

Abstract
This review article features selected examples on the synthesis of functionalized pyrroles that were reported between 2014 and 2019. Pyrrole is an important nitrogen-containing aromatic heterocycle that can be found in numerous compounds of biological and material significance. Given its vast importance, pyrrole continues to be an attractive target for the development of new synthetic reactions. The contents of this article are organized by the starting materials, which can be broadly classified into four different types: substrates bearing π-systems, substrates bearing carbonyl and other polar groups, and substrates bearing heterocyclic motifs. Brief discussions on plausible reaction mechanisms for most transformations are also presented.
Keywords: pyrroles, heterocycles, carbonyls, annulation, aromatic
Graphical Abstract
1. Introduction
Pyrrole is a 5-membered aromatic nitrogen-heterocycle that is ubiquitous in compounds of biological and material significance (Figure 1). For instance, pyrrole is the key structural component in chlorophylls and hemes; both of which are molecules that play a crucial role for the existence of life. Bioactive pharmaceutical compounds and natural products often feature this cyclic motif as their pharmacophore.1,2 Pyrrole can be also found in diverse forms of organic dyes and materials, such as BODIPY, conjugated polymers, optoelectronics, as well as semiconductors.3–6 First detected as a component of a coal tar in 1834,7 some of the pioneering examples on pyrrole synthesis were reported by Knorr, Hantzsch, and Paal at the end of the 19th century.8–10 Despite its prevalence in synthetic chemistries for over 100 years, pyrrole remains an important synthetic target in modern organic chemistry. Interestingly, between 2000 and 2020 over a dozen review articles have been published to summarize the various advancements that have been made to assemble this highly important molecular structure.11–24
Figure 1.
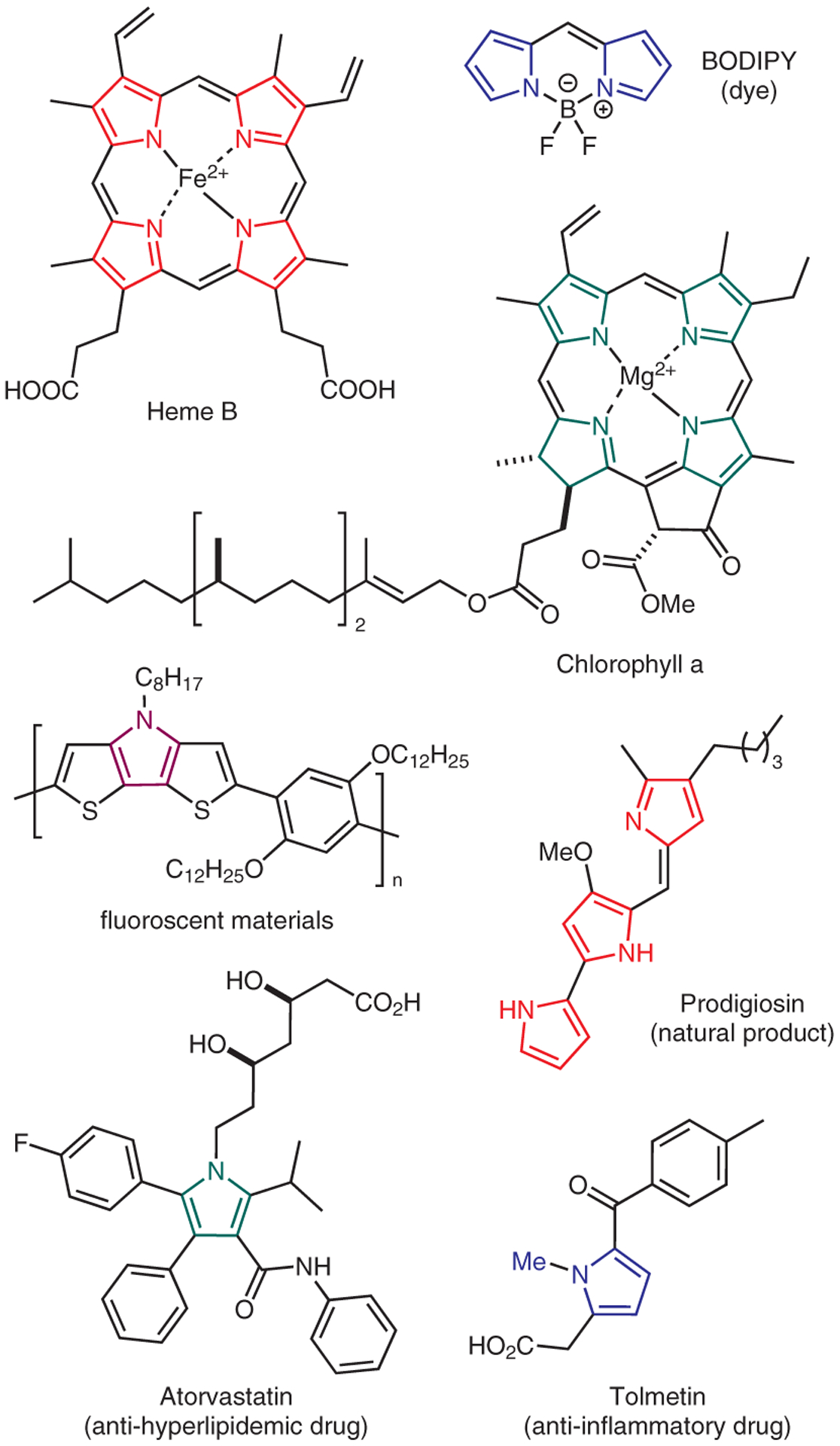
Importance of pyrrole
In this article, we wish to provide a brief survey on selected pyrrole syntheses that were published between 2014–2019. Upon examining literature precedents within this time span, we discerned that the various starting materials used in these new chemistries can be grouped broadly into four different types: substrates bearing π-systems, substrates bearing carbonyl and other polar groups, and substrates bearing heterocyclic motifs (Figure 2). Guided by this classification, we organized the transformations to highlight each type of the starting materials. Most examples will be accompanied by brief discussions on plausible reaction mechanisms. To further aid content arrangement, we also set criteria that the indicated type of starting materials will either form a part of pyrrole ring or contain key functional groups that directly participate in the pyrrole formation. For example, enones are both discussed in Sections 2.1 (Alkenes) and 3.2 (Ketones). The difference is that in Section 2.1, only the alkene portion of enone forms the pyrrole; whereas in Section 3.2, the entire α,β-unsaturation in the enone participates in pyrrole annulation and becomes a part of the ring. Given the broad extent of this subject matter, it is very difficult to dedicate an exclusive section for functional groups that routinely serve as reaction partners in pyrrole synthesis, such as amines, β-enaminones, and azirines. In fact, these compounds will appear in various sections throughout this manuscript.
Figure 2.
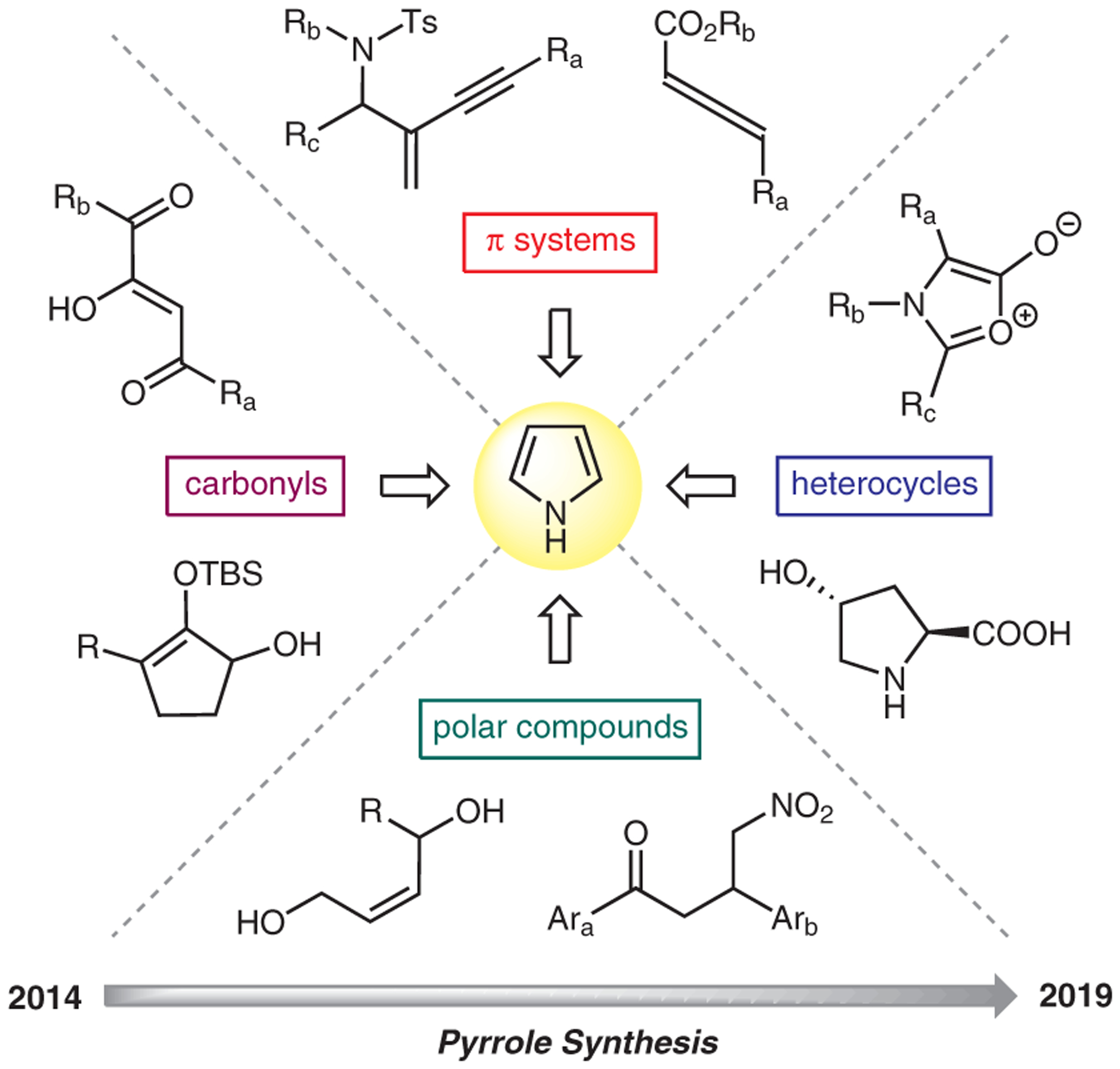
Pyrrole synthesis classification
2. From π-Systems
New chemistries that strategically exploit the reactivity of π-systems as useful starting materials in pyrrole synthesis remain well precedented in the literature. As exemplified in this section, these π-systems include various forms of alkenes, dienes, allenes, and alkynes.
2.1. Alkenes
Substituted alkenes, including those of α,β-unsaturated and allylic systems, are readily employed as starting materials to access pyrrole scaffolds.25,26 For instance, Xu and Zhang reported a silver acetate catalyzed tandem reaction of p-toluenesulfonylmethyl isocyanide (TosMIC) with 2-methyleneindene-1,3-diones 1 in the presence of K2CO3 to access fused pyrrole compounds 2 (Scheme 1).27 The proposed mechanism is via conjugate addition of TosMIC anion 3 to the alkene moiety, leading a 5-membered cyclization to imidoyl intermediate 4. The ensuing cyclization followed by ring opening of the resulting cyclopropanolate 5 and aromatization through elimination of the tosyl group then yielded the pyrrole ring.
Scheme 1.
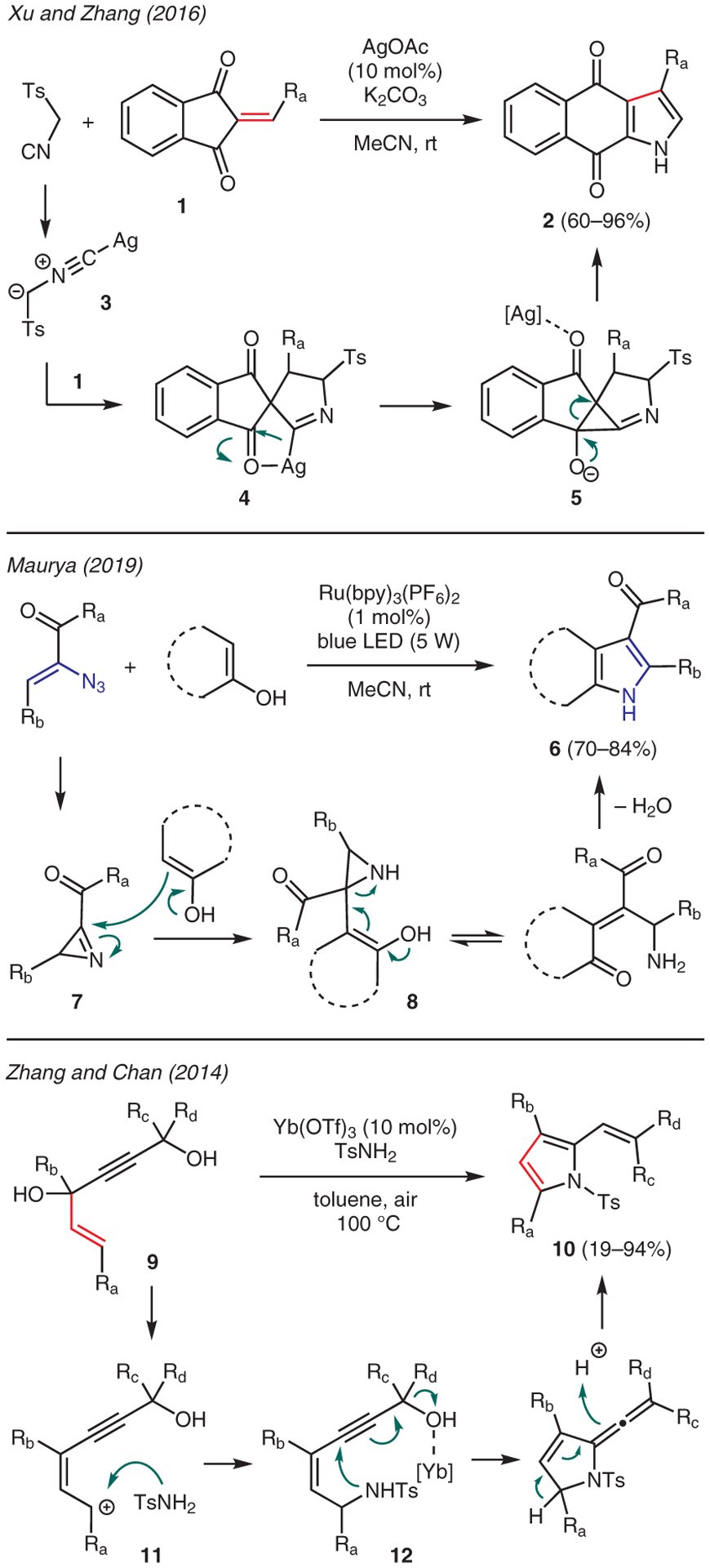
From alkenes
Maurya conveyed the preparation of 2,3-fused pyrroles 6 via the reaction of α-azidoenones with naphthols and related compounds in the presence of catalytic Ru(bpy)3(PF6)2 under blue LED light irradiation (Scheme 1).28 The proposed reaction mechanism commences with photosensitized decomposition of the α-azidoenone to yield reactive 2H-azirine intermediate 7, which is trapped by the enolic reaction partner. Tautomerization of the resulting intermediate 8 enables conjugate aziridine opening, which then allows dehydrative cyclization to produce the pyrrole core.
In another example, Zhang and Chan reported an operationally simple and convenient synthesis of vinyl-substituted pyrroles 10 involving annulation between allylic-propargylic diols 9 and arenesulfonamides in the presence of Yb(OTf)3 (Scheme 1).29 The reaction mechanism is proposed through ionization of the starting material with Yb(III) catalyst to form resonance stabilized carbocation 11. The ensuing capture by the sulfonamide at the less hindered carbon then provides intermediate 12. A second alcohol activation and intramolecular amino cyclization facilitate by the Yb catalyst leads to the vinyl-substituted pyrrole product.
2.2. 1,6-Dienes
The use of 1,6-dienes in pyrrole synthesis has been reported, in which the pyrrole core can be constructed via a ring-closing metathesis methodology. Scheme 2 depicts two examples. Lamaty demonstrated a strategy using nitro-Grela as the ruthenium ring-closing metathesis catalyst to form substituted pyrrolines 14 from β-amino esters 13.30 The ensuing deprotection-aromatization step with NaOt-Bu furnished 2-aryl-1H-pyrrole-3-carboxylates 15. The second example by Wu and Li reported the preparation of N-sulfonyl-and N-acylpyrroles 17 and 18.31 Starting with diallylamines 16, the one-pot protocol involved Grubbs II catalyzed ring-closing metathesis, followed by in situ oxidation of the emerging pyrroline intermediates with a suitable Cu(II) catalyst and O2 as an oxidant.
Scheme 2.
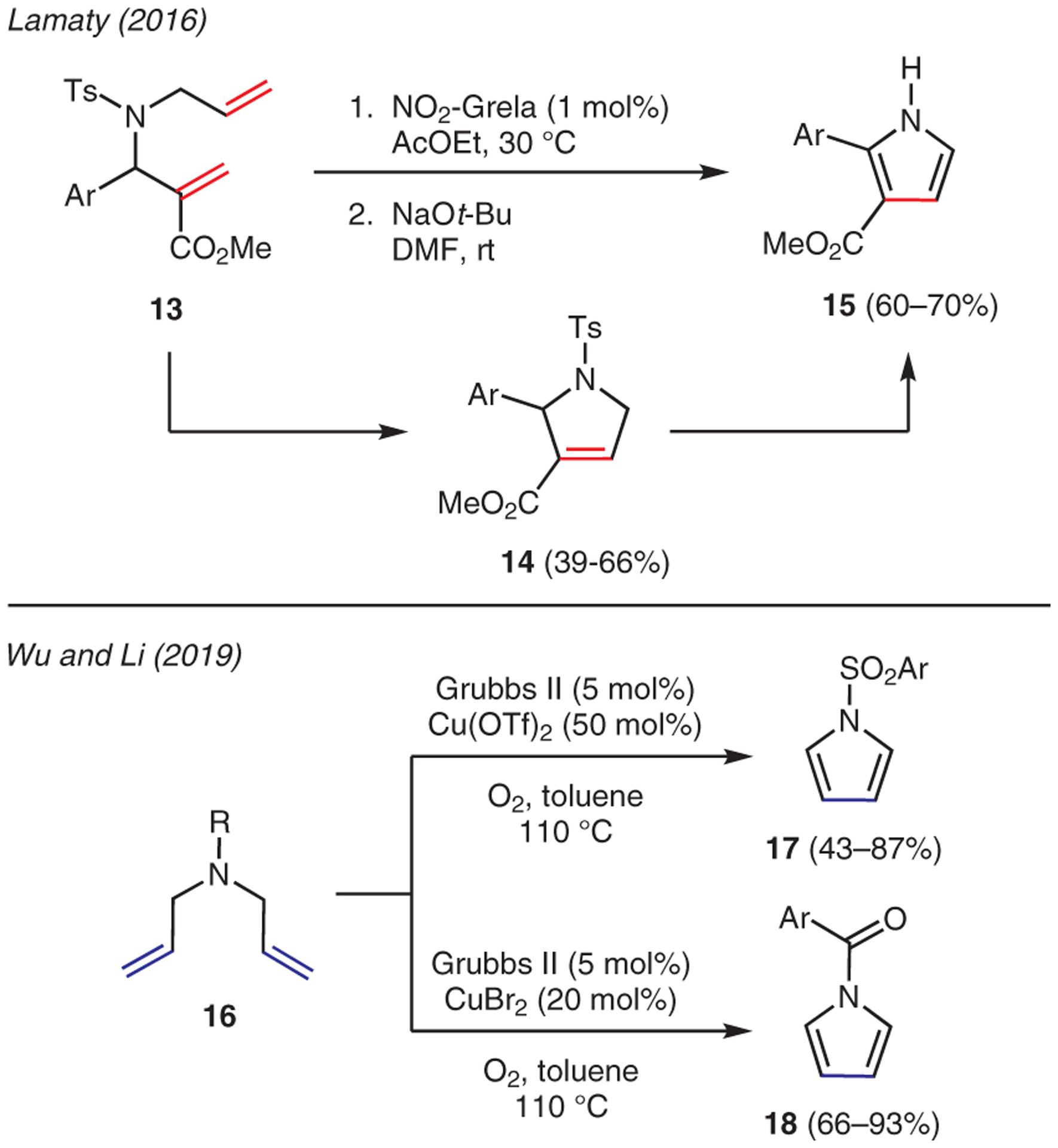
From 1,6-dienes
2.3. Allenes
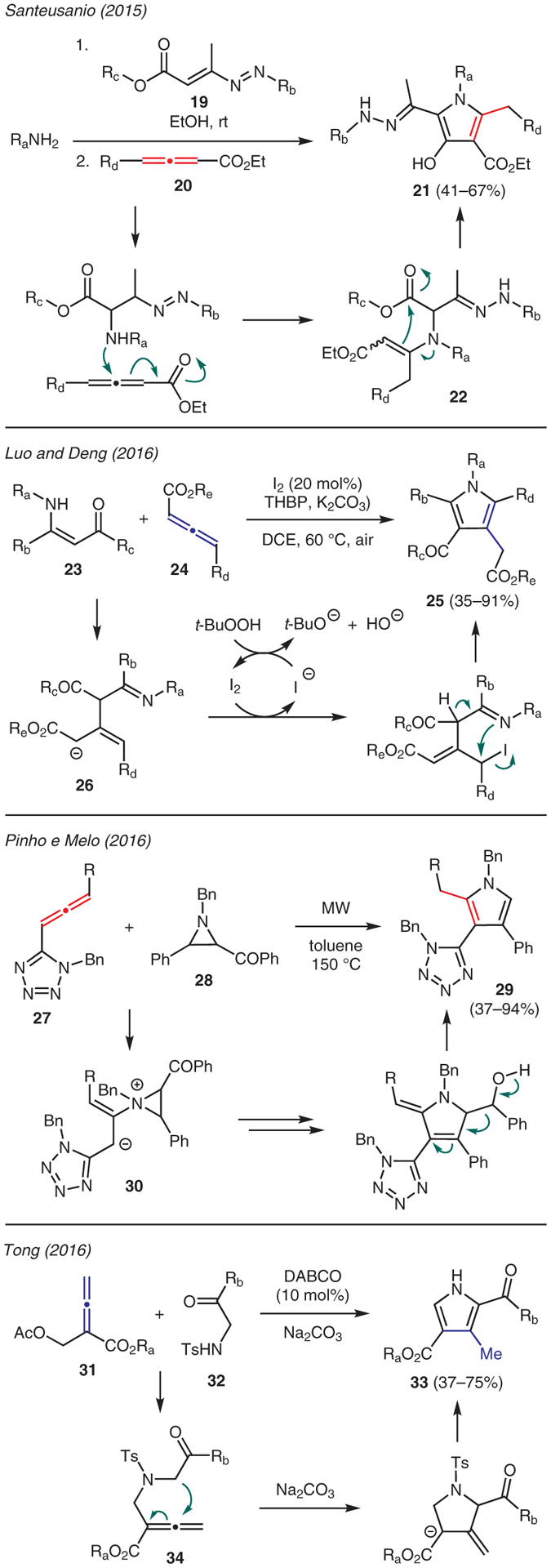
Functionalized pyrroles can be assembled from allenes upon treatment with either amines, enaminones, or aziridines (Scheme 3). For instance, Santeusanio reported the synthesis of pentasubstituted hydroxypyrroles 21 by combining 1,2-diaza-1,3-dienes 19, primary amines, and 2,3-allenoates 20.32 The reaction mechanism is proposed through a conjugate addition of amine to the α-carbon of 19, followed by nucleophilic addition to allenoate 20 to produce intermediate 22. Subsequent intramolecular cyclization then affords the hydroxypyrrole product 21.
Scheme 3.
From allenes
Luo and Deng developed a novel approach to prepare pyrroles 25 using enamines 23 and allenoates 24 in the presence of I2 catalyst and TBHP (Scheme 3).33 This reaction is proposed to occur via a Michael addition between the two starting materials, followed by I2-catalyzed annulation of the resulting anionic intermediate 26 in the presence of TBHP.
Another utility of allenes in pyrrole synthesis was described by Pinho e Melo,34 who treated (1H-tetrazol-5-yl)-allenes 27 and aziridines 28 under microwave irradiation at 150 °C in toluene to form tetrasubstituted pyrroles 29 (Scheme 3). Mechanistically this reaction is proposed to involve formal [3+2] cycloaddition via intermediate 30; subsequent tautomerism and retro-aldol-type fragmentation results in the pyrrole formation.
Tong reported an amine-catalyzed synthesis of pyrroles 33 from β′-acetoxy allenoates 31 and 2-(tosylamino)carbonyl compounds 32 (Scheme 3).35 In this chemistry, the 1,4-addition of DABCO to allenoate, accompanied by elimination of acetate, produces putative intermediate 34. Deprotonation by Na2CO3 then triggers a sequence of [3+2] annulation, followed by 1,2-elimination of the tosyl group and isomerization to generate trisubstituted pyrrole 33.
2.4. Alkynes
A wide collection of reactions have been reported between 2014–2019 that showcase the use of triple bonds as starting materials for pyrroles synthesis.36–41 A number of transition metal catalysts, such as Au, Pd, Cu, Rh, and Ru, have been reported to catalyze reactions between substituted alkynes and amines, enaminones, or oxime to generate pyrroles. In addition, a few reagent-driven syntheses have also been reported.
2.4.1. Gold Catalysis
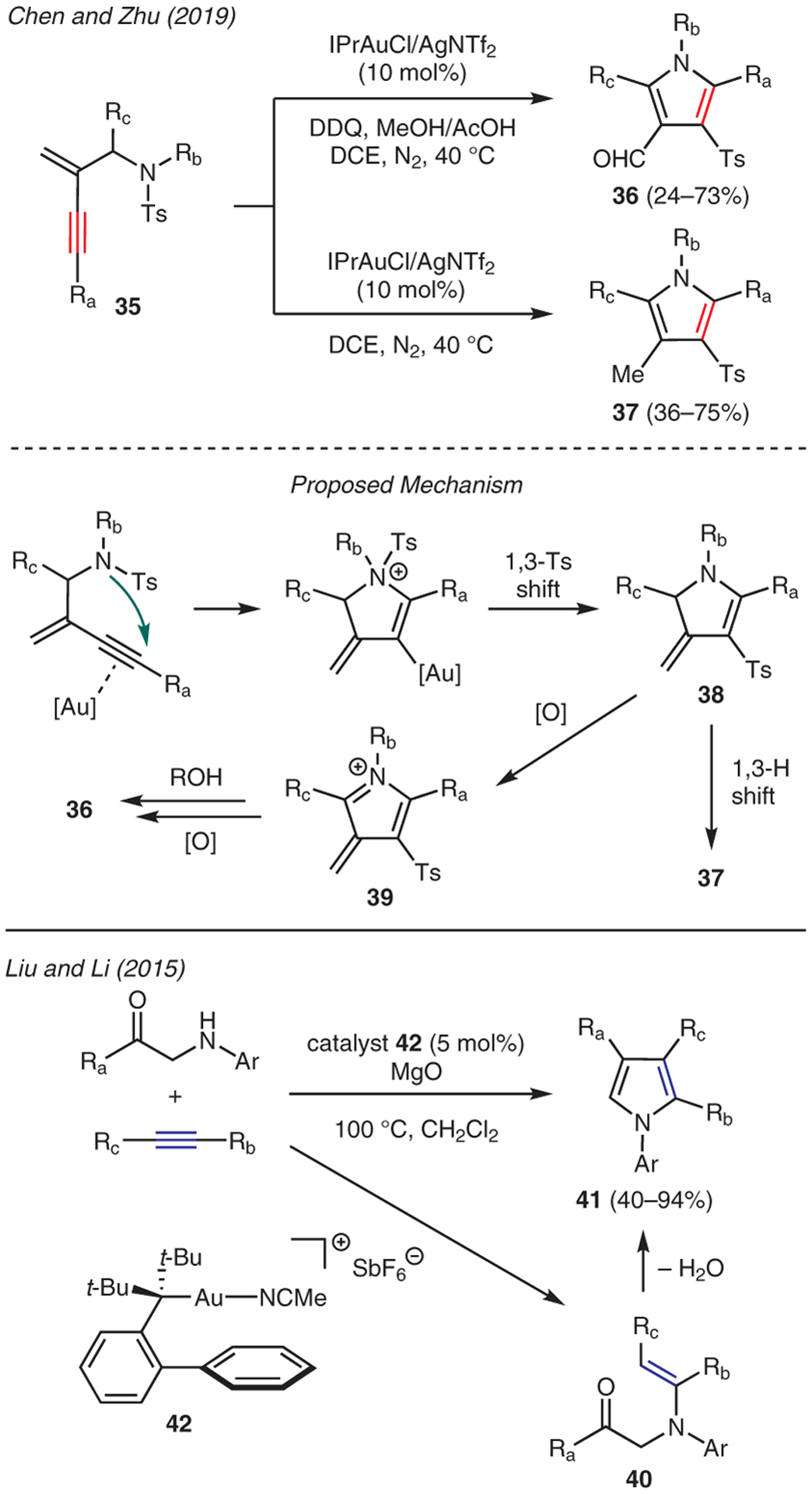
Chen and Zhu developed a synthetic approach toward substituted pyrroles 37 and formylpyrroles 36 from enyne sulfonamides 35 using a dual IPrAuCl/AgNTf2 catalytic system; formylpyrroles 36 were isolated when the reaction was performed in the presence of DDQ (Scheme 4).42 The proposed mechanism commences with gold-catalyzed intramolecular cyclization of enyne sulfonamide, followed by 1,3-Ts shift to form 3-methylene-2,3-dihydropyrrole 38. The ensuing aromatization via 1,3-H shift then afforded pyrrole 37. Interestingly, the introduction of DDQ readily interrupted aromatization of 38, leading to oxidation and thus formation of azafulvenium 39. Nucleophilic addition of MeOH or AcOH, followed by further oxidation and hydrolysis then led to the observed formylpyrrole 36.
Scheme 4.
From alkynes: gold catalysis
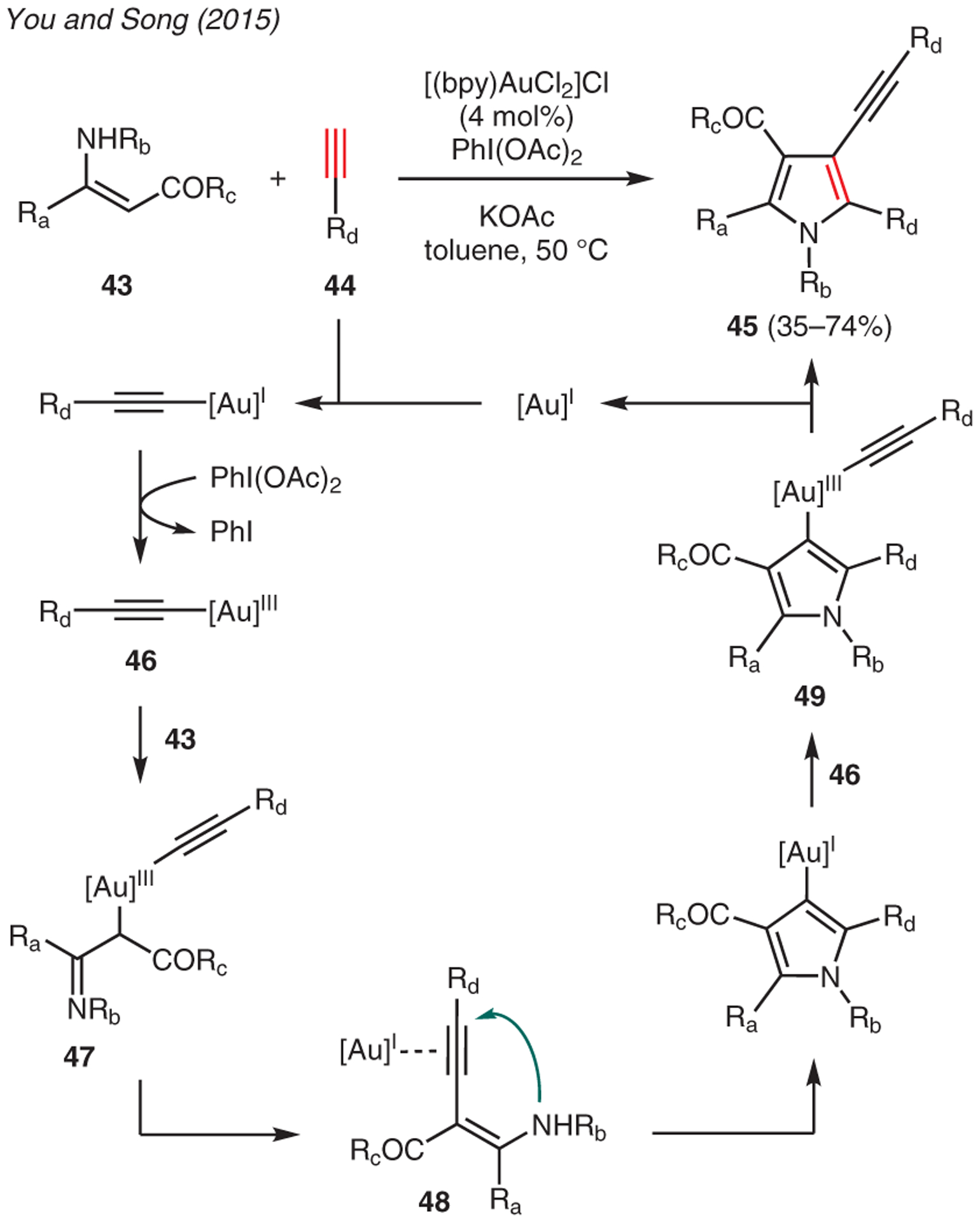
Liu and Li reported the gold-catalyzed hydroamination of electron-deficient alkynes with α-amino ketones using gold catalyst 42 (Scheme 4).43 In this reaction, the conjugate addition adduct 40 is believed to undergo cyclization-condensation, thereby producing the resulting pyrroles 41. The addition of MgO to the reaction mixture facilitated the removal of water byproduct and drove the equilibrium forward. Another example of gold-catalyzed pyrrole synthesis was demonstrated by You and Song (Scheme 5), in which the reaction between β-enaminones 43 and terminal alkynes 44 in the presence of [(bpy)AuCl2]Cl, PhI(OAc)2, and KOAc readily accessed 3-alkynylpyrroles 45.44 Mechanistic investigations suggest this reaction involves the generation of gold(III) intermediate 47 that subsequently undergoes reductive coupling to yield 2-alkynyl-β-enamino ester 48. The ensuing pyrrole cyclization, transmetalation with gold(III) acetylide 46, and reductive elimination of the resulting intermediate 49 then furnishes the 3-alkynylpyrrole motif.
Scheme 5.
From alkynes: gold catalysis
2.4.2. Palladium Catalysis
An efficient three-component reaction of alkyne esters, amines, and alkenes to form 2,3,4-trisubstituted pyrroles 50 in the presence of Pd(II) catalyst and K2S2O8 was conveyed by Zhang and Yi (Scheme 6).45 The reaction mechanism is proposed to involve regioselective alkene migratory insertion of Pd(II)-activated alkene at the β-position of enamine 51, which is generated in situ by the reaction of the alkyne ester and amine, to produce intermediate 52. Facilitated by sulfate radical anion, a sequence of hydrogen abstraction to radical intermediate 53, followed by, in succession, intramolecular radical addition-cyclization and protonolysis then generates dihydropyrrole and HPdOAc. Oxidation by K2S2O8 then affords pyrrole 50 while regenerating the active Pd(II) catalyst via a reductive elimination-oxidation sequence.
Scheme 6.
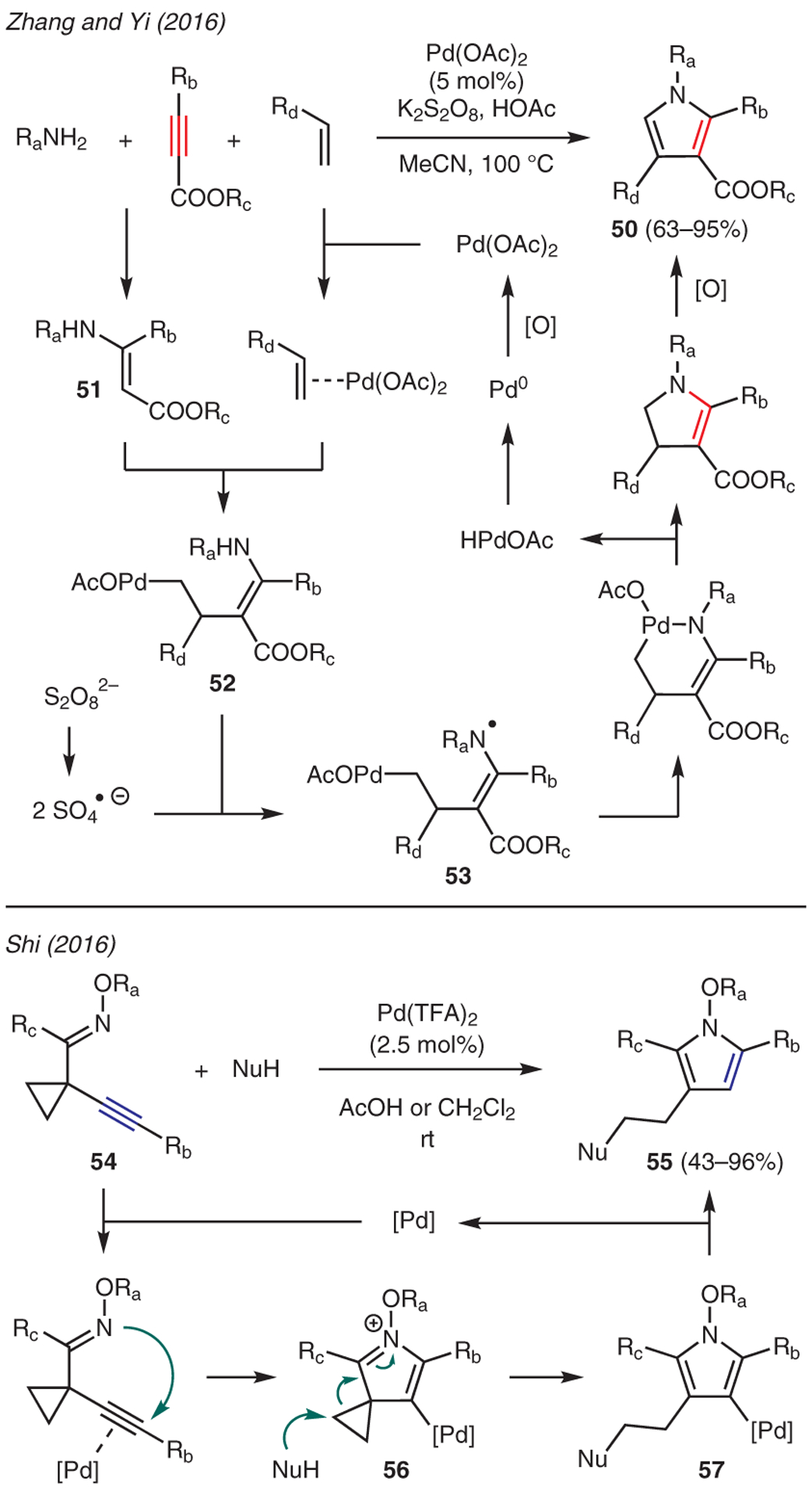
From alkynes: palladium catalysis
Shi reported an efficient Pd(TFA)2-catalyzed reaction for the synthesis of N-OR substituted pyrroles 55 from 1-(alk-1-ynyl)cyclopropyloxime derivatives 54 in the presence of different nucleophiles (Scheme 6).46 This chemistry is believed to occur via activation of the alkyne moiety by Pd(II) to promote intramolecular nucleophilic cyclization to generate N-oxyiminium ion 56. Intermolecular nucleophile-promoted fragmentation of the cyclopropane moiety, followed by protonation of the [Pd]-pyrrole species 57, then affords the final product.
Werz demonstrated the use of PdCl2(PhCN)2 catalyst in the synthesis of substituted pyrroles 59 from internal alkynes 58 (Scheme 7).47 The mechanism of this reaction commences with oxidative addition of the aryl bromide moiety into the Pd(0) source, leading to a syn-migratory insertion into the alkyne C≡C bond to form Pd-vinyl species 60. This is followed by alkene isomerization to 62 via syn-anti transition state 61 and coordination of the amine to the palladium resulting in reductive elimination of intermediate 63. The resulting enamine 64 subsequently undergoes ring-opening isomerization to yield the corresponding 3-aryl-substituted pyrrole.
Scheme 7.
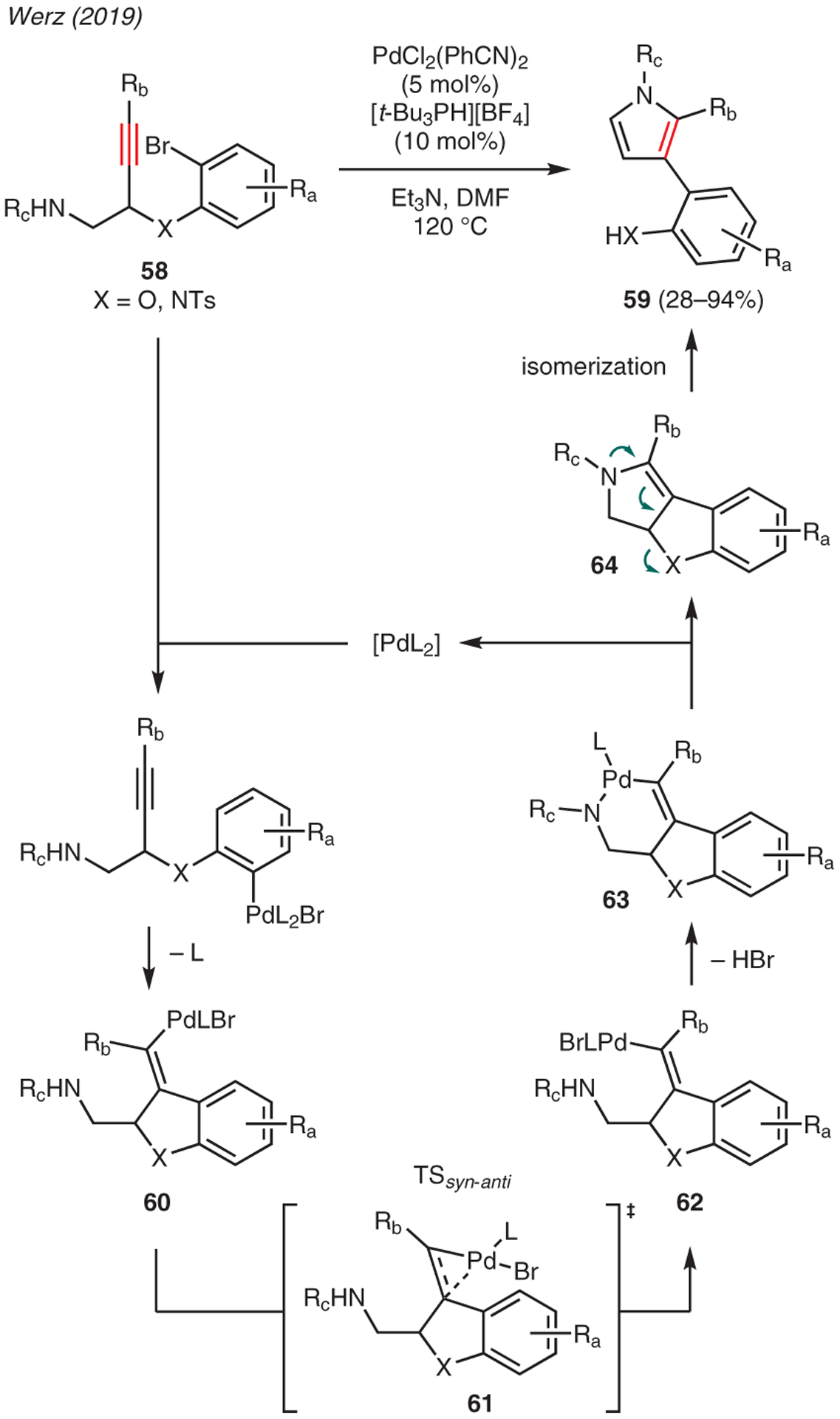
From alkynes: palladium catalysis
2.4.3. Catalysis by Other Transition Metals
Apart from the use of gold and palladium, there are several other metal-catalyzed reactions with alkynes to synthesize various functionalized pyrroles (Scheme 8). For instance, Yin reported the preparation of polysubstituted 2-chloropyrroles 66 from N-furfuryl-β-enaminones, available from starting furfurylamines 65 and alkyne esters, in the presence of catalytic CuCl2.48 Mechanistic studies suggest formation of radical cation 67 through single electron transfer (SET) promoted by the copper(II) catalyst, en route to formation of carbocation 68 via chlorination. The ensuing intramolecular capture by the pendant enamine moiety generates iminium ion 69. A series of deprotonations leads to aromatization and dechlorination to afford pyrrole 70, which forms radical cation 71 in the presence of CuCl2 and undergoes chlorination to carbocation 72; loss of a proton then furnishes the 2-chloropyrrole adduct. It is proposed that O2 serves as a terminal oxidant in the acidic reaction medium, which regenerates CuCl2 from CuCl.
Scheme 8.
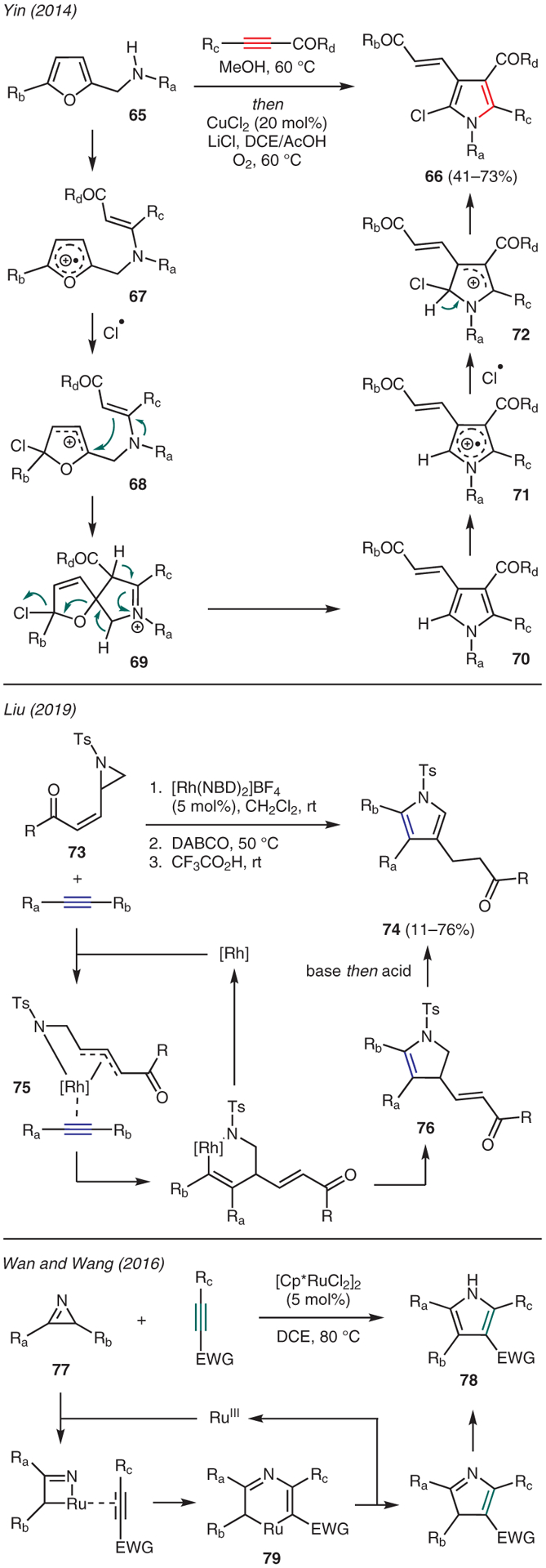
From alkynes: transition-metal catalysis
Liu described the use of [Rh(NBD)2]BF4 catalyst in the reaction of alkynes with 2-vinylaziridines 73 to produce functionalized pyrroles 74 (Scheme 8).49 In this reaction, the proposed mechanism commences with a rhodium-promoted oxidative ring opening of the aziridine, leading to coordination with the alkyne to give intermediate 75, which then undergoes migratory insertion followed by reductive elimination to form dihydropyrrole 76. DABCO then mediates isomerization-aromatization of 76 via a series of proton transfers.
Wan and Wang reported the ruthenium-catalyzed synthesis of polysubstituted pyrroles 78 via intermolecular [3+2] cycloaddition between activated alkynes with 2H-azirines 77.50 The reaction is proposed to proceed through azirine oxidative ring opening and alkyne insertion by the ruthenium catalyst. The resulting intermediate 79 undergoes reductive elimination and aromatization to provide thermodynamically stable pyrrole ring while regenerating the catalyst.
2.4.4. Reagent-Driven Synthesis
Approaches by which the synthesis of pyrroles from alkynes proceeds in the presence of stoichiometric reagents are shown in Scheme 9. For example, Čikotienė described the reaction of 1-(alk-1-ynyl)cyclopropyl imines 80 with various compounds containing polarized covalent bonds (EX) to produce pyrroles 81.51 The mechanism of this reaction is proposed though activation of alkyne with EX, resulting in the fragmentation of the cyclopropyl ring and 5-membered pyrrole cyclization by the imine moiety.
Scheme 9.
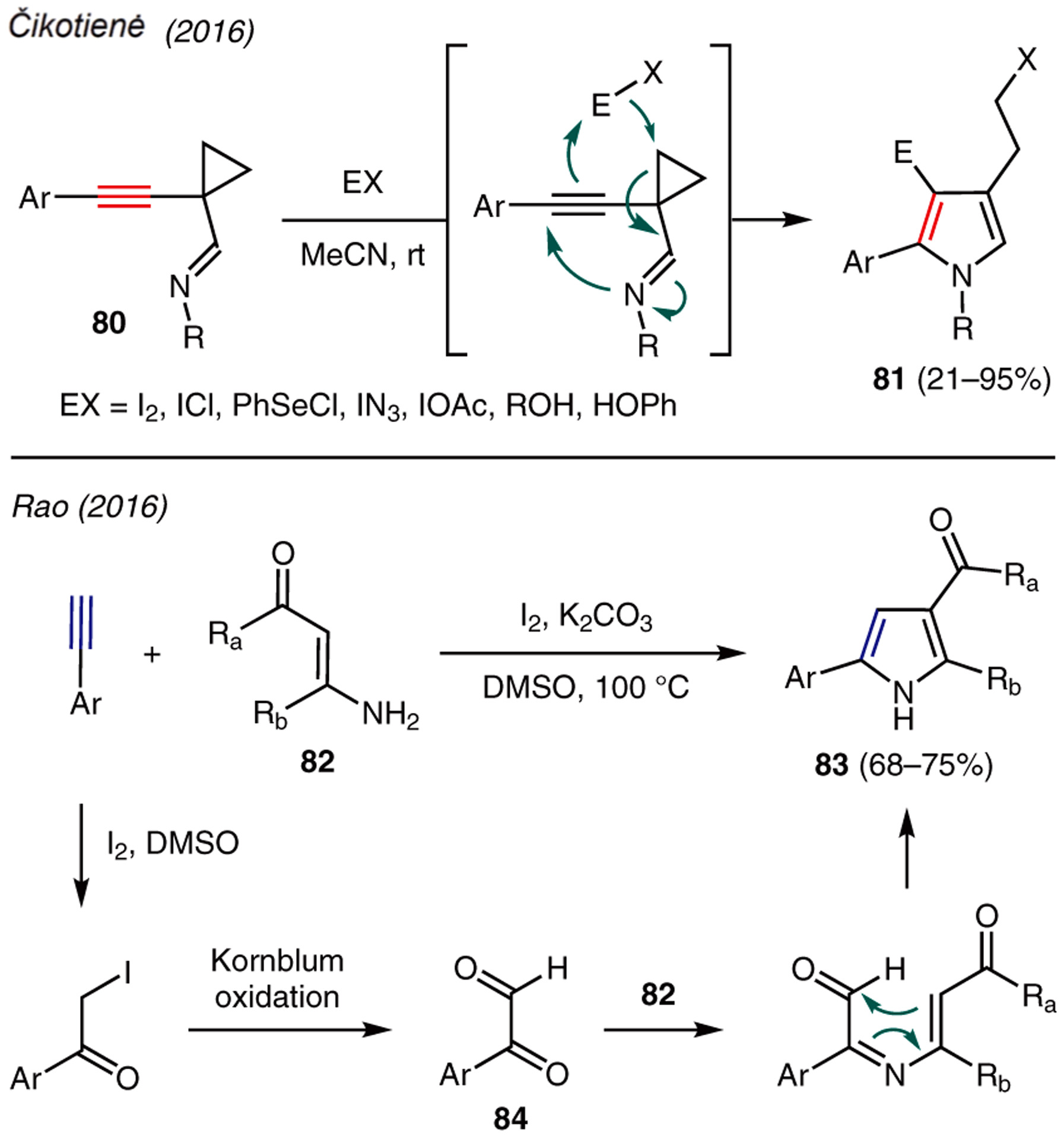
From alkynes: reagent-driven
Rao reported an iodine-mediated synthesis of 2,3,5-trisubstituted pyrroles 83 using arylacetylenes and β-enaminones 82 in the presence of K2CO3 and DMSO (Scheme 9).52 The reaction mechanism begins with the α-iodination of the alkyne, followed by Kornblum oxidation to yield dicarbonyl intermediate 84. The ensuing condensation with β-enaminone, followed by intramolecular cyclization and dehydration then forms the pyrrole ring.
2.5. Propargylic Groups
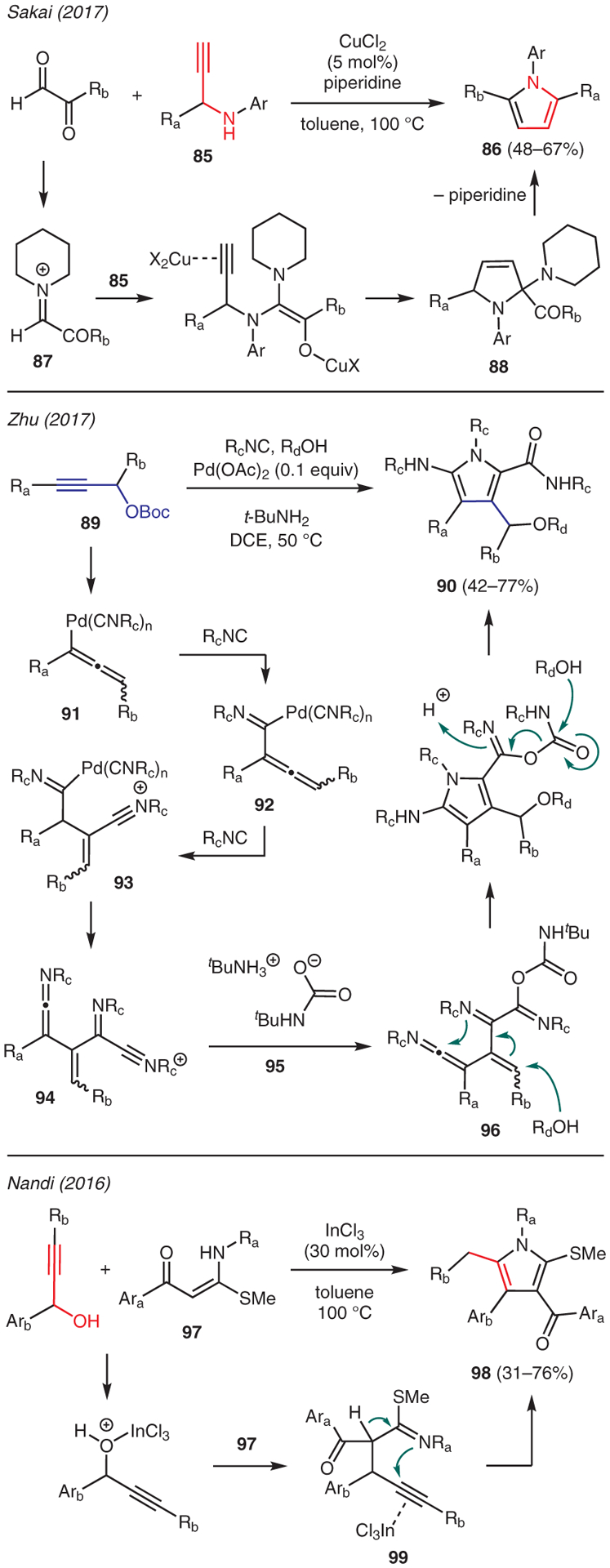
There are numerous examples on the use of propargylic starting materials, such as propargylic amines, carbonates, and alcohols, in pyrrole synthesis.53,54 Scheme 10 depicts representative examples. Sakai reported a [4+1] annulation of propargylamines with either ethyl glyoxalate or phenylglyoxal in the presence of CuCl2 and piperidine to yield 1,2,5-trisubstituted pyrroles 86.55 This reaction is proposed to proceed via iminium intermediate 87 resulting from the condensation of glyoxylate ester (or phenylglyoxal) and piperidine. The ensuing sequence of nucleophilic addition by the propargylamine 85, alkyne activation by CuCl2 to induce 5-endo-dig cyclization, and protonation generates intermediate 88, which undergoes aromatization with loss of piperidine to produce the pyrrole core.
Scheme 10.
From propargylic systems
Zhu developed a multicomponent reaction between propargyl carbonates, alcohols, and isocyanides in the presence of Pd(OAc)2 catalyst and a stoichiometric amount of tert-butylamine for the synthesis of 2-aminopyrroles 90 (Scheme 10).56 The proposed mechanism involves the intermediacy of (σ-allenyl)palladium(II) species 91, which is generated upon decarboxylation of propargylic carbonate 89 promoted by in situ generated Pd(0) isocyanide complex. (σ-Allenyl)palladium(II) species 91 undergoes migratory insertion of isocyanide to give intermediate 92, and this undergoes nucleophilic addition of a second isocyanide resulting in nitrilium ion 93. Subsequent β-hydride elimination from nitrilium ion 93 and a further nucleophilic addition of isocyanide gives nitrilium intermediate 94 which then reacts with carbamate anion 95 to afford advanced intermediate 96. Carbamate anion 95 is produced upon the liberation of CO2 during decarboxylation of 89, which is immediately trapped by t-BuNH2. Intermediate 96 undergoes 1,4-addition of alcohol, followed by cyclization of the resulting enamine onto the ketenimine to form the pyrrole ring. Cleavage of the carbamate group then furnishes the final product 90.
An example of the use of propargylic alcohols was reported by Nandi who demonstrated an efficient synthesis of highly substituted pyrroles in the presence of InCl3 catalyst and α-oxoketene N,S-acetals 97 (Scheme 10).57 This reaction is believed to begin with ionization of the propargylic alcohol by InCl3, leading to nucleophilic substitution by α-oxoketene N,S-acetals 97 to form intermediate 99. The ensuing alkyne activation by InCl3 results in intramolecular 5-membered cyclization, en route to the pyrrole product 98 upon proton transfer.
2.6. Homopropargylic Amines
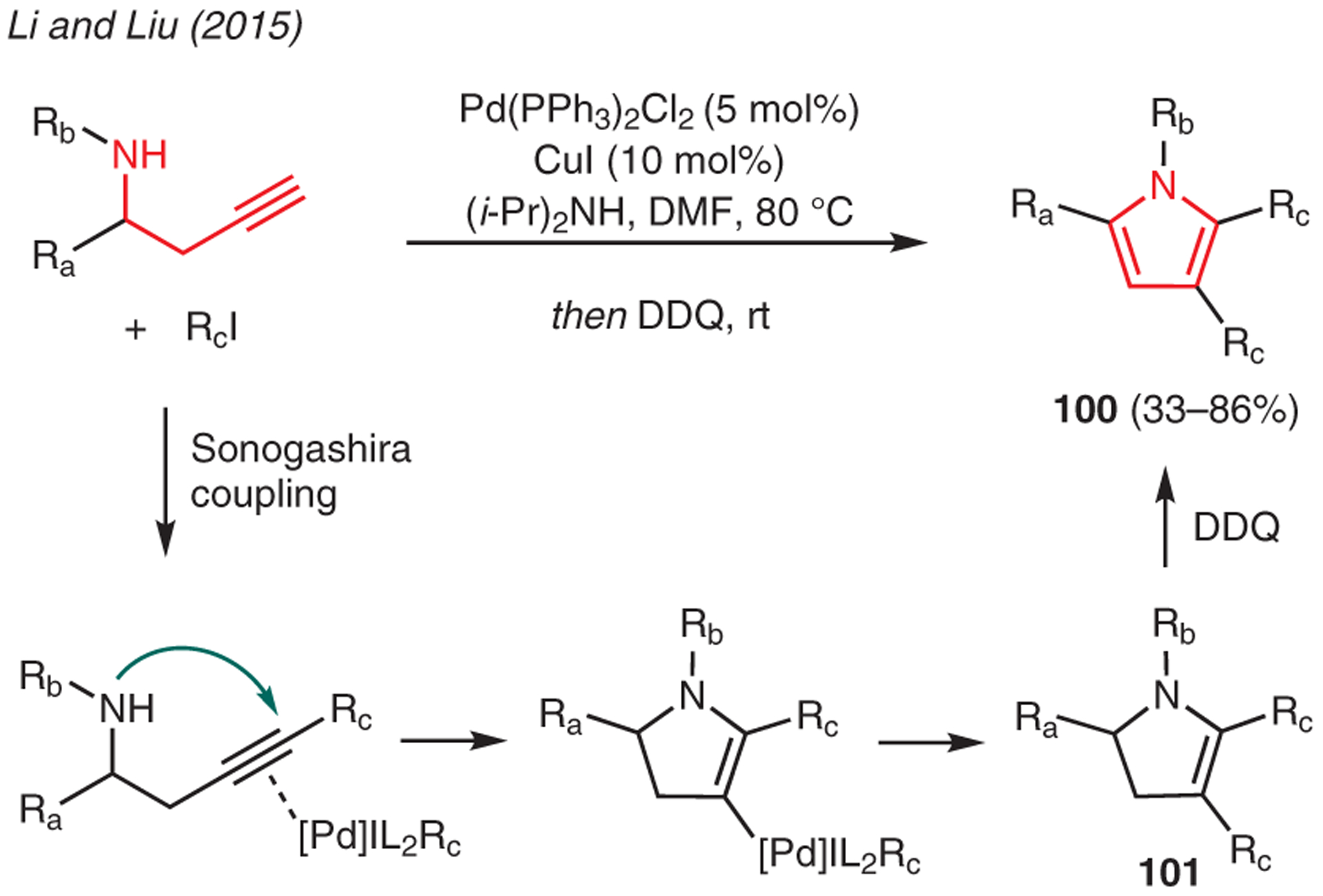
Homopropargylic amines served as an effective substrates for the synthesis of pyrroles.58 As exemplified in Scheme 11, Li and Liu reported the preparation of tetrasubstituted pyrroles 100 via the reaction of homopropargylic amines and aryl iodides in the presence of catalytic Pd(PPh3)2Cl2, CuI, and excess diisopropylamine.59 This multistep cascade reaction is presumed to involve a sequence of Sonogashira coupling, intramolecular hydroamination, and reductive elimination. The final oxidation of the 2-pyrroline intermediate 101 with DDQ then yields the final pyrrole product 100.
Scheme 11.
From homopropargylic amines
3. From Carbonyl Compounds
Carbonyl compounds have been traditionally employed as convenient substrates for pyrrole synthesis. This trend has continued and evidenced by the substantial number of methodologies in the period 2014–2019 that have been developed using these starting materials. The use of nitriles and isonitriles have also been reported.
3.1. Aldehydes
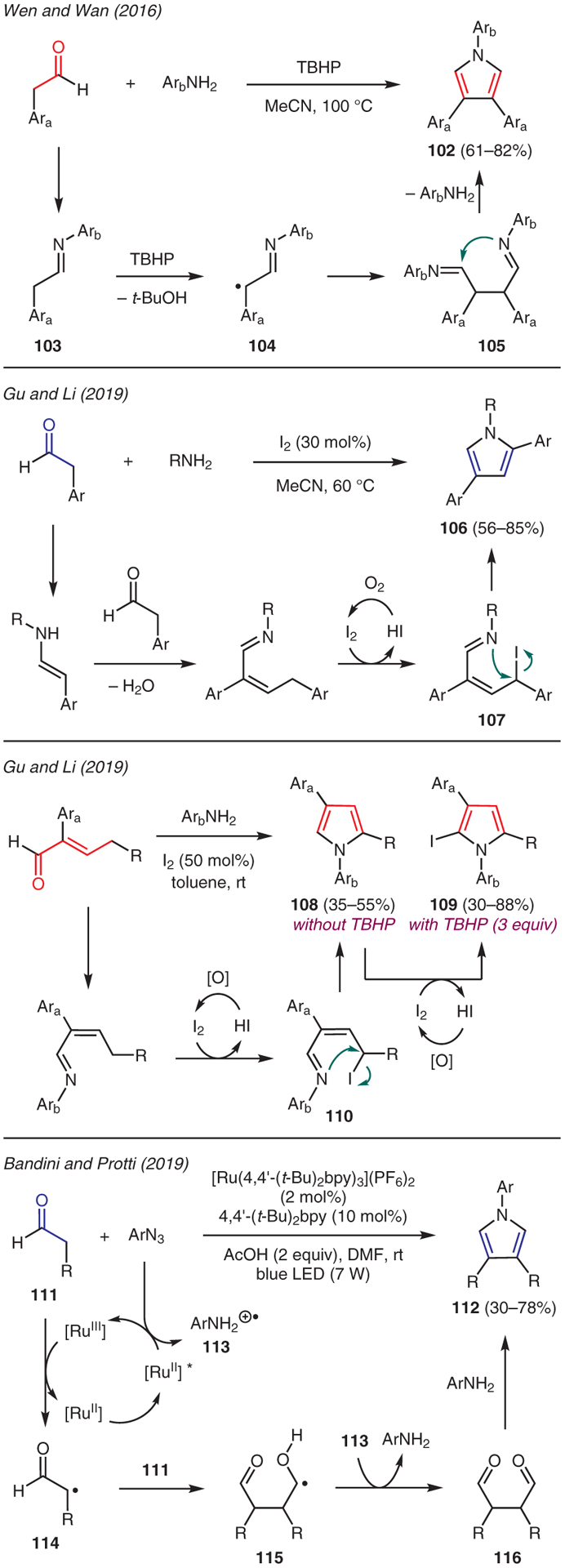
As depicted in Scheme 12, Wen and Wan developed a metal-free cascade reaction of phenylacetaldehydes and anilines, which provided a facile access to 1,3,4-trisubstituted pyrroles upon treatment with the tert-butyl hydroperoxide (TBHP).60 This reaction is proposed to proceed via oxidation of imine 103 to radical intermediate 104, which then dimerizes to 105. A series of tautomerization, intramolecular cyclization, and elimination of one aniline molecule then produces triaryl-substituted pyrroles 102.
Scheme 12.
From aldehydes
Gu and Li described two approaches to the synthesis of pyrroles from aldehydes (Scheme 12). The first method involved treatment of enolizable aldehydes with primary aliphatic amines and catalytic iodine, which served as Lewis acid and mild oxidant.61 The proposed mechanism commences with the Mannich-aldol condensation between the aldehydes and amines, which is then followed by iodination to intermediate 107, thereby enabling intramolecular cyclization to pyrrole 106. In the second method, enolizable α,β-unsaturated aldehydes and aromatic amines were subjected to catalytic iodine to afford N-arylpyrroles 108. Interestingly, the addition of TBHP to this iodine-mediated reaction led to C-2 iodized pyrroles 109. A similar reaction mechanism, involving intramolecular cyclization by the imine moiety in iodized intermediate 110, is proposed. However, the presence of oxidant TBHP readily regenerates molecular iodine from the iodide ion, enabling electrophilic aromatic substitution of the forming pyrroles 108 to furnish 2-iodopyrroles 109.
The emergence of visible-light photoredox catalysis has facilitated the development of new synthetic methods to access functionalized pyrroles from aldehydes. For instance, Bandini and Protti reported condensation of aryl azides and aldehydes promoted by Ru(II) catalysis and blue LED light for the regioselective preparation of 1,3,4-trisubstituted pyrroles 112 (Scheme 12).62 In this reaction, aryl azides served both as a source of nitrogen atom for the pyrrole core as well as a formal stoichiometric oxidant. The proposed mechanism begins by the generation of photoexcited [Ru2+]* complex, which undergoes oxidative quenching by aryl azide to produce anilino radical cation 113 upon protonation of the emerging nitrogen radical intermediate. The transient Ru3+ then oxidizes the aldehyde 111 through SET, followed by deprotonation, to generate α-carbonyl radical 114. The ensuing coupling with another molecule of aldehyde leads to α-hydroxy radical 115, which is oxidized to 1,4-dialdehyde 116 by anilino radical cation 113. Finally, Paal–Knorr condensation between both in situ generated aniline and 1,4-dialdehyde then furnishes pyrrole 112 as the major isomer.
3.2. Ketones
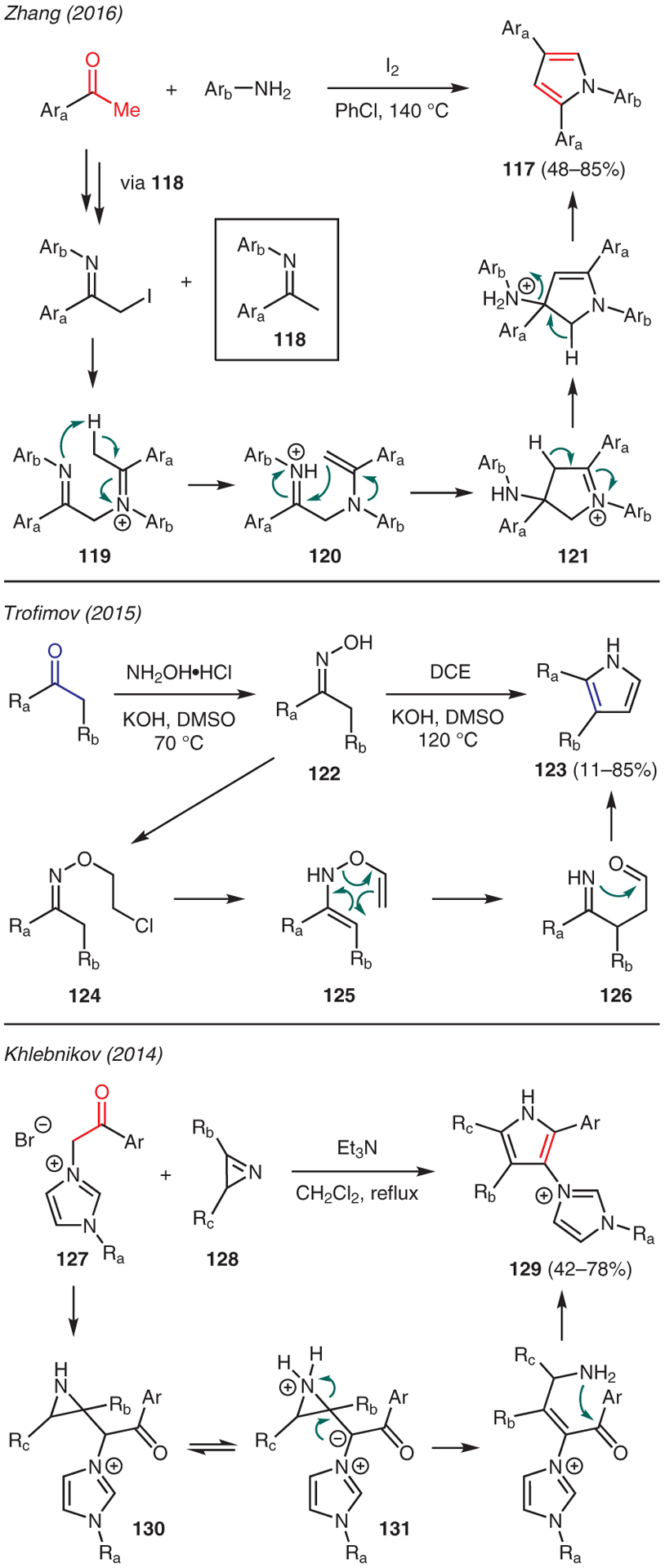
Examples of pyrrole synthesis from simple ketones are showcased in Scheme 13. Using aryl methyl ketones and anilines, Zhang reported a one-pot iodine-mediated cascade condensation-cyclization reaction to prepare 1,2,4-triarylpyrroles 117.63 The mechanism is proposed to involve α-iodination of ketimine intermediate 118, allowing for nucleophilic substitution by another molecule of ketimine resulting in iminium ion 119. Following proton transfer to form enamine 120, intramolecular Mannich-type condensation then takes place to yield cyclic intermediate 121. Aromatization upon extrusion of an aniline moiety gives the pyrrole product 117.
Scheme 13.
From ketones
Trofimov showed that ketones could be converted into pyrroles 123 in a one-pot procedure upon treatment with hydroxylamine hydrochloride and KOH, followed by addition of 1,2-dichloroethane (Scheme 13).64 It is proposed that ketoxime 122 is the putative intermediate in this reaction, which undergoes nucleophilic substitution with 1,2-dichloroethane to form O-(2-chloroethyl) ketoxime 124. The ensuing elimination of chloride to N-vinylketoxime 125 then enables 3,3-sigmatropic rearrangement to iminoaldehyde 126; intramolecular condensation then produces the pyrrole core.
Owing to presence of pyrrole and imidazole units in various medicinally important molecules, Khlebnikov developed a new strategy to synthesize 1-alkyl-3-(1H-pyrrol-3-yl)-1H-imidazol-3-ium bromides starting from 1-alkyl-3-phenacyl-1H-imidazolium bromides 127 and azirine 128 (Scheme 13).65 The mechanism of this reaction is proposed to commence with formation of intermediate 130 through addition of the enolate of 127 to azirine. Upon proton transfer, the ensuing ionization of zwitterion intermediate 131 then promotes ring fragmentation, thus allowing 5-membered cyclization and dehydration to furnish the pyrrole product 129.
Khlebnikov also described another interesting strategy where 1,2,4-tricarbonyl compounds 132 act as Michael acceptors in the presence of 2H-azirines 135 and Cu(OAc)2 catalyst en route to the preparation of pyrrole isomers 133 and 134 (Scheme 14).66 The proposed reaction mechanism commences with a conjugate addition of azirine-metal complex to the tricarbonyl compound. The ensuing intramolecular cyclization of intermediate 136 at two possible carbonyl positions is then followed by azirine fragmentation and aromatization. The resulting two isomeric pyrrole adducts 133 and 134 are easily separable by chromatography.
Scheme 14.
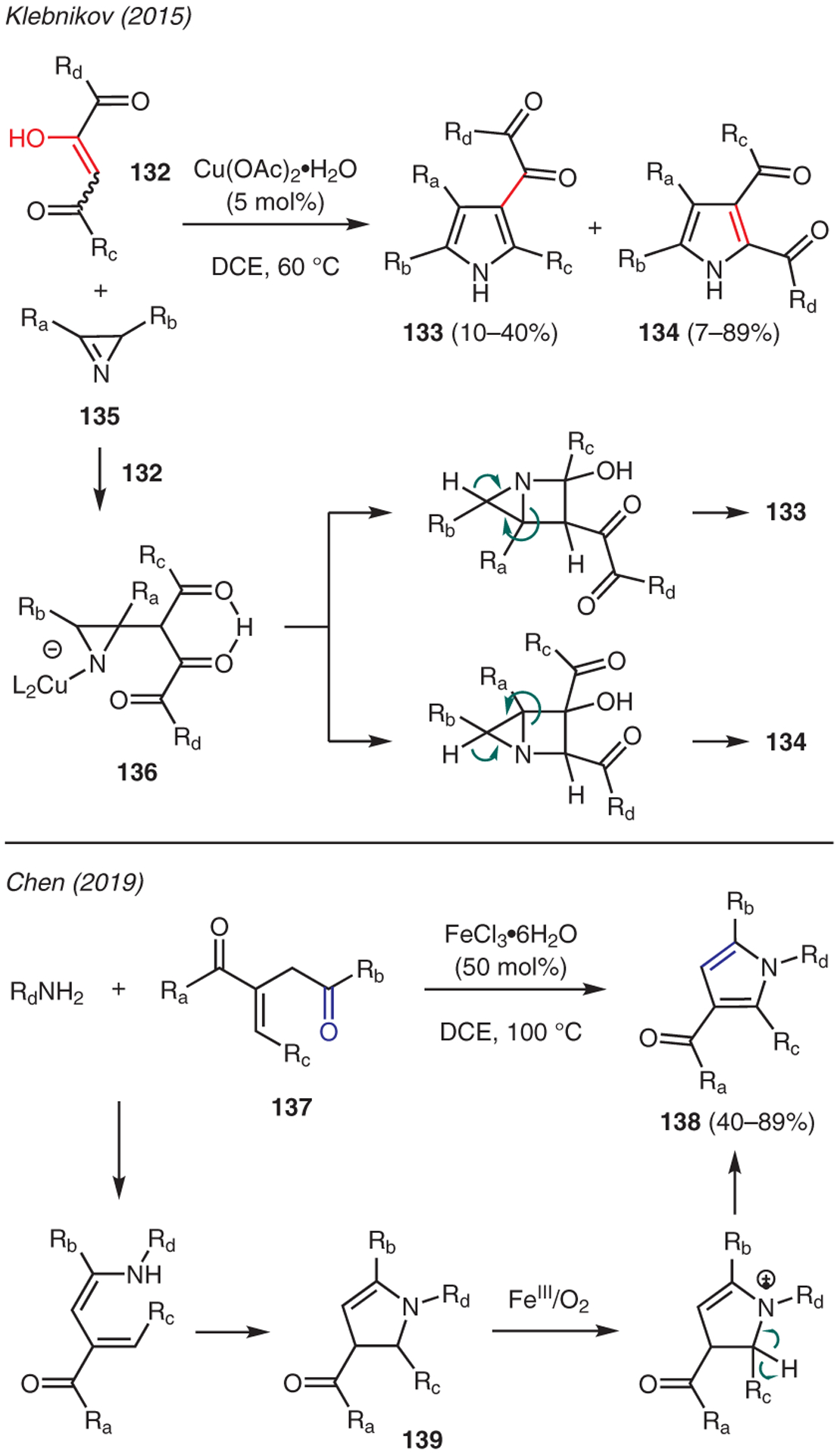
From ketones
Chen reported an FeCl3-mediated synthesis of pyrroles from 3-methylenehexane-2,5-dione 137 and primary amines (Scheme 14).67 This reaction is proposed to proceed via the intermediacy of 2,3-dihydropyrrole intermediate 139, which is generated by reaction of 3-methylenehexane-2,5-dione and primary amine, followed by intramolecular aza-Michael addition of the resulting enamine. Subsequent oxidative aromatization of this intermediate 139 by Fe3+ and O2 affords pyrrole 138.
3.2.1. α-Hydroxy and α-Halo Ketones
Functionalized ketones at the α-carbon have been shown to be a convenient starting point for pyrrole synthesis.68,69 For example, Lei and Lui reported an acid-promoted cross-dehydrative aromatization reaction between α-hydroxyaryl ketones 140 and aryl ketones 141 in the presence of ammonium acetate to access unsymmetrical tetraaryl-substituted pyrroles (Scheme 15).70 This reaction is proposed to involve in situ generation of α-amino ketone 143 upon a sequence of imine formation and tautomerization between the α-hydroxy ketone and ammonium acetate. The ensuing condensation of α-amino ketone 143 with aryl ketone 141, followed by tautomerization to enamine intermediate 144 sets the stage for intramolecular cyclization and aromatization to yield the tetraarylpyrroles 142.
Scheme 15.
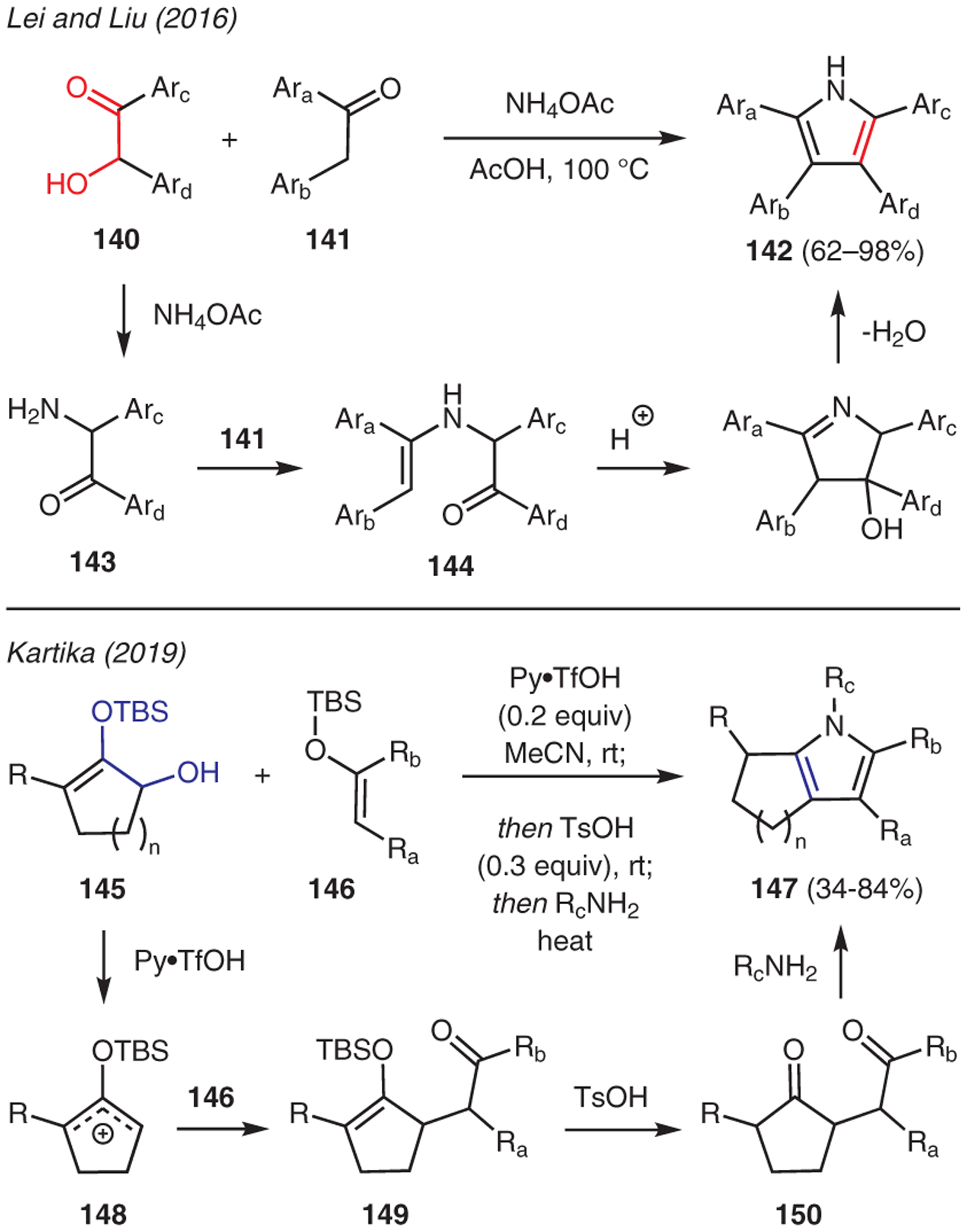
From α-hydroxy ketones
Another example was demonstrated by Kartika, who synthesized pyrrole-derived compounds by subjecting α-hydroxy silyl enol ethers 145 to silylenolates 146, and then primary amines, in the presence of a Brønsted acid catalyst (Scheme 15).71 In this reaction, ionization of α-hydroxy silyl enol ether forms unsymmetrical silyloxyallyl cation 148, which is captured in a regioselective manner by silylenolate 146. The resulting γ-keto silyl enol ether 149 then undergoes protodesilylation under the Brønsted acidic conditions to unmask the 1,4-diketone moiety 150, thus enabling Paal–Knorr condensation with various primary amines to install pyrrole structure 147.
Apart from α-hydroxy ketones, the use of α-halo ketones for pyrrole synthesis has also been documented in the literature (Scheme 16). For instance, Wu reported a photoinduced Hantzsch-type reaction between α-bromo ketones and enaminones to synthesize polysubstituted pyrroles in the presence of catalytic Ir(ppy)3 and triethylamine.72 The mechanism is proposed through photoexcitation of Ir(ppy)3, which generates alkyl radical intermediates 154 from α-bromo ketone 151 via SET. The subsequent coupling of 154 with enaminones 152 leads to amino radical 155, which is oxidized by Ir(ppy)3+ species to ketimine 156. The following intramolecular condensation and dehydration assisted by Et3N generates the pyrrole adduct 153.
Scheme 16.
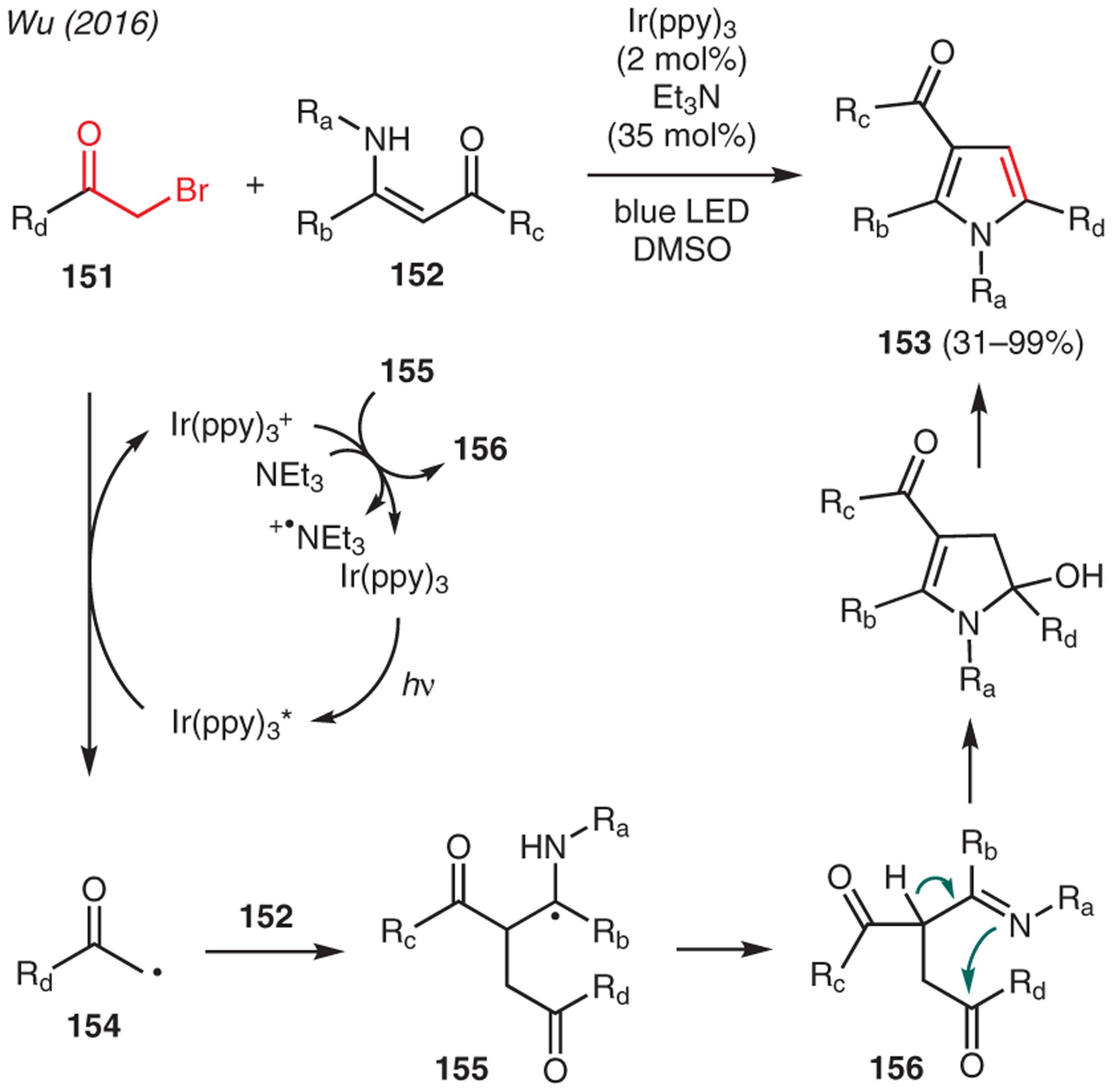
From α-bromo ketones
3.2.2. Enones
Enones are used as versatile precursors to pyrrole synthesis due to their facile reactivity as Michael acceptors.73 As shown in Scheme 17, Opatz showcased cyclocondensation of enones with aminoacetonitriles to form 3,4-dihydro-2H-pyrrole-2-carbonitriles 157.74 Using one-pot protocols, these synthetic intermediates could be converted into either 2,4-disubstituted pyrroles 158 through microwave-mediated dehydrocyanation or to 3,5-disubstituted pyrrole-2-carbonitriles 159 upon oxidation with DDQ. The key mechanistic step in this reaction is believed to involve a 6π-electrocyclic ring closure of pentadienyl anion 160, generated upon condensation of the starting enone and aminoacetonitrile, followed by a loss of a proton.
Scheme 17.
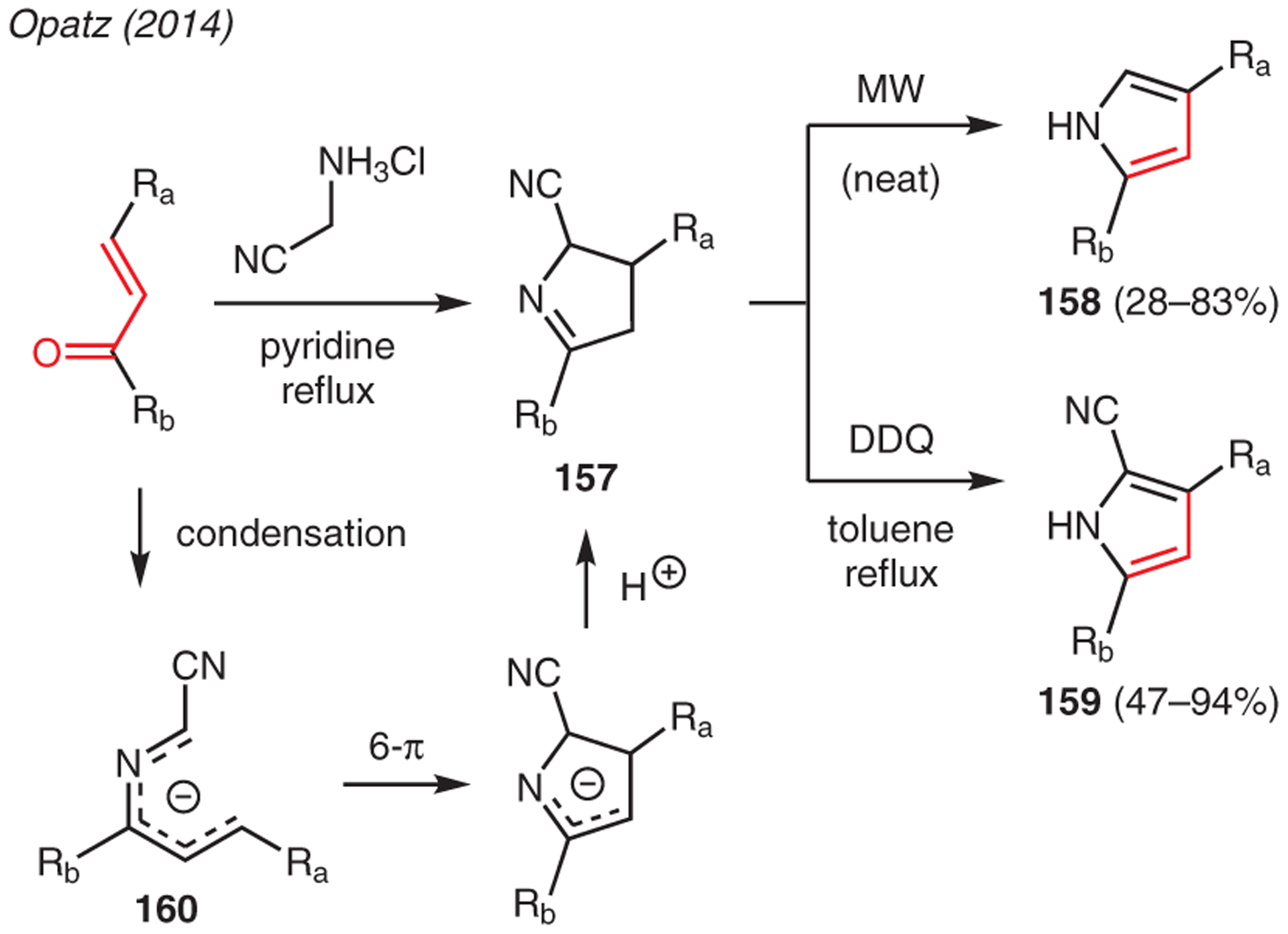
From enones
3.3. Cyanides and Isocyanides
Diederich reported the reaction of nitrile-containing buta-1,3-dienes 161 with amines under microwave conditions toward the synthesis of 2-aminopyrroles 162 (Scheme 18).75 This reaction is proposed to proceed via nucleophilic addition of amine to the nitrile moiety, followed by intramolecular cyclization of the resulting amidine intermediate 163; subsequent tautomerization then constructed the pyrrole product 162.
Scheme 18.
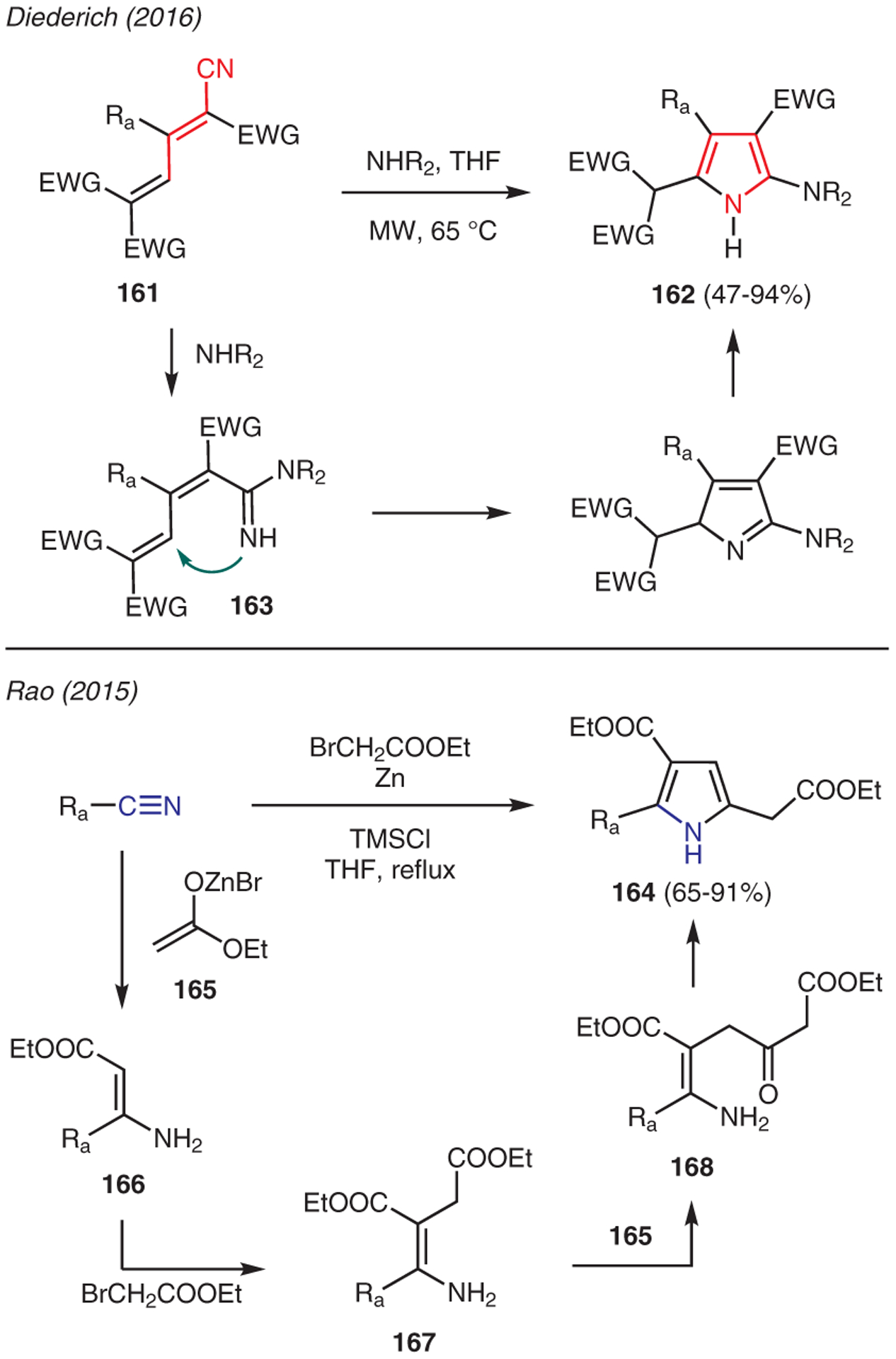
From cyanides
Rao has shown that aliphatic, aromatic, and benzylic nitriles could be converted into 2,3,5-trisubstituted pyrrole diesters 164 via a zinc-mediated pseudo four-component reaction catalyzed by TMSCl (Scheme 18).76 The reaction proceeds by the addition of the zinc enolate of ethyl bromoacetate 165 to the nitrile to form enamino ester 166. Then, C-alkylation with another molecule of ethyl bromoacetate produces diester 167, which undergoes Perkin ester condensation with another molecule of zinc enolate 165 to form key intermediate 168; subsequent dehydrative cyclization results in trisubstituted pyrroles 164.
Examples on the use of isonitriles in pyrrole synthesis are depicted in Scheme 19. Jiang and Wu developed a palladium-catalyzed three-component reaction using aryl halides, isocyanides, and N-tosylhydrazones 169 to assemble substituted 3-aminopyrroles.77 The key mechanistic steps in this reaction begin with oxidative addition of Pd(0) to aryl halides, followed by double isocyanide insertion to form iminopalladium 171. This species is then trapped by in situ generated diazo compound 172, formed as a result of decomposition of N-tosylhydrazones 169, to yield palladium-carbene complex 173. Migratory insertion, followed by β-hydride elimination, leads to intermediate 174 which undergoes intramolecular cyclization to yield 3-aminopyr-roles 170.
Scheme 19.
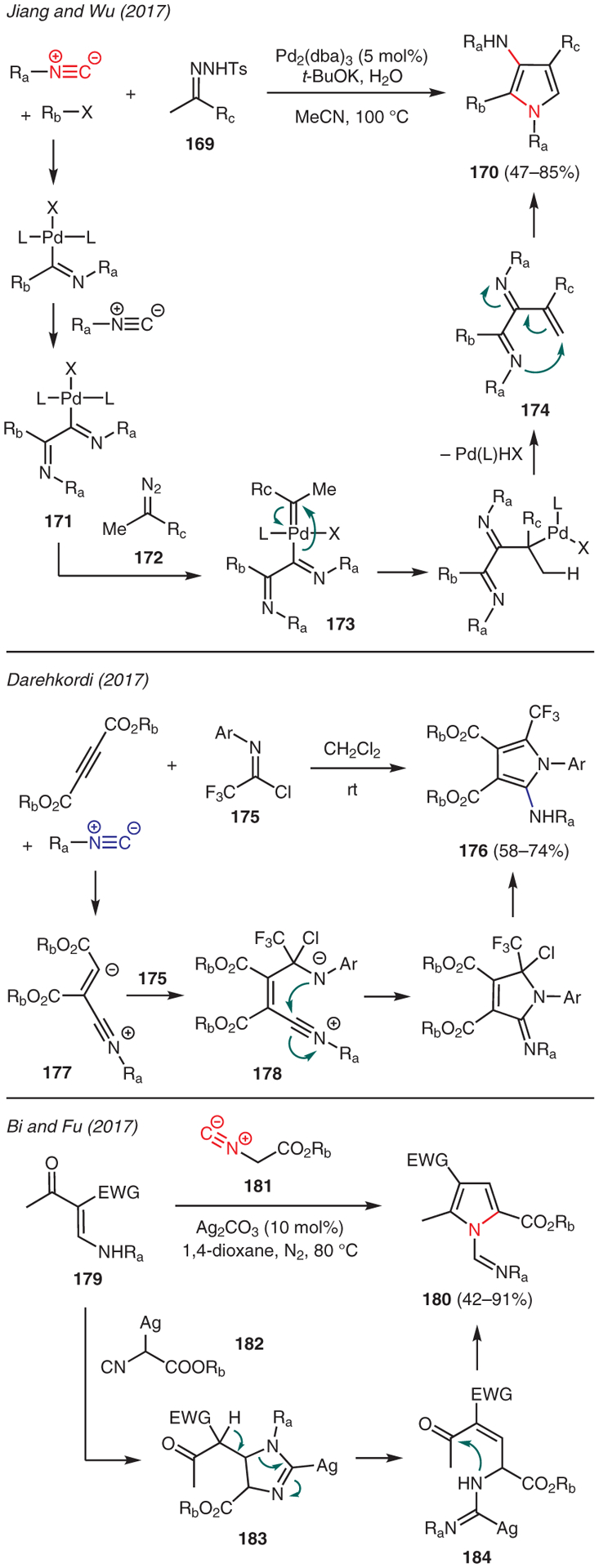
From isocyanides
Darehkordi reported a one-pot multicomponent reaction toward pyrrole synthesis using isocyanides, dialkyl acetylenedicarboxylates, and N-aryl-2,2,2-trifluoroacetimidoyl chlorides 175 (Scheme 19).78 In this example, addition of isocyanide to the acetylenedicarboxylate produces zwitterionic intermediate 177, which then adds to the imidoyl carbon of 175. The resulting tetrahedral intermediate 178 proceeds through intramolecular 5-membered cyclization and aromatization to furnish trifluoromethylated 2-amino-pyrrole 176.
Bi and Fu investigated the silver-catalyzed formal [3+2]-dipolar cycloaddition reactions between β-enaminones 179 and isocyanoacetates for the facile formation of functionalized pyrroles 180 (Scheme 19).79 The mechanism of this reaction involves [3+2] cycloaddition of the imine tautomer of β-enaminone 181 with α-metalated isocyanide 182 to form imidazoline intermediate 183. The enhanced acidity of the proton due to adjacent the electron-withdrawing group in 183 then allows a retro-hetero-Michael addition leading to ring fragmentation of the imidazoline moiety and recyclization to form the pyrrole products 180 via intermediate 184.
3.4. Formamides
Wang demonstrated Cu(OTf)2-catalyzed multicomponent reactions of N,N-disubstituted formamides 185, TMSCN, and aromatic alkenes or alkynes that led to the synthesis of polysubstituted pyrrole-2-carbonitriles 186 (Scheme 20).80 The mechanism commences with the generation of an α-aminonitrile via dicyanation of formamide in the presence of the Cu(II) catalyst. Subsequent release of HCN then yields azomethine ylide intermediate 187, which proceeds to regioselective [3+2] cycloaddition with the dipolarophiles, i.e. alkene or alkyne, to afford intermediate 188. Finally, oxidative dehydroaromatization with DDQ gives the pyrrole structure 186.
Scheme 20.
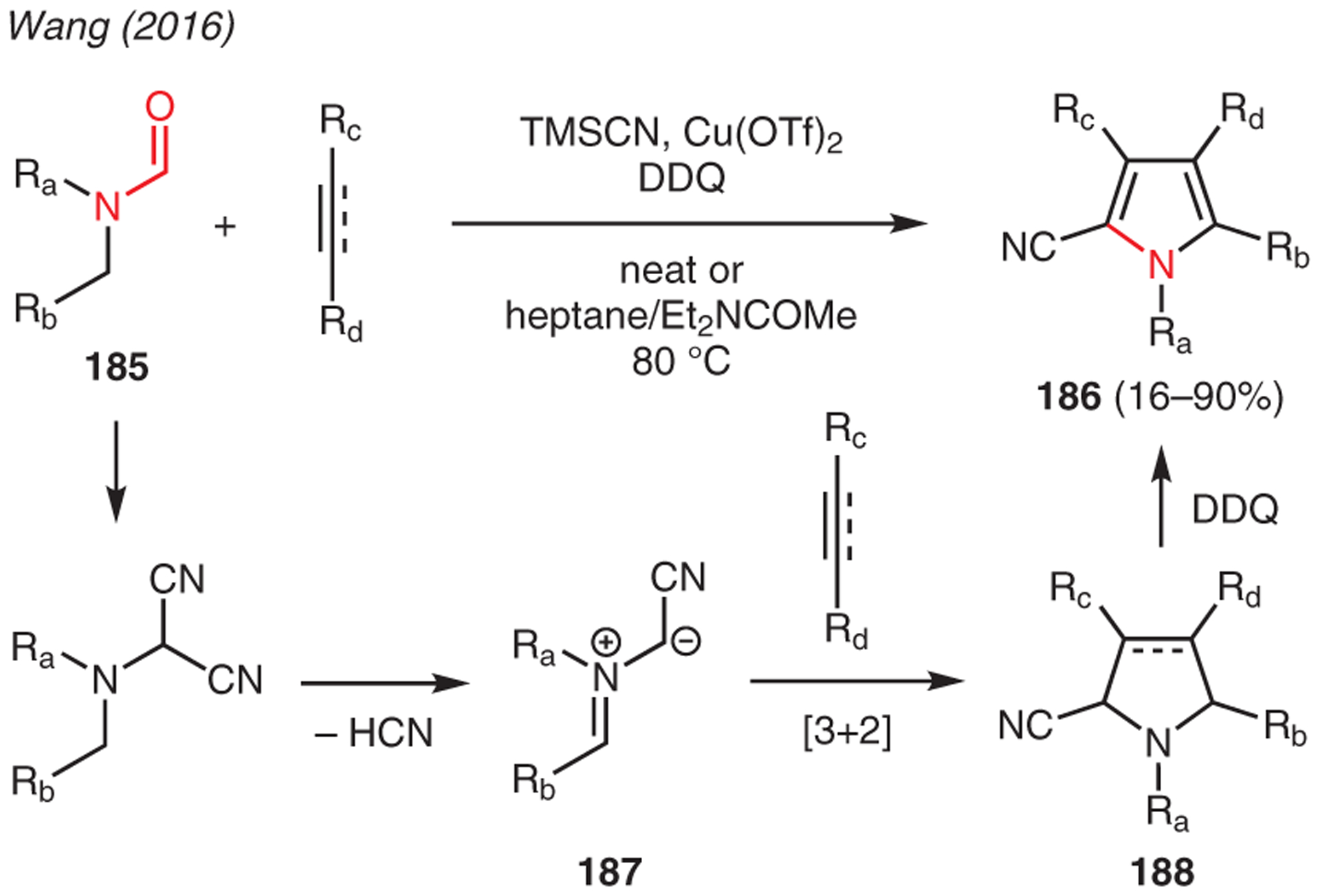
From formamides
3.5. β-Enamines
β-Enamines can be effectively employed in intramolecular cyclization upon self-dimerization to generate pyrroles (Scheme 21). For instance, a simple route to polycarbonyl pyrroles 190 via K2S2O8 promoted oxidative cyclization of enamine 189 was disclosed by Guan and Gao.81 In this chemistry, a radical mechanism is proposed. Initial oxidation of enamine 189 by K2S2O8 produces amino radical cation 191, which dimerizes with a second molecule of 189 to form intermediate 192 that undergoes a sequence of intramolecular C–N bond formation, oxidation, and aromatization upon extrusion of ammonia to yield the pyrrole adduct.
Scheme 21.
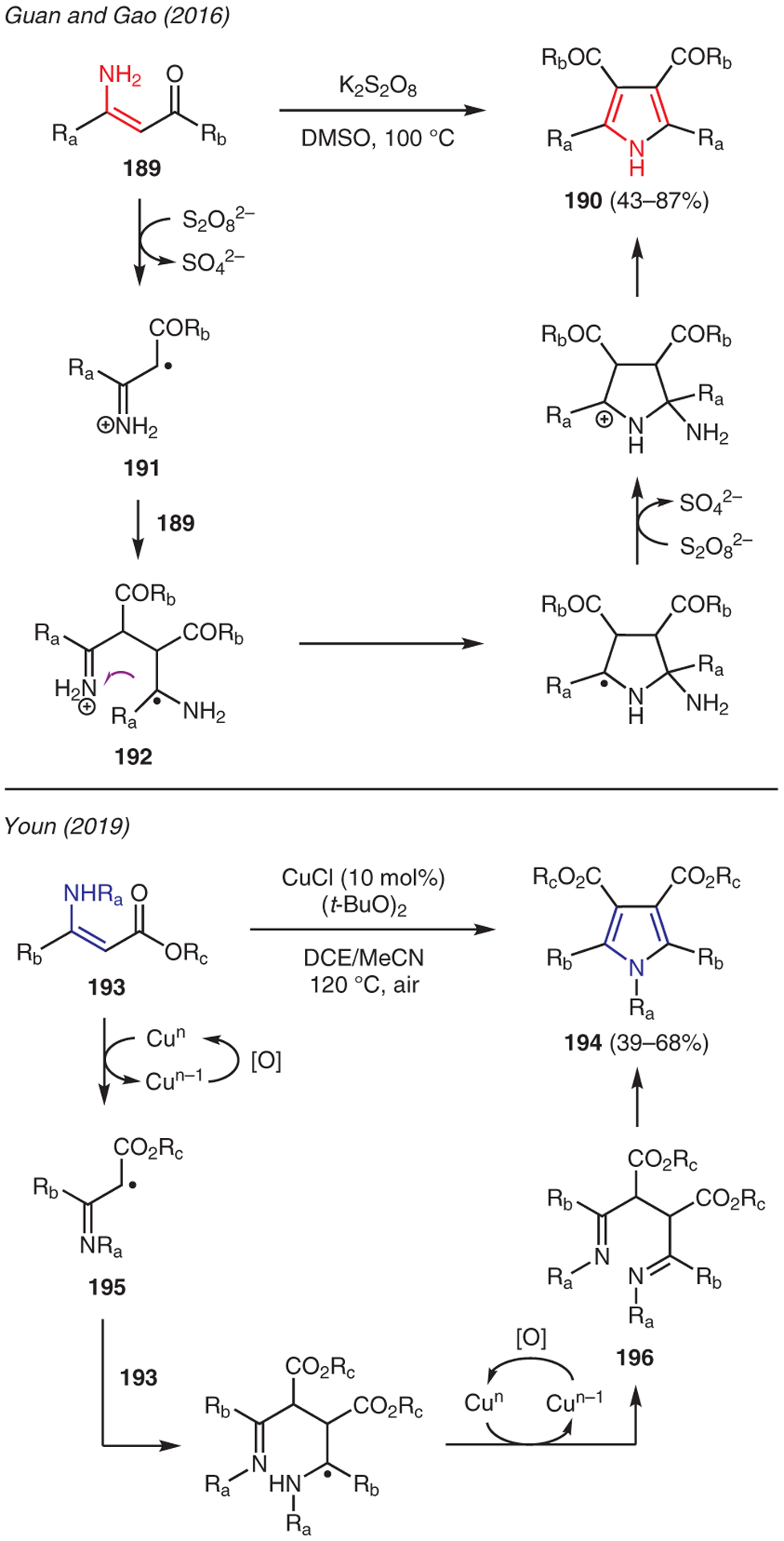
From β-enamines
Youn demonstrated CuCl-catalyzed oxidative annulation reactions using enamine 193 in the presence of di-tert-butyl peroxide (Scheme 21).82 Mechanistic investigations suggest the possible formation of radical intermediate 195 via SET, leading to self-dimerization. A second oxidation then occurs to generate diketimine intermediate 196, which undergoes cyclization to generate pyrrole 194.
3.6. Dicarbonyl Compounds
Various motifs of dicarbonyl compounds have been extensively utilized toward pyrrole synthesis.83–93 As discussed in this section, this class of starting materials have enabled the development of a broad array of new synthetic reactions.
3.6.1. 1,2-Dicarbonyls
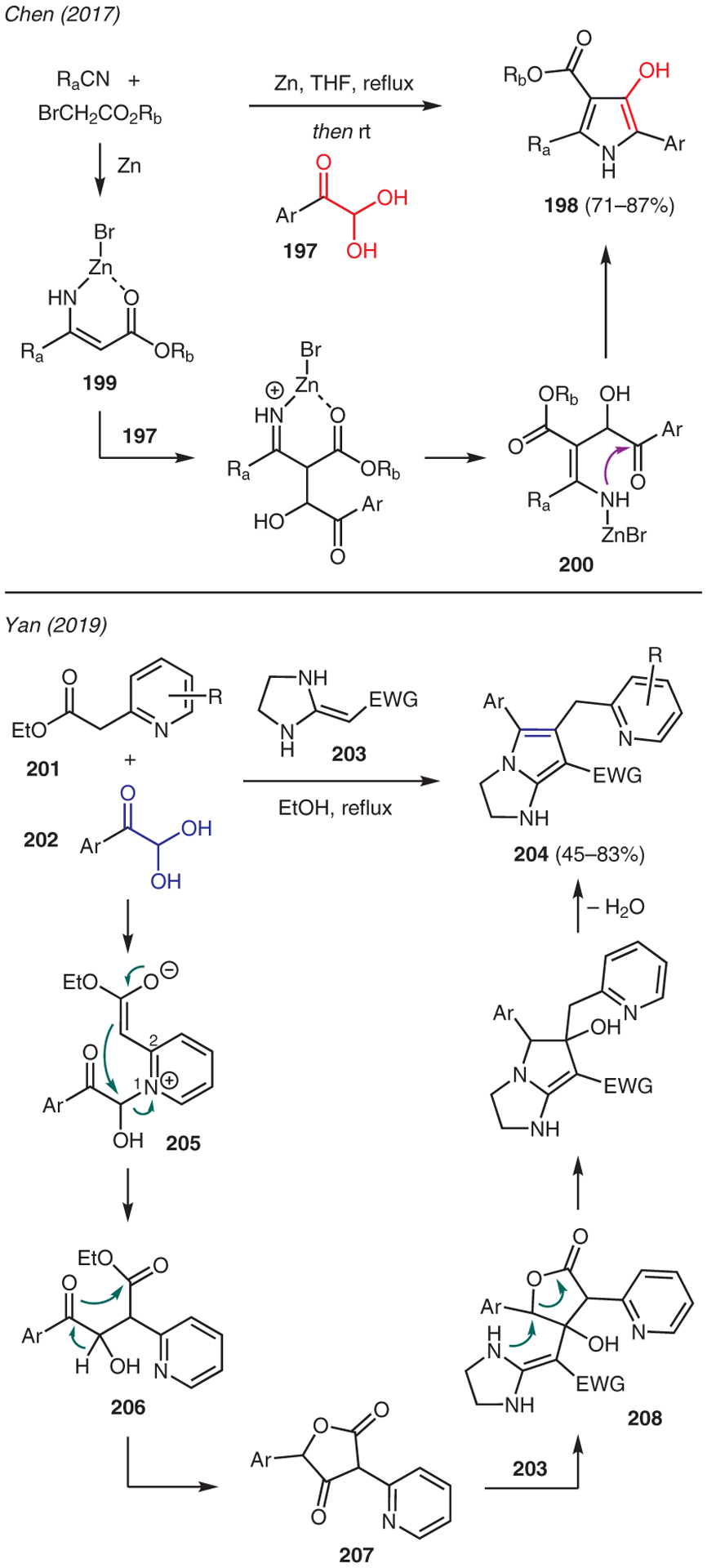
The use of 1,2-dicarbonyl compounds toward pyrrole synthesis has been largely confined to arylgloxals (Scheme 22).94–96 Chen developed a one-pot procedure involving the Blaise reaction between nitriles and α-bromo esters in the presence of zinc, followed by addition of arylglyoxal monohydrates 197.97 This reaction proceeds through formation of β-enaminone 199, which subsequently undergoes nucleophilic addition with the arylglyoxal 197; intramolecular cyclization and dehydration of the resulting intermediate 200 gives the pyrrole 198
Scheme 22.
From 1,2-dicarbonyls
Another example of the use of arylglyoxal is the multicomponent tether catalysis that was reported by Yan.98–101 The protocol in this chemistry involved a simple mixing of ethyl 2-(2-pyridyl)acetates 201, arylglyoxal monohydrates 202, and heterocyclic ketene aminals (HKA) 203 in ethanol at reflux. The mechanism is proposed via pyridinium addition intermediate 205, leading to tethered catalysis intramolecular cyclization to furnish 1,4-keto ester 206. Intra-molecular cyclization-tautomerization to cyclic intermediate 207 and nucleophilic 1,2-addition by HKA 203 gives advanced intermediate 208, which undergoes decarboxylative intramolecular cyclization and dehydration to produce 2-amino-4-(2-pyridylmethyl)pyrrole 204.
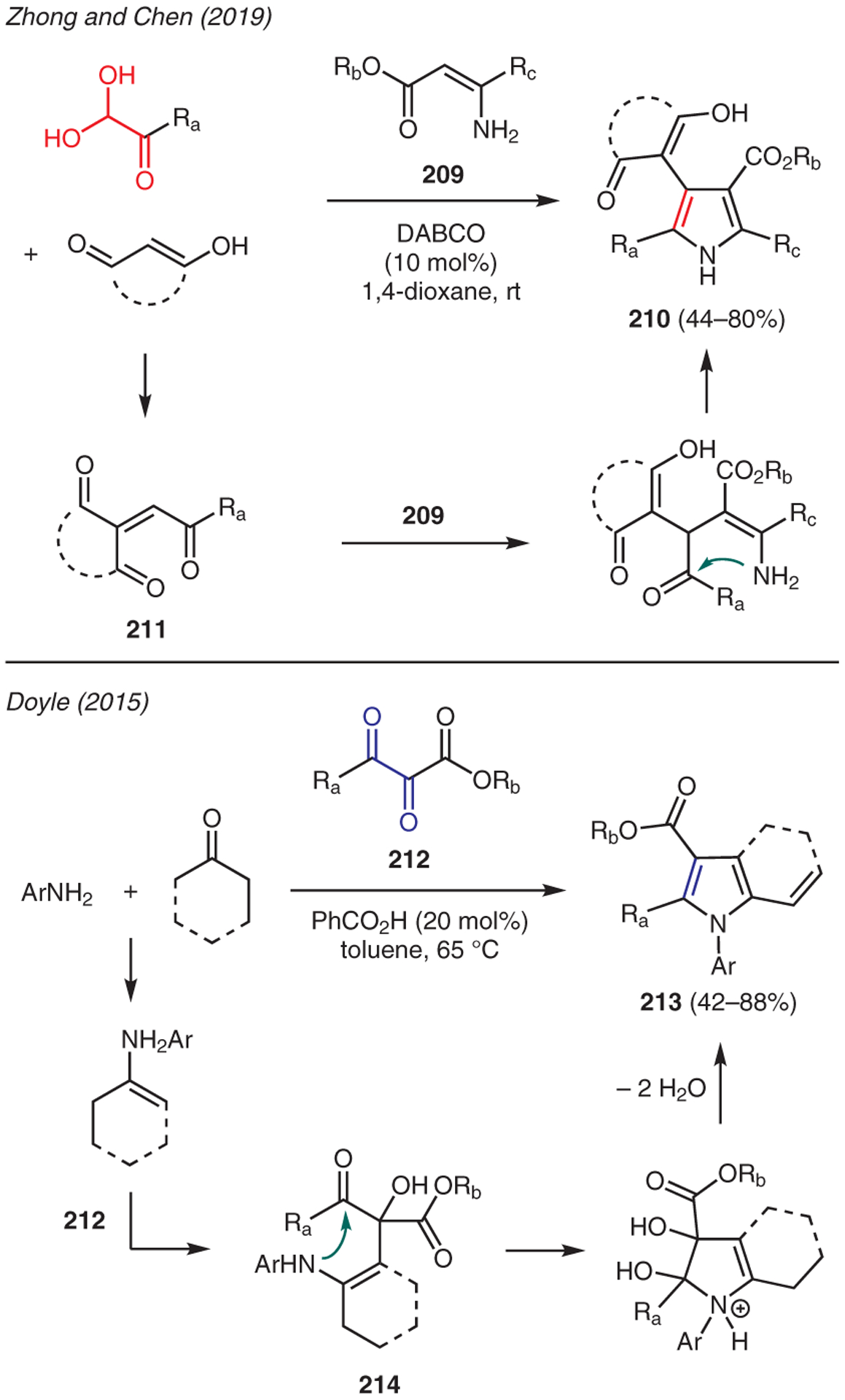
Zhong and Chen reported a DABCO-promoted three-component domino reaction using arylglyoxal monohydrates, enamino esters 209, and 1,3-dicarbonyl compounds to give highly functionalized NH-pyrrole adducts 210 (Scheme 23).102 This reaction commences with the Knoevenagel-type condensation between arylglyoxal and the 1,3-dicarbonyl compound in the presence of catalytic DABCO, thereby producing α,β-unsaturated intermediate 211, which serves as a Michael acceptor to electron-rich β-enaminone 209; cyclization then generates pyrrole 210.
Scheme 23.
From 1,2-dicarbonyls
Doyle developed a unique three-component cascade synthesis of pyrroles 213 by subjecting tricarbonyl compounds 212 to ketones and primary amines (Scheme 23).103 This reaction proceeds through a sequence of enamine addition to the most electrophilic central carbonyl carbon of 212, followed by cyclization of the resulting aldol intermediate 214. Aromatization via dehydration and proton transfer then produce 5-vinylpyrrole product 213.
3.6.2. 1,3-Dicarbonyls
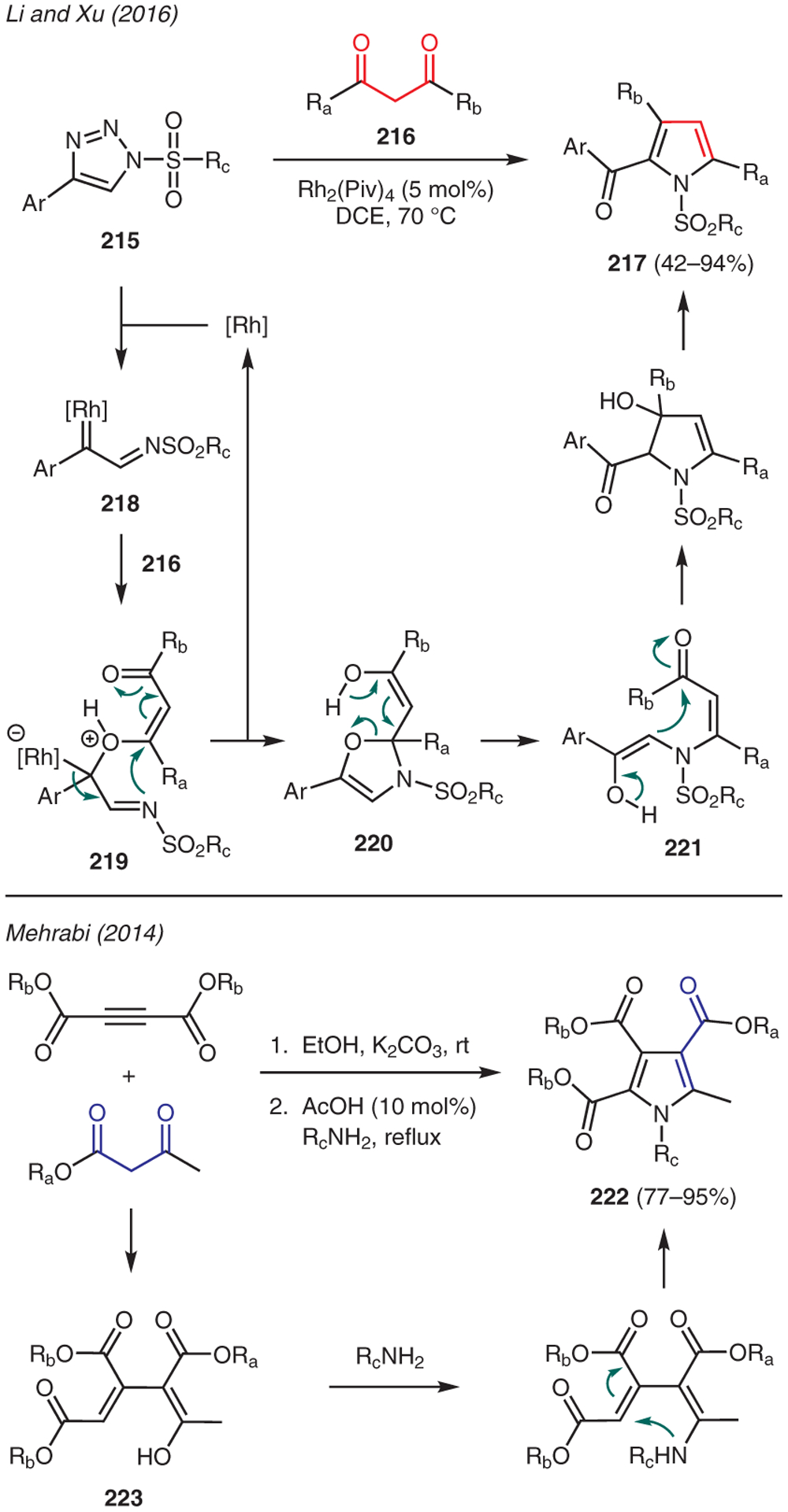
The utility of 1,3-dicarbonyl compounds in pyrrole synthesis is showcased in Scheme 24. For instance, Li and Xu reported the reaction between 1,3-diketones 216 and 1-sulfonyl-1,2,3-triazoles 215 catalyzed by Rh2(Piv)4 to give 2-carbonylpyrroles 217.104 In this chemistry, the Dimroth-type equilibrium of the 1-sulfonyl-1,2,3-triazole to an α-diazo imine enables the generation of α-imino rhodium carbene 218, which is then captured by the enol ether tautomer of the 1,3-diketone 216. The resulting zwitterionic intermediate 219 then proceeds through addition-elimination sequences to produce 221, which undergoes an intramolecular aldol reaction and aromatization to give N-sulfonylpyrrole 217.
Scheme 24.
From 1,3-dicarbonyls
Mehrabi described the synthesis of pentasubstituted pyrroles in a one-pot, two-step protocol using alkyl acetoacetates, dialkyl acetylenedicarboxylates, and amines in the presence of K2CO3.105 This reaction begins with Michael addition of alkyl acetoacetate to dialkyl acetylenedicarboxylate giving 4-hydroxypenta-1,3-diene-1,2,3-tricarboxylate 223. The ensuing condensation with primary amine, followed by cyclization and oxidation by air then yields pyrrole 222.
3.6.3. 1,4-Dicarbonyls
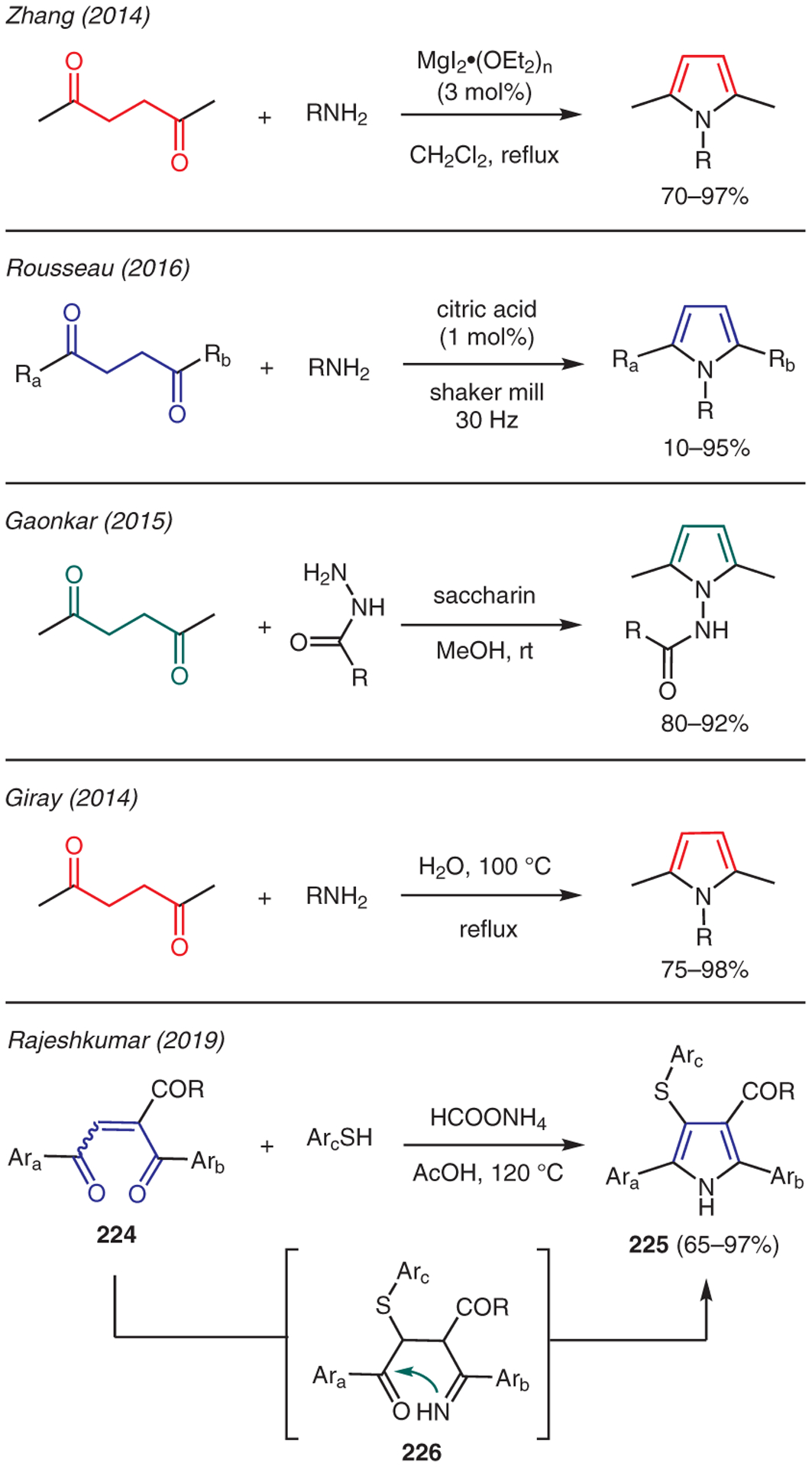
The condensation of primary amines with 1,4-dicarbonyl compounds, the Paal–Knorr reaction, is one of the most well-known approaches to synthesize various N-substituted pyrroles.106 Since 2014, there have been several new advances in this methodology (Scheme 25), particularly those focusing on green chemistry through the use of environmentally benign catalysts, such as MgI2·OEt2 by Zhang,107 citric acid by Rousseau,108 and saccharin by Gaonkar.109 As reported by Giray, an uncatalyzed Paal–Knorr reaction could be also performed in boiling water.110 Rajeshkumar reported the preparation of 4-(arylthio)pyrroles 225 via treatment of 1,4-enediones 224 with arenethiols and ammonium formate.111 The mechanism is proposed to involve Michael addition of arenethiol to 1,4-enedione 224, followed by Paal–Knorr condensation with in situ generated ammonia to form imino-carbonyl intermediate 226, which undergoes intramolecular cyclization and aromatization to generate 4-(arylthio)pyrroles 225.
Scheme 25.
From 1,4-dicarbonyls
The Clauson-Kaas reaction is another well-established methodology to synthesize pyrrole from primary amines and a 1,4-dicarbonyl surrogate in the form of 2,5-dimethoxytetrahydrofuran (DMTHF).112 Recent advancements in this reaction include a report by Chaudhari, who explored alkaline-earth metal salts, like Ca(NO3)2·4H2O, as mild Lewis acid catalysts (Scheme 26).113 A unique variation to the Clauson-Kass reaction was reported by Gu,114 who treated 2-alkoxy-2,3-dihydrofurans 228 with 2-methylindoles and FeCl3, followed by addition of Al(OTf)3 and arylamine, to produce 2-(3-indolyl)pyrroles 229. Both Lewis acids were employed in catalytic amounts. This one-pot, two-step protocol is proposed to proceed via addition of two molecules of indole upon Lewis acid promoted ring fragmentation of dihydrofuran 228 to give intermediate 230. Extrusion of 2-methylindole from 230 gives 231, which undergoes conjugate addition of amine forming intermediate 232; dehydrative cyclization and oxidation forms pyrrole 229.
Scheme 26.
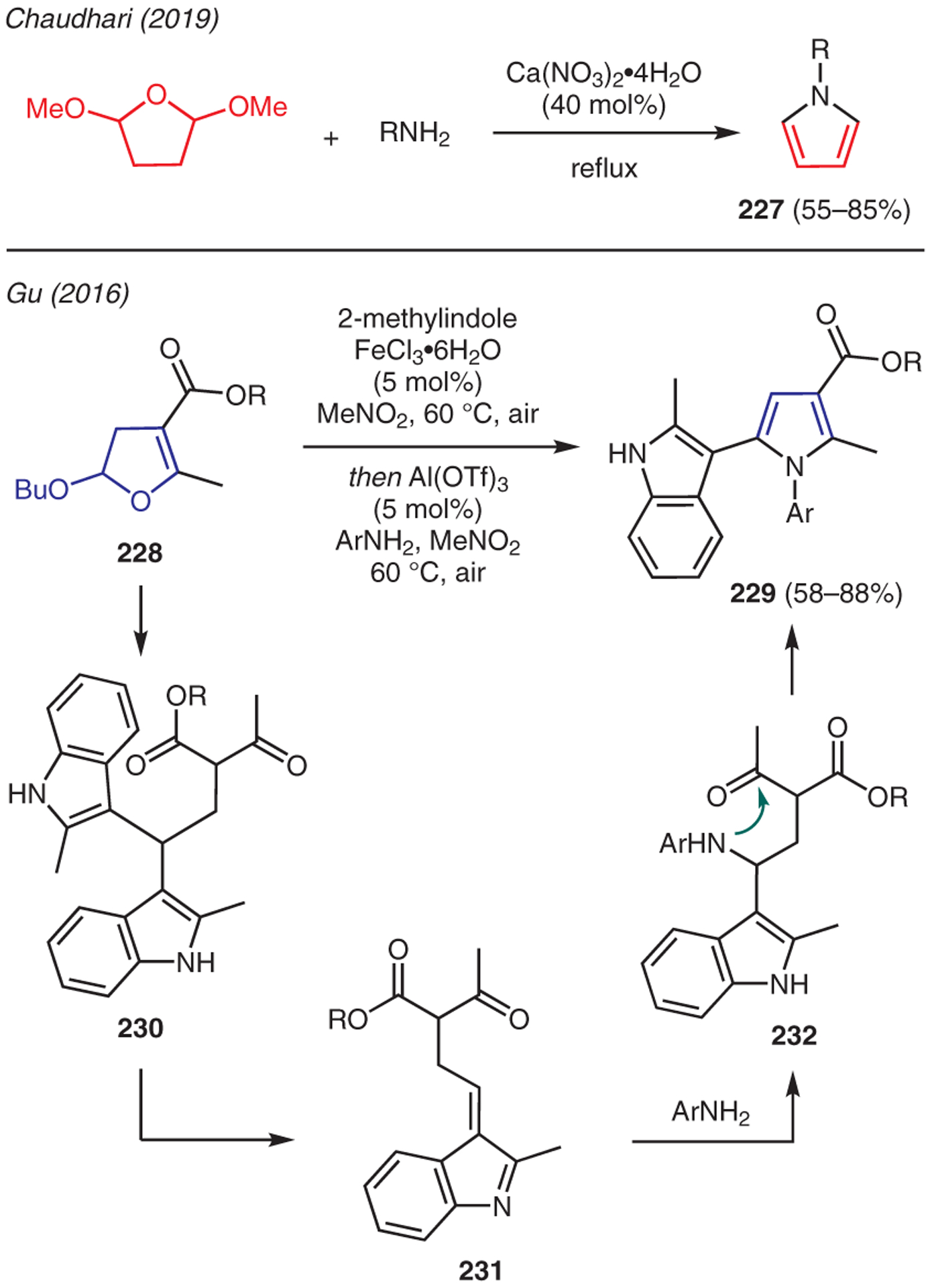
From masked 1,4-dicarbonyls
4. From Polar Compounds
Pyrrole synthesis can be also approached from substrates bearing polar functional groups.115 As discussed in this section, recent reports have demonstrated the effective use of aminols, diols, and organonitro compounds to construct a broad array of substituted pyrroles.
4.1. Aminols
The conversion of aminols into pyrroles via acceptorless dehydrogenative coupling methodology is shown in Scheme 27. In this example, Shimizu reported the synthesis of 2,5-disubstituted pyrroles 235 from 1,2-amino alcohols 233 and secondary alcohols 234 in the presence of heterogeneous carbon-supported platinum as catalyst and a stoichiometric amount of KOt-Bu.116 Mechanistically, the reaction commences with Pt-catalyzed dehydrogenation of the secondary alcohol to give ketone 236, which condenses with the 1,2-amino alcohol 234 to form imine 237. A second Pt-catalyzed dehydrogenation sequence allows the formation of imine aldehyde 238, which undergoes base-catalyzed dehydrative cyclization to furnish substituted pyrrole 235.
Scheme 27.
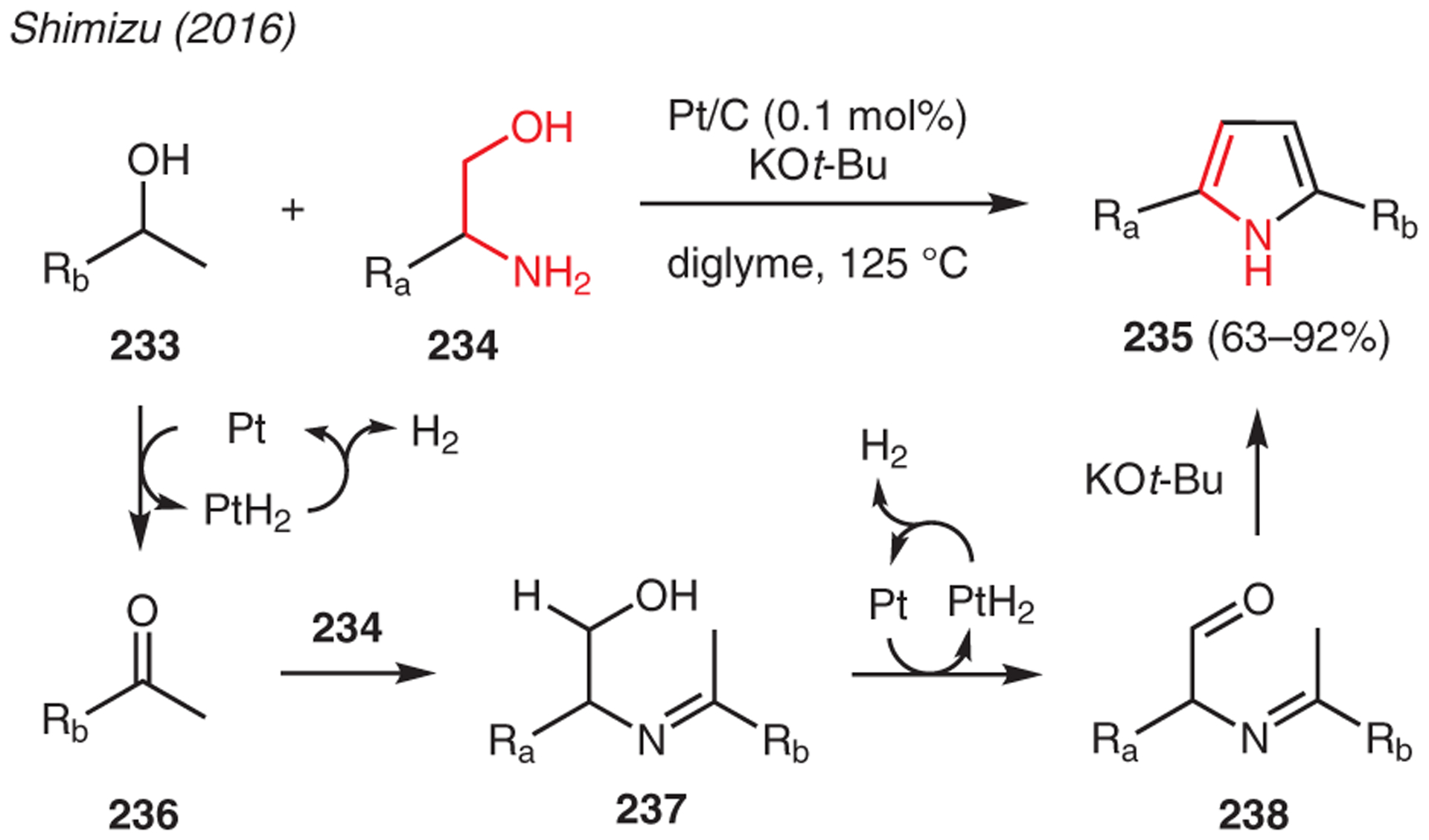
From aminols
4.2. Diols
Banerjee developed a nickel-catalyzed methodology that featured cyclization of butyne-1,4-diols 239 and butene-1,4-diols 241 with various amines to afford N-substituted pyrroles 240 and 242, respectively (Scheme 28).117 In these reactions, it was hypothesized that the nickel catalyst dehydrogenates the alcohol moieties sequentially to allow condensation with the amine, followed by isomerization, cyclization, and dehydration via an acceptorless dehydrogenative coupling pathway. The method serves as an excellent route for the preparation of various N-substituted pyrroles owing to its tolerance to free alcohols, pyridines, benzylic moieties, halides, alkoxy, alkyl, and oxygen heterocycles.
Scheme 28.
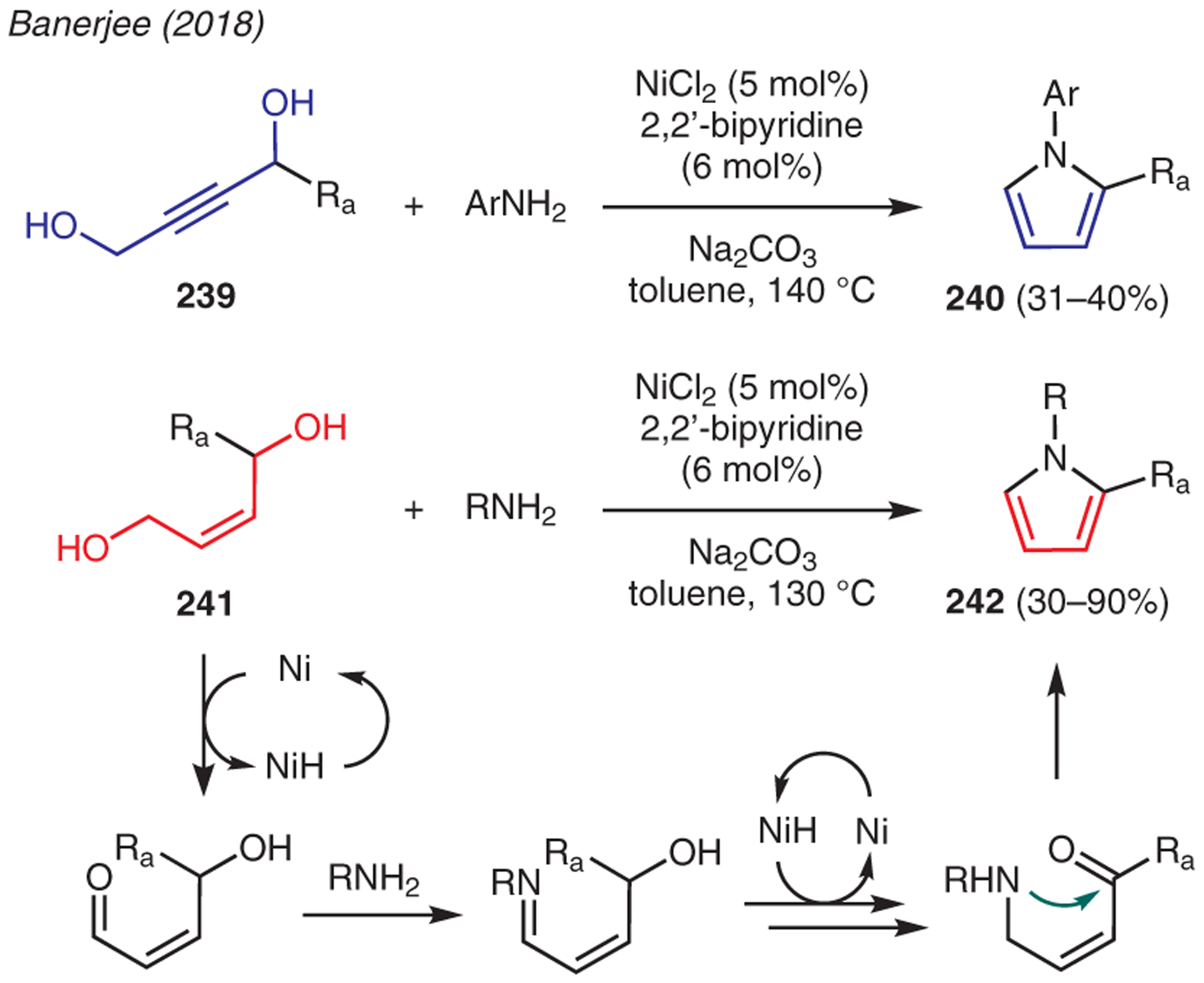
From diols
4.3. Organonitro Compounds
The use of β-nitroalkenes in pyrrole synthesis is showcased in Scheme 29.118,119 For instance, Pal and Rao reported an ultrasound-mediated four-component reaction for the preparation of substituted pyrroles using β-keto esters, benzylamines, aromatic aldehydes, and nitromethane in the presence of Amberlyst-15.120 The mechanism of this reaction is proposed through acid-catalyzed formation of enamine ester 244 from β-keto ester and benzylamine. This intermediate serves as a Michael donor to nitroalkene acceptor 245, which is formed concurrently from aldehyde and nitromethane under the acidic reaction conditions. The resulting addition product 246 proceeds through intramolecular cyclization and aromatization to yield tetrasubstituted pyrrole 243.
Scheme 29.
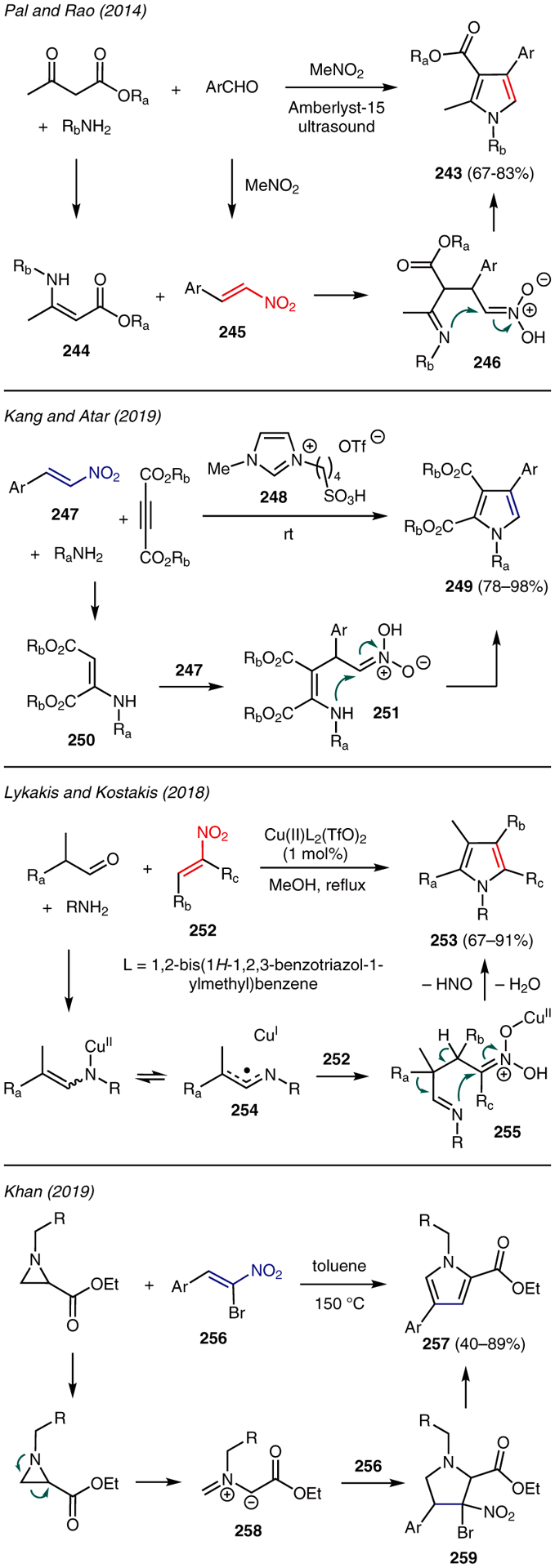
From β-nitroalkenes
A closely related reaction was disclosed by Kang and Atar by a multicomponent reaction involving a mixture of nitroalkenes 247, primary amines, and dialkyl acetylenedicarboxylates using imidazolium-based ionic liquid BAIL 248 (Scheme 29).121 The mechanism proposed involves a BAIL-promoted conjugate addition of the amine to the alkyne resulting in enamine 250. Subsequent reaction of this intermediate with nitroalkene generates 251, which then undergoes a similar cyclization pathway to give pyrrole 249.
Lykakis and Kostakis reported a facile copper(II)-catalyzed reaction between aldehydes, amines, and β-nitroalkenes 252 to yield polysubstituted pyrroles 253 (Scheme 29).122 In this chemistry, the pyrrole framework is proposed to be formed via a radical mechanism involving allylic nitrogen radical intermediate 254, which subsequently adds to nitroalkene 252 at the β-carbon. The resulting aldimine species 255 then undergoes deprotonation, 1,2-migration, cyclization, and aromatization to afford pyrrole 253.
Another example of the use of β-nitroalkenes in pyrrole synthesis was reported by Khan.123 In this work, the reaction of aziridines and β-bromo-β-nitrostyrenes 256 under thermal conditions readily furnished 1,2,4-trisubstituted pyrroles 257 in a regioselective manner (Scheme 29). The proposed mechanism involves the in situ formation of unsymmetrical azomethine ylide 258 as a result of aziridine ring fragmentation upon heating. The ensuing [3+2] cycloaddition with β-bromo-β-nitrostyrene 256 leads to 5-membered intermediate 259, which produces pyrrole 257 upon successive elimination of HBr and HNO2.
Other types of nitro-containing compounds that have been employed as substrates for pyrrole synthesis are depicted in Scheme 30. For instance, Yu and Zhang reported a catalyst-free, three-component, one-pot reaction between α-nitroepoxides 260, primary amines, and dialkyl acetylenedicarboxylates to afford pentasubstituted pyrroles 261.124 The mechanism for this transformation commences with the formation of α-amino ketone 262, which is generated upon nucleophilic ring opening of the α-nitroepoxide by the primary amine, accompanied by the loss of HNO2. Intermolecular conjugate addition of the amino group in 262 to dialkyl acetylenedicarboxylate leads to a sequence of cyclization and condensation to afford the pyrrole adduct.
Scheme 30.
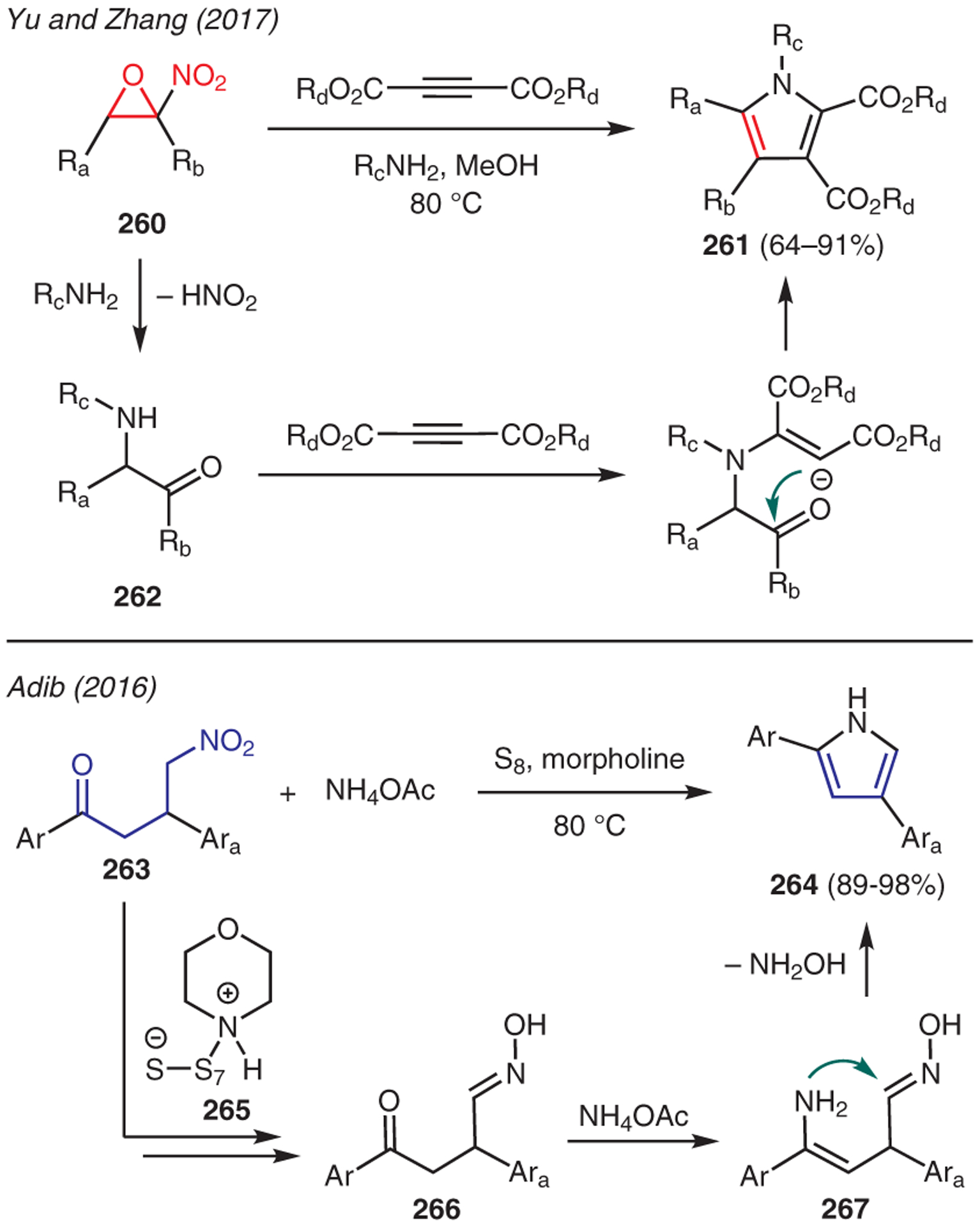
From nitro compounds
Adib disclosed an interesting reaction between 1,3-diaryl-4-nitrobutan-1-one 263 and ammonium acetate in the presence of sulfur and morpholine to give 2,4-diarylpyrroles 264 (Scheme 30).125 The reaction is believed to proceed via the in situ generation of polysulfide morpholinium ion pair 265, which promotes the conversion of 1,3-diaryl-4-nitrobutan-1-one into oxime 266. Condensation of 266 with ammonium acetate gives enamine 267, which undergoes intramolecular cyclization and aromatization with the loss of hydroxylamine to generate pyrrole 264.
5. From Heterocycles
Literature survey on pyrrole synthesis between 2014 and 2019 also revealed interesting strategies in which different classes of heterocycles have been effectively employed as starting materials.126–135 Some examples discussed in this section include münchnones, isoxazoles, carbohydrates, hydroxyprolines, and pyrrolines. It is interesting to note that these heterocyclic motifs readily produce pyrroles that are densely substituted.
5.1. Münchnones
Oxazolium-5-olate systems, commonly referred to as münchnones, are mesoionic compounds that possess excellent reactivity in cycloaddition reactions with dipolarophiles. In fact, the 1,3-dipolar cycloaddition reaction of münchnones has been demonstrated to be one of the most useful approaches to access various substituted pyrroles.136 As shown in Scheme 31, Harrity reported the regioselective synthesis of pyrroles using münchnones and substituted enamines.137 In this chemistry, the 1,3-dipolar cycloaddition of the münchnones 268 and substituted enamines forms 5-membered intermediate 271 upon extrusion of CO2. Subsequent aromatization via elimination of the amino group then generates the pyrrole structures either 269 or 270.
Scheme 31.
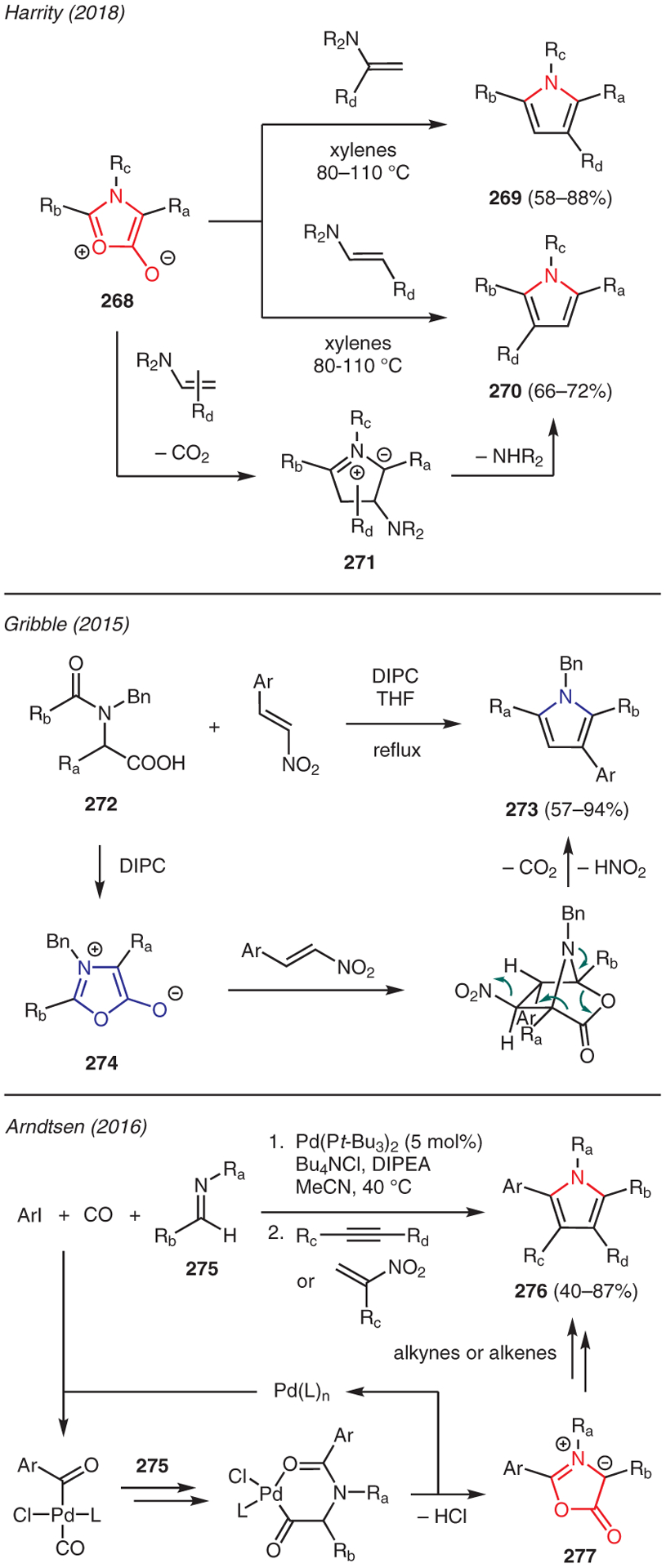
From münchnones
Methodologies that exploited the in situ generation of münchnones towards synthesis of pyrroles have been also reported. For instance, Gribble demonstrated the preparation of münchnones 274 via treatment of N-acylamino acid 272 with N,N-diisopropylcarbodiimide (DIPC). The ensuing 1,3-dipolar cycloaddition with β-nitroalkene then occurred to yield tetrasubstituted pyrrole 273 upon loss of CO2 and HNO2 (Scheme 31).138 The in situ generation of münchnones could be also catalyzed by transition metals. Arndtsen developed a multicomponent reaction using aryl iodides, imines, and CO in the presence of Pd(Pt-Bu3)2, Bu4NCl, and DIPEA to produce transient münchnones 277 (Scheme 31).139,140 This intermediate was then captured by electron-deficient alkynes or alkenes to afford diverse families of highly substituted pyrroles 276 via [3+2] cycloaddition in one synthetic operation.
5.2. Isoxazoles
The utility of isoxazoles as a valuable precursor for the preparation of pyrroles is showcased in Scheme 32. For instance, Kapur reported that exposure of 4-vinylisoxazoles 278 to catalytic RuCl3·xH2O and stoichiometric Cu(OAc)2 furnished trisubstituted pyrroles 279.141 The proposed mechanistic pathway involves activation of the isoxazole nitrogen by the Ru catalyst, enabling ring fragmentation to produce ruthenium-carbene intermediate 280; subsequent 1,5-cyclization then generates the pyrrole ring 279. The role of Cu(OAc)2 in this transformation is not clearly understood. An analogous transformation was also reported by Khlebnikov, in which FeCl2·4H2O catalyst was employed to rearrange 4-vinylisoxazoles 281 to 3-carbonylpyrroles 282 via a putative iron-carbene pathway (Scheme 32).142
Scheme 32.
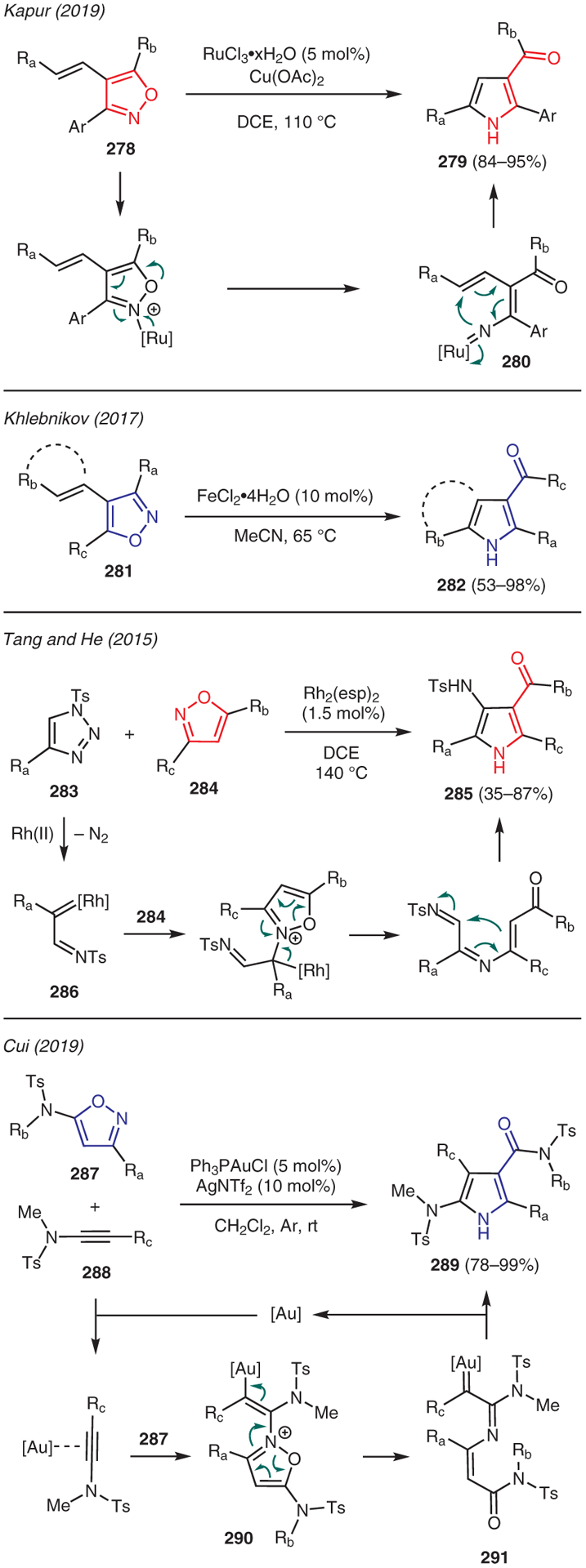
From isoxazoles
Tang and He reported [3+2] cycloaddition of isoxazoles 284 with N-sulfonyl-1,2,3-triazoles 283 in the presence of Rh2(esp)2 catalyst toward the preparation of 3-aminopyrroles 285 (Scheme 32).143 This reaction is proposed to commence with the decomposition of triazole 283 to Rh(II)-azavinylcarbene 286, which is then captured by the isoxazole to yield an isoxazolium ylide intermediate. The ensuing ring-opening to azatriene, recyclization, and aromatization via proton transfer leads to the observed 3-aminopyrrole product 285.
Another example was reported by Cui, who described the synthesis of pyrroles from isoxazoles 287 and internal ynamides 288 using the Ph3PAuCl/AgNTf2 catalytic system (Scheme 32).144 In this chemistry, activation of the internal ynamide with the Au(I) catalyst enables nucleophilic addition of the isoxazole to produce intermediate 290. N–O Bond cleavage and isomerization then leads to acyclic carbenoid intermediate 291, which undergoes intramolecular cyclization to afford pyrrole 289 while regenerating the Au(I) catalyst.
5.3. Carbohydrates
Sugars have been shown to serve as viable substrates for pyrrole synthesis.145 As exemplified in Scheme 33, Koo subjected D-glucose and L-rhamnose to various primary amines in presence of oxalic acid and DMSO.146 This unique transformation begins with N-glycosylation of the amine, followed by tautomerization to enamine 293; dehydration, tautomerization, and a second imine formation gives intermediate 294, which undergoes a cyclization and dehydration sequence to furnish pyrrole-2-carbaldehyde 292.
Scheme 33.
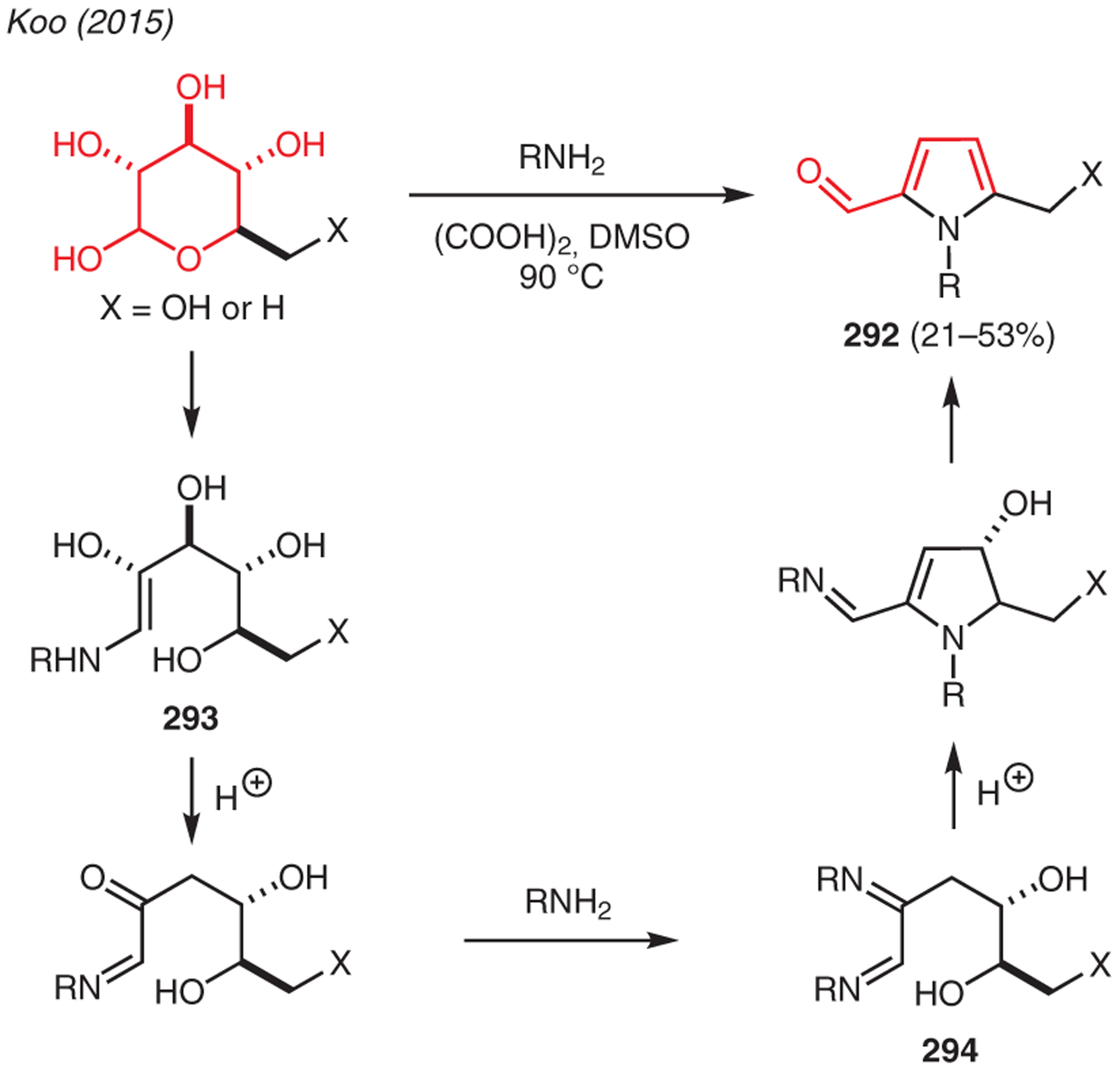
From carbohydrates
5.4. trans-4-Hydroxy-l-prolines
The use of trans-4-hydroxy-l-prolines for the preparation of pyrroles is showcased in Scheme 34. For instance, Roy developed a green synthesis of 3-(pyrrol-1-yl)indolin-2-ones 297 in water.147 In this chemistry, the surfactant additive, i.e. cetyltrimethylammonium bromide (CTAB), formed an aqueous micellar medium, in which isatin 295 and trans-4-hydroxy-l-proline 296 form iminium 298. This species then undergoes decarboxylation, followed by dehydrative aromatization to form pyrrole 297.
Scheme 34.
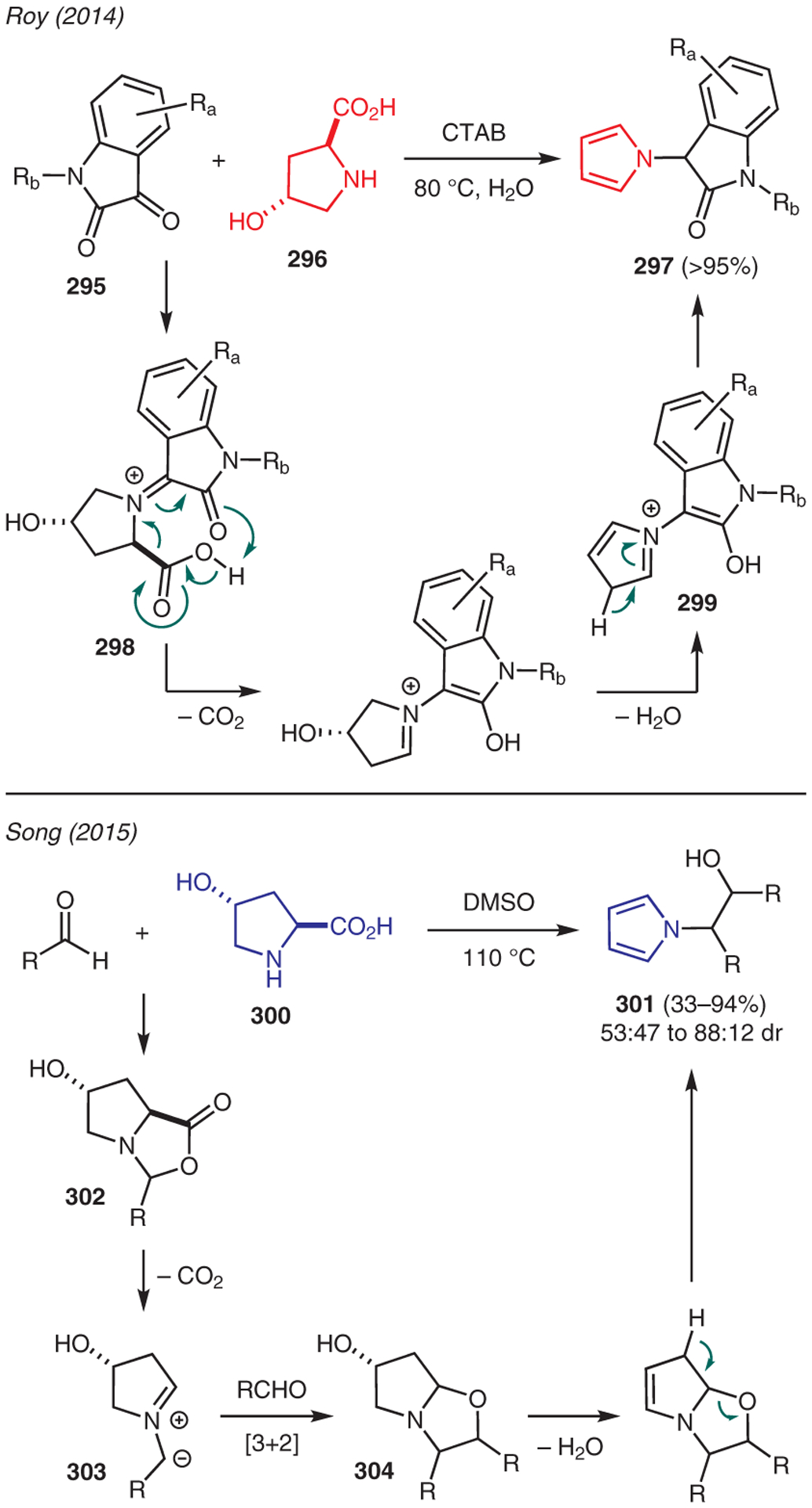
From trans-4-hydroxy-l-prolines
Another example was reported by Song who demonstrated a simple catalyst-free approach to N-(2-hydroxyethyl)pyrroles via a [3+2]-cycloaddition reaction between electron-deficient aldehydes and trans-4-hydroxyprolines 300 (Scheme 34).148 The mechanism proceeds via condensation between the aldehyde and proline to generate oxazolidin-5-one 302, which undergoes decarboxylation to azomethine ylide 303 upon heating. A second molecule of aldehyde captures the ylide 303 via intermolecular [3+2] cycloaddition to produce intermediate 304; dehydrative ring opening and aromatization then produce N-(2-hydroxyethyl)pyrroles 301.
5.5. Pyrrolines
Sun and Xu has developed an interesting synthesis of pyrroles by exploiting the inherent reducing power of 3-pyrrolines (Scheme 35).149,150 For instance, treatment of 2-(3-pyrrolin-1-yl)benzaldehydes 305 with Sc(OTf)3 promoted an intramolecular redox reaction to generate iminium ion intermediate 307 via 1,5-hydride transfer; subsequent isomerization gave the pyrrole 306. Interestingly, the intramolecular redox reaction of 2-(3-pyrrolin-1-yl)benzaldehydes could be also performed in the presence amines and ZnCl2 to afford the amino pyrrole variant 309. In this example, the 1,5-hydride migration occurred upon the generation of iminium ion intermediate 310.
Scheme 35.
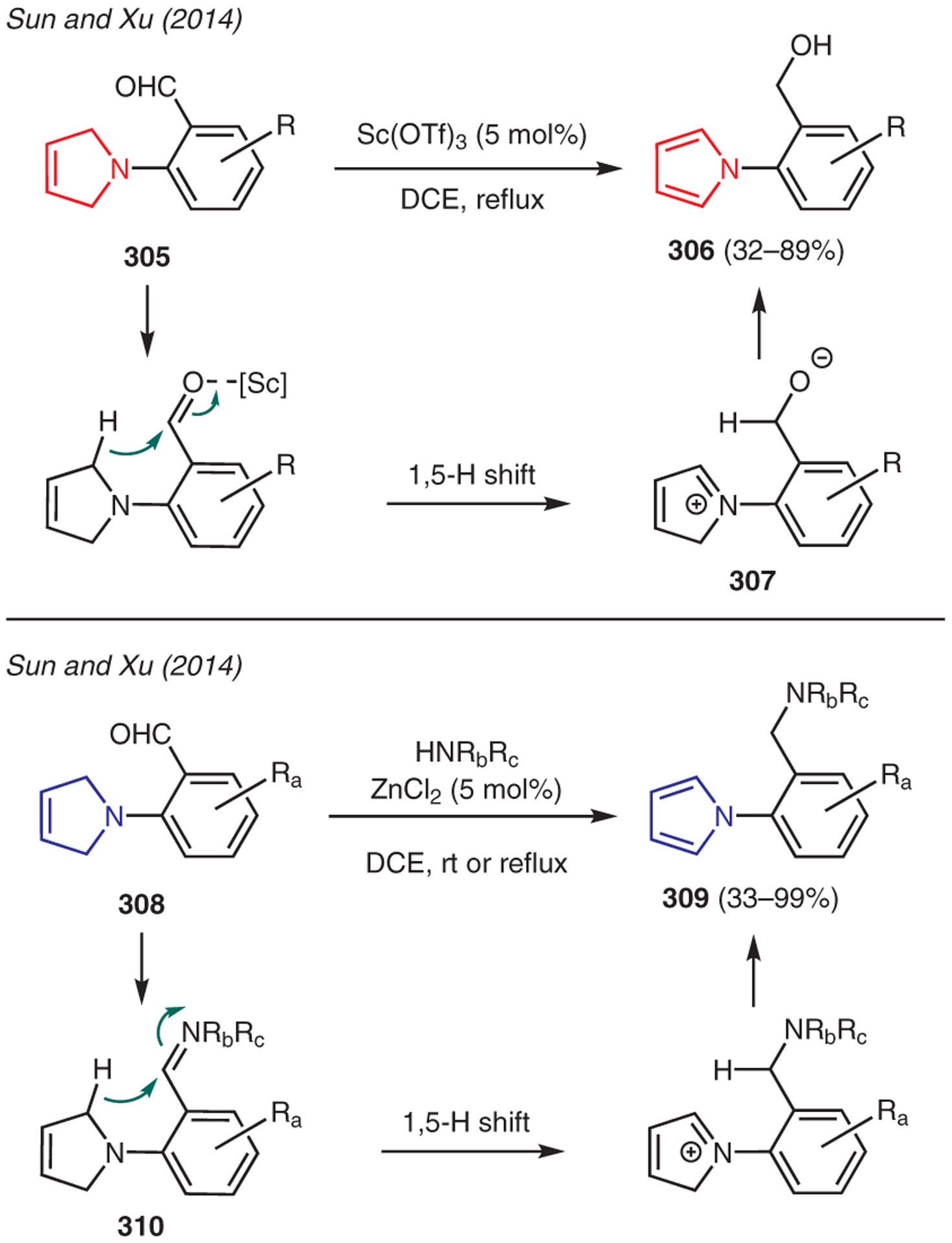
From 3-pyrrolines
6. Summary
This review article summarizes selected examples on pyrrole synthesis that have been developed between 2014 and 2019. Since its discovery, pyrrole has become one of the most important and fascinating aromatic heterocycles. Owing to its vast range of significance and applications in different areas of science, pyrrole will remain an important target in organic synthesis. The interesting structural features within pyrrole, including the challenges in introducing substituents regioselectively at various positions around the ring, will continue to provide inspiration for the discovery of new synthetic technologies as well as chemical reactivities.
Funding Information
Generous financial supports from the National Institute of General Medical Sciences of the National Institutes of Health under Award Number R01GM127649 and Louisiana State University are gratefully acknowledged. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Biographies

Rendy Kartika is an Associate Professor in the Department of Chemistry at Louisiana State University (LSU). Rendy was born in Malang, Indonesia and immigrated to the US in 1998. He earned his B.S. in Chemistry from California State Polytechnic University, Pomona in 2003 and his Ph.D. in organic chemistry from the University of Notre Dame in 2008 under the direction of Prof. Richard Taylor. After a postdoctoral training with Prof. David Spiegel at Yale University, Rendy began his independent academic career as an Assistant Professor at LSU in 2011 and rose the rank of Associate Professor with tenure in 2017. Research in the Kartika lab centers on the development of organic synthetic reactions to assemble complex molecules of biological, pharmaceutical, and industrial importance.

Satish Philkhana is a postdoctoral researcher in the Kartika group at LSU. He earned his Masters degree in organic chemistry from the University of Pune, India and later joined Dr. D. S. Reddy’s group at CSIR-National Chemical Laboratory-Pune, India to pursue his doctoral studies on the synthesis of bioactive natural products and their analogues. Upon graduation, he joined Prof. Jennifer Golden’s group at University of Wisconsin, Madison as a postdoctoral researcher where he worked on the development of new annulation strategies to access quinolinone-type scaffolds. Currently at LSU, he is exploring new stereoselective methods to generate ketones with bis-quaternary stereocenters at the α,α′-positions.

Fatimat Badmus is a 4th year graduate student in the Department of Chemistry at LSU under the supervision of Prof. Rendy Kartika. Fatimat was born in Osun, Nigeria. She earned her B.Sc. degree in chemistry from Obafemi Awolowo University in 2014 and commenced her graduate studies at LSU in 2017. At LSU, Fatimat is working on the development of new organic synthetic reactions that produce biologically significant heterocyclic scaffolds as well as complex structural motifs.

Isaac Dos Reis was born in Florida, USA, and studied chemistry at the State University of New York at Plattsburgh where he received a B.S. in chemistry in 2018. He continued his studies at LSU where he has worked under the instruction of Dr. Rendy Kartika. Currently, he is working on new synthetic methodologies to construct chlorine-containing tetrahydropyrans.
References
- (1).Khajuria R; Dhamb S; Kapoor KK RSC Adv. 2016, 6, 37039. [Google Scholar]
- (2).Bhardwaj V; Gumber D; Abbot V; Dhiman S; Sharma P RSC Adv. 2015, 5, 15233. [Google Scholar]
- (3).Sobenina LN; Vasil’tsov AM; Petrova OV; Petrushenko KB; Ushakov IA; Clavier G; Meallet-Renault R; Mikhaleva AI; Trofimov BA Org. Lett 2011, 13, 2524. [DOI] [PubMed] [Google Scholar]
- (4).Gimenez IF; Alves OL J. Braz. Chem. Soc 1999, 10, 167. [Google Scholar]
- (5).Nizurski-Mann RE; Cava MP Heterocycles 1992, 34, 2003. [Google Scholar]
- (6).D’Silva C; Walker DA J. Org. Chem 1998, 63, 6715. [Google Scholar]
- (7).Runge FF Ann. Phys 1834, 107, 65. [Google Scholar]
- (8).Knorr L Ber. Dtsch. Chem. Ges 1884, 17, 2863. [Google Scholar]
- (9).Hantzsch A Ber. Dtsch. Chem. Ges 1890, 23, 1474. [Google Scholar]
- (10).Paal C Ber. Dtsch. Chem. Ges 1884, 17, 2756. [Google Scholar]
- (11).Arshadi S; Vessally E; Edjlali L; Ghorbani-Kalhor E; Hosseinzadeh-Khanmiri R RSC Adv. 2017, 7, 13198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Azad I; Hassan F; Ahmad N; Khan AR; Nasibullah M; Saquib M; Al-Sehemi AG Orient. J. Chem 2018, 34, 1670. [Google Scholar]
- (13).Belikov MY; Ershov OV Chem. Heterocycl. Compd 2016, 52, 279. [Google Scholar]
- (14).Chelucci G Coord. Chem. Rev 2017, 331, 37. [Google Scholar]
- (15).Estévez V; Villacampa M; Menéndez JC Chem. Soc. Rev 2014, 43, 4633. [DOI] [PubMed] [Google Scholar]
- (16).Gulevich AV; Dudnik AS; Chernyak N; Gevorgyan V Chem. Rev 2013, 113, 3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Keiko NA; Vchislo NV Chem. Heterocycl. Compd 2017, 53, 498. [Google Scholar]
- (18).Leonardi M; Estevez V; Villacampa M; Menendez JC Synthesis 2019, 51, 816. [Google Scholar]
- (19).Mosaad SM; Samar SF Mini-Rev. Org. Chem 2014, 11, 477. [Google Scholar]
- (20).Sharma A; Piplani PJ Heterocycl. Chem 2017, 54, 27. [Google Scholar]
- (21).Shrinivas DJ; Uttam AM; Venkatrao HK; Tejraj MA Curr. Org. Chem 2013, 17, 2279. [Google Scholar]
- (22).Tzankova D; Vladimirova S; Peikova L; Georgieva MJ Chem. Technol. Metall 2018, 53, 451. [Google Scholar]
- (23).Wood JM; Furkert DP; Brimble MA Nat. Prod. Rep 2019, 36, 289. [DOI] [PubMed] [Google Scholar]
- (24).Yurovskaya MA; Alekseyev RS Chem. Heterocycl. Compd 2014, 49, 1400. [Google Scholar]
- (25).Ram RN; Sadanandan S; Kumar Gupta D Adv. Synth. Catal 2019, 361, 5661. [Google Scholar]
- (26).Aquino ED; Leonel G; Gariboti VC; Frizzo CP; Martins MAP; Bonacorso HG; Zanatta NJ Org. Chem 2015, 80, 12453. [DOI] [PubMed] [Google Scholar]
- (27).Zhang L; Zhang X; Lu Z; Zhang D; Xu X Tetrahedron 2016, 72, 7926. [Google Scholar]
- (28).Borra S; Chandrasekhar D; Newar UD; Maurya RA J. Org. Chem 2019, 84, 1042. [DOI] [PubMed] [Google Scholar]
- (29).Zhang X; Teo JW; Ma D-L; Leung C-H; Chan PWH Tetrahedron Lett. 2014, 55, 6703. [Google Scholar]
- (30).Grychowska K; Kubica B; Drop M; Colacino E; Bantreil X; Pawlowski M; Martinez J; Subra G; Zajdel P; Lamaty F Tetrahedron 2016, 72, 7462. [Google Scholar]
- (31).Chen W; Zhang Y-L; Li H-J; Nan X; Liu Y; Wu Y-C Synthesis 2019, 51, 3651. [Google Scholar]
- (32).Perrulli FR; Favi G; De Crescentini L; Attanasi OA; Santeusanio S; Mantellini F Eur. J. Org. Chem 2015, 7154. [DOI] [PubMed] [Google Scholar]
- (33).Wang Y; Jiang C-M; Li H-L; He F-S; Luo X; Deng W-PJ Org. Chem 2016, 81, 8653. [DOI] [PubMed] [Google Scholar]
- (34).Cardoso AL; Henriques MSC; Paixao JA; Pinho e Melo TMVD J. Org. Chem 2016, 81, 9028. [DOI] [PubMed] [Google Scholar]
- (35).Ni C; Wang ML; Tong X Org. Lett 2016, 18, 2240. [DOI] [PubMed] [Google Scholar]
- (36).Mariappan A; Rajaguru K; Muthusubramanian S; Bhuvanesh N Synth. Commun 2016, 46, 805. [Google Scholar]
- (37).Kuruba BK; Vasanthkumar S; Emmanuvel L Tetrahedron 2017, 73, 3093. [Google Scholar]
- (38).Liu Y; Yi X; Luo X; Xi CJ Org. Chem 2017, 82, 11391. [DOI] [PubMed] [Google Scholar]
- (39).George J; Kim HY; Oh K Adv. Synth. Catal 2016, 358, 3714. [Google Scholar]
- (40).Chen D; Shan Y; Li J; You J; Sun X; Qiu G Org. Lett 2019, 21, 4044. [DOI] [PubMed] [Google Scholar]
- (41).Pasko CM; Dissanayake AA; Billow BS; Odom AL Tetrahedron 2016, 72, 1168. [Google Scholar]
- (42).Wu F; Chen L; Wang Y; Zhu S Org. Chem. Front 2019, 6, 480. [Google Scholar]
- (43).Li X; Chen M; Xie X; Sun N; Li S; Liu Y Org. Lett 2015, 17, 2984. [DOI] [PubMed] [Google Scholar]
- (44).Zhang S; Ma Y; Lan J; Song F; You J Org. Biomol. Chem 2015, 13, 5867. [DOI] [PubMed] [Google Scholar]
- (45).Zhang X; Xu X; Chen G; Yi W Org. Lett 2016, 18, 4864. [DOI] [PubMed] [Google Scholar]
- (46).Pan D; Wei Y; Shi M Org. Lett 2016, 18, 3930. [DOI] [PubMed] [Google Scholar]
- (47).Schitter T; Stammwitz S; Jones PG; Werz DB Org. Lett 2019, 21, 9415. [DOI] [PubMed] [Google Scholar]
- (48).Liu J; Zhang X; Peng H; Jiang H; Yin B Adv. Synth. Catal 2015, 357, 727. [Google Scholar]
- (49).Wan S-H; Liu S-T Tetrahedron 2019, 75, 1166. [Google Scholar]
- (50).Li T; Yan H; Li X; Wang C; Wan BJ Org. Chem 2016, 81, 12031. [DOI] [PubMed] [Google Scholar]
- (51).Urbanaitė A; Čikotienė I Eur. J. Org. Chem 2016, 5294. [DOI] [PubMed] [Google Scholar]
- (52).Raju AR; Reddy RV; Rao VM; Naresh VV; Rao AV Tetrahedron Lett. 2016, 57, 2838. [Google Scholar]
- (53).Liu J-Q; Chen X-Y; Shatskiy A; Kärkäs MD; Wang X-SJ Org. Chem 2019, 84, 8998. [DOI] [PubMed] [Google Scholar]
- (54).Tarasova OA; Nedolya NA; Albanov AI; Trofimov BA Synthesis 2019, 51, 3697. [Google Scholar]
- (55).Sakai N; Hori H; Ogiwara Y Eur. J. Org. Chem 2015, 1905. [Google Scholar]
- (56).Qiu G; Wang Q; Zhu J Org. Lett 2017, 19, 270. [DOI] [PubMed] [Google Scholar]
- (57).Nandi GC; Soumini KJ Org. Chem 2016, 81, 11909. [DOI] [PubMed] [Google Scholar]
- (58).Yuan B; Jiang Y; Qi Z; Guan X; Wang T; Yan R Adv. Synth. Catal 2019, 361, 5112. [Google Scholar]
- (59).Wang C; Huang K; Wang J; Wang H; Liu L; Chang W; Li J Adv. Synth. Catal 2015, 357, 2795. [Google Scholar]
- (60).Gao Y; Hu C; Wan J-P; Wen C Tetrahedron Lett. 2016, 57, 4854. [Google Scholar]
- (61).Huang W; Chen S; Chen Z; Yue M; Li M; Gu YJ Org. Chem 2019, 84, 5655. [DOI] [PubMed] [Google Scholar]
- (62).Liu Y; Parodi A; Battaglioli S; Monari M; Protti S; Bandini M Org. Lett 2019, 21, 7782. [DOI] [PubMed] [Google Scholar]
- (63).Xu H; Wang F-J; Xin M; Zhang Z Eur. J. Org. Chem 2016, 925. [Google Scholar]
- (64).Trofimov BA; Mikhaleva AI; Ivanov AV; Shcherbakova VS; Ushakov IA Tetrahedron 2015, 71, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Khlebnikov AF; Tomashenko OA; Funt LD; Novikov MS Org. Biomol. Chem 2014, 12, 6598. [DOI] [PubMed] [Google Scholar]
- (66).Galenko AV; Khlebnikov AF; Novikov MS; Avdontceva MS Tetrahedron 2015, 71, 1940. [Google Scholar]
- (67).Chen X; Yuan JC; Zhou M Synth. Commun 2019, 49, 32. [Google Scholar]
- (68).Farahi M; Davoodi M; Tahmasebi M Tetrahedron Lett. 2016, 57, 1582. [Google Scholar]
- (69).Egorov M; Delpech B; Aubert G; Cresteil T; Garcia-Alvarez MC; Collin P; Marazano C Org. Biomol. Chem 2014, 12, 1518. [DOI] [PubMed] [Google Scholar]
- (70).Wu X; Li K; Wang S; Liu C; Lei A Org. Lett 2016, 18, 56. [DOI] [PubMed] [Google Scholar]
- (71).Malone JA; Toussel CE; Fronczek FR; Kartika R Org. Lett 2019, 21, 3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Lei T; Liu W-Q; Li J; Huang M-Y; Yang B; Meng Q-Y; Chen B; Tung C-H; Wu L-Z Org. Lett 2016, 18, 2479. [DOI] [PubMed] [Google Scholar]
- (73).Lin Z.-q.; Li C.-d.; Zhou Z.-c.; Xue S; Gao J.-r.; Ye Q; Li Y.-j. Synlett 2019, 30, 1442. [Google Scholar]
- (74).Kucukdisli M; Ferenc D; Heinz M; Wiebe C; Opatz T Beilstein J. Org. Chem 2014, 10, 466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Reekie TA; Donckele EJ; Manenti G; Püntener S; Trapp N; Diederich F Org. Lett 2016, 18, 2252. [DOI] [PubMed] [Google Scholar]
- (76).Rao HSP; Desai A Synlett 2015, 26, 1059. [Google Scholar]
- (77).Peng J; Gao Y; Zhu C; Liu B; Gao Y; Hu M; Wu W; Jiang HJ Org. Chem 2017, 82, 3581. [DOI] [PubMed] [Google Scholar]
- (78).Rahmani F; Darehkordi A Synlett 2017, 28, 1224. [Google Scholar]
- (79).Fang G; Liu J; Fu J; Liu Q; Bi X Org. Lett 2017, 19, 1346. [DOI] [PubMed] [Google Scholar]
- (80).Mou X-Q; Xu Z-L; Xu L; Wang S-H; Zhang B-H; Zhang D; Wang J; Liu W-T; Bao W Org. Lett 2016, 18, 4032. [DOI] [PubMed] [Google Scholar]
- (81).Gao P; Wang J; Bai Z-J; Shen L; Yan Y-Y; Yang D-S; Fan M-J; Guan Z-H Org. Lett 2016, 18, 6074. [DOI] [PubMed] [Google Scholar]
- (82).San Jang S; Chang JY; Kang GY; Youn SW Asian J. Org. Chem 2019, 8, 1668. [Google Scholar]
- (83).Bodunov VA; Galenko EE; Sakharov PA; Novikov MS; Khlebnikov AF J. Org. Chem 2019, 84, 10388. [DOI] [PubMed] [Google Scholar]
- (84).Bonacorso HG; Libero FM; Dal Forno GM; Pittaluga EP; Back DF; Horner M; Martins MAP; Zanatta N Tetrahedron Lett. 2016, 57, 4568. [Google Scholar]
- (85).Kan W; Jing T; Zhang X.-h.; Zheng Y.-i.; Chen L; Zhao B Heterocycles 2015, 91, 2367. [Google Scholar]
- (86).Leng J; Xu H; Meng J; Luo X; Deng W-P Tetrahedron 2019, 75, 130709. [Google Scholar]
- (87).Estevez V; Sridharan V; Sabate S; Villacampa M; Menendez JC Asian J. Org. Chem 2016, 5, 652. [Google Scholar]
- (88).Leonardi M; Esteeez V; Villacampa M; Menendez JC Adv. Synth. Catal 2019, 361, 2054. [Google Scholar]
- (89).Bai X; Wang L; Zhang Z; Zhang K; Bu Z; Wu Y; Zhang W; Wang Q Adv. Synth. Catal 2019, 361, 4893. [Google Scholar]
- (90).Kim C-E; Park S; Eom D; Seo B; Lee PH Org. Lett 2014, 16, 1900. [DOI] [PubMed] [Google Scholar]
- (91).Feng J; Wang Y; Li Q; Jiang R; Tang Y Tetrahedron Lett. 2014, 55, 6455. [Google Scholar]
- (92).Ran R-Q; He J; Xiu S-D; Wang K-B; Li C-Y Org. Lett 2014, 16, 3704. [DOI] [PubMed] [Google Scholar]
- (93).Rajasekar S; Anbarasan PJ Org. Chem 2014, 79, 8428. [DOI] [PubMed] [Google Scholar]
- (94).Chechina NV; Kolos NN; Omelchenko IV Chem. Heterocycl. Compd 2019, 55, 1190. [Google Scholar]
- (95).Kolos NN; Chechina NV Chem. Heterocycl. Compd 2019, 55, 1278. [Google Scholar]
- (96).Kolos NN; Zubar VV; Omelchenko IV; Musatov VI Chem. Heterocycl. Compd 2016, 52, 237. [Google Scholar]
- (97).Chen Z; Chen H; Yang X; Chang XQ Synlett 2017, 28, 1463. [Google Scholar]
- (98).Li K; Chen L; Fan Y-X; Wei Y; Yan S-JJ Org. Chem 2019, 84, 11971. [DOI] [PubMed] [Google Scholar]
- (99).Chen X-B; Wang X-Y; Zhu D-D; Yan S-J; Lin J Tetrahedron 2014, 70, 1047. [Google Scholar]
- (100).Chen X-B; Yan S-J; Su A; Liu W; Lin J Tetrahedron 2015, 71, 4745. [Google Scholar]
- (101).Hu L; Wang K-M; Zhao M; Lin X-R; Zhu H-Y; Yan S-J; Lin J Tetrahedron 2014, 70, 4478. [Google Scholar]
- (102).Chang X; Yang X; Chen Z; Zhong W Synlett 2019, 30, 1431. [Google Scholar]
- (103).Sha Q; Arman H; Doyle MP Org. Lett 2015, 17, 3876. [DOI] [PubMed] [Google Scholar]
- (104).Cheng W; Tang Y; Xu Z-F; Li C-Y Org. Lett 2016, 18, 6168. [DOI] [PubMed] [Google Scholar]
- (105).Mehrabi H; Anary-Abbasinejad M; Mirhashemi F Tetrahedron Lett. 2014, 55, 4310. [Google Scholar]
- (106).Hu LF; Luo J; Lu D; Tang Q Tetrahedron Lett. 2018, 59, 1698. [Google Scholar]
- (107).Zhang Y; Weng G; Chen J; Ma D; Zhang X Main Group Met. Chem 2014, 37, 131. [Google Scholar]
- (108).Akelis L; Rousseau J; Juskenas R; Dodonova J; Rousseau C; Menuel S; Prevost D; Tumkevičius S; Monflier E; Hapiot F Eur. J. Org. Chem 2016, 31. [Google Scholar]
- (109).Bhandari N; Gaonkar SL Chem. Heterocycl. Compd 2015, 51, 320. [Google Scholar]
- (110).Akbaşlar D; Demirkol O; Giray S Synth. Commun 2014, 44, 1323. [Google Scholar]
- (111).Rajeshkumar V; Neelamegam C; Anandan S Synthesis 2019, 51, 4023. [DOI] [PubMed] [Google Scholar]
- (112).Zvarych VI; Stasevych MV; Lunin VV; Vovk MV; Novikov VP Chem. Heterocycl. Compd 2016, 52, 421. [Google Scholar]
- (113).Wani RR; Chaudhari HK; Takale BS J. Heterocycl. Chem 2019, 56, 1337. [Google Scholar]
- (114).Liu C; Zhou L; Huang W; Wang M; Gu Y Adv. Synth. Catal 2016, 358, 900. [Google Scholar]
- (115).Midya SP; Landge VG; Sahoo MK; Rana J; Balaraman E Chem. Commun 2018, 54, 90. [DOI] [PubMed] [Google Scholar]
- (116).Siddiki S; Touchy AS; Chaudhari C; Kon K; Toyao T; Shimizu K.-i. Org. Chem. Front 2016, 3, 846. [Google Scholar]
- (117).Singh K; Kabadwal LM; Bera S; Alanthadka A; Banerjee DJ Org. Chem 2018, 83, 15406. [DOI] [PubMed] [Google Scholar]
- (118).Chen J; Chang D; Xiao F; Deng G-JJ Org. Chem 2019, 84, 568. [DOI] [PubMed] [Google Scholar]
- (119).Pachechne LA; Pereira VF; Martins GM; Martendal E; Xavier FR; Mendes SR Tetrahedron Lett. 2019, 60, 151043. [Google Scholar]
- (120).Murthi PRK; Rambabu D; Rao MVB; Pal M Tetrahedron Lett. 2014, 55, 507. [Google Scholar]
- (121).Atar AB; Han E; Sohn DH; Kang J Synth. Commun 2019, 49, 1181. [Google Scholar]
- (122).Andreou D; Kallitsakis MG; Loukopoulos E; Gabriel C; Kostakis GE; Lykakis IN J. Org. Chem 2018, 83, 2104. [DOI] [PubMed] [Google Scholar]
- (123).Kumar V; Awasthi A; Metya A; Khan TJ Org. Chem 2019, 84, 11581. [DOI] [PubMed] [Google Scholar]
- (124).Zhao D; Zhu Y; Guo S; Chen W; Zhang G; Yu Y Tetrahedron 2017, 73, 2872. [Google Scholar]
- (125).Adib M; Ayashi N; Heidari F; Mirzaei P Synlett 2016, 27, 1738. [Google Scholar]
- (126).Xiao Z-F; Ding T-H; Mao S-W; Shah Z; Ning X-S; Kang Y-B Org. Lett 2016, 18, 5672. [DOI] [PubMed] [Google Scholar]
- (127).Galenko EE; Tomashenko OA; Khlebnikov AF; Novikov MS Org. Biomol. Chem 2015, 13, 9825. [DOI] [PubMed] [Google Scholar]
- (128).Kardile RD; Kale BS; Sharma P; Liu RS Org. Lett 2018, 20, 3806. [DOI] [PubMed] [Google Scholar]
- (129).Pusch S; Kowalczyk D; Opatz TJ Org. Chem 2016, 81, 4170. [DOI] [PubMed] [Google Scholar]
- (130).Paternoga J; Opatz T Eur. J. Org. Chem 2019, 7067. [Google Scholar]
- (131).Rostovskii NV; Ruvinskaya JO; Novikov MS; Khlebnikov AF; Smetanin IA; Agafonova AV J. Org. Chem 2017, 82, 256. [DOI] [PubMed] [Google Scholar]
- (132).Galenko EE; Linnik SA; Khoroshilova OV; Novikov MS; Khlebnikov AF J. Org. Chem 2019, 84, 11275. [DOI] [PubMed] [Google Scholar]
- (133).Satish G; Reddy KHV; Ramesh K; Kumar B; Nageswar YVD Tetrahedron Lett. 2014, 55, 2596. [Google Scholar]
- (134).Lei X; Xie H-Y; Chen S-Y; Teng K-S; Wen X; Xu Q-L; Sun H Tetrahedron 2015, 71, 4098. [Google Scholar]
- (135).Zhen L; Lin C; Du H-J; Dai L; Wen X; Xu Q-L; Sun H Tetrahedron 2015, 71, 2839. [Google Scholar]
- (136).Saijo R; Kawase M Eur. J. Org. Chem 2019, 1535. [Google Scholar]
- (137).Kakaawla TKK; Harrity JPA Org. Lett 2018, 20, 201. [DOI] [PubMed] [Google Scholar]
- (138).Lopchuk JM; Song M; Butler B; Gribble GW Synthesis 2015, 47, 2776. [Google Scholar]
- (139).Firoozi N; Torres GM; Arndtsen BA J. Org. Chem 2016, 81, 11145. [DOI] [PubMed] [Google Scholar]
- (140).Torres GM; Quesnel JS; Bijou D; Arndtsen BA J. Am. Chem. Soc 2016, 138, 7315. [DOI] [PubMed] [Google Scholar]
- (141).Kumar P; Kapur M Org. Lett 2019, 21, 2134. [DOI] [PubMed] [Google Scholar]
- (142).Galenko EE; Bodunov VA; Galenko AV; Novikov MS; Khlebnikov AF J. Org. Chem 2017, 82, 8568. [DOI] [PubMed] [Google Scholar]
- (143).Lei X; Li L; He Y-P; Tang Y Org. Lett 2015, 17, 5224. [DOI] [PubMed] [Google Scholar]
- (144).Chen C; Cui SJ Org. Chem 2019, 84, 12157. [DOI] [PubMed] [Google Scholar]
- (145).Shen X; Xia J; Liang P; Ma X; Jiao W; Shao H Org. Biomol. Chem 2015, 13, 10865. [DOI] [PubMed] [Google Scholar]
- (146).Das Adhikary N; Kwon S; Chung W-J; Koo SJ Org. Chem 2015, 80, 7693. [DOI] [PubMed] [Google Scholar]
- (147).Naskar S; Roy S; Sarkar S Synth. Commun 2014, 44, 1629. [Google Scholar]
- (148).Wang J; Shen Q; Zhang J; Song G Tetrahedron Lett. 2015, 56, 2913. [Google Scholar]
- (149).Jiang C-H; Lei X; Zhen L; Du H-J; Wen X; Xu Q-L; Sun H Beilstein J. Org. Chem 2014, 10, 2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (150).Du H-J; Zhen L; Wen X; Xu Q-L; Sun H Org. Biomol. Chem 2014, 12, 9716. [DOI] [PubMed] [Google Scholar]