

Abstract
Cell-penetrating anticancer peptides (Cp-ACPs) are considered promising candidates in solid tumor and hematologic cancer therapies. Current approaches for the design and discovery of Cp-ACPs trust the expensive high-throughput screenings that often give rise to multiple obstacles, including instrumentation adaptation and experimental handling. The application of machine learning (ML) tools developed for peptide activity prediction is importantly of growing interest. In this study, we applied the random forest (RF)-, support vector machine (SVM)-, and eXtreme gradient boosting (XGBoost)-based algorithms to predict the active Cp-ACPs using an experimentally validated data set. The model, CpACpP, was developed on the basis of two independent cell-penetrating peptide (CPP) and anticancer peptide (ACP) subpredictors. Various compositional and physiochemical-based features were combined or selected using the multilayered recursive feature elimination (RFE) method for both data sets. Our results showed that the ACP subclassifiers obtain a mean performance accuracy (ACC) of 0.98 with an area under curve (AUC) ≈ 0.98 vis-à-vis the CPP predictors displaying relevant values of ∼0.94 and ∼0.95 via the hybrid-based features and independent data sets, respectively. Also, the predicting evaluation of Cp-ACPs gave accuracies of ∼0.79 and 0.89 on a series of independent sequences by applying our CPP and ACP classifiers, respectively, which leaves the performance of our predictors better than the earlier reported ACPred, mACPpred, MLCPP, and CPPred-RF. The described consensus-based fusion method additionally reached an AUC of 0.94 for the prediction of Cp-ACP (http://cbb1.ut.ac.ir/CpACpP/Index).
Introduction
Cancer is recognized as a multifaceted disease and the second cause of death in the United States, with the projection of about 2 million new cases and over 500 000 deaths in 2020.1 Despite the significant progress in the development of chemotherapeutics, such treatments give rise to resistant cancer cells and severe side effects,2 which therefore make it indispensable to design and predict novel therapeutics with selective mechanisms.
Anticancer peptides (ACPs) are considered promising to specifically target diseased cells. These molecules are positively charged and made of short to medium-size length of 5–30 amino acids, mainly lysine (Lys) and arginine (Arg).3,4 The ACPs are structurally diverse α-helices and β-sheets often known for their lytic activity on the anionic phosphatidylserine- (PtdSer) and glycoconjugate- (such as mucins and gangliosides) rich cancer plasma membranes.3 The necrotic mechanism of ACPs was, however, suggested to be responsible for the rise of resistant cells through changing the membrane surface glycation pattern by, for instance, omission of sialic acids.5 Because of the lytic activity of these sequences, the release of cancer intracellular contents could additionally result in causing the activation of proinflammatory cascades.6
Cp-ACPs are enabled to selectively cross the cancer cell membrane via the endocytosis and/or the passive pathway and trigger the apoptotic signaling by targeting, e.g., the endoplasmic reticulum (ER) and mitochondrial (mt) membranes.7 We recently showed that the magainin 2 (MG 2) analogue can enter the A549 cells via direct transduction, leading to the mt dysfunction likely in response to ER stress.8 The DNA synthesis inhibition and disruption of the unfolded protein response were also envisaged for the activity behaviors of Cp-ACPs.9 The Lys/tryptophan (Trp)-rich KT2 peptide exhibited clathrin- and caveolae-mediated endocytosis and induced apoptosis by autophagy inhibition in HCT116 colon cells.10 Also, L-K6, a sequence primarily composed of Lys and leucine (Leu), could enter the cytoplasmic space of MCF-7 via clathrin-dependent macropinocytosis, localize in the cell nucleus, and trigger nuclear disruption.11 Also, the anionic azurin truncated domain P28 selectively internalized into the solid tumor cell lines and resulted in P53 activation and proliferation inhibition of cancer cells;12 for more examples, see Table S1 in the Supporting Information. In fact, the design and development of such sequences that specifically promote death signaling is a significant milestone to overcome the challenges associated with membranolytic ACPs.
The valued machine learning (ML) discipline in peptide activity prediction has received growing interest compared with the current costly experimental screenings.13 To date, several ML-based classifiers have been developed for the prediction behaviors of either ACPs or CPPs, most of which utilize compositional-based [e.g., atomic composition (ATC), amino acid composition/index (AAC/AAI), dipeptide composition (DPC)] and physicochemical-based features to encode peptide sequences. In this context, support vector machines (SVMs), random forest (RF), extremely randomized tree (ERT), kernel extreme learning machine (KELM), and neural networks (NNs), as well as different subclasses thereof, such as the long short-term memory (LSTM), are the frequently used algorithms.13−15 Nonetheless, the development of ML-based models capable of simultaneously recognizing both the peptide-cell-penetrating and anticancer characters is certainly of interest to predict and find therapeutics in multidrug-resistant cancer therapy.
In this study, we developed CpACpP(s) that were mainly constructed based on the fusion of two independent binary subclassifiers by utilizing the SVM, RF, and XGBoost, which individually discriminate the anticancer or plasma membrane transducing active sequences from their nonactivated analogues, i.e., non-ACP (NACP) and non-CPP (NCPP) sequences. Various feature sets were used, including AAC, DPC, grouped dipeptide composition (GDPC), the composition of k-spaced amino acid group pairs (CKSAAGPs), pseudo-amino acid composition (PAAC), amphiphilic pseudo-amino acid composition (APAAC), physicochemical properties (PCPs), composition-transition-distribution (CTD), 13C NMR, and their hybrids. The straightforward feature hybridization and the three-layer feature selection algorithm of recursive feature elimination (RFE) were applied on the training data to reduce the model complexity and avoid multicollinearity and/or overfitting.16,17
The independent analysis of Cp-ACPs revealed that our models often outperform the current ACP and CPP predictors, such as the Anticp,18 iACP,19 ACPred,20 and mACPpred,21 as well as the CellPPD,22 MLCPP,23 CPPred-RF,24 and SkipCPP-Pred.25 Taken together, our ML-based models introduce a robust Cp-ACP identification and activity prediction framework that may be of advantage, particularly in the current proteomics investigations, for the development of novel multimodal peptide therapeutics that selectively interfere with cancer cell signaling.26
Results
Amino Acid Analysis
The peptide primary structure, defined by the amino acid composition and composite, could statistically be used for the activity prediction and behaviors of ACPs and/or CPPs.18,23 Accordingly, we presented and analyzed the relevant AAC, DPC, and tripeptide composition (TPC) features for each class of active and nonactive anticancer and cell-penetrating sequences, as well as those having both characters (Figures 1–3).
Figure 1.

Amino acid analysis of active and nonactive ACPs, CPPs, and Cp-ACPs. The p-values were obtained by the Mann–Whitney U test.
Figure 3.

Tripeptide analysis of active and nonactive classes of anticancer, cell-penetrating, and Cp-ACP sequences. The panels represent the most frequently found tripeptides in the active (left panel) and nonactive (right panel) sequences. The p-values were obtained by the Mann–Whitney U test.
AAC analysis exhibited a high frequency of Lys (∼16%), Leu (∼14%), and Arg (∼9%) among the ACPs. Despite being in low abundance, the presence of hydrophobic Phe and Trp was predominantly obvious in the ACP sequences (Figure 1). Even though the most flexible alanine (Ala)/glycine (Gly) and side-chain-branched Ile comprise ∼24% of the active structure, the amino acid frequencies of tiny Ala, Gly, cysteine (Cys), hydrophobic isoleucine (Ile), valine (Val), and basic histidine (His) were almost indifferent between the ACPs and NACPs. The anionic aspartate (Asp), glutamate (Glu), polar asparagine (Asn), glutamine (Gln), serine (Ser), threonine (Thr), tyrosine (Tyr), methionine (Met), and α-helix/β-sheet-unfavored proline (Pro) were primarily found in the NACPs. In comparison, the CPPs were, particularly, Arg-rich (∼20%) and revealed remarkable preference for the cationic Lys residue (∼12%). In contrast to this evidence, we found that the aromatic Trp tilted to active sequences, but rather almost equal contributions were found for the hydrophobic (Phe, Ile, Leu, Met), polar (Asp, Pro, Glu, Ser, Thr, Tyr), Ala, Cys, and His between the active and nonactive anticancer sequences. Also, Asp, Glu, Gly, and Val were primarily observed in the NCPPs.
Intriguingly, the amino acid distribution pattern for Cp-ACPs was relatively biased toward the anticancer sequences in terms of the total occurrence of basic Arg and Lys (∼26%), hydrophobic Leu and Ile (∼19%), and Gly (∼9%) residues (Figure 1). Nonetheless, (i) similar to that of active CPPs, the Arg residue adopted the higher fraction of sequence composition in the Cp-ACPs; and (ii) the distribution of each amino acid in the nonactive Cp-ACPs (NCp-ACPs) showed that the existence chance of the aliphatic Ala, Val, and Met amino acids and the polar Tyr, Thr, Gln, Glu, Asn, Asp, Pro, and His residues increases in the NCp-ACPs relative to their Cp-ACP analogues.
Figure 2 shows the DPC evaluation of the first abundant dipeptides in the active and nonactive sequences. The ACPs displayed dipeptides that are biased toward X1–Y1, where X1/Y1 are Lys and/or Leu, vis-à-vis the NACPs mainly anionic Glu/Asp-rich. Besides, Arg–Arg and the cationic pairs of Ala, Gly, Ile, and Phe were relatively dominant in the ACP sequences. The dipeptide composed of Arg and Lys was not among the active ACPs, whereas Lys–Arg and Arg–Lys, as well as Arg–Arg, Lys–Lys, and the cationic pairs containing Leu, were the most abundant in the CPPs. The presence of His–His, Arg–His, and Trp–Arg was as low as 1%, but nevertheless, they were predominantly found in the active sequences as compared with the NCPPs. Obviously, the NCPPs were characterized by the flexible dipeptides of Leu/Gly, Ala/Gly, and Leu/Ser pairs, as well as Gly–Gly, charged Glu–Leu, and Lys–Ile. We also recognized Arg–Arg, Leu–Leu, Lys–Lys, Lys–Leu, Leu–Lys, Arg–Leu, Ile–Lys, Gly–Gly, Arg–Lys, and Gly–Arg as the first 10 couples with the highest frequency of occurrence among the other pairs in the Cp-ACPs. In this respect, the distribution of dipeptides was relatively comparable to those of both the active ACP and CPP sequences. With the exceptional Lys–Trp and flanked Gly–Arg, the presence of other dipeptide residues, including Trp–Trp, which is most probably found in the active Cp-ACPs,10,27 occurred at the same level. In contrast, DPC analysis revealed that the pair of hydrophobic residues, such as Ala, Leu, Ile, and Val, has a higher frequency in the NCp-ACP sequences.
Figure 2.

Dipeptide analysis of active and nonactive ACPs, CPPs, and Cp-ACPs. The panels compare the most abundant dipeptides in the active (left panel) and nonactive (right panel) sequences. The p-values were obtained by the Mann–Whitney U test.
The TPC analysis was performed over 8000 tripeptides to extract the important functional motifs in the peptide activity interpretation (Figure 3).28 Accordingly, Lys-rich AKK, LAK, KKL, LKK, and KLL were the first five plentiful combinations in the ACPs. In contrast, LAG, AGI, ELV, LVS, and EDL comprised the maximum forms of tripeptides in the NACPs. This evaluation showed that the Arg-based tripeptides have remarkable values in the CPPs, e.g., RRR, KKR, RKK, KRR, and RRQ; nonetheless, the frequency of KKK and KLA even increased in these sequences. Unlike this observation, the mainly hydrophobic-based compositions like LLG, AAL, YLL, GLL, and AAA were dominant in the non-CPPs. Additionally, the Leu-rich cationic tripeptides containing Arg and Lys were among the dominant motifs in the Cp-ACPs. Furthermore, remarkable differences between Cp-ACPs and NCp-ACPs were observed, suggesting that the TPC might be regarded as an effective feature in the discrimination of active and nonactive analogues. These findings suggested that the coexistence of Arg and hydrophobic residues could conspicuously enhance peptide internalization in the cancer cells.27,29 In this regard, our previous investigation demonstrated that Trp-tagging promoted the cellular entry of Arg-rich peptide into the melanoma cells giving rise to no toxic effects.27 In fact, the incorporation of Trp residues within the cationic sequences could enhance the internalization efficiency of peptides through endocytosis.30
Physiochemical Characteristics
The peptides’ PCPs (Figure 4; also see Table S2 in the Supporting Information), most importantly, the amphipathicity, hydrophobicity, and net charge, play significant roles in the determination of anticancer and cell-permeable peptide’s activity behaviors.8,31 Basically, the active sequences displayed a net positive charge ranging from +5 to +6, whereas the NACPs, NCPPs, and NCp-ACPs were among the neutral and weak positively charged analogues.
Figure 4.

Physicochemical characteristics of the active and nonactive sequences. Seaborn 0.10.032 was used to build the plots. Other PCPs of interest are listed in the Supporting Information Table S2.
Given the more amphiphilic nature, strong interaction with and deep penetration into the anionic lipid bilayers are expected for the bioactive peptides, making the plasma membranes prone to disruption, endocytosis, and/or direct translocation.8 This is interestingly in good agreement with the membrane binding affinity of the ACPs, CPPs, and Cp-ACPs as revealed by the increased Boman index (∼1.0–3.5) compared with the nonactive analogues.33 The peptides’ hydrophobic character followed the order of CPPs≫ Cp-ACPs > ACPs > NACPs > Cp-ACPs ≫ NCPPs, suggesting the importance of global hydrophobicity as a discriminatory factor between each individual class of active and nonactive peptides.34
Feature Selection
Methods of feature selection seek to improve the prediction performance of the classification model. In fact, the single feature selection might have specific biases35 and be alleviated and/or compensated through a feature selection algorithm via different strategies. Here, we applied two independent feature selection methods to extract various feature groups and test our ML models against them. Three well-known ML algorithms, SVM, RF, and XGBoost, were exploited to construct the models. The ML algorithms were fed with the well-curated features.36−38
In a straightforward feature combination approach (METH 1), we ensembled different compositional, physicochemical, and 13C NMR-based features. Previous studies suggested that AAC, PACC, and PCP are the most effective features in predicting either ACPs or CPPs.20,23 The AAC displayed the best performance with accuracy in the range of ∼0.95–0.96 [area under curve (AUC) and F1 score ≈ 0.95] through the SVM-, RF-, and XGboost-based ACP predictors (cf. Figures 5 and 6). In this regard, the subclassifiers could reach the highest accuracy of 0.94–0.95 (AUC/F1 score ≈ 0.95/0.94) using the PCP. Comparable results were obtained by the PACC feature; the accuracy, AUC, and F1 score of the predictors were ∼0.94–0.95. On the other hand, the CPP subpredictors presented their best performance with accuracy in the range of 0.87–0.88 and AUC and F1 score ≈ 0.88 when SVM, RF, and XGboost applied the PCP descriptors. The AAC-based subpredictors could also show important achievements with an accuracy of ∼0.83–0.84 (AUC/F1 score ≈ 0.84). Good performance (i.e., accuracy ≈ 0.82–0.83; AUC ≈ 0.83; and F1 score ≈ 0.82) was obtained using the PAAC-based CPP predictors.
Figure 5.

Performance metrics of RF (black), SVM (red), and XGBoost (green) models in response to various feature sets employed for the prediction of ACPs.
Figure 6.

Feature-dependent performance metrics of RF (black), SVM (red), and XGBoost (green) for the prediction of CPPs.
To achieve the highest performance, we could also reach to set the optimized combination of feature groups using the FeatureSelect.17 Accordingly, we created seven hybrids, i.e., HYBR1 of 75-membered features (AAC, APACC, PACC), HYBR2 of 111-membered features (AAC, APACC, PACC, PCP), HYBR3 of 115-membered features (AAC, APACC, PACC, CNMR), HYBR4 of 151-membered features (AAC, APACC, PACC, PCP, CNMR), HYBR5 of 150-membered features (AAC, APACC, CTD, PACC, PCP), HYBR6 of 511-membered features (AAC, APACC, DPC, PACC, PCP), and HYBR7 of 650-membered features (AAC, APAC, CSKAAG, DPC, GDPC, PACC), that help our learners obtain better performance.
Additionally, we performed the RFE algorithm (METH 2) to find the most statistically significant features for our predictions. The RFE consecutively runs in several iterations and removes features of low weights for each learner. In this respect, we managed to obtain 378 features for the ACPs and 128 features for the CPPs. Even though METH 2 considerably uses all feature sets, the results were comparable to those obtained from HYBR2, in nearly all performance metrics (Figures 5 and 6), suggesting that either of the methods could be taken as a competitor to one another. Intriguingly, we highlighted that the features of a different nature could have synergistic effects on improving the models’ performance. In the same sense, the 13C NMR feature set exhibited low performance in the prediction task, yet it played a positive role in improving the performance of our predictors using METH 2 when some of its members are selected and integrated with other attributes.
ACP and CPP Predicting Classifiers
The performance of each learner on both ACP and CPP data is shown in Figures 5 and 6. Each model has been once trained and independently tested against all extracted feature groups to find the impact of each on the performance of our learners. Considering the above-mentioned approaches, the hybrid feature-based classifiers displayed the best prediction accuracy, especially in the CPP-based models, where they showed a steeper increase in all performance metrics. The HYBR2-based SVM, RF, and XGboost often revealed the best performance in terms of accuracy (∼0.97–0.99), with AUC and F1 scores of about 0.98 and 0.98, respectively, for the prediction of ACPs and CPPs (Figures 5 and 6). Given the very low dimension feature space, HYBR2 is preferred over the hybrid features in terms of ML complexity of the models’ train and test processes. Interestingly, although the 13C NMR feature showed low performance from the prediction power’s point of view of both the ACPs and CPPs, the negative impact of this feature group could be spotted in the performance of HYBR3 and 4 feature sets. Notwithstanding, the higher number of features gives a better clue to the ML-based models that enable one to perform more accurate classifications. In this regard, METH 2, which includes the advanced feature selection procedure, allowed investigating the importance of each individual feature in the original feature vector space. In fact, METH 2 utilizes a combination of filter and wrapper selection methods to find those features with a high discriminatory power. Even though HYBR2 (AUC and/or accuracy of 0.99/0.96) and METH 2 (AUC and accuracy of 0.98/0.95 and 0.98/0.94, respectively) showed almost comparable results for the ACP/CPP subpredictors (Figures 5 and 6), the former is described by the smaller size of feature space, resulting in the lower ML time complexity. In contrast, the feature selection by METH 2 could be used in different contexts to obtain statistically important features.
Cp-ACP Data-Set Analysis through ACP/CPP Subclassifiers
Relative to other feature sets, HYBR2, which encompasses a rather low number of features, and METH 2 offer more reliable results to predict Cp-ACPs through our developed ACP and CPP models (Figure 7). The prediction performance (generalization power) of our HYBR2-based and METH 2-based SVM, RF, and XGboost algorithms on the Cp-ACP data set (see Table S1 in the Supporting Information) demonstrated that the performance of our methods was notably better than the current accessible ACP (i.e., Anticp,18 iACP,19 ACPred,20 and mACPpred21) and CPP (i.e., CellPPD,22 MLCPP,23 and CPPred-RF24) tools. Compared with the average of other tested tools, the performance metrics of all tested ACP-based predictors with HYBR2/METH 2 were as follows: recall (0.90/0.59), F1 score (0.90/0.68), accuracy (0.90/0.73), and Matthews correlation coefficient (MCC) (0.79/0.48). Nonetheless, the HYBR2/METH 2-based CPP predictors presented a lower quality in classifying Cp-ACPs on the basis of MCC values (0.60/0.51), yet their predicting power was competitive with the previous tools, namely, recall (0.79/0.64), F1 score (0.79/0.70), and accuracy (0.80/0.75).
Figure 7.

Performance comparison measures of (a) ACP-based and (b) CPP-based models constructed using RF (black), SVM (red), and XGBoost (green) algorithms for prediction of Cp-ACPs. The column bars represent the performance comparison of this study’s ACP and CPP subclassifiers relative to (a) SVM-based Anticp, iACP, ACPred, mACPpred, and (b) prediction tools of CellPPD (SVM), MLCPP (RF), CPPred-RF (RF), and SkipCPP-Pred (RF). See Tables S3 and S4 in the Supporting Information for details of numerical values.
ML-Based Fusion Method for Predicting Cp-ACPs
It was shown that the fusion or ensemble of multiple classifiers’ prediction outputs could improve the final performance.39 Importantly, from this perspective, numerous ensemble-strategy-based models were suggested for the classification of proteins. For instance, EnsDNN, which was developed for the protein–protein interaction (PPI), utilizes a two-hidden-layer neural network to integrate 27 trained independent deep neural networks (DNNs).40 The Rotation Forest was also implemented as the ensemble classifier to predict PPIs.41 In this regard, Nanni et al. additionally applied the weighted sum role as the ensemble approach for the protein classification based on the sum of scores obtained from the pool of SVM classifiers.42 Even though various ensemble predictors, such as iACP-GAEnsC,43 ACPredStackL,14 and StackCPPred,44 are available to predict the ACP and CPP sequences, there is no model that concurrently explores both the anticancer and cell-penetrating properties of peptide sequences. Therefore, we, in this study, proposed a parallel two-layer framework upon which the results from the ACP and CPP predictors, individually built on a separate layer using the same algorithms (i.e., RF, SVM, and XGBoost) and feature selection methods (METH 1: feature hybridization; METH 2: RFE algorithm), were combined to yield a fused decision of binary classifiers on the independent validation test set of Cp-ACPs. Although, for instance, voting, statistical, and fuzzy-logic-based methods could be applied for making the final decision, we used a consensus hard-fusion-based strategy by running a pessimistic “AND” operator for the assessment labeling of each Cp-ACP.
As illustrated in Figure 8, our fusion method for the HYBR2 and METH 2 feature groups managed to achieve average AUCs of 0.95 and 0.94, respectively. Because of the simple nature of hard-fusion applied on the classifier’s decisions, the predictive power for this stage was slightly better than CPP-based machines. In contrast, the performance of our predictors could not exceed that of the ACP-based machines in predicting Cp-ACPs. Overall, the SVM classifier had the highest MCCs of 0.68 and 0.64 using the HYBR2 and METH 2 feature groups, respectively. In comparison, XGBoost presented a competitive MCC of 0.63 on METH 2, but the RF classifier resulted in the lowest quality with an MCC of 0.58 on HYBR2. Taken together, the average precision (PR) values of 0.85 for HYBR2 and 0.83 for METH 2 prove that our simple fusion method could be profoundly considered as a reasonable predictive power in classifying Cp-ACPs. Having said that, we might expect improvements in the results by designing more complex fusion methods and utilizing a higher number of validation data.
Figure 8.

Performance metrics comparison of the intersection of ACP- and CPP-based models in predicting Cp-ACPs.
Discussion
Recent achievements in the design and development of novel therapeutics often failed to control and prevent cancer growth and metastasis as a result of diseased cell heterogeneity and recurrence, as well as the mechanisms of resistance (e.g., drug efflux, inadequate uptake and inactivation, target modification, DNA damage repair).1,2 With regard to this issue, the identification of novel therapeutics that overcome the development of cellular resistance and selective targeting of the neoplastic cells is challenging.
Unlike normal eukaryotic cells, cancer plasma membranes are negatively charged and covered with a higher content of PtdSer and sialic acid-conjugated proteins and lipids and a larger surface area of increased value of microvilli, which are therefore appropriately considered as targets for the action of dominantly cationic and amphipathic peptides (i.e., ACPs, CPPs).3 These sequences are frequently characterized by high occurrences of Arg and Lys, aliphatic Leu/Ile, as well as other aromatic residues, such as Phe and Trp, giving rise to the overall positive charge properties and sufficient hydrophobicity for the peptide molecules’ optimal effects on cancer membranes.3 From the peptide–membrane interaction perspective, however, the impact of each of these residues on the anionic functional entities of the cell surface is distinct at the molecular level. Upon the electrostatic binding of ACPs/CPPs to the cancer membrane and further deep penetration into the hydrophobic interior, the Arg and Lys side chains presumably maintain their positive charge, but nevertheless, the guanidinium group more selectively binds at the anionic lipid bilayer interface than the amino functional moiety. This, however, depends on the sequence length and structure, leading to the compensated free energy required for the direct cellular transduction of relevant peptides.45 The guanidinium side chain additionally tends to multiple hydrogen bonds with the negatively charged molecules of the cell surface. That, as a consequence, promotes the formation of a saddlelike curvature in the plasma membrane and membrane endocytosis in peptides mainly constituting of Arg.46,47 Notwithstanding, the Lys-rich MG 2-derived sequences exhibit either direct membrane transport or endosomal delivery characteristics independent of the lipid-raft structures in the A549 cells, with the eventual peptide localization in the nucleus.8 By comparison, the peptide containing Ile/Leu and Phe/Trp supports hydrophobic contacts with the lipid bilayer core, resulting in an enhanced membrane fluidity and perturbation. Moreover, the presence of these residues in the context of a zipper-like structure may play a pivotal role in the inhibition of cell regulatory protein–protein interaction networks, as well as in the self-assembly-promoted membrane activity of ACPs/CPPs.48−51 Aside from this, the amphipathic indole ring could localize at the polar/apolar lipid interface, resulting in the convenience of membrane selectivity in the Trp-rich peptides.50,51 With the high occurrence of the flexible and tiny residues of Gly and Ala in the Cp-ACPs, the increased plasma membrane interaction and cellular uptake of the peptides are most likely expected.8
Even though recent studies have highlighted the cell membrane-lytic effects of ACPs,52−54 cancer cells characterized by a highly dense sialic acid surface were supposed to become resistant to them.5 The release of proinflammatory cytokines and chemokines is a further consequence of the relevant peptide-induced compromised cell membranes.6 In contrast, Cp-ACPs are known for their selective intracellular activity and are capable of triggering apoptotic signaling via the modulation of protein–protein interaction networks or nucleus DNA fragmentation;8−12 see Table S1 in the Supporting Information for more examples. Because of their distinct anticancer signaling, these peptide molecules would probably not face the emergence of resistant cells.8,55 For instance, the bioactivity of these sequences might probably not be influenced in P53-deficient cells, as well as by the activity of multidrug-resistant efflux pumps,8,55 thus introducing Cp-ACPs as potential alternatives to address the current obstacles in cancer therapy. In this respect, however, the identification and design of Cp-ACP sequences always require rigorous and challenging discovery of peptide/protein libraries using expensive experimental screenings.8 Notwithstanding, there are several ML predicting tools focusing on ACPs or CPPs alone without taking the specific activities of Cp-ACPs into consideration.18−25 It is, therefore, of importance to develop ML-based models to predict such bioactive sequences with novel anticancer functions.
In general, the existing ML-based ACP and CPP predictors are classified into two main groups according to their used learning algorithms.14,15 The first group includes models employing the conventional ML-based algorithms including but not limited to the SVM, RF, ERT, and KELM. For instance, the SVM based on the PseAAC and local alignment kernel was applied to develop ACP classifiers, where, accordingly, four novel anticancer sequences were derived from the HIV-1 P24 protein.56 Likewise, the SVM was used to implement both AntiCP and mACPpred.18,21 Also, Wei et al. developed ACPred-FL through which 40 groups of features from seven different descriptor classes (i.e., binary profile, overlapping property, and twenty-one-Bit features, CTD, AAC, G-gap dipeptide, and adaptive skip dipeptide composition) were fed into the SVM model to predict the model labels.57 Additionally, ACPred-FUSE was built on the RF algorithm based on multiview information composed of the class, probabilistic, and sequential information.58 Last but not least, ACP-DL59 and DeepACP60 are the only deep-learning-based models that can distinguish ACPs. The former employs the recurrent neural network (RNN) with the LSTM method fed with a k-mer sparse matrix and a binary profile encoding the peptide sequence into a two-dimensional (2D) matrix59 while the latter proposes a sequence-based model utilizing the RNN architecture with a bidirectional LSTM for training the prediction model.60 More recently, Charoenkwan et al. proposed iACP-FSCM as a novel computational method to characterize ACPs according to the flexible scoring card method, which could determine the propensity score of local and sequential information in anticancer sequences.61 In this regard, CellPPD was developed using the SVM, fed with various features, such as compositional information, PCP, and binary profile, to classify the CPPs.22 Furthermore, CPPred-RF24 and SkippCPP-Pred25 were two crucial RF-based CPP classifiers. Whereas the former predictor was a two-layer architecture, where the peptide-cell-penetrating characteristics and their uptake efficiency are predicted in the first and second layers, respectively,24 the latter utilized the novel k-skip-2-g feature algorithm, which calculates the composition of two residues with distances ≤ k, to encode the CPP sequences.25 Besides, Manavalan et al. developed MLCPPs, which apply a two-layer prediction design using ERT and RF to predict CPPs and their uptake capability.23 Recently, TargetCPP was constructed based on the gradient boost decision tree capturing four different features, including composite protein sequence representation, split amino acid composition, CTD, and information theory features, to predict CPPs.62 In contrast, the ensemble-learning-based methods are considered as the second group of ACP/CPP classifiers. The examples include iACP-GAEnsC, a genetic algorithm and majority-voting-based ensemble,43 as well as ACPredStackL and StackCPPred, applying a stacking framework to classify the ACP and CPP sequences, respectively.14,44 In the development of ACPredStackL, the k-nearest neighbor (KNN), naïve Bayesian (NB), Light Gradient Boosting Machine (LightGBM), and SVM were utilized as base learners using different features, e.g., AAC and CTD. In the following, the prediction outcome of classifiers was merged and fed to the logistics regression (LR) to create a metamodel for the final prediction of ACPs.14 In a comparable manner, StackCPPred comprised two-level algorithms, where RF, KNN, SVM, LightGBM, and XGBoost were used in the first layer and independently trained based on the residue pairwise energy content matrix as the feature encoding tool, and the SVM, in the second layer, was then trained using the classifiers’ combined prediction outputs.44
Taken together, in this study, we presented an approach that could simultaneously examine both the anticancer and cell-penetrating properties of peptide sequences. To this end, we developed two parallel independent prediction pipelines that identify the ACPs from NACPs, as well as the CPPs from their nonactive analogues, based on the comparable RF, SVM, and XGBoost algorithms, and the same type of feature selection ensembles. We further used a hard-fusion method based on the pessimistic AND operator to integrate the prediction results of the ACP and CPP subclassifiers and assess their combined power for the detection of Cp-ACPs. Most studies provided simple train-test splitting methods or one iteration of cross-validation for the evaluation of trained models.63−66 However, we applied 10 iterations of 10-fold cross-validation with different random seeds to carry out an extremely rigorous assessment of our models. This strategy thus permits one to (i) evaluate the models’ predictive performance and obtain consistently low variance and robust results when tested with diverse combinations of data, (ii) improve the generalization power of our predictors, and (iii) avoid the overfitting problem. Overall, our predictors mostly outperform the current ML-based ACP/CPP models.
The performance of our machines was assessed with an independent data set of 65 Cp-ACPs. To ensure a fair assessment of the models’ performance, these sequences were neither present in the training data nor seen by the predictors. The hard-fusion of the ACP and CPP subclassifiers that was used for the label assessment of each individual peptide candidate exhibited an average AUC of ∼0.94 for the identification of Cp-ACPs in an ensemble setting by our RF-, SVM-, and XGboost-based models. Nevertheless, this simple fusion operator might result in a higher false-negative (FN) rate and probably give rise to the negative evaluation of a number of peptides with the Cp-ACP properties. Thus, it is necessary to develop advanced and intelligent ensemble methods, like the weighted majority voting, and improve the models’ prediction performance. The limited number of experimentally validated Cp-ACP training data set, in this study, is moreover a challenge to build the decision-making systems based on the complex fuzzy integral operator, such as the ordered weighted averaging. Nonetheless, the tolerance of a more false-negative risk for less error in positive prediction is desirable using a pessimistic operation, particularly in laboratory applications. As a result, a peptide candidate that has both anticancer and cell-penetrating properties could certainly be selected with high reliability for further validation in wet-lab experiments. Last but not least, our proposed approach and architecture could potentially be exploited to facilitate the identification of various classes of bioactive sequences with the cell-penetrating property. The web-based CpACpP platform and the package containing the prebuilt models are accessible at http://cbb1.ut.ac.ir/CpACpP/Index.
Experimental Section
Data-Set Preparation
The anticancer and cell-penetrating sequences were separately extracted from the existing databases as mentioned below. Note that we filtered out the databases from those sequences comprising non-natural amino acid residues. These experimentally determined active sequences were taken without considering their activity level. To improve the data statistical significance and the generalization power of our predictors, the CD-HIT program was applied to remove the sequences with a homology of more than 90%.67
ACPs
A 659-membered peptide sequence data set was collected after running the CD-HIT program from the CancerPPD (an anticancer peptide and protein database), APD (antimicrobial peptide database), DADP (database of anuran defense peptides), and DBAASP (database of antimicrobial activity and structure of peptides).68−71 A data set of presumably accepted 657 NACPs was generated using the RandSeq tool (https://web.expasy.org/randseq/), out of which 385 sequences were acquired from the AntiCP data set18 (see Table S5 in the Supporting Information) following the CD-HIT run. The RandSeq tool is able to generate sequences with the user-defined amino acids. Given the different preference and frequency of each amino acid in ACPs vs NACPs, the negative sequences could likely be generated with a higher probability using this tool.
CPPs
CPPsite2.0, which contains around 1850 experimentally validated natural sequences, was used for the analysis.72 The NCPP data set was randomly acquired using the RandSeq tool, as well as the KELM-CPPpred and MLCPP benchmarks23,28 (see Table S6 in the Supporting Information). The collected sequences after the CD-HIT run were, in total, 1180 sequences [CPPs (577) and NCPPs (603)].
Cp-ACPs
This data set contains 65 sequences having both anticancer and cell-permeable properties and was selected from the literature survey (see Table S1 in the Supporting Information). This data set was taken as the independent data set to evaluate and validate our methods. The RandSeq tool was used for the generation of sequences having neither cell-penetrating nor anticancer property. The CD-HIT was further applied to exclude the Cp-ACPs with a similarity >90% in either the ACP or CPP data set. Overall, 150 unique sequences [65 Cp-ACPs and 85 NCp-ACPs] were obtained and used in this work.
Feature Extraction
The AACs, PCPs, and 13C NMR feature-based groups were generated by IFeature, modlAMP, and IAMPE, respectively.36−38
For all of the calculated features in this section, the query peptide is defined as P = P1P2P3, ..., PL, which is composed of a sequence of L amino acid residues. Pi (i = 1, 2, ..., L) denotes the residue at the ith chain position. The feature vectors, their dimension, and a brief description are provided next.
AAC-Based Features
The frequency of naturally occurring amino acids and their 400 dipeptide combinations are formulated in eqs 1 and 2.
| 1 |
| 2 |
 and
and  are the occurrence percentages of the amino
acid (i = 1, 2, ..., 20) and the dipeptide (i = 1, 2, 3, ..., 400), respectively, in the peptide. Ri and Di are the
numbers of residue and dipeptide, respectively. Also, the amino acid
types are classified into five groups based on their aliphaticity
(g1), aromaticity (g2), and charge values, including positive (g3), negative (g4), and neutral
(g5), and employed to calculate the GDPC
vector according to eq 3.
are the occurrence percentages of the amino
acid (i = 1, 2, ..., 20) and the dipeptide (i = 1, 2, 3, ..., 400), respectively, in the peptide. Ri and Di are the
numbers of residue and dipeptide, respectively. Also, the amino acid
types are classified into five groups based on their aliphaticity
(g1), aromaticity (g2), and charge values, including positive (g3), negative (g4), and neutral
(g5), and employed to calculate the GDPC
vector according to eq 3.
| 3 |
Herein, Ngigl is the number of occurrences of the paired amino acids from gi and gl groups in the query peptide (P), where gi and gl are members of {g1,g2,g3,g4,g5}.36
CKSAAGP, which measures the occurrence frequencies of the k-spaced amino acid pairs in the query peptide, is a feature set and calculated according to eq 4. Briefly, a k-spaced amino acid pair is represented as Pi{k}Pj (i, j = 1, 2, ..., 20), where Pi and Pj denote any two residues of 20 amino acids separated by k residues (k = 0, 1, 2, 3, 4, 5).
| 4 |
 is the number
of pair groups of amino acid
types i and l separated by k gaps in the sequence of the query peptide (P).
is the number
of pair groups of amino acid
types i and l separated by k gaps in the sequence of the query peptide (P).
Additional information including the correlation between the residues of a certain distance is accessible in APAAC and PAAC. These feature sets use the hydrophobicity, hydrophilicity, and/or side-chain mass indices to reflect the peptide sequence activity.36
PCP-Based Features
This class of features includes atom-type composition (ATC) and various physicochemical attributes of amino acids that form the peptide sequence. ATC encodes the frequency of each atom containing C, H, N, O, and S in a specific peptide sequence. The PCP feature set can be computed according to eq 5.
| 5 |
where Pi and L are the values of multiple physiochemical properties of amino acid type i and the length of the peptide sequence, respectively. We calculated the fractions of tiny (A, C, D, G, S, T), small (E, H, I, L, K, M, N, P, Q, V), large (F, R, W, Y), aliphatic (I, L, V), aromatic (F, H, W, Y), polar (D, E, R, K, Q, N), charged (D, E, K, H, R), basic (H, K, R), and acidic (D, E) residues. The length of peptides and the isoelectric point (pI) were also considered.73 The dimension of this feature vector is 10.
CTD-Based Features
This feature set contains 237 features (39C, 39T, 139D) characterizing the composition, transition, and distribution of the particular properties, wherein the amino acids are divided into clusters based on particular properties, such as hydrophobicity, normalized van der Waals volume, polarity, polarizability, charge, secondary structure, and solvent accessibility of peptide sequences.38
13C NMR-Based Features
In this respect, the features extracted from the 13C NMR spectra (with the exception of tyrosine), for instance, relative spectral power, slow wave index, and harmonic parameters, were used to cluster the amino acids. We have already demonstrated the discriminating power of this feature vector in the prediction of antimicrobial and biofilm-inhibiting peptides.38,74
Feature Selection
In the first and rather straightforward approach (METH 1), various feature groups, such as AAC, DPC, GDPC, CKSAAGP, PAAC, APAAC, PCP, CTD, and 13C NMR, and seven hybrids thereof were generated and evaluated on the machine learning models. In the second method (METH 2), a multilayer selection procedure was applied with the combination of filter-based and wrapper methods. This feature selection algorithm consists of three layers. We used the Mann–Whitney U test in the first layer to identify the most discriminative features between the positive (class = 1) and negative (class = 0) samples based on their p-values. After this initial filtering, we applied the minimum redundancy maximum relevance (mRMR) method to select the feature subsets according to their importance and similarity.75 This algorithm tends to select a subset of features with higher relevance to dependent variables (target class), though in a lower correlation with each other. Accordingly, the highly correlated features were discarded to avoid overfitting and false predictions. Finally, the RFE was employed to identify the best features from the previous stage candidate list. The RFE is a wrapper selection method that fits a model and removes the weakest features until a specified number of features are achieved. By the end of this selection method, 378 and 128 features were obtained for ACPs and CPPs, respectively.
Machine Learning Model
The SVM, RF, and XGBoost classifiers were utilized to develop the predictor models. The standard GridSearchCV method from the Scikit-learn package,76 taking a cross-validation generator (parameter “cv”), was used and executed in 10 iterations to find the best set of hyperparameters based on the AUC performance metric (Table 1). The optimal parameters were obtained by testing a wide variety of values.
Table 1. Optimal Parameters for the Model Implementation Using the Scikit-Learn Packagea.
| SVM | C = 10 |
| γ = 0.01 | |
| kernel = rbf | |
| RF | number of estimators = 100 |
| max depth = 8 | |
| criterion = entropy | |
| XGBoost | colsample_bytree = 0.2 |
| γ = 0.1 | |
| learning rate = 0.1 | |
| max depth = 3 | |
| number of estimators = 100 | |
| subsample = 1 |
The models were implemented in Python 3.7.76
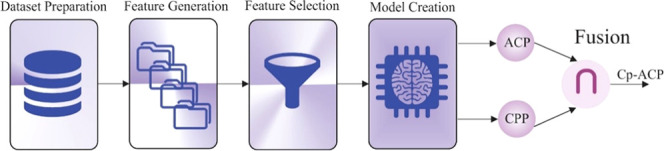
To predict the Cp-ACPs, however, the models were constructed and developed based on two subpredictors. Under this strategy, the active ACPs and CPPs were independently identified, and then, a fusion method through an intersection between the ACP and CPP classifiers’ results was utilized to identify the Cp-ACP sequences (Scheme 1).
Scheme 1. Schematic Representation of the CpACpP Model.

The classification of Cp-ACPs was generated through an intersection between two independently trained ACP and CPP classifiers’ outcomes.
Performance Evaluation
Each independent ACP and CPP was randomly split into 80% training and 20% testing subsets in 10 iterations. The model was trained only on the corresponding 80% training data set, through which the 10-fold cross-validation was applied on them to evaluate the models under the best hyperparameter setting. In this regard, the training set was randomly split into 10 equal subsamples, in which 9 samples were used as the training sets, and the remaining one was used to build the prediction model. In the next step, finally, the first 20% testing subset washeld out from the primary training set and used as independent data to assess the trained models and ensure that they execute fair predictions on the future unseen sequences and meet superior generalization performance. This process was executed 10 times to avoid our predictor overfitting. Finally, the models were tested for the independent hold-out validation of the Cp-ACP data set to independently evaluate the generalization power of each classifier.
Precision (PR), sensitivity (SN), specificity (SP) equal to the true positive rate (TPR), accuracy (ACC), and F1 score were used to quantify the performance of our predicting models. The MCC was also calculated to be used in imbalanced data sets. These measures were defined in terms of true positive (TP), true negative (TN), false positive (FP), and false negative (FN). The calculation of each performance metric is demonstrated in eq 6.
 |
6 |
Furthermore, the receiver operating characteristic (ROC) curve and the area under the ROC curve (AUC; see eq 7) were utilized to assess the predictor’s performance, where false positive rate (FPR) means the fraction of negative instances misclassified as positive.
| 7 |
Data and Software Availability
A ready-to-use package containing all supervised machine learning algorithms including the scripts and instructions, as well as the data sets used in this study (i.e., ACPs, NACPs, CPPs, NCPP, Cp-ACPs, NCp-ACPs), could be found in the Supporting Information. Additionally, an interactive Web-based application that allows access to unpredicted data with our trained machines is accessible at http://cbb1.ut.ac.ir/CpACpP/Index.
Acknowledgments
This work was supported by financial grants from the University of Tehran (M.B. and K.K.) and the Iran National Science Foundation (M.B.; funding no. 97009976).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c02569.
List of practically validated Cp-ACPs, complete list of PCPs of interest for the active and nonactive sequences, performance result comparisons of the proposed ACP- and CPP-based predictors based on the HYBR2 and METH 2 feature sets relative to other classifiers, and the list of nonactive ACP and CPP sequences used for model training (PDF)
Machine learning algorithms including the scripts and instructions, as well as the data sets used in this study (i.e., ACPs, NACPs, CPPs, NCPP, Cp-ACPs, NCp-ACPs) (ZIP)
Author Contributions
F.N., F.F.A., and S.B. equally contributed to the data-set preparation/analysis and machine learning studies. S.B. configured the CpACpP Web server. The manuscript was written and reviewed by all of the authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Siegel R. L.; Miller K. D.; Jemal A. Cancer Statistics, 2020. Ca-Cancer J. Clin. 2020, 70, 7–30. 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- Vasan N.; Baselga J.; Hyman D. M. A View on Drug Resistance in Cancer. Nature 2019, 575, 299–309. 10.1038/s41586-019-1730-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabernet G.; Müller A. T.; Hiss J. A.; Schneider G. Membranolytic Anticancer Peptides. MedChemComm 2016, 7, 2232–2245. 10.1039/C6MD00376A. [DOI] [Google Scholar]
- Gaspar D.; Veiga A. S.; Castanho M. A. R. B. From Antimicrobial to Anticancer Peptides. A Review. Front. Microbiol. 2013, 4, 294 10.3389/fmicb.2013.00294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa K.; Medina S. H.; Schneider J. P.; Klar A. J. S. Glycan Alteration Imparts Cellular Resistance to a Membrane-Lytic Anticancer Peptide. Cell Chem. Biol. 2017, 24, 149–158. 10.1016/j.chembiol.2016.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay J.; Thadhani E.; Samson L.; Engelward B. Inflammation-Induced DNA Damage, Mutations and Cancer. DNA Repair 2019, 83, 102673 10.1016/j.dnarep.2019.102673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciobanasu C.; Kubitscheck U.. Cell-Penetrating Peptides Targeting and Distorting Biological Membranes. In Surface and Interface Science; Wiley, 2020; pp 441–469. [Google Scholar]
- Behzadi M.; Arasteh S.; Bagheri M. Palmitoylation of Membrane-Penetrating Magainin Derivatives Reinforces Necroptosis in A549 Cells Dependent on Peptide Conformational Propensities. ACS Appl. Mater. Interfaces 2020, 12, 56815–56829. 10.1021/acsami.0c17648. [DOI] [PubMed] [Google Scholar]
- Wu D.; Gao Y.; Qi Y.; Chen L.; Ma Y.; Li Y. Peptide-Based Cancer Therapy: Opportunity and Challenge. Cancer Lett. 2014, 351, 13–22. 10.1016/j.canlet.2014.05.002. [DOI] [PubMed] [Google Scholar]
- Maraming P.; Klaynongsruang S.; Boonsiri P.; Peng S. F.; Daduang S.; Leelayuwat C.; Pientong C.; Chung J. G.; Daduang J. The Cationic Cell-Penetrating KT2 Peptide Promotes Cell Membrane Defects and Apoptosis with Autophagy Inhibition in Human HCT 116 Colon Cancer Cells. J. Cell Physiol. 2019, 234, 22116–22129. 10.1002/jcp.28774. [DOI] [PubMed] [Google Scholar]
- Wang C.; Dong S.; Zhang L.; Zhao Y.; Huang L.; Gong X.; Wang H.; Shang D. Cell Surface Binding, Uptaking and Anticancer Activity of L-K6, a Lysine/Leucine-Rich Peptide, on Human Breast Cancer MCF-7 Cells. Sci. Rep. 2017, 7, 8293 10.1038/s41598-017-08963-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T.; Das Gupta T. K.; Beattie C. W. P28, an Anionic Cell-Penetrating Peptide, Increases the Activity of Wild Type and Mutated P53 without Altering Its Conformation. Mol. Pharmaceutics 2013, 10, 3375–3383. 10.1021/mp400221r. [DOI] [PubMed] [Google Scholar]
- Basith S.; Manavalan B.; Hwan Shin T.; Lee G. Machine Intelligence in Peptide Therapeutics: A Next-generation Tool for Rapid Disease Screening. Med. Res. Rev. 2020, 40, 1276–1314. 10.1002/med.21658. [DOI] [PubMed] [Google Scholar]
- Liang X.; Li F.; Chen J.; Li J.; Wu H.; Li S.; Song J.; Liu Q. Large-Scale Comparative Review and Assessment of Computational Methods for Anti-Cancer Peptide Identification. Briefings Bioinf. 2020, bbaa312 10.1093/bib/bbaa312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su R.; Hu J.; Zou Q.; Manavalan B.; Wei L. Empirical Comparison and Analysis of Web-Based Cell-Penetrating Peptide Prediction Tools. Briefings Bioinf. 2020, 21, 408–420. 10.1093/bib/bby124. [DOI] [PubMed] [Google Scholar]
- Bedo J.; Sanderson C.; Kowalczyk A.. An Efficient Alternative to SVM Based Recursive Feature Elimination with Applications in Natural Language Processing and Bioinformatics. In Australian Joint Conference on Artificial Intelligence: Advances in Artificial Intelligence; Sattar A.; Kang B., Eds.; Springer: Berlin, Heidelberg, 2006; pp 170–180. [Google Scholar]
- Masoudi-Sobhanzadeh Y.; Motieghader H.; Masoudi-Nejad A. FeatureSelect: A Software for Feature Selection Based on Machine Learning Approaches. BMC Bioinformatics 2019, 20, 170 10.1186/s12859-019-2754-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyagi A.; Kapoor P.; Kumar R.; Chaudhary K.; Gautam A.; Raghava G. P. In Silico Models for Designing and Discovering Novel Anticancer Peptides. Sci. Rep. 2013, 3, 2984 10.1038/srep02984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W.; Ding H.; Feng P.; Lin H.; Chou K. C. IACP: A Sequence-Based Tool for Identifying Anticancer Peptides. Oncotarget 2016, 7, 16895–16909. 10.18632/oncotarget.7815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaduangrat N.; Nantasenamat C.; Prachayasittikul V.; Shoombuatong W. ACPred: A Computational Tool for the Prediction and Analysis of Anticancer Peptides. Molecules 2019, 24, 1973 10.3390/molecules24101973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boopathi V.; Subramaniyam S.; Malik A.; Lee G.; Manavalan B.; Yang D. C. MACppred: A Support Vector Machine-Based Meta-Predictor for Identification of Anticancer Peptides. Int. J. Mol. Sci. 2019, 20, 1964 10.3390/ijms20081964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautam A.; Chaudhary K.; Kumar R.; Sharma A.; Kapoor P.; Tyagi A.; Raghava G. P. In Silico Approaches for Designing Highly Effective Cell Penetrating Peptides. J. Transl. Med. 2013, 11, 74 10.1186/1479-5876-11-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manavalan B.; Subramaniyam S.; Shin T. H.; Kim M. O.; Lee G. Machine-Learning-Based Prediction of Cell-Penetrating Peptides and Their Uptake Efficiency with Improved Accuracy. J. Proteome Res. 2018, 17, 2715–2726. 10.1021/acs.jproteome.8b00148. [DOI] [PubMed] [Google Scholar]
- Wei L.; Xing P.; Su R.; Shi G.; Ma Z. S.; Zou Q. CPPred-RF: A Sequence-Based Predictor for Identifying Cell-Penetrating Peptides and Their Uptake Efficiency. J. Proteome Res. 2017, 16, 2044–2053. 10.1021/acs.jproteome.7b00019. [DOI] [PubMed] [Google Scholar]
- Wei L.; Tang J.; Zou Q. SkipCPP-Pred: An Improved and Promising Sequence-Based Predictor for Predicting Cell-Penetrating Peptides. BMC Genomics 2017, 18, 742 10.1186/s12864-017-4128-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenson J. M.; Xue V.; Stretz L.; Mandal T.; Reich L. L.; Keating A. E. Peptide Design by Optimization on a Data-Parameterized Protein Interaction Landscape. Proc. Natl. Acad. Sci. U.S.A. 2018, 115, E10342–E10351. 10.1073/pnas.1812939115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duong D. T.; Singh S.; Bagheri M.; Verma N. K.; Schmidtchen A.; Malmsten M. Pronounced Peptide Selectivity for Melanoma through Tryptophan End-Tagging. Sci. Rep. 2016, 6, 24952 10.1038/srep24952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey P.; Patel V.; George N. V.; Mallajosyula S. S. KELM-CPPpred: Kernel Extreme Learning Machine Based Prediction Model for Cell-Penetrating Peptides. J. Proteome Res. 2018, 17, 3214–3222. 10.1021/acs.jproteome.8b00322. [DOI] [PubMed] [Google Scholar]
- Liu X.; Cao R.; Wang S.; Jia J.; Fei H. Amphipathicity Determines Different Cytotoxic Mechanisms of Lysine- or Arginine-Rich Cationic Hydrophobic Peptides in Cancer Cells. J. Med. Chem. 2016, 59, 5238–5247. 10.1021/acs.jmedchem.5b02016. [DOI] [PubMed] [Google Scholar]
- Bechara C.; Pallerla M.; Zaltsman Y.; Burlina F.; Alves I. D.; Lequin O.; Sagan S. Tryptophan within Basic Peptide Sequences Triggers Glycosaminoglycan- Dependent Endocytosis. FASEB J. 2013, 27, 738–749. 10.1096/fj.12-216176. [DOI] [PubMed] [Google Scholar]
- Milletti F. Cell-Penetrating Peptides: Classes, Origin, and Current Landscape. Drug Discovery Today 2012, 17, 850–860. 10.1016/j.drudis.2012.03.002. [DOI] [PubMed] [Google Scholar]
- Waskom M.; Botvinnik O.; Gelbart M.; Ostblom J.; Hobson P.; Lukauskas S.; Gemperline D. C.; Augspurger T.; Halchenko Y.; Warmenhoven J.; Cole J. B.; Ruiter J.; de Vanderplas J.; Hoyer S.; Pye C.; Miles A.; Swain C.; Meyer K.; Martin M.; Bachant P.; Quintero E.; Kunter G.; Villalba S.; Brian; Fitzgerald C.; Evans C. G.; Williams M. L.; O’Kane D.; Yarkoni T.; Brunner T.. mwaskom/seaborn: v0.11.0 (September 2020). https://zenodo.org/record/4019146 (accessed Dec 30, 2020).
- Kourie J. I.; Shorthouse A. A. Properties of Cytotoxic Peptide-Formed Ion Channels. Am. J. Physiol.: Cell Physiol. 2000, 278, C1063–C1087. 10.1152/ajpcell.2000.278.6.C1063. [DOI] [PubMed] [Google Scholar]
- Huang Y.-B.; Wang X.-F.; Wang H.-Y.; Liu Y.; Chen Y. Studies on Mechanism of Action of Anticancer Peptides by Modulation of Hydrophobicity Within a Defined Structural Framework. Mol. Cancer Ther. 2011, 10, 416–426. 10.1158/1535-7163.MCT-10-0811. [DOI] [PubMed] [Google Scholar]
- Neumann U.; Riemenschneider M.; Sowa J. P.; Baars T.; Kälsch J.; Canbay A.; Heider D. Compensation of Feature Selection Biases Accompanied with Improved Predictive Performance for Binary Classification by Using a Novel Ensemble Feature Selection Approach. BioData Min. 2016, 9, 36 10.1186/s13040-016-0114-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z.; Zhao P.; Li F.; Leier A.; Marquez-Lago T. T.; Wang Y.; Webb G. I.; Smith A. I.; Daly R. J.; Chou K.-C.; Song J. IFeature: A Python Package and Web Server for Features Extraction and Selection from Protein and Peptide Sequences. Bioinformatics 2018, 34, 2499–2502. 10.1093/bioinformatics/bty140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller A. T.; Gabernet G.; Hiss J. A.; Schneider G. ModlAMP: Python for Antimicrobial Peptides. Bioinformatics 2017, 33, 2753–2755. 10.1093/bioinformatics/btx285. [DOI] [PubMed] [Google Scholar]
- Kavousi K.; Bagheri M.; Behrouzi S.; Vafadar S.; Fallah Atanaki F.; Teimoori Lotfabadi B.; Ariaeenejad S.; Shockravi A.; Moosavi-Movahedi A. A. IAMPE: NMR Assisted Computational Prediction of Antimicrobial Peptides. J. Chem. Inf. Model. 2020, 60, 4691–4701. 10.1021/acs.jcim.0c00841. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Yang R.; Zhang C. Using a Classifier Fusion Strategy to Identify Anti-Angiogenic Peptides. Sci. Rep. 2018, 8, 14062 10.1038/s41598-018-32443-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.; Yu G.; Xia D.; Wang J. Protein-Protein Interactions Prediction Based on Ensemble Deep Neural Networks. Neurocomputing 2019, 324, 10–19. 10.1016/j.neucom.2018.02.097. [DOI] [Google Scholar]
- Li Y.; Li L.; Wang L.; Yu C.; Wang Z.; You Z. An Ensemble Classifier to Predict Protein–Protein Interactions by Combining PSSM-Based Evolutionary Information with Local Binary Pattern Model. Int. J. Mol. Sci. 2019, 20, 3511 10.3390/ijms20143511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanni L.; Lumini A.; Brahnam S. An Empirical Study of Different Approaches for Protein Classification. Sci. World J. 2014, 2014, 236717 10.1155/2014/236717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akbar S.; Hayat M.; Iqbal M.; Jan M. A. IACP-GAEnsC: Evolutionary Genetic Algorithm Based Ensemble Classification of Anticancer Peptides by Utilizing Hybrid Feature Space. Artif. Intell. Med. 2017, 79, 62–70. 10.1016/j.artmed.2017.06.008. [DOI] [PubMed] [Google Scholar]
- Fu X.; Cai L.; Zeng X.; Zou Q. StackCPPred: A Stacking and Pairwise Energy Content-Based Prediction of Cell-Penetrating Peptides and Their Uptake Efficiency. Bioinformatics 2020, 36, 3028–3034. 10.1093/bioinformatics/btaa131. [DOI] [PubMed] [Google Scholar]
- Herce H. D.; Garcia A. E.; Cardoso M. C. Fundamental Molecular Mechanism for the Cellular Uptake of Guanidinium-Rich Molecules. J. Am. Chem. Soc. 2014, 136, 17459–17467. 10.1021/ja507790z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt N. W.; Lis M.; Zhao K.; Lai G. H.; Alexandrova A. N.; Tew G. N.; Wong G. C. L. Molecular Basis for Nanoscopic Membrane Curvature Generation from Quantum Mechanical Models and Synthetic Transporter Sequences. J. Am. Chem. Soc. 2012, 134, 19207–19216. 10.1021/ja308459j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirama T.; Lu S. M.; Kay J. G.; Maekawa M.; Kozlov M. M.; Grinstein S.; Fairn G. D. Membrane Curvature Induced by Proximity of Anionic Phospholipids Can Initiate Endocytosis. Nat. Commun. 2017, 8, 1393 10.1038/s41467-017-01554-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan R.; Yuan Y.; Zhang Q.; Zhou X. R.; Jia L.; Liu Z.; Yu C.; Luo S. Z.; Chen L. Isoleucine/Leucine Residues at “a” and “d” Positions of a Heptad Repeat Sequence Are Crucial for the Cytolytic Activity of a Short Anticancer Lytic Peptide. Amino Acids 2017, 49, 193–202. 10.1007/s00726-016-2350-9. [DOI] [PubMed] [Google Scholar]
- Hayouka Z.; Mortenson D. E.; Kreitler D. F.; Weisblum B.; Forest K. T.; Gellman S. H. Evidence for Phenylalanine Zipper-Mediated Dimerization in the X-Ray Crystal Structure of a Magainin 2 Analogue. J. Am. Chem. Soc. 2013, 135, 15738–15741. 10.1021/ja409082w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L.; Chou S.; Wang J.; Shao C.; Li W.; Zhu X.; Shan A. Antimicrobial Activity and Membrane-Active Mechanism of Tryptophan Zipper-like β-Hairpin Antimicrobial Peptides. Amino Acids 2015, 47, 2385–2397. 10.1007/s00726-015-2029-7. [DOI] [PubMed] [Google Scholar]
- Bagheri M.; Amininasab M.; Dathe M. Arginine/Tryptophan-Rich Cyclic α/β-Antimicrobial Peptides: The Roles of Hydrogen Bonding and Hydrophobic/Hydrophilic Solvent-Accessible Surface Areas upon Activity and Membrane Selectivity. Chem. - Eur. J. 2018, 24, 14242–14253. 10.1002/chem.201802881. [DOI] [PubMed] [Google Scholar]
- Dhiman N.; Shagaghi N.; Bhave M.; Sumer H.; Kingshott P.; Rath S. N. Selective Cytotoxicity of a Novel Trp-Rich Peptide against Lung Tumor Spheroids Encapsulated inside a 3D Microfluidic Device. Adv. Biosyst. 2020, 4, 1900285 10.1002/adbi.201900285. [DOI] [PubMed] [Google Scholar]
- Miller S. E.; Tsuji K.; Abrams R. P. M.; Burke T. R.; Schneider J. P. Uncoupling the Folding-Function Paradigm of Lytic Peptides to Deliver Impermeable Inhibitors of Intracellular Protein–Protein Interactions. J. Am. Chem. Soc. 2020, 142, 19950–19955. 10.1021/jacs.0c07921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller A. T.; Posselt G.; Gabernet G.; Neuhaus C.; Bachler S.; Blatter M.; Pfeiffer B.; Hiss J. A.; Dittrich P. S.; Altmann K. H.; Wessler S.; Schneider G. Morphing of Amphipathic Helices to Explore the Activity and Selectivity of Membranolytic Antimicrobial Peptides. Biochemistry 2020, 59, 3772–3781. 10.1021/acs.biochem.0c00565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H.; Qin X.; Yang D.; Jiang Y.; Zheng W.; Wang D.; Tian Y.; Liu Q.; Xu N.; Li Z. The Development of Activatable Lytic Peptides for Targeting Triple Negative Breast Cancer. Cell Death Discovery 2017, 3, 17037 10.1038/cddiscovery.2017.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajisharifi Z.; Piryaiee M.; Mohammad Beigi M.; Behbahani M.; Mohabatkar H. Predicting Anticancer Peptides with Chou’s Pseudo Amino Acid Composition and Investigating Their Mutagenicity via Ames Test. J. Theor. Biol. 2014, 341, 34–40. 10.1016/j.jtbi.2013.08.037. [DOI] [PubMed] [Google Scholar]
- Wei L.; Zhou C.; Chen H.; Song J.; Su R. ACPred-FL: A Sequence-Based Predictor Using Effective Feature Representation to Improve the Prediction of Anti-Cancer Peptides. Bioinformatics 2018, 34, 4007–4016. 10.1093/bioinformatics/bty451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao B.; Zhou C.; Zhang G.; Su R.; Wei L. ACPred-Fuse: Fusing Multi-View Information Improves the Prediction of Anticancer Peptides. Briefings Bioinf. 2020, 21, 1846–1855. 10.1093/bib/bbz088. [DOI] [PubMed] [Google Scholar]
- Yi H.-C.; You Z.-H.; Zhou X.; Cheng L.; Li X.; Jiang T.-H.; Chen Z.-H. ACP-DL: A Deep Learning Long Short-Term Memory Model to Predict Anticancer Peptides Using High-Efficiency Feature Representation. Mol. Ther.--Nucleic Acids 2019, 17, 1–9. 10.1016/j.omtn.2019.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L.; Jing R.; Liu F.; Luo J.; Li Y. DeepACP: A Novel Computational Approach for Accurate Identification of Anticancer Peptides by Deep Learning Algorithm. Mol. Ther.--Nucleic Acids 2020, 22, 862–870. 10.1016/j.omtn.2020.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charoenkwan P.; Chiangjong W.; Lee V. S.; Nantasenamat C.; Hasan M. M.; Shoombuatong W. Improved Prediction and Characterization of Anticancer Activities of Peptides Using a Novel Flexible Scoring Card Method. Sci. Rep. 2021, 11, 3017 10.1038/s41598-021-82513-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arif M.; Ahmad S.; Ali F.; Fang G.; Li M.; Yu D. J. TargetCPP: Accurate Prediction of Cell-Penetrating Peptides from Optimized Multi-Scale Features Using Gradient Boost Decision Tree. J. Comput.-Aided Mol. Des. 2020, 34, 841–856. 10.1007/s10822-020-00307-z. [DOI] [PubMed] [Google Scholar]
- Capecchi A.; Cai X.; Personne H.; Köhler T.; van Delden C.; Reymond J.-L. Machine Learning Designs Non-Hemolytic Antimicrobial Peptides. Chem. Sci. 2021, 12, 9221–9232. 10.1039/d1sc01713f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhadra P.; Yan J.; Li J.; Fong S.; Siu S. W. I. AmPEP: Sequence-Based Prediction of Antimicrobial Peptides Using Distribution Patterns of Amino Acid Properties and Random Forest. Sci. Rep. 2018, 8, 1697 10.1038/s41598-018-19752-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrent M.; Andreu D.; Nogués V. M.; Boix E. Connecting Peptide Physicochemical and Antimicrobial Properties by a Rational Prediction Model. PLoS One 2011, 6, e16968 10.1371/journal.pone.0016968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Ding Y.; Wen H.; Lin Y.; Hu Y.; Zhang Y.; Xia Q.; Lin Z. QSAR Modeling and Design of Cationic Antimicrobial Peptides Based on Structural Properties of Amino Acids. Comb. Chem. High Throughput Screening 2012, 15, 347–353. 10.2174/138620712799361807. [DOI] [PubMed] [Google Scholar]
- Huang Y.; Niu B.; Gao Y.; Fu L.; Li W. CD-HIT Suite: A Web Server for Clustering and Comparing Biological Sequences. Bioinformatics 2010, 26, 680–682. 10.1093/bioinformatics/btq003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyagi A.; Tuknait A.; Anand P.; Gupta S.; Sharma M.; Mathur D.; Joshi A.; Singh S.; Gautam A.; Raghava G. P. S. CancerPPD: A Database of Anticancer Peptides and Proteins. Nucleic Acids Res. 2015, 43, D837–D843. 10.1093/nar/gku892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G.; Li X.; Wang Z. APD3: The Antimicrobial Peptide Database as a Tool for Research and Education. Nucleic Acids Res. 2016, 44, D1087–D1093. 10.1093/nar/gkv1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novković M.; Simunić J.; Bojović V.; Tossi A.; Juretić D. DADP: The Database of Anuran Defense Peptides. Bioinformatics 2012, 28, 1406–1407. 10.1093/bioinformatics/bts141. [DOI] [PubMed] [Google Scholar]
- Pirtskhalava M.; Gabrielian A.; Cruz P.; Griggs H. L.; Squires R. B.; Hurt D. E.; Grigolava M.; Chubinidze M.; Gogoladze G.; Vishnepolsky B.; Alekseev V.; Rosenthal A.; Tartakovsky M. DBAASP v.2: An Enhanced Database of Structure and Antimicrobial/Cytotoxic Activity of Natural and Synthetic Peptides. Nucleic Acids Res. 2016, 44, D1104–D1112. 10.1093/nar/gkv1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal P.; Bhalla S.; Usmani S. S.; Singh S.; Chaudhary K.; Raghava G. P. S.; Gautam A. CPPsite 2.0: A Repository of Experimentally Validated Cell-Penetrating Peptides. Nucleic Acids Res. 2016, 44, D1098–D1103. 10.1093/nar/gkv1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manavalan B.; Basith S.; Shin T. H.; Choi S.; Kim M. O.; Lee G. MLACP: Machine-Learning-Based Prediction of Anticancer Peptides. Oncotarget 2017, 8, 77121–77136. 10.18632/oncotarget.20365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallah Atanaki F.; Behrouzi S.; Ariaeenejad S.; Boroomand A.; Kavousi K. BIPEP: Sequence-Based Prediction of Biofilm Inhibitory Peptides Using a Combination of NMR and Physicochemical Descriptors. ACS Omega 2020, 5, 7290–7297. 10.1021/acsomega.9b04119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L.; Liu H. Efficient Feature Selection via Analysis of Relevance and Redundancy. J. Mach. Learn. Res. 2004, 5, 1205–1224. [Google Scholar]
- Pedregosa F.; Varoquaux G.; Gramfort A.; Michel V.; Thirion B.; Grisel O.; Blondel M.; Prettenhofer P.; Weiss R.; Dubourg V.; Vanderplas J.; Passos A.; Cournapeau D.; Brucher M.; Perrot M.; Duchesnay É. Scikit-Learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
A ready-to-use package containing all supervised machine learning algorithms including the scripts and instructions, as well as the data sets used in this study (i.e., ACPs, NACPs, CPPs, NCPP, Cp-ACPs, NCp-ACPs), could be found in the Supporting Information. Additionally, an interactive Web-based application that allows access to unpredicted data with our trained machines is accessible at http://cbb1.ut.ac.ir/CpACpP/Index.