

Abstract
Oligodeoxynucleotides (ODNs) with CpG motifs have potent immunostimulatory effects on many subsets of immune cells. For example, Class B CpG-ODNs, such as ODN1826 induce the phagocytic activity of macrophages by activating the Toll-like receptor 9 signaling pathway. Systemic ODN delivery results in unfavorable pharmacokinetic profiles and can trigger adverse effects. To address this issue, plant virus-like particles (VLPs) are developed for the targeted delivery of ODN1826 to tumor-associated macrophages (TAMs). ODN1826 is encapsulated by the in vitro disassembly and reassembly of Cowpea chlorotic mottle virus (CCMV), producing VLPs that are structurally analogous to the native virus. The encapsulation of ODN1826 in CCMV-derived VLPs promotes ODN uptake by TAMs ex vivo and significantly enhance their phagocytic activity. The antitumor activity of the VLPs in vivo is also evaluated, revealing that the direct injection of ODN1826 VLPs into established tumors induces a robust antitumor response by increasing the phagocytic activity of TAMs in the tumor microenvironment. CCMV encapsulation significantly enhances the efficacy of ODN1826 compared to the free drug, slowing tumor growth and prolonging survival in mouse models of colon cancer and melanoma.
Keywords: biomimetic particle, cancer immunotherapy, CpG, macrophages, nanomedicine, virus particles
1. Introduction
Bacterial DNA and synthetic oligodeoxynucleotides (ODNs) containing CpG motifs have significant immunostimulatory effects on various subsets of immune cells.[1,2] These effects are caused by the presence of unmethylated CpG dinucleotides,[3] which trigger signaling via Toll-like receptor 9 (TLR9).[4,5] Immunotherapy using CpG-ODNs has been successful in the treatment of allergy and infectious diseases,[6,7] and also in the treatment of cancer.[8,9]
Initial studies demonstrated the promising efficacy of CpG-ODNs in several preclinical tumor models, especially hematologic malignancies, such as B cell leukemia and lymphoma.[9] The CpG-ODNs triggered TLR9 signaling and the secretion of proinflammatory cytokines, inducing the activity of CD4+ Th1 cells and eliciting a cytotoxic CD8+ T cell response in vivo.[10] Following these promising preclinical studies, several clinical trials explored the potential of CpG-ODNs as immunoadjuvants delivered as single agents or in combination with cancer vaccines.[11–14] Although these trials failed to demonstrate sufficient antitumor efficacy, they nevertheless provided evidence that TLR9 agonists are well tolerated by cancer patients. Primary adverse effects included local immunostimulation and dose-dependent local reactions, as well as systemic influenza-like symptoms.[5] There is also evidence that prolonged systemic treatment with CpG-ODNs causes liver and kidney toxicity, and may lead to autoimmune disorders.[15]
The success of CpG-ODN therapy is also limited by the presence of nucleases in vivo, which reduce the half-life of ODN drugs and result in unfavorable pharmacokinetic profiles.[16] A second barrier is the strong negative charge carried by CpG-ODNs, which inhibits their interactions with immune cells. Strategies to improve the immunomodulatory properties and efficacy of CpG ODNs include chemical modification with a phosphorothioate backbone,[17] lipids,[18,19] or G-rich DNA ligands[20] to enhance chemical stability, and the encapsulation of ODNs using gold nanoparticles,[21,22] DNA tetrahedra, or hydrogels to enhance delivery.[23–25] Although these approaches can increase the preclinical efficacy of ODNs, they require complex chemical modifications, use synthetic delivery vehicles, or involve the assembly of complex DNA nanostructures.
Virus-like particles (VLPs) are ideal platforms for the targeted delivery of therapeutic nucleic acids, because this is analogous to the natural evolutionary function of viruses.[26] The delivery of CpG-ODNs has already been achieved using VLPs derived from the Hepatitis B virus core antigen and bacteriophage Qβ, leading to more potent immunostimulatory activity and improved pharmacodynamic behavior.[27] For example, vaccination with ODN-loaded VLPs presenting epitope p33 from the Lymphocytic choriomeningitis virus glycoprotein induced the accumulation of peptide-specific CD8+ T cells, protecting mice from a challenge with recombinant Vaccinia virus and eradicating established solid fibrosarcomas.[27]
As an alternative to VLPs derived from animal viruses or bacteriophages, we have developed nanomedical VLP platforms based on plant viruses. These are advantageous because plant viruses are noninfectious in mammals, easy and inexpensive to produce in large quantities by molecular farming in plants, highly immunogenic, rapidly taken up by immune cells (particularly antigen-presenting cells), and some plant viruses can induce antitumor responses when introduced into the tumor microenvironment (TME) as an in situ vaccine.[28–32] Among the diverse applications of plant viruses in drug delivery,[33] Cowpea chlorotic mottle virus (CCMV) is mainly used as a gene delivery vector.[34,35] Its potential for the delivery of CpG-ODNs and its use as an in situ vaccine have not been explored thus far.
Here, we investigated the use of CCMV-based VLPs for the targeted delivery of CpG-ODNs to tumor-associated macrophages (TAMs) in the TME. We focused on the type B CpG ODN1826, which was recently shown to induce the phagocytic activity of macrophages and thus exert antitumor effects, even overcoming the CD47-mediated “don’t eat me” signal on tumor cells.[36] We hypothesized that the encapsulation of CpG-ODNs in the CCMV particle would protect the cargo from the harsh environment and promote uptake by macrophages in the TME, thus enhancing their phagocytic activity. We evaluated the antitumor efficacy of CCMV-encapsulated ODN1826 in two murine tumor models.
2. Results and Discussion
2.1. Disassembly and Reassembly of CCMV to Encapsulate ODNs
CCMV particles were produced by molecular farming and purified by chloroform extraction, poly-ethylene glycol (PEG) precipitation and ultracentrifugation. The encapsulation of ODNs was achieved by pH/salt-dependent disassembly and reassembly (Figure 1A).[37,38] Briefly, wild-type CCMV particles were disassembled by increasing the pH and salt concentration of the buffer, which also precipitated the native viral genomic RNA. The resulting CCMV capsid protein solution was then mixed with therapeutic ODN1826 or the negative control ODN2138 at a 6:1 (w/w) ratio[38] and reassembled in a dilute, low-pH buffer in the presence of Mg2+.[37] After reassembly, the CCMV-ODN VLPs were purified by ultracentrifugation to remove excess ODNs and capsid proteins. If the low-pH buffer contained a high concentration of salts, the CCMV capsid proteins reassembled without nucleic acids to form empty particles (eCCMV).
Figure 1.
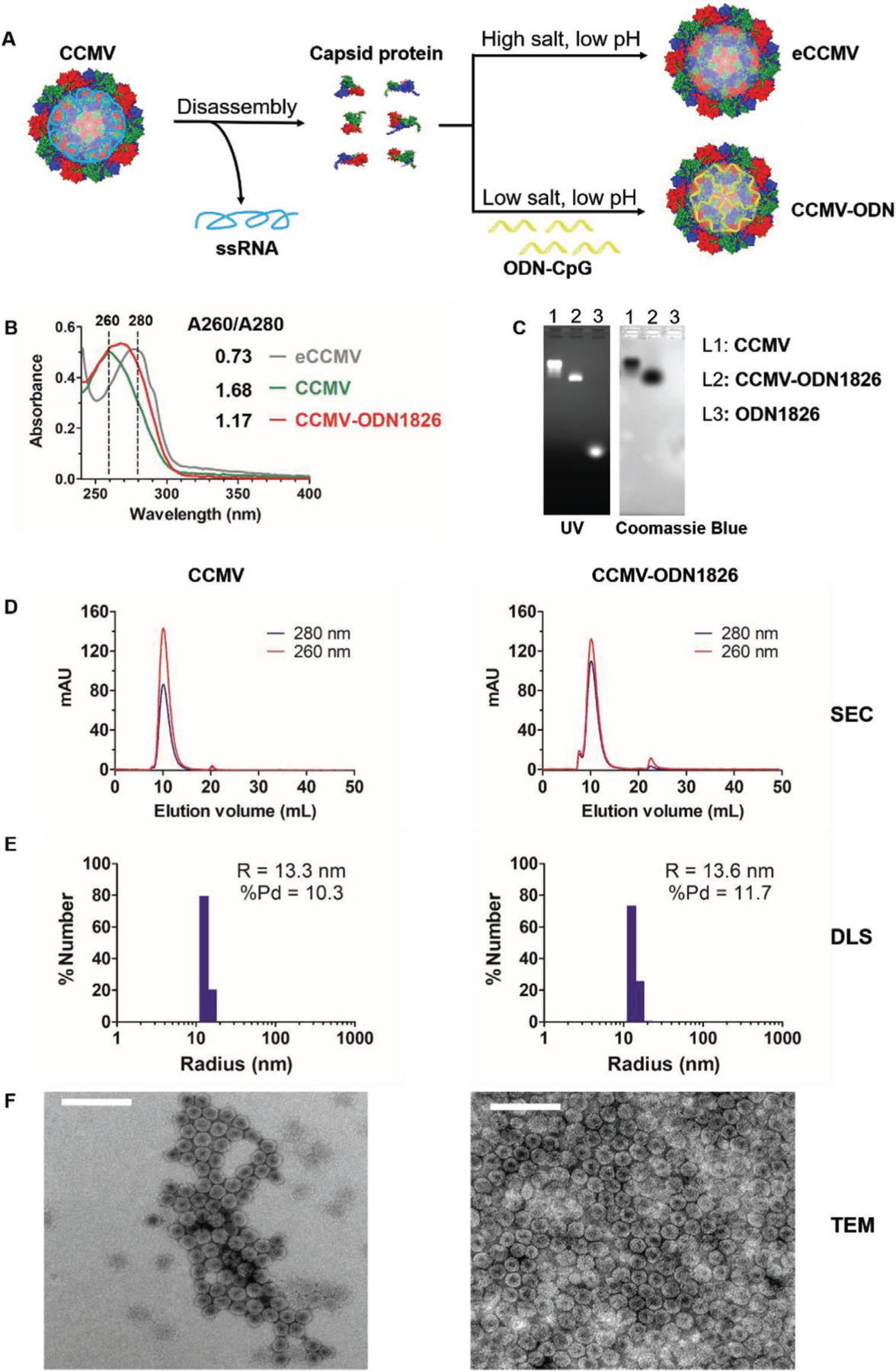
Preparation and characterization of CCMV-derived VLPs containing ODNs. A) Schematic diagram of CCMV disassembly and reassembly. B) Analysis of CCMV, eCCMV, and CCMV-ODN1826 by UV–vis absorbance spectrophotometry. C) Analysis of CCMV, CCMV-ODN1826, and ODN1826 by native agarose gel electrophoresis. D) Analysis of CCMV and CCMV-ODN1826 by size-exclusion chromatography (SEC). E) Analysis of CCMV and CCMV-ODN1826 by dynamic light scattering (DLS). F) Analysis of negatively-stained CCMV and CCMV-ODN1826 by transmission electron microscopy (TEM). Scale bar = 100 nm.
UV–vis absorbance spectrophotometry (Figure 1B) revealed that the A260/A280 ratio of the CCMV-ODN1826 VLPs was 1.17, which is similar to the native CCMV particles (1.68) and thus indicated the successful encapsulation of nucleic acids. In contrast, the A260/A280 ratio of the eCCMV particles was 0.73, reflecting the lack of nucleic acid, which absorbs strongly at 260 nm. The RiboGreen nucleic acid assay indicated that 50 CpG-ODNs were encapsulated per particle. This corresponds to 1100 nucleotides in total, whereas ≈3200 nucleotides of RNA can be packaged (similar to the size of the wild-type CCMV genomic RNA).[38] CCMV therefore appears to package RNA more efficiently than ODNs. The presence of ODNs in the VLPs was further confirmed by native agarose gel electrophoresis (Figure 1C) and the structural integrity of the particles was confirmed by size-exclusion chromatography (SEC) (Figure 1D), with neither method showing any evidence of free ODNs in the VLP preparations. The precise size of the VLPs was determined by dynamic light scattering (DLS) (Figure 1E) and transmission electron microscopy (TEM) (Figure 1F). Both measurements confirmed that CCMV-ODN1826 was structurally similar to wild-type CCMV, revealing an icosahedral structure with a diameter of ≈27 nm. This disassembly and reassembly approach is therefore a rapid and efficient way to prepare CCMV-ODN VLPs.
2.2. In Vitro Stability of CCMV-ODN VLPs
To exclude the possibility that the stability of the CCMV-ODN VLPs is limited to the conditions imposed during the disassembly and reassembly procedure, we tested their stability under physiological conditions. Wild-type CCMV and CCMV-ODN1826 were diluted to 1 mg mL−1 in phosphate buffered saline (PBS, pH 7.4) and incubated at 37 °C. Samples were taken periodically for SEC analysis on a Superose 6 column, which eluted intact particles at 10 mL and capsid proteins at 20 mL. We found that the CCMV-ODN1826 VLPs were stable under physiological conditions, with 90% of the particles remaining intact after 10 d (Figure 2A). The wild-type CCMV particles were less stable than the VLPs under these conditions. Note that eCCMV is unstable under physiological conditions, and these particles were therefore not tested. We also treated CCMV-ODN1826 VLPs with DNase I. There was no evidence of ODN digestion over 2 d, based on the unchanged A260/A280 ratio (Figure 2B). This confirmed that the encapsulated ODNs are resistant to nuclease digestion.
Figure 2.
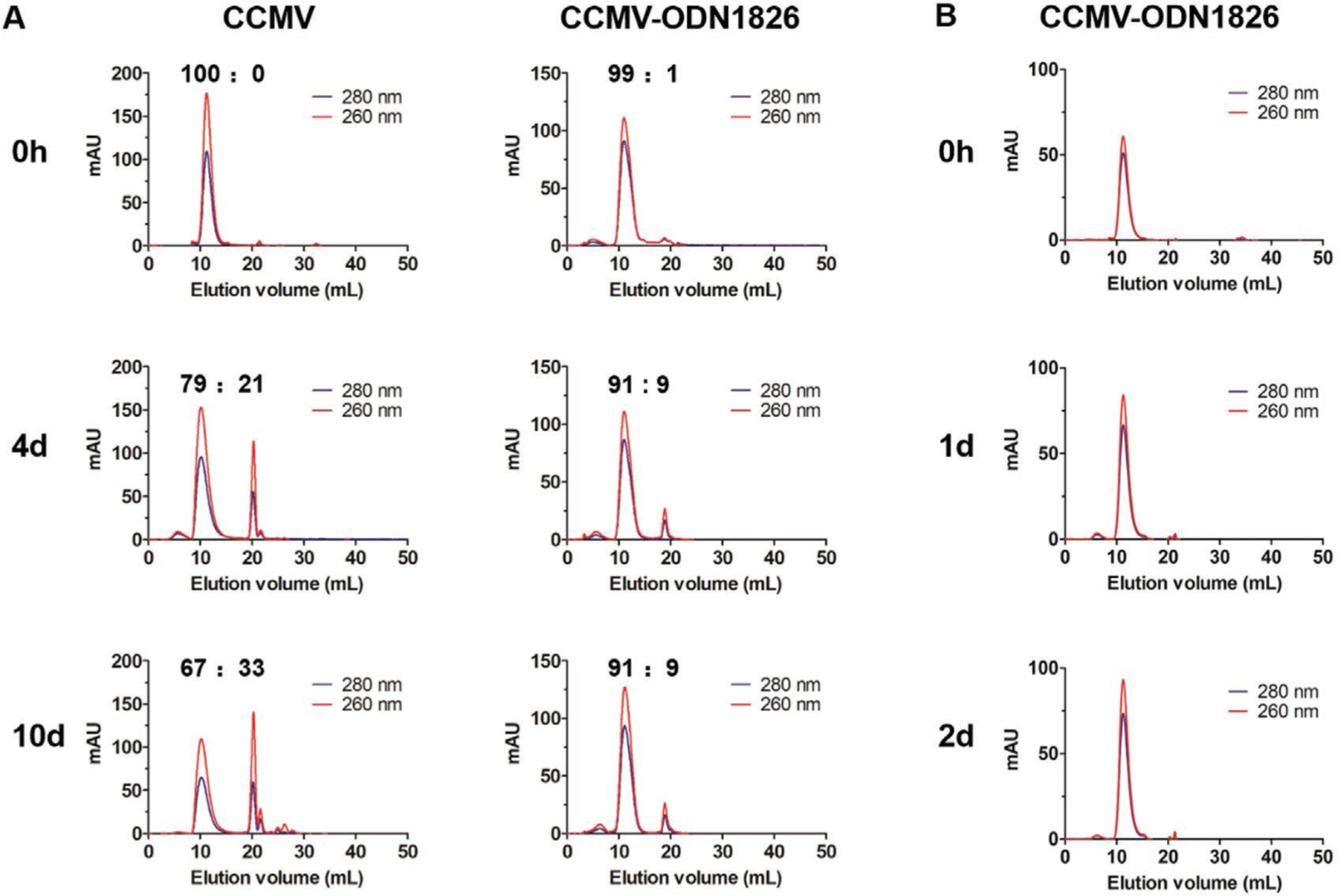
Stability of CCMV and CCMV-ODN VLPs under physiological conditions. A) The stability of CCMV and CCMV-ODN1826 under in vitro physiological conditions (PBS, pH 7.4) over time (0, 4, and 10 days). B) Stability of CCMV-ODN1826 in PBS containing 5 U mL−1 DNase.
2.3. Delivery of ODN1826 to Macrophages by VLPs
Next we evaluated the uptake of CCMV-ODN VLPs by macrophages and cancer cells representing the TME. ODN1826 labeled with cyanine 5 (CY5) was encapsulated during particle reassembly to prepare fluorescent VLPs (CCMV-ODN1826-CY5). Single-cell suspensions derived from murine subcutaneous colon cancer (CT26) were incubated with CCMV-ODN1826-CY5 particles or ODN1862-CY5 in its free form, then harvested and stained with fluorescent antibodies to differentiate TAMs and cancer cells prior to flow cytometry. As shown in Figure 3A, both TAMs and tumor cells were able to take up CCMV-ODN1826-CY5 more efficiently than ODN1826-CY5. However, the encapsulation of ODN1826 increased the proportion of CY5-positive tumor cells from 6.8% to 7.8%, which is a nonsignificant change, but the proportion of CY5-positive TAMs increased significantly from 19% to 25% (Figure 3A,B) resulting in a much higher fold-change in the number of CY5-positive TAMs compared to tumor cells (Figure 3C). We also analyzed the mean fluorescence intensity (MFI) of the TAMs and tumor cells, and observed similar trends (Figure 3D,E). Taken together, these data indicate that encapsulation significantly increases the uptake of ODNs by TAMs but not by tumor cells. This is an important outcome because CpG-ODNs activate immune cells and initiate antitumor responses, but if taken up by cancer cells they can also induce phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt) signaling and therefore promote tumor progression.[39,40] Therefore it is critical that the nanocarrier does not increase the uptake of CpG-ODNs by cancer cells.
Figure 3.
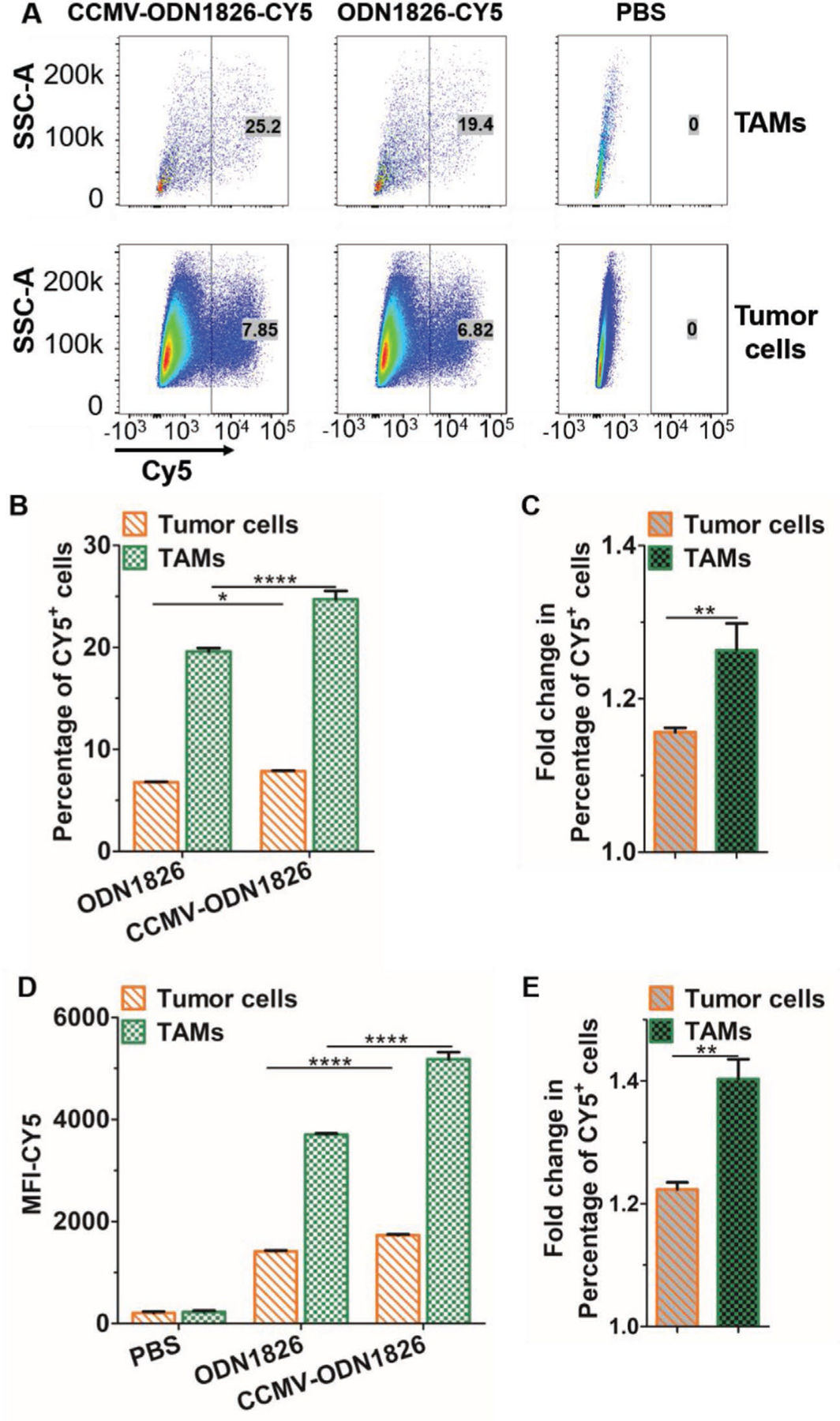
Internalization of ODN1826 and CCMV-ODN1826 by tumor cells and TAMs derived from single-cell suspensions of CT26 tumors. A) Gating strategy for the analysis of macrophages and tumor cells. TAMs were gated as CD45+ CD11b+ F4/80+ cells, and tumor cells were gated as CD45− cells. B) Percentage of CY5+ cells, confirming the uptake of ODN1826 or CCMV-ODN1826. C) Fold-change in the percentage of CY5+ cells following incubation with CCMV-ODN1826 compared to free ON1826. D) Mean fluorescence intensity (MFI-CY5) of tumor cells and TAMs. E) Fold-change in the MFI following incubation with CCMV-ODN1826 compared to free ON1826. Values are means ± stand deviations (n = 3). Statistical significance was determined using a two-tailed Student’s t-test: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
2.4. In Vitro Activation of Macrophages
ODN1826 has been shown to increase the phagocytic activity of macrophages and thus enhance their effects against tumors.[36] Given the ability of CCMV-based VLPs to stabilize ODN1826 and promote its uptake specifically by macrophages in vitro, we anticipated that CCMV-ODN1826 would activate macrophages and induce phagocytosis more efficiently than free ODN1826. To test this hypothesis in vitro, murine colon cancer CT26 cells were labeled with carboxyfluorescein succinimidyl ester (CFSE) and co-cultured with bone-marrow derived macrophages (BMDMs). We then added PBS, eCCMV, wild-type CCMV, free ODN1826, CCMV-ODN1826, or CCMV-ODN2138, the latter containing an inactive control ODN with the same properties as ODN1826 but with the CpG dinucleotides replaced by GpC. As expected, PBS, eCCMV, wild-type CCMV, and CCMV-ODN2138 did not stimulate the BMDMs, whereas ODN1826 or CCMV-ODN1826 increased the number of BMDMs that attacked cancer cells and captured them by phagocytosis (Figure 4A). Importantly, the CCMV-ODN1826 treatment was much more efficacious than the free ODN1826 (Figure 4A).
Figure 4.

Activation of BMDMs by stimulation with ODNs and VLPs. A) In vitro phagocytosis of CFSE-labeled CT26 tumor cells by BMDMs stimulated with free ODN1826, CCMV-ODN1826, or control VLPs (eCCMV, CCMV, or CCMV-ODN2138). BMDMs were gated as F4/80+ cells. B) CT26-luc cell survival following coculture with BMDMs for 48 h in the presence of ODN1826 or the VLPs listed above. C) The iNOS/Arg ratio of BMDMs following stimulation with ODN1826 or the VLPs listed above, with LPS and IL-4 as controls for M1 and M2 polarization, respectively. Values are means ± standard deviations (n = 3). Statistical significance was determined by one-way ANOVA with Tukey’s test: *p < 0.05, **p < 0.01, ***p < 0.001.
To determine whether the phagocytic capacity of the macrophages correlated with antitumor activity, we co-cultured BMDMs with luciferase-labeled CT26 (CT26-luc) cells and treated them with the same reagents listed above. Again, the control treatments did not have a significant effect on the number of surviving cancer cells, whereas both ODN1826 and CCMV-ODN1826 stimulated the antitumor activity of the BMDMs resulting in a decrease in tumor cell survival (Figure 4B). As above, the efficacy of CCMV-ODN1826 was significantly greater than the free ODN1826 (Figure 4B).
In addition to functions such as phagocytosis, immunostimulation often induces the secretion of cytokines, so we also investigated the cytokine profile of BMDMs after stimulation with ODN1826 or CCMV-ODN1826. Both reagents increased the secretion of proinflammatory cytokines, such as IL-12, TNF-α, IL-1β, IL-6, CCL2, and IFN-λ (Figure S1, Supporting Information). Interestingly, CCMV-ODN1826 induced the secretion of less IL-12, IL-6, and CCL2 but more IFN-λ than free ODN1826.
The cytokine profiles induced by ODN1826 and CCMV-ODN1826 were similar to those induced by lipopolysaccharides (LPS), which promote M1 polarization in macrophages and thus trigger proinflammatory and antitumor activity.[41] Immunotherapies that shift the polarization of TAMs toward the M1 phenotype have shown promising efficacy against cancer in preclinical models.[42,43] We therefore tested BMDMs stimulated with ODN1826 or CCMV-ODN1826 by flow cytometry for the presence of inducible nitric oxide synthase (iNOS), an M1 marker, and arginase 1 (Arg1), an M2 marker. LPS was used as a control for M1 polarization and IL4 as a control for M2 polarization, which suppresses inflammation and promotes tumor growth.[44] The ratio of iNOS/Arg reflected the balance of M1/M2 polarization, with LPS producing a high ratio and IL-4 producing a low ratio (Figure 4C). The incubation of BMDMs with the control reagents (eCCMV, CCMV, or CCMV-ODN2138) maintained the M2 polarity of the cells, whereas incubation with ODN1826 or CCMV-ODM1826 increased the iNOS/Arg ratio toward M1 polarization.
Finally, we confirmed that ODN1826 and CCMV-ODN1826 indeed activate and signal through TLR9; this was achieved by assessing NF-κB activation in HEK293 cells expressing a given TLRs (TLR2, 3, 4, 5, 7, 8, 9, and 13) (Figure 5A). Both ODN1826 and CCMV-ODN1826 activated the TLR9 at the same level, but no activation of the other TLRs was observed (Figure 5A). Neither wild-type CCMV nor CCMV-ODN2138 showed any TLR activation, consistent with the assayed antitumor activities of these VLPs and incubated in co-culture of BMDMs and tumor cells (see Figure 4). To validate these observations, we assayed activation of RAW-Blue cells upon incubation with CCMV-ODN and controls. RAW-Blue cells express many pattern recognition receptors (PRRs), TLRs, NOD-like receptors, RIG-I-like receptors, and C-type lectin receptors. Similar to the assays performed using the HEK293 cells, NF-κB and AP.1 activation is assayed to determine whether activation of any of the PRRs is achieved (the assay does not differentiate which pathways are activated). Similar to the results of TLRs activation using the HEK293 cells (Figure 5A), only stimulation using ODN1826 or CCMV-ODN1826 led to signaling in RAW-Blue cells (Figure 5B). Together, these data confirm that ODN1826 in free form or encapsulated in CCMV is effective to activate PRRs and signals through TLR9.
Figure 5.
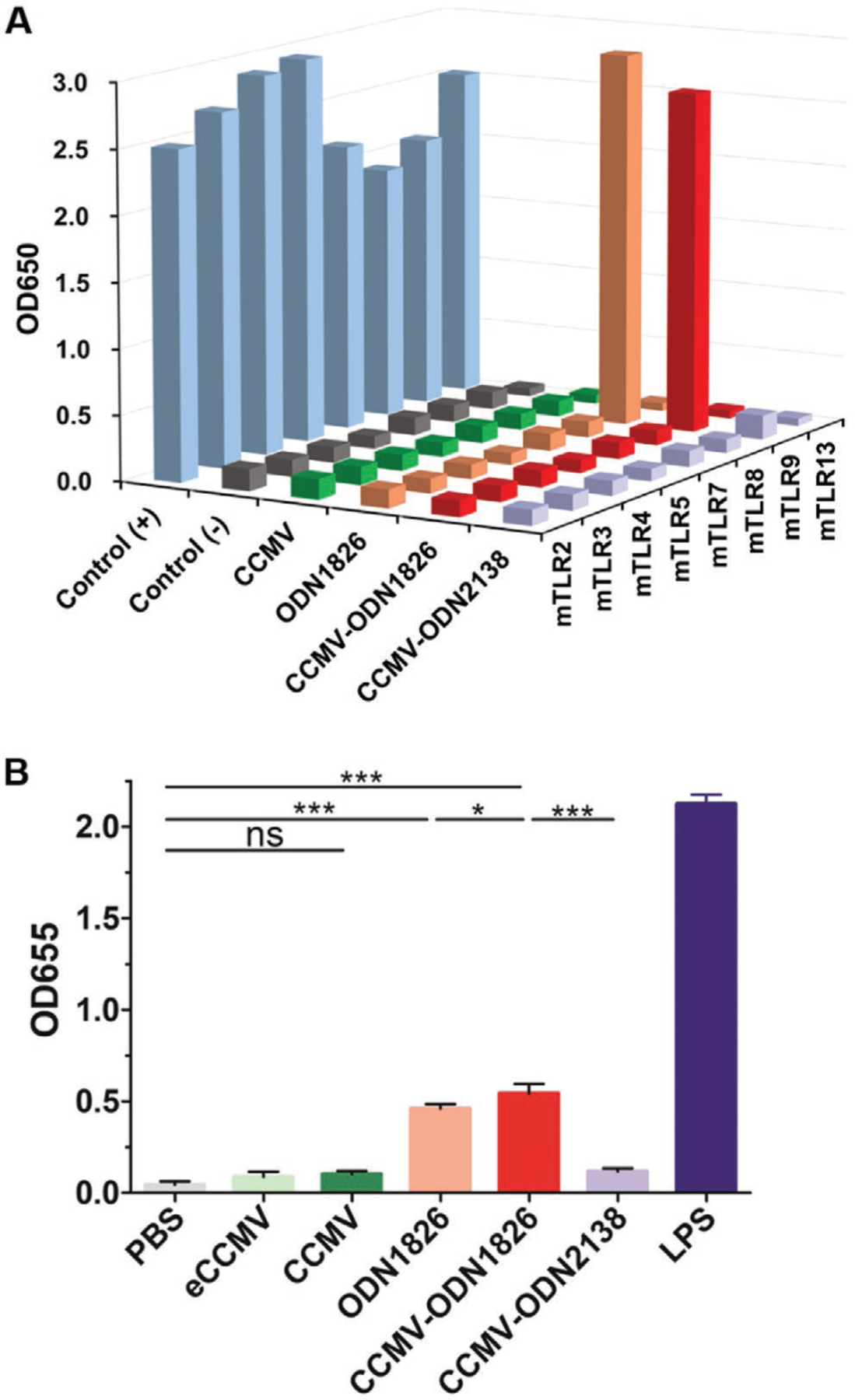
A) NF-κB activation by stimulating HEK293 cells with free ODN1826, CCMV-ODN1826, or control VLPs (eCCMV, CCMV, or CCMV-ODN2138). Averages of three independent experiments are shown. B) Stimulation of PRRs in RAW-Blue cells. RAW-Blue cells were stimulated with free ODN1826, CCMV-ODN1826, or control VLPs (eCCMV, CCMV, or CCMV-ODN2138). NF-κB or AP.1 activation was determined using QUANTI-Blue. Values are means ± standard deviations (n = 4). Statistical significance was determined by one-way ANOVA with Tukey’s test: *p < 0.05, **p < 0.01, ***p < 0.001.
2.5. In Vivo Antitumor Activity
Finally, we investigated whether the in vitro results described above could be replicated in vivo, using the CT26 murine colon cancer model. CT26 cells were subcutaneously (s.c.) inoculated into the right flank of BALB/c mice, and tumor-bearing mice were randomized to one of six treatment groups: i) PBS, ii) eCCMV, iii) wild-type CCMV, iv) CCMV-ODN2138 (negative control), v) ODN1826, and vi) CCMV-ODN1826. All reagents were delivered intratumorally (i.t.) at doses of 100 μg for the VLPs or 10 μg for the free ODN per injection. Treatments were started when the tumor volume reached 30–50 mm3, and three treatments were administered in total at weekly intervals (Figure 6A). Wild-type CCMV, eCCMV, and CCMV-ODN2138 showed no efficacy against CT26 tumors, as shown by the tumor growth curves (Figure 6B,C) and survival data (Figure 6D), which were similar to the PBS group. Treatment with free ODN1826 slowed tumor growth (50% drop in the day 26 mean normalized tumor volume compared to the PBS group) and prolonged median survival from 26 d in the PBS group to 31 d. However, treatment with CCMV-ODN1826 was much more efficacious, significantly inhibiting tumor growth (80% drop in the day 26 mean normalized tumor volume compared to the PBS group) and significantly prolonging median survival from 26 to 42 d.
Figure 6.
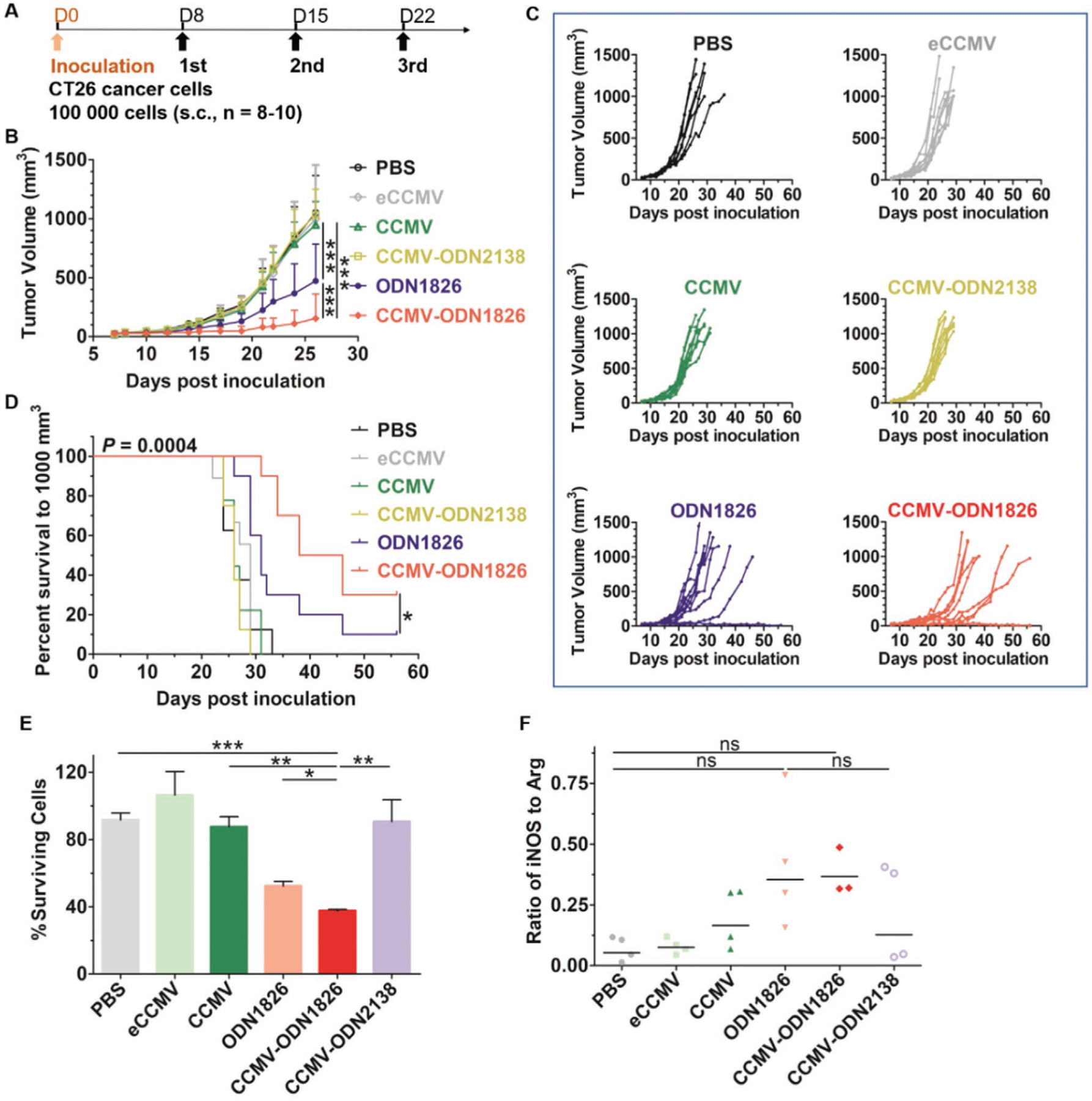
In vivo antitumor efficacy of free ODN1826, CCMV-ODN1826, and control VLPs in a murine model of colon cancer. A) Treatment schedule. B) Growth curves of CT26 tumors, showing mean tumor volumes ± standard deviations (n = 8–10). Statistical significance was determined by two-way ANOVA (***p < 0.001). C) Tumor growth kinetics for each treatment group. D) Survival curves. Statistical significance was determined using the long-rank (Mantel-Cox) test (*p < 0.05). E) CT26-luc cell survival following coculture for 48 h with TAMs in the presence of free ODN1826 or VLPs. F) The iNOS/Arg ratio in TAMs of CT26 tumors after treatment with ODN1826 or VLPs. Values are means ± standard deviations (n = 3–4). Statistical significance was determined by one-way ANOVA with Tukey’s test (ns = not significant, *p < 0.05, **p < 0.01, ***p < 0.001).
To confirm whether ODN1826 increased the phagocytic capacity of TAMs, we isolated the TAMs from the CT26 tumors and co-cultured them ex vivo with CT26-luc cells. The results (Figure 6E) were similar to those reported above for the BMDM cells (Figure 4B). Free ODN1826 stimulated moderate phagocytic activity, whereas CCMV-ODN1826 stimulated substantial phagocytic activity. The ex vivo phagocytic capacity of the TAMs correlated with the tumor growth curves (Figure 6B,E). We also monitored cytokine secretion by the TAMs and found that ODN1826 and CCMV-ODN1826 induced the same palette of proinflammatory cytokines reported above for BMDMs (Figure S1, Supporting Information). We also evaluated M1/M2 polarization in the TME following the treatment of CT26 tumor- bearing mice on day 12 with free ODN1826, CCMV-ODN1826, and the panel of control VLPs. Tumors were harvested after 24 h and single-cell suspensions were prepared. The iNOS/Arg ratio was measured by flow cytometry, and again the results with ex vivo TAMs (Figure 6F) were similar to those reported above for BMDMs (Figure 4F), with both free ODN1826 and CCMV-ODN1826 triggering a slight increase in the iNOS/Arg ratio indicating polarization toward M1. However, the magnitude of the response was not as high as that observed for the BMDMs.
To determine whether CCMV-ODN1826 can enhance the in vivo antitumor activity of macrophages in other tumor models, we carried out a second study using the B16F10 murine melanoma model. B16F10 cancer cells were inoculated intradermally (i.d.). Tumor-bearing mice were treated with free ODN1826, CCMV-ODN1826, or the control VLPs listed above (except eCCMV) following the same schedule as the CT26 tumor model (Figure 7A). Unlike the CT26 model, treatment with wild-type CCMV showed some efficacy against B16F10 melanoma, resulting in slower tumor growth than the PBS treatment and CCMV-ODN2138 control (Figure 7B,C), but there was no significant extension of survival (Figure 7D). Both free ODN1826 and CCMV-ODN1826 showed efficacy in the melanoma model, but the CCMV-ODN1826 treatment was much more efficacious (80% drop in the day 28 mean normalized tumor volume compared to the free ODN1826 group). Encapsulation of the ODN also prolonged survival, with the ODN1826 group showing a median survival of 31 d but the CCMV-ODN1826 group extending that to 39 d. The greater in vivo antitumor efficacy achieved by the encapsulation of ODN1826 for the treatment of melanoma was therefore consistent with that in colon cancer.
Figure 7.
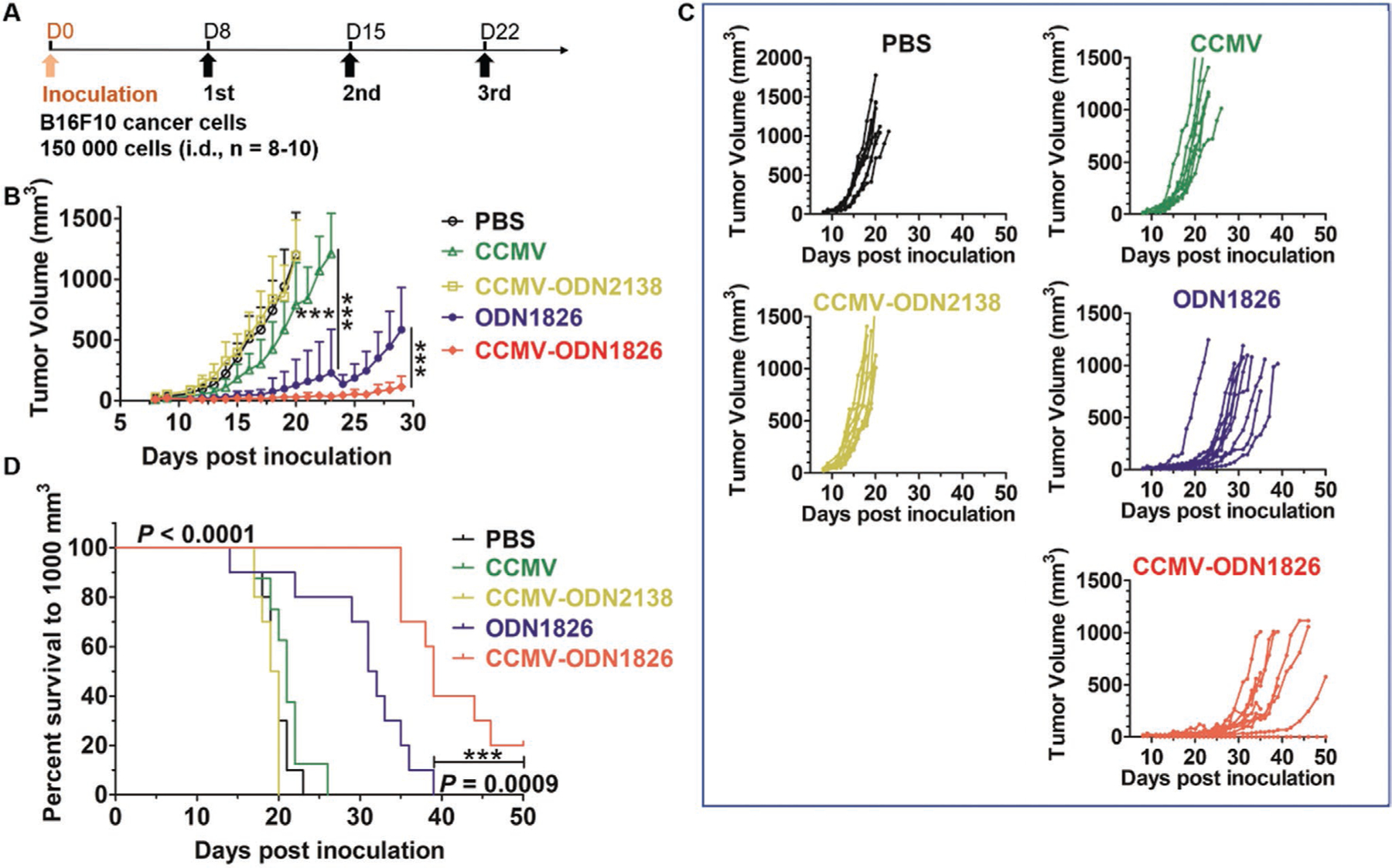
In vivo antitumor efficacy of free ODN1826, CCMV-ODN1826, and control VLPs in a murine model of melanoma. A) Treatment schedule. B) Growth curves of melanoma tumors, showing mean tumor volumes ± standard deviations (n = 8–10). Statistical significance was determined by two-way ANOVA (***p < 0.001). C) Tumor growth kinetics for each treatment group. D) Survival curves. Statistical significance was determined using the long-rank (Mantel-Cox) test (***p < 0.001).
Given the central of role of macrophages within the TME of many tumor types; targeting macrophages with nanotechnologies to shift their polarization toward M1 and therefore antitumor phenotype has been explored using a number of approaches. It has been shown that systemic administration of nanoparticles can induce macrophage polarization with the TME to shift TAMs toward a cancer cell-killing phenotype.[45] Nevertheless, considering the rather low dose of systemically administered nanoparticles that accumulates in tumors;[46] adverse effects may outweigh the clinical benefit. Intratumoral administration has now been recognized in the clinic; as various in situ vaccines, including the oncolytic virotherapy Talimogene laherparepvec (T-VEC), are administered via that route.[47] In our study, we demonstrate that intratumoral injection of CpG-ODNs packaged by CCMV resulted in enhanced antitumor efficacy over free CpG-ODNs. Given the versatile engineering design space of the plant viral nanoparticle platform, this strategy could be applied to deliver various small molecule immunostimulators into the TME.
3. Conclusion
We used the disassembly and reassembly of CCMV to efficiently package ODN1826, which contains immunostimulatory CpG dinucleotides, and found that the potency of the encapsulated ODN1826 was significantly higher than that of the free drug. The reassembled VLPs remained stable under physiological conditions, and were preferentially taken up by innate immune cells (mainly TAMs) in the TME. Encapsulated ODN1826 was a more potent stimulator of TAMs than the free drug, achieving more effective antitumor activity in vitro and in vivo, and exhibiting enhanced therapeutic efficacy for the treatment of murine colon cancer and melanoma. These preclinical results show great promise and indicate that VLPs loaded with CpG-ODNs should be considered for clinical development.
4. Experimental Section
Preparation of CCMV and VLPs:
CCMV was propagated by molecular farming, purified as previously reported[48] and stored in 0.1 m sodium acetate/1 × 10−3 m ethylenediaminetetraacetic acid (EDTA) (pH 4.8). The VLPs were prepared according to the well-established procedures.[35,37] Briefly, CCMV virions were dialyzed against disassembly buffer (0.5 m CaCl2, 50 × 10−3 m Tris-HCl pH 7.5, 1 × 10−3 m EDTA, 1 × 10−3 m dithiolthreitol (DTT), 0.5 × 10−3 m phenylmethylsulfonyl fluoride (PMSF)) at 4 °C for 24 h in a 3.5 kDa molecular weight cut-off (MWCO) Slide-a-Lyzer dialysis cassette (Thermo Fisher Scientific) and centrifuged at 12 000 × g for 30 min at 4 °C to pellet the precipitated viral RNA. The supernatant was then centrifuged at 220 000 × g for 2 h at 4 °C to pellet any nondisassociated virus particles. The supernatant (capsid protein solution) was then dialyzed against protein buffer (1 m NaCl, 20 × 10−3 m Tris pH 7.2, 1 × 10−3 m EDTA, 1 × 10−3 m DTT, 1 × 10−3 m PMSF) as above and stored at 4 °C. The concentration of the CCMV coat proteins was determined by measuring the absorbance at 280 nm (A280) using the extinction coefficient (ε) = 1.27 μL μg−1 cm−1, and the purity was determined by calculating the A280/A260 ratio. Values greater than 1.5 indicated the absence of nucleic acid contamination, and these particles were suitable for reassembly.
The encapsulated ODNs were prepared by mixing coat proteins and ODNs (ODN1826 and ODN2138, Invitrogen) at a 6:1 (w/w) ratio in protein buffer. The mixture was dialyzed against RNA assembly buffer (50 × 10−3 m Tris-HCl pH 7.2, 50 × 10−3 m NaCl, 10 × 10−3 m KCl, 5 × 10−3 m MgCl2, 1 × 10−3 m DTT) and then immediately against virus suspension buffer (50 × 10−3 m sodium acetate, 8 × 10−3 m magnesium acetate, pH 4.5) in each case for at least 6 h at 4 °C using the equipment described above. The eCCMV sample was reassembled under high-salt conditions in the presence of Mg2+ (0.9 m NaCl, 0.1 m sodium acetate pH 4.8, 10 × 10−3 m MgCl2, 0.5 × 10−3 m PMSF). The reassembled VLPs were purified by ultracentrifugation at 220 000 × g for 1 h at 4 °C. All VLPs were diluted in PBS immediately before in vitro and in vivo experiments.
Characterization of VLPs:
The Quant-iT RiboGreen RNA Assay kit (Thermo Fisher Scientific) was used to quantify the encapsulated ODNs using the free ODNs as standards. UV–vis spectra were acquired using a NanoDrop spectrophotometer (Thermo Fisher Scientific) at a concentration 0.5–1 mg mL−1. 5 μg of each VLP preparation was also separated by 1.2% (w/v) agarose gel electrophoresis in 1 × TAE running buffer, and imaged the gels before and after staining with Coomassie Brilliant Blue using the FluorChem R imaging system. SEC analysis was carried out using an AKTA Explorer chromatography system (GE Healthcare) fitted with a Suprose 6 Increase column. ≈100 μg of each VLP preparation was loaded and SEC was carried out at a flow rate 0.5 mL min−1, measuring the absorbance at 260 and 280 nm. A DynaPro NanoStar (Wyatt Technology) was used for the analysis of VLPs by DLS at a concentration 0.3 mg mL−1. VLPs negatively stained with uranyl acetate were observed using a FEI Tecnai F30 transmission electron microscope.
In Vitro Stability Analysis:
Wild-type CCMV and CCMMV-ODN1826 were diluted in PBS (pH 7.4) at a concentration of 1 mg mL−1 and incubated at 37 °C, with 100 μL aliquots taken periodically for SEC analysis and PBS as the elution buffer. The ratio of intact to disassembled particles was determined by measuring the absorbance at 260 nm. For resistance to DNase digestion, CCMV-ODN1826 was diluted in PBS (pH 7.4) at a concentration of 2 mg mL−1. DNase I (Thermo Fisher Scientific) was added at final concentration of 5 U mL−1, then incubated at 37 °C. Aliquots were taken periodically for SEC analysis.
Cells and Mice:
Mouse CT26 (ATCC) and CT26-luc cells (provided by Jeremy Rich, UCSD) were maintained in RPMI 1640 medium (Corning Life Sciences) supplemented with 10% (v/v) fetal bovine serum (Atlanta Biologicals) and 1% (v/v) penicillin/streptomycin (Thermo Fisher Scientific). B16F10 cells (ATCC) were maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Corning Life Sciences) supplemented with 10% (v/v) fetal bovine serum and 1% (v/v) penicillin/streptomycin. The cells were incubated at 37 °C in a 5% CO2 atmosphere. RAW-Blue cells (InvivoGen) were maintained in DMEM supplemented with 10% (v/v) fetal bovine serum, 1% (v/v) penicillin/streptomycin, 100 μg mL−1 Normocin and Zeocin.
BALB/c and C57BL/6 mice were obtained from The Jackson Laboratory and were housed at the Moores Cancer Center (UCSD) in groups with unlimited access to food and water. All mouse studies were performed in compliance with the Institutional Animal Care and Use Committee (IACUC) of UCSD.
Generation of BMDMs:
BMDMs were derived by isolating bone marrow from BALB/c mice. Following treatment with ACK lysis buffer (Thermo Scientific Fisher), bone-marrow cells were cultured in Iscove’s modified Eagle’s medium (Thermo Scientific Fisher) supplemented with 10% (v/v) fetal bovine serum, 2 × 10−3 m GlutaMAX, 10 ng mL−1 gentamicin, and 10 ng mL−1 M-CSF (Peprotech) for 7–10 d. Before experiments, BMDMs were resuspended in the same medium without M-CSF.
Analysis of VLP Internalization by Macrophages and Tumor Cells:
CT26 tumors were harvested and single-cell suspensions were prepared using the Tumor Dissociation Kit (Miltenyi Biotec) with the gentleMACS Octo Dissociator (Miltenyi Biotec). 1 × 106 cells were suspended in RPMI 1640 medium supplemented with 10% (v/v) fetal bovine serum and 1% (v/v) penicillin/streptomycin and the cells were transferred to six-well plates. Then 0.2 μg ODN1826-CY5 or 2 μg CCMV-ODN1826-CY5 were added and the cells were incubated at 37 °C in a 5% CO2 atmosphere for 1 h. The cells were harvested and treated with Fc-block (BioLegend), then stained with Zombie Aqua (BioLegend). The cells were then washed in fluorescence-activated cell sorting (FACS) buffer (2% fetal bovine serum in PBS), and then stained with the following antibodies: Pacific Blue antimouse CD45, FITC antimouse CD11b, and PE antimouse F4/80 (all from BioLegend). After washing with FACS buffer, cells were acquired for immediate analysis using a BD LSR II flow cytometer (BD Biosciences). Data were analyzed using FlowJo v8.6.3 software. Tumor cells were gated as CD45− cells, and TAMs were gated as CD45+ CD11b+ F4/80+ cells.
In Vitro Phagocytosis Assay:
CT26 cells were labeled with 5 × 10−6 m CFSE (Thermo Fisher Scientific) before seeding in transparent 12-well tissue culture plates at 2 × 105 cells per well. BMDMs were gathered as a single-cell suspension and seeded at 2 × 105 cells per well. Then ODN1826 (final concentration 50 μg mL−1) or VLPs (final concentration 500 μg mL−1) were added and incubated the cells for 24 h as above. The cells were then harvested and stained with the following antibodies: Pacific Blue antimouse CD45, APC/Cy7 antimouse CD11b, and PE antimouse F4/80 (all from BioLegend). After washing with FACS buffer, the cells were acquired for immediate analysis as above. Unstimulated BMDMs were gated as CD45+ CD11b+ F4/80+ cells, and phagocytic BMDMs were gated as CD45+ CD11b+ F4/80+ FITC+ cells.
In Vitro Antitumor Assay:
CT26-luc cells were collected as a single-cell suspension and seeded into 96-well tissue culture plates at 5 × 104 cells per well. BMDMs were collected by mechanical detachment and seeded at a 1:1 macrophage-to-tumor-cell ratio. ODN1826 and the VLPs were then added as described above. After coculture for 48 h, tumor cell survival was determined by luciferase assay (Goldbio), with luminescence measured using a Tecan microplate reader. Tumor cell survival was determined by normalizing the luminescence to tumor-only controls. CT26 tumors were collected and single-cell suspensions were prepared as described above. TAMs were isolated using Anti-F4/80 MicroBeads (Miltenyi Biotec). The antitumor assay using TAMs was performed as described above for BMDMs.
M1/M2 Polarization:
BMDMs were collected as single-cell suspensions and seeded into 24-well tissue culture plates at 2 × 105 cells per well. Then ODN1826 (final concentration 50 μg mL−1) or VLPs (final concentration 500 μg mL−1) were added and cocultured the cells for 48 h as above. The cells were harvested and incubated for 10 min at 4 °C with an antimouse CD16/32 antibody (BioLegend) to block Fcγ receptors, washed with FACS buffer and stained with the antibodies against CD45, CD11b, and F40–80 listed above. The cells were then fixed, permeabilized in Cytofix/Cytoperm Plus (BD Biosciences) and stained with PE/Cy7 antimouse iNOS (BD Biosciences) and AF488 antimouse Arg1 (BD Biosciences). After washing, the cells were acquired for immediate analysis by flow cytometry as described above. Macrophages were gated as CD45+ CD11b+ F4/80+ cells. The iNOS/Arg ratio was determined by dividing the number of iNOS+ macrophages by the number of Arg+ macrophages in each sample. For in vivo M1/M2 polarization, CT26 tumors were collected and single-cell suspensions were prepared as above, and the cells were stained and the data analyzed using the procedures described for BMDMs.
Cytokine Assay:
BMDMs were collected as a single-cell suspension and seeded into 24-well tissue culture plates at 2 × 105 cells per well. ODN1826 (final concentration 50 μg mL−1) or the VLPs (final concentration 500 μg mL−1) were added and the cells were incubated for 48 h. The presence of IFN-γ, IL-6, IL-1β, TNF-α, IL-12 p70, and CCL2 secreted by BMDMs was detected in the supernatant using cytokine kits from Thermo Fisher Scientific.
TRLs Activation:
The TLR screening was run by InvivoGen. Briefly, in a 96-well plate (200 μL total volume) containing the appropriate cells (50 000 cells per well), 20 μL of the test article or the positive control ligand was added to the wells. The media added to the wells was designed for the detection of NF-κB induced SEAP expression. After a 20 h incubation the optical density was read at 650 nm on a Molecular Devices SpectraMax 340 PC absorbance detector.
Stimulation of TLRs in RAW-Blue Cells:
RAW-Blue cells were mechanically detached and resuspended in test medium (DMEM supplemented with 10% (v/v) heat-inactivated FBS). Cells were plated on 96-well tissue plate (100 000 cells in 180 μL test medium per well), then 20 μL of ODN or CCMV samples were added to the wells. 1 μg of VLPs per well or 0.1 μg ODN1826 were added. After 20 h incubation, 20 μL of the supernatant from each well were mixed with 180 μL of QUANTI-Blue solution on another 96-well plate. After 3 h incubation, SEAP levels were determined using a Tecan microplate reader (OD655 nm).
In Vivo Antitumor Efficacy:
For the murine colon cancer model, CT26 cells were harvested and resuspended in RPMI-1640 medium at a concentration of 2 × 106 cells mL−1 and mixed 1:1 (v/v) with Matrigel (Corning) at 4 °C. BALB/c mice were injected s.c. with 100 μL of the mixture (1 × 105 cells) into the right flank. Tumors were allowed grow for 8–10 d until the volume reached 30–50 mm3 before randomization and treatments. ODN1826 and the VLPs were administered i.t. with an injection volume of 20 μL. Three treatments in total were administered at intervals of 7 d. Tumors were measured at least every other day using digital calipers. The tumor size (in cubic millimeters) was calculated using the formula: (width2 × length)/2. When the tumor size reached 1000 mm3, the mice were euthanized.
For the murine melanoma model, B16F10 cells were harvested and resuspended in PBS at a concentration of 5 × 106 cells mL−1. C57BL/6 mice were injected i.d. with 30 μL of the mixture (1.5 × 105 cells) into the right flank. Tumors were allowed grow for 8–10 d until the volume reached 30–50 mm3 before randomization, treatments and measurements were carried out as described above.
Statistical Analysis:
Data reported in the figures were analyzed and charts were generated using Prism 5 (GraphPad Software). Statistical significance was determined by two-way or one-way analysis of variance (ANOVA), unless otherwise stated. Survival data were compared using the Mantel-Cox (log-rank) test. In the figures, asterisks represent the following p values: *p < 0.05, **p < 0.01, and ***p < 0.001.
Supplementary Material
Acknowledgements
This research was supported by a grant from the National Institutes of Health, R01 CA224605 to N.F.S. The authors thank Jeremy Rich (UCSD) for providing the CT26-luc cells.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
The ORCID identification number(s) for the author(s) of this article can be found under https://doi.org/10.1002/adfm.201908743.
Contributor Information
Hui Cai, Department of NanoEngineering, University of California-San Diego, La Jolla, CA 92093, USA.
Sourabh Shukla, Department of NanoEngineering, University of California-San Diego, La Jolla, CA 92093, USA.
Nicole F. Steinmetz, Department of NanoEngineering, University of California-San Diego, La Jolla, CA 92093, USA Department of Bioengineering, Department of Radiology, Moores Cancer Center, Center for Nano-ImmunoEngineering, University of California-San Diego, La Jolla, CA 92093, USA.
References
- [1].Wooldridge JE, Ballas Z, Krieg AM, Weiner GJ, Blood 1997, 89, 2994. [PubMed] [Google Scholar]
- [2].Krieg AM, Annu. Rev. Immunol 2002, 20, 709. [DOI] [PubMed] [Google Scholar]
- [3].Krieg AM, Yi AK, Matson S, Waldschmidt TJ, Bishop GA, Teasdale R, Koretzky GA, Klinman DM, Nature 1995, 374, 546. [DOI] [PubMed] [Google Scholar]
- [4].Krieg AM, Oncogene 2008, 27, 161. [DOI] [PubMed] [Google Scholar]
- [5].Krieg AM, Nat. Rev. Drug Discovery 2006, 5, 471. [DOI] [PubMed] [Google Scholar]
- [6].Verthelyi D, Gursel M, Kenney RT, Lifson JD, Liu S, Mican J, Klinman DM, J. Immunol 2003, 170, 4717. [DOI] [PubMed] [Google Scholar]
- [7].Hanagata N, Int. J. Nanomed 2017, 12, 515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Jahrsdorfer B, Weiner GJ, Update Cancer Ther. 2008, 3, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Adamus T, Kortylewski M, Contemp. Oncol 2018, 22, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Krieg AM, J. Clin. Invest 2007, 117, 1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Valmori D, Souleimanian NE, Tosello V, Bhardwaj N, Adams S, O’Neill D, Pavlick A, Escalon JB, Cruz CM, Angiulli A, Angiulli F, Mears G, Vogel SM, Pan L, Jungbluth AA, Hoffmann EW, Venhaus R, Ritter G, Old LJ, Ayyoub M, Proc. Natl. Acad. Sci. USA 2007, 104, 8947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Molenkamp BG, van Leeuwen PA, Meijer S, Sluijter BJ, Wijnands PG, Baars A, van den Eertwegh AJ, Scheper RJ, de Gruijl TD, Clin. Cancer Res 2007, 13, 2961. [DOI] [PubMed] [Google Scholar]
- [13].Smith DA, Conkling P, Richards DA, Nemunaitis JJ, Boyd TE, Mita AC, de La Bourdonnaye G, Wages D, Bexon AS, Cancer Immunol. Immunother 2014, 63, 787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Chan E, Kwak EL, Hwang J, Heiskala M, de La Bourdonnaye G, Mita M, Cancer Chemother. Pharmacol 2015, 75, 701. [DOI] [PubMed] [Google Scholar]
- [15].Krieg AM, Nucleic Acid Ther. 2012, 22, 77. [DOI] [PubMed] [Google Scholar]
- [16].Mutwiri GK, Nichani AK, Babiuk S, Babiuk LA, J. Controlled Release 2004, 97, 1. [DOI] [PubMed] [Google Scholar]
- [17].Sands H, Gorey-Feret LJ, Cocuzza AJ, Hobbs FW, Chidester D, Trainor GL, Mol. Pharmacol 1994, 45, 932. [PubMed] [Google Scholar]
- [18].Liu H, Kwong B, Irvine DJ, Angew. Chem., Int. Ed 2011, 50, 7052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Liu H, Moynihan KD, Zheng Y, Szeto GL, Li AV, Huang B, Van Egeren DS, Park C, Irvine DJ, Nature 2014, 507, 519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Dalpke AH, Zimmermann S, Albrecht I, Heeg K, Immunology 2002, 106, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Wei M, Chen N, Li J, Yin M, Liang L, He Y, Song H, Fan C, Huang Q, Angew. Chem., Int. Ed 2012, 51, 1202. [DOI] [PubMed] [Google Scholar]
- [22].Lin AY, Almeida JP, Bear A, Liu N, Luo L, Foster AE, Drezek RA, PLoS One 2013, 8, e63550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Li J, Pei H, Zhu B, Liang L, Wei M, He Y, Chen N, Li D, Huang Q, Fan C, ACS Nano 2011, 5, 8783. [DOI] [PubMed] [Google Scholar]
- [24].Nishikawa M, Ogawa K, Umeki Y, Mohri K, Kawasaki Y, Watanabe H, Takahashi N, Kusuki E, Takahashi R, Takahashi Y, Takakura Y, J. Controlled Release 2014, 180, 25. [DOI] [PubMed] [Google Scholar]
- [25].Zhang L, Zhu G, Mei L, Wu C, Qiu L, Cui C, Liu Y, Teng IT, Tan W, ACS Appl. Mater. Interfaces 2015, 7, 24069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Rohovie MJ, Nagasawa M, Swartz JR, Bioeng. Transl. Med 2017, 2, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Storni T, Ruedl C, Schwarz K, Schwendener RA, Renner WA, Bachmann MF, J. Immunol 2004, 172, 1777. [DOI] [PubMed] [Google Scholar]
- [28].Wen AM, Steinmetz NF, Chem. Soc. Rev 2016, 45, 4074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lizotte PH, Wen AM, Sheen MR, Fields J, Rojanasopondist P, Steinmetz NF, Fiering S, Nat. Nanotechnol 2016, 11, 295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Wang C, Steinmetz NF, Adv. Healthcare Mater 2019, 8, 1801288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Wang C, Beiss V, Steinmetz NF, J. Virol 2019, 21, e00129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Cai H, Wang C, Shukla S, Steinmetz NF, Adv. Sci 2019, 6, 1802281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Sokullu E, Soleymani Abyaneh H,Gauthier MA, Pharmaceutics 2019, 11, 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Azizgolshani O, Garmann RF, Cadena-Nava R, Knobler CM, Gelbart WM, Virology 2013, 441, 12. [DOI] [PubMed] [Google Scholar]
- [35].Lam P, Steinmetz NF, Biomater. Sci 2019, 7, 3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Liu M, O’Connor RS, Trefely S, Graham K, Snyder NW, Beatty GL, Nat. Immunol 2019, 20, 265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Lavelle L, Michel JP, Gingery M, J. Virol. Methods 2007, 146, 311. [DOI] [PubMed] [Google Scholar]
- [38].Comas-Garcia M, Garmann RF, Singaram SW, Ben-Shaul A, Knobler CM, Gelbart WM, J. Phys. Chem. B 2014, 118, 7510. [DOI] [PubMed] [Google Scholar]
- [39].Gao M, Ha T, Zhang X, Wang X, Liu L, Kalbfleisch J, Singh K, Williams D, Li C, J. Infect. Dis 2013, 207, 1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Mantovani A, Allavena P, Sica A, Balkwill F, Nature 2008, 454, 436. [DOI] [PubMed] [Google Scholar]
- [41].Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P, Nat. Rev. Clin. Oncol 2017, 14, 399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, Huhn RD, Song W, Li D, Sharp LL, Torigian DA, O’Dwyer PJ, Vonderheide RH, Science 2011, 331, 1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Rolny C, Mazzone M, Tugues S, Laoui D, Johansson I, Coulon C, Squadrito ML, Segura I, Li X, Knevels E, Costa S, Vinckier S, Dresselaer T, Akerud P, De Mol M, Salomaki H, Phillipson M, Wyns S, Larsson E, Buysschaert I, Botling J, Himmelreich U, Van Ginderachter JA, De Palma M, Dewerchin M, Claesson-Welsh L, Carmeliet P, Cancer Cell 2011, 19, 31. [DOI] [PubMed] [Google Scholar]
- [44].Chanmee T, Ontong P, Konno K, Itano N, Cancers 2014, 6, 1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Reichel D, Tripathi M, Perez JM, Nanotheranostics 2019, 3, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Dai Q, Wilhelm S, Ding D, Syed AM, Sindhwani S, Zhang Y, Chen YY, MacMillan P, Chan WCW, ACS Nano 2018, 12, 8423. [DOI] [PubMed] [Google Scholar]
- [47].Conry RM, Westbrook B, McKee S, Norwood TG, Hum. Vaccines Immunother 2018, 14, 839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Ali A, Roossinck MJ, J. Virol. Methods 2007, 141, 84. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.