

Abstract

Peptide nucleic acids (PNAs) are analogues of DNA with a neutral acyclic polyamide backbone containing nucleobases attached through a t-amide link on repeating units of aminoethylglycine (aeg). They bind to complementary DNA or RNA in a sequence-specific manner to form duplexes with higher stablity than DNA:DNA and DNA:RNA hybrids. We have recently explored a new type of PNA termed bimodal PNA (bm-PNA) designed with two nucleobases per aeg repeating unit of PNA oligomer and attached at Cα or Cγ of each aeg unit through a spacer sidechain. We demonstrated that Cγ-bimodal PNA oligomers with mixed nucleobase sequences bind concurrently two different complementary DNAs, forming double duplexes, one from each t-amide and Cγ face, sharing a common PNA backbone. In such bm-PNA:DNA ternary complexes, the two duplexes show higher thermal stability than individual duplexes. Herein, we show that Cγ(S/R)-bimodal PNAs with homothymines (T8) on a t-amide face and homocytosine (C6) on a Cγ-face form a conjoined pentameric complex consisting of a triplex (bm-PNA-T8)2:dA8 and two duplexes of bm-PNA-C6:dG6. The pentameric complex [dG6:Cγ(S/R)-bm-PNA:dA8:Cγ(S/R)-bm-PNA:dG6] exhibits higher thermal stability than the individual triplex and duplex, with Cγ(S)-bm-PNA complexes being more stable than Cγ(R)-bm-PNA complexes. The conjoined duplexes of Cγ-bimodal PNAs can be used to generate novel higher-order assemblies with DNA and RNA. The Cγ(S/R)-bimodal PNAs are shown to enter MCF7 and NIH 3T3 cells and exhibit low toxicity to cells.
Introduction
Peptide nucleic acids (PNAs) are acyclic DNA analogues with an achiral backbone composed of repeating units of aminoethyl glycine (aeg) (Figure 1a) in which the nucleobases (A/T/C/G) are linked to each aeg unit via a tertiary amide group.1,2 The interbase distance in PNA matches that in DNA/RNA (Figure 1b), allowing the PNA strand to form canonical base pairing with complementary DNA/RNA strands, leading to stable duplexes in a sequence-specific manner.3,4 The stability of PNA:DNA/RNA duplexes is higher than DNA:DNA/RNA duplexes and the sequence fidelity imparts a unique property to PNA strands, which can invade DNA duplexes.5 The high avidity of PNA for complementary DNA/RNA has been employed in various applications for DNA/RNA diagnostics6 and antisense therapeutics.7 The simplicity of the PNA structure, ease of synthesis, and its remarkable properties broaden its scope for new applications through backbone modification and conjugation with ligands that recognize cells for use as putative gene regulatory agents.8,9 The chemical substitutions at Cα and Cγ on the aeg-PNA backbone do not significantly impede its hybridization with complementary DNA/RNA.8,10 The introduction of cationic alkylamino, guanidino, and polyethylene glycol substituents at Cα or Cγ of aeg-PNA improved its binding to DNA and cell penetration.10,11 One class of modifications that constrains the aeg backbone is by intraresidue cyclization to five-membered cyclopentyl, proline/pyrrolidine rings or six-membered cyclohexyl moieties, which introduces conformational pre-organization, leading to preferential hybridization with DNA or RNA.8b,12
Figure 1.

(a) aeg-PNA, (b) DNA, (c) Cα(R)-bm-triazole-PNA, (d) Cγ(S)-bm-PNA, (e) Cγ(R)-bm-PNA, (f) Cα(S)-bm-PNA:DNA double duplex, and (g) Cγ(S/R)-bm-PNA:DNA double duplex. B = T/A/G/C.
We have recently designed and introduced “bimodal PNAs” as new generation PNA analogues in which each aminoethylglycine unit carries two nucleobases with the capability to simultaneously bind two complementary DNA/RNA strands.13,14 In addition to the base linked to a t-amide sidechain as in standard PNA (Figure 1a), a second nucleobase is attached either at Cα via a triazole linker (Figure 1c)13 or conjugated at Cγ via amide linkage (Figure 1d,e).14bm-PNAs with mixed composition of base sequences (A/G/T/C) on both faces form double duplexes as demonstrated for both Cα-triazolyl13a and Cγ(S/R)-amide-linked bimodal PNA complexes.14 The double duplexes share a common bimodal PNA backbone that participates in the formation of PNA:DNA duplexes from both faces (Figure 1f,g). The bimodal PNAs possess chirality on the backbone at Cα or Cγ, and in the case of Cγ-bimodal PNAs, the S-stereomeric duplexes have higher stability than R-stereomeric duplexes.14 Previous attempts to design similar DNA structures with two nucleobases per nucleotide unit on the DNA backbone by linking a second nucleobase at C4/C2 of sugar residues or at C5 of pyrimidine base (double-headed DNA) destabilized the derived DNA:DNA duplexes and the additional nucleobase failed to base-pair with DNA to form a second duplex.15 The structural simplicity of the acyclic aeg-PNA backbone allows facile hosting of two nucleobases per repeating aeg unit unlike the other DNA analogues. The bimodal PNAs designed by us are conceptually quite different from DNA/PNA oligomers having synthetic Janus bases that recognize two strands of DNA from two faces of bases,16 and these have recently found interesting applications.17 In our bm-PNAs, the backbone itself acquires Janus character, with natural nucleobases involved in standard base pairing.
It is well known that polypyrimidine PNAs form stable PNA2:DNA triplexes with complementary polypurine DNA1,5 through base pairing by one strand of PNA via Watson–Crick hydrogen bonds and the second PNA strand via Hoogsteen hydrogen bonds with complementary DNA. In this context, we immediately extended the versatility of new bimodal Cα-triazole PNAs (Figure 1c) having oligothymine on the t-amide face that formed a (bm-triazole-PNA-Tn)2:dAn triplex, allowing the base sequence linked to Cα-triazole on each bm-PNA strand to form a duplex with DNA, resulting in the generation of triplex of duplexes.13b In the chiral trimeric double duplexes [DNA:Cγ(S/R)-bm-PNA:DNA], the S-double duplexes are more stable than R-double duplexes.14 Combining these two concepts, this paper reports on the assembly of pentameric complexes from Cγ(S/R)-amide bimodal PNAs composed of homothymine on the t-amide face and homocytosine on the Cγ-amide face upon binding to complementary DNA strands dA8 and dG6 (Figure 2). The results not only demonstrate the generality of formation of higher-order assemblies from Cα/Cγ-bimodal PNAs but also examine the S/R-stereochemical effects of the Cγ-sidechain on the stability of the higher-order PNA:DNA assembly. Further, we also report on the relative cell permeation abilities and toxicities of Cγ(S/R)-bm-PNAs in comparison to Cγ(S/R)-iso-PNA and aeg-PNA.
Figure 2.

H-bonded pentameric triplex of duplexes that constituted from homothyminyl-homocysteinyl Cγ(S/R)-bm-PNA and DNA strands. Strands 1, 3, and 5 are DNA, and strands 2 and 4 are bimodal PNA.
The target Cγ(S)-bm-PNA 1 and Cγ(S)-bm-PNA 2 (Figure 3) have oligothymine T8 linked via a t-amide sidechain as in standard PNA with each aeg unit carrying a second nucleobase cytosine (C) on the Cγ-sidechain in both S and R stereo dispositions. The control Cγ(S/R)-iso-PNAs (PNA 3 and PNA 4) are analogues of standard aeg-PNA oligomers but bear nucleobases only on the Cγ-sidechain, with no nucleobases on the t-amide sidechain. The four target PNA oligomers were synthesized to examine the relative stereochemical effects of Cγ(S/R) substituents on derived assemblies from bimodal PNAs, although it is generally known that Cγ(S)-substituted aeg-PNAs are better for hybridization with DNA.8e,11a The two-carbon spacer at Cγ for linking the base C was chosen due to easy availability of the starting material and provided a concept of proof rather than from any structural or functional considerations. The Cγ(S/R)-iso-PNAs provide independent evidence on the base pairing ability of nucleobases linked at Cγ.
Figure 3.

Structures of Cγ(S/R)-bm-PNAs (1 and 2) and Cγ(S/R)-iso-PNAs (3 and 4).
Results and Discussion
The synthesis of target bimodal PNAs (Figure 4) was done on a solid phase employing the Cγ(S)-bm-PNA-T monomer 1 and Cγ(R)-bm-PNA-T monomer 2, which were synthesized by following reported procedures.6b,14 The Cγ(S/R)-bm-PNA monomers 1 and 2 have t-amide-linked nucleobases on the aeg backbone with orthogonally protected amino groups (NH-Boc on the backbone and Fmoc on the Cγ-ethylamino sidechain). The synthesis of PNA oligomers Cγ(S)-iso-PNA 3 and Cγ(R)-iso-PNA 4 was done by using the protected monomer blocks 3 and 4 (Figure 4) synthesized as per the reported procedures.14
Figure 4.

Structures of protected monomers used in the solid-phase synthesis of PNA oligomers. 1. Cγ (S)-bm-PNA-T; 2. Cγ(R)-bm-PNA-T; 3. Cγ(S)-iso-PNA-T; 4. Cγ(R)-iso-PNA-T. eam, ethylamino; aeg, aminoethylglycyl.
Solid-Phase Synthesis of Cγ(S/R)-bimodal and iso-PNA Oligomers
The bimodal PNAs Cγ(S)-bm-PNA 1 and Cγ(R)-bm-PNA 2 with identical base sequences but different S/R stereochemistries of sidechain at Cγ (Figure 3) were synthesized on 4-methyl-benzhydryl amine (MBHA) resin using standard solid-phase synthesis protocols.18 In these PNAs, both C and N termini contain unsubstituted aeg-PNA units to avoid the steric hindrance in synthesis and DNA hybridization reactions. The MBHA resin was functionalized with l-lysine and synthesis was done from the C-terminus to the N-terminus using the protected monomers Cγ(S)-bm-PNA-T 1 and Cγ(R)-bm-PNA-T 2 (Scheme 1). First coupling with the aeg-PNA-T monomer (5) was followed by deprotection of the NHBoc group with TFA to generate a free NH2 group on resin needed for the next coupling with Cγ(S)-bm-PNA-T (1) or Cγ(R)-bm-PNA-T (2) monomers. It was followed by deprotection and subsequent coupling with the appropriate monomer (1 or 2) and repetition of the steps in every cycle. After six such cycles, the resin bound product A (Scheme 1) was obtained. This was deprotected at the N-terminus to yield B (Scheme 1), followed by final coupling with the aeg-PNA-T monomer 5 to obtain the product C [Cγ(S)-aeg-aem6-PNA-T8] (Scheme 1) that has six Fmoc-protected ethylamine sidechains at Cγ with S or R stereochemistry. This was treated with piperidine in DMF (reagent c) to achieve single-step deprotection of all Cγ-NHFmoc groups to generated free amino groups on the Cγ-aminoethyl sidechain as in product D. A global coupling reaction of Cγ-ethyl amino groups on the solid phase with cytosinyl-N1-acetic acid 6 using the coupling agents HBTU, HOBt, and DIEA gave the resin-bound bimodal Cγ(S)-bm-PNA 1 oligomer E (Scheme 1). The coupling reactions were done under microwave conditions to enhance the efficiency of reactions. A similar protocol as in Scheme 1 was also used for the synthesis of Cγ(R)-bm-PNA 2, Cγ(S)-iso-PNA 3, and Cγ(R)-iso-PNA 4 oligomers from monomers 2, 3, and 4, respectively, followed by global coupling with cytosinyl-1-acetic acid 6. All the synthesized Cγ(S/R)-bm-PNA and Cγ(S/R)-iso-PNA oligomers were cleaved from the resin, purified by RP-HPLC, and characterized by mass spectral data as shown in Table 1 and the Supporting Information (S4–S7).
Scheme 1. Common Protocol for Synthesis of Cγ(S/R)-bm-PNA Oligomers from Monomers 1 and 2 and Cγ(S/R)-iso-PNA Oligomers from Monomers 3 and 4.
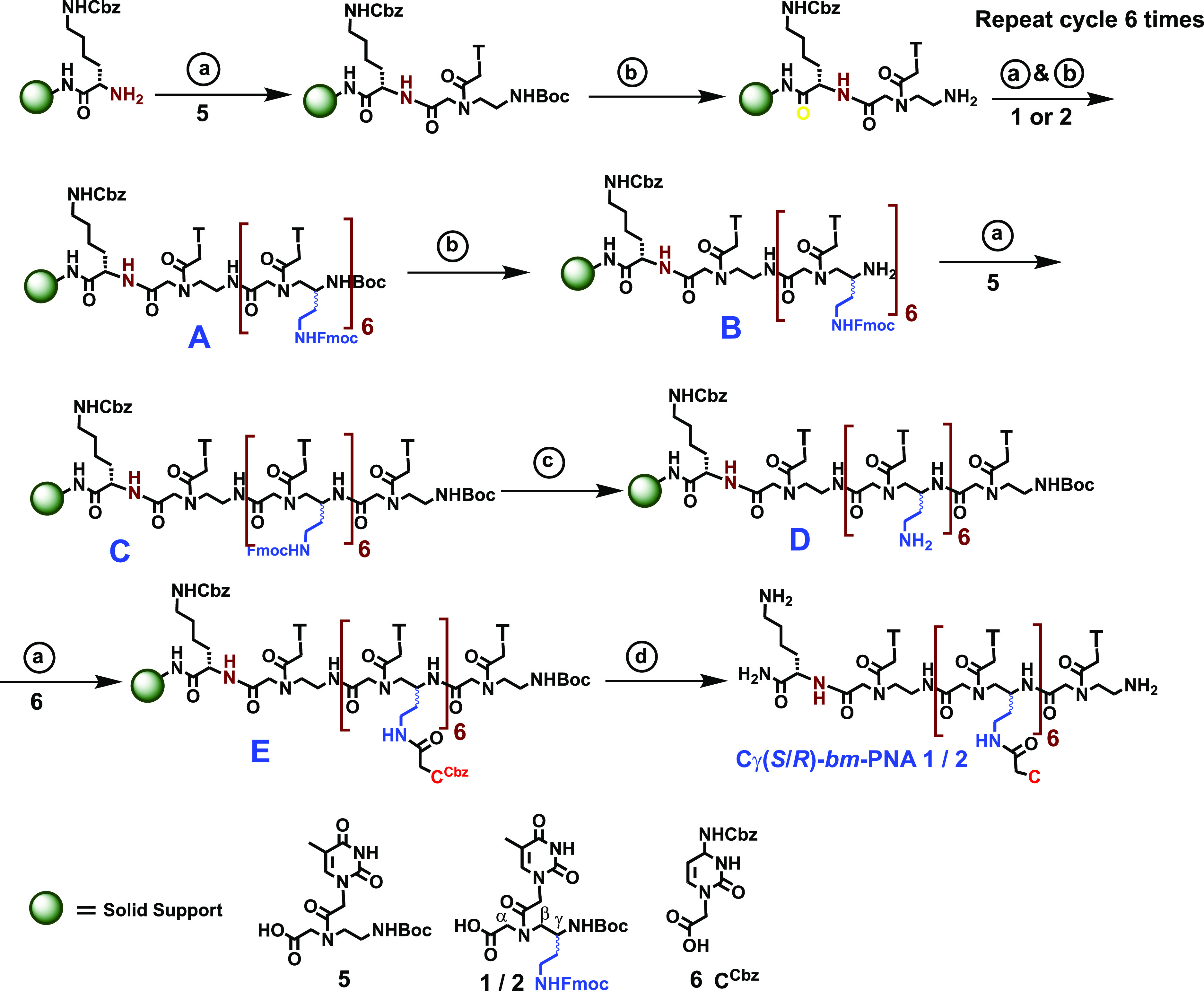
The wavy line represents the Cγ sidechain with S or R stereochemistry. Typical coupling reaction: microwave, 25 W; 5 min; rt, 6 h. Resin, 25 mg (∼0.2 mmol/g); monomers (12 mg, 3 equiv): (a) (i) HOBt (3 mg, 3 equiv), HBTU (6 mg, 3 equiv), DIPEA (6 μL), DMF. (b) (i) 50% TFA in DCM; (ii) neutralization with 10% DIPEA in DCM. (c) 20% piperidine in DMF. (d) (ii) TFA-TFMSA, thioanisole.
Table 1. HPLC and MALDI-TOF Spectral Data of the Synthesized Oligomersa.
| entry | PNA oligomers | HPLC Rt | mol. formula | calcd. mass | obs. mass |
|---|---|---|---|---|---|
| 1 | Cγ(S)-bm-PNA 1 | 13.4 | C142H187N59O45 | 3440.42 [M + H]+ | 3441.88 |
| 2 | Cγ(R)-bm-PNA 2 | 13.8 | C142H187N59O45 | 3440.42 [M + H]+ | 3441.55 |
| 3 | Cγ(S)-iso-PNA 3 | 13.6 | C102H155N43O29 | 2446.19 [M + H]+ | 2446.79 |
| 4 | Cγ(R)-iso-PNA 4 | 13.8 | C102H155N43O29 | 2446.19 [M + H]+ | 2446.74 |
| 5 | aeg-PNA-C65 | 11.6 | C66H93N33O19 | 1675.73 [M + Na]+ | 1675.15 |
| 6 | aeg-PNA-T86 | 13.9 | C94H127N35O33 | 2296.92 [M + Na]+ | 2296.39 |
| 7 | Cf-Cγ(S)-bm-PNA 7 | 16.5 | C164H201N59O51 | 3812.49 [M + H]+ | 3812.47 |
| 8 | Cf-Cγ(R)-bm-PNA 8 | 16.7 | C164H201N59O51 | 3812.49 [M + H]+ | 3812.76 |
| 9 | Cf-Cγ(S)-iso-PNA 9 | 16.8 | C124H169N43O35 | 2861.08 [M + K]+ | 2861.99 |
| 10 | Cf-Cγ(R)-iso-PNA 10 | 16.9 | C124H169N43O35 | 2844.26 [M + Na]+ | 2844.97 |
| 11 | Cf-aeg-PNA-C611 | 16.3 | C87H103N33O25 | 2009.78 [M + H]+ | 2009.80 |
| 12 | Cf-aeg-PNA-T812 | 17.0 | C115H137N35O39 | 2670.94 [M + K]+ | 2670.87 |
Rt, retention time (min) on the C18 column. For HPLC conditions, see Experimental Section. Observed mass by MALDI-TOF.
Thermal Stability of Cγ(S/R)-bm and iso(S/R)-PNA:DNA Duplexes
The bimodal Cγ(S)-bm-PNA 1, Cγ(S)-bm-PNA 2, Cγ(S)-iso-PNA 3, and Cγ(R)-iso-PNA 4 oligomers were individually hybridized with DNA 1 (dG6) or DNA 2 (dA8) that are complementary to Cγ-amide (C6) and t-amide side (T8) base sequences, respectively. The Cγ(S)-iso-PNAs 3 and 4 cannot form t-amide complexes with DNA 2 as they lack nucleobases at the t-amide side and can form only duplexes from the Cγ-side. The thermal stability of various bm-PNA:DNA and iso-PNA:DNA complexes was obtained by recording UV absorbance at 260 nm as a function of temperature.19 The melting temperature (Tm) of the PNA:DNA hybrids corresponds to the midpoint of the corresponding UV–T plots (Figure 5) and has been confirmed from the peak of the first derivative curve since it provides a more accurate estimation of the Tm value.19d
Figure 5.

Structure and UV–T plots of Cγ-duplexes of Cγ(S/R)-iso/bm-PNA with DNA 1 (dG6). (A) Cγ(S)-iso-PNA 3:dG6; (B) Cγ(R)-iso-PNA 4:dG6; (C) Cγ(S)-bm-PNA 1:dG6; (D) Cγ(R)-bm-PNA 2:dG6 . Red curve, melting curve; blue dotted curve, first derivative plot. Numbers in figures indicate Tm’s. Sodium cacodylate (10 mM), NaCl (10 mM), pH 7.2.
Duplexes from Cγ(S/R)-iso/bm-PNA:DNA dG6
The single sigmoidal transitions seen for Cγ(S)-iso-PNA:dG6 (Figure 5A) and Cγ(R)-iso-PNA:dG6 duplexes (Figure 5B) clearly indicate that the new isomeric PNA structures carrying homocytosine only on the Cγ-sidechain can form a perfect duplex with complementary DNA 1 (dG6). The Cγ(S)-iso-PNA 3:dG6 duplex (5A) has thermal stability (Tm = 44.3 °C), which is higher than that of the Cγ(R)-iso-PNA 4:dG6 duplex (5B) (Tm = 38.7 °C) with ΔTm(S-R) of +5.6 °C. The designed Cγ(S/R)-bm-PNAs also formed corresponding duplexes Cγ(S)-bm-PNA 1:dG6 (5C) and Cγ(R)-bm-PNA 2:dG6 (5D) with Tm’s of 68.2 and 48.3 °C, respectively (Figure 5C,D). We have earlier shown that Cα-bm-PNA-Cn forms only a duplex with dGn at pH 7.0 through the Jobs plot experiment and is supported by its characteristic CD spectra. Thus, the present data suggested that (i) the nucleobase sequence on the Cγ-sidechain (iso-PNA) can form a duplex with DNA, similar to t-amide-linked nucleobases in standard aeg-PNA and (ii) the presence of unpaired nucleobases on the t-amide side (bm-PNA) significantly enhanced the stability of Cγ-amide duplexes: the Cγ(S)-bm-PNA 1:DNA 1 duplex (5C) stabilized by +23.9 °C compared to Cγ(S)-iso-PNA 3:dG6 duplex (5A) and Cγ(R)-bm-PNA 2:dG6 duplex (5D) is 9.6 °C more stable than Cγ(R)-iso-PNA 4:dG6 duplex (5B), and (iii) the Cγ(S)-duplexes from both iso-PNA and bm-PNA are more stable than Cγ(R)-duplexes.
Triplexes from Cγ(S/R)-bm-PNA:DNA
The homothyminyl Cγ(S/R)-bm-PNAs complexed with dA8 (DNA 2) to form bm-PNA2:dA8 triplexes (Figure 6A,B) from the T8 on the t-amide side similar to that of standard PNA2:DNA triplexes.1,4,19e A single transition was seen with a Tm of 58.8 °C for the Cγ(S)-bm-PNA 1 triplex (6A), which is higher than the Tm of 43.6 °C for the Cγ(R)-bm-PNA 2 triplex (6B). Again, the S-triplex was significantly more stable than the R-triplex by +15.2 °C. The results with triplexes are consistent with literature precedence on duplexes that Cγ(S) substitution stabilizes PNA:DNA duplexes better than Cγ(R)-substitution.11
Figure 6.

Structure and UV–T plots of Cγ-triplexes of Cγ(S/R)-iso/bm-PNA with DNA 2 (dA8). (A) [Cγ(S)-bm-PNA-T 3]2:dG6 and (B) [Cγ(R)-bm-PNA-T 4]2:dG6. Red curve, melting curve; blue dotted curve, first derivative plot. Numbers in figures indicate Tm’s. Sodium cacodylate (10 mM), NaCl (10 mM), pH 7.2.
Triplex of Duplexes from Cγ(S/R)-bm-PNA:DNA Complexes
The Cγ(S/R)-bm-PNAs were individually hybridized with two complementary DNAs by stoichiometric additions of DNA 1 (dG6, complementary to the Cγ-amide side) and DNA 2 (dA8, complementary to the t-amide side). Figure 7A,B shows the possible composite complexes of triplex of duplex from Cγ(S/R)-bm-PNA:DNA 1:DNA 2 along with their melting profiles. In contrast to single sigmoidal melting curves noticed for individual duplexes (Figure 5) and triplexes (Figure 6), the pentameric complexes exhibited double sigmoidal curves, indicating the presence of two distinct melting transitions. The Cγ(S)-pentameric complex dG6:Cγ(S)-bm-PNA 1:dA8:Cγ(S)-bm-PNA 1:dG6 (7A) exhibited two Tm’s of 57.4 and 77.9 °C (Figure 7A), which are higher than the corresponding single Tm’s of the individual duplex (Figure 5A) and triplex (Figure 6A). Similarly, the Cγ(R)-pentenary complex dG6:Cγ(R)-bm-PNA 2:dA8:Cγ(R)-bm-PNA 2:dG6 (7B) exhibited two discernible transitions with distinct Tm’s of 42.1 and 63.8 °C (Figure 7B) that are higher than the corresponding Tm’s of the individual duplex (Figure 5B) and triplex (Figure 6B). Once again, the Tm’s of the Cγ(S)-pentameric complex were higher than those of the Cγ(R)-pentameric complex. The double sigmoidal pattern of the UV–T plot is consistent with similar composite complexes noticed earlier.13,14 This indicated the melting process to be biphasic (three-state), originating from a sequential melting process as in DNA triplexes19a rather than a simultaneous disassociation of both PNA strands from DNA as in PNA2:DNA melting.19b,19e The latter possibility should lead to only a single transition representing true melting of the pentenary complex. Since the two Tm’s in each Cγ(S/R) bimodal PNA:DNA complex are higher than individual duplexes/triplexes, the melting processes of the duplex and triplex are coupled to each other, mutually enhancing their stability.
Figure 7.

Structures of pentameric triplex of duplexes and UV–T plots of Cγ(S/R)-bm-PNA:DNA 1:DNA 2 complexes. (A) dG6:Cγ(S)-bm-PNA 1:dA8:Cγ(S)-bm-PNA 1:dG6. (B) dG6:Cγ(R)-bm-PNA 2:dA8:Cγ(R)-bm-PNA 2:dG6. Buffer: sodium cacodylate (10 mM), NaCl (10 mM), pH 7.2.
Mismatch Cγ(S/R)-bm-PNA:DNA Duplexes
The sequence fidelity in the complementation of base pairing from the Cγ-sidechain in bm-PNA:DNA duplexes was examined by determining the Tm’s of duplexes with mismatched DNA. Cγ(S)-bm-PNA 1 and Cγ(R)-bm-PNA 2 were individually hybridized with mismatched DNA 1m (5′-GGTGGG-3′) and DNA 2m (5′-AAAACAAA-3′) that carry a single C:T base mismatch on the Cγ-side and T:C t-amide side sequences (Supporting Information, S8–S10). The comparative UV–Tm plots for perfect and mismatched complexes are shown in Figure 8A, and it is seen that single base mismatches destabilized all complexes (Figure 8B). The Cγ-side mismatch duplexes in iso-PNAs [Cγ(S/R)-iso-PNA:DNA 1m] were destabilized by 6.1 and 4.4 °C, respectively, compared to respective perfect duplexes (S, 5A) and (R, 5B). The destabilization was much larger in the bm-PNA duplexes [Cγ(S/R)-bm-PNA:DNA 1m], 22.1 and 8.1 °C, compared to respective perfect duplexes (S, 5C) and (R, 5D) (Figure 8B). The corresponding t-amide mismatch triplexes with DNA 2m showed lower stability for [Cγ(S)-bm-PNA 1]2:DNA 2m by 19.6 °C compared to the Tm of perfect triplex 6A and 4.5 °C for [Cγ(R)-bm-PNA 2]2:DNA 2m compared to the Tm of perfect triplex 6B. In the pentameric mismatched triplex of duplexes, the two transitions collapsed into single transition with Tm’s of 58.5 °C for Cγ(S)-bm-PNA 1 (S, 7A) and 38.4 °C for Cγ(R)-bm-PNA 2 (R, 7B) complexes, leading to lowering of Tm by 19.4 and 25.4 °C, respectively (Figure 8). Thus, in mismatched bimodal complexes, both DNA strands clearly dissociate in a single step, similar to that in the PNA2:DNA triplex.19b,19e It should be pointed out that compared to a single mismatch in an individual duplex, the triplexes have two mismatches and the triplex of duplexes have four mismatch pairs (Supporting Information, S8–S10). The degree of destabilization observed in mismatched duplexes of S-bm-PNA was similar to destabilization per base mismatch seen in unmodified PNA:DNA duplexes.1,19 The mismatched duplex stability results substantiate the co-existence of duplex/triplex sharing a common bimodal PNA backbone, mutually conferring higher stability on each other without loss of any sequence specificity.
Figure 8.

Comparative (A) Tm’s of duplexes, triplexes, and triplex of duplexes and (B) ΔTm of mismatch complexes. Labels on the x axis correspond to complexes shown in Figures 5 and 7 and this figure. The numbers in panel (B) indicate the destabilization (ΔTm) of mismatched duplexes in °C.
CD Spectra of Cγ-bm-PNA:DNA Duplexes
The formation of duplexes and triplexes from Cγ(S/R)-bm-PNA and complementary DNA was supported by the CD spectra (Figure 9) that resembled the characteristic profiles of PNA:DNA duplexes,20 with minor changes in relative intensities of bands. The CD spectra of triplex [Cγ(S)-bm-PNA 1]2:dA8 showed a major positive band at 220 nm with a shoulder and a lower intensity double hump pattern with positive bands at 265 and 282 nm (Figures 6A and 9). This is characteristic of the (PNA-T8)2:poly dAn triplex composed from right-handed helices.20a The CD spectra of the duplex Cγ(S)-bm-PNA 1:dG6 showed a positive band in the region around 260 nm, accompanied by another positive band around 210 nm (Figures 5C and 9). The corresponding duplex and triplex from Cγ(R)-bm-PNA 2 exhibited a similar CD profile as shown for 5D and 6B for the duplex and triplex, respectively.
Figure 9.

CD spectra of Cγ(S/R)-bm-PNA:DNA complexes. 5C duplex: dCγ(S)-bm-PNA 1:dG6; 6A triplex: [Cγ(S)-bm-PNA 1]2:dA8; 7A triplex of duplex: dG6:Cγ(S)-bm-PNA 1:dA8:Cγ(S)-bm-PNA 1:dG6. 5D, 6B, and 7B are the corresponding complexes of Cγ(R)-bm-PNA 1. Structures of complexes are as shown in Figures 5–7. Sodium cacodylate (10 mM), NaCl (10 mM), pH 7.2.
The triplex of duplexes from Cγ(S/R)-bm-PNA (Figures 7A,B and 9) showed prominent positive bands at 260–262 nm and 217–220 nm. The Cγ(S)-bm-PNA 1 complex 7A exhibited a relatively higher intensity band at 260 nm, perhaps due to a better stacking of bases compared to that in the Cγ(R)-bm-PNA 2 complex 7B. The handedness of two stereomeric Cγ(S/R)-bm-PNA:DNA duplexes remain the same as both of them have l-lysine at the C-terminus and the CD spectra are dominated by DNA contribution with well-established right-handedness of DNA:PNA duplexes.20 The CD profile of triplex of duplexes 7A and 7B shows the composite spectra of the t-amide triplex and Cγ-duplex. The fact that these complexes also showed two transitions with enhanced Tm’s of both the triplex and duplex components suggested CD to be an inherent composite of the triplex of duplex and not a result of the simple additive spectra of the isolated triplex and duplex.
Order of Formation of Triplexes and Duplexes in Forming a Triplex of Duplex
The observed conformation of triplex of duplexes can be attained by two paths depending on the order of assembly of the duplex and triplex (Figure 10A). In path I, Cγ(S)-bm-PNA 1 was mixed with a stoichiometric amount of the complementary dA8 (Figure 10, step a) and kept for equilibration for 10 min to obtain the [Cγ(S)-bm-PNA 1]2:dA8 triplex (6A). This was followed by stoichiometric addition of dG6 (Figure 10A, step b) to result in the triplex of duplex dG6:Cγ(S)-bm-PNA 1:dA8:Cγ(S)-bm-PNA 1:dG6. The formation of products after each of steps a and b was followed by recording of the CD spectra. The formation of the PNA2:DNA triplex [Cγ(S)-bm-PNA 2]2:dA8 after step a was supported by the characteristic CD profile (Figure 10B, curve a) identical to that of triplex 6A in Figure 9. The CD spectra of the product obtained after step b (Figure 10B, curve b) were the same as those of the triplex of duplex 7A in Figure 9.
Figure 10.

(A) Schematic representation of stepwise addition of DNA 1 and DNA 2 to Cγ(S/R)-bm-PNA by two paths. Path I: Triplex formation after step a followed by its duplex formation with dG6. Path II: Duplex formation with dG6 in step c followed by its triplexation with dA8 to yield a triplex of duplex. (B, C) CD spectra of products; curve a: triplex 6A after step a; curve b, triplex of duplex 7A after step b; curve c, duplex 5C after step c; curve d, triplex of duplex 7A after step d. (D) Overlap of CD spectra of compounds in paths I (red) and II (blue). Sodium cacodylate (10 mM), NaCl (10 mM), pH 7.2.
The experiment was repeated (path II) by reversing the order of DNA additions to Cγ(S)-bm-PNA 1 by first adding of dG6 (Figure 10A, step c) to first yield the duplex Cγ(S)-bm-PNA 1:dG6 (5C) followed by addition of dA8 (Figure 10, step d) to yield the same final complex dG6:Cγ(S)-bm-PNA 1:dA8:Cγ(S)-bm-PNA 1:dG6. It is seen that the CD spectra of the initial product after step c was characteristic of the duplex (Figure 10B, curve c), same as those of 5C (Figure 9) and CD of the product dG6:Cγ(S)-bm-PNA 1:dA8:Cγ(S)-bm-PNA 1:dG6 (Figure 10B, curve d) after step d was that of the triplex of duplex (7A). The final products from both experiments were identical in terms of spectral bands and intensities (Figure 10C). A similar experiment with Cγ(R)-bm-PNA 2 also provided identical CD spectra from both paths (Figure S5). This experiment indicated that the order in which the duplex and triplex are formed on either face of Cγ-bm-PNA does not affect the conformational state of final triplex of duplexes.
Cell Penetration and Cytotoxicity Studies
To explore the efficiency of cell penetration, Cγ-bm-PNA and Cγ-iso-PNA were tagged with 5(6)-carboxyfluorescein dye at the N-terminus (Supporting Information, Schemes S3 and S4) to obtain fluorescent Cf-Cγ(S)-bm-PNA 7, Cf-Cγ(R)-bm-PNA 8, Cf-Cγ(S)-iso-PNA 9, and Cf-Cγ(R)-iso-PNA 10. Cellular uptake properties of fluorescent Cf-Cγ(S/R)-bm-PNAs and Cf-Cγ(S/R)-iso-PNAs along with control Cf-aeg-PNA-C611 and Cf-aeg-PNA-T812 were examined individually in MCF7 and NIH 3T3 cell lines by confocal microscopy. Figure 11 shows the confocal laser scanning images of live cells incubated separately with fluorescently labeled PNAs (4 μM) for 24 h. All panels in rows of Figure 11A,C show DAPI-stained images of treated MCF7 and NIH 3T3 cells, respectively, while the rows of Figure 11B,D show MCF7 and NIH 3T3 cells respectively treated with Cf-PNAs as shown on top of each of the columns. It is generally observed that Cγ-bm-PNA, iso-PNA, and aeg-PNA enter the cell and are localized around the nucleus irrespective of the type of modification and cell line used. Further, NIH 3T3 cells seem to have better uptake of all PNAs in comparison to that in MCF7 cells, and the amount of uptake of PNAs depends on the type of modification. Unsubstituted aeg-PNAs showed better uptake than bm-PNA and iso-PNAs.
Figure 11.

Cell uptake confocal fluorescence images of bm-PNA, iso-PNA, and aeg-PNA in MCF7 cells (A) DAPI-stained and (B) treated with Cf-PNA and NIH 3T3 cells (C) DAPI-stained and (D) treated with Cf-PNAs. The individual PNAs are shown on top of each column of panels.
Flow Cytometry Studies
The cell penetrating ability of bm-PNA, iso-PNA, and aeg-PNA oligomers were quantitatively estimated by a fluorescence-activated cell sorter (FACS) as shown in Figure 12. MCF7 and NIH 3T3 cells were cultured as mentioned in the Experimental Section. The cells were treated with 5(6)-carboxyfluorescein-functionalized bm-PNAs [Cf-Cγ(S)-bm-PNA 7 and Cf-Cγ(R)-bm-PNA 8], iso-PNAs [Cf-Cγ(S)-iso-PNA 9, and Cf-Cγ(R)-iso-PNA 10], and aeg-PNAs (Cf-aeg-PNA-C611 and Cf-aeg-PNA-T812) at 4 μM concentrations of each for 24 h. The results show that Cf-Cγ(S)-bm-PNA 7 showed higher uptake than Cf-Cγ(R)-bm-PNA 8 in both cell lines and a similar trend was observed in the case of iso-PNAs: Cf-Cγ(S)-iso-PNA 9 is better than Cf-Cγ(R)-iso-PNA 10. The Cf-aeg-PNA-T812 showed maximum uptake among all the PNAs. The results are in good agreement with that seen by confocal microscopy.
Figure 12.

Percentage cell uptake of aeg-PNA, Cγ(S/R)-iso-PNAs, and Cγ(S/R)-bm-PNAs measured from FACS in (A) MCF7 and (B) NIH 3T3 cells.
Cell Cytotoxicity Assay
The cytotoxicity of bimodal PNAs [Cγ(S)-bm-PNA 1 and Cγ(R)-bm-PNA 2], iso-PNAs (Cγ(S)-iso-PNA 3 and Cγ(R)-iso-PNA 4), and aeg-PNA (aeg-PNA-C65 and aeg-PNA-T86) was assessed by standard 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Figure 13). Both MCF7 and NIH 3T3 cells were incubated with individual PNAs at the concentration of 4 μM for 24 h, prior to treatment with MTT. The percent cell viability results indicated that in MCF7 cells, all PNAs were nontoxic to the cells (>100%), except for Cγ(S)-iso-PNA 3 and aeg-PNA-T86 (<75% viability, Figure 13A). In NIH 3T3 cells, all PNAs are nontoxic to the cell (>100% viability), except Cγ(S)-iso-PNA 3 and Cγ(R)-iso-PNA 4 (<75% viability, Figure 13B). It is not unusual to see cell viability >100% in MTT assays as it is based on the cell population and the UV absorption of formazan, which is a product of enzymatic reaction and is cell-dependent. The compounds may also cause cells (including dead cells) to metabolize MTT more than the control and may induce cell proliferation, leading to >100% viability. Thus, although the uptake of bm-PNAs was not as good as the iso- and aeg-PNAs, they were less toxic than the other PNAs.
Figure 13.

Cell viability measured using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay for (A) MCF7 cells and (B) NIH 3T3 cells after treatment with 4 μM bimodal PNAs (Cγ(S)-bm-PNA 1 and Cγ(R)-bm-PNA 2), iso-PNA (Cγ(S)-iso-PNA 3 and Cγ(R)-iso-PNA 4), and aeg-PNA (aeg-PNA-C65 and aeg-PNA-T86) for 12 h. The data shown are the average of three measurements. The error bars represent standard deviations. Control experiments do not have any added PNAs.
Discussion
The UV-melting data on (S/R)-iso-PNA:DNA hybrids suggested the formation of stable duplexes from new analogue iso-PNAs that lack the t-amide-linked base as in standard PNA. The designed bimodal Cγ(S/R)-bm-PNAs bind DNAs complementarily from each side to yield the corresponding binary Cγ-amide duplexes (bm-PNA-C6:dG6) and t-amide triplexes (bm-PNA-T8)2:dA8. The formation of bm-PNA-C5:dG6 duplexes at pH 7.0 rather than the possible C+G:C triplexes (that needs acidic pH) is supported by our previously reported Jobs plot showing 1:1 stoichiometry14 and the CD profiles characteristic of duplexes (Figure 9). The homomeric C sequence linked to Cγ in bm-PNAs is different compared to dCn since it is attached via a flexible spacer chain rather than by a rigid glycosidic bond as in dCn. The formation of dCn:dGn duplexes at pH 7.0 also has literature precedence.21
In the presence of both complementary DNAs dA8 and dG6, Cγ(S/R)-bm-PNAs hybridize on both sides of the backbone to generate triplex of duplexes (dG6:C6-bm-PNA T8:dA8:T8-bm-PNA-C5:dG6). This pentameric complex that constituted from two strands of Cγ-bm-PNA and three strands of DNA (2xdG6 and 1xdA8) contains two duplexes and one triplex with a shared backbone. The complex shows biphasic double transition with both Tm’s higher compared to that of independent duplex T8-bm-PNA-C6:dG6 and the triplex (C5-bm-PNA-T8)2:dA8. Thus, Cγ(S/R)-bm/iso-PNA pentameric assemblies are significantly more stable than the corresponding isolated Cγ(S/R)-bm/iso-PNA duplexes and triplexes. The UV-melting profiles of Cγ(S/R)-bm-PNA duplexes of triplex observed in this work are consistent with that we recently reported for Cα(R)-bm-triazole-PNA13 and Cγ(S/R)-bm-amide-PNA.14 The Cγ(S)-stereomeric complexes (duplex, triplex, and triplex of duplex) are more stable than Cγ(R)-stereomeric complexes, consistent with the previous literature.11 The S-configuration at Cγ is derived from l-amino acids and it is well established that this will favor right-handedness in the derived PNA, which is more conducive to bind with right-handed DNA to form a duplex. This preferred handedness is due to helical induction occurring in PNA from the C-terminal direction to the N-terminal direction dictated by the chiral l-amino acid and is sterically driven. The d-amino acid-derived PNA has R-configuration at Cγ and the induced left-handed helix forms a less stable duplex with right-handed DNA (entropic destabilization), although the overall duplex is right-handed.11 The CD spectra show characteristic patterns of a PNA:DNA duplex and PNA2:DNA triplex for the bimodal PNA complexes, and the triplex of duplex exhibits characteristic composite CD, distinctly different from the constituent duplex and triplex. The order of assembly to yield the triplex of duplexes (first triplex and then duplex or vice versa) does not matter, and the assembly always leads to the same final complex. This validates the general design principles of bimodal PNAs to anchor a second nucleobase on the aeg-PNA backbone to concurrently bind and create two complementary DNA/RNA strands, forming double duplexes or triplex of duplexes that are structurally coupled on a shared PNA backbone. Since the duplexes show sequence specificity as indicated from destabilized mismatch complexes, the base pairing of nucleobases on Cγ-sidechains with complementary DNA is likely to be the stronger canonical Watson–Crick (WC) type, although Hoogsteen H-bonding cannot be ruled out from present results. It is well known that mechanistic variations in base pairing are possible in PNA:DNA interactions, depending on the choice of nucleobases.22
The two-step melting of a pentameric complex with higher Tm’s suggests a sequential melting mechanism as in DNA triplexes19a rather than a single-step simultaneous dissociation as in PNA2:DNA triplexes.19b,19c The complete dissociation of a ternary complex is perhaps preceded by a pre-melting transition involving a partial unstacking of bases and backbone conformational change as proposed in our earlier report.14 The model also suggests that when two duplexes or duplex/triplex coexist in a complex with a shared backbone, they exert mutual stability on each other. The synergistic stabilizations observed in co-existing duplex/triplex complexes perhaps arise from a favorable conformational pre-organization provided by the first duplex formation to facilitate association of the second complementary strand and need further investigations.
Conclusions
Cγ-bimodal PNAs that have a second nucleobase linked at Cγ in addition to that on the t-amide sidechain in each aeg unit provide opportunity for concurrent binding of two complementary DNA/RNA sequences to from conjoined duplexes (double duplex) or a triplex of duplex with sharing of PNA backbone.13,14 Cγ-iso-PNA oligomers are analogues of standard aeg-PNA with bases present only on the Cγ-sidechain and devoid of t-amide-linked bases. The orthogonally protected stereomeric monomers (1 and 2) along with cytosinyl N1-acetic acid 6 were used for solid-phase synthesis of bimodal Cγ(S/R)-bm-PNA having T8 on the t-amide side and C5 on the Cγ-sidechain. It is demonstrated that homopyrimidinyl CγS/R-bm-PNA-T8 can form a PNA2:DNA triplex and seeds higher-order assembly by formation of duplexes on either side of the triplex to form a triplex of double duplex. The Cγ(S/R)-bimodal PNAs are shown to enter MCF7 and NIH 3T3 cells to localize in the cytoplasm around the nucleus with less efficiency than aeg-PNAs and are nontoxic.
The Cα/Cγ-bm-PNAs as designed by us open up avenues for studying entirely new types of PNA:DNA complexes. Based on the choice of the sequences on either side (polypurines/polypyrimidines/Gn/Cn), the Cα/Cγ-bimodal PNAs have potential to generate fused duplexes, triplexes, tetraplexes, and even extended two-dimensional assemblies. The enhanced stability and augmented molecular recognition properties of the bimodal PNAs can be harnessed to rationally design complex supramolecular PNA nanoassemblies with defined functions tailored for various applications in biotechnology as well as materials science.23 Among the probable biological applications, bimodal PNAs can be used to specifically target two genes or microRNA structures simultaneously either to probe biological processes or to modulate them for therapeutics.24 They also have the potential to rationally engineer stabilization of hairpin structures in DNA/RNA in a “paper-clip” fashion.25 Further, bimodal PNAs can be used like DNA stapler strands in a DNA origami to expand the repertoire of programmable folding in nucleic acid nanotechnology.26
Experimental Section
The chemicals used were of laboratory or analytical grade. All the solvents used were distilled or dried to carry out different reactions. The Cγ(S/R)-bm-PNA-T monomers (1 and 2) and Cγ(S/R)-iso-PNA monomers (3 and 4) were synthesized as per earlier reported procedures.14 The UV–Visible spectrophotometric studies were done on a PerkinElmer Lambda 45 double beam UV–Vis spectrophotometer. PNA oligomers synthesized by solid-phase protocols on MBHA resin were purified using a reversed-phase HPLC system equipped with a semipreparative BEH130 C18 (10 × 250 mm) column. Their identity was established by MALDI-TOF/TOF with 2,5-dihydroxybenzoic acid or α-cyano-4-hydroxycinnamic acid as a matrix. The DNA oligonucleotides were commercially obtained from Integrated DNA Technologies (IDT). Salts and reagents used in buffer preparation such as NaCl, sodium cacodylate, etc., were obtained from Sigma-Aldrich. The pH of the buffer solutions was adjusted using HCl (Sigma-Aldrich). The solid-phase synthesis was carried out using established Boc and Fmoc protocols.27,28
Solid-Phase Synthesis of bm-PNA, iso-PNA, and aeg-PNA Oligomers and Their Fluorescent Derivatives
The Cγ-iso/bm-PNA oligomers were synthesized on MBHA resin (50 mg) using both Boc and Fmoc protocols.18,27 The solid-phase synthesis was carried out in a reactor with a sintered glass bottom and the loading value of the resin was 0.20 mmol/g. The deprotection of the N-t-Boc group from the resin-bound lysine with 50% TFA in DCM (3 × 15 min) was followed by washing with DCM and DMF (3 × 10 mL) to give a TFA salt of amine, which was neutralized using 10% N,N-diisopropylethylamine (DIPEA) in DCM (3 × 10 min) to liberate free amine. After washing with DCM and DMF (3 × 10 mL), the free amine on the main chain was coupled with appropriate individual monomers 1–4 using (i) HOBt (3 equiv, 5.0 mg), HBTU (3 equiv, 11.5 mg), and DIPEA (3 equiv, 6.0 μL) in DMF. The deprotection, washing, and coupling cycles were repeated until the assembly of individual Cγ(S/R)-iso/bm-PNA oligomers was completed (Supporting Information, Schemes S1 and S2). At the end of the assembly, global deprotection of all sidechain NHFmoc groups was done using 20% piperidine in DMF. The liberated sidechain amine groups were coupled with cytosine-1-acetic acid (18 equiv, 55 mg) using HOBt (18 equiv, 30 mg), HBTU (18 equiv, 70 mg), and DIPEA (18 equiv, 35 μL ) in DMF (500 μL) under microwave conditions to obtain the resin-linked PNA Cγ-iso/bm-PNA oligomers (Table 1, Cγ(S/R)-iso/bm-PNA 1–PNA 4). The unsubstituted aeg-PNA-C6 (PNA 5) and aeg-PNA-T8 (PNA 6) were synthesized by Boc chemistry following reported protocols.28
The corresponding fluorescent PNA oligomers Cf-Cγ(S/R)-bm-PNA and Cf-Cγ(S/R)-iso-PNA (Table 1) required for cell permeation studies were synthesized (Supporting Information, Scheme S3 and S4) after the final coupling of bm-PNA and iso-PNA oligomers at the N-terminus. The deprotection of terminal NHBoc on iso/bm-PNAs linked to resin was done using 50% TFA in DCM (3 × 15 min), followed by washing with DCM and DMF (3 × 10 mL). This gave TFA salt of N-terminus amine, which was neutralized using 10% N,N-diisopropylethylamine (DIPEA) in DCM (3 × 10 min) to liberate free amine. This was coupled with 5/6-carboxy fluorescein (3 equiv, 11.3 mg) using HOBt (3 equiv, 5.0 mg), HBTU (3 equiv, 11.5 mg), and DIPEA (3 equiv, 6.0 μL) in DMF to obtain Cf-Cγ-iso/bm-PNAs (Table 1, PNA 7–PNA 10). The unmodified control Cf-aeg-PNA-C6 and Cf-aeg-PNA-T8 (Table 1, PNA 11 and PNA 12) for cell permeation studies were synthesized from resin-linked aeg-PNA (PNA 6) using similar reactions.
Cleavage of PNA Oligomers from Solid Support
The MBHA resin (10 mg) after assembly of bm-PNA oligomers was stirred with thioanisole (20 μL) and 1,2-ethanedithiol (8 μL) in an ice bath for 10 min. Cold TFA (200 μL) was added. TFMSA (16 μL) was added slowly with stirring and the reaction mixture was stirred for another 1.5 to 2 h at room temperature. The resin was removed by filtration under reduced pressure and washed twice with TFA, and the filtrate was evaporated on a rotary evaporator at ambient temperature. The concentrated filtrate was transferred to a microfuge tube and the PNAs were precipitated with cold diethyl ether. The crude PNAs were isolated by centrifugation and the precipitate was dissolved in MilliQ water, filtered, and purified by HPLC.
The purification of PNAs was carried out on an Agilent HPLC system with a semipreparative BEH130 C18 (10 × 250 mm) Phenomenex column using water and acetonitrile as solvents with compositions A [0.1% TFA in CH3CN:H2O (5:95)] and B [0.1% TFA in CH3CN:H2O (1:1)]. The gradient for elution was 100% A to 100% B in 20 min, with a flow rate of 2 mL/min. The HPLC elutions were monitored at 220 and 254 nm wavelengths.
UV–Temperature Absorbance Measurements
UV-melting experiments were carried out on a Varian Cary 300 UV spectrophotometer equipped with a Peltier heating system. The samples were prepared in sodium cacodylate buffer (10 mM) and NaCl (10 mM) at pH 7.2. Calculated amounts of respective oligonucleotides in the stoichiometric ratio (1:1, duplex) were mixed to achieve a final concentration of 2 μM for each strand. The samples were annealed by heating the mixture at 90 °C for 10 min, followed by slow cooling to room temperature over 8–10 h and then refrigeration for 24 h. The samples (500 μL) were transferred to a quartz cell and equilibrated at room temperature for 5 min. The absorbance at 260 nm was recorded in steps from 20 to 90 °C with a temperature increment of 0.5 °C. Each melting experiment was repeated at least thrice. The normalized absorbance at 260 nm plotted as a function of the temperature was fitted by the Boltzmann function sigmoidal curve for one-face binding and the biphasic dose–response curve for two-face binding, with an R2 value in the range of 0.96 to 0.99. The Tm was determined from the first derivative of normalized absorbance with respect to temperature and was accurate to ±1.0 °C. The data were processed using OriginPro 8.5. The concentrations of all oligonucleotides were calculated on the basis of the absorbance at 260 nm from the molar extinction coefficients of the corresponding nucleobases: T = 8.8 cm2/μmol, C = 6.6 cm2/μmol, G = 11.7 cm2/μmol, and A = 13.7 cm2/μmol as per the literature.18
Circular Dichroic Spectra
CD spectra were recorded on a JASCO J-815 spectropolarimeter. The calculated amounts of bm-PNA oligomers and the complementary DNA were mixed together in the stoichiometric ratio (1:1 for the duplex) in sodium cacodylate buffer (10 mM) containing NaCl (10 mM) at pH 7.2 to achieve a final strand concentration of 10 μM for each strand. The samples were annealed by heating at 90 °C for 10 min, followed by slow cooling to room temperature during a period of 8–10 h and refrigeration at 4 °C for 12 h. The CD spectra of bm-PNA:DNA complexes were recorded with samples in a 2 mm cell at a temperature of 10 °C, scanning from 300 to 200 nm using a resolution of 0.1 nm, bandwidth of 1 nm, sensitivity of 2 m deg., response of 2 s, and a scan speed of 50 nm/min. The final spectra are shown as addition of three scans.
Cellular Uptake Studies
MCF7 and NIH 3T3 (2 × 104 cells per well) cells were individually seeded on eight-well chamber slides and allowed to grow at 37 °C in a 5% CO2 atmosphere in DMEM medium containing 10% fetal bovine serum and 0.1% streptomycin for 24 h. The fluorescent PNAs bm-PNAs (Cf-Cγ(S)-bm-PNA 7 and Cf-Cγ(R)-bm-PNA 8), iso-PNAs (Cf-Cγ(S)-iso-PNA 9 and Cf-Cγ(R)-iso-PNA 10), and aeg-PNAs (Cf-aeg-PNA-C611 and Cf-aeg-PNA-T812) (4 μM) were added for 4 and 24 h, respectively. Later, the cells were washed three times with PBS buffer, and nuclei were stained with DAPI reagent (2 μg/mL). Fluorescence measurements were performed using excitation with an argon laser (I = 405 and 488 nm), and the emission was collected at 450–500 nm (for blue) and 490–550 nm (for green).
Flow Cytometry Studies
Both MCF-7 and NIH 3T3 (0.24 × 106 cells per well) cells were seeded on 24-well plates and allowed to grow at 37 °C in a 5% CO2 atmosphere in DMEM medium containing 10% fetal bovine serum and 0.1% streptomycin until they reached 80% confluency. After that, the fluorescent PNAs bm-PNAs (Cf-Cγ(S)-bm-PNA 7 and Cf-Cγ(R)-bm-PNA 8), iso-PNAs (Cf-Cγ(S)-iso-PNA 9 and Cf-Cγ(R)-iso-PNA 10), and normal control aeg-PNAs (Cf-aeg-PNA-C611 and Cf-aeg-PNA-T812) at 4 μM concentration were added and incubated for further 24 h. Upon incubation, wells were washed three times with PBS buffer to remove any excess PNAs. Finally, cells were trypsinized and resuspended in 0.5 mL of PBS for FACS measurements. Fluorescence with respect to the control (no PNA) was measured to quantify the cell uptake.
Cell Cytotoxicity Assay
MCF7 and NIH 3T3 (15,000 cells/well) cells were separately seeded in 96-well plates and allowed to grow overnight at 37 °C in a 5% CO2 atmosphere in DMEM medium containing 10% fetal bovine serum and 0.1% streptomycin. The cells were then individually treated with aeg/iso/bm-PNAs (Table 1, PNA 1–PNA 7) and incubated for 24 h. The media were then replaced with fresh DMEM media (100 μL) containing the MTT reagent (10 μL). After 4 h, DMSO (100 μL) was added and incubated further for 15 min. The absorbance was measured at 540 nm and the percentage viability was calculated, considering untreated cells as 100% viable.
Acknowledgments
P.B. thanks CSIR-New Delhi for a research fellowship and K.N.G. acknowledges DST, New Delhi, Government of India, for a research grant (EMR/2016/007601).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c02451.
Schemes for synthesis of Cγ-bm-PNAs and iso-PNAs, HPLC and MALDI-TOF spectral data of all PNA oligomers, and UV–Tm study of mismatch DNA with bm-PNAs (PDF)
Author Contributions
P.B. carried out the chemical synthesis and biophysical studies and with P.J. did the cell uptake experiments.
The authors declare no competing financial interest.
Supplementary Material
References
- Nielsen P.; Egholm M.; Berg R.; Buchardt O. Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science 1991, 254, 1497–1500. 10.1126/science.1962210. [DOI] [PubMed] [Google Scholar]
- Egholm M.; Buchardt O.; Nielsen P. E.; Berg R. H. Peptide Nucleic Acids (PNA). Oligonucleotide analogs with an achiral peptide backbone. J. Am. Chem. Soc. 1992, 114, 1895–1897. 10.1021/ja00031a062. [DOI] [Google Scholar]
- Egholm M.; Nielsen P. E.; Buchardt O.; Berg R. H. Recognition of guanine and adenine in DNA by cytosine and thymine containing peptide nucleic acids (PNA). J. Am. Chem. Soc. 1992, 114, 9677–9678. 10.1021/ja00050a068. [DOI] [Google Scholar]
- Egholm M.; Buchardt O.; Christensen L.; Behrens C.; Freier S. M.; Driver D. A.; Berg R. H.; Kim S. K.; Norden B.; Nielsen P. E. PNA hybridizes to complementary oligonucleotides obeying the Watson–Crick hydrogen-bonding rules. Nature 1993, 365, 566–568. 10.1038/365566a0. [DOI] [PubMed] [Google Scholar]
- a Nielsen P. E.; Christensen L. Strand displacement binding of a duplex forming homopurine PNA to a homopyrimidine duplex DNA target. J. Am. Chem. Soc. 1996, 118, 2287–2288. 10.1021/ja953125q. [DOI] [Google Scholar]; b Demidov V. V.; Yavnilovich M. V.; Belotserkovskii B. P.; Frank-Kamenetskii M. D.; Nielsen P. E. Kinetics and mechanism of polyamide ‘peptide’ nucleic acid binding to duplex DNA. Proc. Natl. Acad. Sci. U. S. A. 1995, 92, 2637–2641. 10.1073/pnas.92.7.2637. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Nielsen P. E. Peptide nucleic acid. A molecule with two identities. Acc. Chem. Res. 1999, 32, 624–630. 10.1021/ar980010t. [DOI] [Google Scholar]
- a Nielsen P. E. PNA technology. Mol. Biotechnol. 2004, 26, 233–248. 10.1385/MB:26:3:233. [DOI] [PubMed] [Google Scholar]; b Jain D. R.; Ganesh K. N. Clickable Cγ-azido(methylene/butylene) peptide nucleic acids and their clicked fluorescent derivatives: synthesis, DNA hybridization properties, and cell penetration studies. J. Org. Chem. 2014, 79, 6708–6714. 10.1021/jo500834u. [DOI] [PubMed] [Google Scholar]; c D’Agata R.; Giuffrida M.; Spoto G. Peptide nucleic acid-based biosensors for cancer diagnosis. Molecules 2017, 22, 1951–1966. 10.3390/molecules22111951. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Vilaivan T. Fluorogenic PNA probes. Beilstein J. Org. Chem. 2018, 14, 253–281. 10.3762/bjoc.14.17. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Saarbach J.; Sabale P. M.; Winssinger N. Peptide nucleic acid (PNA) and its applications in chemical biology, diagnostics and therapeutics. Curr. Opin. Chem. Biol. 2019, 52, 112–124. 10.1016/j.cbpa.2019.06.006. [DOI] [PubMed] [Google Scholar]; f Economos N. G.; Oyaghire S.; Quijano E.; Ricciardi A. S.; Saltzman W. M.; Glazer P. M. Peptide nucleic acids and gene editing: Perspectives on structure and repair. Molecules 2020, 25, 735–736. 10.3390/molecules25030735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Good L.; Nielsen P. E. Progress in developing PNA as a gene-targeted drug. Antisense Nucleic Acid Drug Dev. 1997, 7, 431–437. 10.1089/oli.1.1997.7.431. [DOI] [PubMed] [Google Scholar]; b Doyle D. F.; Braasch D. A.; Janowski B. A.; Corey D. R. Inhibition of gene expression inside cells by peptide nucleic acids: Effect of mRNA target sequence, mismatched bases, and PNA length. Biochemistry 2001, 40, 53–64. 10.1021/bi0020630. [DOI] [PubMed] [Google Scholar]; c Lundin K. E.; Good L.; Stromberg R.; Graslund A.; Smith C. I. E. Biological activity and biotechnological aspects of peptide nucleic acid. Adv. Genet. 2006, 56, 1–51. 10.1016/S0065-2660(06)56001-8. [DOI] [PubMed] [Google Scholar]; d Quijano E.; Bahal B.; Ricciard A.; Saltzman W. M.; Glazer P. M. Therapeutic peptide nucleic acids: Principles, limitations, and opportunities. Yale J. Biol. Med. 2017, 90, 583–598. [PMC free article] [PubMed] [Google Scholar]; e Montazersaheb S.; Saeid Hejazi M.; Charoudeh H. N. Potential of peptide nucleic acids in future therapeutic applications. Adv. Pharm. Bull. 2018, 8, 551–563. 10.15171/apb.2018.064. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Lee H. T.; Kim S. K.; Yoon J. W. Antisense peptide nucleic acids as a potential anti-infective agent. J. Microbiol. 2019, 57, 423–430. 10.1007/s12275-019-8635-4. [DOI] [PubMed] [Google Scholar]; g Gambari R.; Gasparello J.; Finotti A. Peptide nucleic acid-based targeting of microRNAs: possible therapeutic applications for glioblastoma. J. Cancer Metastas. Treat. 2019, 5, 55–62. [Google Scholar]; h Smith C. I. E.; Zain R. Therapeutic oligonucleotides: State of the art. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 605–630. 10.1146/annurev-pharmtox-010818-021050. [DOI] [PubMed] [Google Scholar]
- a Hyrup B.; Egholm M.; Nielsen P. E.; Wittung P.; Norden B.; Buchardt O. Structure-Activity studies of the binding of modified peptide nucleic acids (PNAs) to DNA. J. Am. Chem. Soc. 1994, 116, 7964–7970. 10.1021/ja00097a002. [DOI] [Google Scholar]; b Kumar V. A.; Ganesh K. N. Conformationally constrained PNA analogues: Structural evolution toward DNA/RNA binding selectivity. Acc. Chem. Res. 2005, 38, 404–412. 10.1021/ar030277e. [DOI] [PubMed] [Google Scholar]; c Govindaraju T.; Kumar V. A.; Ganesh K. N. (SR/RS)-Cyclohexanyl PNAs: Conformationally preorganized PNA analogues with unprecedented preference for duplex formation with RNA. J. Am. Chem. Soc. 2005, 127, 4144–4145. 10.1021/ja044142v. [DOI] [PubMed] [Google Scholar]; d Kumar V.; Ganesh K. Structure-editing of nucleic acids for selective targeting of RNA. Curr. Top. Med. Chem. 2007, 7, 715–726. 10.2174/156802607780487722. [DOI] [PubMed] [Google Scholar]; e Corradini R.; Sforza S.; Tedeschi T.; Totsingan F.; Manicardi A.; Marchelli R. Peptide nucleic acids with a structurally biased backbone. Updated review and emerging challenges. Curr. Top. Med. Chem. 2011, 11, 1535–1554. 10.2174/156802611795860979. [DOI] [PubMed] [Google Scholar]; f Moccia M.; Adamo M. F. A.; Saviano M. Insights on chiral, backbone modified peptide nucleic acids: Properties and biological activity. Artif. DNA PNA XNA 2014, 5, e1107176 10.1080/1949095X.2015.1107176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Koppelhus U.; Nielsen P. E. Cellular delivery of peptide nucleic acid (PNA). Adv. Drug Delivery Rev. 2003, 55, 267–280. 10.1016/S0169-409X(02)00182-5. [DOI] [PubMed] [Google Scholar]; b Równicki M.; Wojciechowska M.; Wierzba A. J.; Czarnecki J.; Bartosik D.; Gryko D.; Trylska J. Vitamin B12 as a carrier of peptide nucleic acid (PNA) into bacterial cells. Sci. Rep. 2017, 7, 7644. 10.1038/s41598-017-08032-8. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Gasparello J.; Manicardi A.; Casnati A.; Corradini R.; Gambari R.; Finotti A.; Sansone F. Efficient cell penetration and delivery of peptide nucleic acids by an argininocalix[4]arene. Sci. Rep. 2019, 9, 3036–3045. 10.1038/s41598-019-39211-4. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Ellipilli S.; Murthy R. V.; Ganesh K. N. Perfluoroalkylchain conjugation as a new tactic for enhancing cell permeability of peptide nucleic acids (PNAs) via reducing the nanoparticle size. Chem. Commun. 2016, 521–524. 10.1039/C5CC05342K. [DOI] [PubMed] [Google Scholar]; e Bhingardeve P.; Madhanagopal B. R.; Naick H.; Jain P.; Manoharan M.; Ganesh K. N. Receptor-specific delivery of peptide nucleic acids conjugated to three sequentially linked N-acetyl galactosamine moieties into hepatocytes. J. Org. Chem. 2020, 85, 8812–8824. 10.1021/acs.joc.0c00601. [DOI] [PubMed] [Google Scholar]
- a Mitra R.; Ganesh K. N. PNAs grafted with (α/γ, R/S)-aminomethylene pendants: Regio and stereospecific effects on DNA binding and improved cell uptake. Chem. Commun. 2011, 47, 1198–1200. 10.1039/C0CC03988H. [DOI] [PubMed] [Google Scholar]; b Mitra R.; Ganesh K. N. Aminomethylene Peptide nucleic acid (am-PNA): synthesis, regio–/ stereospecific DNA binding, and differential cell uptake of (α/γ, R/S) am-PNA analogues. J. Org. Chem. 2012, 77, 5696–5704. 10.1021/jo300860f. [DOI] [PubMed] [Google Scholar]; c Jain D. R.; Ganesh K. N. Influence of pendant chiral Cγ-(alkylideneamino/guanidino) cationic sidechains of PNA backbone on hybridization with complementary DNA/RNA and cell permeability. J. Org. Chem. 2014, 79, 9567–9577. 10.1021/jo501639m. [DOI] [PubMed] [Google Scholar]; d Ellipilli S.; Palvai S.; Ganesh K. N. Fluorinated peptide nucleic acids with fluoroacetyl side chain bearing 5-(F/CF3)-uracil: Synthesis and cell uptake studies. J. Org. Chem. 2016, 81, 6364–6373. 10.1021/acs.joc.6b01009. [DOI] [PubMed] [Google Scholar]
- a Dragulescu-Andrasi A.; Rapireddy S.; Frezza B. M.; Gayathri C.; Gil R. R.; Ly D. H. A simple gamma-backbone modification preorganizes peptide nucleic acid into a helical structure. J. Am. Chem. Soc. 2006, 128, 10258–10267. 10.1021/ja0625576. [DOI] [PubMed] [Google Scholar]; b Ishizuka T.; Yoshida J.; Yamamoto Y.; Sumaoka J.; Tedeschi T.; Corradini R.; Stefano Sforza S.; Komiyama M. Chiral introduction of positive charges to PNA for double-duplex invasion to versatile sequences. Nucleic Acids Res. 2008, 36, 1464–1471. 10.1093/nar/gkm1154. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Manicardi A.; Corradini R. Effect of chirality in gamma-PNA: PNA interaction, another piece in the picture. Artif DNA PNA XNA. 2014, 5, e1131801 10.1080/1949095X.2015.1131801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Govindaraju T.; Kumar V. A.; Ganesh K. N. (1S,2R/1R,2S)-cis-cyclopentyl PNAs (cpPNAs) as constrained PNA analogues: synthesis and evaluation of aeg-cpPNA chimera and stereopreferences in hybridization with DNA/RNA. J. Org. Chem. 2004, 69, 5725–5734. 10.1021/jo049442+. [DOI] [PubMed] [Google Scholar]; b Govindaraju T.; Madhuri V.; Kumar V. A.; Ganesh K. N. Cyclohexanyl Peptide Nucleic Acids (chPNAs) for preferential RNA binding: effective tuning of dihedral angle β in PNAs for DNA/RNA discrimination. J. Org. Chem 2006, 71, 14–21. 10.1021/jo051227l. [DOI] [PubMed] [Google Scholar]; c Zheng H.; Saha M.; Appella D. H. Synthesis of Fmoc-protected (S,S)-trans-cyclopentane diamine monomers enables the preparation and study of conformationally restricted peptide nucleic acids. Org. Lett. 2018, 20, 7637–7640. 10.1021/acs.orglett.8b03374. [DOI] [PubMed] [Google Scholar]; d Suparpprom C.; Nuanyai T.; Pansuwan H.; Vilaivan T.; Wanichwecharungruang S.; Ditmangklo B.; Palaga T.; Pan-In P.; Jiangchareon B.; Vilaivan C. Hydrophilic and cell-penetrable pyrrolidinyl peptide nucleic acid via post-synthetic modification with hydrophilic side chains. Bioconjugate Chem. 2017, 28, 2284–2202. [DOI] [PubMed] [Google Scholar]
- a Gupta M. K.; Madhanagopal B. R.; Datta D.; Ganesh K. N. Structural Design and Synthesis of bimodal PNA that simultaneously binds two complementary DNAs to form fused double duplexes. Org. Lett. 2020, 22, 5255–5260. 10.1021/acs.orglett.0c01950. [DOI] [PubMed] [Google Scholar]; b Gupta M. K.; Madhanagopal B. R.; Ganesh K. N. Peptide nucleic acid with double face: homothymine-homocytosine bimodal Cα-PNA (bm-Cα-PNA) forms a double duplex of the bm-PNA2:DNA Triplex. J. Org. Chem. 2021, 86, 414–428. 10.1021/acs.joc.0c02158. [DOI] [PubMed] [Google Scholar]
- Bhingardeve P.; Madhanagopal B. R.; Ganesh K. N. Cγ (S/R)-Bimodal peptide nucleic acids (Cγ-bm-PNA) synchronously bind two different DNA strands to form coupled double duplexes with enhanced stability. J. Org. Chem. 2020, 85, 13680–13693. 10.1021/acs.joc.0c01853. [DOI] [PubMed] [Google Scholar]
- a Kumar P.; Sorinas A. F.; Nielsen L. J.; Slot M.; Skytte K.; Nielsen A. S.; Jensen M. D.; Sharma P. K.; Vester B.; Petersen M.; Nielsen P. Double-coding nucleic acids: Introduction of a nucleobase sequence in the major groove of the DNA duplex using double-headed nucleotides. J. Org. Chem 2014, 79, 8020. 10.1021/jo501151w. [DOI] [PubMed] [Google Scholar]; b Mick H.; Julie S.; Sharma P. K.; Pawan K.; Nielsen R. B; Michael P.; Nielsen P. Base-pairing properties of double-headed nucleotides. Chemistry, A European Journal 2019, 25, 7387–7395. [DOI] [PubMed] [Google Scholar]
- a Branda N.; Kurz G.; Lehn J.-M. Janus wedges: A new approach towards nucleobase-pair recognition. Chem. Commun. 1996, 2443–2444. 10.1039/cc9960002443. [DOI] [Google Scholar]; b Hang Z.; Wen H.; Xiaohua W.; Yong Q.; Zhihua X.; Yang H. Synthesis of a complete Janus-type guanosine-cytosine base and its 2′-deoxyribonucleoside. Chem. Lett. 2011, 40, 684–686. [Google Scholar]; c Chen H.; Meena; McLaughlin L. M. A Janus-wedge DNA triplex with A-W1-T and G-W2-C base triplets. J. Am. Chem. Soc. 2008, 130, 13190–13191. 10.1021/ja804607v. [DOI] [PubMed] [Google Scholar]; d Shin D.; Tor Y. Bifacial nucleoside as a surrogate for both T and A in duplex DNA. J. Am. Chem. Soc. 2011, 133, 6926–6929. 10.1021/ja201397e. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Artigas G.; Marchan V. Synthesis of janus compounds for the recognition of G-U mismatched nucleobase pairs. J. Org. Chem. 2013, 78, 10666–10677. 10.1021/jo401684j. [DOI] [PubMed] [Google Scholar]
- a Xia X.; Piao X.; Bong D. Bifacial peptide nucleic acid as an allosteric switch for aptamer and ribozyme function. J. Am. Chem. Soc. 2014, 136, 7265–7268. 10.1021/ja5032584. [DOI] [PubMed] [Google Scholar]; b Thadke S. A.; Hridya V. M.; Perera J. D. R.; Gil R. R.; Mukherjee A.; Ly D. H. Shape selective bifacial recognition of double helical DNA. Comm. Chem. 2018, 1, 79. 10.1038/s42004-018-0080-5. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Thadke S. A.; Perera J. D. R.; Hridya V. M.; Bhatt K.; Shaikh A. Y.; Hsieh W.-C.; Chen M.; Gayathri C.; Gil R. R.; Rule G. S.; Mukherjee A.; Thornton C. A.; Ly D. H. Design of bivalent nucleic acid ligands for recognition of RNA-repeated expansion associated with Huntington’s disease. Biochemistry 2018, 57, 2094–2108. 10.1021/acs.biochem.8b00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Bentin T.; Hansen G. I.; Nielsen P. E. In Methods in Molecular Biology; Nielsen P. E., Ed.; Humana Press Inc.: Totowa, NJ, 2002; Vol. 208, pp. 91–109. [DOI] [PubMed] [Google Scholar]; b Nielsen P. E., Ed. Peptide Nucleic Acids Protocols and Applications, 2nd ed.; Horizon Bioscience: Norfolk, UK, 2004. [Google Scholar]
- a Plum G. E.; Park Y.-W.; Singleton S. F.; Dervan P. B.; Breslauer K. J. Thermodynamic characterization of the stability and the melting behavior of a DNA triplex: A spectroscopic and calorimetric study. Proc. Natl. Acad. Sci. U. S. A. 1990, 87, 9436–9440. 10.1073/pnas.87.23.9436. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ratilainen T.; Holmén A.; Tuite E.; Nielsen P. E.; Nordén B. Thermodynamics of sequence-specific binding of PNA to DNA. Biochemistry 2000, 39, 7781–7791. 10.1021/bi000039g. [DOI] [PubMed] [Google Scholar]; c Krupnik O. V.; Guscho Y. A.; Sluchanko K. A.; Nielsen P. E.; Lazurkin Y. S. Thermodynamics of the melting of PNA2/DNA triple helices. J. Biomol. Struct. Dynamics 2001, 19, 535–542. 10.1080/07391102.2001.10506761. [DOI] [PubMed] [Google Scholar]; d Mergny J. -L.; Lacroix L. Analysis of thermal melting curves. Oligonucleotides 2003, 13, 515–537. 10.1089/154545703322860825. [DOI] [PubMed] [Google Scholar]; e Jasiński M.; Miszkiewicz J.; Feig M.; Trylska J. Thermal stability of peptide nucleic acid complexes. J. Phys. Chem. B 2019, 123, 8168–8177. 10.1021/acs.jpcb.9b05168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kim S. K.; Nielsen P. E.; Egholm M.; Buchardt O.; Berg R. H.; Norden B. J. Am. Chem. Soc. 1993, 115, 6477–6481. 10.1021/ja00068a001. [DOI] [Google Scholar]; b Corradini R.; Tedeschi T.; Sforza S.; Marchelli R.. Electronic Circular Dichroism of Peptide Nucleic Acids and Their Analogues. In Comprehensive Chiroptical Spectroscopy, Volume 2: Applications in Stereochemical Analysis of Synthetic Compounds, Natural Products, and Biomolecules. Berova N.; Polavarapu P. L.; Eds Nakanishi K.; Woody R. W.. John Wiley & Sons, Inc, 2012; Vol. 2, pp. 581–617. [Google Scholar]
- Marck C.; Thiele D. Poly(dG).poly(dC) at neutral and alkaline pH: the formation of triple stranded poly(dG).poly(dG).poly(dC). Nucleic Acids Res. 1978, 5, 1017–1028. 10.1093/nar/5.3.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Wittung P.; Nielsen P.; Norden B. Extended DNA-recognition repertoire of peptide nucleic acid (PNA): PNA-dsDNA triplex formed with cytosine-rich homopyrimidine PNA. J. Am. Chem. Soc. 1997, 36, 7973–7979. [DOI] [PubMed] [Google Scholar]; b Hashem G. M.; Wen J.-D.; Do Q.; Gray D. M. Evidence from CD spectra and melting temperatures for stable Hoogsteen-paired oligomer duplexes derived from DNA and hybrid triplexes. Nucleic Acids Res. 1999, 27, 3371–3379. 10.1093/nar/27.16.3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Berger O.; Gazit E. Molecular self-assembly using peptide nucleic acids. Biopolymers 2017, 108, e22930 10.1002/bip.22930. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Manicardi A.; Rozzi A.; Korom S.; Corradini R. Building on the peptide nucleic acid (PNA) scaffold: a biomolecular engineering approach. Supramol. Chem. 2017, 784. 10.1080/10610278.2017.1371720. [DOI] [Google Scholar]
- Cadoni E.; Manicardi A.; Madder A. PNA-based micro-RNA detection methodologies. Molecules 2020, 25, 1296–1321. 10.3390/molecules25061296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kan L.-S.; Pasternack L.; Wey M.-T.; Tseng Y.-Y.; Huang D.-H. The “paperclip” triplex: understanding the role of apex residues in tight turns. Biophys. J. 2006, 91, 2552–2563. 10.1529/biophysj.106.084137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Hong F.; Zhang F.; Liu Y.; Yan H. DNA Origami: Scaffolds for creating higher order structures. Chem. Rev. 2017, 117, 12584–12640. 10.1021/acs.chemrev.6b00825. [DOI] [PubMed] [Google Scholar]; b Bila H.; Kurisinkal E. E.; Bastings M. M. C. Engineering a stable future for DNA-origami as a biomaterial. Biomater. Sci. 2019, 7, 532–541. 10.1039/C8BM01249K. [DOI] [PubMed] [Google Scholar]
- Koch T.PNA synthesis by Boc chemistry In Peptide Nucleic Acids: Protocols and Applications, 2nd ed.; Nielsen P. E., Ed.; Horizon Scientific: Norfolk, UK, 2004; pp. 37e59. [Google Scholar]
- Debaene F.; DaSilva J. A.; Pianowski Z.; Duran F. J.; Winssinger N. Expanding the scope of PNA-encoded libraries: divergent synthesis of libraries targeting cysteine, serine and metallo-proteases as well as tyrosine phosphatases. Tetrahedron 2007, 63, 6577–6586. 10.1016/j.tet.2007.03.033. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.