

Supplemental Digital Content is Available in the Text.
Key Words: myocardial infarction, ACAP2, miR-532, cardiomyocytes, apoptosis
Abstract:
CircRNA ACAP2 and miR-532 both promotes the apoptosis of cardiomyocytes, which contributes to myocardial infarction (MI). Therefore, ACAP2 and miR-532 may interact with each other to participate in MI. Plasma samples from both patients with MI (n = 65) and healthy controls (n = 65) were subjected to RNA extractions and real-time quantitative polymerase chain reaction to analyze the expression of ACAP2, mature miR-532, and premature miR-532. Correlations among them were analyzed by Pearson's correlation coefficient. Expression of both mature miR-532 and premature miR-532 in cardiomyocytes with ACAP2 overexpression was analyzed by real-time quantitative polymerase chain reaction to study the effects of ACAP2 overexpression on the maturation of miR-532. The role of ACAP2 and miR-532 in regulating the apoptosis of cardiomyocytes induced by hypoxia was analyzed by cell apoptosis assay. In this study, we found that ACAP2 and mature miR-532 were both upregulated in plasma from patients with MI. ACAP2 and mature miR-532 were inversely correlated, whereas ACAP2 and premature miR-532 were not significantly correlated. In cardiomyocytes, overexpression of ACAP2 increased the expression of mature miR-532, but not premature miR-532. Cell apoptosis analysis showed that ACAP2 and miR-532 overexpression promoted the apoptosis of cardiomyocytes induced by hypoxia treatment. In addition, miR-532 inhibitor reduced the effects of ACAP2 overexpression. ACAP2 is overexpressed in MI and may promote the maturation of miR-532 to induce the apoptosis of cardiomyocyte.
INTRODUCTION
Myocardial infarction (MI), commonly known as heart attack, is the death or damage of heart tissues caused by the formation of blood clot that blocks coronary artery, which is responsible for the transportation of oxygen and blood to the heart.1,2 Irreversible death of heart cells will occur if restoration of blood does not occur within 20–40 minutes after the sudden blockage.1,2 MI is correlated with high mortality rate. It is estimated that more than 10% of patients with MI will die before hospitalization.3,4 Even worse, only less than half of the survivors can live longer than 10 years.3,4 Open heart surgery, medications, and interventional procedures may help to improve MI patients' life quality, whereas no cure is currently available.5,6
The treatment and prevention of MI require the development of novel therapeutic approaches. Studies on the molecular mechanism of MI have showed that nearly all aspects of the development and progression of MI require the involvement of multiple molecular players.7,8 Some molecular factors, such as TNF-related apoptosis-inducing ligand has been proven to be potential molecular targets to treat MI.9,10 However, safe and effective targets for molecular targeted MI therapy remain lacking. Circular RNAs (circRNAs) are covalently closed RNA transcripts that participate in human diseases mainly by regulating gene expression rather that encoding protein products.11 In effect, circRNAs are potential targets to treat human diseases including MI.12 However, the function of most circRNAs in MI remains unclear. Previous studies have reported that circRNA ACAP2 and miR-532 both promotes the apoptosis of cardiomyocytes, which contributes to MI,13,14 suggesting their involvement in MI and the existence of crosstalk between them. It has been well established that premature miRNAs are mainly localized to nucleus, and the maturation of miRNAs, which produces functional mature miRNAs, requires the transportation of premature miRNAs from nucleus to cytoplasm.15 This study was therefore performed to study the interaction between ACAP2 and miR-532 in MI, with a focus on the maturation of miR-532.
MATERIALS AND METHODS
Research Subjects
Chengdu First People's Hospital Ethics Committee approved this study. At this hospital, we enrolled both patients with MI (n = 65) and healthy controls (n = 65) from July 2018 to July 2020 to serve as research subjects. Both groups included 38 men and 27 women, with a mean age of 56.6 ± 5.9 years. Patients with MI were diagnosed through the following criteria: (1) biomarkers in serum (increased troponin I or T and increased creatine kinase MB) and (2) ECG signs (pathologic Q waves and ST segment elevation). Healthy controls received systemic physiological examination at the physiological center of the aforementioned hospital, and all physiological functions of controls were within normal range. Patients with MI complicated with other severe diseases (such as diabetes, severe infections, metabolic disorders, cancers, heart diseases, etc), and the ones with initiated therapy were excluded. Healthy controls with a history of MI were also excluded. All patients with MI and healthy controls signed informed consent.
Preparation of Plasma and Cardiomyocytes
Both patients with MI (before therapy) and healthy controls were subjected to the extraction of blood (2 mL) after overnight fasting. The 2 mL blood was transferred to tubes containing 0.2 mL citric acids, followed by centrifuging the mixture for 20 minutes at 1200g to separate plasma samples. In liquid nitrogen, plasma samples were kept before the subsequent experiments.
The cell model used in this study was human cardiomyocyte cell line AC16 from Sigma-Aldrich (USA). Cell culture medium as composed of 12% fetal bovine serum and 88% dulbecco's modified eagle medium medium (pH = 7.5). Antibiotics (1% penicillin and streptomycin) were also added. Normal cell culture conditions were 5% CO2, 95% humidity, and 37°C. To study the effects of hypoxia on gene expression, AC16 cells were cultivated under hypoxic condition (1% O2, 5% CO2, and 94% N2) for 24, 48, 72, and 96 hours before the subsequent experiments. No obvious changes in pH value (indicated by the medium color) were observed under hypoxic conditions.
Transient Transfections
AC 16 cells were transfected with ACAP2 expression vector, miR-532 inhibitor, mimic of miR-532, or ACAP2 siRNA. All transfections were performed using lipofectamine 2000 (Invitrogen). ACAP2 expression vector was established with pcDNA3.1 vector (Invitrogen) as backbone. Mimic of miR-532 and negative control (NC) miRNA, ACAP2 siRNA, as well as miR-532 inhibitor and NC inhibitor were both from Invitrogen. NC experiments (empty pcDNA3.1 vector-transfected cells, NC inhibitor, NC siRNA, or NC miRNA-transfected cells) and C experiments (untransfected cells) were included in each transfection. Before the subsequent experiments, transfected cells were cultivated in fresh medium for 48 hours.
Preparation of RNA Samples
RNAzol (Sigma-Aldrich) was used for RNA extractions from both plasma samples and AC16 cells. Genomic DNA in all RNA samples was digested by DNase I (Invitrogen) for 90 minutes at 37°C. RNA integrity was analyzed by 5% urea–PAGE gel electrophoresis. The purity of RNA samples were analyzed by determining the OD260/280 ratios. RNA samples with an OD260/280 ratio close to 2.0 were considered to be pure RNA.
Real-Time Quantitative Polymerase Chain Reaction
RNA samples with satisfactory integrity and purity were used as template to prepare cDNA samples through reverse transcriptions (RTs), which were also performed using the SS-IV-RT system (Invitrogen). SYBR Green Real-Time polymerase chain reaction (PCR) Master mix (Toyobo) was used to perform all qPCRs with 18S rRNA as internal control to determine premature miR-532 and ACAP2 expression.
All-in-One miRNA qRT-PCR Detection Kit (GeneCopoeia) was used to determine the expression of mature miR-532. To measure the levels of mature miR-532 expression, poly (A) was added, followed by using poly (T) as reverse primer to perform RTs and qPCRs with U6 as the internal control. Primer sequences were 5′-GAATGGGATTCGAGACCTG-3′ (forward) and 5′-TTCTTCCAAAGCTGCCTGT-3′ (reverse) for ACAP2; 5′-GAATGGGATTCGAGACCTG-3′ (forward) and 5′-TTCTTCCAAAGCTGCCTGT-3′ (reverse) for premature miR-532; and 5′-CTACCACATCCAAGGAAGCA-3′ (forward) and 5′-TTTTCGTCACTACCTCCCCG-3′ (reverse) for human 18S rRNA; 5′-CATGCCTTGAGTGTAGGACC-3′ (forward) for mature miR-532. Universal reverse primer and U6 primers were from the kit.
Ct values of target genes were normalized to endogenous controls based on the 2-ΔΔCT method. The sample with the biggest ΔCT value was set to value “1,” and all other samples were normalized to this sample to calculate relative expression levels.
Cell Apoptosis Assay
At 48-h post-transfection, cell apoptosis assay was performed to analyze the effects of transfections on cell apoptosis. In brief, AC16 cells were cultivated in a 6-well cell culture plate with 12,000 cells in 2 mL medium per well. Cells were cultivated under hypoxic conditions (1% O2, 5% CO2, and 94% N2) for further 48 hours, followed by washing with ice-cold PBS. After that, cells were resuspended in binding buffer, followed by staining with Annexin V conjugated with fluorescein isothiocyante (FITC) and propidium iodide (PI) for 20 minutes in dark. Finally, cell apoptosis was analyzed with BD FACS Flow Cytometry (Becton). FlowJo software version 8.8.6 (Tree Star Inc.) was used for data analysis.
Statistical Analysis
In vitro cell experiments, including overexpression assays, hypoxic treatment assays, and cell apoptosis assays, were performed in 3 independent (performed at different times) biological replicates, and data were expressed as mean ± SD values. Comparisons between 2 groups of participants were performed by the Mann–Whitney test. Comparisons among multiple independent transfection groups were analyzed by the Kruskal–Wallis test followed by the post hoc Dunn test. Pearson's correlation coefficient was used to analyze correlations. Comparisons between different time points of the same group were performed with repeated measures ANOVA. Differences were statistically significant when P < 0.05.
RESULTS
Analysis of the Expression of Plasma ACAP2 and miR-532 in Patients with MI and Controls
Compared with the control group, the MI group exhibited significantly upregulated ACAP2 (Fig. 1A, P < 0.001) and mature miR-532 (Fig. 1B, P < 0.001). By contrast, no significantly altered expression of premature miR-532 was observed between MI and control groups (Fig. 1C). Therefore, patients with MI exhibited altered expression of ACAP2 and mature miR-532 in plasma.
FIGURE 1.
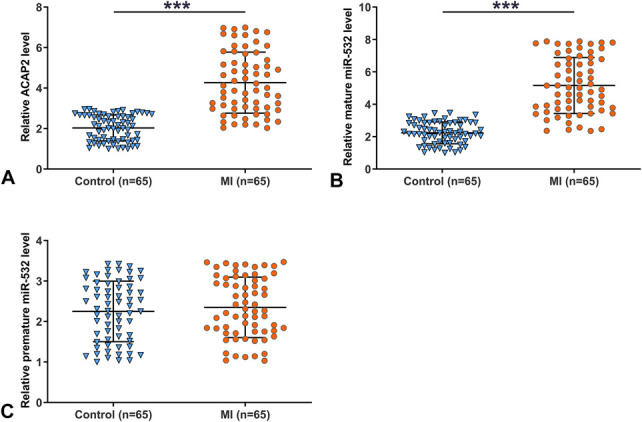
Patients with MI exhibited altered expression of ACAP2 and mature miR-532 in plasma. RNA isolations and RT-qPCRs were performed to determine the expression of ACAP2 (A), mature miR-532 (B), and premature miR-532 (C) in both MI and control plasma samples. Three technical replicates were performed on each sample. The values presented here were average values of 3 technical replicates. The arbitrary unit was used for all values. ***P < 0.001.
Analysis of the Correlations Between ACAP2 and miR-532 Cross MI and Control Plasma Samples
Correlations between ACAP2 and mature miR-532 or premature miR-532 across MI plasma samples were analyzed by Pearson's correlation coefficient. It was observed that ACAP2 and mature miR-532 were inversely and significantly correlated (Fig. 2A), whereas ACAP2 and premature miR-532 were not significantly correlated (Fig. 2B). It is worth noting that no significant correlation between ACAP2 and mature (see Fig. 1A, Supplemental Digital Content 1, http://links.lww.com/JCVP/A657) or premature (see Fig. 1B, Supplemental Digital Content 2, http://links.lww.com/JCVP/A657) miR-532 across control samples. Therefore, ACAP2 may be correlated with the maturation of miR-532 in MI.
FIGURE 2.
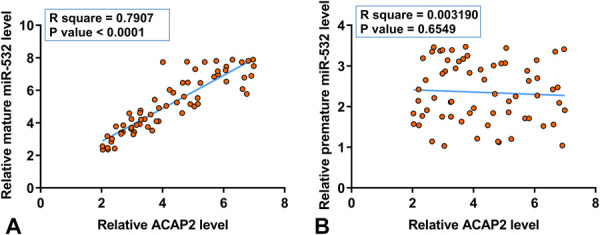
ACAP2 and mature miR-532 were inversely correlated cross MI plasma samples. Correlations between ACAP2 and mature miR-532 (A) or premature miR-532 (B) across MI plasma samples were analyzed by Pearson's correlation coefficient. All values subjected to correlation analysis were average values of 3 technical replicates of RT-qPCR. The arbitrary unit was used for all values.
The Effects of Hypoxia on the Expression of ACAP2 and Mature miR-532 in AC16 Cells
To study the effects of hypoxia on the expression of ACAP2, mature miR-532, and premature miR-532, AC16 cells were cultivated under hypoxic condition (1% O2, 5% CO2, and 94% N2) for 24, 48, 72, and 96 hours, followed by RNA isolations and RT-qPCRs to determine gene expression. It was observed that hypoxia upregulated the expression of ACAP2 (Fig. 3A, P < 0.05) and mature miR-532 (Fig. 3B, P < 0.05) in AC16 cells. By contrast, hypoxia treatment failed to significantly alter the expression of premature miR-531 (Fig. 3C). Therefore, hypoxic treatment can increase the expression of ACAP2 and mature miR-532 in AC16 cells.
FIGURE 3.
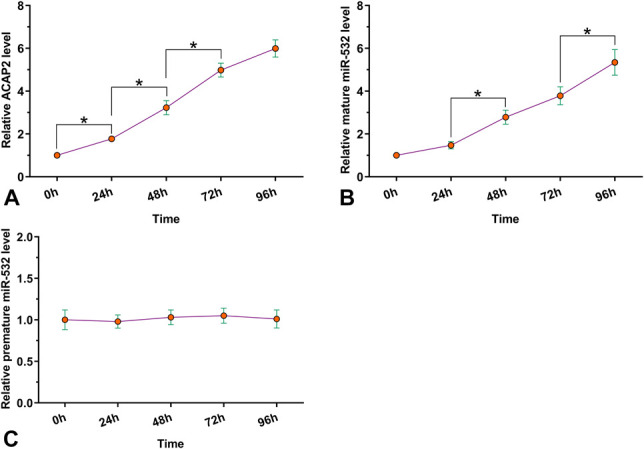
Hypoxia upregulated the expression of ACAP2 and mature miR-532 in AC16 cells. AC16 cells were cultivated under hypoxic condition (1% O2, 5% CO2, and 94% N2) for 24, 48, 72, and 96 hours, followed by RNA isolations and RT-qPCRs to determine the expression of both ACAP2 and miR-532 (mature and premature) through RT-qPCR. Hypoxia treatment was performed in 3 biological replicates, and values presented here were mean ± SD values. The arbitrary unit was used for all values. *P < 0.05.
The Role of ACAP2 in the Maturation of miR-532 in AC16 Cells
To determine the effects of ACAP2 overexpression on the maturation of miR-532, AC16 cells were transfected with ACAP2 expression vector or miR-532 mimic, followed by the determination of ACAP2 and miR-532 expression every 24 until 96 hours. It was observed that ACAP2 and miR-532 were significantly overexpressed between 24 and 96 hours (Fig. 4A, P < 0.05). It was observed that overexpression of ACAP2 decreased the expression of mature miR-532 (Fig. 4B, P < 0.05), but not premature miR-532 (Fig. 4C, P < 0.05). In addition, overexpression of miR-532 failed to significantly alter the expression of ACAP2 (Fig. 4D). Therefore, ACAP2 overexpression suppressed the maturation of miR-532 in AC16 cells.
FIGURE 4.

ACAP2 overexpression suppressed the maturation of miR-532 in AC16 cells. AC16 cells were transfected with ACAP2 expression vector or miR-532 mimic, followed by the determination of ACAP2 and miR-532 expression every 24 until 96 hours (A). The effects of the overexpression of ACAP2 on the expression of mature miR-532 (B) and premature miR-532 (C), and the effects of miR-532 overexpression on the expression of ACAP2 were analyzed by RT-qPCR (D). Each overexpression experiment performed in 3 biological replicates, and values presented here were mean ± SD values. The arbitrary unit was used for all values. *P < 0.05.
The Role of ACAP2 and miR-532 in the Hypoxia-induced Apoptosis of AC16 Cells
The effects of transfections on the apoptosis of AC16 cells induced by hypoxic treatment were analyzed by cell apoptosis assay. ACAP2 and miR-532 overexpression promoted the apoptosis of cardiomyocytes induced by hypoxia treatment. By contrast, miR-532 inhibitor and ACAP2 siRNA silencing suppressed cell apoptosis. In addition, miR-532 inhibitor reduced the effects of ACAP2 overexpression (Fig. 5, P < 0.05). Therefore, ACAP2 increased hypoxia-induced apoptosis of AC16 cells through miR-532.
FIGURE 5.
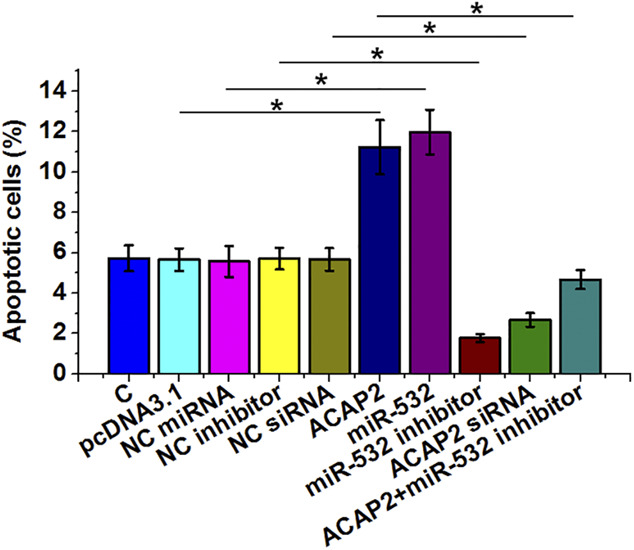
ACAP2 increased hypoxia-induced apoptosis of AC16 cells through miR-532. The effects of transfections on the apoptosis of AC16 cells induced by hypoxic treatment (1% O2, 5% CO2, and 94% N2) for further 48 hours were analyzed by cell apoptosis assay, which was performed by annexin V-FITC and PI staining and flow cytometry. Each cell apoptosis experiment was performed in 3 biological replicates, and values presented here were mean ± SD values. The arbitrary unit was used for all values. *P < 0.05.
DISCUSSION
In this study, we analyzed the involvement of ACAP2 and miR-532 in MI and explored the interactions between them. We found that ACAP2 and mature miR-532 were both upregulated in MI. In addition, ACAP2 overexpression in MI may promote the maturation of miR-532 in AC16 cells to increase hypoxia-induced cell apoptosis.
In a recent study, Liu et al13 reported that ACAP2 was upregulated in a MI rat model and it could sponge miR-29 to induce the apoptosis of cardiomyocytes, thereby promoting disease progression. However, the expression pattern of in ACAP2 patients and its function are unknown. In this study, we showed that ACAP2 was upregulated in patients with MI. In addition, overexpression of ACAP2 in AC16 cells was observed after hypoxic treatment. Under hypoxic treatment, ACAP2 overexpression increased the apoptosis of AC16 cell apoptosis. Therefore, the upregulation of ACAP2 in MI is likely induced by hypoxia, and hypoxia-inducible ACAP2 may promote the apoptosis of AC16 cells to aggregate disease condition.
Interestingly, miR-532 was upregulated in the diabetic heart and promoted cardiomyocyte apoptosis to promote disease progression.14 Interestingly, we observed that mature miR-532, but not premature miR-532, was overexpressed in MI. Therefore, the development of MI may accelerate the maturation of miR-532. Consistently, in this study, we showed that miR-532 overexpression increased the apoptosis of AC16 cells induced by hypoxia, whereas miR-532 inhibitor suppressed cell apoptosis. Therefore, inhibition of miR-532 may serve as a potential target for the treatment of MI. However, animal model experiments and clinical trial experiments are needed to further verify our hypothesis. In addition, miR-532 may have multiple downstream targets. Therefore, the application of miR-532 as a target for the treatment of MI requires careful analysis of the safety.
Interestingly, in this study, we showed that ACAP2 overexpression increased the expression of mature miR-532, but not premature miR-532 in AC16 cells. Therefore, the increased maturation of miR-532 in AC16 cells may be caused by the upregulation of ACAP2. However, the mechanism of the maturation of miR-532 induced by ACAP2 is unknown. Considering that the maturation of miR-532 requires the transportation of premature miR-532 from the nucleus to cytoplasm, ACAP2 may suppress the movement of miR-532 to decrease the production of mature miR-532.
CONCLUSIONS
In conclusion, ACAP2 is overexpressed in MI and it may promote the maturation of miR-532 to induce hypoxia-induced apoptosis of cardiomyocytes.
Supplementary Material
ACKNOWLEDGMENTS
The authors express their gratitude for those who have critically reviewed this article and those who give us help during this experiment. This article is available on a preprint server that can be accessed at https://www.researchsquare.com/article/rs-96943/v1.
Footnotes
Supported by the National Nature Science Foundation of China (Grant No. 30700312).
The authors report no conflicts of interest.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Web site (www.jcvp.org).
Guarantor of integrity of the entire study: J. Zhang. Study concepts: J. Zhang. Study design: Y. Tang. Definition of intellectual content: J. Zhang. Experimental studies: J. He. Data acquisition: Z. Zhang. Data analysis: F. Liu. Statistical analysis: J. Zhang. Manuscript review: J. Zhang.
This study was approved by the Ethics Committee of Chengdu First People's Hospital Ethics Committee. All procedures followed the guideline of this hospital. Availability of data and materials: The data sets used analyzed during the current study are available from the corresponding author on reasonable request.
Contributor Information
Yanrong Tang, Email: ng1179@163.com.
Jing Zhang, Email: jw6450@163.com.
Jing Wang, Email: ia7569@163.com.
Jiyun He, Email: kz8787@163.com.
Zhenzhen Zhang, Email: iw1551@163.com.
Fuqiang Liu, Email: hp7815@163.com.
REFERENCES
- 1.Reed GW, Rossi JE, Cannon CP. Acute myocardial infarction. Lancet. 2017;389:197–210. [DOI] [PubMed] [Google Scholar]
- 2.Schultze JL. Myocardial infarction cell by cell. Nat Immunol. 2019;20:7–9. [DOI] [PubMed] [Google Scholar]
- 3.Al-Shaar L, Li Y, Rimm EB, et al. Physical activity and mortality among male survivors of myocardial infarction. Circulation. 2019;140(suppl 1):A15962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chatterjee P, Maddox KE. US national trends in mortality from acute myocardial infarction and heart failure: policy success or failure? JAMA Cardiol. 2018;3:336–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spath NB, Mills NL, Cruden NL. Novel cardioprotective and regenerative therapies in acute myocardial infarction: a review of recent and ongoing clinical trials. Future Cardiol. 2016;12:655–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gluckman TJ, Wilson MA, Chiu ST, et al. Case rates, treatment approaches, and outcomes in acute myocardial infarction during the coronavirus disease 2019 pandemic. JAMA Cardiol. 2020;7:e203629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Curley D, Plaza BL, Shah AM, et al. Molecular imaging of cardiac remodelling after myocardial infarction. Basic Res Cardiol. 2018;113:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lindsey ML, Ma Y, Flynn ER, et al. Identifying the molecular and cellular signature of cardiac dilation following myocardial infarction. Biochim Biophys Acta Mol Basis Dis. 2019;1865:1845–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saxena A, Russo I, Frangogiannis NG. Inflammation as a therapeutic target in myocardial infarction: learning from past failures to meet future challenges. Transl Res. 2016;167:152–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Y, Zhang H, Wang Z, et al. Blocking the death checkpoint protein TRAIL improves cardiac function after myocardial infarction in monkeys, pigs, and rats. Sci Transl Med. 2020;12:eaaw3172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ebbesen KK, Kjems J, Hansen TB. Circular RNAs: identification, biogenesis and function. Biochim Biophys Acta. 2016;1859:163–168. [DOI] [PubMed] [Google Scholar]
- 12.Devaux Y, Creemers EE, Boon RA, et al. Circular RNAs in heart failure. Eur J Heart Fail. 2017;19:701–709. [DOI] [PubMed] [Google Scholar]
- 13.Liu X, Wang M, Li Q, et al. CircRNA ACAP2 induces myocardial apoptosis after myocardial infarction by sponging miR-29. Minerva Med. 2020. doi:10.23736/s0026-4806.20.06600-8 [epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 14.Chandrasekera DN, Neale JPH, van Hout IV, et al. Upregulation of microRNA-532 enhances cardiomyocyte apoptosis in the diabetic heart. Apoptosis. 2020;25:388–399. [DOI] [PubMed] [Google Scholar]
- 15.Treiber T, Treiber N, Meister G. Regulation of microRNA biogenesis and its crosstalk with other cellular pathways. Nat Rev Mol Cel Biol. 2019;20:5–20. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.