

Abstract
AMD3100 (plerixafor) is a vital component of many clinical and pre-clinical transplant protocols, facilitating harvest of hematopoietic stem and progenitor cells through mobilization into the peripheral blood circulation. Repeat mobilization with AMD3100 is also necessary for many patients with suboptimal first stem cell collection or those requiring repeat transplantation. In this study we investigated the mobilization efficacy of repeated AMD3100 dosages in the non-human primate and humanized mouse models. In non-human primates we demonstrate effective mobilization after the first AMD3100 administration but significantly poorer response in CD34+ and hematopoietic stem cell-enriched CD90+ cells with subsequent doses of the drug. A similar loss of efficacy with repeated administration was found in immunodeficient mice engrafted with human CD34+ cells, in whom the total human white cell population and particularly human hematopoietic stem and progenitor cells mobilized significantly less effectively following a second AMD3100 administration when compared to first dose. Together, our results are expected to inform future mobilization protocols for the purposes of peripheral blood hematopoietic stem cell extraction or for applications in which hematopoietic stem cells must be made accessible for in vivo-delivered gene targeting agents.
Keywords: Hematopoietic Stem Cells, Hematopoietic Stem Cell Mobilization, Hematopoietic Stem Cell Transplantation, Anemia, Sickle Cell
Introduction
Hematopoietic stem and progenitor cell (HSPC) transplantation constitutes the gold standard treatment for many malignant and non-malignant conditions.1 Mobilization of HSPCs from the bone marrow (BM) niche into the peripheral blood (PB) circulation is a critical part of most autologous and allogeneic HSPC transplant protocols, facilitating HSPC collection from the circulation by apheresis.2,3 This avoids invasive BM harvest, with inherent risks of infection, bleeding, structural damage, and the need for a general anaesthetic.4,5 A further advantage to transplantation of PB HSPCs rather than BM-derived cells is more rapid hematopoietic reconstitution.5,6
The most frequently administered HSPC mobilization agent is the pro-inflammatory cytokine granulocyte colony stimulating factor (G-CSF).2,7,8 AMD3100 (Plerixafor) was first approved for clinical use as a second mobilization agent in 2008, providing an additional or alternative option for patients and donors. AMD3100’s mechanism of action is primarily through reversible, competitive antagonism of the cytokine receptor CXCR4 expressed on the HSPC cell surface, interrupting the bond with its ligand CXCL12 in the BM. Interaction with a second receptor, CXCR7, for which AMD3100 acts as an allosteric agonist, is also likely to contribute to its mobilization property.9,10 In addition, AMD3100 administration has been demonstrated to reverse the CXCL12 gradient across the BM niche in mice, which may contribute to the egress of HSPCs from the BM into the PB circulation.11 AMD3100 is most frequently used alongside G-CSF, since both molecules synergize to increase numbers of circulating HSPCs and offer better chance of successful harvest.12,13 AMD3100 has also been used as a single mobilization agent in patients and healthy donors, thereby circumventing the side-effects and potentially severe complications associated with G-CSF administration.14–17 These include bone pain, ‘flu-like symptoms, and splenic rupture.18,19 AMD3100 administration, on the other hand, is usually associated with minimal side-effects and a more rapid mobilization profile than G-CSF.15,20,21 In sickle cell disease patients, single agent AMD3100 is the only safe and effective mobilization regimen available to facilitate ex vivo genome modification strategies, since G-CSF can precipitate life-threatening sickle cell crises.22–24
Mobilization of HSPCs into the PB is increasingly important in the field of ex vivo- as well as in vivo-genome engineering. Since hematopoietic stem cells are responsible for long-term repopulation of all hematopoietic cell lineages, targeting these cells for genetic manipulation by genome editing or gene transfer therapy results in modification of their entire progeny.25 This approach offers the promise of long-term therapeutic alteration within all blood cell types and hence cure of a vast array of hematological and immunological disorders.26 Ex vivo genetic manipulation of autologous HSPCs prior to reinfusion is being trialled in many preclinical applications and is already under clinical investigation for the treatment of various conditions including sickle cell disease, β-thalassemia and severe combined immunodeficiency.27–29 There is now growing interest in in vivo genome engineering of HSPCs, which in most cases also relies on mobilization of these cells into the periphery to facilitate exposure to agents such as retroviral vectors or nanoparticles.30–33
A single mobilization episode results in inadequate HSPC collection in many patients, requiring remobilization at a future date.2 Sickle cell disease patients mobilized for HSPC collection and gene therapy treatment are predicted to require repeat mobilization in one third of cases.22 Remobilization may also be employed at the point of disease relapse for second or subsequent autologous transplants.1 It is therefore important to establish the efficacy of repeated AMD3100 administration for HSPC mobilization.
In this study, we investigated the effect of repeated AMD3100 administration on HSPC mobilization in two key preclinical transplant models: the non-human primate (NHP) and the humanized mouse model. The NHP model has been extensively established by our group and has the advantage of a high degree of systemic uniformity and genetic homology to humans, allowing more direct translation to the clinic.34–36 In this model we further described the hematopoietic stem cell-enriched CD34+CD90+CD45RA− (CD90+) subset as a novel target for genome editing to reactivate fetal hemoglobin for the cure of β-hemoglobinopathies.27,34,37 While these studies used G-CSF-primed BM-derived HSPCs for gene modification and transplantation, we sought to implement a mobilization regimen based on AMD3100 alone followed by PB stem cell collection, since this more closely reproduces standard clinical practice and is currently the gold standard for sickle cell disease patients.22,24
To complement these findings in NHPs and directly examine the response of first and subsequent dosages of AMD3100 in human HSPCs, we additionally used the mouse xenotransplantation model. Non-obese diabetic (NOD)-severe combined immunodeficiency (SCID)-common γ chain−/− (NSG) mice engrafted with human CD34+ cells are commonly used as a preclinical model to study HSPC transplantation and multilineage engraftment.38–40 In addition, this model has been employed for the evaluation of in vivo gene therapy strategies involving HSPCs mobilized with G-SCF and AMD3100.41,42 Together our data demonstrate reduced mobilization of HSPCs with repeated AMD3100 administration in both the NHP and humanized mouse models.
Results
Reduced CD34+ HSPC and hematopoietic stem cell (HSC)-enriched CD90+ PB mobilization upon repeat ADM3100 mobilization in NHPs
We first investigated kinetics of mononuclear cell (MNC) mobilization (taken to be reported total white cell count) in healthy adult rhesus macaques (Macaca mulatta) receiving a first dose (n=5), a second dose (n=3) and a third dose (n=1) of AMD3100. Peak MNC count in the PB was found at 2–3 hours post AMD3100 dose, a little earlier than previously reported (Fig 1A).43
Figure 1. :
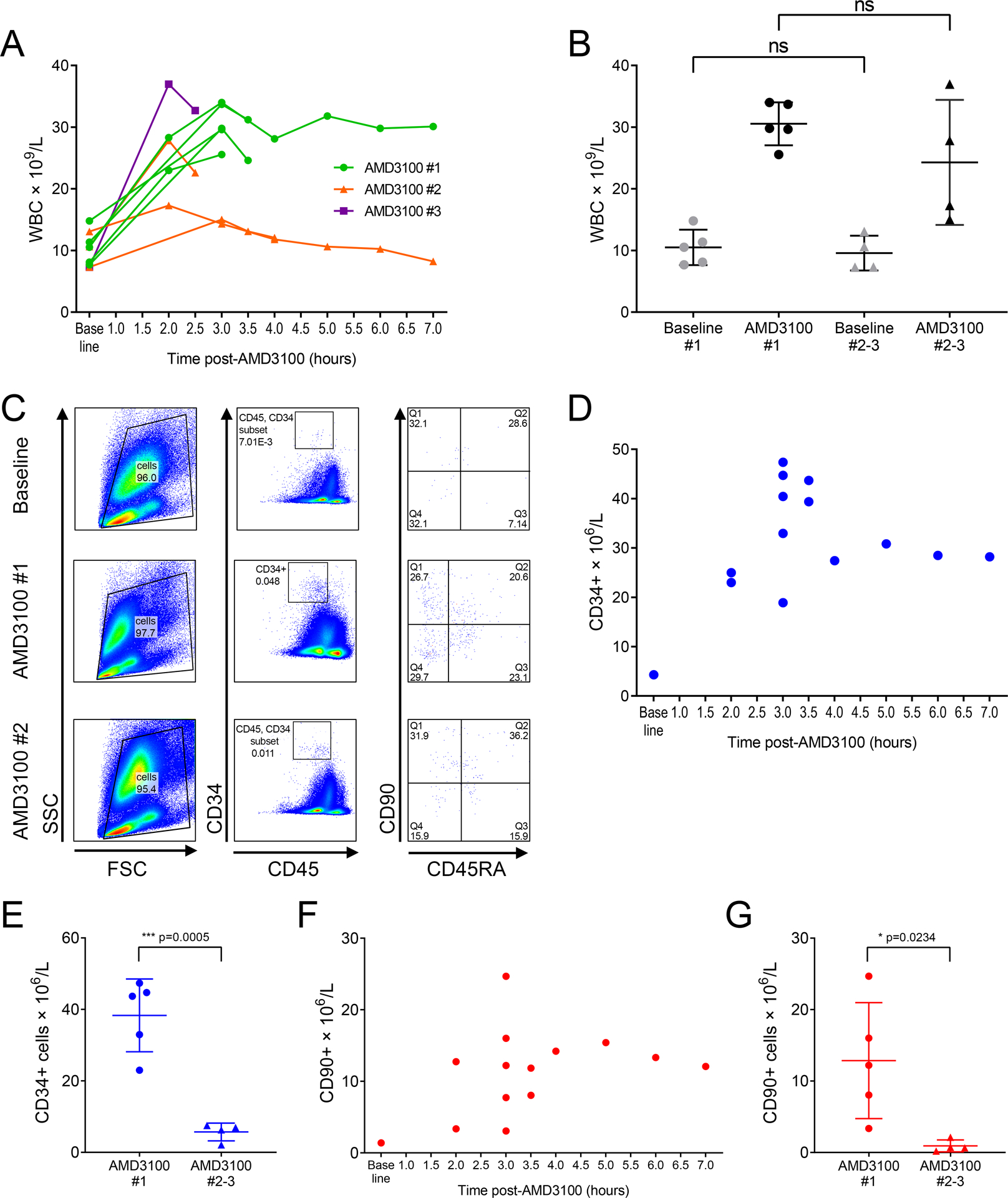
Mononuclear cell counts (MNC), CD34+ HSPCs and HSC-enriched CD90+ subpopulation counts in NHPs treated with one or repeated AMD3100 doses.
A: Longitudinal analysis of MNC count in PB of each NHP treated with 1, 2 or 3 AMD3100 doses. B: MNC counts measured pre-AMD3100 (baseline) and at the peak post-AMD3100 administration in all animals following first and subsequent AMD3100 doses (ns = non-significant). C: Representative example of flow cytometric gating strategy for the CD34+ and CD90+ populations. D: Kinetics of CD34+ cell mobilization post first dose AMD3100. E: Peak CD34+ cell mobilization post-AMD3100 in all animals following first and subsequent AMD3100 doses. F: Kinetics of CD90+ cell mobilization post first dose AMD3100. G: CD90+ count within bulk CD34+ population at point of peak CD34+ mobilization in all animals following first and subsequent AMD3100 doses. In B, E and G, bars show mean and SD.
Mean baseline MNC count prior to the first dose of AMD3100 was 10.5 × 109/l (within normal range) and rose significantly to 30.5 × 109/l after treatment (n=5, p=0.0003). Mean baseline MNC count prior to the second or third dose of AMD3100 was 9.6 × 109/l, and rose to 24.3 × 109/l after treatment (n=4, p>0.05). There was no significant difference between baseline MNC counts before first vs subsequent dose AMD3100, or between peak MNC count following first vs subsequent doses (Fig 1B). Where analysis was restricted to the results pertaining to first vs second dose AMD3100 only in animals receiving at least 2 doses (n=3), mean baseline MNC count prior to first dose AMD3100 was 11.3 × 109/l and rose significantly to 31.0 × 109/l (p= 0.0152). Prior to second dose AMD3100 baseline MNC count was 10.3 × 109/l and rose to 20.0 × 109/l (p>0.05). Again, there was no significant difference between baseline or peak MNC count when comparing first to second dose AMD3100. In summary, these data demonstrate successful increase in the MNC count in NHPs following first AMD3100, with non-significantly reduced response on repeated treatments.
Kinetics of CD34+ cell mobilization with first dose AMD3100 are consistent with those reported by other groups (Fig 1C, Fig 1D). In contrast to MNCs, peak CD34+ count in PB was significantly reduced with repeated AMD3100 dosages. Mean CD34+ cell count following the first dose of AMD3100 for all animals was 38.3 × 106/l (n=5), whereas CD34+ count measured after each subsequent dose was significantly reduced by nearly 7-fold with a mean of 5.7 × 106/l (n=4, p=0.0005, Fig 1E). Where results for animals receiving first and second dose AMD3100 only were taken, peak CD34+ cell count after first dose was 45.2 × 106/l and after second dose was 5.5 × 106/l (n=3, p=0.0011).
We were also interested to determine if similar results apply to our recently described HSC-enriched immunophenotype CD34+CD90+CD45RA−.37 The timing of CD90+ mobilization appears to follow a similar pattern to that of the total CD34+ population (Fig 1C, Fig 1F).43,44 Similarly to bulk CD34+ counts, we observed a significant decrease in the number of CD90+ cells mobilized after the second or third AMD3100 doses relative to the first dose. PB CD90+ count at time of peak CD34+ mobilization was 12.9 × 106/l (n=5) after the first dose and was reduced to 0.9 × 106/l (n=4) after subsequent dosages (p=0.0234, Fig 1G). Where results for animals receiving first and second dose AMD3100 only were taken, peak CD90+ cell count were 15.0 × 106/l and 0.5 × 106/l respectively (p>0.05).
Therefore, while total MNC count demonstrated only a non-significant trend towards reduced response with repeated doses of AMD3100, HSPC mobilization (both bulk CD34+ cells and HSC-enriched CD90+ cells) was significantly poorer with second and subsequent dosages in comparison to first AMD3100 administration.
Reduced human HSPC mobilization with repeated AMD3100 administration in the humanized mouse model
Our observation that HSPCs fails to re-mobilize with repeat AMD3100 in NHPs was unexpected, so we sought to further investigate this phenomenon in human HSPCs. For this, we used the NSG mouse transplantation model. NSG mice were engrafted with human CD34+ for 20 weeks, and subsequently received a first, then a second dose of AMD3100, and the effect on human HSPC mobilization was measured by PB analysis at 1.5 hours after dosing. Among these, a selected number of animals also received a third dose of AMD3100. Human engraftment, as reflected by the frequency of human CD45+ (hCD45+) cells, remained stable during the course of the experiment (Fig 2A–B). Time between each AMD3100 administration episode was 3–4 weeks.
Figure 2. :
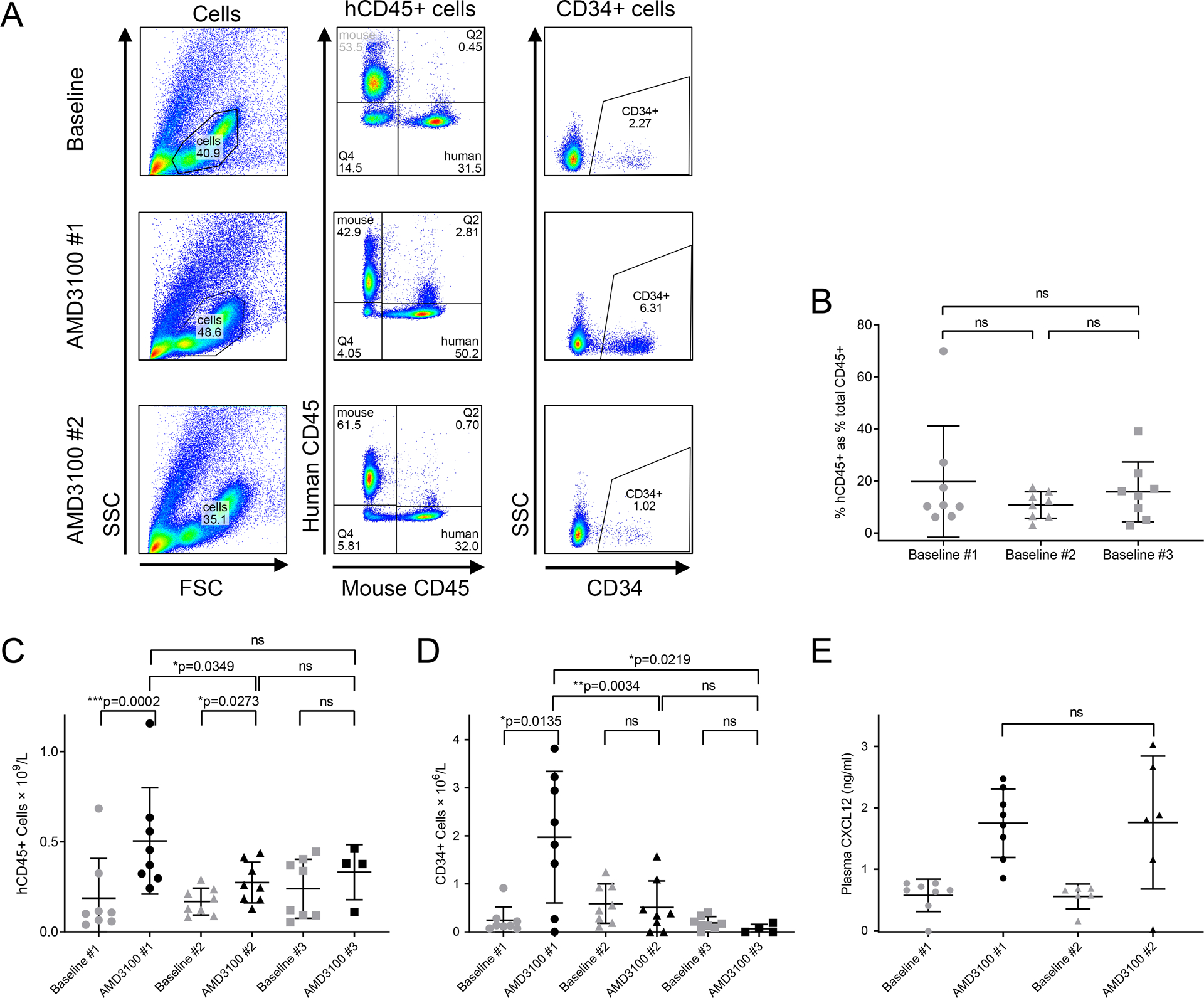
Human CD45+ and CD34+ HSPC counts, and plasma CXCL12 levels in humanized mice treated with one or repeated AMD3100 doses.
A: Representative example of flow cytometric gating strategy utilized in humanized mice. B: Human engraftment, calculated as human CD45+ cell percentage of total CD45+ cells (mean, SD). C: Absolute hCD45+ cell count in peripheral blood pre- and post-AMD3100 doses 1–3 (mean, SD). D: Absolute CD34+ cell count in peripheral blood pre- and post-AMD3100 doses 1–3 (mean, SD). E: Plasma CXCL12 levels measured in peripheral blood pre- and post-AMD3100 doses 1–2 (mean, SD).
The response of total human MNCs was assessed first, as defined by the hCD45+ cell population, by comparing baseline to peak levels, and a reduced count in PB was found after each sequential AMD3100 dosing. After the first dose, mean human MNC count rose from 0.19 × 109/l to 0.50 × 109/l (n=8, p=0.0002). After second dose, the increase was less significant, from 0.17 × 109/l to just 0.27 × 109/l (n=8, p=0.0349). Following a third dose of AMD3100, there was no significant increase in circulating human MNCs when comparing baseline 0.24 × 109/l (n=8) to peak count 0.33 × 109/l (n=4, p>0.05). Furthermore, we noted a significant decrease in the amplitude of mobilized hCD45+ cell count between first and second doses (n=8, p=0.0349) (Fig 2C).
Even more importantly, the response of human HSPCs, as defined by CD34+ cells, was also reduced with repeated doses of AMD3100. With a first dose, mean CD34+ cell count rose from 0.24 × 106/l to 1.97 × 106/l (p=0.0135) whereas counts with the second and third doses did not change significantly (from 0.59 × 106/l to 0.51 × 106/l with second dose and from 0.19 × 106/l to 0.07 × 106/l with third dose). The amplitude of mobilization of human HSPCs following second or third dose AMD3100 was also significantly lower than after first dose (p=0.0034 and p=0.0219 respectively) (Fig 2D).
A second humanized NSG mouse cohort was used to confirm the loss of efficacy of repeated AMD3100 dosages in this model. Results confirmed findings of poorer hCD45+ and CD34+ cell mobilization with a second dose of AMD3100 when compared to first dose (n=7, p=0.0065 and p=0.0225 respectively). (Suppl figure 1).
In summary, total human MNCs mobilized more poorly in response to subsequent doses of AMD3100 in comparison to first dose and CD34+ cells also failed to mobilize with repeat doses after a good response to first dose was demonstrated. Therefore, the reduction in AMD3100 efficacy with repeated dosing first noted in the NHPs was confirmed to also apply to human HSPCs.
AMD3100 reverses the CXCL12 gradient across the BM in humanized mice and is not impacted by repeated dosing
One hypothesis for the mechanism behind the loss of AMD3100 efficacy with repeated dosing is a failure to re-establish the CXCL12 gradient reported in mice following the first AMD3100 exposure, after each subsequent dose.11 To investigate this hypothesis, we measured plasma CXCL12 pre- and post-AMD3100 treatment in several engrafted mice after the first and second dosing. Consistent with AMD3100 reversing the CXCL12 gradient across the BM, we found that plasma CXCL12 levels increased after administration (Fig2E). There was no statistical difference in this increase in CXCL12 after repeat AMD3100 dosage when compared to first dose, thus ruling out this possible mechanism to explain the reduced AMD3100 efficacy in this context.
Discussion
We have presented results demonstrating significantly poorer mobilization of HSPCs in NHPs following repeated administration of AMD3100 in comparison to first dose. This is the case for the CD34+ cell population as a whole, the most commonly used marker for HSPCs and therefore the measure of mobilization adequacy employed routinely in the clinic and in preclinical NHP transplant protocols. Arguably of even greater consequence, we also observed reduced mobilization of the HSC-enriched CD90+ subset with repeat AMD3100 dosage. Single dose AMD3100 mobilization was previously documented in NHPs,45,46 this data adds to the available literature by characterizing the response to repeat AMD3100 administration in a cohort of rhesus macaques. Given the importance of the NHP model in preclinical investigation, including HSPC transplant and genome engineering studies, these results are noteworthy.
Failure of human HSPCs to remobilize with repeated AMD3100 doses in the humanized mouse model was demonstrated in 2 separate animal cohorts. Response of the total human mononuclear cell population was also reduced with subsequent doses. Since this is the most commonly used pre-clinical animal model for the study of human hematopoiesis, the inability to effectively remobilize HSPCs in these experimental animals with AMD3100 is also highly significant and will impact upon preclinical protocols for the investigation of a multitude of conditions.
The loss of AMD3100 activity on repeated administration in these 2 valuable preclinical models is of greatest significance where therapeutic interventions for sickle cell disease are being investigated. This is because AMD3100 is the only clinically available mobilization agent which can safely be used in this patient population. Limitations imposed by an inability to reuse this drug effectively in experimental animals may result in protocols utilizing AMD3100 alone being abandoned. This would introduce a further level of variance between preclinical and clinical protocols for the treatment of sickle cell disease including modified autologous transplantation and in vivo genome engineering.
It was previously documented that the clinical response to G-CSF may be reduced on repeated exposure. Whilst there are some conflicting data relating to remobilization in patients, where healthy donors were given repeated doses of G-CSF with up to a year between doses, efficacy of remobilization was reduced, demonstrating a long-lasting deficit in remobilization.47,48 This is the first study to demonstrate a similar deficit in remobilization with repeat AMD3100 dosing, many weeks after the initial exposure. This loss of efficacy is highly significant in practical as well as statistical terms.
One human study in healthy volunteers reported adequate CD34+ cell mobilization with a repeated dose of AMD3100, at least 2 weeks after initial dose.49 One significant difference between this study and ours is the AMD3100 dosage used. This clinical study used doses of only 0.24 mg/kg or 0.48 mg/kg whereas significantly higher doses of 1mg/kg (NHPs) and 5mg/kg (immunodeficient mice) were administered in our experiments. It is likely therefore that a higher proportion of the potentially AMD3100-sensitive cells were mobilized in our preclinical models than in the clinical study of healthy donors. We hypothesize therefore that the reservoir of potentially AMD3100-sensitive cells remaining in the BM after first mobilization was greater in the healthy donor study than in the NHPs or mice, allowing for successful mobilization of a different cell population upon second AMD3100 administration in the volunteer donors. This reservoir of mobilizable cells was proportionally more depleted in the preclinical models leaving fewer potentially AMD3100-sensitive cells to be mobilized upon repeat dosing of this drug, resulting in failure of HSPC mobilization on subsequent administration. It may be that if higher AMD3100 doses are used in the clinic then a similar depletion of potentially mobilizable cells would occur after first administration, and failure of remobilization on repeated dosing would also be seen.
Furthermore, pathological BM such as that of sickle cell disease or thalassemia patients cannot be assumed to respond to repeated AMD3100 administration in the same way as that of a healthy volunteer. Defective hematopoietic support of BM stromal cells from β-thalassemia patients has been demonstrated,50 and, anecdotally, patients with β-thalassemia have been found to remobilize poorly with repeated AMD3100 dosing (C Samuelson, personal communications). Further clinical studies are required to investigate the response of patients in this scenario, particularly those with the types of hematological pathology most likely to require repeated AMD3100 administration, such as the β-hemoglobinopathy populations.
We have presented data showing poor response to repeated AMD3100 administration in humanized mice up to 4 weeks after initial dose, and in NHPs up to 14 weeks after first exposure. It is unclear how long this effect would last and further studies are recommended to investigate this in the preclinical models described. This study has investigated the effect of repeated AMD3100 when dosed as a single mobilization agent. It must be established whether the same drop in efficacy with repeat AMD3100 dosing is also observed where this drug is combined with G-CSF, as is often the case in clinical practice. This also warrants detailed investigation, as the implications of such would be even more far-reaching. For example, the American Society for Blood and Marrow Transplantation (ASBMT) recommends that in patients failing to mobilize adequately during a first episode, a repeat mobilization attempt can begin only 2–4 weeks following the first.2 As many of these patients would have received AMD3100 during their first attempt and are then likely to receive it also during repeat treatment, it is important to establish whether a fall-off in AMD3100 efficacy in this scenario is also a concern.
The mechanism for reduced HSPC mobilization in response to repeat AMD3100 has not yet been elucidated. One hypothesis investigated in our studies was that this may be due to a reduction in the plasma CXCL12 response to repeat AMD3100 dosing – a response demonstrated in BALB/c mice by Redpath et al.11 However, our results have shown no significant difference in the rise in plasma CXCL12 concentration with second as compared to first dose AMD3100, thereby ruling out this hypothesis. Of note, this is the first report of plasma CXCL12 response to AMD3100 in humanized NSG mice. Redpath also presented data showing a simultaneous drop in BM CXCL12 level as the plasma concentration rises, following AMD3100 administration. As measurement of BM CXCL12 levels would have required early necropsy of study animals, this was not carried out in this study but an alternative hypothesis as to the cause of AMD3100 inefficacy with repeated dose is a reduction in the response to AMD3100 of the BM milieu, including a possible deficit of CXCL12 response. Future investigations should also include further cellular analysis of HSPCs exposed to AMD3100, including detailed examination of CXCR4 and CXCR7 activation, endocytosis and recycling.
In conclusion, we have presented evidence of a failure of NHPs and humanized mice to remobilize HSPCs on repeat administration of AMD3100. This has significant implications for pre-clinical transplant and HSPC genome engineering protocols. It remains to be proven whether the pathological BM of patients with hematological disorders will similarly fail to respond to repeat mobilization attempts with this drug and clinical studies in this area are warranted.
Materials and Methods
Ethics and animal welfare statements
All experimental animal protocols employed in these studies were granted approval by the Institutional Animal Care and Use Committee (IACUC) of the Fred Hutchinson Cancer Research Center and University of Washington. NHPs were treated under protocol #3235-01 and NSG mice under protocol #1864. All animal management conforms to recommendations of the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health.51
Rhesus macaques were housed at the UW National Primate Research Center (WaNPRC) under conditions approved by the American Association for the Accreditation of Laboratory Animal Care and in accordance with Animal Welfare Act regulations. Animals were cared for and monitored by accredited animal technicians and veterinarians. They were administered sedation prior to blood draws and were fasted appropriately prior to this. All procedures and routine care were conducted according to WaNPRC standard operating procedures.
NSG mice were bred in-house under approved protocols and in pathogen-free housing conditions. Blood draws (by retro-orbital puncture), tail vein and subcutaneous injections were carried out by appropriately trained animal technicians according to center protocols.
AMD3100 mobilization
NHPs underwent baseline blood sampling prior to AMD3100 (AMD3100 octahydrochloride hydrate, Sigma-Aldrich, MO, US) administration then received a first dose of AMD3100 at 1mg/kg SQ resuspended in sterile H20 at a concentration of 5mg/ml. PB draws were taken at a minimum of 2 time points between 2–3.5 hours post-dose into anticoagulated blood collection tubes. 1 animal was sedated for a longer period of time and had additional blood samples drawn: hourly blood draws from 2–7 hours post dose. Samples were taken for investigation of complete blood count (CBC) and for fluorescent activated cell sorting (FACS).
Some of these same animals were given a second administration of AMD3100 6–14 weeks after the first, with identical dosing protocol. Blood draws were again taken at a minimum of 2 timepoints 2–3.5 hours post dose. 2 animals had additional blood draws taken: 1 also had blood drawn at 4 hours post dose and 1 had hourly blood draws from 2–7 hours post dose. PB samples were processed as described below. One animal received a third dose of AMD3100 4 weeks after the second dose with dosing regimen and post-dose testing carried out as above.
2 cohorts of NSG mice were humanized in adulthood, at the age of 6–8 weeks. Humanization was carried out by a single tail vein injection of human G-CSF-mobilized, CD34+-enriched PB MNCs via the tail vein, at 0.5 – 1.5 ×106 cells per mouse, resuspended in phosphate-buffered saline with 1% heparin (APP Pharmaceuticals, IL, US) to a total volume of 200ul, following 275 cGy irradiation.
The first cohort received first dose AMD3100 20 weeks post humanization, followed by second dose and for surviving mice a third dose was also given. The second cohort received first dose AMD3100 9 weeks post humanization followed by a second dose. The time interval between doses was 3–4 weeks in every case. Each dose consisted of SQ injection of 5mg/kg AMD3100 made up to a total of 100ul with sterile H2O. Following each dose blood was drawn 1.5 hours post AMD3100, to coincide with expected peak of mobilization, for CBC and FACS analysis.52
CBC and flow cytometric analysis of PB
Aliquots of NHP blood underwent CBC testing by the UW Medicine Laboratories using the Sysmex XN 9000 Hematology Automation Line (Sysmex, IL, US) with MNC reported as total white cell count. Samples for FACS testing first underwent lysis of red blood cells by application of hemolytic buffer (150mM Ammonium chloride, 12mM Sodium bicarbonate, and 500mM EDTA in water). Remaining cells were stained with anti-CD45-V450 (clone D058-1283, BD Biosciences, CA, US); anti-CD34-APC (clone 563, BD Biosciences); anti-CD45RA-APC Cy7 (clone HI100, BD Biosciences) and anti-CD90-PE (clone 5E10, Biolegend, CA, US). FACS was carried out on the BD FACSAria II or BD FACSCanto II flow cytometers.
PB from the NSG mouse cohorts was analysed for total MNC count using the Element HT5 Veterinary Hematology Analyser (Heska, CO, US). Remaining blood was spun down and plasma extracted. Cells then underwent red cell lysis using BD Pharm Lyse™ buffer (BD Biosciences) followed by staining for FACS analysis. Antibody panel utilized for staining included: anti-human CD45 (hCD45)-PerCP or anti-human CD45-PerCP- Cy™5.5 (both clone 2D1); anti-mouse CD45-V500 (clone 30-F11) and anti-CD34-APC (clone 563) (all BD Biosciences). CD34+ gating was informed by results of an FMO sample processed with each cohort of blood samples, stained with anti-hCD45 and anti-mCD45 but not anti-CD34 (clones as above).
Post-acquisition analysis of flow cytometric data was carried out using FlowJo Version 9.
Statistical analysis
For NHP samples, the percentage of CD34+ cells was defined by CD34-high/CD45-low fluorescence within the MNC population (MNCs first being defined by forward and side scatter). Within the bulk CD34+ cell population, the proportion of the hematopoietic stem cell-enriched subpopulation as defined by CD34+CD90+CD45RA− was determined. At each blood draw timepoint, absolute circulating CD34+ count was calculated by multiplication of CD34+ fraction of MNCs by the total circulating MNC count. Absolute CD90+ cell response was determined by multiplication of CD90+ fraction of CD34+ cells by absolute CD34+ cell number.
For mouse samples, total circulating mononuclear cells and total circulating CD34+ cells were determined at each time point by multiplication of human proportion of CD45+ cells by total MNC count (taken to be total white cell count reported on CBC), and by multiplication of fraction of CD34+ cells by human CD45+ count, respectively.
Presentation of graphical data utilized GraphPad Prism 7 for Windows (Prism). Excel 2016 for Windows and Prism were used for statistical analysis. Descriptive statistics are presented along with comparison of results related to first and subsequent AMD3100 doses by 2-tailed t-test. Paired t-test is applied where each value has an appropriate paired result; unpaired t-test is applied in every other case. p values of <0.05 are taken to be significant (*) and p values of <0.001 are taken to be highly significant (**).
Plasma CXCL12
Plasma CXCL12 levels were measured pre- and post-AMD3100 dosing for first and second doses in the first mouse cohort. Blood samples were spun down at 1700rpm for 15 mins and plasma extracted by pipetting. Plasma from each blood draw was frozen to allow simultaneous testing of all samples for CXCL12 level by ELISA assay using Mouse CXCL12/SDF-1 alpha Quantikine ELISA Kit (R+D Systems, MN, US) with analysis of optical density carried out on the Epoch microplate spectrophotometer (Biotek, VT, US).
Supplementary Material
{kind=link}
Acknowledgments
This work was supported by grants from the National Heart, Lung, and Blood Institute (R01 HL136135 and R01 HL147324), from the National Institute of Allergy and Infectious and Infectious Diseases (R01 AI135953-01, U19 AI096111 and UM1 AI126623), NIH, Bethesda, MD, and charitable support to C.S. from the British Society for Haematology, the UK Thalassaemia Society and Sheffield Hospitals Charity. H.-P.K. is a Markey Molecular Medicine Investigator and received support as the inaugural recipient of the José Carreras/E. Donnall Thomas Endowed Chair for Cancer Research and the Fred Hutch Endowed Chair for Cell and Gene Therapy. C.S. would like to thank Dr Andrew Chantry, Professor John Snowden and Dr Josh Wright for their support of her research fellowship. The authors thank Helen Crawford for her assistance in formatting the figures.
Footnotes
Conflicts of interest: H.-P.K. is consulting for Rocket Pharma, Homology Medicines, CSL Behring, Vor Biopharma, and Magenta Therapeutics. Other authors have no competing interests.
References
- 1.Majhail NS, Farnia SH, Carpenter PA, Champlin RE, Crawford S, Marks DI, Omel JL, Orchard PJ, Palmer J, Saber W and others. Indications for Autologous and Allogeneic Hematopoietic Cell Transplantation: Guidelines from the American Society for Blood and Marrow Transplantation. Biol Blood Marrow Transplant 2015;21(11):1863–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Duong HK, Savani BN, Copelan E, Devine S, Costa LJ, Wingard JR, Shaughnessy P, Majhail N, Perales MA, Cutler CS and others. Peripheral blood progenitor cell mobilization for autologous and allogeneic hematopoietic cell transplantation: guidelines from the American Society for Blood and Marrow Transplantation. Biol Blood Marrow Transplant 2014;20(9):1262–73. [DOI] [PubMed] [Google Scholar]
- 3.Naldini L Genetic engineering of hematopoiesis: current stage of clinical translation and future perspectives. EMBO Mol Med 2019;11(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Siddiq S, Pamphilon D, Brunskill S, Doree C, Hyde C, Stanworth S. Bone marrow harvest versus peripheral stem cell collection for haemopoietic stem cell donation in healthy donors. Cochrane Database Syst Rev 2009(1):Cd006406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ho AD. Evolution of Peripheral Blood Stem Cell Transplantation. Methods Mol Biol 2019;2017:1–10. [DOI] [PubMed] [Google Scholar]
- 6.Holtick U, Albrecht M, Chemnitz JM, Theurich S, Skoetz N, Scheid C, von Bergwelt-Baildon M. Bone marrow versus peripheral blood allogeneic haematopoietic stem cell transplantation for haematological malignancies in adults. Cochrane Database Syst Rev 2014(4):Cd010189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bernitz JM, Daniel MG, Fstkchyan YS, Moore K. Granulocyte colony-stimulating factor mobilizes dormant hematopoietic stem cells without proliferation in mice. Blood 2017;129(14):1901–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Demetri GD, Griffin JD. Granulocyte colony-stimulating factor and its receptor. Blood 1991;78(11):2791–808. [PubMed] [Google Scholar]
- 9.Martin C, Bridger GJ, Rankin SM. Structural analogues of AMD3100 mobilise haematopoietic progenitor cells from bone marrow in vivo according to their ability to inhibit CXCL12 binding to CXCR4 in vitro. Br J Haematol 2006;134(3):326–9. [DOI] [PubMed] [Google Scholar]
- 10.Kalatskaya I, Berchiche YA, Gravel S, Limberg BJ, Rosenbaum JS, Heveker N. AMD3100 is a CXCR7 ligand with allosteric agonist properties. Mol Pharmacol 2009;75(5):1240–7. [DOI] [PubMed] [Google Scholar]
- 11.Redpath AN, Francois M, Wong SP, Bonnet D, Rankin SM. Two distinct CXCR4 antagonists mobilize progenitor cells in mice by different mechanisms. Blood Adv 2017;1(22):1934–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DiPersio JF, Micallef IN, Stiff PJ, Bolwell BJ, Maziarz RT, Jacobsen E, Nademanee A, McCarty J, Bridger G, Calandra G. Phase III prospective randomized double-blind placebo-controlled trial of plerixafor plus granulocyte colony-stimulating factor compared with placebo plus granulocyte colony-stimulating factor for autologous stem-cell mobilization and transplantation for patients with non-Hodgkin’s lymphoma. J Clin Oncol 2009;27(28):4767–73. [DOI] [PubMed] [Google Scholar]
- 13.DiPersio JF, Stadtmauer EA, Nademanee A, Micallef IN, Stiff PJ, Kaufman JL, Maziarz RT, Hosing C, Fruehauf S, Horwitz M and others. Plerixafor and G-CSF versus placebo and G-CSF to mobilize hematopoietic stem cells for autologous stem cell transplantation in patients with multiple myeloma. Blood 2009;113(23):5720–6. [DOI] [PubMed] [Google Scholar]
- 14.Chen Y-B, Le-Rademacher J, Brazauskas R, Kiefer DM, Hamadani M, DiPersio JF, Litzow MR, Craig M, Horwitz ME, Artz AS and others. Plerixafor alone for the mobilization and transplantation of HLA-matched sibling donor hematopoietic stem cells. Blood advances 2019;3(6):875–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Greef GE, Braakman E, van der Holt B, Janssen J, Petersen E, Vucinic V, Thuss N, Grootes M, Cornelissen JJ. The feasibility and efficacy of subcutaneous plerixafor for mobilization of peripheral blood stem cells in allogeneic HLA-identical sibling donors: results of the HOVON-107 study. Transfusion 2019;59(1):316–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Flomenberg N, Comenzo RL, Badel K, Calandra G. Plerixafor (Mozobil) alone to mobilize hematopoietic stem cells from multiple myeloma patients for autologous transplantation. Biol Blood Marrow Transplant 2010;16(5):695–700. [DOI] [PubMed] [Google Scholar]
- 17.Schroeder MA, Rettig MP, Lopez S, Christ S, Fiala M, Eades W, Mir FA, Shao J, McFarland K, Trinkaus K and others. Mobilization of allogeneic peripheral blood stem cell donors with intravenous plerixafor mobilizes a unique graft. Blood 2017;129(19):2680–2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pulsipher MA, Chitphakdithai P, Miller JP, Logan BR, King RJ, Rizzo JD, Leitman SF, Anderlini P, Haagenson MD, Kurian S and others. Adverse events among 2408 unrelated donors of peripheral blood stem cells: results of a prospective trial from the National Marrow Donor Program. Blood 2009;113(15):3604–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuendgen A, Fenk R, Bruns I, Dommach M, Schutte A, Engers R, Hunerliturkoglu A, Haas R, Kobbe G. Splenic rupture following administration of pegfilgrastim in a patient with multiple myeloma undergoing autologous peripheral blood stem cell transplantation. Bone Marrow Transplant. Volume 38. England 2006. p 69–70. [DOI] [PubMed] [Google Scholar]
- 20.Dugan MJ, Maziarz RT, Bensinger WI, Nademanee A, Liesveld J, Badel K, Dehner C, Gibney C, Bridger G, Calandra G. Safety and preliminary efficacy of plerixafor (Mozobil) in combination with chemotherapy and G-CSF: an open-label, multicenter, exploratory trial in patients with multiple myeloma and non-Hodgkin’s lymphoma undergoing stem cell mobilization. Bone Marrow Transplant 2010;45(1):39–47. [DOI] [PubMed] [Google Scholar]
- 21.Stewart DA, Smith C, MacFarland R, Calandra G. Pharmacokinetics and pharmacodynamics of plerixafor in patients with non-Hodgkin lymphoma and multiple myeloma. Biol Blood Marrow Transplant 2009;15(1):39–46. [DOI] [PubMed] [Google Scholar]
- 22.Esrick EB, Manis JP, Daley H, Baricordi C, Trebeden-Negre H, Pierciey FJ, Armant M, Nikiforow S, Heeney MM, London WB and others. Successful hematopoietic stem cell mobilization and apheresis collection using plerixafor alone in sickle cell patients. Blood Adv 2018;2(19):2505–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fitzhugh CD, Hsieh MM, Bolan CD, Saenz C, Tisdale JF. Granulocyte colony-stimulating factor (G-CSF) administration in individuals with sickle cell disease: time for a moratorium? Cytotherapy 2009;11(4):464–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hsieh MM, Tisdale JF. Hematopoietic stem cell mobilization with plerixafor in sickle cell disease. Haematologica. Volume 103. Italy: 2018. p 749–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dominici M, Pritchard C, Garlits JE, Hofmann TJ, Persons DA, Horwitz EM. Hematopoietic cells and osteoblasts are derived from a common marrow progenitor after bone marrow transplantation. Proc Natl Acad Sci U S A 2004;101(32):11761–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cornu TI, Mussolino C, Cathomen T. Refining strategies to translate genome editing to the clinic. Nature Medicine 2017;23(4):415–423. [DOI] [PubMed] [Google Scholar]
- 27.Humbert O, Radtke S, Samuelson C, Carrillo RR, Perez AM, Reddy SS, Lux C, Pattabhi S, Schefter LE, Negre O and others. Therapeutically relevant engraftment of a CRISPR-Cas9-edited HSC-enriched population with HbF reactivation in nonhuman primates. Sci Transl Med 2019;11(503). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Magrin E, Miccio A, Cavazzana M. Lentiviral and genome-editing strategies for the treatment of beta-hemoglobinopathies. Blood 2019;134(15):1203–1213. [DOI] [PubMed] [Google Scholar]
- 29.Mamcarz E, Zhou S, Lockey T, Abdelsamed H, Cross SJ, Kang G, Ma Z, Condori J, Dowdy J, Triplett B and others. Lentiviral Gene Therapy Combined with Low-Dose Busulfan in Infants with SCID-X1. N Engl J Med 2019;380(16):1525–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li C, Psatha N, Sova P, Gil S, Wang H, Kim J, Kulkarni C, Valensisi C, Hawkins RD, Stamatoyannopoulos G and others. Reactivation of gamma-globin in adult beta-YAC mice after ex vivo and in vivo hematopoietic stem cell genome editing. Blood 2018;131(26):2915–2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Humbert O, Chan F, Rajawat YS, Torgerson TR, Burtner CR, Hubbard NW, Humphrys D, Norgaard ZK, O’Donnell P, Adair JE and others. Rapid immune reconstitution of SCID-X1 canines after G-CSF/AMD3100 mobilization and in vivo gene therapy. Blood Adv 2018;2(9):987–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bahal R, Ali McNeer N, Quijano E, Liu Y, Sulkowski P, Turchick A, Lu Y-C, Bhunia DC, Manna A, Greiner DL and others. In vivo correction of anaemia in β-thalassemic mice by γPNA-mediated gene editing with nanoparticle delivery. Nature communications 2016;7:13304–13304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Richter M, Stone D, Miao C, Humbert O, Kiem HP, Papayannopoulou T, Lieber A. In Vivo Hematopoietic Stem Cell Transduction. Hematol Oncol Clin North Am 2017;31(5):771–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Humbert O, Peterson CW, Norgaard ZK, Radtke S, Kiem HP. A Nonhuman Primate Transplantation Model to Evaluate Hematopoietic Stem Cell Gene Editing Strategies for beta-Hemoglobinopathies. Mol Ther Methods Clin Dev 2018;8:75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peterson CW, Wang J, Norman KK, Norgaard ZK, Humbert O, Tse CK, Yan JJ, Trimble RG, Shivak DA, Rebar EJ and others. Long-term multilineage engraftment of autologous genome-edited hematopoietic stem cells in nonhuman primates. Blood 2016;127(20):2416–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Radtke S, Perez AM, Venkataraman R, Reddy S, Haworth KG, Humbert O, Kiem HP, Peterson CW. Preparation and Gene Modification of Nonhuman Primate Hematopoietic Stem and Progenitor Cells. J Vis Exp 2019(144). [DOI] [PubMed] [Google Scholar]
- 37.Radtke S, Adair JE, Giese MA, Chan YY, Norgaard ZK, Enstrom M, Haworth KG, Schefter LE, Kiem HP. A distinct hematopoietic stem cell population for rapid multilineage engraftment in nonhuman primates. Sci Transl Med 2017;9(414). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Radtke S, Haworth KG, Kiem HP. The frequency of multipotent CD133(+)CD45RA(−)CD34(+) hematopoietic stem cells is not increased in fetal liver compared with adult stem cell sources. Exp Hematol 2016;44(6):502–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Haworth KG, Ironside C, Norgaard ZK, Obenza WM, Adair JE, Kiem HP. In Vivo Murine-Matured Human CD3(+) Cells as a Preclinical Model for T Cell-Based Immunotherapies. Mol Ther Methods Clin Dev 2017;6:17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Radtke S, Humbert O, Kiem HP. Mouse models in hematopoietic stem cell gene therapy and genome editing. Biochem Pharmacol 2020;174:113692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Richter M, Saydaminova K, Yumul R, Krishnan R, Liu J, Nagy EE, Singh M, Izsvak Z, Cattaneo R, Uckert W and others. In vivo transduction of primitive mobilized hematopoietic stem cells after intravenous injection of integrating adenovirus vectors. Blood 2016;128(18):2206–2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang H, Georgakopoulou A, Psatha N, Li C, Capsali C, Samal HB, Anagnostopoulos A, Ehrhardt A, Izsvak Z, Papayannopoulou T and others. In vivo hematopoietic stem cell gene therapy ameliorates murine thalassemia intermedia. J Clin Invest 2019;129(2):598–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kean L, Sen S, Onabajo O, Singh K, Robertson J, Stempora L, Bonifacino A, Metzger M, Promislow D, Mattapallil J and others. Significant mobilization of both conventional and regulatory T cells with AMD3100. Blood 2011;118:6580–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Larochelle A, Krouse A, Metzger M, Orlic D, Donahue RE, Fricker S, Bridger G, Dunbar CE, Hematti P. AMD3100 mobilizes hematopoietic stem cells with long-term repopulating capacity in nonhuman primates. Blood 2006;107(9):3772–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Haynes LD, Coonen J, Post J, Brunner K, Bloom D, Hematti P, Kaufman DB. Collection of hematopoietic CD34 stem cells in rhesus macaques using Spectra Optia. J Clin Apher 2017;32(5):288–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Donahue RE, Jin P, Bonifacino AC, Metzger ME, Ren J, Wang E, Stroncek DF. Plerixafor (AMD3100) and granulocyte colony-stimulating factor (G-CSF) mobilize different CD34+ cell populations based on global gene and microRNA expression signatures. Blood 2009;114(12):2530–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fiala MA, Park S, Slade M, DiPersio JF, Stockerl-Goldstein KE. Remobilization of hematopoietic stem cells in healthy donors for allogeneic transplantation. Transfusion 2016;56(9):2331–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Velier M, Granata A, Bramanti S, Calmels B, Furst S, Legrand F, Harbi S, Faucher C, Devillier R, Blaise D and others. A matched-pair analysis reveals marginally reduced CD34+ cell mobilization on second occasion in 27 related donors who underwent peripheral blood stem cell collection twice at the same institution. Transfusion 2019;59(11):3442–3447. [DOI] [PubMed] [Google Scholar]
- 49.Pantin J, Purev E, Tian X, Cook L, Donohue-Jerussi T, Cho E, Reger R, Hsieh M, Khuu H, Calandra G and others. Effect of high-dose plerixafor on CD34(+) cell mobilization in healthy stem cell donors: results of a randomized crossover trial. Haematologica 2017;102(3):600–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Crippa S, Rossella V, Aprile A, Silvestri L, Rivis S, Scaramuzza S, Pirroni S, Avanzini MA, Basso-Ricci L, Hernandez RJ and others. Bone marrow stromal cells from β-thalassemia patients have impaired hematopoietic supportive capacity. The Journal of clinical investigation 2019;129(4):1566–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Animals NRCUCftUotGftCaUoL. Guide for the Care and Use of Laboratory Animals. 8th ed. Washington DC, US: National Academies Press (US); 2011. [Google Scholar]
- 52.Redpath AN, François M, Wong SP, Bonnet D, Rankin SM. Two distinct CXCR4 antagonists mobilize progenitor cells in mice by different mechanisms. Blood Adv 2017;1(22):1934–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.