

Abstract
Viruses are abundant, diverse and ancestral biological entities. Their diversity is high, both in terms of the number of different protein families encountered and in the sequence heterogeneity of each protein family. The recent increase in sequenced viral genomes constitutes a great opportunity to gain new insights into this diversity and consequently urges the development of annotation resources to help functional and comparative analysis. Here, we introduce PHROG (Prokaryotic Virus Remote Homologous Groups), a library of viral protein families generated using a new clustering approach based on remote homology detection by HMM profile-profile comparisons. Considering 17 473 reference (pro)viruses of prokaryotes, 868 340 of the total 938 864 proteins were grouped into 38 880 clusters that proved to be a 2-fold deeper clustering than using a classical strategy based on BLAST-like similarity searches, and yet to remain homogeneous. Manual inspection of similarities to various reference sequence databases led to the annotation of 5108 clusters (containing 50.6 % of the total protein dataset) with 705 different annotation terms, included in 9 functional categories, specifically designed for viruses. Hopefully, PHROG will be a useful tool to better annotate future prokaryotic viral sequences thus helping the scientific community to better understand the evolution and ecology of these entities.
INTRODUCTION
Viruses are key players in most ecosystems as they actively participate to the regulation of microbial communities (1) and are important vectors for horizontal gene transfer (2). Viruses infecting prokaryotes out-number eukaryotic viruses in some ecosystems (3) and represent the great majority of the viruses found in viral metagenomes (4). Viral genomes and metagenomes highlight (i) the recurrent presence of virus groups, especially in similar ecosystems, recently termed ‘virus operational taxonomic units’ (vOTUs; 5), (ii) the large number of different vOTUs found in most ecosystems (4) and (iii) the dominance in most vOTUs of genes not similar to any known genes. The growing amount of viral sequences produced nowadays, especially from metagenomes, calls for a need in developing resources to help assigning functions to viral proteins in order to improve functional and comparative analyses. As determining information for newly identified genes in sequences relies on finding an annotated homologous sequence using similarity searches, a set of well-annotated proteins that can be used for future work is particularly important. In order to build such a reference set of proteins, a classical way is to organize these proteins into homologous groups and to annotate these groups. Several methods have been used to cluster viral proteins into either homologous or orthologous groups: (i) an approach based on the identification of genome-specific best hits that are joined to form clusters of orthologs has been used for the pVOGs database (Prokaryotic Virus Orthologous Groups; (6)), (ii) a similar approach based on best-hit triangles has been implemented recently in eggNOG (evolutionary genealogy of genes: non-supervised orthologous groups; (7)), which integrates an additional step of in-paralogs detection and the identification of fused genes and (iii) clustering-based approaches have also been used to compute groups of homologous viral proteins (8–10). Despite using different clustering strategies, all these methods rely on similarity search results generated by BLAST (11) or faster equivalent such as MMseqs (12) or DIAMOND (13), and none integrate remote homology detection. Yet, viruses are known to have distant evolutionary relationships, resulting in distant sequence similarities not always captured by sequence comparison tools. This can be illustrated by the Microviridae family, whose members all encode homologous major capsid and replication initiation proteins in their ∼5 kb genomes. As the origin of the family is ancient, homologs are difficult to detect using sequence similarity search tool such as BLAST. For instance, for two distantly related Microviridae, Spiroplasma phage 4 and Enterobacteria phage phiX174 belonging respectively to the Gokushovirinae and Bullavirinae sub-families respectively, the best BLASTp hit between their capsid proteins only exhibit a bit-score of 38.5 (corresponding to an E-value of 0.14 on a ∼1 million protein database) and an alignment coverage below 30% for the two proteins. These values are much below any reasonable thresholds on the bit-score, E-value or coverage to detect homology. Thus, no clustering methods will group these two proteins together even though they are distant yet true homologs. Recent developments in remote homology detection software (14,15) coupled to the great wealth of genomic sequences could help tackle the problem of distant homology of viral proteins, yet to our knowledge, no clustering method uses remote homology detection in an automatic way for viral proteins.
Here, we propose such a procedure and clustered viral proteins into homologous groups, in two steps: (i) proteins are first gathered based on similarity search results (score >30 and coverage >80%) and (ii) HMM profiles generated for each protein cluster are then compared to each other and grouped (coverage >60%, probability >90 %) into super-clusters hereafter termed PHROGs (Prokaryotic Virus Remote Homologous Groups). These PHROGs were then annotated using annotation transfer, coupled with a careful manual curation. An interface has been developed, allowing to browse the data either by PHROG or by viral genome.
MATERIALS AND METHODS
Two datasets of archaeal and bacterial viruses
First, all known sequences of viruses infecting Bacteria or Archaea were assembled in a dataset. To this end, 2318 reference sequences of viruses infecting prokaryotes were retrieved in RefSeqVirus genomes (as of April 2018). Viruses included in the pVOGs database (6) and complete virus genomes from GenBank were also downloaded. As one virus can be present in these three databases with different identifiers (RefSeq, pVOGs and GenBank), all these viruses were compared to each other using BLASTn and viruses almost identical were removed (>99.9 identity percent and coverage >97%). The resulting 4975 completely sequenced viruses (2315 from RefSeqVirus, 686 from pVOGs and 1986 from GenBank) will be refered as RefVirus from here on. To this dataset, 12 498 previously published curated viral sequences derived from cultivated microbial isolates were added (9). This last dataset is composed of both integrated proviruses (>10 kb) and circular episomes, and is here termed ProVirus. These (pro)viruses were found in 5492 microbial genomes. In total, 496 859 proteins from complete viral genomes (i.e. RefVirus) and 442 005 proteins from (pro)viruses (i.e. ProVirus) were collected.
Generating the viral homologous groups or PHROGs
Protein clustering using similarity searches (Figure 1, Panel B1 and B2)
Figure 1.

Overview figure of the dataset, the clustering procedure, results and website main page. (A) Protein sets of reference viruses from the NCBI (RefVirus) and (pro)viruses detected using VirSorter (ProVirus) were collected. It should be noted that Provirus proteins (green) were not initially annotated. (B) The four steps of the clustering procedure: (i) the protein network built from pairwise sequence similarities, each dot/vertex representing a protein (green for ProVirus proteins, red for annotated RefVirus proteins and pink for unannotated RefVirus), linked by edges if the two proteins are similar. (ii) Protein clusters are identified by applying MCL on to this network, and clusters are depicted as gray circle. (iii) Clusters (and singletons) are compared using protein profiles and edges are drawn for pairs of protein clusters that are similar. (iv) This network of clusters is here again clustered into PHROGs, depicted as dark brown and blue for unannotated and annotated PHROGs. For example, PHROG2 is made of two protein clusters and one singleton. (C) Description of the number of annotated and unannotated PHROGs and singletons, with the number and origin of proteins involved (red and green for RefVirus and ProVirus, respectively). (D) The PHROGs Web site main page, where users can search for PHROGs or viruses of interest.
The 938 864 proteins were compared to each other using MMseqs (12). To be further considered, a protein pair should have (i) at least a local alignment with a bit-score >30 and (ii) >80% of the residues of each protein should be involved in at least one alignment found between the two proteins (i.e. coverage >80%). Using each protein pair and the lowest E-value found in a local alignment between two proteins, the proteins were clustered with MCL (inflation 2.0; (16)).
Grouping the protein clusters using remote homology detection (Figure 1, Panel B3 and B4)
For each of the 63 673 clusters containing at least two proteins, a multiple alignment was built using ClustalOmega (17; 5 guide-tree/HMM iterations) and an HMM profile was computed for each alignment using hhmake of the HHsuite toolkit (version 2.0.16; (18)). All these profiles were then compared to each other using the hhsearch command. The 85 024 singletons were also compared to the 63 673 cluster profiles. To be further considered, a cluster pair should have a hit (i) with a probability >90%, (ii) that involves at least 60% of the two HMM profiles (same thresholds when comparing singletons to clusters). Based on these rules, singletons and clusters were then clustered with MCL (inflation 2.0; (16)). This resulted in placing 868 340 of the initial 938 864 protein sequences into 38 880 ‘super-clusters’ containing at least two proteins, hereafter named PHROGs. Only 7.5% of the protein dataset remained as singleton, or ORFan (70 524 proteins). Multiple sequence alignments and HMM profiles were computed for all the 38 880 PHROGs containing at least two proteins using ClustalOmega (17) and HHsuite (18). Each multiple alignment was processed in order to consider as gaps the columns made of >50% of gaps (saved in a2m format). HMM profiles were generated for each of the masked alignment and these profiles were compared to each other using hhsearch (18). Results are accessible through the website and all files are downloadable as zipped archives.
Comparing PHROGs to the standard clustering method
To estimate the performance of our procedure, the same set of proteins were clustered using a classical approach. Sequence similarities were searched for all protein pairs using MMseqs and clustered using Markov clustering algorithm (here the MCL software) (12,16). This clustering procedure is similar to the first step of the PHROG clustering described above, except that a less stringent coverage threshold was used (coverage ≥50 % for the two proteins). Two complementary metrics were used to analyze the differences between the two methods: the number of proteins clustered and the size of the clusters (Figure 3), and the identity percent and coverage between proteins grouped in each cluster (Figure 4). To this end, multiple alignments of clusters generated with the classical approach were also computed using ClustalOmega. For all protein pairs inside these multiple alignments and the PHROGs, the percent identity and the coverage were calculated as being, respectively, (i) the number of identical amino acids in the two aligned proteins divided by the length of the smallest of the two proteins and (ii) the proportion of amino acids of one protein that is aligned to any amino acid (not to a gap) of the other protein. Subsamples of 1000 values were taken to draw each boxplot of Figure 4.
Figure 3.

Cumulated number of clustered proteins. For example, point a means that for the standard clustering procedure, ∼234 000 proteins are in clusters that contain at least 200 proteins, whereas for the PHROG procedure, ∼390 000 proteins are in clusters >200 (point b). The inset at the top right is a zoom of the left part of the curve. The 5 largest PHROGs are highlighted by a cross at the bottom right (the two largest PHROGs at the bottom right gather 5795 and 5879 proteins).
Figure 4.

Identity percent (A) and coverage (B) for protein pairs in the same clusters. The clusters were separated according to interval of size, the first value «3» representing clusters that contain 3, 4, 5 or 6 proteins, «7» being clusters containing between 7 and 19 proteins, and the last interval «2000» being clusters >2000 proteins. Using the multiple alignments of each cluster, (i) the identity percent between two proteins is the number of amino acids that are identical in the two aligned proteins divided by the length of the smallest of the two proteins, and (ii) the coverage is the proportion of amino acids of one protein that is aligned to any amino acid (not to a gap) of the other protein. Subsamples of 1000 values where taken to draw each boxplot.
Coverage threshold values chosen for the two steps of the clustering procedure are critical parameters, since relaxed values come with the risk of aggregating heterogeneous functions (due to the presence of protein fusions), and stringent values limit cluster sizes. Based on a subset of 50 000 randomly extracted proteins, three thresholds on coverage (80%, 50%, no coverage threshold, named respectively ‘1-step C80’, ‘1-step C50’,‘1-step C0’) were tested for the first step of the clustering procedure (the 50 threshold corresponding to the standard procedure described above). Then, results from the 80 % threshold on coverage were further considered and used as a basis for the second step of the clustering and 5 thresholds were tested for the coverage between HMM pairs (80%, 60%, 40%, 20% and no threshold, named respectively ‘2-step C80 C80’, ‘2-step C80 C60’, ‘2-step C80 C40’, ‘2-step C80 C20’, ‘2-step C80 C0’), the combination of C80 and C60 being the ones retained for building PHROGs.
Functional annotation of PHROGs
NCBI protein annotations (RefVirus) were first automatically curated (no upper case except in gene names, correction of typos, etc.). Phages for which all protein annotations was only an uninformative list (‘hypothetical protein GP1’, ‘hypothetical protein GP2’, etc.) were considered as unannotated. These protein annotations were first combined to determine an annotation for PHROGs. To refine this annotation, the 38 880 PHROGs were compared to different databases. PHROG profiles were compared to Pfam domains (version of jan 2018; 19) and UNICLUST (20) and individual viral protein were compared to proteins in KEGG Orthologous groups (KOs; version of jan 2018) using MMseq (bit-score>50, coverage >50%). Manual curation of the collected annotations and similarities allowed to extract a single annotation per PHROG.
Colocalization of genes according to their functions
For each pair of functional annotations, the number of their respective proteins that are neighbors on the genomes were determined. Neighbors were here defined as the two proteins encoded right before and after the protein considered, and only if they are on the same strand. As some annotations gather many more proteins than others, a colocalization score between two annotations was computed. The hypergeometric formula was first used to calculate the probability P that two annotations (noted here annotation A and annotation B) that each have nA and nB proteins (among the whole dataset of n proteins), would have k pairs of proteins that are neighbors by chance.
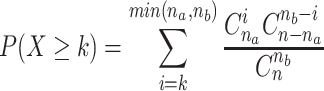 |
This probability represents the statistical significance of an observed number of neighbor protein pairs for two annotations. This probability was also computed considering each PHROG and not each annotation and PHROG neighbors are reported on the Web site. To draw a graph of the most significant relationships, all P values were then corrected by the total number of annotations pairs T and finally converted into a significance score : S(A,B) = − log(P×T) (as in 21). Annotation pairs with significance scores >1 are considered as significantly colocalized (scores are capped to 1000). A network of neighboring annotations was generated using the igraph library in R, in which annotations were joined by an edge with a weight equal to their significance score. For visualizing purposes, only the 112 more frequently retrieved annotations (used for >650 proteins) were considered. When neighbor genes have the same annotations, these annotations are drawn as squares. Annotations were placed using the Fruchterman-Reingold layout algorithm. Annotations were gathered into modules using community structure detection based on edge betweenness.
RESULTS
Description of the dataset
All prokaryote-infecting viral sequences (complete genomes or not) from existing sequence databases were compiled. Overall, to the 4975 viruses infecting a prokaryote from the RefSeq, pVOGs and GenBank databases were added the 12 498 high-confidence viral sequences identified by the VirSorter tool (9,22) as proviruses or circular viral genomes in microbial genomes. The viruses considered here were mostly dsDNA viruses and infected 35 different bacterial and archaeal classes and 410 genera. Yet, viruses infecting bacteria belonging to the Proteobacteria class represented as much as 63% of the 17 473 total viral genomes or contigs, the Escherichia genus alone representing as much as 29% of this set (Figure 2).
Figure 2.

Number of viruses considering their viral family and the class of their host (when not specified, viral families have a dsDNA genome). The size of the balloons are proportional to the number of viruses and the color reflects the proportion of proviruses.
Analysis of the clustering characteristics and comparison to a standard clustering procedure
Viral proteins were clustered using either our two-step procedure or a more classical approach (see Materials and Methods). The main improvement provided by the PHROGs strategy was a reduction by nearly two-fold in the number of clusters (at least 2 proteins) compared to the standard procedure (38 880 and 57 073, respectively) and a concomitant increase in the average size of the protein clusters (22.3 and 15.2, respectively). Unsurprisingly, the number of proteins left as singletons was comparable for the PHROG and standard procedures (70 524 and 67 430, respectively) and represented <8% of the 938 864 proteins. The largest clusters were composed of 5879 and 2267 proteins, respectively, with 19 PHROGs being >2000, whereas only 2 clusters made with the classical approach had such a large size. All along, highly populated protein families were more frequent among PHROGs than traditional clusters (see Figure 3).
Considering the classical clustering approach (Figure 4), the average identity percent inside clusters tended to decrease with the cluster size, from 79.7% for small clusters (<7 proteins) to 56.6% for large ones (>2000 proteins). This trend was dramatically accentuated for the PHROG procedure, as the average identity went down from 79.8 to 27.2%. The average coverage remained high for all cluster sizes for the classical approach (98.3% in average) whereas it dropped for the PHROG procedure from 99% to 80.7% in average, for small and large clusters, respectively (Figure 4). This shows that more distant homologs are gathered with the PHROG procedure. We verified that these large clusters maintained a good consistency, with an acceptable average coverage: over 95.8% of the proteins inside a large PHROG covered >50% of any other protein of the PHROG. As expected, the percent identity were quite low within these large clusters, with 66.8% of the protein pairs having an amino acid percent identity <30%. Coming back to the Microviridae protein example mentioned in the Introduction, capsids of viruses of the two known sub-families, Gokushovirinae and Bullavirinae, were grouped into two separate clusters by the first step of our method, as well as by the classical method. Indeed, using pairwise sequence alignment (MMseqs), all Gokushovirinae capsid protein sequences were similar to each other but no similarities were detected between these and Bullavirinae capsid sequences. An HMM profile was built for these two groups of capsids and the two HMMs were clearly identified as similar by HHsearch (probability of 99.5, E-value of 8.4 × 10–18, score of 176.8, covering >400 positions of the two HMMs). Thus, the two clusters of capsid proteins were gathered by the second step of our method, resulting in PHROG_514 that contains capsid protein for all members of this phage family.
We further studied the effect of coverage thresholds on clusters and found that all procedures based on only one clustering step gave comparable results, regardless of the coverage threshold (Supplementary Figure S1A). This parameter however had a major impact on the second clustering step (Supplementary Figure S1A). Indeed, for ‘1-step C80’, ‘1-step C50’ and ‘1-step C0’, the largest clusters were of 71, 77 and 90 proteins respectively, whereas the 2-step procedures built clusters composed of 151, 164, 269, 465 and 343 proteins when coverage thresholds of the second step were decreased from 80% to 0, by 20% steps, using a coverage of 80% in the first step. Considering the average identity percent for protein pairs inside clusters, the use of HMM comparisons allowed to group proteins with significantly more remote homology (Supplementary Figure S1B). Yet, procedures involving thresholds lower than 50 % on the first or second step (‘1-step C0’, ‘2-step C80 C40’, ‘2-step C80 C20’ and ‘2-step C80 C0’) of course resulted in the non negligible presence of protein pairs that cover each other <50% inside clusters (Supplementary Figure S1C). Thus, the combination of a threshold of 80% and 60% on coverage for the two steps of the clustering allows to detect remote homologies (<30 id%) and to build large clusters without grouping proteins that do not cover themselves significantly.
The whole protein dataset, when compared by a sequence-to-sequence comparison tool (MMseqs, bit-score>50), resulted in 238.3 millions of protein pairs. Considering only the 195.7 million pairs with a coverage >50%, the standard method built 57 073 clusters (178.5 millions of protein pairs inside these clusters). The 38 880 PHROGs created by our clustering strategy resulted in 463.1 millions of protein pairs inside these PHROGs, so over twice as much the number of pairs identified by BLAST-like searches.
Functional annotation of PHROGs
To determine a putative function for each PHROG, protein annotation from NCBI complete viral genomes (RefVirus) inside each PHROG were confronted, harmonized and transferred. At the onset, as many as 20 872 different annotations were found for the 496 859 proteins from RefVirus. For example, PHROG_2 is made of proteins annotated under 107 different designations such as ‘terminase large subunit’, ‘terminase’, ‘terminase DNA packaging enzyme large subunit’, ‘phage terminase large subunit’, ‘large terminase protein’, ‘large terminase subunit’, ‘TerL’, ‘terminase, large subunit’, ‘large subunit terminase’, ‘large terminase’, (etc…) as well as some ‘hypothetical protein’. This PHROG was then annotated as ‘terminase large subunit’. To further improve these annotations, different sources of information were collected and manually inspected to refine each PHROG annotation: (i) 11 810 PHROGs contain at least one Pfam domain, (ii) 6710 PHROGs were attributed at least one GO term via a similarity to UNICLUST proteins, (iii) 1788 PHROGs were similar to at least one KEGG Ortholog group (KO). Presence of proteins annotated unambiguously in the ACLAME database (23) were also retrieved. Finally, 5108 of the 38 880 PHROGs were functionally annotated using a set of 705 annotations. Nine functional categories were then defined to cover these 705 annotations : ‘head and packaging’, ‘connector’, ‘tail’, ‘DNA, RNA and nucleotide metabolism’, ‘integration and excision’, ‘lysis’, ‘transcription regulation’, ‘moron, auxiliary metabolic gene and host takeover’, ‘other’ (Table 1 and Figure 5). Even though the number of annotated PHROG seems low (5108 of the 38 880 PHROGs), large PHROGs are more often annotated than small ones and thus, these 5108 annotated PHROGs contain 475 493 proteins (50.6% of the total protein dataset, including singletons) (Table 1). These annotated PHROGs contain as many as 68 478 ‘hypothetical protein’ from RefVirus that can now be annotated with their PHROG’s annotation. On the contrary, 11 660 RefVirus proteins initially considered as annotated have lost there annotation (i.e. are in unannotated PHROGs; Figure 1C). This is due to PHROGs (i) containing only uninformative protein annotations (e.g. protein annotations such as ‘prophage Lp1 protein 30’, ‘DUF1642 domain containing protein’, ‘similar to P2 orf80’, etc…) or (ii) containing protein with non congruent annotations and in both cases, information collected using KEGG, UNICLUST or Pfam did not help to define an annotation. For example, PHROG_358 contains RefVirus proteins annotated as ‘virion structural protein’, ‘virulence associated protein’, ‘baseplate protein’, ‘neck protein’, ‘tail protein’ (etc.), has no similarity with KEGG or UNICLUST proteins, is only similar to a Pfam domain of unknown function (DUF4815) and is similar to five unannotated PHROGs and to one ‘virion structural protein’ PHROG but on a very small portion. Thus, this PHROG was attributed to ‘unknown function’.
Table 1.
Number of PHROGs (and proteins inside these PHROGs) annotated in the nine functional categories. The first line represent the whole dataset and the hierarchy in the following lines is represented by indentations. A color was attributed to each of the nine functional category

|
Figure 5.

For each annotation term, the number of PHROGs with this annotation and the number of proteins in these PHROGs, each term being colored according to its functional category.
Further gaining some functional clues using (often short) similarities between PHROGs
We then performed iterative HMM alignments, with relaxed parameters, to extend annotation possibilities. For this, columns of the multiple sequence alignments with >50% gaps were considered as insertion states, allowing to get rid of spurious ends and insertions present in only a handful of proteins. A total of 26 177 new connections were detected between 12 791 PHROGs (E-value < 0.001 and no threshold on the coverage), and in 25% of cases, non annotated PHROGs were joined to annotated ones. These PHROGs were not clustered together in the first place, for several reasons. First, similarities had to cover at least 60% of the two initial protein clusters (Figure 1B(iii)) to build PHROGs and thus, local similarities existing between protein clusters were considered insufficient for aggregation. Moreover, MCL clustering (Figure 1B(iii)) implies the presence of more edges within members of PHROGs than between different PHROGs, but links between clusters (and thus PHROGs) exist. Finally, PHROGs are larger and more informative than the protein clusters they are built upon and thus homologies even more distant can be detected when comparing PHROGs than when comparing initial clusters. In any case, similarities between PHROGs can provide hints, especially for unannotated PHROGs similar to annotated ones. Among annotated PHROGs, 3409 different PHROGs were similar to at least one other annotated PHROG and 52.5% of the 10 752 similarities involved PHROGs with the same annotation (83.4% with the same functional category). Moreover, among the 824 similarities for which the two PHROGs reciprocally covered each other by >60% of their length, 64.7% were involving PHROGs with the same annotation. This suggests that information provided by these more distant links could be of relevance for exploring the function of unannotated PHROGs. Indeed, among PHROGs of ‘unknown function’, for which gaining some information is essential, 3037 (48 704 proteins) were linked to annotated PHROGs (8179 links). These information are indicated in the lower right panel of each PHROG individual page on the Web page.
Further gaining some functional clues using colocalization of annotations
To further help in the annotation of PHROGs, a score of colocalization was computed and was significant for 1731 annotation pairs. For example, the 5122 and 13 119 genes in PHROGs annotated as ‘head scaffolding protein’ and ‘major head protein’ were adjacent 4690 times on the different viral genomes and the link had the maximum weight of 1000. Colocalized annotation terms often involved annotations from the same functional category (Figure 6). Some of them corresponded to known functional interactions, such as ‘integrase’ and ‘excisionase’ (24), ‘SbcC-like subunit of palindrome specific endonuclease’ and ‘SbcD-like subunit of palindrome specific endonuclease’ (25), ‘RecT-like ssDNA annealing protein’ and ‘exonuclease’ (for a review see 26) or ‘Sak4-like ssDNA annealing protein’ and ‘single strand DNA binding protein’ (27). Other, less expected colocalizations, such as ‘UvsX-like recombinase’ and ‘2OG-Fe(II) oxygenase’ or ‘gam-like host nuclease inhibitor’ and ‘Kil protein for bacterial septation inhibition’, might suggest new avenues of investigations.
Figure 6.

Each vertex represents an annotation and two annotations are linked if considered as significantly colocalized (see Materials and Methods section). Edges were attributed a weight equal to their significance score. Only the 112 most frequent annotations (used for >650 proteins) are displayed. Annotations for which genes were colocalized with genes having the same annotation are drawn as squares. Among these 112, the 21 annotations not significantly colocalized to any other are displayed on the right.
DISCUSSION
No ORFans anymore?
The PHROG database includes 17 473 genomes (or genome fragments) of viruses infecting a broad range of prokaryotic hosts (410 prokaryotic genera). The great majority of the 938 864 proteins encoded within these genomes could be assigned to a PHROG, and only 7.5% of them remained as singletons. In accordance with previous studies, these 70 524 ORFans were shorter than the other proteins, with an average length of 125.3 amino acids compared to 216.1 for all proteins. It should be noted that our cutoffs for PHROG inclusion were stringent, in particular in terms of alignment coverage (>60%), and lowering this threshold would have allowed to affiliate 23 937 more of these 70 524 singletons. These singletons similar to a PHROG might correspond to protein fusions or to proteins for which the gene prediction software wrongly predicted the beginning or end. For the remaining 46 587 proteins (4.96% of the whole dataset), a non-negligible fraction of these ORFans might not be bona fide genes but false predictions, sometimes due to errors in genomes, as single nucleotide insertions or deletions lead to spurious gene calling. Moreover, for the viruses found in RefSeq and GenBank (RefVirus), various gene prediction programs were used to annotate their genomes over the years and these programs display small differences in the genes they predict for a given genome. Thus, <5% of the proteins can be considered as real ORFans, a value much lower than what is usually described. Their great number in genomes are often described as being a specificity of viruses, with approximately 30% of a phage proteins being described to be such ORFans (28). As already mentioned, the viruses considered here were not randomly sampled, as for example viruses infecting Escherichia represent 29% of the current dataset. Yet, 25 viral families infecting 410 prokarotic genera were present and we can thus assume that ORFans are rare in viruses and that their over-representation in newly described viruses is only due to a lack of closely related reference in databases. This proportion of ORFans much smaller than previously thought helps to draw this picture on the genetic diversity in prokaryotic viruses: viral protein families are numerous and internally genetically diverse, yet these families are conserved in related viruses.
The two steps of the clustering procedure and the search for remote homology
The standard procedure usually implemented to build protein clusters consists in comparing all proteins using a sequence similarity search tool and then applying a Markov clustering algorithm to define groups (9,16). As many as 47% more clusters were defined by such a classically used procedure, compared to PHROG, which leads to an over-estimation of the number of viral protein families and thus of viral genetic diversity. The clusters delineated by the standard procedure being of a smaller size than PHROGs, the average protein coverage and identity percent inside clusters were greater for this classical procedure. Most protein pairs inside large PHROGs (>2000 proteins) had an identity percent below 30%, a threshold described as the twilight zone under which protein pairs are historically considered to have non-similar structure (29). Yet, the coverage percentages inside PHROGs were high, even for large PHROGs, and combined to the threshold of 90% on the probability of HHsearch, indicate that protein inside these PHROGs are bona fide homologs. The low amino acid percentages likely reflect the high diversity of each viral protein family and not the fact that the grouped protein have a non similar structure. Indeed, studies on particular viral protein families have shown that some proteins sharing as little as 10% amino acid identity still belonged to the same functional category (30). Thus, PHROGs are larger than traditional clusters and the coverage between proteins inside each PHROG is still good. To explain this result, we believe that the first step of the PHROG procedure, although similar to the standard clustering procedure, builds very homogeneous protein clusters as the very stringent threshold (80% of coverage for the two proteins) prevents transitivity issues. Then, the second step based on the detection of distant homologies, allows to securely connect remote homologs.
More viral proteins are now annotated thanks to the annotation of PHROGs
About 13.8% of the RefVirus proteins that were ‘hypothetical protein’ are now in annotated PHROGs. Even more proteins are newly annotated thanks to PHROGs, as a not negligible (but difficult to estimate) fraction of proteins considered as annotated in RefVirus are not informative (e.g. ‘similar to P2 orf80’). These proteins were not detected by our script as ‘hypothetical protein’ even though no real information on their putative function is known. Another positive improvement (also difficult to quantify) is the correction of existing annotations, that could be misleading, partial or wrong. For example, 3 proteins in PHROG_2, annotated as ‘terminase large subunit’, were initially annotated as ‘type I secretion target domain-containing protein’ or ‘Mu portal protein gp28 TerL’ and annotating these 3 proteins as ‘terminase large subunit’ feels more informative and legitimate. Furthermore, 263 777 proteins of the ProVirus set are now annotated in a uniform way.
Hints for unannotated PHROGs similar to annotated ones
At present, PHROG annotations are not transferred to unannotated PHROGs that are collected when an additional HHsearch comparison of PHROG profiles is run (no threshold on coverage). However, these results are shown on each PHROG web page and should provide valuable hints. Indeed, more than half of the time, annotated PHROGs similar to other annotated PHROGs have the same annotations. The two most frequent cases are PHROGs ‘tail length tape measure protein’ similar to PHROGs with the same annotation (1044 different PHROG pairs) and ‘tail fiber protein’ PHROGs similar to other ‘tail fiber protein’ PHROGs (929 cases). These two cases are coherent as (i) ‘tape measure’ have different lengths by nature and (ii) ‘tail fiber proteins’ being the point of contact with bacteria, they are prone to natural selection, and therefore highly variable in essence. For PHROGs similar to a PHROG with a different annotation, the annotations are often coherent, one being more specific than the other such as ‘tail protein’ PHROGs similar to ‘tail fiber protein’ PHROGs (433 cases). Some PHROGs from different categories are also linked because they share a functional domain, such as ‘tail length tape measure protein’ (‘tail’ category) and ‘endolysin’ (‘lysis’ category) (65 cases). These similarity results should help predict in the future the function of the 3037 unannotated PHROGs (48 704 proteins) that are similar to an annotated PHROG.
CONCLUSION
Due to the ancient origin of viruses, their gene families have a long evolutionary history and often encompass distant homologs not detected by standard sequence comparison tools such as BLAST. To be able to identify these distant homologs, our clustering strategy involved the use of HMM comparison tools as these are much more sensitive. The clusters built here proved to be larger while remaining cohesive, leading to an increase in annotated proteins. In addition, a special effort was made to standardize these annotations. Annotations of PHROG families were performed based on Refseq and several other databases, and manually curated by experts, adding a real value to the PHROG database. These annotations will be updated in the future, as new phage functions are discovered, or when experts provide ‘Suggestions’ through the dedicated page of the web site. Hopefully, PHROG will constitute a useful tool for scientists to better characterize new viral sequences, especially the millions of proteins derived from metagenomes, bringing useful insights on the nature of viruses infecting microbes.
DATA AVAILABILITY
All results are accessible on the PHROG website available at https://phrogs.lmge.uca.fr/.
Sequences, multiple alignments, HMM profiles and annotations are downloadable as zipped archives on this website. An online manual helps users compare their protein datasets to the PHROG profiles.
Supplementary Material
Contributor Information
Paul Terzian, Université Clermont Auvergne, CNRS, LMGE, F-63000 Clermont-Ferrand, France; Université Fédérale de Toulouse, INRAE, BioinfOmics, Genotoul Bioinformatics facility, 31326, Castanet-Tolosan, France.
Eric Olo Ndela, Université Clermont Auvergne, CNRS, LMGE, F-63000 Clermont-Ferrand, France.
Clovis Galiez, Univ. Grenoble Alpes, CNRS, Grenoble INP, LJK, 38000 Grenoble, France.
Julien Lossouarn, Université Paris-Saclay, INRAE, AgroParisTech, Micalis Institute, 78350, Jouy-en-Josas, France.
Rubén Enrique Pérez Bucio, Université Clermont Auvergne, CNRS, LMGE, F-63000 Clermont-Ferrand, France; Univ. Grenoble Alpes, CNRS, Grenoble INP, LJK, 38000 Grenoble, France; Centro de Ciencias Genómicas, Universidad Nacional Autónoma de México, Av. Universidad s/n, Apdo. Postal 565-A, Cuernavaca, Morelos, CP62210, Mexico.
Robin Mom, Université Clermont Auvergne, CNRS, LMGE, F-63000 Clermont-Ferrand, France; Université Clermont Auvergne, INRAE, PIAF, 63000 Clermont-Ferrand, France.
Ariane Toussaint, Cellular and Molecular Microbiology, IBMM-DBM, Université libre de Bruxelles, 6041 Gosselies, Belgium.
Marie-Agnès Petit, Université Paris-Saclay, INRAE, AgroParisTech, Micalis Institute, 78350, Jouy-en-Josas, France.
François Enault, Université Clermont Auvergne, CNRS, LMGE, F-63000 Clermont-Ferrand, France.
SUPPLEMENTARY DATA
Supplementary Data are available at NARGAB Online.
FUNDING
H2020 European Research Council [685778]. Funding for open access charge: Institut national de recherche pour l’agriculture, l’alimentation et l’environnement.
Conflict of interest statement. None declared.
REFERENCES
- 1. Breitbart M., Rohwer F.. Here a virus, there a virus, everywhere the same virus. Trends Microbiol. 2005; 13:278–284. [DOI] [PubMed] [Google Scholar]
- 2. Suttle C.A. Marine viruses — major players in the global ecosystem. Nat. Rev. Microbiol. 2007; 5:801–812. [DOI] [PubMed] [Google Scholar]
- 3. Reyes A., Haynes M., Hanson N., Angly F.E., Heath A.C., Rohwer F., Gordon J.I.. Viruses in the faecal microbiota of monozygotic twins and their mothers. Nature. 2010; 466:334–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gregory A.C., Zayed A.A., Conceição-Neto N., Temperton B., Bolduc B., Alberti A., Ardyna M., Arkhipova K., Carmichael M., Cruaud C.et al.. Marine DNA viral macro- and microdiversity from pole to pole. Cell. 2019; 177:1109–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Roux S., Adriaenssens E.M., Dutilh B.E., Koonin E.V., Kropinski A.M., Krupovic M., Kuhn J.H., Lavigne R., Brister J.R., Varsani A.et al.. Minimum information about an uncultivated virus genome (MIUVIG). Nat. Biotechnol. 2019; 37:29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Grazziotin A.L., Koonin E.V., Kristensen D.M.. Prokaryotic Virus Orthologous Groups (pVOGs): a resource for comparative genomics and protein family annotation. Nucleic Acids Res. 2017; 45:D491–D498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Huerta-Cepas J., Szklarczyk D., Heller D., Hernández-Plaza A., Forslund S.K., Cook H., Mende D.R., Letunic I., Rattei T., Jensen L.J.et al.. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019; 47:D309–D314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li L., Stoeckert C.J., Roos D.S.. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003; 13:2178–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Roux S., Hallam S.J., Woyke T., Sullivan M.B.. Viral dark matter and virus–host interactions resolved from publicly available microbial genomes. Elife. 2015; 4:e08490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Soares S.C., Geyik H., Ramos R.T., de Sá P.H., Barbosa E.G., Baumbach J., Figueiredo H.C., Miyoshi A., Tauch A., Silva A.et al.. GIPSy: Genomic island prediction software. J. Biotechnol. 2015; 232:2–11. [DOI] [PubMed] [Google Scholar]
- 11. Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J.. Basic local alignment search tool. J. Mol. Biol. 1990; 215:403–410. [DOI] [PubMed] [Google Scholar]
- 12. Steinegger M., Söding J.. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 2017; 35:1026–1028. [DOI] [PubMed] [Google Scholar]
- 13. Buchfink B., Xie C., Huson D.H.. Fast and sensitive protein alignment using DIAMOND. Nat. Methods. 2015; 12:59–60. [DOI] [PubMed] [Google Scholar]
- 14. Remmert M., Biegert A., Hauser A., Söding J.. HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods. 2012; 9:173–175. [DOI] [PubMed] [Google Scholar]
- 15. Steinegger M., Meier M., Mirdita M., Vöhringer H., Haunsberger S.J., Söding J.. HH-suite3 for fast remote homology detection and deep protein annotation. BMC Bioinformatics. 2019; 20:473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Enright A.J., Van Dongen S., Ouzounis C.A.. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002; 30:1575–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sievers F., Wilm A., Dineen D., Gibson T.J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J.et al.. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011; 7:539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Söding J. Protein homology detection by HMM–HMM comparison. Bioinformatics. 2005; 21:2144. [DOI] [PubMed] [Google Scholar]
- 19. Finn R.D., Coggill P., Eberhardt R.Y., Eddy S.R., Mistry J., Mitchell A.L., Potter S.C., Punta M., Qureshi M., Sangrador-Vegas A.et al.. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 2016; 44:D279–D285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mirdita M., von den Driesch L., Galiez C., Martin M.J., Söding J., Steinegger M.. Uniclust databases of clustered and deeply annotated protein sequences and alignments. Nucleic Acids Res. 2017; 45:D170–D176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bolduc B., Jang H.B., Doulcier G., You Z.Q., Roux S., Sullivan M.B.. vConTACT: an iVirus tool to classify double-stranded DNA viruses that infect Archaea and Bacteria. PeerJ. 2017; 5:e3243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Roux S., Enault F., Hurwitz B.L., Sullivan M.B.. VirSorter: mining viral signal from microbial genomic data. PeerJ. 2015; 3:e985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Leplae R., Lima-Mendez G., Toussaint A.. ACLAME: A CLAssification of Mobile genetic Elements, update 2010. Nucleic Acids Res. 2010; 38:D57–D61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cho E.H., Gumport R.I., Gardner J.F.. Interactions between integrase and excisionase in the phage lambda excisive nucleoprotein complex. J. Bacteriol. 2002; 184:5200–5203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Käshammer L., Saathoff J.H., Lammens K., Gut F., Bartho J., Alt A., Kessler B., Hopfner K.P.. Mechanism of DNA End Sensing and Processing by the Mre11-Rad50 Complex. Mol. Cell. 2019; 76:382–394. [DOI] [PubMed] [Google Scholar]
- 26. Caldwell B.J., Bell C.E.. Structure and mechanism of the Red recombination system of bacteriophage λ. Prog. Biophys. Mol. Biol. 2019; 147:33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hutinet G., Besle A., Son O., McGovern S., Guerois R., Petit M.A., Ochsenbein F., Lecointe F.. Sak4 of Phage HK620 Is a RecA remote homolog with single-strand annealing activity stimulated by its cognate SSB protein. Front. Microbiol. 2018; 9:743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Frost L.S., Leplae R., Summers A.O., Toussaint A.. Mobile genetic elements: the agents of open source evolution. Nat. Rev. Microbiol. 2005; 3:722–732. [DOI] [PubMed] [Google Scholar]
- 29. Rost B. Twilight zone of protein sequence alignments. Protein. Eng. 1999; 12:85–94. [DOI] [PubMed] [Google Scholar]
- 30. Lopes A., Tavares P., Petit M.A., Guérois R., Zinn-Justin S.. Automated classification of tailed bacteriophages according to their neck organization. BMC Genomics. 2014; 15:1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All results are accessible on the PHROG website available at https://phrogs.lmge.uca.fr/.
Sequences, multiple alignments, HMM profiles and annotations are downloadable as zipped archives on this website. An online manual helps users compare their protein datasets to the PHROG profiles.