

Abstract
Purpose:
Ocular coloboma is caused by failure of optic fissure closure during development and recognized as part of the microphthalmia, anophthalmia, and coloboma (MAC) spectrum. While many genes are known to cause colobomatous microphthalmia, relatively few have been reported in coloboma with normal eye size.
Methods:
Genetic analysis including trio exome sequencing and Sanger sequencing was undertaken in a family with two siblings affected with bilateral coloboma of the iris, retina, and choroid.
Results:
Pathogenic variants in MAC genes were excluded. Trio analysis identified compound heterozygous donor splice site variants in CDON, a cell-surface receptor known to function in the Sonic Hedgehog pathway, c.928+1G>A and c.2650+1G>T, in both affected individuals. Heterozygous missense and truncating CDON variants are associated with dominant holoprosencephaly (HPE) with incomplete penetrance and Cdon−/− mice display variable HPE and coloboma. A homozygous nonsense allele of uncertain significance was recently identified in a consanguineous patient with coloboma and a second molecular diagnosis.
Conclusions:
We report the first compound heterozygous variants in CDON as a cause of isolated coloboma. CDON is the first HPE gene identified to cause recessive coloboma. Given the phenotypic overlap, further examination of HPE genes in coloboma is indicated.
Keywords: coloboma, CDON, recessive, splicing, dual diagnosis
Graphical Abstract
INTRODUCTION
Coloboma is a gap in one or more tissues in the eye, typically caused by failure of optic fissure closure during development. In many cases, additional ocular anomalies such as microphthalmia or anophthalmia (in the contralateral eye) are also present, leading to the designation of MAC spectrum. While more than 82 genes have been associated with MAC spectrum disorders (1), only a subset have been reported to cause nonsyndromic/mildly syndromic coloboma with normal eye size, including heterozygous variants in YAP1, MAB21L2, and ABCB6, and recessive variants in SALL2 (2–5) while variants in other MAC genes such as SOX2, OTX2, PAX6, and ALDH1A3 have occasionally been reported in patients with coloboma with normal eye size (6). Heterozygous variants in RAX, BMP7, GDF6, and GDF3 may play a role in isolated coloboma with incomplete penetrance, since many alleles are also seen in control populations (1). Most cases with isolated coloboma remain unexplained genetically.
CDON (OMIM:608707) is a cell-surface receptor known to function in the Sonic Hedgehog (SHH; OMIM:600725) pathway (7, 8). Heterozygous missense and nonsense variants have been reported in multiple patients with holoprosencephaly (HPE) and pituitary stalk interruption syndrome, with incomplete penetrance noted in many families (9–13). In addition to variable strain-specific holoprosencephaly, careful examination of ocular structures in Cdon−/− knockout mice revealed multiple ocular anomalies including failure of optic fissure closure resulting in coloboma. More recently, homozygous variants in CDON and MAPRE2 (OMIM:605789) were identified in a single child of a consanguineous couple affected with retinal coloboma, developmental delay, dysmorphic features, pyloric stenosis, and circumferential skin creases; the authors note that while the majority of features were consistent with the MAPRE2 variant, the CDON variant may have contributed to the coloboma. However, since previously reported cases with MAPRE2 variants displayed microphthalmia (14), a role for this gene in coloboma could not be ruled out.
In this study, we present confirmation of the role of CDON in human coloboma through identification of the first compound heterozygous donor splice site variants in two siblings with ocular coloboma.
METHODS
This human study was approved by the Institutional Review Board of Children’s Wisconsin and adhered to the tenets of the Declaration of Helsinki. Written informed consent was obtained for every participant.
Ocular images were taken using a fundus camera (Topcon; Tokyo, Japan) or a widefield fundus camera (Optos; Dunfermline, Scotland).
Trio exome sequencing was completed on the proband and both parents by Psomagen (Rockville, MD) as previously described (15). Data was analyzed with SNP and Variation Suite (Golden Helix; Bozeman, MT) using gnomAD tracts for general population frequency and dbNSFP for in silico predictions (16, 17). Constraint data was accessed directly from gnomAD (https://gnomad.broadinstitute.org, (16)). Predicted effect on splicing was analyzed using Human Splicing Finder at https://www.genomnis.com/access-hsf (18), MaxEntScan at http://hollywood.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq.html (19), and/or Alternative Splice Site Predictor at http://wangcomputing.com/assp/index.html (20). Confirmation of variants in the proband and parents and co-segregation analysis in three siblings was completed through Sanger sequencing using the following primer pairs: F- GACTATTCTGCCTTTAGTTGTC and R- GCAATAACACATACACCATTCC (c.2650+1G>T) and F-CACACAGATATAGGCTAGATTCTTCC and R-ATGCCCACAGGTTTGCTAAG (c.928+1G>A).
RESULTS
The proband (Patient 1A) is a 17-year-old male with bilateral coloboma and a 47,XYY karyotype. Ophthalmologic exam identified bilateral inferior coloboma of the iris, retina, and choroid (Figure 1A and B) and severe myopia (−24.50 OD; −21.50 OS). Visual acuity was 20/150 OD and 20/100 OS. Physical exam at 15 years of age identified mildly dysmorphic facial features with elongated face, malar flattening, long columella, narrow nasal bridge, upslanting palpebral fissures, overfolded/pinched outer helix and possible velopharyngeal insufficiency along with shortened arms and brachydactyly. He has normal growth with weight of 61.2 kg (50–75%ile) and height of 162.5 cm (10th centile). Lumbar vertebral hypoplasia resulting in kyphosis was treated with a brace. He has a history of conductive hearing loss due to eustachian tube dysfunction, hypotonia, and developmental delay as a child but current cognitive abilities appear to be in the low normal range. Brain MRI was performed at 7 months of age with normal results.
Figure 1: Ocular images from affected patients.
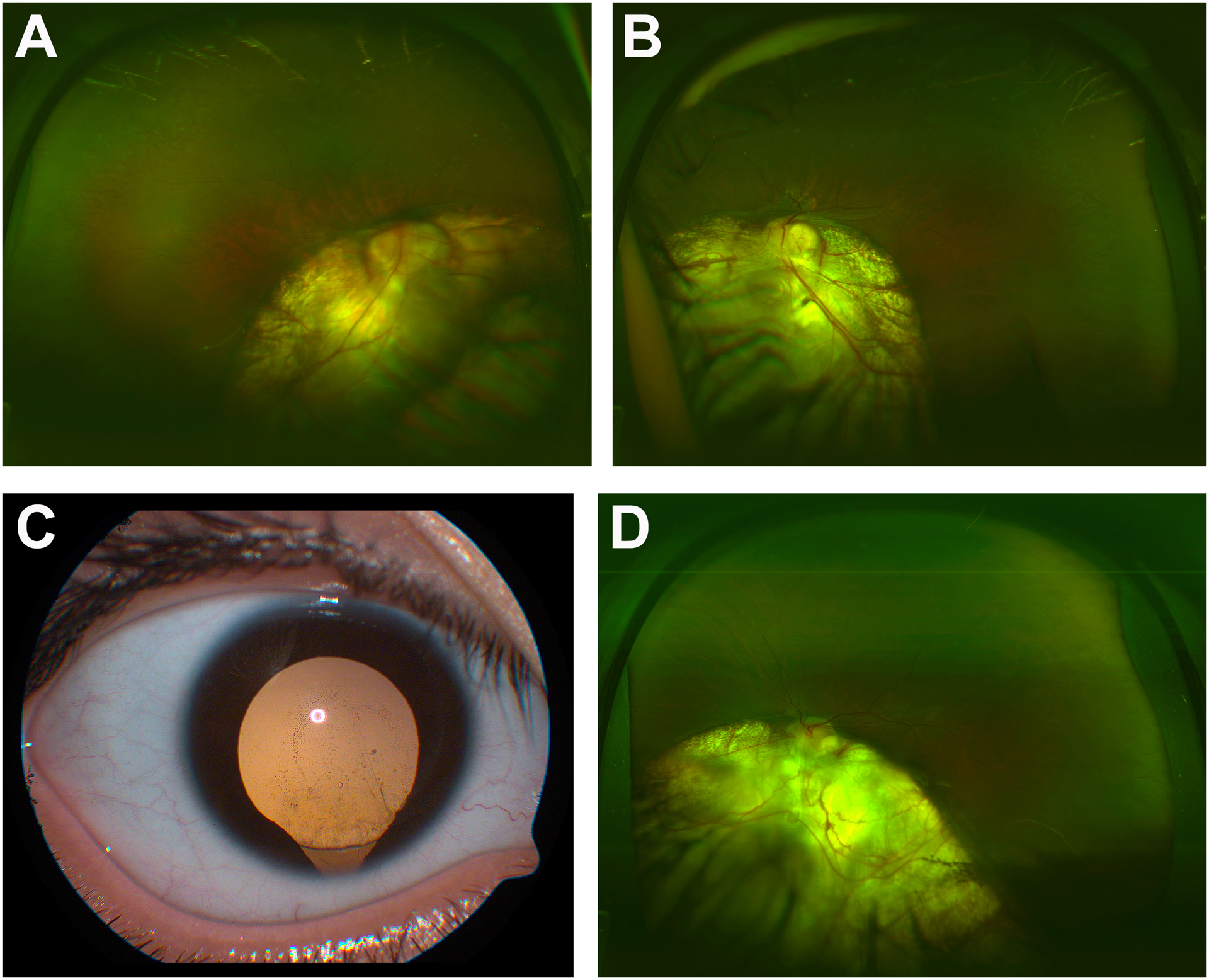
Fundus photos of the right eye (A) and left eye (B) of Patient 1A showing large inferonasal chorioretinal colobomas involving both optic nerves. Anterior segment photo (C) of Patient 1B (right eye at 6 years old) showing inferior coloboma of the iris and zonules and fundus photo (D) (left eye at 14 years old) showing large inferonasal chorioretinal coloboma adjacent to the inferior pole of the optic nerve. The right eye had a similar appearance prior to the development of a retinal detachment.
Both parents are from Mexico and there is no consanguinity. Patient 1A has six siblings (Figure 2A); only his oldest sister (19 years old; Patient 1B) has similar ocular findings of bilateral inferior coloboma of the iris (Figure 1C), retina, and choroid (Figure 1D) and high myopia (−9.25 OS). Visual acuity was no perception of light OD due to a chronic retinal detachment and 20/150 OS. At 6 years of age she was discovered to have a rhegmatogenous retinal detachment in her right eye which failed multiple attempts at surgical repair. She has no other health concerns and received services in school for visual impairment but is reported to have normal intelligence.
Figure 2: Pedigree and sequencing results.
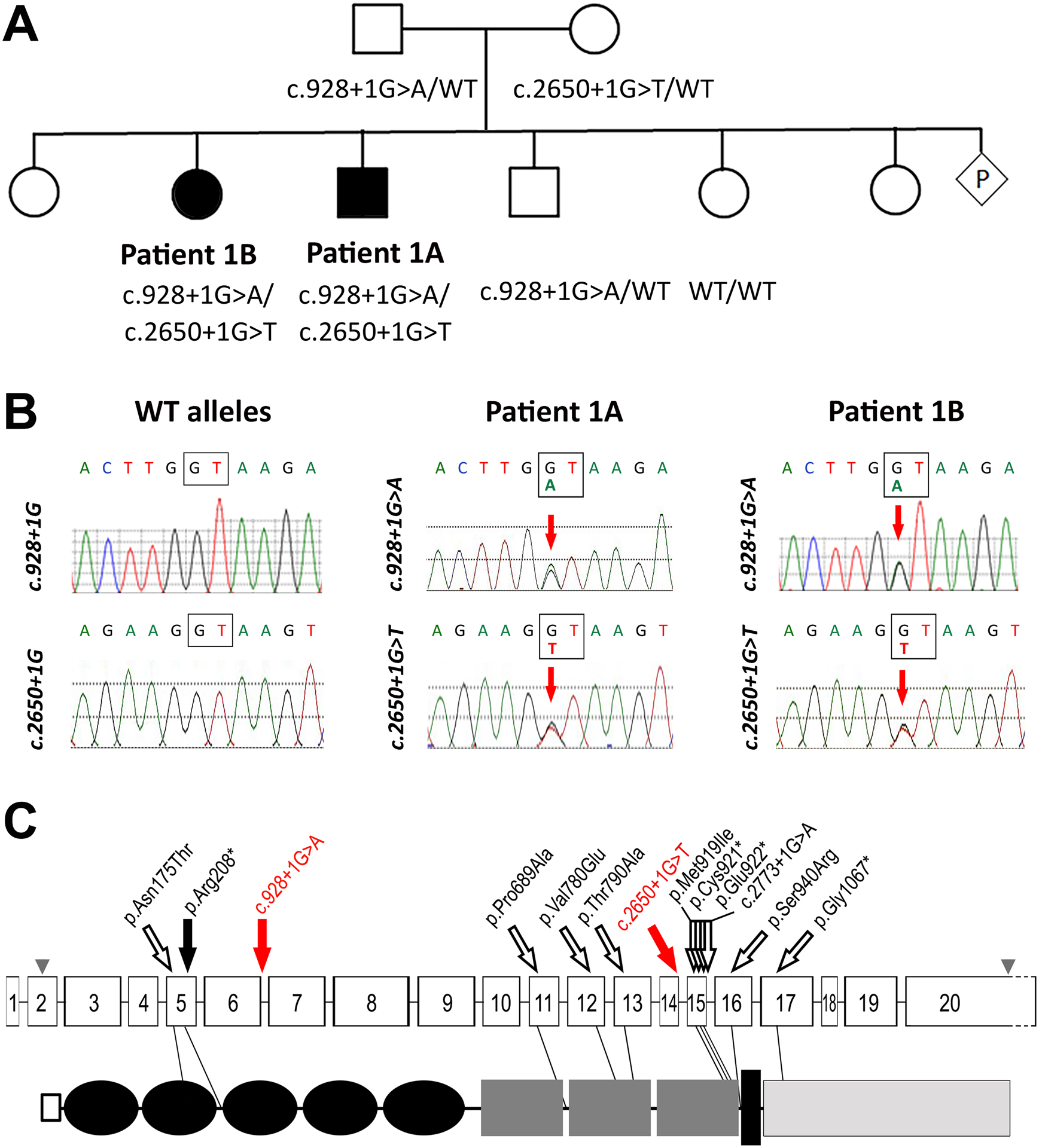
Pedigree of Patient 1A (A) including genotyping results for tested family members. Filled in symbols indicate individuals with coloboma; WT- wild type. Sanger sequencing chromatograms (B) showing compound heterozygous variants in both affected siblings and WT alleles at same position. Schematic of the CDON gene (top) and protein (bottom) structure (C) showing variants identified in this family (red solid arrows), the previously reported recessive variant (black solid arrow), and heterozygous HPE alleles (hollow black arrows) indicating their position in regard to CDON exons (numbered boxes) and known protein domains including signal peptide (hollow square: 1–25), Ig-like C2 (black ovals: 29–114, 120–20, 225–303, 310–396, 405–516), Fibronectin type-III (dark gray rectangles: 579–677, 723–821, 826–926), transmembrane (black rectangle: 964–984), and cytoplasmic (light gray rectangle: 985–1287). Accession numbers for CDON gene: NM_016952.4; CDON protein: NP_058648.4; domain position according to UniProt Q4KMG0; grey arrowheads indicate initiation and stop codons in exons 2 and 20 respectively.
Analysis of exome data did not identify any causative variants in known MAC genes. Trio analysis identified compound heterozygous donor splice site variants in CDON (NM_016952.4): c.928+1G>A inherited from the father and c.2650+1G>T inherited from the mother (Figure 2B). Neither variant is present in >250,000 alleles in gnomAD and both are predicted to abolish splicing at the variant site through alteration of the donor splice site; Human Splicing Finder (HSF) predicts a decrease in HSF splicing score from 90.06 to 62.92 for the first variant and from 99.86 to 72.72 for the second (strong splicing score is >80) and a decrease in MaxEnt score from 8.85 to 0.66 and 11 to 2.49 (score >3.5 predicts splicing), respectively. Previous studies have shown that in the absence of a strong cryptic donor site in the region, exon-skipping is the typical outcome for 5’ canonical splice site variants (21, 22). Exon-skipping resulting from the c.928+1G>A variant would result in deletion of 288 nucleotides, causing an in-frame deletion of 96 amino acids (p.Arg214_Glu310delinsGln) with loss of the IG-like C2-type 3 domain and likely disruption of protein conformation. Alternatively, the nearest strong cryptic donor splice site is upstream, with splicing predicted to occur after nucleotide c.732 (HSF score 59.54 but MaxEnt score of 8.9), and would be expected to result in frameshift with early truncation p.(Val245Asnfs*13) likely resulting in nonsense-mediated decay. Exon-skipping resulting from the c.2650+1G>T variant would lead to deletion of 106 nucleotides with subsequent protein truncation due to frameshift (p.Tyr849Valfs*7), again likely subject to nonsense-mediated decay; there were no strong cryptic donor sites identified within the region surrounding this variant.
Co-segregation analysis revealed that the affected sister (Patient 1B) is also compound heterozygous while the two tested unaffected siblings were not (one is a heterozygous carrier the other does not have either variant) (Figure 2A). Heterozygous CDON variants have been previously reported in patients with holoprosencephaly and pituitary stalk interruption syndrome (Table 1; Figure 2C); recently, a single child of a consanguineous couple affected with retinal coloboma, developmental delay, dysmorphic features, pyloric stenosis, and circumferential skin creases has been identified to carry homozygous variants in CDON and MAPRE2 (Table 1).
Table 1.
Summary of recessive and dominant CDON alleles and associated phenotypes.
| Patient/Ref | Zygosity | DNA change | Protein change | gnomAD | Inheritance# | Brain | Eye | Other |
|---|---|---|---|---|---|---|---|---|
| RECESSIVE ALLELES | ||||||||
| Berkun 2019 | homozygous | c.622C>T | p.Arg208* | 3/251202 | Biparental | MRI WNL | B optic nerve coloboma; U retinal coloboma | Features consistent with MAPRE2 variant |
| Patient 1A (this study) | compound heterozygous | c.928+1G>A | altered splicing | NP | Paternal | MRI WNL | B inferior coloboma of iris, retina, and choroid; severe myopia; iris pigment on lens capsule | Features consistent with 47,XYY karyotype |
| c.2650+1G>T | altered splicing | NP | Maternal | |||||
| Patient 1B (this study) | compound heterozygous | c.928+1G>A | altered splicing | NP | Paternal | - | B inferior coloboma of iris, retina, and choroid; high myopia; R retinal detachment; cataract | - |
| c.2650+1G>T | altered splicing | NP | Maternal | |||||
| DOMINANT HPE ALLELES | ||||||||
| Roessler 2018 | heterozygous | c.524A>C | p.Asn175Thr | 2/251132 | Maternal | alobar HPE | hypotelorism | cleft lip, maxillary hypoplasia |
| Bae 2011* | heterozygous | c.2051C>G | p.Thr684Ser | 2023/282638 (10 hom) | Maternal | HPE | - | - |
| Bae 2011 | heterozygous | c.2065C>G | p.Pro689Ala | 1/251290 | Unknown | ACC, global delay | hypotelorism | growth hormone deficiency |
| Bae 2011* | heterozygous | c.2071G>A | p.Val691Met | 30/282664 | Unknown | HPE | - | microcephaly |
| Bae 2011 | heterozygous | c.2339T>A | p.Val780Glu | NP | Unknown | HPE-like | - | - |
| Bae 2011 | heterozygous | c.2368A>G | p.Thr790Ala | NP | De novo | ACC, alobar HPE | hypotelorism, optic tracts with single cerebral artery | cleft lip/palate, hepatic choelestatis and polysplenia |
| Roessler 2018* | heterozygous | c.2462G>A | p.Arg821His | 143/251266 | Unknown | HPE | - | - |
| Jones 2016 | heterozygous | c.2757G>C | p.Met919Ile | NP | Paternal (hypotelorism) | absent vomar | hypotelorism | single central incisor, CED, hypoplastic midface/ nose; |
| Roessler 2018 | heterozygous | c.2763C>A | p.Cys921* | NP | Unknown | HPE | U | U |
| Bashamboo 2016 | heterozygous | c.2764G>T | p.Glu922* | NP | Maternal (strabismus) | PSIS | - | hepatic disease |
| Roessler 2018 | heterozygous | c.2773+1G>A | altered splicing | NP | Maternal | HPE | hypotelorism | single central incisor, hypoplastic midface/nose |
| Bae 2011 | heterozygous | c.2818A>C | p.Ser940Arg | 4/251400 | Unknown | alobar HPE | - | - |
| Roessler 2018 | heterozygous | c.3199G>T | p.Gly1067* | NP | Unknown | HPE | U | U |
ACC: Agenesis of corpus callosum; CED: congenital elbow dislocation; HPE: Holoprosencephaly; PSIS: pituitary stalk interruption syndrome, U: unknown; -: not present; Bold: alleles identified in this study;
and italics: likely benign alleles based on in silico/allele frequency;
parental phenotype (if present) is indicated in parenthesis.
DISCUSSION
Holoprosencephaly (HPE), caused by failure of the developing brain to separate into distinct hemispheres, is frequently associated with midline craniofacial anomalies and is characterized by a continuum in clinical severity (23). Both genetic and environmental etiologies have been identified and variable expressivity is common within families with genetic diagnosis (23, 24). Many genetic variants are inherited from unaffected parents and up to 1/3 of obligate carriers are clinically unaffected, leading to the hypothesis that HPE is multiple-hit disorder, requiring a combination of multiple genetic and/or environmental exposures (25, 26). Ophthalmological anomalies including coloboma and microphthalmia are frequently present in patients with HPE (27) and heterozygous variants in HPE loci SHH and HPE8 have also been reported in individuals with coloboma without HPE (28–30).
CDON is the first HPE gene identified to cause coloboma with a recessive inheritance pattern, in contrast to the dominant pattern observed in HPE. In addition to the previously reported homozygous nonsense allele of uncertain significance (31), we identified the first compound heterozygous splicing variants in CDON. All three recessive coloboma alleles are expected to result in loss-of-function: the nonsense variant c.622C>T p.Arg208* is located in exon 5 and the donor splice site variants affect the donor splice sites of exons 6 and 14. While developmental delay was reported in two of the three patients with recessive alleles, both had alternative genetic diagnoses associated with developmental delay (see below). In addition, brain MRI in both patients with delay was normal with no features of HPE identified. While heterozygous HPE alleles are typically missense variants, three nonsense variants in exons 15 and 17 (of 20) have also been reported (9–13). It is interesting to note that these three HPE truncation alleles occur later in the gene compared to the recessive loss-of-function variants associated with coloboma; however, since all three occur >50 nucleotides from the final intron, they would still be expected to be subject to nonsense-mediated decay, so the significance of this distribution is unclear. Since many of the reported heterozygous HPE alleles (truncating and missense) are inherited from an unaffected or mildly affected parent, several variants are too common in the general population to represent highly penetrant dominant alleles (Table 1), and data from gnomAD does not support loss-of-function intolerance (in haploinsufficiency model) for this gene (pLI=0 and o/e=0.97), it is possible that some or all heterozygous HPE variants in CDON represent high-risk alleles, similar to other HPE genes, and require a second genetic or environmental factor to cause HPE. Two studies looked at other HPE genes in patients with CDON variants and identified additional rare variants of uncertain significance in other HPE genes in most patients (9, 13). Additionally, a mouse model supports a synergistic effect for genetic and environmental factors in HPE. The 129S6/SvEvTac (129S6) background confers both low penetrance for HPE microforms with Cdon−/− and resistance to ethanol teratogenesis. In 129S6 mice homozygous for the Cdontm1Rsk allele (which deletes the first coding exon of Cdon, including the start codon (32)), in utero ethanol exposure in combination with the loss of Cdon resulted in a synergistic high rate of HPE, with HPE-related phenotypes present in 75% of embryos (33). While no features of HPE were reported in either family with recessive CDON alleles, the possibility that they also represent HPE risk alleles could not be ruled out.
Dual genetic diagnoses were present in two out of three patients with recessive CDON variants. In the previously published consanguineous family, a homozygous MAPRE2 variant was also identified, providing a genetic diagnosis for congenital circumferential skin creases for the patient. Interestingly, microphthalmia has been reported in previous cases with MAPRE2 variants (14), suggesting that variants in both genes may affect the eye in this patient. In the current family, the ocular phenotype in the two affected siblings was very similar, with high myopia and large iris and chorioretinal colobomas in both eyes. In addition, Patient 1B developed a retinal detachment in one eye at age 6. Patient 1A is also affected with non-ocular features that are consistent with his 47,XYY molecular diagnosis, in addition to the compound heterozygous CDON alleles; his affected sister (Patient 1B) with the same CDON genotype and no additional pathogenic alleles displays only ocular findings, suggesting that recessive CDON variants result in isolated ocular anomalies and the additional systemic features in Patient 1A result from the second genetic diagnosis (47,XYY). Given the rarity of Mendelian disorders, it was initially expected that dual diagnosis would be rare; the increased utilization of exome/genome sequencing has surprisingly shown that 4–7% of cases are found to have two molecular diagnosis (34, 35). Therefore, in cases of apparent phenotypic expansion, it is important to consider the possibility of a second genetic condition.
The frequent co-occurrence of HPE and coloboma and overlapping genetic etiology between the two conditions suggests that similar pathways are involved in both brain developmental and optic fissure closure. Analysis of other HPE genes in patients with non-syndromic coloboma may identify additional causative variants in this relatively unexplained condition.
ACKNOWLEDGEMENTS
We are grateful to patients and their family for participation in this study. This work was supported by NIH grants R21HD099701 and R01EY025718 as well as funds provided by the Children’s Research Institute Foundation at Children’s Hospital of Wisconsin (EVS) and 1UL1RR031973 from the Clinical and Translational Science Award (CTSA) program.
Footnotes
CONFLICT OF INTEREST:
The authors have no conflict of interest to declare.
DATA AVAILABILITY:
There are no other data associated with this manuscript.
References
- 1.Reis LM, Semina EV. Conserved genetic pathways associated with microphthalmia, anophthalmia, and coloboma. Birth defects researchPart C, Embryo today : reviews. 2015;105(2):96–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Williamson KA, Rainger J, Floyd JA, Ansari M, Meynert A, Aldridge KV, et al. Heterozygous loss-of-function mutations in YAP1 cause both isolated and syndromic optic fissure closure defects. American Journal of Human Genetics. 2014;94(2):295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deml B, Kariminejad A, Borujerdi RH, Muheisen S, Reis LM, Semina EV. Mutations in MAB21L2 result in ocular coloboma, microcornea and cataracts. PLoS genetics. 2015;11(2):e1005002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang L, He F, Bu J, Zhen Y, Liu X, Du W, et al. ABCB6 mutations cause ocular coloboma. American Journal of Human Genetics. 2012;90(1):40–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kelberman D, Islam L, Lakowski J, Bacchelli C, Chanudet E, Lescai F, et al. Mutation of SALL2 causes recessive ocular coloboma in humans and mice. Human molecular genetics. 2014;23(10):2511–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Williamson KA, FitzPatrick DR. The genetic architecture of microphthalmia, anophthalmia and coloboma. European journal of medical genetics. 2014;57(8):369–80. [DOI] [PubMed] [Google Scholar]
- 7.Zhang W, Kang JS, Cole F, Yi MJ, Krauss RS. Cdo functions at multiple points in the Sonic Hedgehog pathway, and Cdo-deficient mice accurately model human holoprosencephaly. Dev Cell. 2006;10(5):657–65. [DOI] [PubMed] [Google Scholar]
- 8.Tenzen T, Allen BL, Cole F, Kang JS, Krauss RS, McMahon AP. The cell surface membrane proteins Cdo and Boc are components and targets of the Hedgehog signaling pathway and feedback network in mice. Dev Cell. 2006;10(5):647–56. [DOI] [PubMed] [Google Scholar]
- 9.Bae GU, Domene S, Roessler E, Schachter K, Kang JS, Muenke M, et al. Mutations in CDON, encoding a hedgehog receptor, result in holoprosencephaly and defective interactions with other hedgehog receptors. Am J Hum Genet. 2011;89(2):231–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Karaca E, Harel T, Pehlivan D, Jhangiani SN, Gambin T, Coban Akdemir Z, et al. Genes that Affect Brain Structure and Function Identified by Rare Variant Analyses of Mendelian Neurologic Disease. Neuron. 2015;88(3):499–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bashamboo A, Bignon-Topalovic J, Rouba H, McElreavey K, Brauner R. A Nonsense Mutation in the Hedgehog Receptor CDON Associated With Pituitary Stalk Interruption Syndrome. J Clin Endocrinol Metab. 2016;101(1):12–5. [DOI] [PubMed] [Google Scholar]
- 12.Jones GE, Robertson L, Maniyar A, Shammas C, Phelan MM, Vasudevan PC, et al. Microform holoprosencephaly with bilateral congenital elbow dislocation; increasing the phenotypic spectrum of Steinfeld syndrome. Am J Med Genet A. 2016;170(3):754–9. [DOI] [PubMed] [Google Scholar]
- 13.Roessler E, Hu P, Marino J, Hong S, Hart R, Berger S, et al. Common genetic causes of holoprosencephaly are limited to a small set of evolutionarily conserved driver genes of midline development coordinated by TGF-beta, hedgehog, and FGF signaling. Hum Mutat. 2018;39(10):1416–27. [DOI] [PubMed] [Google Scholar]
- 14.Isrie M, Breuss M, Tian G, Hansen AH, Cristofoli F, Morandell J, et al. Mutations in Either TUBB or MAPRE2 Cause Circumferential Skin Creases Kunze Type. Am J Hum Genet. 2015;97(6):790–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reis LM, Sorokina EA, Thompson S, Muheisen S, Velinov M, Zamora C, et al. De Novo Missense Variants in WDR37 Cause a Severe Multisystemic Syndrome. Am J Hum Genet. 2019;105(2):425–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu X, Jian X, Boerwinkle E. dbNSFP v2.0: a database of human non-synonymous SNVs and their functional predictions and annotations. Human mutation. 2013;34(9):E2393–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Desmet FO, Hamroun D, Lalande M, Collod-Beroud G, Claustres M, Beroud C. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37(9):e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yeo G, Burge CB. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol. 2004;11(2–3):377–94. [DOI] [PubMed] [Google Scholar]
- 20.Wang M, Marin A. Characterization and prediction of alternative splice sites. Gene. 2006;366(2):219–27. [DOI] [PubMed] [Google Scholar]
- 21.Talerico M, Berget SM. Effect of 5’ splice site mutations on splicing of the preceding intron. Mol Cell Biol. 1990;10(12):6299–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anna A, Monika G. Splicing mutations in human genetic disorders: examples, detection, and confirmation. J Appl Genet. 2018;59(3):253–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Calloni SF, Caschera L, Triulzi FM. Disorders of Ventral Induction/Spectrum of Holoprosencephaly. Neuroimaging Clin N Am. 2019;29(3):411–21. [DOI] [PubMed] [Google Scholar]
- 24.Roessler E, Muenke M. The molecular genetics of holoprosencephaly. Am J Med Genet C Semin Med Genet. 2010;154C(1):52–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ming JE, Muenke M. Multiple hits during early embryonic development: digenic diseases and holoprosencephaly. Am J Hum Genet. 2002;71(5):1017–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mercier S, Dubourg C, Garcelon N, Campillo-Gimenez B, Gicquel I, Belleguic M, et al. New findings for phenotype-genotype correlations in a large European series of holoprosencephaly cases. J Med Genet. 2011;48(11):752–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pineda-Alvarez DE, Solomon BD, Roessler E, Balog JZ, Hadley DW, Zein WM, et al. A broad range of ophthalmologic anomalies is part of the holoprosencephaly spectrum. American journal of medical geneticsPart A. 2011;155A(11):2713–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schimmenti LA, de la Cruz J, Lewis RA, Karkera JD, Manligas GS, Roessler E, et al. Novel mutation in sonic hedgehog in non-syndromic colobomatous microphthalmia. American journal of medical geneticsPart A. 2003;116A(3):215–21. [DOI] [PubMed] [Google Scholar]
- 29.Ginocchio VM, De Brasi D, Genesio R, Ciccone R, Gimelli S, Fimiani F, et al. Sonic Hedgehog deletion and distal trisomy 3p in a patient with microphthalmia and microcephaly, lacking cerebral anomalies typical of holoprosencephaly. Eur J Med Genet. 2008;51(6):658–65. [DOI] [PubMed] [Google Scholar]
- 30.Piccione M, Serra G, Consiglio V, Di Fiore A, Cavani S, Grasso M, et al. 14q13.1–21.1 deletion encompassing the HPE8 locus in an adolescent with intellectual disability and bilateral microphthalmia, but without holoprosencephaly. Am J Med Genet A. 2012;158A(6):1427–33. [DOI] [PubMed] [Google Scholar]
- 31.Berkun L, Slae M, Mor-Shaked H, Koplewitz B, Eventov-Friedman S, Harel T. Homozygous variants in MAPRE2 and CDON in individual with skin folds, growth delay, retinal coloboma, and pyloric stenosis. Am J Med Genet A. 2019;179(12):2454–8. [DOI] [PubMed] [Google Scholar]
- 32.Cole F, Krauss RS. Microform holoprosencephaly in mice that lack the Ig superfamily member Cdon. Curr Biol. 2003;13(5):411–5. [DOI] [PubMed] [Google Scholar]
- 33.Hong M, Krauss RS. Cdon mutation and fetal ethanol exposure synergize to produce midline signaling defects and holoprosencephaly spectrum disorders in mice. PLoS Genet. 2012;8(10):e1002999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Balci TB, Hartley T, Xi Y, Dyment DA, Beaulieu CL, Bernier FP, et al. Debunking Occam’s razor: Diagnosing multiple genetic diseases in families by whole exome sequencing. Clinical genetics. 2017. [DOI] [PubMed] [Google Scholar]
- 35.Reis LM, Tyler RC, Weh E, Hendee KE, Schilter KF, Phillips JA 3rd, et al. Whole exome sequencing identifies multiple diagnoses in congenital glaucoma with systemic anomalies. Clinical genetics. 2016;90(4):378–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
There are no other data associated with this manuscript.