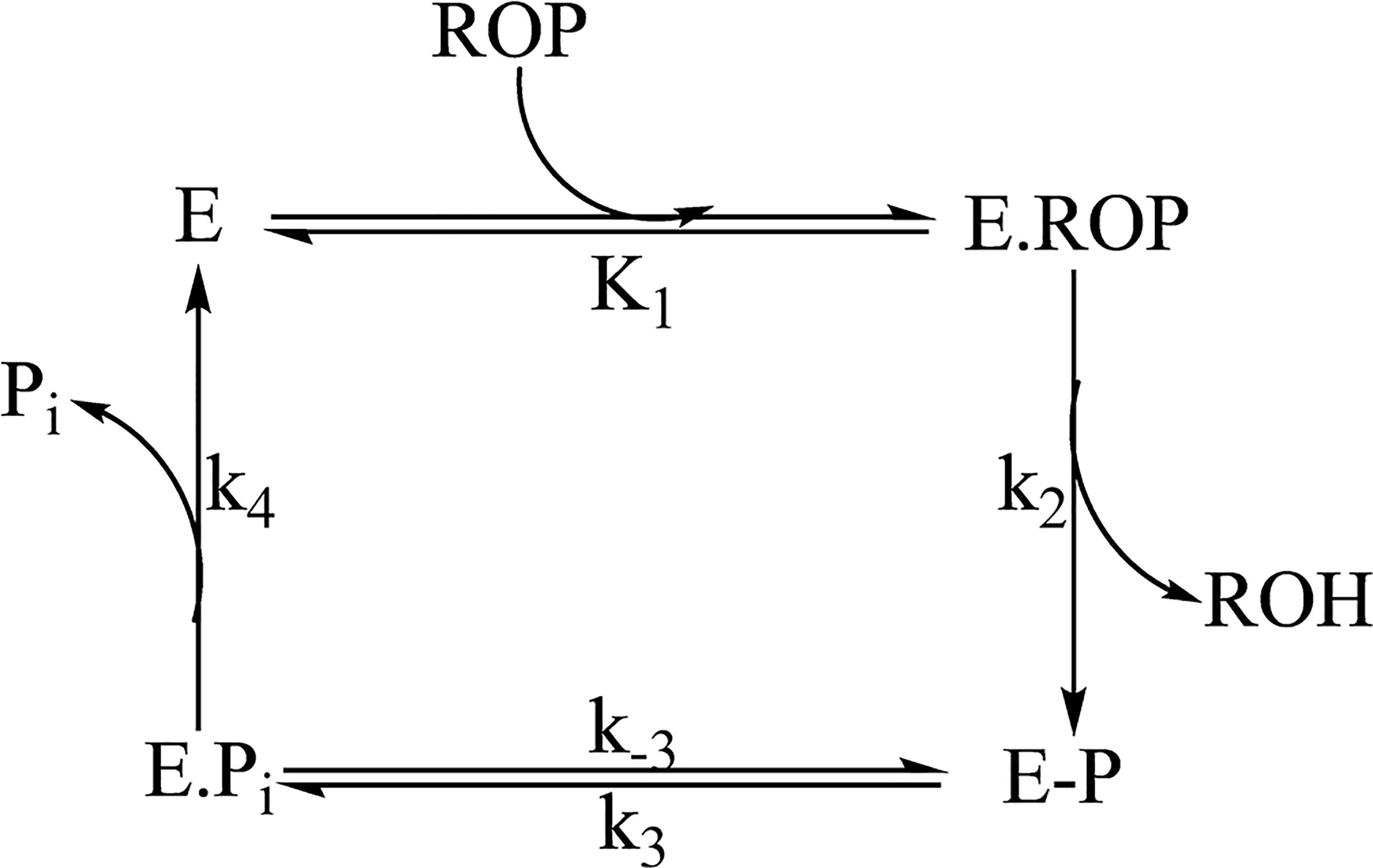
Schematic Representation of the AP-Catalyzed Phosphorylation of 4-Methylumbelliferyl Phosphatea
aE, ROP, ROH, E-P, and E·Pi represent the enzymes, substrate 4-methylumbelliferyl phosphate, product 4-methylumbelliferone, covalently phosphorylated enzyme, and noncovalent enzyme phosphate complex, respectively.
Although employing the temperature dependence of KIEs to study H transfer and quantum tunneling are now widely established,6,40 only a few studies have been performed to understand the effect of temperature on heavy enzyme KIEs.11,12,41,42 Pudney and co-workers have provided insights into the temperature dependence of heavy enzyme KIEs in the pentaerythritol tetranitrate reductase (PETNR) enzyme.41 The authors used unlabeled (light PETNR), 13C-, 15N-, and 2H-labeled (heavy PETNR), and 15N-labeled PETNR ([15N]-PETNR) and determined the temperature dependence of KIEs using NADH and NADPH cofactors in the stopped-flow experiments. Although the enzyme KIEs were temperature-independent for the NADH substrate, it increased as a function of temperature (5–35 °C) for the NADPH substrate. As mentioned above, the single-turnover rate constants of APs were measured at 5 °C due to the limitation in the stopped-flow instrument that had a dead time of 1.25 ms. We cannot exclude the possibility that heavy enzyme rate effects might be seen at experimentally inaccessible temperatures.
Although most enzymes exhibit catalytic turnover times on the millisecond time scale, the enzyme–substrate complex passes through a series of successive events before product release. Many of those steps take place on a time scale faster than that of catalysis. The sequential transformation among these events requires specific atomic rearrangement and bond distortion of both the substrate and the enzyme. The lifetime of the chemical events, namely, transition-state lifetime, or barrier crossing is in the femtosecond43 range and requires coherent dynamic conditions to optimize multiple geometric and distance constraints that provide electrostatic interactions to minimize the barrier crossing energy.44 The energy for barrier crossing fluctuates significantly as atomic distances change on the 0.1 Å scale, within the atom-to-atom distance on the femtosecond time scale. An altered atomic mass in catalytic site amino acids alters the nature of these fluctuations. The rapid (femtosecond) intramolecular dynamics creating the transition state are localized to the catalytic site, as femtosecond transition-state modes cannot equilibrate through the enzyme on the femtosecond time scale of the transition state. Disrupting intramolecular dynamics by the altered protein mass is expected to affect barrier crossing if protein dynamics are directly linked to transition-state formation. In at least one case, E. coli dihydrofolate reductase, the transition-state vibrational dynamics are caused by reactants, making the chemical step independent of the mass of the protein.45 In most other cases, experimental links between the enzyme protein mass and the probability of barrier crossing are readily measured. Here, the zinc ions are directly involved in the reaction coordinate, and their mass is not altered in the heavy enzyme constructs. The independent rate of the chemical step from protein mass indicates a reaction coordinate dominated by the zinc–reactant interactions, and the secondary contacts of zinc ions to the mass of the altered protein are sufficiently uncoupled to prevent mass effects on the chemical steps.
Previous studies of isotopically labeled enzymes indicate that specific dynamics–catalysis relationships depend on the detailed nature of the transition state. Perturbation of the protein vibrational dynamics by isotopic substitution can influence the transition state in several ways. (i) Enzymatic heavy atom substitution impedes the transition-state barrier crossing by altering the optimal donor and acceptor distance at the transition state. The diminished protein dynamics alters the optimal donor–acceptor distance (DAD) to decrease the probability of barrier crossing. For example, transition path sampling (TPS) suggests that the minimal DAD distance for native PNP is attained at the transition state but for heavy PNP several femtoseconds after transition-state formation.18 Although TPS data have not yet been reported for other enzymatic systems explored by heavy protein effects (including AR,46 PETNR,47 and HIV-1 protease48), asynchronization between optimal DAD at the TS could also play a role in experimental observations of the diminished rate for heavy enzymes. (ii) Enzymatic heavy atom substitution facilitates the transition-state barrier crossing reaching the optimal DAD more frequently to result in inverse heavy enzyme effects, where an increased mass increases the frequency of the chemical step.16 Although this is a rare phenomenon, in the case of light and heavy F159Y PNP, the optimal DAD forms the transition state more frequently in heavy F159Y PNP to increase the probability of barrier crossing and the experimental observation of inverse KIE.18 (iii) Protein dynamics at the active site does not influence the transition-state barrier crossing. In this case, the protein dynamics is orthogonal to the reaction coordinate and has no effect on the DAD at the TS. In the case of the Glu258Asp/Leu261Ala double mutant of PNP, the heavy enzyme did not alter the DAD at the TS.49 This observation was explained by the participation of the substrate (q) and protein (s) degrees of freedom in the reaction coordinate, RC[ξ(s,q)].50 If the RC can be exclusively defined in terms of substrate vibrational coordinates, femtosecond protein dynamics provides a secondary engagement in transition-state formation. The current literature reports most, but not all, enzymatic systems with femtosecond coupling of protein dynamics into transition-state barrier crossing. However, this phenomenon is not present in the AP-catalyzed reaction. Similar examples include the EcDHFR, TmDHFR, and FDH proteins.12
CONCLUSIONS
Recent studies establish that enzymatic heavy atom substitution provides an experimental tool for reporting on the dynamic link between fast catalytic site motions and formation of the transition state. Most recent studies show significant (≤30%) changes of different kinetic parameters, often in the chemical step(s). A foundational report on deuterated AP showed a decrease in its kcat by a factor of 80% compared to that of native (natural abundance isotope) AP. We optimized the expression and purification methods to produce native AP, [2H]AP, and [2H,13C,15N]AP and characterized both steady-state and chemical step parameters with multiple substrates. We report that the steady-state kinetic constants and rate of chemistry are indistinguishable among three APs with distinct isotopic masses. Comparison of the kinetic properties of enzymes prepared under different expression conditions requires quantitation beyond normal steady-state kinetics. Our results demonstrate that the nucleophilic attack of serine on the phosphomonoester and its subsequent hydrolysis by a Zn2+-activated water are uncoupled from the local catalytic site femtosecond motions perturbed by an altered protein mass. The lack of heavy enzyme effects for AP implicates the participation of the catalytic site zinc ions as a dominating step in transition-state formation rather than the heavy protein links to catalytic site reactants.
Earlier reports of a large normal enzymatic isotope effect for deuterium-labeled AP relied on comparison of steady-state kinetic constants, where the isotope effect was observed in the turnover number, kcat. Differences in purity or specific catalytic activity are reflected in kcat values and are subject to error. Heavy enzyme isotope effects require quantitative analysis, including methods independent of enzyme concentration. Thus, pre-steady-state methods with pseudo-first-order kinetics are advised. Therefore, differences in the purity or specific catalytic activity of the natural and deuteron-AP enzymes reported earlier are likely sources of error in the previous study.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Dr. Daniel Herschlag and Dr. Fanny Sunden of Stanford University for providing an expression plasmid for E. coli alkaline phosphatase. The authors also thank Dr. Rajesh K. Harijan for helpful suggestions and help in preparing Figure 1.
Funding
This research was supported by National Institutes of Health Grant GM041916.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.biochem.0c00917.
ESI-MS analysis of native AP, [2H]AP, and [2H,13C,15N]APs and analysis of the significance between kcat values for native AP, [2H]AP, and [2H,13C,15N]AP (PDF)
Accession Codes
AP, UniProt entry P00634.
Contributor Information
Ananda K. Ghosh, Department of Biochemistry, Albert Einstein College of Medicine, Bronx, New York 10461, United States.
Vern L. Schramm, Department of Biochemistry, Albert Einstein College of Medicine, Bronx, New York 10461, United States.
REFERENCES
- (1).Hammes-Schiffer S, and Benkovic SJ (2006) Relating Protein Motion to Catalysis. Annu. Rev. Biochem. 75, 519–541. [DOI] [PubMed] [Google Scholar]
- (2).Silva RG, Murkin AS, and Schramm VL (2011) Femtosecond dynamics coupled to chemical barrier crossing in a born-oppenheimer enzyme. Proc. Natl. Acad. Sci. U. S. A. 108, 18661–18665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Nagel ZD, and Klinman JP (2010) Update 1 of: Tunneling and Dynamics in Enzymatic Hydride Transfer. Chem. Rev. 110, PR41–PR67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Kamerlin SC, and Warshel A (2010) At the dawn of the 21st century: Is dynamics the missing link for understanding enzyme catalysis? Proteins: Struct., Funct., Genet. 78, 1339–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Hammes-Schiffer S, and Benkovic SJ (2006) Relating Protein Motion to Catalysis. Annu. Rev. Biochem. 75, 519–541. [DOI] [PubMed] [Google Scholar]
- (6).Kohen A (2015) Role of Dynamics in Enzyme Catalysis: Substantial versus Semantic Controversies. Acc. Chem. Res. 48, 466–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Mandel AM, Akke M, and Palmer AG III (1995) Backbone Dynamics ofEscherichia coliRibonuclease HI: Correlations with Structure and Function in an Active Enzyme. J. Mol. Biol. 246, 144–163. [DOI] [PubMed] [Google Scholar]
- (8).Boehr DD, Dyson HJ, and Wright PE (2006) An NMR Perspective on Enzyme Dynamics. Chem. Rev. 106, 3055–3079. [DOI] [PubMed] [Google Scholar]
- (9).Bandaria JN, Dutta S, Nydegger MW, Rock W, Kohen A, and Cheatum CM (2010) Characterizing the dynamics of functionally relevant complexes of formate dehydrogenase. Proc. Natl. Acad. Sci. U. S. A. 107, 17974–17979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Meadows CW, Ou R, and Klinman JP (2014) Picosecond-Resolved Fluorescent Probes at Functionally Distinct Tryptophans within a Thermophilic Alcohol Dehydrogenase: Relationship of Temperature-Dependent Changes in Fluorescence to Catalysis. J. Phys. Chem. B 118, 6049–6061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Wang Z, Singh P, Czekster CM, Kohen A, and Schramm VL (2014) Protein Mass-Modulated Effects in the Catalytic Mechanism of Dihydrofolate Reductase: Beyond Promoting Vibrations. J. Am. Chem. Soc. 136, 8333–8341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Ranasinghe C, Guo Q, Sapienza PJ, Lee AL, Quinn DM, Cheatum CM, and Kohen A (2017) Protein Mass Effects on Formate Dehydrogenase. J. Am. Chem. Soc. 139, 17405–17413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Kholodar SA, Ghosh AK, and Kohen A (2017) Chapter 3: Measurement of Enzyme Isotope Effects. In Methods in Enzymology (Harris ME, and Anderson VE, Eds.) pp 43–83, Academic Press. [DOI] [PubMed] [Google Scholar]
- (14).Ghosh AK (2018) Exploring the mechanism and dynamics of thymidylate synthase. Ph.D. Thesis, The University of Iowa, Iowa City, IA. [Google Scholar]
- (15).Wang Z, Chang EP, and Schramm VL (2016) Triple Isotope Effects Support Concerted Hydride and Proton Transfer and Promoting Vibrations in Human Heart Lactate Dehydrogenase. J. Am. Chem. Soc. 138, 15004–15010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Harijan RK, Zoi I, Antoniou D, Schwartz SD, and Schramm VL (2018) Inverse enzyme isotope effects in human purine nucleoside phosphorylase with heavy asparagine labels. Proc. Natl. Acad. Sci. U. S. A. 115, E6209–e6216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Suarez J, and Schramm VL (2015) Isotope-specific and amino acid-specific heavy atom substitutions alter barrier crossing in human purine nucleoside phosphorylase. Proc. Natl. Acad. Sci. U. S. A. 112, 11247–11251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Harijan RK, Zoi I, Antoniou D, Schwartz SD, and Schramm VL (2017) Catalytic-site design for inverse heavy-enzyme isotope effects in human purine nucleoside phosphorylase. Proc. Natl. Acad. Sci. U. S. A. 114, 6456–6461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Rokop S, Gajda L, Parmerter S, Crespi HL, and Katz JJ (1969) Purification and characterization of fully deuterated enzymes. Biochim. Biophys. Acta 191, 707–715. [DOI] [PubMed] [Google Scholar]
- (20).O’Brien PJ, and Herschlag D (2002) Alkaline Phosphatase Revisited: Hydrolysis of Alkyl Phosphates. Biochemistry 41, 3207–3225. [DOI] [PubMed] [Google Scholar]
- (21).Lassila JK, Zalatan JG, and Herschlag D (2011) Annu. Rev. Biochem. 80, 669–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Peck A, Sunden F, Andrews LD, Pande VS, and Herschlag D (2016) Tungstate as a Transition State Analog for Catalysis by Alkaline Phosphatase. J. Mol. Biol. 428, 2758–2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Sun L, Martin DC, and Kantrowitz ER (1999) Rate-determining step of Escherichia coli alkaline phosphatase altered by the removal of a positive charge at the active center. Biochemistry 38, 2842–2848. [DOI] [PubMed] [Google Scholar]
- (24).Schwartz JH, and Lipmann F (1961) Phosphate incorporation into alkaline phosphatase of E. coli. Proc. Natl. Acad. Sci. U. S. A. 47, 1996–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Holtz KM, and Kantrowitz ER (1999) The mechanism of the alkaline phosphatase reaction: insights from NMR, crystallography and site-specific mutagenesis. FEBS Lett. 462, 7–11. [DOI] [PubMed] [Google Scholar]
- (26).Coleman JE (1992) Structure and Mechanism of Alkaline Phosphatase. Annu. Rev. Biophys. Biomol. Struct. 21, 441–483. [DOI] [PubMed] [Google Scholar]
- (27).Bobyr E, Lassila JK, Wiersma-Koch HI, Fenn TD, Lee JJ, Nikolic-Hughes I, Hodgson KO, Rees DC, Hedman B, and Herschlag D (2012) High-Resolution Analysis of Zn2+ Coordination in the Alkaline Phosphatase Superfamily by EXAFS and X-ray Crystallography. J. Mol. Biol. 415, 102–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Holtz KM, Stec B, and Kantrowitz ER (1999) J. Biol. Chem. 274, 8351–8354. [DOI] [PubMed] [Google Scholar]
- (29).Bale JR, Huang CY, and Chock PB (1980) Transient kinetic analysis of the catalytic cycle of alkaline phosphatase. J. Biol. Chem. 255, 8431–8436. [PubMed] [Google Scholar]
- (30).Bloch W, and Schlesinger MJ (1973) The phosphate content of Escherichia coli alkaline phosphatase and its effect on stopped flow kinetic studies. J. Biol. Chem. 248, 5794–5805. [PubMed] [Google Scholar]
- (31).Lev S, Kaufman-Francis K, Desmarini D, Juillard PG, Li C, Stifter SA, Feng CG, Sorrell TC, Grau GE, Bahn YS, and Djordjevic JT (2017) Pho4 Is Essential for Dissemination of Cryptococcus neoformans to the Host Brain by Promoting Phosphate Uptake and Growth at Alkaline pH. mSphere 2, n/a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Xu X, and Kantrowitz ER (1993) Binding of magnesium in a mutant Escherichia coli alkaline phosphatase changes the rate-determining step in the reaction mechanism. Biochemistry 32, 10683–10691. [DOI] [PubMed] [Google Scholar]
- (33).Lazdunski C, and Lazdunski M (1969) Zn2+ and Co2+-alkaline phosphatases of E. coli. A comparative kinetic study. Eur. J. Biochem. 7, 294–300. [DOI] [PubMed] [Google Scholar]
- (34).Stec B, Hehir MJ, Brennan C, Nolte M, and Kantrowitz ER (1998) Kinetic and X-ray structural studies of three mutant E. coli alkaline phosphatases: insights into the catalytic mechanism without the nucleophile Ser102. J. Mol. Biol. 277, 647–662. [DOI] [PubMed] [Google Scholar]
- (35).Ruiz-Pernia JJ, Luk LY, Garcia-Meseguer R, Marti S, Loveridge EJ, Tunon I, Moliner V, and Allemann RK (2013) Increased dynamic effects in a catalytically compromised variant of Escherichia coli dihydrofolate reductase. J. Am. Chem. Soc. 135, 18689–18696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Hollfelder F, and Herschlag D (1995) The nature of the transition state for enzyme-catalyzed phosphoryl transfer. Hydrolysis of O-aryl phosphorothioates by alkaline phosphatase. Biochemistry 34, 12255–12264. [DOI] [PubMed] [Google Scholar]
- (37).Hengge AC, Edens WA, and Elsing H (1994) Transition-state structures for phosphoryl-transfer reactions of p-nitrophenyl phosphate. J. Am. Chem. Soc. 116, 5045–5049. [Google Scholar]
- (38).Bale JR, Chock PB, and Huang CY (1980) The nature of negative cooperativity in alkaline phosphatase. J. Biol. Chem. 255, 8424–8430. [PubMed] [Google Scholar]
- (39).Halford SE (1972) Escherichia coli alkaline phosphatase. Relaxation spectra of ligand binding. Biochem. J. 126, 727–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Nagel ZD, and Klinman J. P. J. N. c. b. (2009) Nat. Chem. Biol. 5, 543–550. [DOI] [PubMed] [Google Scholar]
- (41).Pudney CR, Guerriero A, Baxter NJ, Johannissen LO, Waltho JP, Hay S, and Scrutton NS (2013) Fast Protein Motions Are Coupled to Enzyme H-Transfer Reactions. J. Am. Chem. Soc. 135, 2512–2517. [DOI] [PubMed] [Google Scholar]
- (42).Egawa T, Deng H, Chang E, and Callender R (2019) Effect of Protein Isotope Labeling on the Catalytic Mechanism of Lactate Dehydrogenase. J. Phys. Chem. B 123, 9801–9808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Schwartz SD, and Schramm V. L. J. N. c. b. (2009) Nat. Chem. Biol. 5, 551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Phillipson PE (1968) On the possible importance of relaxation processes in enzyme catalysis. J. Mol. Biol. 31, 319–321. [DOI] [PubMed] [Google Scholar]
- (45).Luk LYP, Javier Ruiz-Pernía J, Dawson WM, Roca M, Loveridge EJ, Glowacki DR, Harvey JN, Mulholland AJ, Tuñón I, Moliner V, and Allemann RK (2013) Proc. Natl. Acad. Sci. U. S. A. 110, 16344–16349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Toney MD, Castro JN, and Addington TA (2013) Heavy-enzyme kinetic isotope effects on proton transfer in alanine racemase. J. Am. Chem. Soc. 135, 2509–2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Pudney CR, Guerriero A, Baxter NJ, Johannissen LO, Waltho JP, Hay S, and Scrutton NS (2013) Fast protein motions are coupled to enzyme H-transfer reactions. J. Am. Chem. Soc. 135, 2512–2517. [DOI] [PubMed] [Google Scholar]
- (48).Kipp DR, Silva RG, and Schramm VL (2011) Mass-dependent bond vibrational dynamics influence catalysis by HIV-1 protease. J. Am. Chem. Soc. 133, 19358–19361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Zoi I, Suarez J, Antoniou D, Cameron SA, Schramm VL, and Schwartz SD (2016) Modulating Enzyme Catalysis through Mutations Designed to Alter Rapid Protein Dynamics. J. Am. Chem. Soc. 138, 3403–3409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Swiderek K, Javier Ruiz-Pernia J, Moliner V, and Tunon I (2014) Heavy enzymes–experimental and computational insights in enzyme dynamics. Curr. Opin. Chem. Biol. 21, 11–18. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.