

Abstract
Immune checkpoint therapy (ICT) has the potential to treat cancer by removing the immunosuppressive brakes on T cell activity. However, ICT benefits only a subset of patients because most tumors are “cold”, with limited pre-infiltration of effector T cells, poor immunogenicity, and low-level expression of checkpoint regulators. It has been previously reported that Cowpea mosaic virus (CPMV) promotes the activation of multiple innate immune cells and the secretion of pro-inflammatory cytokines to induce T cell cytotoxicity, suggesting that immunostimulatory CPMV could potentiate ICT. Here it is shown that in situ vaccination with CPMV increases the expression of checkpoint regulators on Foxp3−CD4+ effector T cells in the tumor microenvironment. It is shown that combined treatment with CPMV and selected checkpoint-targeting antibodies, specifically anti-PD-1 antibodies, or agonistic OX40-specific antibodies, reduced tumor burden, prolonged survival, and induced tumor antigen-specific immunologic memory to prevent relapse in three immunocompetent syngeneic mouse tumor models. This study therefore reveals new design principles for plant virus nanoparticles as novel immunotherapeutic adjuvants to elicit robust immune responses against cancer.
Keywords: Cowpea mosaic virus, immune checkpoint therapy, immunotherapy
Dedicated to the memory of Marian K. Shaughnessy, who passed away on February 24, 2020, after battling ovarian cancer
1. Introduction
Immune checkpoint therapy (ICT) involves the administration of drugs that either inhibit negative regulators of the immune response against cancer cells, such as programmed cell death protein 1 (PD-1), or act as agonists for positive regulators such as tumor necrosis factor receptor superfamily, member 4 (TNFRSF4 or OX40). However, the objective response rate to single-agent ICT is only ≈30% for most types of cancer.[1–3] Patients with immunogenic tumors are more likely to respond to ICT because their tumors are characterized by a high antigen burden, the pre-infiltration of effector T cells, and high-level expression of negative checkpoint regulators such as cytotoxic-T-lymphocyte-associated protein 4 (CTLA-4) and PD-1.[4,5] To pre-sensitize the immune system and generate abundant effector T cells against tumors, conventional adjuvants and complementary approaches, such as adoptive T cell therapy, radiation therapy, and chemotherapy, have been combined with ICT.[6–9] However, these approaches did not achieve the expected synergistic benefits and long-lasting systemic protection in preclinical/clinical tumor models.
We previously reported that in situ vaccination with Cowpea mosaic virus (CPMV), a plant virus that does not infect mammalian cells, promotes the activation of multiple innate immune cells and triggers their repolarization from pro-tumor to antitumor phenotypes. This immunostimulation within the tumor leads to the secretion of cytokines in the tumor microenvironment and the induction of CD8+ cytotoxic T lymphocyte (CTL) responses in multiple murine tumor models.[10–12] CPMV is particularly useful as an adjuvant because it remodels the suppressive tumor microenvironment by increasing the number of tumor-infiltrating lymphocytes (TILs), inducing the killing of tumor cells, which thus release a full range of tumor-associated and neo- antigens. We therefore hypothesized that CPMV in situ vaccination combined with ICT could improve the objective response rate by increasing the number of tumor antigen-specific TILs and thereby achieving a stronger response to lower systemic doses of ICT, thus improving antitumor efficacy while mitigating the impact of adverse events associated with ICT.
To identify checkpoint-targeting drugs suitable for combination therapy with CPMV, we characterized the expression of different immune checkpoint regulators after CPMV treatment, focusing on PD-1 and OX40 as representative negative and positive regulators, respectively. PD-1 is expressed on the surface of immune cells, especially activated T cells,[13] and interacts with programmed death ligand 1 (PD-L1), which is abundantly expressed on multiple tumor cells. The engagement of PD-1 by PD-L1 causes the inhibition or exhaustion of activated immune cells and thus suppresses the antitumor response.[14] Antibodies that block PD-1/PD-L1 interactions allow the differentiation of CD8+ T cells into CTLs that help to eradicate the tumor. In contrast, OX40 is a costimulatory molecule that augments antitumor responses.[15] OX40 is a member of the TNF receptor superfamily and is expressed on CD4+ and CD8+ effector T cells as well as regulatory T cells. When engaged by its ligand OX40L, it triggers the NF-κB signaling pathway, leading to T cell clonal expansion and activation.[16,17] Therefore, agonistic OX40-specific antibodies can promote T cell activation and elicit a stronger antitumor immune response. Although inhibitory anti-PD-1 antibodies and agonistic OX40-specific antibodies can mediate tumor suppression in several tumor models, their efficacy is poor in tumors with low immunogenicity.[18] In this study, we characterized the expression of PD-1 and OX40 in response to CPMV treatment in order to increase the immunogenicity of tumors, and then assessed the effectiveness of combination therapy using mouse models of ovarian cancer, colon cancer, and melanoma.
2. Results
2.1. In Situ Vaccination with CPMV Upregulates the Expression of PD-1 and OX40 on Tumor Infiltrated Effector T Cells In Vivo
To explore the expression of PD-1 and OX40 after CPMV treatment, luciferase-labeled ID8-Defb29/Vegf-A (ID8-Defb29/Vegf-A-luc) ovarian tumor cells were implanted into C57BL/6 mice via the intraperitoneal (i.p.) route, which resulted in formation of peritoneal tumors and ascetic fluid. The presence of ascites correlates with the peritoneal spread and metastasis of ovarian cancer and indicated advanced stages of ovarian cancer.[19–21] The establishment of ascites/tumors was confirmed by luminescence detection 35 days post inoculation (dpi). At this point the mice were injected (i.p.) with 100 μg CPMV in PBS and peritoneal lavage was carried out 24 h later to collect T cells. A single dose of CPMV significantly increased the expression of PD-1 and OX40 in the total CD4+ T cell population from the tumor microenvironment. However, the upregulation of PD-1 was mostly restricted to Foxp3−CD4+ effector T cells whereas the upregulation of OX40 was mostly restricted to Foxp3+CD4+ regulatory T cells (Figure S1, Supporting Information). Multiple CPMV treatments (five doses, weekly injection starting from 7 dpi) were also carried out, and the expression of both checkpoint proteins was measured 48 h after the last treatment. As described for the single treatment, the upregulation of PD-1 on CD4+ T cells was mostly observed among the effector T cell population, whereas the upregulation of OX40 mostly observed among the regulatory T cells (Figure 1a,b). We also found that PD-1 expression was significantly upregulated in the total CD8+ T cell population (p < 0.0001) and specifically among CD44+CD8+ effector T cells (p < 0.005). OX40 showed a similar profile among CD8+ T cells but with much weaker expression than PD-1 (Figure 1c,d). These results indicate that in situ vaccination with CPMV can sensitize the tumor to both OX40 agonists and PD-1 inhibitors. In this tumor model, we found that PD-1 was potentially a better target for combination therapy than OX40 because CPMV increased the expression of PD-1 on both CD4+ and CD8+ effector T cells to a greater extent than OX40.
Figure 1.
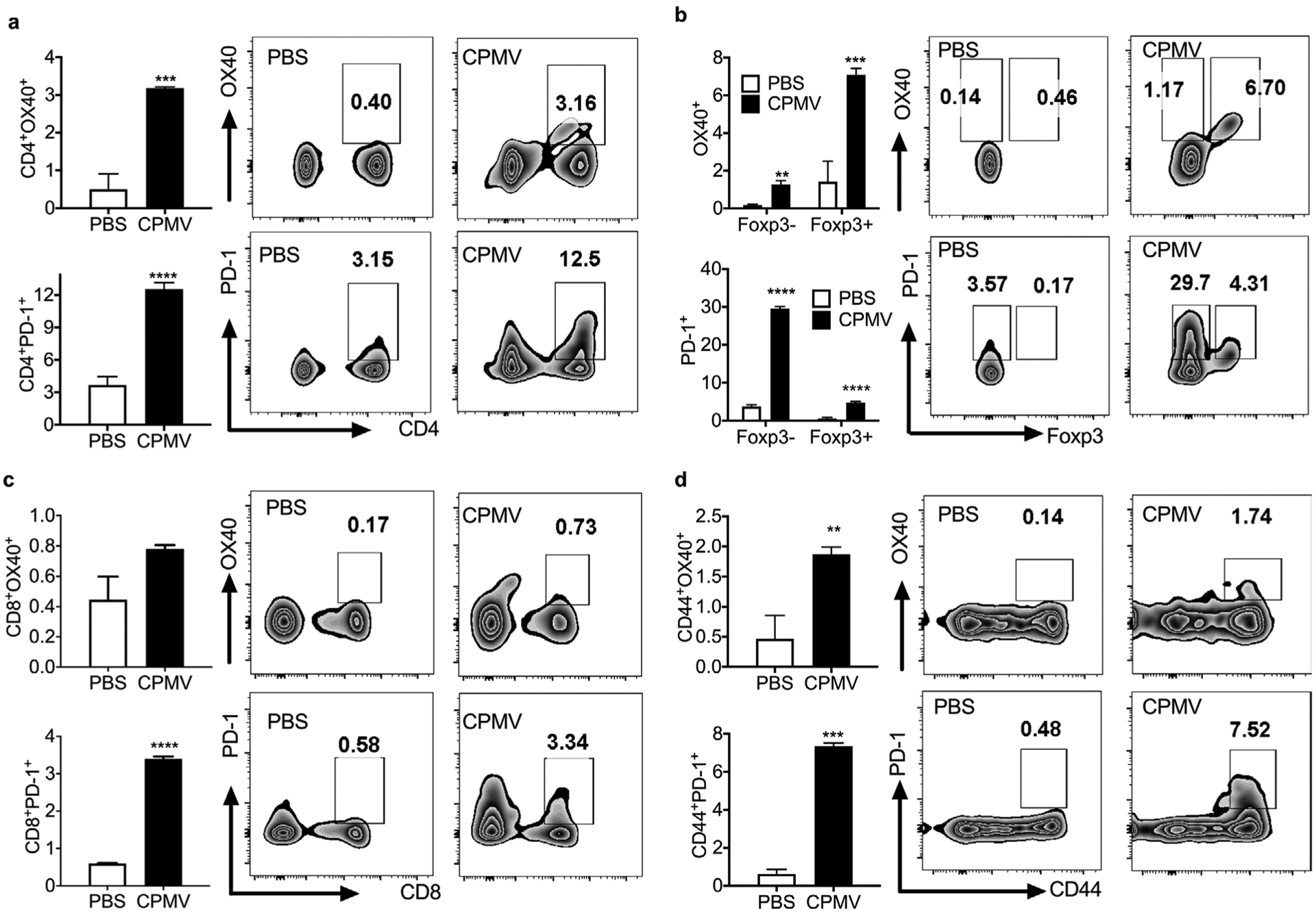
Repetitive administration of CPMV induces the expression of OX40 and PD-1 on CD4+ and CD8+ T cells. C57BL/6 mice were inoculated (i.p.) with 2 × 106 ID8-Defb29/Vegf-A-luc cells followed by five weekly injections (i.p.) of 100 μg CPMV. Cells collected from peritoneal washes carried out 48 h after the last treatment were analyzed by flow cytometry. a) Percentages and representative FACS plots of OX40+CD4+ and PD-1+CD4+ T cells gated on CD3+ T cells. b) Percentages and representative FACS plots showing PD-1 and OX40 expression on Foxp3− effector T cells and Foxp3+ regulatory T cells gated on CD3+CD4+ T cells. c) Percentages and representative FACS plots of OX40+CD8+ and PD-1+CD8+ T cells gated on CD3+ T cells. d) Percentages and representative FACS plots of CD44+OX40+ and CD44+PD-1+ subsets gated on CD3+CD8+ T cells. Data are means ± SEM (n = 3). Statistical significance was calculated using a paired t-test (*p < 0.05, **p < 0.01, ***p < 0.0005, ****p < 0.0001).
2.2. Combined CPMV and PD-1 Inhibitor Treatment Synergistically Prevents Ovarian Tumor Growth in the Peritoneal Cavity Following Re-Challenge
Given the ability of CPMV to induce the expression of PD-1 and (to a lesser extent) OX40 in the microenvironment of ovarian tumors, we hypothesized that the antitumor responses to CPMV could be augmented by combining CPMV treatment with a PD-1 inhibitor or OX40 agonist antibody. As above, we implanted 2 × 106 ID8-Defb29/Vegf-A ovarian cancer cells into C57BL/6 mice and administered 100 μg of CPMV in PBS 7 dpi, this time with or without 50 μg of the antagonistic (PD-1) or agonistic (OX40) antibody (Figure 2a). Control mice were injected with PBS alone. Six doses were injected at weekly intervals, and tumor growth was monitored by measuring body weight which reflected tumor-associated ascites accumulation in this model (Figure 2b). There was no difference in tumor growth between the solo ICT groups and PBS controls, indicating that neither the rat anti-mouse PD-1 IgG2a (clone RMP1–14, BioXCell) nor the rat anti-mouse OX40 IgG1 (clone OX86, BioXCell) alone were sufficient to suppress tumor growth. As expected, ≈20% of the mice treated with CPMV alone survived compared to none of the control mice at 100 dpi, which is consistent with our previous studies.[10] Tumor growth was suppressed in both combination therapy groups, with ≥80% of the mice surviving until 100 dpi, whereas all mice in the control and solo ICT groups died by 70 dpi (Figure 2c). However, only mice treated with CPMV and the PD-1 inhibitor achieved 100% survival when re-challenged with the initial dose of 2 × 106 ID8-Defb29/Vegf-A tumor cells (Figure 2d).
Figure 2.
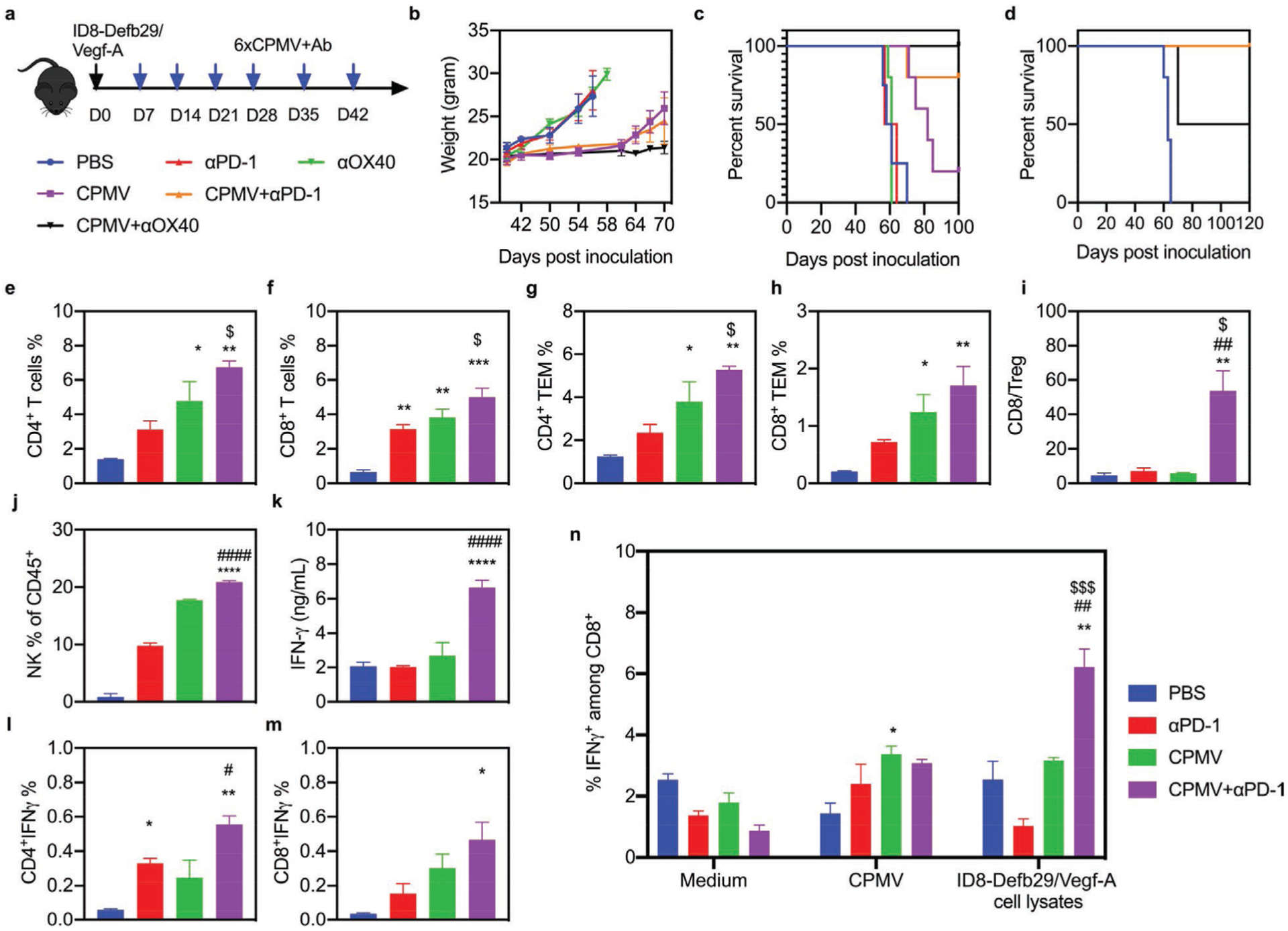
Combined CPMV and PD-1 inhibitor treatment synergistically enhances immunotherapeutic efficacy in a model of ovarian cancer. a) Schematic of the treatment strategy and dosing regimen. C57BL/6 mice were inoculated (i.p.) with 2 × 106 ID8-Defb29/Vegf-A cells followed by six weekly injections (i.p.) of 50 μg antibody (PD-1 antagonist or OX40 agonist), 100 μg CPMV, the combination, or PBS as a control (n = 5). b) Body weight was measured to monitor tumor growth. c) Survival curves of the treatment groups. d) Survival curves of the combination therapy groups following tumor re-challenge at 100 dpi. e–n) C57BL/6 mice were inoculated (i.p.) with 2 × 106 ID8-Defb29/Vegf-A-luc cells followed by two i.p. doses (21 and 28 dpi) of the PD-1 inhibitor antibody (100 μg), CPMV (100 μg), or the combination, and spleens and peritoneal wash/ascites were collected 2 days after the second dose. e–j) Percentages of CD4+ and CD8+ infiltrated T cells (and their subsets) and NK cells among CD45+ cells determined by flow cytometry. k) IFNγ levels in the supernatant of peritoneal wash/ascites. l–m) Percentages of total CD4+ and CD8+ infiltrated T cells staining positive for IFNγ. n) Splenocytes were cultured in fresh medium, CPMV suspensions, or ID8-Defb29/Vegf-A cell lysates for 24 h. Percentage of intracellular IFNγ was measured in CD8+ T cells by flow cytometry. Data are means ± SEM (n = 3). Statistical significance was calculated by one-way ANOVA: * versus PBS; # versus CPMV monotherapy; $ versus antibody monotherapy (*p < 0.05; **p < 0.01; ***p < 0.0005; ****p < 0.0001).
2.3. Combined CPMV and PD-1 Inhibitor Treatment Promotes the Repolarization of Myeloid Cells and the Activation of Cytotoxic Lymphocytes
To evaluate how the combination of CPMV in situ vaccination and PD-1 inhibition affects the behavior of immune cells, we injected mice with two doses (21 and 28 dpi) of the PD-1 inhibitor alone (100 μg), CPMV alone (100 μg), the combined reagents, or PBS as a control, and collected peritoneal wash/ascites from mice 2 days after the second treatment. The solo CPMV treatment and combination therapy significantly (p < 0.0001) increased the total number of TILs compared to the control and solo PD-1 inhibitor groups (Figure S2b, Supporting Information). However, the TILs isolated from mice treated with CPMV alone contained a significantly higher proportion of granulocytic myeloid-derived suppressive cells (G-MDSCs: CD11b+Ly6G+MHCII−CD86−) and monocytic myeloid-derived suppressive cells (M-MDSCs: CD11b+Ly6G−Ly6C+MHCII−SSClow) compared to the control group, as well as higher proportions of type 1 neutrophils (N1/TINs: CD11b+Ly6G+MHCII+CD86+), type 1 macrophages (M1: CD11b+F4/80+MHCII+CD86+), and dendritic cells (DCs: CD11b+CD11c+). The combination therapy also resulted in a high proportion of N1, M1, and DCs, but the proportion of MDSCs and type 2 macrophages (M2: CD11b+F4/80+MH CII−CD86−) was lower compared to the solo CPMV group (Figure S2c–h, Supporting Information). Further subset analysis revealed that the combination treatment increased the total number of CD4+ and CD8+ T cells and their effector memory subsets (CD44+CD62L−) in the peritoneal wash/ascites, as well as the CD8+/regulatory T cell ratio and proportion of natural killer (NK) cells, compared to the other groups (Figure 2e–j). The level of secreted interferon γ (IFNγ) was significantly higher in the peritoneal wash supernatant following combination therapy, and intracellular staining indicated that both CD4+ and CD8+ T cells in this group secreted more IFNγ (Figure 2k–m). The combination therapy therefore appeared to recruit multiple antitumor immune cell types while depleting the immunosuppressive cell populations.
To determine whether the CPMV plus PD-1 inhibitor combination could induce a systemic immune response to prevent metastasis, we collected splenocytes from the different treatment groups and pulsed them with fresh medium, a CPMV suspension, or ID8-Defb29/Vegf-A tumor cell lysates (Figure 2n and Figure S3, Supporting Information). After 24 h, we observed a marked increase in the number of tumor-specific IFNγ-secreting CD8+ effector T cells in the spleens of mice from the combination therapy group compared to the control, CPMV monotherapy and solo ICT groups. These results showed that the CPMV plus PD-1 inhibitor combination elicited long-term immune system activation by generating systemic tumor-specific T cells targeting the ID8-Defb29/Vegf-A tumor cells.
Finally, we observed the depletion of CD45− tumor cells in the peritoneal wash/ascites of the combination therapy group, reduced by 23-fold compared to the control group, 10-fold compared to the solo ICT group, and 4-fold compared to the CPMV monotherapy group (Figure S2i, Supporting Information). We also found that the expression of PD-L1 in tumor cells was significantly increased by CPMV treatment alone compared to the PBS control (Figure S2j, Supporting Information), suggesting the antitumor effect of CPMV may be hindered by the interaction between PD-1 on cytotoxic T cells and PD-L1 on tumor cells.[22] PD-1 blocking antibodies therefore appear to compensate for the limitation of CPMV.
2.4. CPMV Upregulates OX40 Expression on Foxp3−CD4+ T Cells and Combination Therapy with an OX40 Agonist is Efficacious in a Model of Colon Cancer
Next we used a CT26 solid tumor model of peritoneal colon carcinomatosis to determine the immunomodulatory potential of CPMV in situ vaccination as described above. Accordingly, BALB/c mice were inoculated (i.p.) with luciferase-labeled CT26 (CT26-luc) tumor cells and injected (i.p.) with CPMV. Analysis of the peritoneal wash revealed that CPMV significantly (p < 0.0005) increased the expression of OX40 on Foxp3−CD4+ effector T cells, whereas the level of PD-1 significantly (p < 0.01) declined (Figure S4a,b, Supporting Information). We also found that CPMV induced the expression of PD-1 and OX40 on CD44+CD8+ effector T cells to a similar degree (Figure S4c,d, Supporting Information). As described above for the ovarian tumor model, we sought to demonstrate the synergistic antitumor efficacy of the combination of CPMV and ICT in colon tumors derived from CT26-luc cells. We therefore i.p. treated CT26-luc tumor-bearing mice with two doses (7 and 14 dpi) of CPMV combined with either the inhibitory anti-PD-1 antibody or the agonistic OX40-specific antibody, as well as setting up the corresponding monotherapy and PBS control groups (Figure 3a). In the control group, the tumor burden increased rapidly and all mice were euthanized by 19 dpi. Mice treated with either antibody alone had smaller tumors than the control group when assessed at 17 dpi (p < 0.05) but they did not survive any longer than the control group (Figure 3b–d). Treatment with CPMV alone or CPMV plus the PD-1 inhibitor achieved a comparable benefit, extending survival by approximately 1 week compared to controls (p < 0.05). However, the CPMV plus OX40 agonist achieved significantly more potent antitumor effects than other groups and prevented tumor growth in animals for up to 50 days (p = 0.0067 versus PBS; Figure 3d). These results indicated that the expression of immune checkpoint molecules on Foxp3−CD4+ effector T cells can help to predict the potency of specific combination therapies.
Figure 3.
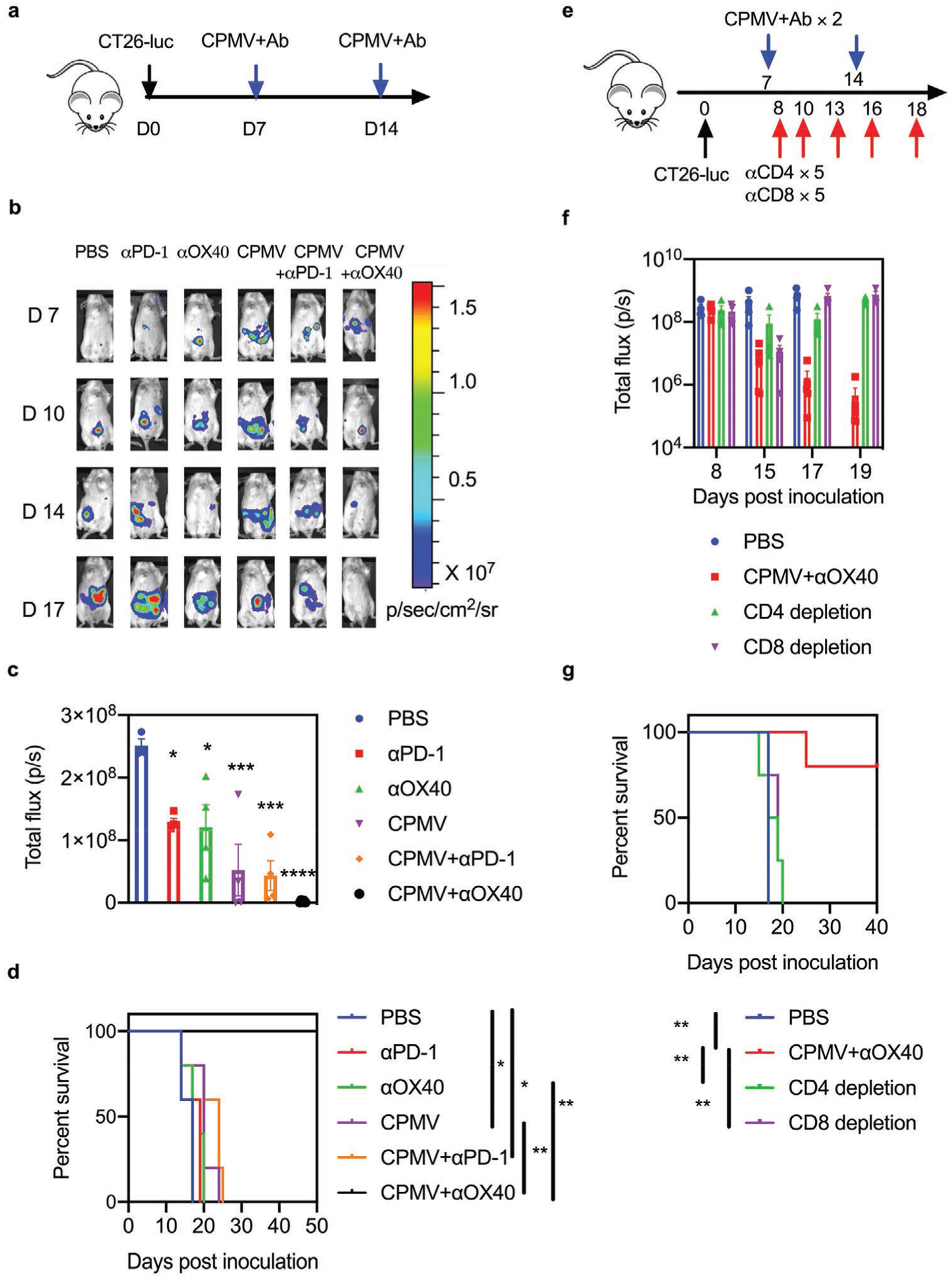
Combined CPMV and OX40 agonist treatment induces systemic antitumor effects in a CT26 colon tumor model. a) Schematic of the treatment strategy and dosing regimen. BALB/c mice were inoculated (i.p.) with 5 × 105 CT26-luc cells followed by two weekly injections (i.p.) of 50 μg antibody (PD-1 antagonist or OX40 agonist), 50 μg CPMV, the combination, or PBS as a control (n = 5). b) IVIS images showing the growth of luc+ CT26 tumors in the different treatment groups. c) The average luciferase expression of tumor cells 17 dpi in the different treatment groups. Data are means ± SEM (n = 3–5). Statistical significance was calculated by one-way ANOVA: * versus PBS; *p < 0.05; ***p < 0.0005; ****p < 0.0001. d) Survival curves of the treatment groups. Statistical significance was calculated using a log-rank Mantel-Cox test: *p < 0.05, **p < 0.01. e) Schematic of the T cell depletion strategy using a CT26-luc colon tumor model. f) The average luciferase expression of tumor cells from different treatment groups in the T cell depletion study: PBS (blue), CPMV+OX40 agonist (red), CD4-specific antibody (100 μg, green), CD8-specific antibody (100 μg, purple). Data are means ± SEM (n = 4–5). g) Survival rate of each treatment group in the T cell depletion study. Statistical significance was calculated using a log-rank Mantel-Cox test: **p < 0.01.
Flow cytometry revealed that the CPMV plus OX40 agonist combination recruited the highest proportion of TILs, antitumor innate immune cell types, and T cells in the peritoneal wash of CT26 tumor-bearing mice, while depleting the populations of immunosuppressive cells (Figures S5 and S6, Supporting Information). Intriguingly, the OX40 agonist monotherapy exclusively increased the total CD4+ and effector memory CD4+ T cell counts and the expression of IFNγ in CD4+ T cells, whereas CPMV monotherapy exclusively increased those factors in CD8+ T cells. In contrast, both CD4+ and CD8+ T cells were activated by the CPMV plus OX40 agonist combination therapy (Figure S6, Supporting Information). To determine which components of the immune system are responsible for the therapeutic efficacy of the combination therapy, we examined the effect on CT26-luc tumor-bearing mice pre-treated with neutralizing antibodies against CD4 and CD8 to remove specific subpopulations (Figure 3e–g and Figure S7, Supporting Information). Notably, the depletion of either CD8+ or CD4+ T cells significantly (p < 0.01) abrogated the tumor suppression achieved by the combination therapy, indicating that the combination of CPMV plus OX40 agonist induced the priming of both CD8+ and CD4+ T cells, resulting in potent antitumor efficacy (Figure 3g).
2.5. Combined CPMV and OX40 Agonist Treatment Induces Profound Tumor Regression and Achieves Long-Term Survival in a B16F10 Melanoma Model
To challenge our combination immunotherapy and selection strategy in an aggressive and immunosuppressive solid tumor model, we implanted 2.5 × 105 B16F10 melanoma cells intradermally (i.d.) in the flanks of syngeneic C57BL/6 mice, and administered CPMV monotherapy, antibody monotherapy (PD-1 inhibitor or OX40 agonist), combination therapy, or PBS as a control, with two doses injected directly into the tumor 10 and 17 dpi (Figure 4a). We collected tumors from the mice on day 18 and phenotyped the T cells. We found that T cells from the PBS and CPMV monotherapy groups expressed similar levels of PD-1 and OX40 on effector CD8+ T cells, but Foxp3–CD4+ T cells from the CPMV monotherapy group were significantly more abundant compared to the PBS control (Figure S8, Supporting Information). Both antibody monotherapies delayed tumor growth and prolonged the survival of the animals to 31 dpi, whereas CPMV monotherapy and the combination of CPMV plus PD-1 inhibitor prolonged survival to 43 dpi. However, the combination of CPMV plus OX40 agonist prolonged survival to day 80. Notably, two of three treated mice in the latter group achieved complete tumor regression (Figure 4b,c and Figure S9, Supporting Information) and these animals were fully protected against re-challenge with the same initial dose of B16F10 tumor cells (Figure 4d).
Figure 4.
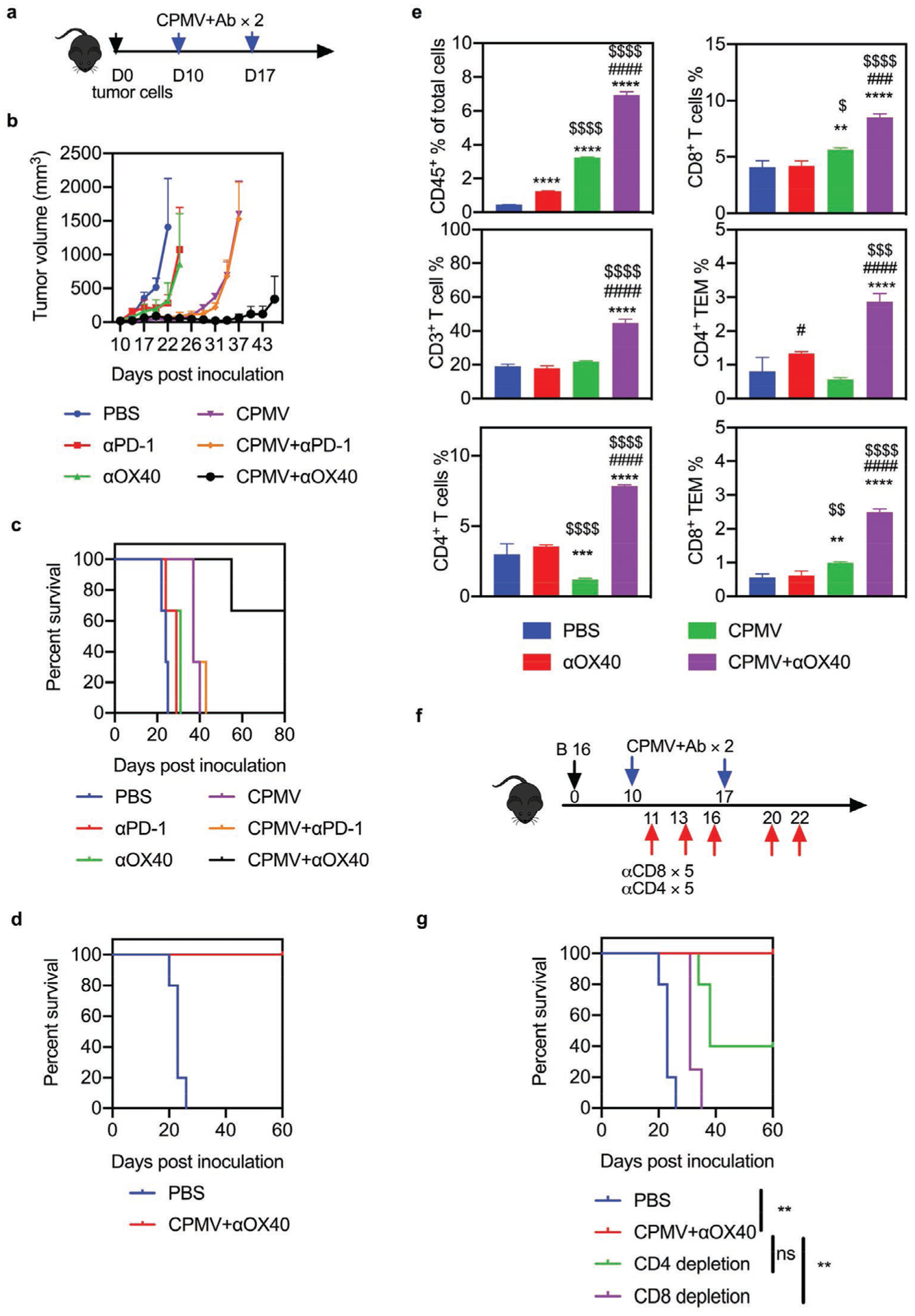
Combined CPMV and OX40 agonist treatment induces systemic antitumor effects in a B16F10 dermal melanoma model. a) Schematic of treatment strategy and dosing regimen. C57BL/6 mice were inoculated intradermally (i.d.) with 2.5 × 105 B16F10 cells on the right flank and followed by two doses (directly into the resulting tumor) of 100 μg antibody (PD-1 antagonist or OX40 agonist), 100 μg CPMV, the combination, or PBS as a control. b) Average tumor growth curve of mice receiving PBS (blue), PD-1 inhibitor (100 μg, red), OX40 agonist (100 μg, green), CPMV (100 μg, purple), CPMV+PD-1 inhibitor (orange), or CPMV+OX40 agonist (black). Data are means ± SEM (n = 3). c) Survival rate of each treatment group. d) Survival curves of combination therapy groups following tumor re-challenge. Data are means ± SEM (n = 5 for control, n = 2 for CPMV+OX40 agonist). e) Percentage of CD45+ leukocytes among total cells, the percentages of CD3+, CD4+, and CD8+ T cells among CD45+ cells, and the percentage of CD44+CD62L− effector memory T cells among CD45+ cells. Data are means ± SEM (n = 3). Statistical significance was calculated by one-way ANOVA: * versus PBS; # versus CPMV; $ versus ICT (*p < 0.05; **p < 0.01; ***p < 0.0005; ****p < 0.0001). f) Schematic of the T cell depletion strategy using a B16 dermal melanoma tumor model. g) Survival rate of each treatment group in the T cell depletion study. Data are means ± SEM (n = 4–5). Statistical significance was calculated using a log-rank Mantel–Cox test. **p < 0.01. ns: no significant difference.
The total number of infiltrating leukocytes and lymphocytes in the tumors of the combination therapy groups were significantly higher than in the control group (15.8-fold for the CPMV plus OX40 agonist combination and 2.35-fold for the CPMV plus PD-1 inhibitor combination, Figure 4e). Furthermore, tumor infiltration by CD4+ and CD8+ T cells (and their effector memory subsets) was significantly higher in the combination therapy groups compared to any monotherapy (Figure 4e and Figure S10, Supporting Information). These results suggested that the CPMV plus OX40 agonist combination elicits a robust antitumor response and establishes effective long-term immunity in the B16F10 melanoma model by amplifying the CD4+ and CD8+ TIL populations.
Finally, we conducted T cell depletion experiments to determine the functions of the infiltrated CD4+ and CD8+ T cells (Figure 4f). Tumor growth was significantly faster in mice injected with a CD8-specific antibody than in mice receiving the combination treatment. However, two of the five mice injected with the CD4-specific antibody survived the tumor challenge (Figure 4g). These results showed that the systemic antitumor response caused by CPMV and the OX40 agonist in the B16F10 melanoma model mainly required the presence of CD8+ T cells, with CD4+ T cells playing a lesser role. The B16F10 melanoma model therefore demonstrated that in situ vaccination with CPMV upregulated OX40 but not PD-1 expression on Foxp3−CD4+ effector T cells and the combination of CPMV and an OX40 agonist restored T-cell activation and achieved more potent antitumor effects than either ICT or CPMV alone.
3. Discussion and Conclusion
We previously reported that in situ vaccination with CPMV triggers a broad antitumor response, including the recruitment and repolarization of immune cells and the secretion of cytokines.[10–12] Here, we found that CPMV enhances the expression of immune checkpoint regulators on Foxp3−CD4+ effector T cells in three tumor models. Specifically, CPMV induced the expression of PD-1 in the ID8-Defb29/Vegf-A ovarian tumor model, and OX40 in the CT26 colon carcinomatosis and B16F10 melanoma models. Combination therapy comprising CPMV and antibodies targeting the induced checkpoint regulators increased tumor immunogenicity and suppressed the proliferation of immunosuppressive cell types, resulting in prolonged survival benefits and long-lasting CTL memory against tumor re-challenge. The combination of CPMV and ICT generated a synergistic antitumor effect that significantly prolonged survival compared with solo CPMV or ICT treatment groups in all three tumor models.
ICT has been used for the successful treatment of multiple solid tumors, but many patients do not respond to therapy or subsequently relapse.[23] Several factors may limit the efficacy of ICT, including insufficient tumor immunogenicity, inability to overcome the immunosuppressive microenvironment, and inability to modulate immune checkpoint signaling.[24,25] The promise of in situ CPMV immunotherapy lies in its ability to remodel the immunosuppressive tumor microenvironment into a pro-inflammatory state, suggesting that in situ CPMV vaccination combined with ICT should achieve a targeted antitumor response. We hypothesized that the upregulation of PD-1 and OX40 expression on CD4+ and CD8+ effector T cells following in situ CPMV vaccination would help to determine the most suitable ICT strategy, allowing different checkpoint-modulating drugs to be combined with CPMV for the treatment of different types of tumor.[26] The in vivo therapeutic efficacy of the combination treatments we tested in the current study appeared to confirm this hypothesis.
In the ID8-Defb29/Vegf-A ovarian tumor model, single doses of CPMV upregulated PD-1 expression on Foxp3−CD4+ effector T cells whereas multiple doses upregulated both PD-1 and OX40. Combination therapy comprising CPMV plus a PD-1 inhibitor or OX40 agonist showed greater therapeutic efficacy than any monotherapy, but only the CMPV plus PD-1 inhibitor combination ensured 100% survival following a tumor re-challenge. The PD-1 inhibitor alone increased the population of IFNγ-secreting CD4+ T cells in peritoneal wash/ascites while depleting the population of pro-tumor M2 macrophages, whereas CPMV monotherapy promoted the accumulation of both immunostimulatory and immunosuppressive cell types. In contrast, CPMV plus the PD-1 inhibitor not only limited the infiltration of immunosuppressive cell types,[27,28] but also led to the accumulation of antitumor immune cells in peritoneal wash/ascites, increasing the immunogenicity of the tumor microenvironment and boosting the therapeutic response.[25,29] We previously showed that repeated doses of CPMV upregulated IFNγ secretion in the peritoneal cavity of ID8-Defb29/Vegf-A ovarian tumor-bearing mice,[10] and this Th1 cytokine may increase PD-L1 expression on cancer cells.[30,31] We therefore evaluated the levels of PD-L1 on tumor cells (CD45−) in the ID8-Defb29/Vegf-A ovarian tumor microenvironment and showed CPMV alone significantly upregulated PD-L1 expression in cancer cells whereas CPMV plus the PD-1 inhibitor reduced PD-L1 expression by at least 50% compared to other groups. These results suggest that a PD-1-blocking antibody combined with CPMV could effectively counteract the immunosuppression elicited by solo CPMV treatments.
CPMV upregulated OX40 but not PD-1 expression on Foxp3−CD4+ effector T cells in the CT26 colon carcinomatosis and B16F10 melanoma models. The in vivo efficacy studies in both models confirmed that only the combination of CPMV plus the OX40 agonist significantly reduced the tumor burden and achieved better survival compared to monotherapy. This combination was also the only treatment that promoted the recruitment of antigen presenting cells (such as DCs, M1, and N1 cells) that subsequently triggered a T cell response.[32,33] In addition, we observed that CPMV monotherapy exclusively increased the infiltration of CD8+ T cells but had a negligible effect on CD4+ T cells in both tumor models. In contrast, the OX40 agonist monotherapy promoted the infiltration of total CD4+ T cells and the level of IFNγ secreted by these cells was higher compared to other treatment groups. CD8+ cytotoxic T cells are thought to be critical for antitumor immunity, but CD4+ T cells are required to induce antitumor effects by supporting the activation and recruitment of CD8+ T cells.[34,35] The combination therapy therefore incorporates the ability of OX40 activation to drive the proliferation of CD4+ T cells, including cytokine production and immune memory formation,[36] and the ability of CPMV to convert the tumor microenvironment from a cold to a hot state, thus significantly enhancing the infiltration and activation of both CD4+ and CD8+ effector T cells and the attenuation of immunosuppressive cells. The synergistic effects of these processes lead to profound antitumor immunity.
Oncolytic virus immunotherapy is an attractive concept because oncolytic viruses replicate selectively within cancer cells, ultimately causing lysis and the release of tumor-associated antigens that induce antitumor immunity.[37,38] In contrast, CPMV does not replicate within and kill cancer cells directly, and thus forms the basis of a novel class of immunostimulatory adjuvants. We previously demonstrated that both RNA-containing wild-type CPMV and RNA-free virus-like particle (VLP or eCPMV) are potent in situ vaccines in treating various tumor models.[10–12] The potential immunomodulation mainly relies on the multivalent (nucleo)capsid architecture of CPMV/eCPMV, which can be recognized by pattern recognizing receptors (PRRs) and Toll-like-receptors (TLRs) on various immune cells.[39,40] CPMV/eCPMV primes innate immune responses to kill tumor cells and release tumor associated antigens in the tumor microenvironment and then initiate tumor-specific adaptive immunity. We demonstrated that CPMV is more potent than eCPMV. The encapsidated viral RNA of CPMV acts as a TLR7/8 agonist and therefore further enhances the immunomodulatory behavior, such as enhancement of antigen-processing capacity of APCs; and therefore CPMV showed stronger immunogenicity and can further promote tumor regression compared to eCPMV.[41,42] Meanwhile, the lack of genomic RNA makes eCPMV more bio-safe and potential to add functional secondary payload. The common and unique properties of CPMV and eCPMV platforms provide us high flexibility and selectivity in developing different biomedical applications.
In summary, our study shows that in situ CPMV vaccination upregulates the expression of at least two immune checkpoint regulators on CD4+ effector T cells in different tumor models. By selecting these checkpoint regulators as targets for ICT in combination with CPMV, the combined treatment recruits CD4+ and CD8+ immune cells and promotes their activation and antitumor potential. Furthermore, the combination therapy leads to tumor-specific immune responses against different cancers and induces long-lasting protection against tumor re-challenge. These findings suggest that the therapeutic effects achieved by in situ vaccination with CPMV can be harnessed to drive systemic antitumor immunity, providing a strong rationale for the clinical testing of CPMV, or other plant viruses combined with specific checkpoint-targeting drugs.
4. Experimental Section
CPMV Preparation and Therapeutic Antibodies:
Wild-type CPMV was produced in house as previously described.[43] Briefly, black-eyed pea plants (Vigna unguiculata) were inoculated with CPMV (0.1 mg mL−1) in 10 mm potassium phosphate buffer (pH 7.0) and propagated for 3 weeks. The virus concentration in plant extracts was determined by UV/vis spectroscopy (ε260 nm = 8.1 mg−1 mL cm−1), and virus integrity was determined by size exclusion chromatography. Monoclonal rat antibodies against the mouse proteins OX40/CD134 (clone OX86; rat IgG1) and PD-1/CD279 (clone RMP1–14; rat IgG2a) were purchased from BioXCell.
Cell lines and Cell Culture:
B16F10 cells (American Type Culture Collection) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM, Thermo Fisher Scientific), supplemented with 10% (v/v) fetal bovine serum (FBS, Atlanta Biologicals) and 1% (v/v) penicillin-streptomycin (Thermo Fisher Scientific). CT26-luc cells (a gift from Dr. Jeremy Rich, UCSD) were cultured in complete medium (RPMI 1640 containing 10% (v/v) FBS and 1% (v/v) penicillin-streptomycin). ID8-Defb29/Vegf-A cells were transfected with luciferase as previously described,[42] and maintained in RPMI 1640 medium supplemented with 10% (v/v) FBS and 1% (v/v) Pen/Strep, 2 mm l-glutamine, 1 mm sodium pyruvate, and 0.05 mm 2-mercaptoethanol. Both cell lines were maintained at 37 °C, 5% CO2, below 50% confluence, and early-passage cultures were used in the experiments.
Tumor Inoculation and Animal Studies:
All experiments were conducted in accordance with UCSD’s Institutional Animal Care and Use Committee and involved female C57BL/6 or BALB/c mice (The Jackson Laboratory) 6–8 weeks of age. For the dermal melanoma tumor model, 2.5 × 105 B16F10 cells were suspended in 50 μL PBS and were injected (i.d.) into the right flank of C57BL/6 mice on day 0. CPMV (100 μg), antibody (50 μg), or both reagents were administered by intratumoral injection in 30 μL PBS. Tumor volumes were measured using a digital caliper. The tumor volume (mm3) was calculated as follows: (long diameter × short diameter2)/2. Animals were euthanized when the tumor volume exceeded 1500 mm3. For the ovarian tumor model, 2 × 106 ID8-Defb29/Vegf-A cells per 200 μL PBS were injected (i.p.) into C57BL/6 mice. The mice were monitored weekly for signs of tumor progression, including abdominal distension, weight, circumference, and other morbidity indicators. CPMV (100 μg), antibody (50 μg), or both reagents were administered weekly by injection (i.p.) in 200 μL PBS for six treatments in total. Mice were euthanized when their weight reached 35 g or when moribund. For the colon tumor model, 5 × 105 CT26-luc cells per 200 μL PBS were injected (i.p.) into BALB/c mice. CPMV (50 μg), antibody (50 μg), or both reagents were administered on days 7 and 14 by injection (i.p.) in 200 μL PBS (it should be noted that 100 μg CPMV alone was effective in the treatment of CT26 tumors; we lowered the dose to investigate the combination therapy). Tumor growth was monitored by body weight and total bioluminescence imaging, based on the i.p. injection of 100 mg kg−1 luciferin (Thermo Fisher Scientific) followed by analysis in an IVIS Spectrum Imaging System (PerkinElmer). Total bioluminescence was determined using Living Image software (PerkinElmer). Regions of interest were quantified as average radiance (photons s−1). Mice were euthanized when their weight reached 24 g or when moribund.
Quantification of IFNγ:
Peritoneal cavity washes or cell culture supernatants were tested by enzyme-linked immunosorbent assay (ELISA) to detect IFNγ (BioLegend) according to the manufacturer’s instructions.
Flow Cytometry:
Fresh tumor cells were excised from mice, processed into single-cell suspensions and washed in cold PBS containing 1 mm EDTA, and then resuspended in staining buffer (PBS containing 2% (v/v) FBS, 1 mm EDTA, 0.1% (w/v) sodium azide). Fc receptors were blocked using anti-mouse CD16/CD32 (Biolegend) for 15 min and then tested with the following fluorescence-labeled antibodies (BioLegend) for 30 min at 4 °C: CD45 (30-F11), CD11b (M1/70), CD86 (GL-1), major histocompatibility complex class II (MHCII, M5/114.15.2), Ly6G (1A8), CD11c (N418 A), F4/80 (BM8), Ly6C (HK1.4), NK1.1 (PK136), CD4 (GK1.5), CD3ε (145–2V11 A), CD8α (53–6.7), CD44 (IM7), CD62L (MEL-14), and isotype controls. For intracellular cytokine staining, single-cell suspensions were made from the spleens or tumors of treated mice. Splenocytes (106 cells mL−1) were co-cultured with freeze–thaw tumor lysates (106 cells mL−1) or CPMV (0.1 mg mL−1) for 48 h and treated with brefeldin A (10 mg mL−1) for the last 5 h at 37 °C. Following staining for surface antibodies as described above, the cells were fixed in 3% paraformaldehyde, permeabilized with 0.1% (w/v) saponin, and incubated with anti-Foxp3 (MF-14, BioLegend) or anti-IFNγ (XMG1.2, BioLegend) for 30 min in 0.1% (w/v) saponin. Cells were washed twice and resuspended in staining buffer for data acquisition. Flow cytometry was carried out using a BD LSRII cytometer (BD Biosciences), and the data were analyzed using FlowJo software (Tree Star). OneComp eBeads (eBiosciences) were used as compensation controls.
Depletion of CD4+ and CD8+ T Cells:
For the CT26-luc colon tumor model, monoclonal antibodies (BioXCell) specific for CD4 (clone GK1.5, rat IgG2b), and CD8 (clone 53–6.7, rat IgG2a) were injected on the first day after treatment, and on days 2, 5, 8, and 10 thereafter, each at a dose of 0.1 mg per injection. For the B16F10 melanoma tumor model, the same antibodies at the same dose were injected on days 1, 3, 6, and 10 after the first treatment.[26,44]
Statistical Analysis:
All results are expressed as means ± SEM (n = 3 – 5) as indicated. Student’s t-test was used to compare the statistical difference between two groups, and one-way or two-way analysis of variance (ANOVA) with Sidak’s or Tukey’s multiple comparison tests were used to compare three or more groups (*p < 0.05, **p < 0.01, ***p < 0.0005, ****p < 0.0001). Survival rates were analyzed using the log-rank (Mantel–Cox) test (**p < 0.01). All statistical tests were performed using GraphPad Prism v7.0 (GraphPad Software).
Supplementary Material
Acknowledgements
This work was supported in part by a grant from the National Institutes of Health, NCI Alliance for Cancer Nanotechnology (U01CA218292 to N.F.S.) and a gift by Michael R. Shaughnessy. Jooneon Park (UCSD) is thanked for help with monitoring animals.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Chao Wang, Department of NanoEngineering, University of California San Diego, La Jolla, CA 92093, USA.
Nicole F. Steinmetz, Department of NanoEngineering, Bioengineering, Radiology, Moores Cancer Center, Center for Nano-Immunoengineering, University of California San Diego, La Jolla, CA 92093, USA
Reference
- [1].Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, Horn L, Drake CG, Pardoll DM, Chen L, Sharfman WH, Anders RA, Taube JM, McMiller TL, Xu H, Korman AJ, Jure-Kunkel M, Agrawal S, McDonald D, Kollia GD, Gupta A, Wigginton JM, Sznol M, Engl N. J. Med 2012, 366, 2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Topalian SL, Sznol M, McDermott DF, Kluger HM, Carvajal RD, Sharfman WH, Brahmer JR, Lawrence DP, Atkins MB, Powderly JD, Leming PD, Lipson EJ, Puzanov I, Smith DC, Taube JM, Wigginton JM, Kollia GD, Gupta A, Pardoll DM, Sosman JA, Hodi FS, J. Clin. Oncol 2014, 32, 1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].McDermott DF, Drake CG, Sznol M, Choueiri TK, Powderly JD, Smith DC, Brahmer JR, Carvajal RD, Hammers HJ, Puzanov I, Hodi FS, Kluger HM, Topalian SL, Pardoll DM, Wigginton JM, Kollia GD, Gupta A, McDonald D, Sankar V, Sosman JA, Atkins MB, J. Clin. Oncol 2015, 33, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, West AN, Carmona M, Kivork C, Seja E, Cherry G, Gutierrez AJ, Grogan TR, Mateus C, Tomasic G, Glaspy JA, Emerson RO, Robins H, Pierce RH, Elashoff DA, Robert C, Ribas A, Nature 2014, 515, 568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Champiat S, Ferté C, Lebel-Binay S, Eggermont A, Soria JC, OncoImmunology 2014, 3, e27817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Sato-Kaneko F, Yao S, Ahmadi A, Zhang SS, Hosoya T, Kaneda MM, Varner JA, Pu M, Messer KS, Guiducci C, Coffman RL, Kitaura K, Matsutani T, Suzuki R, Carson DA, Hayashi T, Cohen EEW, JCI Insight 2017, 2, e93397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Escorcia FE, Postow MA, Barker CA, Cancer J 2017, 23, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Pfirschke C, Engblom C, Rickelt S, Cortez-Retamozo V, Garris C, Pucci F, Yamazaki T, Poirier-Colame V, Newton A, Redouane Y, Lin YJ, Wojtkiewicz G, Iwamoto Y, Mino-Kenudson M, Huynh TG, Hynes RO, Freeman GJ, Kroemer G, Zitvogel L, Weissleder R, Pittet MJ, Immunity 2016, 44, 343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Melero I, Berman DM, Aznar MA, Korman AJ, Gracia JLP, Haanen J, Nat. Rev. Cancer 2015, 15, 457. [DOI] [PubMed] [Google Scholar]
- [10].Wang C, Fiering SN, Steinmetz NF, Adv. Ther 2019, 2, 1900003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Murray AA, Wang C, Fiering SN, Steinmetz NF, Mol. Pharmaceutics 2018, 15, 3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Lizotte PH, Wen AM, Sheen MR, Fields J, Rojanasopondist P, Steinmetz NF, Fiering SN, Nat. Nanotechnol 2016, 11, 295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, Gupta R, Tsai JM, Sinha R, Corey D, Ring AM, Connolly AJ, Weissman IL, Nature 2017, 545, 495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Freeman GJ, ong AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne MC, Horton HF, Fouser L, Carter L, Ling V, Bowman MR, Carreno BM, Collins M, Wood CR, Honjo T, J. Exp. Med 2000, 192, 1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Marin-Acevedo JA, Dholaria B, Soyano AE, Knutson KL, Chumsri S, Lou Y, J. Hematol. Oncol 2018, 11, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Gramaglia I, Jember A, Pippig SD, Weinberg AD, Killeen N, Croft M, J. Immunol 2000, 165, 3043. [DOI] [PubMed] [Google Scholar]
- [17].Rogers PR, Song J, Gramaglia I, Killeen N, Croft M, Immunity 2001, 15, 445. [DOI] [PubMed] [Google Scholar]
- [18].Guo Z, Wang X, Cheng D, Xia Z, Luan M, Zhang S, PLoS One 2014, 9, e89350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Roby KF, Taylor CC, Sweetwood JP, Cheng Y, Pace JL, Tawfik O, Persons DL, Smith PG, Terranova PF, Carcinogenesis 2000, 21, 585. [DOI] [PubMed] [Google Scholar]
- [20].Scarlett UK, Cubillos-Ruiz JR, Nesbeth YC, Martinez DG, Engle X, Gewirtz AT, Ahonen CL, Conejo-Garcia JR, Cancer Res. 2009, 69, 7329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Cubillos-Ruiz JR, Engle X, Scarlett UK, Martinez D, Barber A, Elgueta R, Wang L, Nesbeth Y, Durant Y, Gewirtz AT, Sentman CL, Kedl R, Conejo-Garcia JR, J. Clin. Invest 2009, 119, 2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Teng MWL, Ngiow SF, Ribas A, Smyth MJ, Cancer Res. 2015, 75, 2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Hoos A, Nat. Rev. Drug Discovery 2016, 15, 235. [DOI] [PubMed] [Google Scholar]
- [24].Sharma P, Allison JP, Science 2015, 348, 56. [DOI] [PubMed] [Google Scholar]
- [25].Gajewski TF, Schreiber H, Fu YX, Nat. Immunol 2013, 14, 1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Sagiv-Barfi I, Czerwinski DK, Levy S, Alam IS, Mayer AT, Gambhir SS, Levy R, Sci. Transl. Med 2018, 10, eaan4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Allavena P, Sica A, Garlanda C, Mantovani A, Immunol.l Rev 2008, 222, 155. [DOI] [PubMed] [Google Scholar]
- [28].Tiemessen MM, Jagger AL, Evans HG, van Herwijnen MJC, John S, Taams LS, Proc. Natl. Acad. Sci. U. S. A 2007, 104, 19446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Gajewski TF, Semin. Oncol 2015, 42, 663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Boyerinas B, Jochems C, Fantini M, Heery CR, Gulley JL, Tsang KY, Schlom J, Cancer Immunol. Res 2015, 3, 1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Abiko K, Matsumura N, Hamanishi J, Horikawa N, Murakami R, Yamaguchi K, Yoshioka Y, Baba T, Konishi I, Mandai M, Br. J. Cancer 2015, 112, 1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Liao JB, Disis ML, Gynecol. Oncol 2013, 130, 667. [DOI] [PubMed] [Google Scholar]
- [33].Fridlender ZG, Sun J, Mishalian I, Singhal S, Cheng G, Kapoor V, Horng W, Fridlender G, Bayuh R, Worthen GS, Albelda SM, PLoS One. 2012, 7, e31524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Laidlaw BJ, Craft JE, Kaech SM, Nat. Rev. Immunol 2016, 16, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Phares TW, Stohlman SA, Hwang M, Min B, Hinton DR, Bergmann CC, J. Virol 2012, 86, 2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Aspeslagh S, Postel-Vinay S, Rusakiewicz S, Soria JC, Zitvogel L, Marabelle A, Eur. J. Cancer 2016, 52, 50. [DOI] [PubMed] [Google Scholar]
- [37].Kaufman HL, Kohlhapp FJ, Zloza A, Nat. Rev. Drug Discovery 2015, 14, 642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Lichty BD, Breitbach CJ, Stojdl DF, Bell JC, Nat. Rev. Cancer 2014, 14, 559. [DOI] [PubMed] [Google Scholar]
- [39].Kawai T, Akira S, Nat. Immunol 2010, 11, 373. [DOI] [PubMed] [Google Scholar]
- [40].Bachmann MF, Jennings GT, Nat. Rev. Immunol 2010, 10, 787. [DOI] [PubMed] [Google Scholar]
- [41].Albakri MM, Veliz FA, Fiering SN, Steinmetz NF, Sieg SF, Immunology 2020, 159, 183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Wang C, Beiss V, Steinmetz NF, J. Virol 2019, 93, e00129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Wellink J, in Plant Virology Protocols: From Virus Isolation to Transgenic Resistance (Eds: Foster GD, Taylor SC), Humana Press, Totowa, NJ: 1998, pp. 205–209. [Google Scholar]
- [44].Liu Z, Ravindranathan R, Kalinski P, Guo ZS, Bartlett DL, Nat. Commun 2017, 8, 14754. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.