

Abstract
Traditional sampling methods for the study of poultry gut microbiota preclude longitudinal studies as they require euthanasia of birds for the collection of caecal and ileal contents. Some recent research has investigated alternative sampling methods to overcome this issue. The main goal of this study was to assess to what extent the microbial composition of non-invasive samples (excreta, litter and poultry dust) are representative of invasive samples (caecal and ileal contents). The microbiota of excreta, dust, litter, caecal and ileal contents (n = 110) was assessed using 16S ribosomal RNA gene amplicon sequencing. Of the operational taxonomic units (OTUs) detected in caecal contents, 99.7% were also detected in dust, 98.6% in litter and 100% in excreta. Of the OTUs detected in ileal contents, 99.8% were detected in dust, 99.3% in litter and 95.3% in excreta. Although the majority of the OTUs found in invasive samples were detected in non-invasive samples, the relative abundance of members of the microbial communities of these groups were different, as shown by beta diversity measures. Under the conditions of this study, correlation analysis showed that dust could be used as a proxy for ileal and caecal contents to detect the abundance of the phylum Firmicutes, and excreta as a proxy of caecal contents for the detection of Tenericutes. Similarly, litter could be used as a proxy for caecal contents to detect the abundance of Firmicutes and Tenericutes. However, none of the non-invasive samples could be used to infer the overall abundance of OTUs observed in invasive samples. In conclusion, non-invasive samples could be used to detect the presence and absence of the majority of the OTUs found in invasive samples, but could not accurately reflect the microbial community structure of invasive samples.
Introduction
The large and complex communities of bacteria that inhabit the mucosa and lumen of the intestinal tract play an important role in host health, nutrition and physiology [1, 2]. Caecal microbiota has been widely investigated in birds because of its role in fermentation and digestion of complex carbohydrates such as cellulose, starch, and resistant polysaccharides. This digestion results in the production of short-chain fatty acids which influence bird health and performance [3–5]. Caeca also play a major role in water absorption [6–8]. The ileum is important for nutrient digestion and absorption but its microbiota is less diverse [9, 10]. Caecal microbiota is qualitatively and quantitatively the most diverse and functionally the most complex in birds compared to other parts of the intestinal tract [11]. Sampling of caecal contents or other sections of the intestinal tract is labour-intensive and requires the bird to be sacrificed which may add variability in studies due to animal to animal microbiota variation [4]. Additionally, longitudinal studies are not possible when using invasive sampling as samples can only be collected at a single time-point from each bird. Thus, some research effort has been devoted to identifying and characterising non-invasive samples as an alternative to intestinal samples. This would contribute both to reduce the noise in the data, as the same birds would be sampled, and decrease the number of birds required [12], which is in line with the current framework for humane animal research.
The microbiota found in commonly used non-invasive samples, such as cloacal swabs and excreta are not stable and abrupt temporal fluctuations occur due to the cyclic emptying of the different regions of the gastrointestinal tract [13]. Overall, Firmicutes is the most dominant phylum in the chicken intestine [14]. Lactobacillus is the primary bacterial taxa found in the crop, gizzard, duodenum and ileum while Bifidobacterium and Enterobacter are also commonly detected in the crop [15, 16]. Clostridium, Ruminococcus, Streptococcus, Candidatus, Arthromitus (phylum Firmicutes) and Escherichia and Enterococcus (phylum Proteobacteria) have also been reported in the ileum [16–20]. The most abundant phyla found in the caecum are Firmicutes followed by Bacteroidetes while Proteobacteria and Archaea are present in lesser amounts [21–24]. The most dominant bacterial taxa in the caecum within the phylum Firmicutes were found to be Clostridiaceae, Lactobacillus, Ruminococcus, Faecalibacterium, Bacteroidaceae, Clostridium, Sporobacter, Oscillospira, Acetanaaerobacterium, Subdoligranulum and Pseudobutyrivibrio [18, 19, 25, 26]. Lactobacillus, Clostridium, Faecalibacterium, Ruminococcus, Bacillus, Eubacterium (phylum Firmicutes) are predominant in cloaca and excreta [3, 27, 28]. Although excreta has been shown to contain microbial taxa present in the caecum, there are quantitative differences in the microbial composition between sample types [4].
Microbial communities of cloacal swabs have been shown to be representative of litter samples, and to a lesser extent representative of ileum samples, while cloacal swabs are not representative of caecal contents [4, 29]. No difference in bacterial genera has been reported between cloacal swabs and ileal contents at day 2, 7 and 14 while Faecalibacterium, Blautia and Enterococcus have been found to be highly abundant in excreta compared to ileal contents at day 35 [30]. Caecal droppings have been suggested as a useful alternative to study caecal microbiota composition longitudinally as no difference in bacterial genera has been reported between these sample types, except for a higher abundance of facultative anaerobic bacteria such as Lactobacillus in caecal droppings [12, 31]. The collection of caecal droppings is, however, more labour-intensive than cloacal swabs or excreta as chickens produce a caecal dropping after 7–8 excreta droppings [32].
Non-invasive population-level samples have also been explored for their ability to evaluate the gut microbiota [31]. The microbiota of boot sock samples, which are a mixture of excreta and litter, was reflective of caecal microbiota composition early in the grow-out period (days 0 to 14) but not at later stages [31]. The management, quality and type of litter, which is a mixture of a bedding material (e.g. wood shavings, straw) and bird excreta, have been demonstrated to directly influence the gut microbiota composition in a reciprocal manner [33]. Lactobacillus has been shown to be a predominant bacterial taxa in the ileal mucosa of chickens raised on fresh pine shavings while unclassified groups of Clostridiales were dominant in the ileal mucosa of chickens raised on reused litter [33].
Poultry house dust could potentially be used as a non-invasive sample type, at the population level, for monitoring flock microbiota. Poultry dust is a mixture of dried excreta, exfoliated skin and feathers, feed, litter and microorganisms that become aerosolised during human and chicken movement in the poultry house [34]. Excreta is the major component of poultry house dust in turkey [35] and meat chicken flocks [34, 36]. A recent study by Luiken et al. [37] that investigated antimicrobial resistant bacteria in farmers and livestock, showed that the microbiota of settled poultry house dust are associated with the microbiota of chicken excreta, providing evidence that this approach could be useful for the study of gut microbiota. In addition, previous studies on molecular testing of poultry dust for population-level monitoring of viruses, protozoa, and bacteria in commercial flocks have shown that dust samples collected at any location of the poultry house could be used to detect microorganisms of interest even at relative low levels of infection (approximately 20% of infected birds) [38–44], suggesting that this may be a practical sample type for population-level monitoring of pathogens and perhaps gut microbiota. This study aimed to compare non-invasive samples (dust, excreta, and litter) to invasive (caecal and ileal contents) sampling methods for studying microbiota composition. Samples were collected from Eimeria and Clostridium perfringens challenged and unchallenged birds; however, no difference in microbial communities between challenged and unchallenged birds was observed. Therefore, the data obtained from both groups were combined and analysed together to test the following propositions. 1) The comparison of the microbial communities of non-invasive samples (dust, excreta and litter) with invasive samples (ileal and caecal contents) will show similarities in the detected operational taxonomic units but quantitative differences in their abundance; and 2) the comparison of microbial communities of non-invasive samples will show similarities in the detected operational taxonomic units and their abundance.
Methodology
Sample collection
Samples used in this study were collected as part of a previous experiment [45] that was approved by the University of New England Animal Ethics Committee (AEC18-059). Briefly, 972 day-old Ross cockerels (Aviagen breeder hatchery, Goulburn, New South Wales, Australia) were assigned into a ‘challenged’ or an ‘unchallenged room’ containing 72 equal sized floor pens for rearing chickens. Birds were randomly allocated to a group of 13–14 birds into each of the floor pens (120 × 75 cm) covered with fresh wood shavings at approximately 7 cm depth on arrival [45]. The facility underwent a deep clean before placement of chickens, to minimise the microbial load from previous use. On day 9, birds in the challenged room were orally gavaged 2,500 sporulated oocysts of Eimeria brunetti, and 5,000 sporulated oocysts each of E. maxima and E. acervulina (Eimeria Pty Ltd., Ringwood, Victoria, Australia) in 1 mL phosphate buffer solution. Birds in the unchallenged room were given 1 mL sterile phosphate buffer solution. Challenged birds were orally gavaged with 1 ml of approximately 108 colony forming units per ml of C. perfringens suspension in thioglycolate broth on day 14 with a repeat dose on day 15 for the induction of subclinical necrotic enteritis [45]. Unchallenged birds received 1 mL of sterile thioglycolate broth each day.
Birds were fed with starter diet (24.6% CP) until day 7, which was composed of wheat, sorghum, and soybean meal (3,000 kcal/kg). After day 7, birds were fed with pelleted diets isoenergetic at 3,080 kcal/kg for the grower diet (7–21 days) and 3,100 kcal/kg (days 21–35) for the finisher diet. Two pens in which chickens were fed with an industry-standard diet (S1 Table), were selected from both challenged and unchallenged rooms for sample collection in this study.
Excreta swab and litter samples from the selected pens were collected at days 0, 7, 13, 16, 21, 28 and 35. For the collection of fresh excreta, a clean white paper was placed on the floor of the selected pens and fresh excreta was immediately sampled by rolling a flocked swab (FLOQSwabs, Copan, Brescia, Italy) until all its surface was covered with excreta from one individual bird per pen, total of 28 samples. Two excreta samples were discarded due to poor quality DNA making a total of 26 excreta samples analysed in this study. Dust samples were collected using two dust plates (surface area of 520 cm2 each) suspended at approximately 1.5 m height in each room as previously described [46], total of 24 samples. Briefly, the dust plates were installed at chick placement and dust settled on the plates was scraped using a tissue into a ziplock bag at days 7, 13, 16, 21, 28 and 35. After each dust collection, the plates were repositioned until the next collection day. The settled dust was dry and there was enough accumulated material for testing (at least 50 mg dust) in each sampling day.
Swabs of ileal and caecal contents were collected from two birds per pen shortly after euthanasia on days 16 and 35, total of 32 samples, and were immediately placed on ice until arrival to the laboratory. Litter was collected from nine different locations per pen using a spatula and pooled together (approximately 4 g/pen) to form a single sample per pen, total of 28 samples. The litter samples were collected without entering the pen. Once transported to the laboratory, each pooled litter was dried in an oven at 36°C for up to 7 days and was ground, using an ultra-centrifugal grinder (ZM 200, Retsch, Germany), to pass through a 2 mm sieve, and thoroughly mixed. All samples were stored at -20°C until further analysis.
DNA extraction and 16S rRNA sequencing
DNA extraction was performed using DNeasy PowerSoil Pro Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions, with minor modifications. Briefly, all samples (swabs of excreta, ileal and caecal contents, 10 mg dust, 50 mg excreta or 50 mg litter) were added to powerbead pro tubes, resuspended in 800 μl of lysis buffer and homogenised for 5 min at maximum speed in a bullet blender (Model BBY24M-CE, Sigma Aldrich, St. Louis, USA) with subsequent heating of the homogenised suspension at 90°C for 10 min. A negative control (no sample added) was used in each DNA extraction batch. DNA concentration of each sample was measured using a Nanodrop before proceeding to the library generation step. Amplicons of the V3-V4 region of 16S rRNA genes, targeting the 343–806 bp region, were produced using forward (ACTCCTACGGGAGGCAGCAG) and reverse (GGACTACHVGGGTWTCTAAT) primers. The primers also contained barcodes, spacer sequences, and Illumina sequencing linkers [47]. Water was used as a negative control for each PCR amplification in a 96 well plate. The sequencing of the amplicons was performed on the Illumina MiSeq platform using 2 × 300 bp paired-end reads and sequence data were processed in QIIME [48]. Paired-end sequences were combined using Fastq-Join algorithm allowing no mismatch within overlapped regions. The reads with the Phred quality scores of 20 or above were used for analysis. The operational taxonomic unit (OTUs) were picked at 97% similarity using UCLUST [49]. Chimera sequences were checked using Pintail [50] and taxonomy assignment was performed against the GreenGenes database (v 2013_8) using QIIME default parameters [51]. After quality trimming, rare sequences that represented Operational Taxonomic Units (OTUs) present at less than 0.01% of the total sequences, were removed from further analysis. The data set consisted of 2,668,239 sequences, with an average of 24,257 sequences per sample. The sequence data is publicity available at the MG-RAST database under a project ID mgm4926609.3 and library ID mgl839711.
Statistical analysis
Hellinger transformation was used for normalizing microbial community composition data before statistical analyses, which were performed using Calypso software [52]. Alpha diversity was calculated using the Shannon index, which is a measure of species richness and evenness, and Chao1, which also measures microbial richness but gives more weight to low abundance species compared to Shannon [53]. The difference in microbial community composition between the samples types were visualized using principal coordinate analysis (PCoA) using Bray–Curtis distance metric, as a quantitative measure of beta diversity (abundance of OTUs), and Jaccard distance metric, as a qualitative measure (presence/absence of OTUs) [54]. Hierarchical clustering was performed on Bray–Curtis and Jaccard distance metrics to visualize the cluster of different samples and the output was shown as dendrograms. Homogeneity of multivariate dispersion was tested using permutational analyses of multivariate dispersions (PERMDISP) on Bray–Curtis and Jaccard distance metrics [55]. The overall difference in microbial community between the sample types for the challenged and unchallenged groups was investigated using permutational multivariate analysis of variances (PERMANOVA) on Bray–Curtis and Jaccard distance metrics using the Adonis function. Venn diagrams were constructed to visualise common bacterial taxa and linear discriminant analysis effect size (LEfSe) was performed to find the most discriminatory features between the sample types. Spearman rho ranked order correlation coefficient was performed to investigate if non-invasive samples (dust, excreta and litter) could serve as a proxy to study the microbiota of invasive sampling methods (ileal and caecal contents). Correlation coefficient values < 0.1 were considered as negligible correlation, 0.10–0.39 as weak, 0.40–0.69 as moderate, 0.70–0.89 as strong and > 0.9 were considered very strong correlation [56]. Non-invasive samples (dust, excreta and litter) were considered as a proxy for invasive samples (caecal and ileal contents) if the correlation of the microbial abundance between non-invasive and invasive samples was strong (≥ 70).
Results
There was no significant difference in the microbial communities between challenged and unchallenged birds for any sample type (S2 Table), therefore the data of both groups were combined and analysed together. Because of the low number of samples collected for each age group, samples collected after challenge with Eimeria spp. were combined. The results of the study are presented below.
Microbial alpha diversity
Overall, the highest microbial diversity was observed in dust samples and the lowest was observed in excreta and ileal contents using both Shannon and Chao1 alpha-diversity indices (Fig 1). Shannon diversity was similar in caecal contents and litter (P = 0.21), and the diversity in those samples were higher than ileal contents and excreta (P < 0.001) (Fig 1). Chao1 diversity was higher in litter compared to excreta, ileal and caecal contents (P < 0.001) and was similar among the latter samples (Fig 1). The number of OTUs found in caecal, ileal and excreta was lesser than previous studies [57–59] because of the stricter quality control and trimming steps applied in this study for removing sequences of low quality and low abundance OTUs.
Fig 1. Alpha diversity assessed by Shannon diversity (A) and Chao1 richness (B).
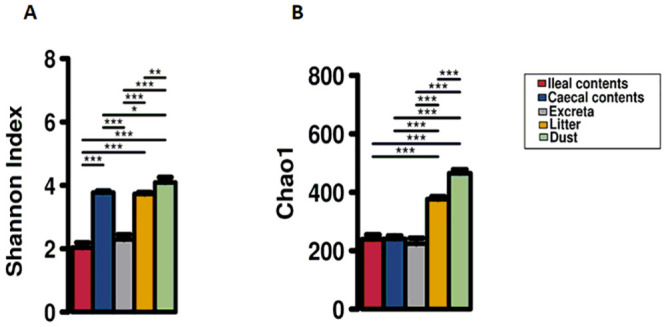
*Denotes significant difference at the P < 0.05 level, ** denotes significant difference at P < 0.01 level, *** denotes significant difference at the P < 0.001 level between sample types connected by a line.
Microbial community structure
The microbial composition of ileal and caecal contents, excreta, litter, and dust samples at phylum and genus level are shown in Fig 2. There was a clear differentiation of the phylogenetic structure of each sample type at phylum and genus levels (Fig 2).
Fig 2. Abundance at the phylum level and top 20 most abundant genera for each sample type (caecal and ileal contents, dust, excreta and litter).
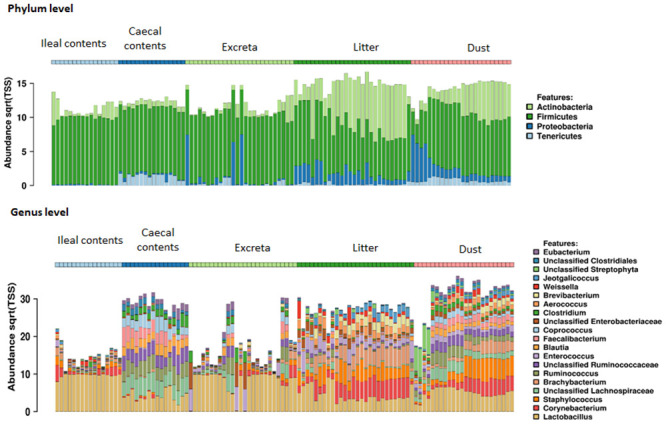
The clustering by sample type was also evident in the PCoA plots in which most samples tended to cluster together based on their source, with some interspersion between sample sources, particularly among ileal contents and excreta (Fig 3, S1 Fig). Microbiota of all sample types contained microbial taxa of phyla Firmicutes, Actinobacteria, Proteobacteria and Tenericutes (Fig 2), with Firmicutes the most abundant phyla in all sample types. Lactobacillus was the most dominant genera in ileal contents, dust, excreta and litter samples while unclassified Lachnospraceae was the most dominant genera in caecal contents (Fig 2).
Fig 3. Principal coordinates analysis (PCoA) using Bray-Curtis (A) and Jaccard distance metric (B) showing the difference in the microbial community between caecal and ileal contents, dust, excreta and litter samples.
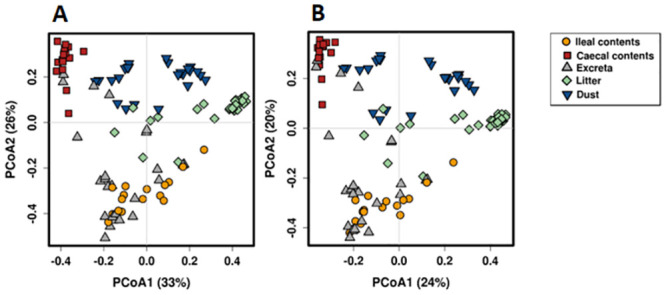
Overall, the microbial communities of each sample type were significantly different from each other in both Adonis Bray-Curtis and Jaccard diversity measures on pair-wise comparisons (Table 1).
Table 1. Difference in the microbial community between the two groups measured using permutational multivariate analysis of variances (PERMANOVA) on Bray–Curtis and Jaccard distance metrics using the Adonis function.
| Comparison | Adonis Bray-Curtis | Adonis Jaccard | ||||
|---|---|---|---|---|---|---|
| R-square | P-value | PERMDISP (P-value) | R-square | P-value | PERMDISP (P-value) | |
| Caecal contents vs dust | 0.41 | <0.001 | 0.54 | 0.32 | <0.001 | 0.73 |
| Caecal contents vs litter | 0.61 | <0.001 | 0.43 | 0.45 | <0.001 | 0.56 |
| Caecal vs ileal contents | 0.58 | <0.001 | 0.005 | 0.43 | <0.001 | 0.005 |
| Caecal contents vs excreta | 0.32 | <0.001 | <0.001 | 0.25 | <0.001 | <0.001 |
| Ileal contents vs dust | 0.41 | <0.001 | 0.09 | 0.31 | <0.001 | 0.07 |
| Ileal contents vs litter | 0.38 | <0.001 | 0.19 | 0.28 | <0.001 | 0.18 |
| Ileal contents vs excreta | 0.06 | 0.02 | 0.06 | 0.05 | 0.02 | 0.04 |
| Dust vs excreta | 0.27 | <0.001 | <0.001 | 0.22 | <0.001 | <0.001 |
| Dust vs litter | 0.25 | <0.001 | 0.81 | 0.21 | <0.001 | 0.78 |
| Litter vs excreta | 0.31 | <0.001 | 0.001 | 0.25 | <0.001 | 0.73 |
PERMDISP = permutational multivariate dispersions test calculated using Bray-Curtis and Jaccard distance metrics. R-square is the proportion of the variance explained by the group. Bolded values signify statistical difference at a P<0.05 level.
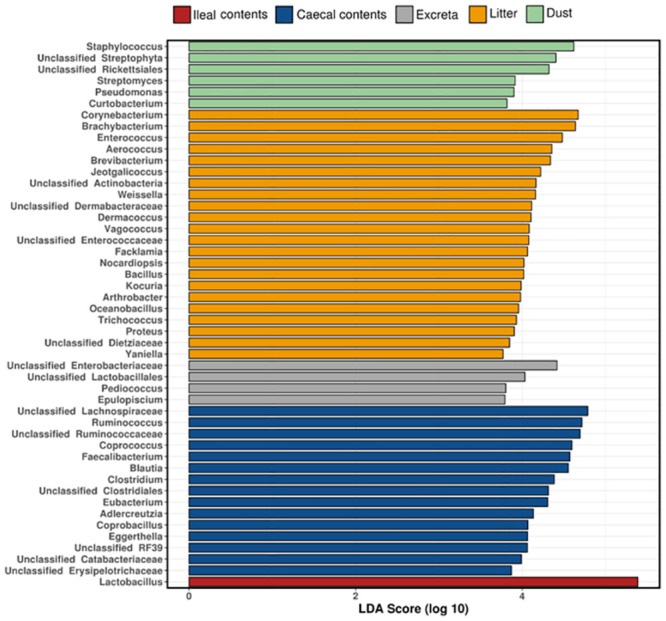
The elements of the microbiota that are most responsible for differentiating the composition of the different sample types are shown in Fig 4.
Fig 4. Genera that differentiate sample types identified using linear discriminant analysis effect size (LEfSe).
The proportion of operational taxonomic units (OTUs) shared by invasive and non-invasive samples
The number of OTUs in dust (n = 526), litter (n = 521) and excreta (n = 500) was higher than in ileal (n = 427) and caecal contents (n = 359) (Fig 5A, S3 Table). Although the number of OTUs were higher in non-invasive samples compared to invasive samples, most of the OTUs found in caecal and ileal contents were detected in dust, litter and excreta. A total of 99.7% of OTUs detected in caecal contents were also detected in dust, while 98.6% were detected in litter and 100% in excreta (Fig 5A, S4 Table). Lactobacillus helveticus (otu_256176) was detected in caecal contents but not in dust [abundance sqrt (TSS) = 0.01, occurrence = 12%]. Lachnospiraceae (otu_884077), Catabacteriaceae (otu_1033420), RF39 (otu_764779), Lachnospiraceae (otu_351809) and Blautia (otu_961773) were detected in caecal contents but not in litter [abundance sqrt (TSS) = 0.24–0.40, occurrence = 38–94%].
Fig 5. Venn diagram showing common bacterial OTUs between sample types A) across all OTUs and in the B) top 100 most abundant OTUs.
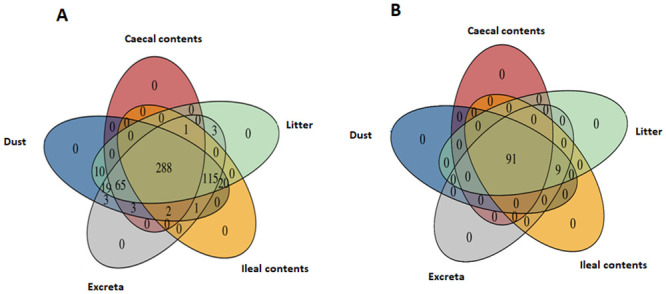
A total of 99.8% of OTUs detected in ileal contents were also detected in dust, 99.3% in litter and 95.3% in excreta (Fig 5A). Lactobacillus helveticus (otu_256176) was detected in ileal contents but not in dust [abundance sqrt (TSS) = 0.1, occurrence = 38%]. Similarly, Blautia (otu_961773), Lachnospiraceae (otu_884077) and Propionibacterium acnes (otu_522210) were detected in ileal contents but not detected in litter [abundance sqrt (TSS) = 0.004, occurrence = 6%]. There were 20 OTUs detected in ileal contents but not in excreta (S5 Table); however, they were not part of the ileal core OTUs as their occurrence and abundance was relatively low [abundance sqrt (TSS) = 0.003–0.03, occurrence = 6–31%]. The OTUs found in dust, excreta and litter but absent in ileal or caecal contents are presented in S5 Table.
Dust, excreta and litter shared 92% of total detected OTUs among them. A total of 99.2% of OTUs detected in excreta and litter were also detected in dust (Fig 5A). The OTUs detected in excreta and litter but absent in dust were Actinobacteria (otu_216320), Clostridium (otu_306483), Lactobacillus helveticus (otu_256176) and Enterococcus faecalis (otu_115349) [abundance sqrt (TSS) = 0.003–0.12].
When the microbial composition between the sample types were compared across top 100 OTUs, 91% of the total detected OTUs were shared between dust, litter and caecal contents while 100% were shared between litter, dust and ileal contents (Fig 5B).
Correlation of microbial composition between invasive samples and non-invasive samples
The overall microbial abundance of non-invasive samples (excreta, dust, litter) was weakly to moderately correlated (Spearman rs < 70) with invasive samples (caecal and ileal contents) (S6 Table). Overall, there was a moderate positive correlation of OTUs abundance between caecal contents and dust (rs = 0.41; P < 0.001), and caecal contents and excreta (rs = 0.58; P < 0.001), while a moderate negative correlation was found between caecal contents and litter (rs = -0.46; P < 0.001). Similarly, moderate positive correlation of the OTUs abundance was found between ileal contents and excreta (rs = 0.49; P < 0.001), and ileal contents and litter (rs = 0.55; P < 0.001), while a weak positive correlation was found between ileal contents and dust (rs = 0.34; P < 0.001).
No phylum was significantly positively correlated in abundance between ileal contents and excreta (P > 0.05) (S7 Table). Tenericutes was the only phylum showing a strong positive correlation between caecal contents and excreta (rs = 0.78, P < 0.001). Strong positive correlations between caecal contents and dust, and caecal contents and litter samples were found for the phylum Firmicutes (rs = 0.83–0.85, P < 0.001) while negative correlations between caecal contents and dust, and caecal contents and litter were found for Actinobacteria and Proteobacteria (rs = -0.71 –-0.82, P < 0.001). Strong positive correlation between caecal contents and litter was also found for the phylum Tenericutes (rs = 0.81, P < 0.001) (S7 Table). Similarly, strong positive correlation between ileal contents and dust, and ileal contents and litter was found for Firmicutes (rs = 0.75–0.77, P < 0.001) and negative correlation was found for Tenericutes, Proteobacteria and Actinobacteria (rs = -0.50 –-0.85, P < 0.001). The correlation of the microbiota at the genus level between non-invasive samples and invasive samples are shown in Table 2.
Table 2. Spearman’s rank correlation between the relative abundance of genera of non-invasive samples (dust, excreta and litter) and invasive samples (ileal and caecal contents).
| Genera | Spearman’s rank correlation | |||||
|---|---|---|---|---|---|---|
| Caecal contents and litter | Caecal contents and dust | Ileal contents and dust | Ileal contents and litter | Caecal contents and excreta | Ileal contents and excreta | |
| Acinetobacter | -0.80 | -0.88 | -0.85 | -0.75 | -0.12 | 0.25 |
| Adlercreutzia | 0.84 | 0.74 | -0.83 | -0.38 | 0.71 | -0.31 |
| Aerococcus | -0.84 | -0.84 | -0.68 | -0.75 | -0.28 | 0.28 |
| Arthrobacter | -0.80 | -0.88 | -0.88 | -0.8 | -0.12 | -0.12 |
| Bacillus | -0.79 | -0.67 | -0.57 | -0.76 | 0.05 | 0.37 |
| Blautia | 0.83 | 0.8 | -0.85 | -0.38 | 0.81 | -0.18 |
| Brachybacterium | -0.83 | -0.8 | -0.57 | -0.76 | -0.20 | 0.26 |
| Brevibacterium | -0.84 | -0.85 | -0.63 | -0.77 | -0.25 | 0.27 |
| Burkholderia | -0.12 | -0.19 | -0.05 | 0.06 | NA | 0.2 |
| Clostridium | 0.83 | 0.68 | -0.77 | -0.52 | 0.55 | -0.48 |
| Coprobacillus | 0.8 | 0.4 | -0.86 | -0.39 | 0.78 | -0.25 |
| Coprococcus | 0.83 | 0.81 | -0.85 | -0.37 | 0.73 | -0.17 |
| Corynebacterium | -0.83 | -0.77 | -0.42 | -0.63 | -0.18 | 0.36 |
| Curtobacterium | -0.55 | -0.79 | -0.68 | -0.33 | NA | 0.47 |
| Dermacoccus | -0.78 | -0.52 | -0.37 | -0.71 | -0.22 | 0.11 |
| Eggerthella | 0.92 | 0.84 | -0.64 | -0.2 | 0.76 | -0.35 |
| Enterococcus | -0.83 | -0.85 | -0.75 | -0.77 | -0.57 | -0.24 |
| Epulopiscium | -0.24 | -0.52 | -0.52 | -0.24 | -0.22 | -0.22 |
| Eubacterium | 0.83 | 0.72 | -0.85 | -0.47 | 0.62 | -0.35 |
| Facklamia | -0.78 | -0.77 | -0.41 | -0.64 | -0.03 | 0.47 |
| Faecalibacterium | 0.75 | 0.36 | -0.85 | -0.62 | 0.71 | -0.21 |
| Jeotgalicoccus | -0.78 | -0.83 | -0.55 | -0.64 | -0.02 | 0.33 |
| Klebsiella | -0.85 | -0.88 | -0.86 | -0.84 | -0.18 | 0.15 |
| Kocuria | -0.68 | -0.52 | -0.42 | -0.62 | -0.12 | 0.16 |
| Lactobacillus | 0.06 | -0.3 | 0.85 | 0.83 | -0.61 | 0.25 |
| Nocardiopsis | -0.85 | -0.79 | -0.64 | -0.81 | NA | 0.41 |
| Oceanobacillus | -0.75 | -0.23 | -0.23 | -0.75 | NA | NA |
| Pediococcus | -0.34 | -0.73 | -0.28 | 0.21 | -0.55 | -0.05 |
| Propionibacterium | NA | -0.31 | -0.21 | 0.2 | -0.12 | 0.05 |
| Proteus | -0.85 | -0.76 | -0.77 | -0.85 | -0.21 | -0.23 |
| Pseudomonas | -0.5 | -0.88 | -0.88 | -0.5 | -0.12 | -0.12 |
| Ruminococcus | 0.83 | 0.69 | -0.85 | -0.52 | 0.67 | -0.22 |
| Salinicoccus | -0.8 | -0.75 | -0.45 | -0.37 | -0.21 | 0.32 |
| Serratia | -0.55 | -0.67 | -0.67 | -0.55 | -0.22 | -0.22 |
| Staphylococcus | -0.84 | -0.85 | -0.66 | -0.68 | -0.26 | 0.31 |
| Streptomyces | -0.6 | -0.88 | -0.87 | -0.56 | -0.18 | -0.03 |
| Trichococcus | -0.78 | -0.73 | -0.56 | -0.7 | -0.22 | 0.25 |
| Unclassified Actinobacteria | -0.85 | -0.86 | -0.68 | -0.8 | -0.37 | -0.06 |
| Unclassified Aerococcaceae | -0.55 | -0.61 | -0.58 | -0.51 | NA | 0.2 |
| Unclassified Bacilli | -0.79 | -0.64 | -0.33 | -0.54 | -0.20 | 0.25 |
| Unclassified Catabacteriaceae | 0.92 | 0.53 | -0.79 | -0.2 | 0.87 | -0.29 |
| Unclassified Clostridiaceae | -0.08 | -0.48 | -0.25 | 0.15 | -0.29 | -0.09 |
| Unclassified Clostridiales | 0.79 | 0.6 | -0.85 | -0.71 | 0.74 | -0.16 |
| Unclassified Dermabacteraceae | -0.8 | -0.79 | -0.62 | -0.72 | -0.29 | 0.11 |
| Unclassified Dietziaceae | -0.65 | -0.67 | -0.54 | -0.53 | 0.05 | 0.38 |
| Unclassified Enterobacteriaceae | -0.72 | -0.75 | -0.85 | -0.84 | 0.18 | -0.57 |
| Unclassified Enterococcaceae | -0.84 | -0.86 | -0.49 | -0.71 | -0.65 | -0.03 |
| Unclassified Erysipelotrichaceae | 0.72 | 0.11 | -0.82 | -0.48 | 0.62 | -0.38 |
| Unclassified Lachnospiraceae | 0.83 | 0.83 | -0.85 | -0.3 | 0.73 | -0.22 |
| Unclassified Lactobacillales | -0.84 | -0.85 | -0.58 | -0.7 | -0.58 | -0.06 |
| Unclassified Planococcaceae | 0.52 | 0.03 | -0.76 | -0.24 | 0.61 | -0.12 |
| Unclassified RF39 | 0.84 | 0.49 | -0.83 | -0.45 | 0.80 | -0.29 |
| Unclassified Rickettsiales | -0.53 | -0.88 | -0.81 | -0.01 | -0.43 | 0.23 |
| Unclassified Ruminococcaceae | 0.83 | 0.72 | -0.85 | -0.72 | 0.81 | -0.32 |
| Unclassified Streptophyta | -0.6 | -0.88 | -0.74 | 0.17 | -0.51 | 0.54 |
| Vagococcus | -0.85 | -0.76 | -0.67 | -0.84 | NA | 0.29 |
| Weissella | -0.81 | -0.74 | -0.41 | -0.46 | -0.30 | 0.32 |
| Yaniella | -0.58 | -0.31 | -0.14 | -0.51 | -0.12 | 0.15 |
Positive correlated taxa are coloured red, while negatively associated taxa are coloured yellow and grey indicates non-significant association. Significance was set a P<0.05 level. NA indicates that a particular genus was absent in both sample types.
Discussion
In this study, the microbial community structures of non-invasive samples (dust, excreta and litter) were different from invasive samples (caecal and ileal contents), particularly on the abundance of the detected OTUs. However, the majority of OTUs detected in the caecal and ileal contents were also detected in non-invasive samples. This suggests that dust, litter or excreta could be used to determine the presence/absence of most of OTUs present in invasive samples. Similarly, the microbial community structure of non-invasive samples was also different between sample types but the majority of OTUs were detected in all non-invasive sample types.
The results partially support the proposition that compared to invasive samples, the microbial communities of non-invasive samples will show similarities in the detected OTUs but quantitative differences in abundance of the OTUs. Although the majority of the OTUs detected in invasive samples were also detected in non-invasive samples, a higher number of unique OTUs were detected in dust, excreta and litter compared to caecal and ileal contents as reflected in differences in beta-diversity measures. The microbiota of the gut is primarily composed of anaerobic bacteria [60] whereas litter and dust samples contain both aerobic bacteria and the nucleic acid remnants of obligate anaerobes, similarly to excreta. This could be a reason for the higher number of OTUs in dust, excreta and litter compared to ileal and caecal contents. When comparing the number of OTUs that were shared between different sample types, 98–100% OTUs detected in caecal contents and 95–99.8% of OTUs detected in ileal contents were present in non-invasive samples, suggesting that non-invasive samples could be used to identify presence and absence of most of the OTUs of invasive samples. The majority of OTUs detected in ileal or caecal contents that were not detected in non-invasive samples were of low abundance. This is similar to [61], which demonstrated that ~99% of the OTUs found in caecal samples were present in excreta and ~87% of the OTUs found in ileal samples were present in excreta, but higher than [62], reporting that ~60% of the OTUs found in caecal swabs were present in the cloacal swabs. Differences among studies could be attributed due to differences in sample size, sampling techniques, age of the birds, DNA extraction methods, feed, breed or other factors.
In this study, abundance of bacterial phylum Firmicutes in caecal contents and dust, caecal contents and litter, ileal contents and dust, and ileal contents and litter were strongly and positively correlated while the abundance of the phylum Tenericutes in caecal contents and excreta, and caecal contents and litter were strongly positively correlated. This indicates that, under the conditions of this study, dust and litter could be used as a proxy for ileal and caecal contents to study the abundance of the phylum Firmicutes, and excreta as a proxy for caecal contents to study the abundance of the phylum Tenericutes. Similarly, litter could also be used as a proxy for caecal contents to study the abundance of phylum Tenericutes but not necessarily to investigate the abundance of specific OTUs. None of the non-invasive sample types could be used as a proxy to map the overall microbial composition of invasive samples. Similar to our findings, previous research has shown that arbitrarily collected excreta and cloacal swabs cannot be used as a reference to accurately monitor the microbiota of caecum or ileum [12, 63]. Contrarily, when caecal droppings were sampled, there was a correlation between the microbiota of this sample type and the caecum microbiota [12, 31].
The results partially support the second proposition that the comparison of microbial communities of non-invasive samples will show similarities in the detected OTUs and their abundance. A higher number of unique OTUs was detected in the dust compared to excreta and litter, as depicted by beta diversity measures. The difference in substrate requirements for growth and multiplication of different bacterial species [64] may influence species distribution and abundance level of the same bacterium in different sample types. When comparing the number of OTUs shared between dust and excreta, and litter and dust, 99.2% of the total OTUs found in excreta and litter were detected in the dust. The OTUs exclusive to excreta and litter had low abundance.
The similarity of OTUs shared between non-invasive samples highlights health concerns as it is well-known that aerosolised poultry dust and litter can pose serious issues for farm workers and birds [65]. Culture of settled poultry dust collected from meat chickens and laying hen farms has found an average total bacteria of 3.2 × 109 colony forming units (cfu)/g dust, and actinomycetes at 5.5 × 106 cfu/g, and fungi at 1.2 × 106 cfu/g [65]. Commonly cultivated bacteria in settled dust were Bacillus, Clostridia, Corynebacterium, Enterobacter, Flavobacterium, Pseudomonas, Staphylococcus and E. coli; in addition several biological toxins, allergens and odorous gases have been also detected [65]. Likewise, poultry litter can harbour high levels of culturable potential pathogens such as Salmonella and Campylobacter [66] in addition to antibiotic resistance genes that may persist in horticultural soils amended with poultry litter [67]. A previous study using a culture-based methodology has shown a differential capacity of aerosolization and survival of bacteria present in litter samples of four commercial meat chicken farms during summer and winter grow-outs, which was affected by the environment temperature and relative humidity [66]. Although Campylobacter was detected at high levels in poultry litter (107 most probable number [mpn]/g), it could not be isolated from aerosols inside the poultry house, while Salmonella (103–105 mpn/g) could be cultured from aerosols and settled dust and E. coli was readily isolated in litter and aerosols (102–105 cfu/g). It appears, however, that the survival of these microorganisms outside of tunnel-ventilated meat chicken houses is limited to 20 m beyond the exhaust fan [68].
In this study, no difference in the microbial community structure between challenged and unchallenged groups was observed. The challenge of birds induced subclinical necrotic enteritis as previously described [45] and the lack of difference is unexpected as several studies have reported marked differences in the microbiota of challenged birds in similar experiments [69–71]. A possible explanation for this is the small sample size of the current study which reduced the power to detect changes between groups. In the current study, 18–20 samples were collected for each sample type after inoculation with Eimeria spp and C. perfringens or sham-inoculation (5 sampling times, 2–4 samples per sampling time). Although the use of 8–10 samples per treatment (challenged or control group) per sample type would provide sufficient power for differentiation of the microbiota between treatments [69–71], combining the samples from birds of different ages is likely to have increased the variation in the microbiota of the chickens. Additionally, the microbial composition from samples collected at later stages, i.e., at d 35 is unlikely to differ between treatments as the birds recovered from subclinical necrotic enteritis soon after challenge. All these factors may have contributed to the lack of difference between treatments. A larger sample size should be used and combination of samples from different age should be avoided in future studies to reduce issues of individual bird variation in microbiota studies. Functional analysis of the microbiota would provide additional insights into the capabilities of the microbial communities of different sample types.
In conclusion, dust, excreta, and litter could not be used to map the overall microbial composition of caecal and ileal contents; however, they could be used to detect the presence and absence of the majority of the OTUs found in caecal and ileal contents. This could potentially be useful for screening poultry flocks for bacterial taxa of interest such as pathogenic bacteria or for investigating the effects of microbial feed additives using specific molecular assays or changes in microbial community structure after management interventions. This study provides insight on the use of non-invasive sample types to monitor intestinal microbiota. Further investigation on a larger sample size would be useful to determine the relationship between the microbiota of diverse sample types and their relationship with gut health and production parameters.
Supporting information
(DOCX)
PERMDISP = permutational multivariate dispersions test calculated using Bray-Curtis and Jaccard distance metrics. R-square is the proportion of the variance explained by the group. Cumulative data from days 13–35 were used (n = 92 samples). The challenged group received 2,500 sporulated oocyst of E. brunetti, and 5,000 sporulated oocysts of E. maxima and E. acervulina on day 9 and 108 colony forming units of C. perfringens on days 14 and 15. The unchallenged group received 1 ml sterile phosphate buffer solution on day 9 and sterile broth on day 14 and 15.
(DOCX)
(XLSX)
(XLSX)
(XLSX)
(DOCX)
(DOCX)
(DOCX)
Acknowledgments
We thank Sarbast Qassim for technical support during sample analysis.
Data Availability
All relevant data are within the manuscript and its Supporting information files.
Funding Statement
Poultry Hub Australia (grant 18-424) partially funded this research (PFG). There was no additional external funding received for this study. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. Matthew Hilliar, who was a PhD candidate at University of New England at the time in which the study was carried out, is currently employed by Turosi. The funder provided support in the form of salaries for author MH during the write up of this study, but did not have any additional role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. The specific roles of this author are articulated in the ‘author contributions’ section.
References
- 1.Díaz-Sánchez S, Perrotta AR, Rockafellow I, Alm EJ, Okimoto R, Hawken R, et al. Using fecal microbiota as biomarkers for predictions of performance in the selective breeding process of pedigree broiler breeders. PloS One. 2019;14(5):e0216080. doi: 10.1371/journal.pone.0216080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang M, Hong G, Jin Y, Li Y, Li G, Hou X. Mucosal-associated microbiota other than luminal microbiota has a close relationship with diarrhea-predominant irritable bowel syndrome. Frontiers in cellular and infection microbiology. 2020;10:606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stanley D, Hughes RJ, Moore RJ. Microbiota of the chicken gastrointestinal tract: influence on health, productivity and disease. Applied microbiology and biotechnology. 2014;98(10):4301–10. doi: 10.1007/s00253-014-5646-2 [DOI] [PubMed] [Google Scholar]
- 4.Stanley D, Geier MS, Chen H, Hughes RJ, Moore RJ. Comparison of fecal and cecal microbiotas reveals qualitative similarities but quantitative differences. BMC microbiology. 2015;15(1):51. doi: 10.1186/s12866-015-0388-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mead G. Microbes of the avian cecum: types present and substrates utilized. Journal of Experimental Zoology. 1989;252(S3):48–54. doi: 10.1002/jez.1402520508 [DOI] [PubMed] [Google Scholar]
- 6.Clench MH, Mathias JR. The avian cecum: a review. The Wilson Bulletin. 1995:93–121. [Google Scholar]
- 7.Gasaway WC, White RG, Holleman DF. Digestion of dry matter and absorption of water in the intestine and cecum of rock ptarmigan. The Condor. 1976;78(1):77–84. [Google Scholar]
- 8.Obst BS, Diamond JM. Interspecific variation in sugar and amino acid transport by the avian cecum. Journal of Experimental Zoology. 1989;252(S3):117–26. doi: 10.1002/jez.1402520519 [DOI] [PubMed] [Google Scholar]
- 9.Thaiss CA, Zeevi D, Levy M, Segal E, Elinav E. A day in the life of the meta-organism: diurnal rhythms of the intestinal microbiome and its host. Gut microbes. 2015;6(2):137–42. doi: 10.1080/19490976.2015.1016690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gong J, Si W, Forster RJ, Huang R, Yu H, Yin Y, et al. 16S rRNA gene-based analysis of mucosa-associated bacterial community and phylogeny in the chicken gastrointestinal tracts: from crops to ceca. FEMS microbiology ecology. 2007;59(1):147–57. doi: 10.1111/j.1574-6941.2006.00193.x [DOI] [PubMed] [Google Scholar]
- 11.Ducatelle R, Goossens E, De Meyer F, Eeckhaut V, Antonissen G, Haesebrouck F, et al. Biomarkers for monitoring intestinal health in poultry: present status and future perspectives. Veterinary Research. 2018;49(1):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pauwels J, Taminiau B, Janssens G, De Beenhouwer M, Delhalle L, Daube G, et al. Cecal drop reflects the chickens’ cecal microbiome, fecal drop does not. Journal of microbiological methods. 2015;117:164–70. doi: 10.1016/j.mimet.2015.08.006 [DOI] [PubMed] [Google Scholar]
- 13.Deusch S, Tilocca B, Camarinha-Silva A, Seifert J. News in livestock research—use of Omics-technologies to study the microbiota in the gastrointestinal tract of farm animals. Computational and Structural Biotechnology Journal. 2015;13:55–63. doi: 10.1016/j.csbj.2014.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wei S, Morrison M, Yu Z. Bacterial census of poultry intestinal microbiome. Poultry science. 2013;92(3):671–83. doi: 10.3382/ps.2012-02822 [DOI] [PubMed] [Google Scholar]
- 15.Saxena S, Saxena V, Tomar S, Sapcota D, Gonmei G. Characterisation of caecum and crop microbiota of Indian indigenous chicken targeting multiple hypervariable regions within 16S rRNA gene. British poultry science. 2016;57(3):381–9. doi: 10.1080/00071668.2016.1161728 [DOI] [PubMed] [Google Scholar]
- 16.Yeoman CJ, Chia N, Jeraldo P, Sipos M, Goldenfeld ND, White BA. The microbiome of the chicken gastrointestinal tract. Animal Health Research Reviews. 2012;13(1):89. doi: 10.1017/S1466252312000138 [DOI] [PubMed] [Google Scholar]
- 17.Munyaka P, Nandha N, Kiarie E, Nyachoti C, Khafipour E. Impact of combined β-glucanase and xylanase enzymes on growth performance, nutrients utilization and gut microbiota in broiler chickens fed corn or wheat-based diets. Poultry Science. 2016;95(3):528–40. doi: 10.3382/ps/pev333 [DOI] [PubMed] [Google Scholar]
- 18.Wang L, Lilburn M, Yu Z. Intestinal microbiota of broiler chickens as affected by litter management regimens. Frontiers in microbiology. 2016;7:593. doi: 10.3389/fmicb.2016.00593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jin L, Ho Y, Abdullah N, Kudo H, Jalaludin S. Studies on the intestinal microflora of chicken under tropical condition. Asian-Australasian Journal of Animal Sciences. 1997;10(5):495–504. [Google Scholar]
- 20.Lu J, Idris U, Harmon B, Hofacre C, Maurer JJ, Lee MD. Diversity and succession of the intestinal bacterial community of the maturing broiler chicken. Applied and environmental microbiology. 2003;69(11):6816–24. doi: 10.1128/AEM.69.11.6816-6824.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dibner J, Richards J. Antibiotic growth promoters in agriculture: history and mode of action. Poultry Science. 2005;84(4):634–43. doi: 10.1093/ps/84.4.634 [DOI] [PubMed] [Google Scholar]
- 22.Shakouri M, Iji P, Mikkelsen LL, Cowieson A. Intestinal function and gut microflora of broiler chickens as influenced by cereal grains and microbial enzyme supplementation. Journal of animal physiology and animal nutrition. 2009;93(5):647–58. doi: 10.1111/j.1439-0396.2008.00852.x [DOI] [PubMed] [Google Scholar]
- 23.Oakley BB, Lillehoj HS, Kogut MH, Kim WK, Maurer JJ, Pedroso A, et al. The chicken gastrointestinal microbiome. FEMS microbiology letters. 2014;360(2):100–12. doi: 10.1111/1574-6968.12608 [DOI] [PubMed] [Google Scholar]
- 24.Chen J, Tellez G, Richards JD, Escobar J. Identification of potential biomarkers for gut barrier failure in broiler chickens. Frontiers In Veterinary Science. 2015;2:14. doi: 10.3389/fvets.2015.00014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gong J, Forster RJ, Yu H, Chambers JR, Sabour PM, Wheatcroft R, et al. Diversity and phylogenetic analysis of bacteria in the mucosa of chicken ceca and comparison with bacteria in the cecal lumen. FEMS microbiology letters. 2002;208(1):1–7. doi: 10.1111/j.1574-6968.2002.tb11051.x [DOI] [PubMed] [Google Scholar]
- 26.Qu A, Brulc JM, Wilson MK, Law BF, Theoret JR, Joens LA, et al. Comparative metagenomics reveals host specific metavirulomes and horizontal gene transfer elements in the chicken cecum microbiome. PloS one. 2008;3(8):e2945. doi: 10.1371/journal.pone.0002945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lim S, Cho S, Caetano-Anolles K, Jeong SG, Oh MH, Park BY, et al. Developmental dynamic analysis of the excreted microbiome of chickens using next-generation sequencing. Journal of molecular microbiology and biotechnology. 2015;25(4):262–8. doi: 10.1159/000430865 [DOI] [PubMed] [Google Scholar]
- 28.Sekelja M, Rud I, Knutsen S, Denstadli V, Westereng B, Naes T, et al. Abrupt temporal fluctuations in the chicken fecal microbiota are explained by its gastrointestinal origin. Applied and environmental microbiology. 2012;78(8):2941–8. doi: 10.1128/AEM.05391-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Johnson TJ, Youmans BP, Noll S, Cardona C, Evans NP, Karnezos TP, et al. A consistent and predictable commercial broiler chicken bacterial microbiota in antibiotic-free production displays strong correlations with performance. Applied and environmental microbiology. 2018;84(12). doi: 10.1128/AEM.00362-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnson JS, Spakowicz DJ, Hong B-Y, Petersen LM, Demkowicz P, Chen L, et al. Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nature communications. 2019;10(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kers JG, Fischer EA, Stegeman JA, Smidt H, Velkers FC. Comparison of different invasive and non-invasive methods to characterize intestinal microbiota throughout a production cycle of broiler chickens. Microorganisms. 2019;7(10):431. doi: 10.3390/microorganisms7100431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Williams R. Epidemiological studies of coccidiosis in the domesticated fowl (Gallus gallus): III. The influence of the fowl’s defaecation pattern on the excretion patterns of Eimeria tenella and E acervulina oocysts. Applied parasitology. 1995;36(4):279–89. [PubMed] [Google Scholar]
- 33.Cressman MD, Yu Z, Nelson MC, Moeller SJ, Lilburn MS, Zerby HN. Interrelations between the microbiotas in the litter and in the intestines of commercial broiler chickens. Applied and Environmental Microbiology. 2010;76(19):6572–82. doi: 10.1128/AEM.00180-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Just N, Duchaine C, Singh B. An aerobiological perspective of dust in cage-housed and floor-housed poultry operations. Journal of Occupational medicine and toxicology. 2009;4(1):1–8. doi: 10.1186/1745-6673-4-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Feddes JJ, Zuidhof MJ, Cook H. Characterization of airborne dust particles in turkey housing. 1992. [Google Scholar]
- 36.Ahaduzzaman M, Milan L, Morton CL, Gerber PF, Walkden-Brown SW. Characterization of poultry house dust using chemometrics and scanning electron microscopy imaging. Poultry Science. 2021:101188. doi: 10.1016/j.psj.2021.101188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luiken RE, Van Gompel L, Bossers A, Munk P, Joosten P, Hansen RB, et al. Farm dust resistomes and bacterial microbiomes in European poultry and pig farms. Environment International. 2020;143:105971. doi: 10.1016/j.envint.2020.105971 [DOI] [PubMed] [Google Scholar]
- 38.Bindari YR, Kheravii SK, Morton C, Wu S-B, Walkden-Brown SW, Gerber PF. Molecular detection of Eimeria species and Clostridium perfringens in poultry dust and pooled excreta of commercial broiler chicken flocks differing in productive performance. Veterinary Parasitology. 2021:109361. doi: 10.1016/j.vetpar.2021.109361 [DOI] [PubMed] [Google Scholar]
- 39.Assen AM, Stillman M, Alfirevich S, Gerber PF, Groves PJ, Walkden-Brown SW. Assessment of A20 infectious laryngotracheitis vaccine take in meat chickens using swab and dust samples following mass vaccination in drinking water. Veterinary Microbiology. 2020;251:108903. doi: 10.1016/j.vetmic.2020.108903 [DOI] [PubMed] [Google Scholar]
- 40.Ahaduzzaman M, Walkden-Brown SW, Gerber PF, Keerqin C, Kumar A, Musigwa S, et al. Detection and quantification of Clostridium perfringens and Eimeria spp. in poultry dust using real-time PCR under experimental and field conditions. Avian Diseases. 2020. [DOI] [PubMed] [Google Scholar]
- 41.Walkden-Brown SW, Islam A, Groves PJ, Rubite A, Sharpe SM, Burgess SK. Development, application, and results of routine monitoring of Marek’s disease virus in broiler house dust using real-time quantitative PCR. Avian diseases. 2013;57(2s1):544–54. [DOI] [PubMed] [Google Scholar]
- 42.Ahaduzzaman M, Groves PJ, Walkden-Brown SW, Gerber PF. A molecular based method for rapid detection of Salmonella spp. in poultry dust samples. MethodsX. 2021;8:101356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ahaduzzaman M, Groves PJ, Sharpe SM, Williamson SL, Gao YK, Nguyen TV, et al. A practical method for assessing infectious laryngotracheitis vaccine take in broilers following mass administration in water: Spatial and temporal variation in viral genome content of poultry dust after vaccination. Vet Microbiol. 2020;241:108545. Epub 2020/01/14. doi: 10.1016/j.vetmic.2019.108545 . [DOI] [PubMed] [Google Scholar]
- 44.Assen AM, Walkden-Brown SW, Stillman M, Alfirevich S, Gerber PF. Comparison of tracheal and choanal cleft swabs and poultry dust samples for detection of Newcastle disease virus and infectious bronchitis virus genome in vaccinated meat chicken flocks. PLoS One. 2021;16(4):e0247729. Epub 2021/04/17. doi: 10.1371/journal.pone.0247729 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hilliar M, Keerqin C, Girish C, Barekatain R, Wu S-B, Swick R. Reducing protein and supplementing crystalline amino acids, to alter dietary amino acid profiles in birds challenged for subclinical necrotic enteritis. Poultry Science. 2020. doi: 10.1016/j.psj.2019.11.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nguyen TV, Ahaduzzaman M, Campbell DL, Groves PJ, Walkden-Brown SW, Gerber PF. Spatial and temporal variation of Marek’s disease virus and infectious laryngotracheitis virus genome in dust samples following live vaccination of layer flocks. Veterinary microbiology. 2019;236:108393. doi: 10.1016/j.vetmic.2019.108393 [DOI] [PubMed] [Google Scholar]
- 47.Fadrosh DW, Ma B, Gajer P, Sengamalay N, Ott S, Brotman RM, et al. An improved dual-indexing approach for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq platform. Microbiome. 2014;2(1):6. doi: 10.1186/2049-2618-2-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nature Methods. 2010;7(5):335–6. doi: 10.1038/nmeth.f.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26(19):2460–1. doi: 10.1093/bioinformatics/btq461 [DOI] [PubMed] [Google Scholar]
- 50.Ashelford KE, Chuzhanova NA, Fry JC, Jones AJ, Weightman AJ. At least 1 in 20 16S rRNA sequence records currently held in public repositories is estimated to contain substantial anomalies. Applied and Environmental Microbiology. 2005;71(12):7724–36. doi: 10.1128/AEM.71.12.7724-7736.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Applied and Environmental Microbiology. 2006;72(7):5069–72. doi: 10.1128/AEM.03006-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zakrzewski M, Proietti C, Ellis JJ, Hasan S, Brion M-J, Berger B, et al. Calypso: a user-friendly web-server for mining and visualizing microbiome–environment interactions. Bioinformatics. 2017;33(5):782–3. doi: 10.1093/bioinformatics/btw725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim B-R, Shin J, Guevarra RB, Lee JH, Kim DW, Seol K-H, et al. Deciphering diversity indices for a better understanding of microbial communities. Journal of Microbiology and Biotechnology. 2017;27(12):2089–93. doi: 10.4014/jmb.1709.09027 [DOI] [PubMed] [Google Scholar]
- 54.Goodrich JK, Di Rienzi SC, Poole AC, Koren O, Walters WA, Caporaso JG, et al. Conducting a microbiome study. Cell. 2014;158(2):250–62. doi: 10.1016/j.cell.2014.06.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Anderson MJ. Distance-based tests for homogeneity of multivariate dispersions. Biometrics. 2006;62(1):245–53. doi: 10.1111/j.1541-0420.2005.00440.x [DOI] [PubMed] [Google Scholar]
- 56.Schober P, Boer C, Schwarte LA. Correlation coefficients: appropriate use and interpretation. Anesthesia & Analgesia. 2018;126(5):1763–8. doi: 10.1213/ANE.0000000000002864 [DOI] [PubMed] [Google Scholar]
- 57.Ngunjiri JM, Taylor KJ, Abundo MC, Jang H, Elaish M, Kc M, et al. Farm stage, bird age, and body site dominantly affect the quantity, taxonomic composition, and dynamics of respiratory and gut microbiota of commercial layer chickens. Applied and environmental microbiology. 2019;85(9):e03137–18. doi: 10.1128/AEM.03137-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hou Q, Kwok L-Y, Zheng Y, Wang L, Guo Z, Zhang J, et al. Differential fecal microbiota are retained in broiler chicken lines divergently selected for fatness traits. Scientific reports. 2016;6(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Trudeau S, Thibodeau A, Côté J-C, Gaucher M-L, Fravalo P. Contribution of the broiler breeders’ fecal microbiota to the establishment of the eggshell microbiota. Frontiers in microbiology. 2020;11:666. doi: 10.3389/fmicb.2020.00666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pan D, Yu Z. Intestinal microbiome of poultry and its interaction with host and diet. Gut microbes. 2014;5(1):108–19. doi: 10.4161/gmic.26945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yan W, Sun C, Zheng J, Wen C, Ji C, Zhang D, et al. Efficacy of fecal sampling as a gut proxy in the study of chicken gut microbiota. Frontiers in microbiology. 2019;10:2126. doi: 10.3389/fmicb.2019.02126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Andreani NA, Donaldson CJ, Goddard M. A reasonable correlation between cloacal and cecal microbiomes in broiler chickens. Poultry Science. 2020;99(11):6062–70. doi: 10.1016/j.psj.2020.08.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Videvall E, Strandh M, Engelbrecht A, Cloete S, Cornwallis CK. Measuring the gut microbiome in birds: comparison of faecal and cloacal sampling. Molecular ecology resources. 2018;18(3):424–34. doi: 10.1111/1755-0998.12744 [DOI] [PubMed] [Google Scholar]
- 64.Apajalahti J, Kettunen A, Graham H. Characteristics of the gastrointestinal microbial communities, with special reference to the chicken. World’s Poultry Science Journal. 2004;60(2):223–32. [Google Scholar]
- 65.Skóra J, Matusiak K, Wojewódzki P, Nowak A, Sulyok M, Ligocka A, et al. Evaluation of microbiological and chemical contaminants in poultry farms. International Journal of Environmental Research and Public Health. 2016;13(2):192. doi: 10.3390/ijerph13020192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chinivasagam HN, Tran T, Maddock L, Gale A, Blackall PJ. Mechanically ventilated broiler sheds: a possible source of aerosolized Salmonella, Campylobacter, and Escherichia coli. Applied and environmental microbiology. 2009;75(23):7417–25. Epub 2009/10/02. doi: 10.1128/AEM.01380-09 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cook KL, Netthisinghe AM, Gilfillen RA. Detection of pathogens, indicators, and antibiotic resistance genes after land application of poultry litter. J Environ Qual. 2014;43(5):1546–58. Epub 2015/01/21. doi: 10.2134/jeq2013.10.0432 . [DOI] [PubMed] [Google Scholar]
- 68.Chinivasagam HN, Tran T, Maddock L, Gale A, Blackall PJ. The aerobiology of the environment around mechanically ventilated broiler sheds. Journal of Applied Microbiology. 2010;108(5):1657–67. doi: 10.1111/j.1365-2672.2009.04571.x [DOI] [PubMed] [Google Scholar]
- 69.Lin Y, Xu S, Zeng D, Ni X, Zhou M, Zeng Y, et al. Disruption in the cecal microbiota of chickens challenged with Clostridium perfringens and other factors was alleviated by Bacillus licheniformis supplementation. PloS one. 2017;12(8):e0182426. doi: 10.1371/journal.pone.0182426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stanley D, Keyburn AL, Denman SE, Moore RJ. Changes in the caecal microflora of chickens following Clostridium perfringens challenge to induce necrotic enteritis. Veterinary microbiology. 2012;159(1–2):155–62. doi: 10.1016/j.vetmic.2012.03.032 [DOI] [PubMed] [Google Scholar]
- 71.Wu S-B, Stanley D, Rodgers N, Swick RA, Moore RJ. Two necrotic enteritis predisposing factors, dietary fishmeal and Eimeria infection, induce large changes in the caecal microbiota of broiler chickens. Veterinary microbiology. 2014;169(3–4):188–97. doi: 10.1016/j.vetmic.2014.01.007 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX)
PERMDISP = permutational multivariate dispersions test calculated using Bray-Curtis and Jaccard distance metrics. R-square is the proportion of the variance explained by the group. Cumulative data from days 13–35 were used (n = 92 samples). The challenged group received 2,500 sporulated oocyst of E. brunetti, and 5,000 sporulated oocysts of E. maxima and E. acervulina on day 9 and 108 colony forming units of C. perfringens on days 14 and 15. The unchallenged group received 1 ml sterile phosphate buffer solution on day 9 and sterile broth on day 14 and 15.
(DOCX)
(XLSX)
(XLSX)
(XLSX)
(DOCX)
(DOCX)
(DOCX)
Data Availability Statement
All relevant data are within the manuscript and its Supporting information files.