

Abstract
Purpose.
The TRK pathway controls appetite, balance, and pain sensitivity. While these functions are reflected in the on-target adverse effects (AEs) observed with TRK inhibition, these AEs remain under-recognized, and pain upon drug withdrawal has not previously been reported. As TRK inhibitors are approved by multiple regulatory agencies for TRK or ROS1 fusion-positive cancers, characterizing these AEs and corresponding management strategies is crucial.
Methods.
Patients with advanced or unresectable solid tumors treated with a TRK inhibitor were retrospectively identified in a search of clinical databases. Among these patients, the frequency, severity, duration, and management outcomes of AEs including weight gain, dizziness or ataxia, and withdrawal pain were characterized.
Results.
Ninety-six patients with 15 unique cancer histologies treated with a TRK inhibitor were identified. Weight gain was observed in 53% (95% CI, 43%-62%) of patients and increased with time on TRK inhibition. Pharmacologic intervention, most commonly with GLP-1 analogs or metformin, appeared to result in stabilization or loss of weight. Dizziness, with or without ataxia, was observed in 41% (95% CI, 31%-51%) of patients with a median time to onset of 2 weeks (range, 3 days to 16 months). TRK inhibitor dose reduction was the most effective intervention for dizziness. Pain upon temporary or permanent TRK inhibitor discontinuation was observed in 35% (95% CI, 24%-46%) of patients; this was more common with longer TRK inhibitor use. TRK inhibitor re-initiation was the most effective intervention for withdrawal pain.
Conclusions.
TRK inhibition-related AEs including weight gain, dizziness, and withdrawal pain occur in a substantial proportion of patients receiving TRK inhibitors. This safety profile is unique relative to other anticancer therapies and warrants careful monitoring. These on-target toxicities are manageable with pharmacologic intervention and dose modification.
Keywords: TRK inhibitors, NTRK fusion, toxicity management
INTRODUCTION
Tyrosine kinase inhibitors with potent anti-TRK activity are approved by several regulatory agencies for the treatment of TRK or ROS1 fusion-positive cancers based on positive results in early phase trials[1–3]. Larotrectinib is a selective TRK inhibitor that is active in adult and pediatric patients with TRK fusion-positive cancers. It leads to high response rates and durable disease control[3], and is currently approved in the United States, Canada, Brazil, and Europe. Entrectinib is a multikinase inhibitor with potent activity against TRK, ROS1, and ALK that is active in TRK fusion-positive cancers and ROS1 fusion-positive lung cancers[4] and is thus approved in the United States and Japan. Next-generation TRK inhibitors such as selitrectinib, a selective TRK inhibitor, and repotrectinib, a multikinase inhibitor with activity against TRK and ROS1, are in early phase trials[1, 5–7].
On-target adverse events are occasionally observed with drugs that have potent anti-TRK activity[1, 3, 4]. Given that TRK receptors (TRKA/B/C) are intimately involved in the development and maintenance of the nervous system, the consequences of TRK inhibition are largely neurological[8–14]. In preclinical studies and congenital syndromes, decreased TRK pathway signaling results in hyperphagia and abnormal movement[14, 15]. Consistently, weight gain and dizziness have been reported in prospective trials of TRK inhibitors[3, 4]. As TRK inhibition also results in decreased nociception[16, 17], TRK inhibitor therapy withdrawal could result in heightened pain sensitivity, a phenomenon that has not systematically been reported.
While the overall side-effect profile of TRK inhibitors has been reported in prospective trials[3, 18], further characterizing the adverse events that are likely emerge secondary to TRK inhibition and defining potential management strategies for these adverse events is an unmet need. To address this, we report the frequency, clinical course, and management outcomes of common adverse events likely mediated by TRK inhibition: weight gain, dizziness, and withdrawal pain.
METHODS
Study design.
Data on patients treated in the Early Drug Development Service of Memorial Sloan Kettering Cancer Center between January 1st, 2013 and April 1st, 2019 were retrospectively analyzed with Institutional Review Board approval. Patients were eligible for inclusion if they fulfilled the following criteria: pathologic evidence of a solid tumor, advanced or unresectable disease, and treatment with at least one dose of a tyrosine kinase inhibitor with potent anti-TRK activity (defined as an IC50 < 20 nM for TRKA, TRKB, and TRKC)[1]. Demographics, toxicity assessments, and adverse event (AE) management were obtained from electronic records and pharmacy databases.
Adverse events.
AEs were graded according to the Common Terminology Criteria for Adverse Events (CTCAE) version 4.0[19]. This analysis focused on treatment-emergent AEs likely to be mediated by TRK inhibition: paresthesias, weight gain, dizziness with or without ataxia, and pain with temporary or permanent TRK inhibitor withdrawal. Any treatment-emergent AE within these 3 categories was captured and characterized in terms of grade and duration. For all AEs, grade 3 was the maximum grade as per CTCAE v4.0.
Paresthesia was defined as: grade 1, mild symptoms; grade 2, moderate symptoms that limit instrumental activities of daily living (ADL); or grade 3, severe symptoms that limit self-care ADL[19].
Weight gain was defined as: grade 1, 5% to < 10% increase from baseline for patients ≥ 18 years old and 5% to < 10% increase compared to growth curves in patients younger than 18; grade 2, 10% to < 20% increase from baseline or growth curves; or grade 3, ≥ 20% from baseline or growth curves. Baseline weight was compared to ideal body weight[19].
Dizziness was defined as: grade 1, mild unsteadiness or sensation of movement; grade 2, moderate unsteadiness or sensation of movement that limits instrumental ADL; or grade 3, severe unsteadiness or sensation of movement that limits self-care ADL[19]. Orthostatic hypotension related to drug-induced dizziness was defined as a reduction of systolic blood pressure of ≥ 20 mm Hg or a reduction of diastolic blood pressure of ≥ 10 mm Hg within 3 minutes of standing[19].
Withdrawal pain was defined as pain experienced during temporary (i.e. for surgery or radiation) or permanent (i.e. switch to a new therapy or a complete discontinuation of therapy) TRK inhibitor discontinuation. Withdrawal pain was classified as: grade 1, not limiting instrumental ADL; grade 2, limiting instrumental ADL; or grade 3, limiting self-care ADL[19].
Statistics.
Descriptive statistics were used to summarize demographics and the frequency, duration, and management of TRK inhibitor-mediated AEs. The association between patient- or treatment-related factors and the frequency of select AEs was assessed using univariate logistic regression analyses. Patient-related factors considered include baseline body mass index (BMI) for weight gain; history of brain metastasis, brain radiation, neurological AEs at baseline, seizures, and psychotropic medication use for dizziness and ataxia; and history of opioid use and TRK inhibitor duration for withdrawal pain. Two-sided P-values were calculated by Fisher’s exact test. A P-value < 0.05 was considered significant. All statistical analyses were performed using GraphPad Prism 7 (GraphPad Software, Inc).
RESULTS
Patients.
Ninety-six patients received a TRK inhibitor for the treatment of advanced cancer. The median age was 52 years (range, 5-81 years) and 51% of patients were women (Table 1). Fifteen unique cancer diagnoses were represented, the most common of which was non-small cell lung cancer (45%), followed by gastrointestinal cancers (10%), salivary gland cancer (8%), and sarcoma (8%). The most common genomic alterations involved NTRK1/2/3 (44%), ROS1 (26%), and ALK (26%). Patients received either a TRK inhibitor alone (83%) or 2 (16%) or 3 TRK inhibitors in sequence (1%).
Table 1. Demographics.
Clinicopathologic features of the study population (n=96). Categorical data are summarized as n (%) and continuous as median (range).
| Age* (years) | 52 (5-81) |
| Female sex | 49 (51%) |
| Histology | |
| Lung | 43 (45%) |
| Gastrointestinal | 10 (10%) |
| Salivary | 8 (8%) |
| Sarcoma | 8 (8%) |
| Thyroid | 6 (6%) |
| Melanoma | 6 (6%) |
| Primary brain tumor | 5 (5%) |
| Neuroblastoma | 5 (5%) |
| Other | 7 (7%) |
| Genomic alteration | |
| NTRK fusion | 39 (41%) |
| ROS1 fusion | 24 (25%) |
| Other** | 29 (30%) |
| Unknown | 4 (4%) |
| TRK inhibitor | |
| First-generation TKI | 81 (84%) |
| Other TKI | 30 (31%) |
| TRK inhibitor duration(months) | 6 (1-42) |
| TRK inhibitors received | |
| 1 | 80 (83%) |
| 2 or 3 in sequence | 16 (17%) |
| Baseline BMI*** | |
| < 30 | 64 (72%) |
| ≥ 30 | 25 (28%) |
| History of brain metastases | 34 (35%) |
| ≥ 2 lines prior systemic therapy | 54 (56%) |
7 patients were < 18years old.
Other alterations included NTRK mutation (N = 1), NTRK amplification (N = 2), ROS1 mutation (N = 1), and ALK fusion/mutation (N = 25).
Data are provided for adult patients only.
Overall safety profile.
The frequency of paresthesias, weight gain, dizziness, and withdrawal pain are summarized in Figure 1. Paresthesias were observed in 18% (17 of 96; 95% CI, 12%-29%) of patients. The frequencies of grade 1 and 2 paresthesia were 17% (16 of 96) and 1% (1 of 96), respectively; most cases were described as involving a perioral distribution and/or a “sunburn” sensation and largely improved after the first month of therapy. No cases required dose modification or escalating doses of gabapentin/pregabalin. As these symptoms were mild and did not require substantial intervention, this series focused in more detail on the AEs of weight gain, dizziness, and withdrawal pain as outlined below.
Figure 1. Neurologic adverse events observed with TRK inhibition.
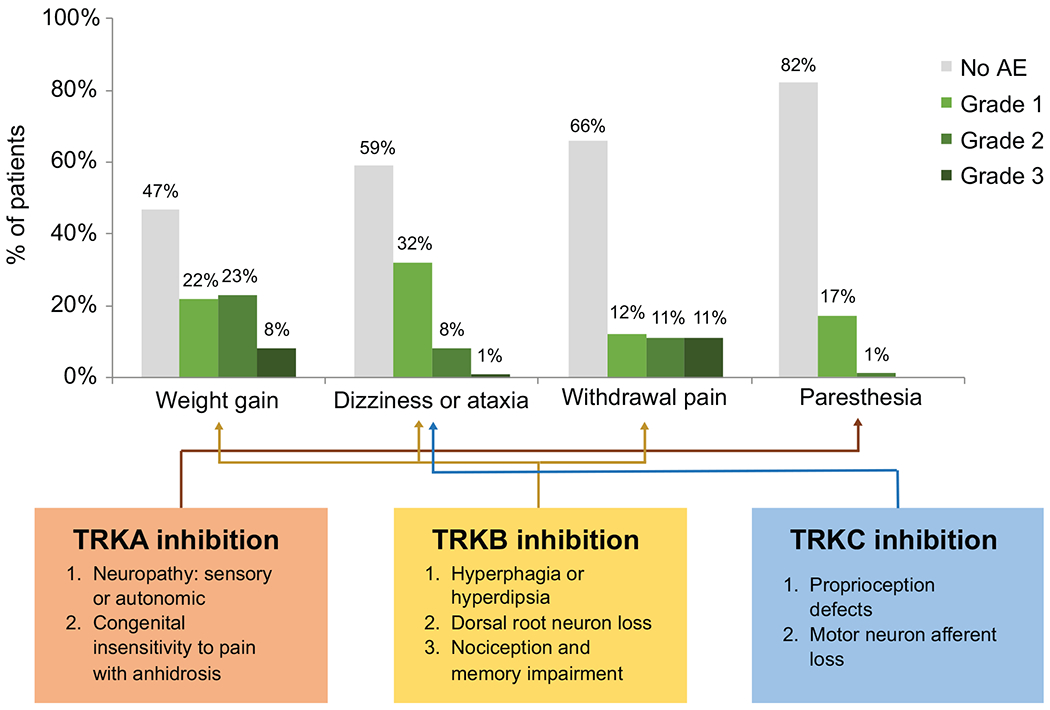
The frequency of paresthesias, weight gain, dizziness with or without ataxia, and withdrawal pain are summarized. The frequency of each adverse event is shown by the worst grade the patient experienced during therapy. For withdrawal pain, only patients who had dose interruptions and were at risk for this event were included (N = 81); all 96 patients were included in the other analyses. The effects of TRKA, TRKB, or TRKC loss in preclinical models that predict the emergence of these toxicities is summarized below.
Weight gain.
Weight gain ≥ grade 1 was observed in 53% (51 of 96; 95% CI, 45%-64%) of patients. The median time to onset of weight gain was 1 month (range, 0.2-33.4 months) and the frequency and severity of weight gain increased with time on therapy (Figure 2A). The frequencies of grade 1, grade 2, and grade 3 weight gain were 22% (21 of 96), 23% (22 of 96), and 8% (8 of 96), respectively (Figure 1). The majority (86%, 44 of 51) of patients who gained weight had a baseline weight higher than their ideal body weight (Figure 2B–D). Most patients (72%, 64 of 96) had a BMI < 30 (non-obese) (Table 1). A BMI ≥ 30 (obese) prior to TRK inhibitor initiation was not associated with a significantly greater incidence of weight gain (P = 0.62 among patients ≥ 18 years) (Table S1). Confounding factors such as worsening fluid retention (e.g. lower extremity edema) before the onset of maximal weight gain were not observed. Consistently, increased adipose tissue was the predominant radiologic finding with chronic TRK inhibitor use (Supplemental Figure 1).
Figure 2. Weight gain.
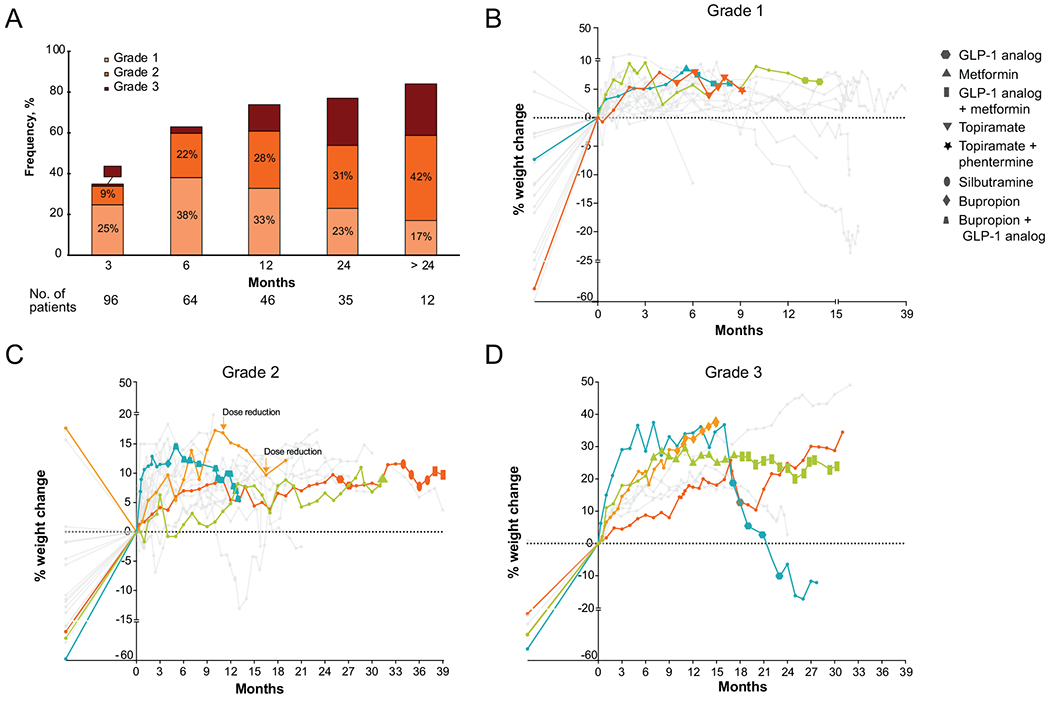
A, cumulative frequency of weight gain by worst grade over time on TRK inhibitor therapy. B-D, pattern of weight gain on therapy, separated by severity; B, grade 1; C, grade 2; D, grade 3. Points on the left side of the y-axis represent the patient’s ideal body weight plotted in reference to baseline weight, i.e. points below 0 indicate baseline weight prior to TRK inhibitor therapy is above ideal body weight. Weight trends for patients for whom pharmacologic intervention or dose reduction was recommended are plotted with colored lines, and size of data points indicates duration of treatment with corrective pharmacological intervention. TRK inhibitor dose was reduced in 1 patient with grade 2 weight gain (C).
Pharmacologic management was initiated in 10 of 51 patients (20%) who experienced weight gain. The following agents were administered as a single agent or in combination: GLP-1 analogs such as liraglutide or exenatide in 7 patients, metformin in 3, bupropion in 2, and topiramate, sibutramine, and phentermine in 1 patient each. Three patients received therapy with 2 agents after a single agent was considered ineffective: in 2, a GLP-1 analog was added to metformin, and in the other, liraglutide was added to bupropion. Overall, 8 of the 10 patients who received pharmacological interventions lost weight or stopped gaining weight, although the percentage of weight loss was generally modest: no weight loss or < 5% weight loss in 3, 5% to 10% weight loss in 2, 10-15% weight loss in 1, and > 15% in 2. Dose reduction secondary to weight gain (without concomitant pharmacologic intervention) was deemed necessary in only 1 patient whose weight gain improved from grade 2 to grade 1 after the first dose reduction and remained grade 1 after a second dose reduction (Figure 2C). While diet and exercise are cornerstones of management and were recommended in all patients with weight gain, specific dietary changes and the amount of exercise patients engaged in were not captured in the medical record. As such, the contribution of these factors could not be assessed systematically, particularly considering that there were patients who lost weight without pharmacologic intervention or dose modification in Figures 2B–D.
Dizziness.
Dizziness was observed in 41% (39 of 96; 95% CI, 31%-51%) of patients; 6 patients with dizziness developed ataxia concurrently. The frequencies of grade 1,2, and 3 dizziness were 32% (30 of 96), 8% (8 of 96), and 1% (1 of 96), respectively (Figure 1). The 6 patients with concurrent ataxia had moderate to severe symptoms (5 with grade 2 and 1 with grade 3). The median time to onset was 2 weeks (range, 3 days to 16 months) (Figure 3A). The median duration of dizziness was 5 months (range, 1 day to 40 months) from symptom onset. Of the 10 patients who received 2 or 3 TRK inhibitors in sequence, dizziness and ataxia were only observed with the second or third TRK inhibitor in 50% (5) of patients (Table S2).
Figure 3. Dizziness.
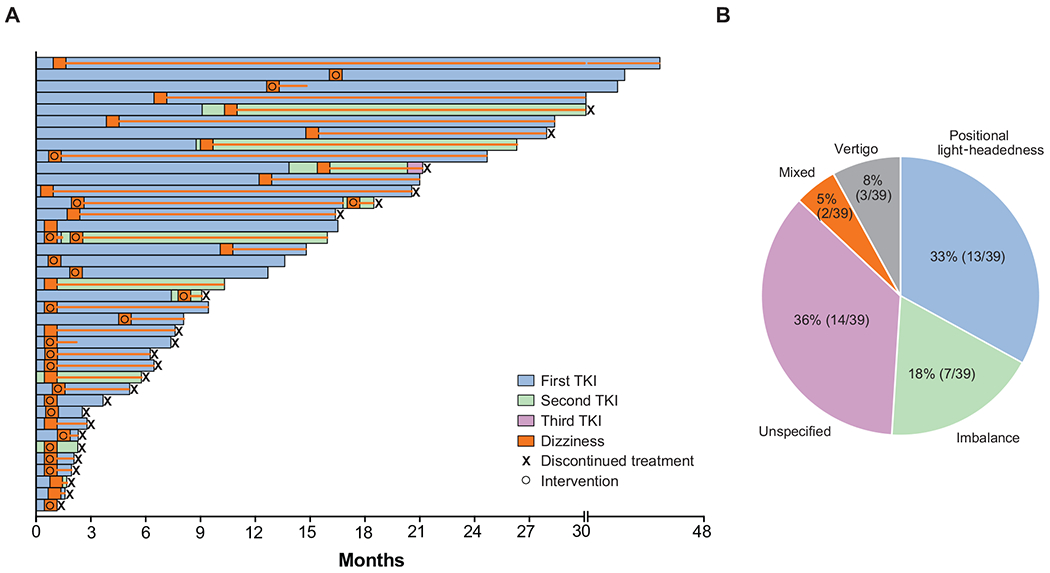
A. Swimmer’s plot of time to treatment discontinuation for all patients who developed dizziness with or without ataxia on TRK inhibitor therapy (N = 39). Each bar represents an individual patient. The orange box indicates the time of dizziness onset and the orange line indicates ongoing dizziness. B. Pie chart showing the frequency distribution of the categories of dizziness that patients experienced.
Dizziness was described as positional light-headedness in 33% (13 of 39), imbalance in 18% (7 of 39), vertigo in 8% (3 of 39), or mixed (positional light-headedness, imbalance, and/or vertigo) in 5% (2 of 39) of patients (Figure 3B). Dizziness was associated with orthostatic hypotension in 21% (8 of 39) of patients, none of whom showed signs of frank volume depletion. Among potential risk factors for increased neurotoxicity, including neurological symptoms present at baseline, brain metastasis/radiation history, seizure history, and psychotropic use, only having a dizziness, ataxia, peripheral sensory neuropathy, or headache at baseline was significantly associated with dizziness/ataxia (P < 0.05) (Table S1).
Intervention was required in 21 of 39 patients (54%, Figure 3A): pharmacologic intervention alone in 12 patients, dose reduction alone in 6 patients, and pharmacologic intervention and dose reduction in 3 patients. The most effective intervention was dose reduction (5 for orthostatic hypotension and 4 for ataxia) which led to symptom resolution in 7 of 9 patients (78%). Treatment was not discontinued secondary to dizziness or ataxia for any patient, nor was hospitalization required.
Pharmacologic intervention alone resulted in modest benefit. Meclizine was administered to 12 patients (3 with positional light-headedness, 3 with imbalance, and 1 with vertigo), and led to improvement in symptoms in 5 (45%), including 3 with qualitative improvement within the same grade and 2 for whom it lowered severity from grade 2 to grade 1. Of the 8 patients with orthostatic hypotension, 4 received midodrine and/or fludrocortisone. All 4 had minor symptom improvement within the same grade and 3 had improvement of standing blood pressure (increase by 8 mm Hg systolic and 4 mm Hg diastolic). Only a subsequent dose reduction in all 4 patients resulted in resolution or substantial symptom improvement from grade 2 or 3 to grade 1.
Withdrawal pain.
Withdrawal pain was observed in 34% (28 of 81) of patients who discontinued TRK inhibitor therapy during their course (Figure 1). Discontinuation was temporary (i.e. for surgery, radiation or switch to a different TRK inhibitor) in 83% (23 of 28) of patients and permanent (e.g. to switch to another therapy) in 45% (13 of 28); 28% (8 of 28) of patients who temporarily discontinued therapy later permanently discontinued therapy. Symptoms were described as full-body ache, muscle pain, and/or allodynia, occasionally accompanied by a headache; concurrent flares of pre-existing cancer pain, when present, were not observed. The median time to onset was 2 days (range, 1-6 days). The most severe degree of withdrawal pain was grade 1, 2, and 3 in 12% (10 of 81), 11% (9 of 81), and 11% (9 of 81) of patients, respectively (Figures 1 and 4). Hospitalization for the management of withdrawal pain was not required for any patient.
Figure 4. Withdrawal pain.
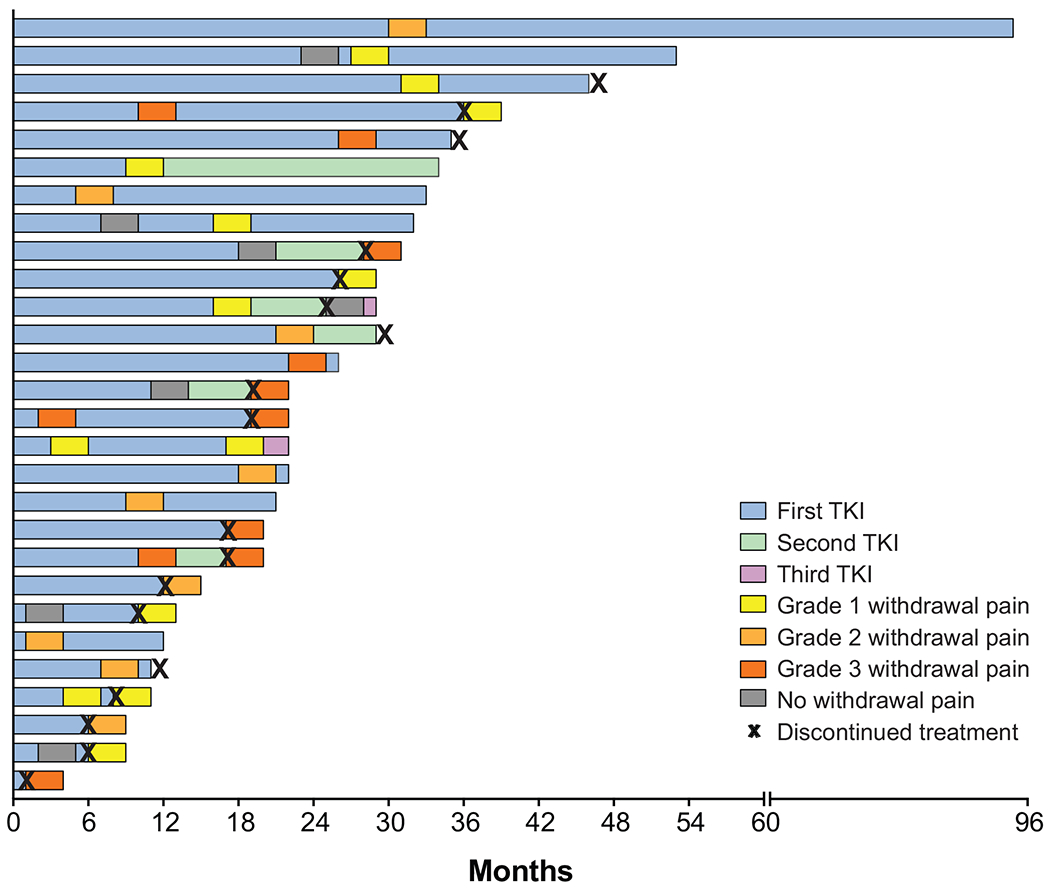
Swimmer’s plot of time to treatment discontinuation for all patients who developed withdrawal pain on TRK inhibitor therapy (N = 28). Each bar represents an individual patient.
In patients who permanently discontinued therapy, the median duration of withdrawal pain was 14 days (range, 10-26 days). In patients who temporarily discontinued therapy, the median duration of flare was 3 days (range, 1-7 days) which was consistent with the median duration of the drug hold which was 3 days (range, 1-7 days). Patients who had been on a TRK inhibitor for longer than 6 months (32 of 96, 33%) were more likely to experience withdrawal pain (63% vs 13%; P < 0.05). Taking opioids for cancer-related pain immediately prior to TRK inhibitor interruption did not affect the risk of withdrawal pain (P = 0.43) (Table S1).
Pharmacologic intervention was required in 46% (13 of 28) of patients. Of these 13 patients who received opioid or non-opioid analgesics without resuming the TRK inhibitor, benefit was only observed in 3 (23%): 2 had qualitative improvement in pain within the same grade and only 1 had complete pain resolution. Three patients received gabapentin; no benefit was observed. In contrast, all patients that resumed TRK inhibitor use (15 resumed the original TKI, 4 switched to a new TKI) achieved resolution of withdrawal pain within a median of 1 day from TRK inhibitor initiation. Many noted that the pain completely resolved within a few hours of their first dose.
Given this observation, the utility of a tapering TRK inhibitor dose was investigated in 2 patients who were to permanently discontinue therapy; both had a history of severe withdrawal pain with previous temporary interruptions. For the first patient, the dose was tapered rapidly (50% decrease every 2 days) with minimal impact; the patient had the same grade of withdrawal pain (grade 3) as with prior interruptions (Figure 4). Thus, with the second patient, the dose was tapered more slowly (25% every 7 days). This resulted in a much lower severity of withdrawal pain (grade 1) compared to prior pain flares (grade 3).
DISCUSSION
The neurological consequences of TRK inhibitor therapy remain underrecognized. This is unsurprising as these agents entered clinical testing in 2012 and were first approved in 2018[20–22]. This series provides a comprehensive description of the frequency and time course of the major on-target adverse consequences of TRK inhibition or TRK inhibitor withdrawal, recognizing that other less-common neurologic AEs such as cognitive impairment, mood disorders, sleep disturbance, and dysarthria have been reported prospectively[2–4]. In particular, it is the first to systematically describe the syndrome of pain flares that patients can experience with drug interruption or discontinuation. The outcomes of treatment strategies to address these events are characterized, providing a springboard for creating formal guidelines that, to date, do not exist.
Weight gain, which is not a commonly observed adverse effect of anticancer therapy, was observed in > 50% of patients in this series. This finding is not unexpected as the TRKB pathway regulates appetite centers[14, 15, 23], and TRKB loss was known to result in hyperphagia in mice well before TRK inhibitors entered clinical testing. In our series, the frequency and severity of weight gain increased over time, highlighting the need to recognize this complication early. This pattern is consistent with weight gain resulting from adipose accumulation; similarly, adipose hypertrophy has been observed in mice with decreased TRKB signaling[14].
The management of weight gain secondary to TRK inhibition was largely informed by approaches developed in other settings, such as antipsychotic-induced weight gain. While diet and exercise remain management cornerstones, these are not routinely quantified or described in the medical record and are challenging to evaluate, as is evidenced by this series. Furthermore, these may not be sufficient, calling for a systematic approach to pharmacological intervention. FDA-approved medications for weight management include phentermine-topiramate, liraglutide, naltrexone-bupropion, lorcaserin, and orlistat (Table 2)[24, 25]. While liraglutide is the only GLP-1 receptor agonist FDA-approved for weight loss, other members of this diabetes drug class, including semaglutide, dulaglutide, lixisenatide, and exenatide, have similar or greater effects and have been used off-label[26]. Metformin, while not FDA-approved for weight management, also appears to cause modest weight loss[27]. Guidelines do not offer a clear order of preference, underscoring the need to develop care algorithms. We recommend that (1) weight be monitored serially, (2) pharmacologic management be considered for ≥ grade 2 weight gain (particularly if ideal body weight is exceeded), (3) dose reduction be considered for persistent ≥ grade 2 weight gain despite maximal supportive care, and (3) difficult cases be referred to an endocrine or weight loss clinic. Further studies and long-term follow-up are warranted to define the role of pharmacological management and other interventions.
Table 2. Supportive medication.
Agents approved by the US FDA or recommended in clinical guidelines for the management of weight gain, dizziness, and pain24–27,30–34.
| Adverse event | Agent(s) | Mechanism of action | Dose and schedule |
|---|---|---|---|
| Weight gain | Liraglutide | GLP-1 analog | 0.6 to 3.0 mg 1x/day |
| Orlistat | Inhibits fat absorption | 60 to 120 mg 3x/day | |
| Phentermine/topiramate combination | Increases norepinephrine release; GABA receptor agonist | 3.75/23 to 15/92 mg 1x/day | |
| Lorcaserin | 5-HT2C receptor agonist | 10 mg 2x/day | |
| Naltrexone/bupropion combination | μ-opioid receptor antagonist; dopamine and norepinephrine reuptake inhibitor | 8/90 to 16/180 mg 1x or 2x/day | |
| Metformin | Modulates hypothalamic appetite regulatory centers | 500 to 2000 mg 1x/day | |
| Dizziness (ataxia or vertigo) | Meclizine | H1 histamine receptor antagonist, suppresses vestibular stimulation, anticholinergic | 25 to 50 mg 1x/day |
| Scopolamine | Antagonizes histamine and serotonin | 1 patch every 3 days | |
|
Dizziness (orthostasis) |
Midodrine | α1 adrenergic receptor agonist, increases vascular tone | 5 to 10 mg 3x/day |
| Fludrocortisone | Mineralocorticoid | 0.05-0.2 mg 1x/day | |
| Droxidopa | Metabolized to norepinephrine, induces vasoconstriction | 100 mg 3x/day (1.8 g/day maximum) | |
| Withdrawal pain | Non-steroidal anti-inflammatory agents | COX-1/COX-2 inhibitors | Per agent/label |
| Opioids | Opioid receptor agonists | Per label | |
| Gabapentin/pregabalin | GABA analog | Per label | |
Dizziness was observed in approximately 40% of patients, a subset of whom also had ataxia. These changes in proprioception are similarly linked to TRKB/C inhibition, as mice with TRKB ligand loss develop severe ataxia secondary to cerebellar insufficiency, and TRKC knockout mice exhibit abnormal movements and postures secondary to proprioceptive insufficiency, similar to motion sickness[8, 12, 28, 29]. Further, several patients in this series had orthostatic hypotension without signs of frank volume depletion, providing the first indication that TRK inhibition could result in clinically significant autonomic dysfunction. Thus, the root cause of dizziness should be investigated to distinguish central from autonomic effects. While antihistamines can be used for vestibular/proprioceptive insufficiency, and mineralocorticoids or vasopressors for orthostasis[30–34], these agents did not lead to dramatic improvement in several patients, and dose reduction may be necessary. We recommend that (1) “dizziness” be characterized more concretely in practice and trials, (2) patients be matched to appropriate agents based on this characterization (Table 2), (3) dose reduction be considered for intolerable dizziness unresponsive to pharmacologic management, and (4) refractory cases be referred to neurology.
TRK inhibitor withdrawal pain has not been reported elsewhere. Pain flares can be highly disturbing to patients and providers and are often misattributed to other etiologies. The exact mechanism of this phenomenon is unclear, although TRK inhibition likely modulates pain thresholds, leading to increased sensitivity when withdrawn[35]. Consistently, TRKA loss can result in congenital insensitivity to pain[17]. TRKA-mediated signaling results in a rapid increase in the expression of the transient receptor potential vanilloid I (TRPV1) channel, an important nociceptive mediator; TRK inhibitor withdrawal may result in rebound nociceptive TPRV1 expression[36, 37].
Interestingly, the most effective method to address withdrawal pain was resumption of TRK inhibition. Of patients who received opioid or non-opioid analgesics, < 20% had pain improvement and < 10% had complete pain resolution. In contrast, all patients had complete and rapid pain resolution with TRK inhibitor re-administration, and gradual dose tapering appeared to reduce withdrawal pain in one patient, whereas rapid dose reduction in a separate patient did not. We thus recommend (1) avoiding or minimizing TRK inhibitor interruption, (2) adjunctively managing pain (Table 2) when discontinuation is necessary, and (3) considering a slow tapering dose schedule for patients who need to permanently discontinue therapy.
In conclusion, on-target consequences of TRK inhibition such as weight gain, dizziness with or without ataxia, and withdrawal pain occur in a substantial proportion of patients and should be carefully monitored in the clinic. These are manageable with pharmacologic intervention and/or dose modification.
Supplementary Material
Highlights:
TRK inhibitors have a unique on-target side effect profile.
Weight gain, dizziness/ataxia, and paresthesias should be monitored on therapy.
In addition, withdrawal pain can occur in patients who temporarily or permanently discontinue treatment.
These side effects are manageable with pharmacologic intervention and/or dose modification..
ACKNOWLEDGMENTS
The authors thank Jessica Moore, MS, staff editor at Memorial Sloan Kettering Cancer Center, for editing the manuscript.
FUNDING
This research was supported by the National Cancer Institute of the US National Institutes of Health (P30 CA008748, 1R01CA226864-01A1) and by Nonna’s Garden Foundation.
DISCLOSURES
JF has received honoraria from Genentech. AL has received research funding from Bristol Myers Squibb and NantOmics and holds equity in Sanofi. MO has served on advisory boards for PharmaMar, Novartis, and Targeted Oncology, and received travel expenses from Bristol Myers Squibb and Merck. YRM-G has received travel expenses from AstraZeneca. BTL holds two institutional patents (US62/685,057, US62/514,661), and has served on advisory boards for Genentech (subsidiary of Roche), Eli Lilly, Guardant Health, Hengrui Therapeutics, Mersana Therapeutics, and Thermo Fisher Scientific, received travel expenses from MORE Health and Resolution Bioscience, and received research funding from Amgen, AstraZeneca, BioMedValley Discoveries, Daiichi Sankyo, Genentech, GRAIL, Guardant Health, Eli Lilly, Hengrui Therapeutics, Illumina, and MORE Health. JJH has received consulting fees from Bristol Myers Squibb, Cytomx, Eli Lilly, Eisai, Exelixis, Imvax, QED, and research funding from Bristol Myers Squibb. GI has received research funding from Janssen, Mirati Therapeutics, and Novartis, and consulting fees from Mirati Therapeutics. MMG has received honoraria from Bayer and Flatiron Health, served on advisory boards for Bayer, Boehringer Ingelheim, Epizyme, Daiichi Sankyo, Karyopharm, and Springworks Therapeutics, served on a speakers’ bureau for Amgen, and received travel expenses from Epizyme. AS has received research funding from Eli Lilly, Kura Oncology, Merus, Northern Biologics, and Surface Oncology. DMH owns stock in Fount Therapeutics, has served on advisory boards for ArQule, AstraZeneca, Bayer, Boehringer Ingelheim, Chugai Pharma, CytomX, Debiopharm, Fount Therapeutics, Genentech, Eli Lilly, Janssen, Jazz Pharma, Pfizer, and Puma Biotechnology, received research funding from AstraZeneca, Bayer, Loxo Oncology, and Puma Biotechnology, and received travel expenses from Chugai Pharma and Genentech; he is currently employed by and holds equity in Loxo Oncology, a subsidiary of Eli Lilly. AD has received honoraria and/or served on advisory boards for 14ner/Elevation Oncology, Abbvie, ArcherDX, AstraZeneca, Axis Pharma, Bayer, Beigene, BergenBio, Blueprint Medicines, Exelixis, Helsinn, Hengrui Therapeutics, Loxo Oncology (subsidiary of Eli Lilly), Monopteros, MORE Health, Remedica, Roche (subsidiaries Genentech and Ignyta), Pfizer, Takeda (subsidiaries Ariad and Millennium), TP Therapeutics, Tyra Biosciences, and Verastem; and received research funding from Exelixis, Foundation Medicine GlaxoSmithKline, Pfizer, PharmaMar, Teva, and Taiho. All research funding was provided to the institution. All other authors report no financial relationships.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Cocco E, Scaltriti M, Drilon A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat Rev Clin Oncol 2018; 15: 731–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Demetri GD, Paz-Ares L, Farago AF et al. Efficacy and safety of entrectinib in patients with NTRK fusion-positive tumours: Pooled analysis of STARTRK-2, STARTRK-1, and ALKA-372-001. Annals of Oncology 2018; 29. [Google Scholar]
- 3.Drilon A, Laetsch TW, Kummar S et al. Efficacy of Larotrectinib in TRK Fusion-Positive Cancers in Adults and Children. N Engl J Med 2018; 378: 731–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Drilon A, Siena S, Ou SI et al. Safety and Antitumor Activity of the Multitargeted Pan-TRK, ROS1, and ALK Inhibitor Entrectinib: Combined Results from Two Phase I Trials (ALKA-372-001 and STARTRK-1). Cancer Discov 2017; 7: 400–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hyman D, Kummar S, Farago A et al. Phase I and expanded access experience of LOXO-195 (BAY 2731954), a selective next-generation TRK inhibitor (TRKi). Cancer Res 2019; 79: CT127. [Google Scholar]
- 6.Drilon A, Ou SI, Cho BC et al. Repotrectinib (TPX-0005) Is a Next-Generation ROS1/TRK/ALK Inhibitor That Potently Inhibits ROS1/TRK/ALK Solvent-Front Mutations. Cancer Discov 2018; 8: 1227–1236. [DOI] [PubMed] [Google Scholar]
- 7.Drilon A, Nagasubramanian R, Blake JF et al. A Next-Generation TRK Kinase Inhibitor Overcomes Acquired Resistance to Prior TRK Kinase Inhibition in Patients with TRK Fusion-Positive Solid Tumors. Cancer Discov 2017; 7: 963–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Snider WD. Functions of the neurotrophins during nervous system development: what the knockouts are teaching us. Cell 1994; 77: 627–638. [DOI] [PubMed] [Google Scholar]
- 9.Barbacid M The Trk family of neurotrophin receptors. J Neurobiol 1994; 25: 1386–1403. [DOI] [PubMed] [Google Scholar]
- 10.Smeyne RJ, Klein R, Schnapp A et al. Severe sensory and sympathetic neuropathies in mice carrying a disrupted Trk/NGF receptor gene. Nature 1994; 368: 246–249. [DOI] [PubMed] [Google Scholar]
- 11.Klein R, Smeyne RJ, Wurst W et al. Targeted disruption of the trkB neurotrophin receptor gene results in nervous system lesions and neonatal death. Cell 1993; 75: 113–122. [PubMed] [Google Scholar]
- 12.Klein R, Silos-Santiago I, Smeyne RJ et al. Disruption of the neurotrophin-3 receptor gene trkC eliminates la muscle afferents and results in abnormal movements. Nature 1994; 368: 249–251. [DOI] [PubMed] [Google Scholar]
- 13.Crowley C, Spencer SD, Nishimura MC et al. Mice lacking nerve growth factor display perinatal loss of sensory and sympathetic neurons yet develop basal forebrain cholinergic neurons. Cell 1994; 76: 1001–1011. [DOI] [PubMed] [Google Scholar]
- 14.Lin JC, Tsao D, Barras P et al. Appetite enhancement and weight gain by peripheral administration of TrkB agonists in non-human primates. PLoS One 2008; 3: e1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yeo GS, Connie Hung CC, Rochford J et al. A de novo mutation affecting human TrkB associated with severe obesity and developmental delay. Nat Neurosci 2004; 7: 1187–1189. [DOI] [PubMed] [Google Scholar]
- 16.Indo Y, Tsuruta M, Hayashida Y et al. Mutations in the TRKA/NGF receptor gene in patients with congenital insensitivity to pain with anhidrosis. Nat Genet 1996; 13: 485–488. [DOI] [PubMed] [Google Scholar]
- 17.Greco A, Villa R, Fusetti L et al. The Gly571Arg mutation, associated with the autonomic and sensory disorder congenital insensitivity to pain with anhidrosis, causes the inactivation of the NTRK1/nerve growth factor receptor. J Cell Physiol 2000; 182: 127–133. [DOI] [PubMed] [Google Scholar]
- 18.Doebele RC, Drilon A, Paz-Ares L et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: integrated analysis of three phase 1-2 trials. Lancet Oncol 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Common Terminology Criteria for Adverse Events (CTCAE). In. 2010.
- 20.Administration USFaD. FDA approves larotrectinib for solid tumors with NTRK gene fusions. In. 2018.
- 21.Administration USFaD. FDA approves entrectinib for NTRK solid tumors and ROS-1 NSCLC. In. 2019.
- 22.Agency EM. First ‘histology-independent’ treatment for solid tumours with a specific gene mutation. In. EMA Press office; 2019. [Google Scholar]
- 23.Rios M, Fan G, Fekete C et al. Conditional deletion of brain-derived neurotrophic factor in the postnatal brain leads to obesity and hyperactivity. Mol Endocrinol 2001; 15: 1748–1757. [DOI] [PubMed] [Google Scholar]
- 24.Igel LI, Kumar RB, Saunders KH, Aronne LJ. Practical Use of Pharmacotherapy for Obesity. Gastroenterology 2017; 152: 1765–1779. [DOI] [PubMed] [Google Scholar]
- 25.Apovian CM, Aronne LJ, Bessesen DH et al. Pharmacological management of obesity: an endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2015; 100: 342–362. [DOI] [PubMed] [Google Scholar]
- 26.Holst JJ. Incretin therapy for diabetes mellitus type 2. Curr Opin Endocrinol Diabetes Obes 2019. [DOI] [PubMed] [Google Scholar]
- 27.Flory J, Lipska K. Metformin in 2019. JAMA 2019; 321: 1926–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen AI, Nguyen CN, Copenhagen DR et al. TrkB (tropomyosin-related kinase B) controls the assembly and maintenance of GABAergic synapses in the cerebellar cortex. J Neurosci 2011; 31: 2769–2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Richardson CA, Leitch B. Phenotype of cerebellar glutamatergic neurons is altered in stargazer mutant mice lacking brain-derived neurotrophic factor mRNA expression. J Comp Neurol 2005; 481: 145–159. [DOI] [PubMed] [Google Scholar]
- 30.Low PA, Singer W. Management of neurogenic orthostatic hypotension: an update. Lancet Neurol 2008; 7: 451–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cohen B, DeJong JM. Meclizine and placebo in treating vertigo of vestibular origin. Relative efficacy in a double-blind study. Arch Neurol 1972; 27: 129–135. [DOI] [PubMed] [Google Scholar]
- 32.Chobanian AV, Volicer L, Tifft CP et al. Mineralocorticoid-induced hypertension in patients with orthostatic hypotension. N Engl J Med 1979; 301: 68–73. [DOI] [PubMed] [Google Scholar]
- 33.Davies IB, Bannister RG, Sever PS, Wilcox CS. Fludrocortisone in the treatment of postural hypotension: altered sensitivity to pressor agents [proceedings]. Br J Clin Pharmacol 1978; 6: 444P–445P. [PubMed] [Google Scholar]
- 34.Lanier JB, Mote MB, Clay EC. Evaluation and management of orthostatic hypotension. Am Fam Physician 2011; 84: 527–536. [PubMed] [Google Scholar]
- 35.Hirose M, Kuroda Y, Murata E. NGF/TrkA Signaling as a Therapeutic Target for Pain. Pain Pract 2016; 16: 175–182. [DOI] [PubMed] [Google Scholar]
- 36.Indo Y Molecular basis of congenital insensitivity to pain with anhidrosis (CIPA): mutations and polymorphisms in TRKA (NTRK1) gene encoding the receptor tyrosine kinase for nerve growth factor. Hum Mutat 2001; 18: 462–471. [DOI] [PubMed] [Google Scholar]
- 37.Zhang X, Huang J, McNaughton PA. NGF rapidly increases membrane expression of TRPV1 heat-gated ion channels. EMBO J 2005; 24: 4211–4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.