

SUMMARY
Alcohol-associated liver disease (ALD) is a global health issue and leads to progressive liver injury, comorbidities, and increased mortality. Human-relevant preclinical models of ALD are urgently needed. Here, we leverage a triculture human Liver-Chip with biomimetic hepatic sinusoids and bile canaliculi to model ALD employing human-relevant blood alcohol concentrations (BACs) and multimodal profiling of clinically relevant endpoints. Our Liver-Chip recapitulates established ALD markers in response to 48 h of exposure to ethanol, including lipid accumulation and oxidative stress, in a concentration-dependent manner and supports the study of secondary insults, such as high blood endotoxin levels. We show that remodeling of the bile canalicular network can provide an in vitro quantitative readout of alcoholic liver toxicity. In summary, we report the development of a human ALD Liver-Chip as a powerful platform for modeling alcohol-induced liver injury with the potential for direct translation to clinical research and evaluation of patient-specific responses.
Graphical abstract
In brief
Nawroth et al. develop an advanced human Liver-Chip model of alcohol-associated liver disease (ALD) with direct translation to clinical research. The chip recapitulates early critical events of ALD, such as lipid accumulation and oxidative stress, mimics disease worsening due to leaky gut syndrome, and models recovery following alcohol abstinence.
INTRODUCTION
Fatty liver disease (FLD), or hepatic steatosis, is a growing global health problem that affects up to 30% of the general population in Western countries (Asrani et al., 2019; Bedogni et al., 2014; Wong et al., 2019). The disease is classified based on the causative trigger as either alcohol-associated liver disease (ALD) or diet-induced non-alcoholic fatty liver disease (NAFLD) (Bedogni et al., 2014), which share several genetic susceptibility markers (Hotta et al., 2010; Meroni et al., 2018) and progress from simple hepatic steatosis to steatohepatitis and liver fibrosis and finally to irreversible cirrhosis and hepatocellular carcinoma (Chacko and Reinus, 2016; Magdaleno et al., 2017; Orman et al., 2013; Rehm et al., 2009; Scaglioni et al., 2011; Toshikuni et al., 2014). In Western countries, approximately half of all deaths from liver cirrhosis are due to ALD resulting from alcohol abuse (Magdaleno et al., 2017; Rehm et al., 2009; Singal et al., 2018). ALD is also a co-factor in the progression of chronic viral hepatitis, NAFLD, iron overload, and other liver diseases (Crabb et al., 2020).
Currently, diagnosis of ALD relies on a combination of history and clinical and laboratory findings. Because alcohol intake is the established cause of clinical alcohol-associated steatohepatitis (ASH), patients presenting with hepatic steatosis and a history of alcohol intake above 20 g/d for women and 30 g/d for men are commonly diagnosed with ALD/ASH (Bedogni et al., 2014). To improve diagnostics and develop effective treatments, it is important to develop faithful models of the human liver responses to disease-relevant challenges. However, the great majority of the experimental studies on FLD have employed animal models that display aspects of the liver disease phenotype but do not capture the spectrum of the metabolic, inflammatory, and fibrotic responses found in human patients (Bertola, 2018; Santhekadur et al., 2018). Similarly, in vitro models such as hepatocyte sandwich culture and liver spheroids may recapitulate several of the features of liver diseases but are missing the dynamics of the tissue microenvironment and cell-cell interactions. Common models have been unable to recapitulate systemic effects induced by alcohol in humans, such as compromised intestinal epithelial barrier function and increased gut permeability (“leaky gut”), a response subject to significant species-dependent variability (Bishehsari et al., 2017). Leaky gut and the ensuing inflammatory response due to the escape of bacterial endotoxins to the portal and systemic circulation are thought to directly contribute to the development of ASH (Bishehsari et al., 2017).
We hypothesized that a microphysiological system (MPS) of the human liver (Liver-Chip) using clinically relevant blood-alcohol concentrations (BACs) could recapitulate alcohol-induced and endotoxin-mediated hepatic tissue injury and hence provide a platform for studying ALD/ASH. Ethanol-induced alterations in hepatic cytochrome P450 CYP2E1 and their role in the mechanisms driving liver damage (Seitz and Stickel, 2006) can be studied in human MPS, whereas in commonly used cell lines (e.g., HepG2, HuH7 cells) or the standard sandwich primary hepatocyte culture, the expression of these enzymes is not maintained beyond the early time of the culture (Jang et al., 2019).
Here, we advanced our previously published Liver-Chip for drug toxicity screening (Jang et al., 2019) with additional capabilities to recapitulate early, critical events in human ALD using alcohol at concentrations as those found in the blood of patients. We demonstrate the ability of the ALD Liver-Chip to model recovery of early injury through alcohol abstinence, as well as the worsening of the phenotype in a two-hit model of co-exposure to alcohol and bacterial endotoxin. We optimized the extracellular matrix (ECM) scaffolding to achieve robust formation of a biomimetic BC network, and we propose that ethanol-induced disruptions of the BC network are a sensitive and early marker of toxicity.
Together, we demonstrate that our ALD Liver-Chip recapitulates early events in human-relevant alcohol-induced steatosis and remodeling that may be missed by other in vitro systems.
RESULTS
Development of the Liver-Chip for modeling ALD/ASH
To model human ALD, we continuously perfused an advanced version of our recently reported Liver-Chip, an organotypic MPS using primary human cells (Jang et al., 2019), with ethanol doses within clinically relevant BACs, followed by multimodal phenotyping and functional analysis. The Liver-Chip is made of polydimethylsiloxane (PDMS) and contains an upper channel and a lower channel separated by a porous membrane to allow for cell-cell interactions. The tri-culture configuration used in this study includes primary hepatocytes cultured in the upper channel and primary liver sinusoidal endothelial cells (LSECs) and Kupffer cells cultured in the lower channel (Figure 1A). The co-culture of primary hepatocytes with LSECs is essential for sustained hepatocyte physiological functions over time, and Kupffer cells modulate important inflammatory responses (Bale et al., 2015; Jang et al., 2019). We further aimed to recreate the extensive bile canaliculi (BC) network that is established between neighboring hepatocytes in the liver. In mice, changes to the BC network structure are an early and sensitive indicator of liver drug toxicity (Meyer et al., 2017). BC network structure is also altered in human patients with NAFLD (Segovia-Miranda et al., 2019). We reasoned that alcohol-induced toxicity would generate similar remodeling suitable for digital pathology. However, in contrast to rodent hepatocytes, human hepatocytes do not readily form biomimetic bile canalicular networks in vitro but usually exhibit disconnected BC pockets (Abe et al., 2009; Deharde et al., 2016; Nakakariya et al., 2012; Reif et al., 2015; Swift et al., 2010). Inspired by the recent finding that hepatocyte polarization and BC lumen formation are driven by symmetric mechanical anchoring to the matrix (Li et al., 2016), we probed whether optimizing ECM scaffold composition, thickness, and homogeneity in the Liver-Chip could improve BC network integrity (see STAR Methods and Figure S1). We tested five different scaffolding designs consisting of Matrigel and collagen-I-based gels (“ECM-A” to “ECM-D” in Figure S1G) and used a digital pathology approach to quantify the MRP2-stained BC network branching density, overall area fraction (porosity), and branching radius, which describe BC network integrity (Meyer et al., 2017) (Figure S1H). We chose for the Liver-Chip the matrix protocols “ECM-C” and “ECM-D,” which reproducibly elicited the best BC metrics throughout the chip channel (Figure 1C).
Figure 1. Development of the ALD Liver-Chip.
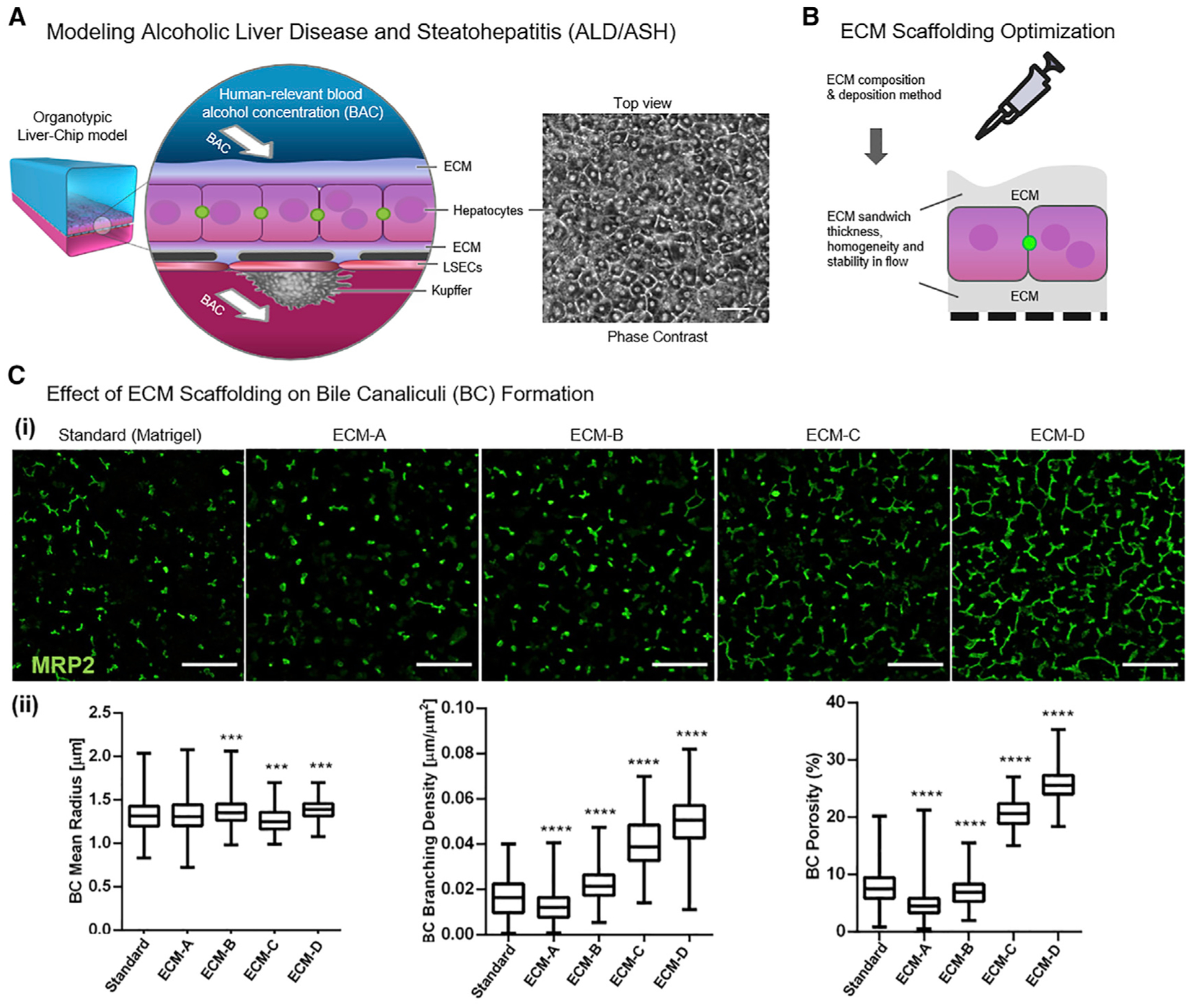
(A) Approach for modeling human ALD/ASH by exposing the organotypic Liver-Chip to human-relevant blood-alcohol concentrations (BACs). Scale bar, 50 μm.
(B) Different ECM composition and deposition methods were tested in the Liver-Chip to improve ECM scaffold thickness, homogeneity, and stability under flow.
(C) Optimization of BC network integrity in the Liver-Chip. Scale bar, 100 μm. (i) Representative images of BC networks (MRP2, green) of the triculture Liver-Chip as a function of ECM conditions. (ii) Effects of different ECM conditions on the radius, branching density, and area fraction of BC networks. Biomimetic hepatobiliary architecture (condition ECM-C and ECM-D) is characterized by higher branching density, higher porosity, and more narrowly distributed mean radius compared to control (standard). Data are from one experiment with n = 3 chips per condition. Data represent median ± (minimum and maximum), ***p < 0.001; ****p < 0.0001 versus control (branching density and porosity, Kruskal-Wallis and Dunnett’s multiple comparisons test; radius, Kolmogorov–Smirnov (KS) test and Bonferroni correction for multiple comparisons test).
Hepatic intracellular lipid accumulation, or steatosis, is the key histological finding of both diet- and alcohol-induced FLD (Scaglioni et al., 2011; Toshikuni et al., 2014). To confirm that the Liver-Chip is sufficiently sensitive to ethanol exposure for modeling ALD-like hepatocyte injury, we measured lipid accumulation upon short-term (48-h-long) treatment with ethanol. During the treatment, hepatocyte morphology and albumin levels were monitored routinely, which we had found to be a more sensitive method than lactate dehydrogenase activity (LDH) assays for detecting early liver toxicity effects. No evidence of significant liver toxicity effects was found. As a positive control for induction of steatosis, we exposed the chips to fatty acids, a reproducible experimental method for induction of the steatotic phenotype (Breher-Esch et al., 2018). Ethanol concentrations ranging from 0.08% to 0.16% were chosen to mimic the BAC of human patients after alcohol consumption (Elgammal et al., 2015). A BAC of 0.08% is the upper limit for legal driving in the United States and United Kingdom. Based on flow and material properties of chip and perfusion system, the expected loss of ethanol from the medium during the treatment duration was minimal (see STAR Methods). To increase the sensitivity of the Liver-Chip to ethanol in a reproducible manner, we adjusted the glucose, insulin, and cortisol concentrations of the cell culture medium toward more physiological ranges (see STAR Methods). Steatosis was induced following treatment with either ethanol (at BACs ranging from 0.08% to 0.16%) or oleic acid (1 μg/mL) for 48 h as visualized by AdipoRed staining (Figure 2A, i). The average lipid droplet size increased with rising ethanol concentrations (Figure 2A, ii), indicating the sensitivity of the Liver-Chip to operate within a range of clinically relevant BACs, as required for disease modeling (Bala et al., 2016; Takahashi and Fukusato, 2014). Triglyceride (TG) storage did not change significantly, likely due to the short treatment window.
Figure 2. Assessment of liver toxicity, metabolic changes, and polyploidy in the ALD/ASH Liver-Chip.
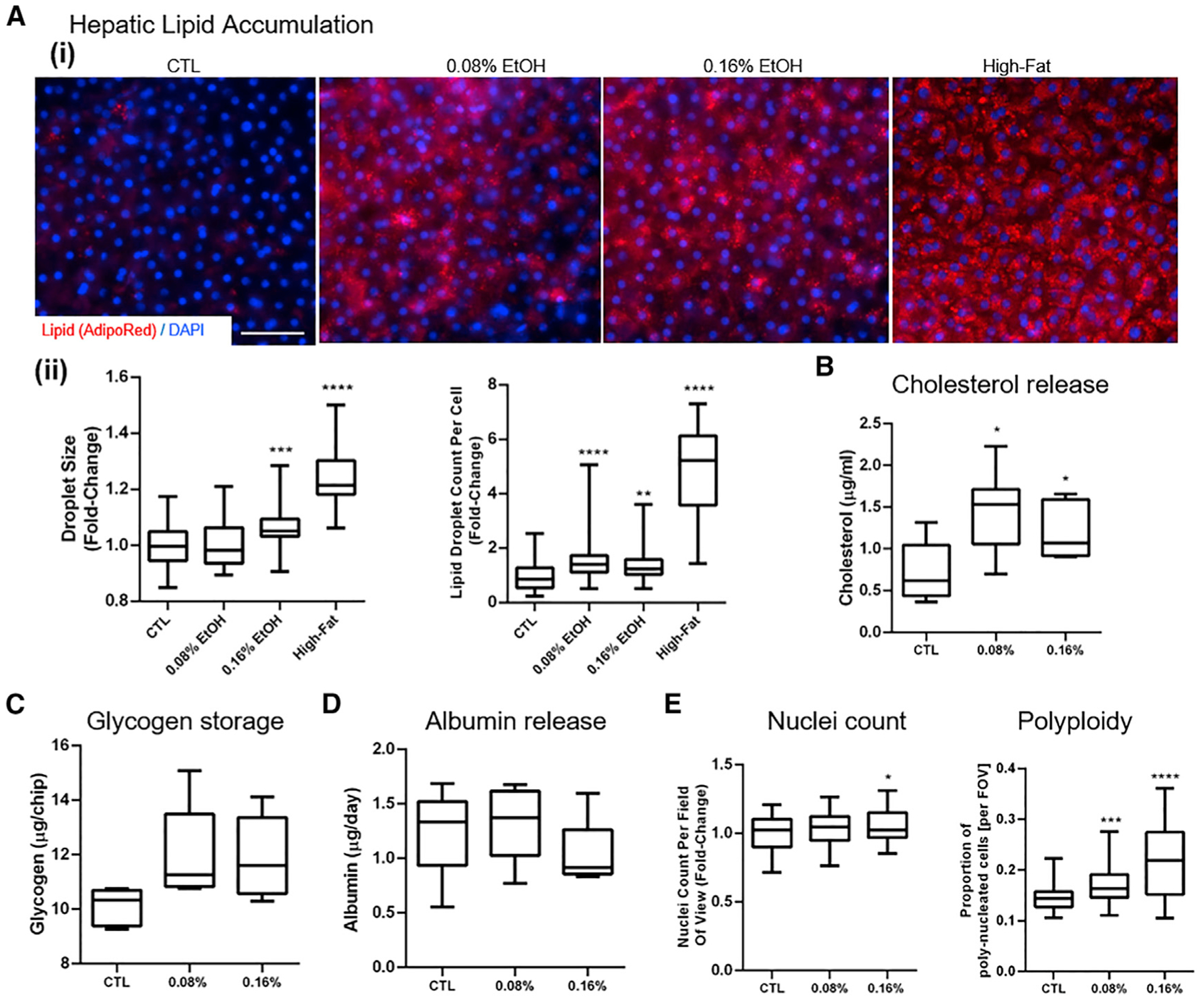
(A) Hepatic lipid accumulation. (i) Lipid droplet accumulation in hepatocytes visualized using AdipoRed staining after administration of fat (oleic acid 1 μg/mL; positive control) or ethanol (0.08% and 0.16%) for 48 h. Scale bar, 50 μm. (ii) Number of lipid droplets per cell and lipid droplet size (projected area).
(B–D) Quantitative analysis of hepatic functional markers in the Liver-Chip after 48 h of exposure to physiologically relevant BACs. Fluorometric assessment using ELISA of cholesterol levels in the effluent (B), glycogen storage in cell lysate (C), and albumin release (D).
(E) Nuclei count per field of view and proportion of hepatocytes with multiple nuclei per cell (polyploidy). Data represent median ± (minimum and maximum). *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001 versus control (Kruskal-Wallis and Dunnett’s multiple comparisons test).
Next, we assessed whether the lipid accumulation observed in the Liver-Chip in response to ethanol treatment was associated with signs of metabolic dysregulation, although this usually requires more prolonged exposure to the challenge (Tsukamoto, 2015). The 48-h exposure to human-relevant BACs resulted in a significant increase in cholesterol levels in the effluent (Figure 2B), and to a lesser degree, in the cell lysates (Figure S2A). We confirmed this finding with hepatocytes from two different donors, which showed that despite the donor-to-donor variability in baseline cholesterol release (Figure S3A), there was no significant difference in response to ethanol (Figure S3C). Glycogen storage, measured in cell lysates, showed a slight, but not significant, increase upon exposure to 0.08% ethanol (Figure 2C), while no changes were detected in glucose release (Figure S2B), except with toxic ethanol concentrations above 0.32% (data not shown). Similarly, albumin release was not affected by this short-term exposure to ethanol (Figure 2D), in line with clinical data in patients where such changes develop over time and reflect severe deterioration of liver function (Dyson et al., 2014; Petersen et al., 2017). Exposure to ethanol had a minimal effect on the number of hepatocytes nuclei counted per field of view but increased polyploidy, thought of as a marker of tissue stress (Zhang et al., 2018) (Figure 2E). High-fat treatment did not have this effect (Figure S2C).
To assess for changes in gene expression underpinning the above phenotypic treatment responses, we performed RNA sequencing (RNA-seq) analysis and identified the differentially expressed genes (DEGs) and the associated stratification, based on the magnitude of differences between ethanol-exposed and control chips (Figure S4A). Ethanol-exposed chips (at concentrations of either 0.08% or 0.16%) had 123 DEGs (red dots, adjusted p value < 0.05 and |log2 fold change| > 1), of which 87 were upregulated and 36 were downregulated (Figure S4B). Pathway analysis of the DEGs using the Kyoto Encyclopedia of Genes and Genomes (KEGG) bioinformatics database revealed changes in the expression of genes implicated in functions consistent with the readouts discussed above (Figure S4C). Relevant to lipid metabolism, we found upregulation of three genes related to cholesterol transport, NR0B2 (Smalling et al., 2013), APOA4 (Kang et al., 2017), and APOA1 (Castera et al., 2019); and another seven genes related to cholesterol biosynthesis, MVK, MVD, and HMGCS1 (Iravani et al., 2018), FDFT1 (Stättermayer et al., 2014), HMGCR (Min et al., 2012), FDPS (Tommasi and Besaratinia, 2019), and CYP51A1 (Kakehashi et al., 2017); along with downregulation of AKR1D1 (Nikolaou et al., 2019), involved in cholesterol catabolism (Figure 3A). Glycolysis/gluconeogenesis, steroid hormone biosynthesis, and insulin resistance/type II diabetes were also among the key pathways affected by ethanol treatment (Figure 3B). Further, ethanol induced significant changes in the expression of members of the alcohol dehydrogenase (ADH) gene family, including ADH1C (Aljomah et al., 2015), ADH1B (Li et al., 2018), and ADH1A, all involved in alcohol metabolism (Li et al., 2018). Notably, alcohol induced the expression of CYP2E1, critical for ethanol oxidation and the associated induction of oxidative stress (Figure 3C), a major pathogenic mechanism of ALD (Caro and Cederbaum, 2004). Further, exposure to ethanol affected the expression of clinically relevant genes involved in bile acid production and processing, including the major BC transporters ABCB4 (MDR3) (Farrell et al., 2012), ABCB1 (MDR1) (Pirola and Sookoian, 2018), ABCC3 (MRP-3) (Atilano-Roque et al., 2016), SLC10A1 (NTCP) (Brunt, 2017), ABCB11 (BSEP) and ABCC2 (MRP2) (Arab et al., 2017), and ADCY8 (Liang et al., 2018); along with genes associated with bile acid processing, such as SLC27A5, involved in fatty acid elongation (Benedict and Zhang, 2017); CYP7A1 (Chiang, 2018; Liu et al., 2017), which regulates the overall bile acid production rate; and CYP27A1 (Chiang, 2017), associated with the bile acids synthesis alternative pathway (Figure 3D). Our data show that the ALD Liver-Chip recapitulated the impact of ethanol treatment on genes involved in the metabolism of alanine, aspartate, and glutamate. In line with previous data showing the link between FLD and DNA damage in hepatocytes (Seitz and Stickel, 2006), our analysis revealed upregulation of POLE and POLD2 (Kuttippurathu et al., 2016), both involved in DNA replication and repair, as well as RAD51 and FANCB, which are expressed at DNA damage sites and implicated in homologous recombination (Nomura et al., 2007; Owada et al., 2018) (Figure 3E). Further, we saw upregulation of E2F1 (Denechaud et al., 2016) and CCNB1 (Gentric et al., 2015) and downregulation of KIF14 (Yang et al., 2013) and CCNE2 (Natarajan et al., 2017), all participating in cell-cycle regulation (Figure 3F). As several of the DNA-damage-associated genes identified in our data have not yet been implicated in the pathogenesis of FLD, this suggests the potential of the ALD Liver-Chip platform for identification of unknown mechanisms involved in alcohol-induced damage. Lastly, ethanol exposure led to the dysregulation of several markers of oxidative stress in the Liver-Chip, such as genes of the metallothionein family (MT1) (Ruttkay-Nedecky et al., 2013; Si and Lang, 2018; Waller-Evans et al., 2013) (Figures S4C and 3G) and altered the expression of DUSP1 (Ye et al., 2019) (Figure 3G). While some of the gene expression changes shown in Figure 3 are nonsignificant or present with |log2 fold change| < 1, they are listed for context, as they are part of known pathways involved in ALD.
Figure 3. Gene expression profiling of the Liver-Chip induced by physiologically relevant ethanol concentrations.
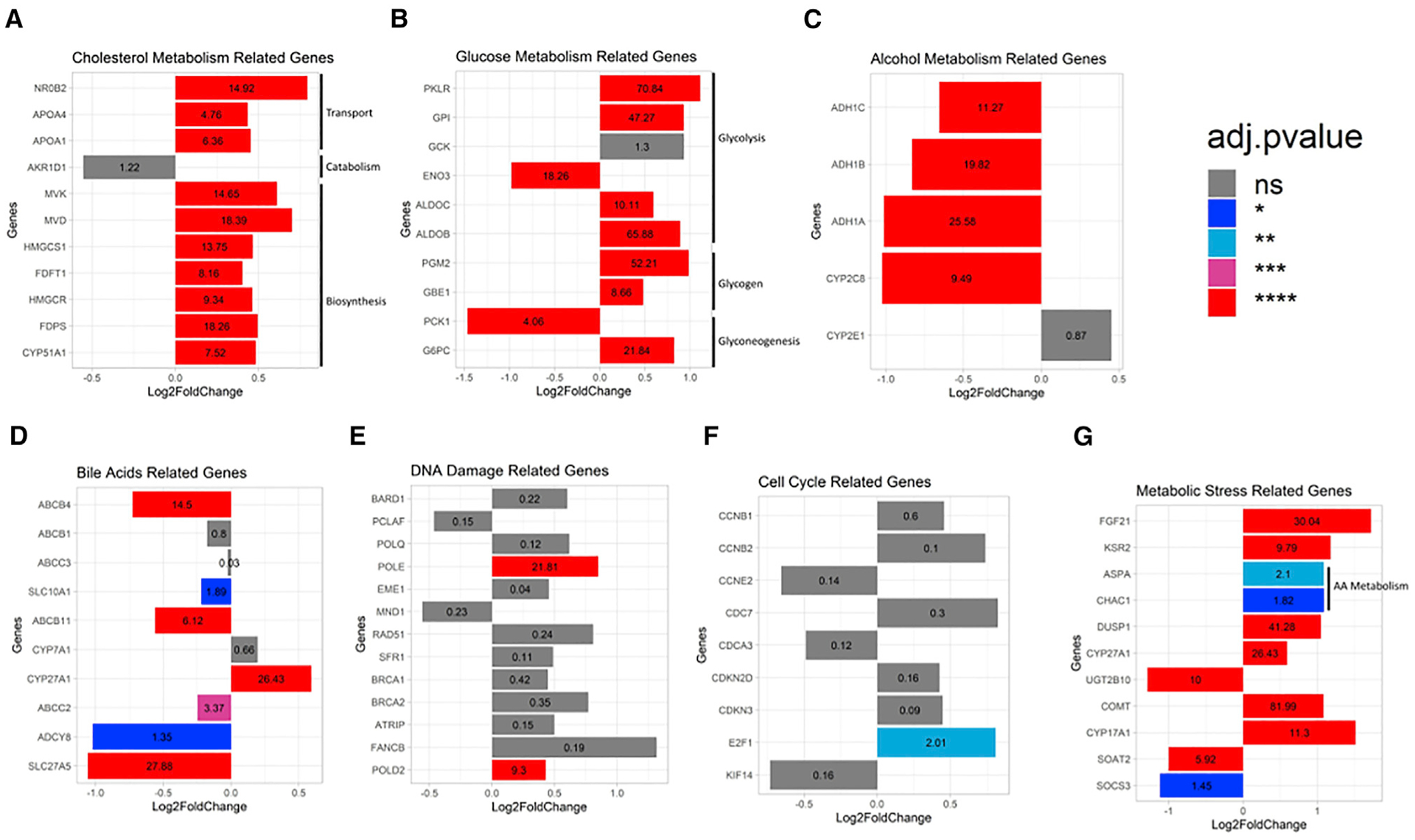
(A–G) Differential gene expression analysis in hepatocytes from ethanol-treated (exposed for 48 h at either 0.08% or 0.16%; see STAR Methods) and control Liver-Chip revealed significant differences in the expression of genes related to alcohol metabolism (A), cholesterol metabolism (B), glucose metabolism (C), bile acid production and maintenance (i.e., cholestasis) (D), DNA damage (E), cell-cycle regulation (F), and oxidative and metabolic stress (G). The adjusted p value is listed within each bar, with statistical significance indicated by bar color and *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. Data are from one experiment with two to five chips per condition.
Modeling the two-hit hypothesis for ALD/ASH development
Having established that the Liver-Chip responds to ethanol, we next explored the possibility of modeling the two-hit hypothesis, which states that simple steatosis induced by alcohol requires a second insult for progression to ASH (Tsukamoto et al., 2009). Ethanol consumption compromises the intestinal barrier function, resulting in increased permeability to intestinal endotoxins, such as bacterial lipopolysaccharide (LPS), from the gut to the liver via the portal vein (Bishehsari et al., 2017). The endotoxins are thought to act as the critical second hit in the progression of ALD to inflammation (ASH) (Szabo and Bala, 2010). Therefore, we tested whether co-exposure of the Liver-Chip to ethanol and LPS would worsen steatosis and the oxidative stress in the ALD Liver-Chip. We found that the size of hepatic lipid droplets was increased after 48 h of exposure to 0.08% ethanol + LPS (Figure 4A). We also found a dose-dependent increase in mitochondrial reactive oxygen species (ROS) events in response to ethanol, which was further exacerbated upon co-exposure to ethanol and LPS. Treatment with LPS alone had no effect, highlighting the specificity of the response (Figure 4B). These results are in line with our RNA-seq data showing the effects of alcohol on genes related to DNA damage and the cell cycle and are consistent with reported disease mechanisms. Induction of CYP450 by ethanol and free fatty acids and the consequential oxidative stress rampage have been implicated in the increased DNA damage in hepatocytes and the associated progression to hepatocellular carcinoma (HCC) (Seitz and Stickel, 2006).
Figure 4. Modeling the two-hit hypothesis for ALD/ASH and alcohol abstinence in the ALD Liver-Chip.
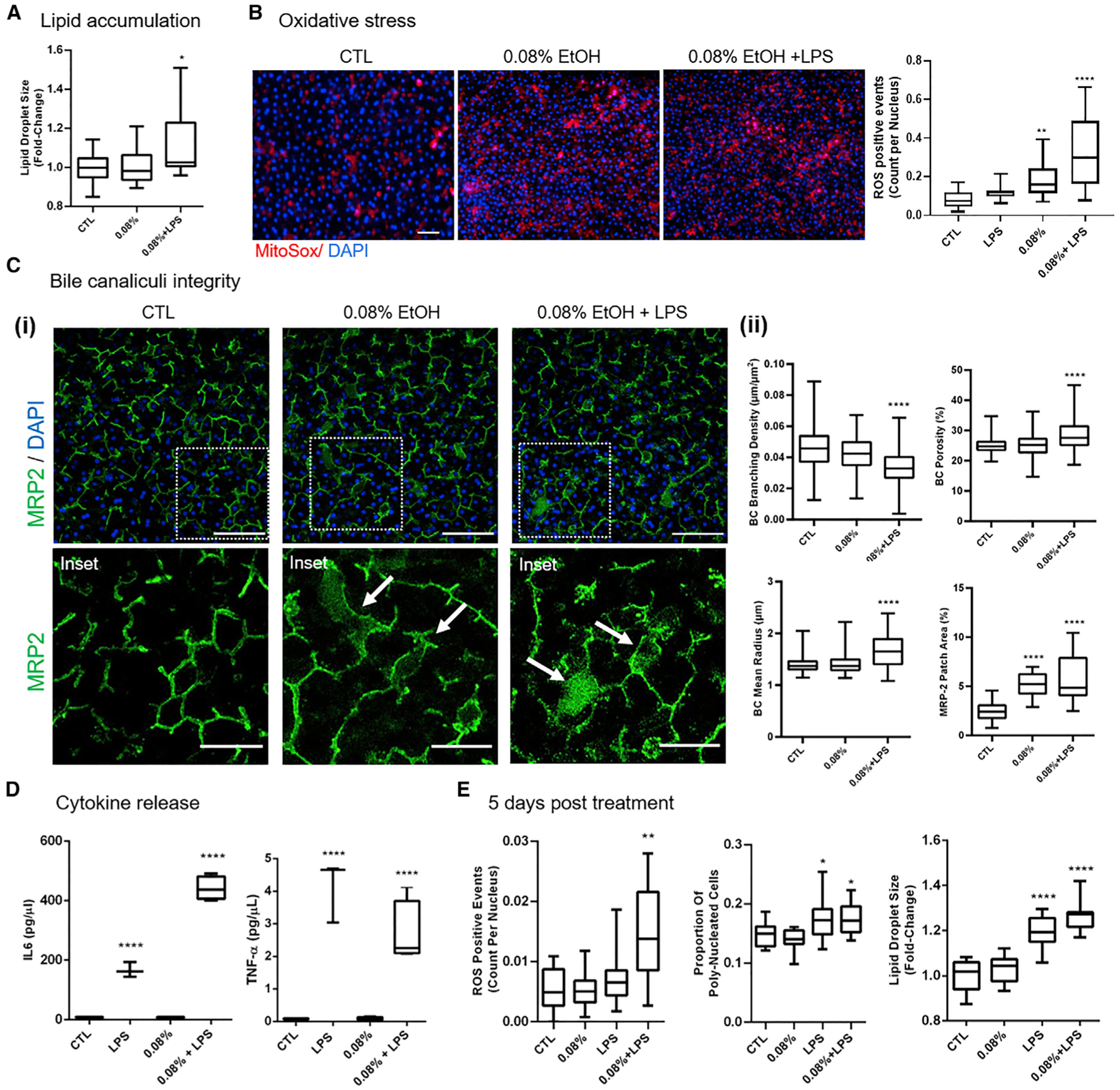
Data were collected after 48 h of exposure to 0.08% ethanol or ethanol + LPS.
(A) Quantification of mean lipid droplet size in the hepatocytes. Scale bar, 100 μm.
(B) Representative images of MitoSox staining in the hepatocytes (left) and quantification of ROS events (right).
(C) BC network changes in responses to treatment. (i) Representative images of changes in MRP2 BC staining (green) showing large MRP2-positive patches in the treatment conditions (arrows). Main scale bar, 100 μm; inset scale bar, 50 μm. (ii) Quantification of the changes in BC radius, branching density, and porosity, as well as the relative area of MRP2 patches.
(D) Release of IL-6 and TNF-α as measured by multiplexed immunoassays. Data are from two (48 h ethanol) or one (48 h high-fat diet; 5 days of recovery) independent experiments; minimally n = 2 chips per condition, five to eight images per chip where applicable. Data represent median ± (minimum and maximum). *p < 0.05; **p < 0.01; ****p < 0.0001 versus control (Kruskal-Wallis and Dunnett’s multiple comparisons test).
(E) Quantification of mean lipid droplet size and frequency of oxidative stress events and polyploidy in hepatocytes after 48 h of exposure of the Liver-Chip to either ethanol or ethanol + LPS followed by 5 days of recovery without exposure to ethanol.
Inflammatory cytokines are significantly elevated in patients with alcoholism with advanced liver disease (Urbaschek et al., 2001) and have been proposed as therapeutic targets, as inhibition of tumor necrosis factor α (TNF-α) action was protective against alcohol-induced liver injury in mouse models (Hines and Wheeler, 2004). Therefore, we assessed the secretion of cytokines in response to ethanol and LPS, as our Liver-Chip model contains Kupffer cells, which are a major source of proinflammatory cytokines (Jang et al., 2019). Whereas treatment with LPS robustly increased the release of the proinflammatory cytokines interleukin-6 (IL-6) and TNF-α, co-treatment with 0.08% ethanol + LPS resulted in further increase in the production of IL-6 (Figure 4D), similar to findings from clinical and experimental studies demonstrating its role in disease progression (Bird et al., 1990; Remmler et al., 2018; Urbaschek et al., 2001). The cytokine rise is most likely linked to oxidative stress (Figure 4B) and the associated cell injury or other mechanisms as previously described (Schmidt-Arras and Rose-John, 2016).
Patients with ALD frequently manifest clinical and/or histologic evidence of cholestasis (Tung and Carithers, 1999), defined as decrease in bile secretion or obstruction of bile flow through the intra- or extrahepatic bile transport network. Currently, the mechanisms underlying the alcohol-induced cholestasis remain poorly understood. Our RNA-seq data revealed altered expression of BC-related genes in response to ethanol treatment, indicative of potential damage to BC integrity, a known finding in cholestatic livers (Jansen et al., 2017). We have recently shown the ability of our human Liver-Chip to recapitulate in vivo findings on drug-induced destruction of BC transporters (Jang et al., 2019). In the current study, the Liver-Chip was optimized to develop biomimetic BC networks, allowing us to directly measure the presence of cholestasis-associated changes, such as BC dilation (Chung et al., 2002; Fickert et al., 2002; Popper, 1981). For this, we quantitatively measured the MRP2-stained BC network to assess alcohol-induced cholestatic structural changes at the level of the canaliculi (Figure 4C, i). In the Liver-Chips exposed to 0.08% ethanol, we identified large patches of MRP2 expression (Figure 4C, ii), which could either indicate cholestatic protrusions of the BC network (Jansen et al., 2017) or abnormal MRP2 internalization, as described in human cholestatic livers (Chai et al., 2015). These MRP2 patches were significantly expanded in those with 0.08% ethanol + LPS together, consistent with the more severe hepatic injury (Figure 4C, ii). Further, in the latter the average BC radius increased throughout the BC network, whereas the branching density was decreased. BC dilation, leading to reduced biliary clearance (Meyer et al., 2017), together with the loss of connectivity indicate a worsening of hepatic damage, in line with the two-hit hypothesis for the pathogenesis of ASH/NASH.
Modeling of hepatocyte recovery following abstinence from alcohol with the Liver-Chip
Clinical data indicate the potential of the steatotic liver to repair the associated hepatocyte injury following timely abstinence from alcohol or with diet modifications (Menon et al., 2001). Thus, we assessed the sensitivity of the “alcoholic” Liver-Chip to respond to withdrawal from ethanol or ethanol + LPS. Treatment of the Liver-Chip for 48 h with ethanol or ethanol + LPS as above was followed by a 5-day-long treatment-free “recovery” period. By the end of the recovery period, oxidative stress, polyploidy, and lipid droplet size normalized in ethanol-only-treated chips, whereas this was not the case for the ethanol + LPS condition (Figure 4E). Although it is possible that a longer recovery period might lead to recovery of all treatment groups, these data suggest that the ALD Liver-Chip shows an insult-dependent recovery, which parallels the lack of repair of hepatic injury in patients with more advanced alcoholic disease and indicates the potential of the Liver-Chip to model recovery from ethanol-induced damage in a clinically relevant way.
DISCUSSION
Here, we leveraged a human Liver-Chip to enable the preclinical modeling of progressive alcohol-induced liver injury, a major health concern that is on the rise globally. We chose the Liver-Chip as it supports a complex co-culture system containing the main cell types found in the liver. Overwhelming evidence from the literature indicates that non-parenchymal cells are heavily involved in driving ALD (Cohen and Nagy, 2011). This implies that responses of monocultures to ethanol are unlikely to reflect the response of the multi-cell models, which is also driving the growing interest in using precision cut liver slices with multi-cell-type architecture for studying ethanol toxicity (Klassen et al., 2008). Applying a short-term experimental design to probe the system’s sensitivity, we showed that the ALD Liver-Chip model recapitulates critical disease signs such as intracellular accumulation of lipids, development of oxidative stress, and cholesterol synthesis dysregulation upon exposure to alcohol for 48 h (Lieber, 2004; Menon et al., 2001). Our study also suggested that changes to the biomimetic BC network are a sensitive maker of liver injury, a finding corroborated by altered gene expression of BC transporters. This rapid and multimodal response of the chip to ethanol could facilitate fast go/no-go decisions in drug development studies.
The ALD Liver-Chip recapitulated known effects of LPS on hepatocytes, demonstrating the model’s sensitivity to the two-hit injury described in ASH (Tsukamoto et al., 2009). In extension, our results suggest that chronic alcohol intake coupled with systemic inflammation worsens the liver damage and may significantly compromise recovery in ALD/ASH, similar to the responses of patients in advanced disease stages (Zhou and Zhong, 2017). In animal experiments, administration of antibiotics to reduce endotoxemia or inactivation of Kupffer cells with gadolinium chloride helped prevent liver injury (Thurman, 1998). The ALD Liver-Chip model could be useful as a platform to determine human relevancy of proposed mechanisms for new therapeutic approaches and the role of LPS in ASH progression, support patient-specific responses using donor-specific cells, and probe the progression of alcoholic FLD to fibrosis and cirrhosis (Lieber, 2004). Here, it is worth mentioning that the optimized ECM scaffold (Figures 1B and S1) supports the embedding and co-culture of hepatic stellate cells to generate a quad-culture Liver-Chip (data not shown), as needed for modeling fibrosis and progression of alcohol-induced steatosis.
In summary, the ALD Liver-Chip provides a promising platform toward modeling human ALD and study clinically relevant metabolic events, including ethanol metabolism, lipogenesis, biliary function, and oxidative stress.
Limitations of study
The present article was submitted for consideration in September 2019, and reviewers’ comments were received in October 2020. While we resolved the majority of the reviewers’ concerns, we were unable to conduct additional studies due to the consequences of the coronavirus disease 2019 (COVID-19) pandemic in our operations, leading to three main limitations of the present study. First, a future full characterization of tight junctions, cell-cell contacts, and bile acid distribution would be very desirable to better understand the mechanisms and context of the altered BC architecture. Second, while our previous study strongly suggested that tricultures containing hepatocytes, LSECs, and Kupffer cells are essential for modeling multiple mechanisms of action (Jang et al., 2019), it would be informative to study the effect of ethanol on various mono- and co-culture configurations in order to further elucidate the role of the different cell types in ALD. Lastly, the 48-h window was likely too short to see significant changes in TG storage and other metabolic responses, and it limited the changes in gene expression detected by RNA-seq. Further, the recovered lysate from the Liver-Chip was not sufficient to conduct western blots and investigate the associated protein-level changes. In closing, our data on the ALD Liver-Chip’s responses to ethanol is robust and clinically relevant. Nonetheless, the platform’s value to the community would benefit from additional studies on hepatocyte remodeling, the role of individual cell types, longer-term exposure to ethanol, and changes in protein expression.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Janna Nawroth (jnawroth@gmail.com).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The RNA-seq data generated during this study are available at the NCBI Gene Expression Omnibus (GEO) database repository (GEO: GSE175396).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cryopreserved primary human hepatocytes from two healthy donors, Donors G and Q, were purchased from Triangle Research Labs (Lonza) and GIBCO (Thermo Fisher Scientific). Cells where cultured under standard human cell culture conditions (37°C, 5% CO2). Donor Q: Lot QHum15063, male, 6 years of age, ethnicity unknown; donor G: Lot HU8305, male, 53 years of age, Caucasian.
METHOD DETAILS
Liver-Chip culture
Liver-Chip culture was established according to our previously published protocol (Jang et al., 2019). Briefly, primary human hepatocytes (3.5 million cells/mL) from two healthy donors, Donors G and Q, were cultured on one side of a porous membrane (pore size ~7 microns) in the Emulate dual-channel microfluidic chip (Kasendra et al., 2018). Extracellular matrix sandwiching and composition in the Liver-Chip was optimized to support BC integrity (Figures 1B, 1C, and S1; STAR Methods sections below). On the other side of the membrane (the lower chamber of the chip) human primary LSECs (3 million cells/mL) and Kupffer cells (0.5 million cells/mL) were seeded to mimic the hepatic sinusoid architecture (Figure 1A). The two cell compartments were perfused independently, and flow rates were optimized to enable optimal survival and maturation of the different cell types (Jang et al., 2019). The seeded hepatocytes were overlaid with ECM as described in section below and maintained in William’s E Medium (WEM) containing Glutamax (GIBCO), ITS+ (Corning), dexamethasone (1μM, Sigma-Aldrich), ascorbic acid (0.05 mg/ml, Sigma-Aldrich), fetal bovine serum (Sigma-Aldrich), and penicillin/streptomycin (Sigma-Aldrich), and incubated at 37°C, 5% CO2. The vascular channel of the Liver-Chip was maintained with human endothelial media (Emulate, Inc.). Two days after seeding, the Liver-Chips were connected to the Human Emulation System (Emulate, Inc.) and both chip channels were perfused at 30 μL/h to provide a continuous supply of fresh medium for the duration of the experiments. Note that the hepatocytes were protected from the shear stress generated by this flow through the optimized gel coating, as discussed in the section on ECM optimization. Meanwhile, at 30 μL/h, the endothelial and Kupffer cells in the bottom channel experience a shear stress of ca. 0.008 dyn/cm2, which is lower than the average normal fluid shear level of 4 dyn/cm2 reported for microvasculature (Topper and Gimbrone, 1999). Hence, no shear-mediated damage of either parenchymal or non-parenchymal cells is expected.
In order to increase the sensitivity required for stimulus-dependent metabolic readouts, the traditional hepatocyte cell culture media was modified at day 5 in culture to a lower, more physiological relevant, concentration glucose of 1 g/L (~5.5 mmol/L) DMEM (GIBCO, Cat#11054001) (Wannamethee et al., 1999), and supplemented with non-essential amino acids solution (NEAA, 1:200 dilution), glutaMAX 1:100 dilution (Thermo Fisher), ascorbic acid (0.05 mg/ml, Sigma-Aldrich), dexamethasone (50nM, Sigma-Aldrich) and penicillin/streptomycin (Sigma-Aldrich). Moreover, ITS+ premix (Corning 1:100 dilution) was replaced by ITS (GIBCO, 1:500 dilution) in order to reduce the insulin concentration from 10 to 2 μg/ml, a more physiological relevant concentration (Wannamethee et al., 1999), and the FBS supplement was removed.
Optimization of Liver-Chip ECM for BC network formation
We had observed that the chip ports were frequently site of highly branched bile canalicular networks compared to cholestatic pockets in the chip channels (Figure S1A). BC formation and integrity has recently been shown to be strongly dependent on the presence of a stable extracellular matrix that affords the symmetric mechanical anchoring needed for hepatocyte polarization and elongated BC lumen formation (Figure S1B) (Li et al., 2016). Indeed, ECM gel thickness was consistently greater in the ports than in the channels (Figure S1C), where shear stress was found to significantly remodel and remove ECM gels depending on their composition. Matrigel-based scaffolds commonly used for human hepatocyte sandwich culture fared worse than collagen-I based gels which exhibited greater initial thickness and in-flow stability; however, gels of both compositions displayed great inhomogeneity of gel thickness across and within chips (Figures S1C and S1D). Based on these preliminary findings, we tested whether improved deposition of collagen-I based gels to create thick, flow-stable, and homogeneous ECM sandwich scaffolds throughout the hepatic channel could improve BC network formation. Consequently, iteratively modified the original ECM coating protocol (Jang et al., 2019) and optimized the planar deposition of various collagen-I based gels to achieve homogeneous and robust thickness in the chip through pressured injection of liquid into the channels filled with gelled ECM (Figure S1E). We also developed a luminal scaffold design based on viscous fingering (Figure S1F). Five different combinations of ECM composition and deposition strategy were tested for their effect on BC network formation (Figure S1G), where “Standard” denotes the original protocol (Jang et al., 2019). Briefly, for “Standard” ECM condition, top and bottom channels were coated by incubating with 100 μg/mL rat tail collagen-I (Corning) and 25 μ/mL fibronectin (GIBCO) overnight at 37°C. Hepatocytes were seeded as described above. After seeding, Matrigel® prepolymer solution was prepared on ice and injected into the top channel, which was then incubated at 37°C overnight. The following day, the top channel was gently flushed with warm medium. In conditions ECM-A, -B, -C, and -D, the hepatocytes are sandwiched between two 3D-ECM gel of different compositions, including Collagen-I (FibriCol®), Fibronectin (GIBCO), Collagen-IV (Sigma), and the collagen cross-linking agent, microbial transglutaminase (MTG) (Modernist Pantry LLC). The underlying gel was prepared by injecting the gel prepolymer solution after surface activation and incubated overnight at 37°C. The next day, the gel was then flushed twice with 100 μL of warm medium at 187.5 μL/s flow (Eppendorf Xplorer automatic pipet), generating a 3D-ECM on the membrane with a thickness of 30–50 μm (data not shown). Afterward, hepatocytes were seeded as described above. One day after the hepatocyte seeding, the same method was used to prepare the overlaying gel in conditions ECM-A, -B, and -C. In condition ECM-D, the overlay was prepared via viscous fingering method (Bischel et al., 2012). Briefly, 5 mg/mL of bovine collagen-I (FibriCol®) was injected into the top channel and a pipette tip filled with 200 μL of warm medium was immediately injected into the channel inlet to apply hydrostatic pressure. This will cause an interface instability between medium and prepolymerized collagen-I, enabling the medium to flow through the middle part of the prepolymerized collagen-I, thereby creating a lumen (Figure S1F). The chips were then immediately moved into humidified incubator to promote gelation of the lumen formed thick collagen-I ECM on top of the hepatocyte monolayer. ECM-C was used in the experiments in this study.
To assess and confirm ECM scaffold formation during the optimization procedure, we stained the gels with a fluorescent dye: Briefly, 1 mg/mL of N-hydroxysuccinimide (NHS) ester dye (Atto 488 NHS Ester, Sigma Aldrich) was mixed with 50 mM borate buffer (pH 9) in 1:500 ratio. Directly after ECM formation, the prepared staining solution was injected into the top channel and the chips were incubated for 25 minutes at room temperature in the dark. The top channel was then rinsed three times with PBS prior to fluorescence imaging.
To assess the effect of each ECM protocol on BC network formation, we quantified the MRP-2 stained BC network topology (see method sections on IF staining and Quantification). Optimal conditions thus identified were ECM-C and ECM-D, and these protocols were then used for ALD Liver-Chip studies.
Ethanol and high-fat treatment
To model FLD, cell culture medium was supplemented at day 7 in culture either with ethanol (0.08% or 0.16%, i.e., 80 mg/ml to 160 mg/ml, or 17.4 mM to 34 mM), with or without lipopolysaccharides (LPS, 1 μg/ml), or fatty acid (oleic acid, 1 μg/ml). The Liver-Chip was maintained for 48h in treatment medium and either assayed or allowed to recover for 5 days in basal medium. During treatment, liver toxicity was routinely monitored by albumin quantification and morphology assessment, which we had found to be a more sensitive method for detecting early liver toxicity effects than LDH assays.
Robustness of ethanol dosing
Minimal loss of ethanol from the medium is expected due to the material properties of the system. Though the inlet reservoirs allow ethanol and water vapor to off-gas, dissolving ethanol in water drastically decreases ethanol’s vapor pressure, which means that at the low ethanol/water ratios used in this study ethanol’s vapor pressure is much lower than that of water (Liu et al., 2008). Therefore, the usual slow rate of water evaporation in the reservoir is still faster than that of ethanol, such that the concentration of ethanol would not be significantly impacted. Further, the primary reservoir and conduit material that contacts the fluid is COC and is known not to allow significant permeation of gas (Ochs et al., 2014).
The diffusion coefficient of ethanol through PDMS, the most porous material in our microfluidic system, is on the order of D = 5*10−8 cm2/s (Lan et al., 2016). Assuming steady-state flux, we can apply Fick’s First law along the length of the Chip to compute the diffusive , with wall thickness dx = 600 μm and internal channel surface area A = 0.28 cm2. This model predicts a 2.8% loss of the initially dosed concentration C of ethanol; e.g., the 0.08% ethanol dose in the chip would be reduced to 0.077%.
During the initial onset of flow, absorption of ethanol into PDMS also contributes to total loss. However, the partition coefficient of ethanol in PDMS is on the order of 2.98 (Qiu et al., 2019) which is reasonably low. We expect an effective system equilibrium to be achieved after less than 20h during the 48h study. This initial loss to the PDMS at early time points would also primarily impact cellular exposure concentrations at the outlet of the chip channel rather than the inlet since the loss occurs along the length of the chip.
Taken together, we conclude that the ethanol dosing protocol is robust, as changes to the ethanol concentrations due to loss are expected to occur only briefly and locally over the course of the treatment.
Immunofluorescence staining and imaging
Cells on the Liver-Chips were washed 3 times in 1X PBS then fixed with 4% paraformaldehyde for 20 minutes at room temperature (RT). Chips were then washed 3 times with cold 1X PBS and blocked using 1% bovine serum albumin (BSA) in 1X PBS for 30 minutes to 2h at RT. Cells were permeabilized using a 1X PBS solution containing 1% saponin and 10% serum matching the species of the secondary antibody for 30min at RT. Cells were then washed 3 times in 1X PBS and blocked again in a 1X PBS solution containing 1% BSA for 2h to overnight at 4°C. All incubations with primary antibodies against MRP2 (1:50, Abcam) were carried out in this blocking buffer overnight at 4°C. This was followed by a two-hour incubation with secondary antibodies (Cell Signaling, Danvers, MA, USA) and DAPI in the blocking buffer at RT. Images were acquired with either an Olympus fluorescence microscope (IX83) or a Zeiss confocal microscope (LSM880). The fields of view were chosen randomly but fell always within the same three locations (near inlet, center, and near outlet) as to span the length of the chip channel. Two images were taken per region, providing six images per chip sample for analysis.
Live cell staining and imaging
Liver-Chips were stained in the upper channel with AdipoRed (1:40 dilution in PBS, Lonza) to visualize lipid droplet accumulation, Tetramethylrhodamine, methyl ester, perchlorate (TMRM) (0.1μM in hepatocyte medium, Thermo Fisher) to visualize active mitochondria, and MitoSox® (5uM in hepatocyte medium, Thermo Fisher) to visualize cellular oxidative stress, and cholyl-lysyl-fluorescein (CLF, Corning) to visualize bile canaliculi. Each staining solution was prepared and added to the upper channel, incubated for 15–30 minutes at 37°C, and washed three times before imaging. NucBlue (Thermo Fisher) staining was used to identify cell nuclei during live imaging. The stained chips were imaged using either an Olympus fluorescence microscope (IX83) or Zeiss confocal microscope (LSM880) and were de-blurred with Olympus cellSens software.
RNA extraction and sequencing
The cells from both top and bottom channel were separately lysed from the Liver-Chip according to the Protocol for Emulate Organ-Chips: Cell Lysis for RNA Isolation (EP161 v1.0). In brief, we used PureLink RNA Mini Kit lyse buffer (Thermo Fischer #12183018) to directly lyse the cells while still adhering to the chip, collected the lysate, and immediately frozen in dry ice. RNA sequencing was performed by GENEWIZ.
QUANTIFICATION AND STATISTICAL ANALYSIS
Image analysis
Analyses of lipid droplet accumulation, ROS events, and nuclei were conducted using the software tool ICY (de Chaumont et al., 2012), ImageJ-Fiji (Schindelin et al., 2012), CellProfiler (McQuin et al., 2018), and MATLAB (MATLAB, MathWorks Inc., Natick, MA). For ROS measurements, the histogram of the fluorescent images was adjusted to remove the background signal, followed by quantification of ROS events in the region of interest (ROI) based on minimum and maximum size and fluorescent intensity using the batch processing tool in ICY. ImageJ-Fiji was used to preprocess the AdipoRed images for analysis of lipid droplet accumulation. Here, the AdipoRed channel was median filtered, corrected for illumination, and then filtered with a Laplacian filter to emphasize droplet edges and remove larger background structures. The DAPI channel was filtered with an adaptive contrast enhancement algorithm (CLAHE) and then thresholded, followed by multiple dilation and erosion steps to yield a binary image in which nuclei in very close proximity, which likely belong to a single poly-nucleated cells, are merged. These preprocessed images were further processed in CellProfiler where a pipeline first automatically segmented the fields of view into estimated cell boundaries using the nuclei as reference points, followed by thresholding and detection of lipid droplets in the AdipoRed channel within each estimated cell boundary. Using CellProfiler modules as well as MATLAB scripts, we then computed mean droplet size (i.e., the projected area of the droplet in μm2) and the number of droplets per cell. Values of treatment groups were normalized to the median values of the associated control group in order to express fold-change values and thereby mitigate baseline variability due to donor-to-donor and cell batch variability. Furthermore, we computed the proportion of poly-nucleated cells by binning the detected nuclei according to their perimeter, which revealed two distinct populations, i.e., single, well separated nuclei, indicating mononucleated cells, and closely neighboring nuclei fused during thresholding, indicating poly-nucleated cells.
For measuring bile canalicular network properties, MRP-2 stained chips were imaged at 40x using a confocal point-scanning microscope (Zeiss LSM880, Airyscan). Fields of views were randomly chosen along the entire length of the channel to catch heterogeneity caused by ECM deposition or erosion in flow. In each field of view, a z stack was recorded and combined using maximal intensity projection in order to fully capture the bile canalicular network. In subsequent image analysis, each field of view was first segmented into 16 sub-windows, and analyzed for three quantitative metrics: Porosity, branching density, and average radius (Figure S1H). The area fraction taken up by the BC network, which is a measure of porosity, was determined using an ImageJ-Fiji macro that filters and thresholds the signal, followed by the ImageJ particle analysis which detects the area occupied by BC elements (Area BC). BC porosity was then computed as the ratio of Area BC to the total area of the field of view. Average radius and branching density were determined by applying the ImageJ-Fiji plugin “ridge detection” (Steger, 1998) to detect and measure the radius and length of all BC segments in each window. Then, we computed the average radius measured in each window as well as the branching density, defined as the summed length of the BC branches in the sub-window divided by the area of the sub-window.
Cell lysing
Cells in the Liver-Chip were lysed according to the protocol for Emulate Organ-Chips (Cell Lysis for Protein Extraction (EP135 v1.0)). In brief, we used Tris lysis buffer (MSD, #R60TX-3]) to directly lyse the cells while still adhering to the chip, collected the lysate, and performed downstream assays.
Albumin quantification assay
Albumin secretion was quantified in Liver-Chip effluent collected from the top channel using the Human Albumin SimpleStep ELISA® Kit (Abcam, #ab179887) according to the manufacturer’s protocol.
Cholesterol quantification assay
Cholesterol was quantified in Liver-Chip effluent according to manufacturer’s protocol for fluorometric detection (Amplex Red Cholesterol Assay Kit, Thermo Fisher). Medium in the top channel was changed to standard hepatocyte medium without FBS prior to the experiment. The following sample quantification was used to determine amount of cholesterol in the hepatocytes channel effluent: Net effluent cholesterol = [cholesterol from effluent] μg/ml MINUS [Cholesterol from dosing medium] μg/ml. The same quantification method was used to determine the cholesterol concentration of the hepatocytes cell lysate as described above.
Glycogen quantification assay
Hepatocytes in the top channel of the Liver-Chip were lysed as described above, then diluted at a range of 1:500 to 1:1000. Glycogen levels were determined using a standard assay according to manufacturer’s instructions for fluorometric detection (Abcam, #ab65620).
Glucose quantification assay
Glucose was quantified in Liver-Chip effluent collected from the top channel. Sample concentration were adjusted be within a 50 mg/dl to 200 mg/dl range. Glucose was quantified using a standard kit according to manufacturer’s instruction (Abcam, # ab65333).
RNA-seq/pathway analysis
The RNaseq dataset consisted of 2 vehicle samples, 3 samples treated with 0.08% alcohol, and 2 samples treated with 0.16% alcohol. Since differential gene expression analysis between the 0.08% and 0.16% groups yielded no significant differentially expressed genes, we pooled the samples of the two ethanol treatment groups together and constructed a single larger “ethanol-treated” group (consisting of 5 samples) which we compared to the vehicle group.
To remove poor quality adaptor sequences and nucleotides, the sequence reads were trimmed using Trimmomatic v.0.36. Next, using the STAR (Spliced Transcripts Alignment to a Reference) aligner v.2.5.2b, we mapped the trimmed reads to the Homo sapiens reference genome GRCh38 (available on ENSEMBL). Using the generated BAM files and the feature Counts from the v.1.5.2 subread package, we calculated the unique gene hit counts. Of note, only unique reads that fell within exon regions were counted. We prepared a strand-specific library; therefore, the reads were strand-specifically counted. Using the gene hit counts table, we filtered out genes with very low expression across the samples. The remainder were used for differential gene expression (DGE) analysis. For the DGE analysis, we used the “DESeq2” R package (Love et al., 2014) (Bioconductor), which normalizes the gene expression data and RNA composition using the “median-of-ratios” method (Anders and Huber, 2010; Love et al., 2014). In order to select the differentially expressed genes, we applied the following thresholds: adjusted p value < 0.05 and |log2FoldChange| > 1. Of the 57,500 genes annotated in the genome, 123 were found to have significant differential expression between the vehicle (n = 2) and the ethanol exposed chips (n = 5). More specifically, 87 (36) genes were found to be significantly upregulated (downregulated) in the ethanol exposed chips (0.08% and 0.16%). These 123 differentially expressed genes were used for the KEGG pathway analysis.
Cytokine quantification assay
Liver-Chip bottom channel (containing Kupffer cells) effluent cytokine levels were measured using the U-PLEX® Biomarker Group 1 Human Assays (MSD® Cat No. K15067L) according to the manufacturer’s instructions.
Statistical analysis
As indicated in the figure legends, one-way ANOVA, Sidak’s and Dunnett’s multiple comparisons tests were used for comparing the mean values of parametric data, and the Mann-Whitney U test or Kruskal-Wallis tests followed by Dunnett’s multiple comparisons test was used for comparing the median value of nonparametric data. As noted in figures, the KS-test was occasionally used to test for differences in distributions. All statistical analyses were performed using Prism v6, 7 or 8 (GraphPad). All data was collected from at least 2 independent experiments with at least 3 chips per condition and imaged at 5–10 fields of view per chip (where applicable), unless stated otherwise in the figure legends.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| MRP2 | Abcam | ab172630 |
| Chemicals, peptides, and recombinant proteins | ||
| AdipoRed | Lonza | PT-7009 |
| TMRM | Thermo Fisher | T668 |
| MitoSox | Thermo Fisher | M36008 |
| Cholyl-lysyl-fluorescein (CLF) | Corning | 451041 |
| Critical commercial assays | ||
| PureLink RNA Mini Kit lyse buffer | Thermo Fischer | 12183018 |
| Human Albumin SimpleStep ELISA® Kit | Abcam | ab179887 |
| Amplex Red Cholesterol Assay Kit | Thermo Fischer | A12216 |
| Glycogen Assay Kit | Abcam | ab65620 |
| Glucose Assay Kit | Abcam | ab65333 |
| Deposited data | ||
| RNA-sequencing data | Illumina HiSeq | GEO: GSE175396 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE175396 |
| Experimental models: cell lines | ||
| Cryopreserved primary human hepatocytes from 1 healthy donor | Triangle Research Labs (Lonza) | QHum15063 |
| Cryopreserved primary human hepatocytes from 1 healthy donor | GIBCO (Thermo Fisher). | HU8305 |
| Software and algorithms | ||
| MATLAB | MathWorks | https://www.mathworks.com/products/matlab.html |
| ImageJ-Fiji | https://imagej.nih.gov/ij/ | Schindelin et al., 2012 |
| CellProfiler | https://cellprofiler.org/ | McQuin et al., 2018 |
| ICY | http://icy.bioimageanalysis.org/ | de Chaumont et al., 2012 |
| RStudio Team (2021). Integrated Development Environment for R. “DESeq2” R package (Bioconductor) | https://www.rstudio.com/. | Love et al., 2014 |
| Trimmomatic v.0.36 | The USADEL LAB http://www.usadellab.org/cms/ | Bolger et al., 2014 |
| STAR (Spliced Transcripts Alignment to a Reference) v.2.5.2b | https://github.com/alexdobin/STAR | Dobin et al., 2013 |
| Other | ||
| Emulate Organ-Chip System (Zoe, Orb and Chip-S1) | Emulate Inc. | https://www.emulatebio.com/our-technology |
Highlights.
The Liver-Chip models early critical events of ALD
Key readouts include markers of steatosis, cholestasis, and oxidative stress
The chip recapitulates second-hit injury through circulating endotoxins (leaky gut)
The chip also models injury recovery following abstinence from alcohol
ACKNOWLEDGMENTS
We thank Josiah Sliz for the discussions and technical advice on microfluidic transport mechanisms. This study was supported by the National Institute of on Alcohol Abuse and Alcoholism of the National Institutes of Health (award numbers R43AA026473 and 1R43AA026473-01) and Emulate.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109393.
DECLARATION OF INTERESTS
All authors except S.L. and E.S. are current or former employees or interns of Emulate and may hold patents related to this work or equity interests in Emulate.
REFERENCES
- Abe K, Bridges AS, and Brouwer KLR (2009). Use of sandwich-cultured human hepatocytes to predict biliary clearance of angiotensin II receptor blockers and HMG-CoA reductase inhibitors. Drug Metab. Dispos 37, 447–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aljomah G, Baker SS, Liu W, Kozielski R, Oluwole J, Lupu B, Baker RD, and Zhu L (2015). Induction of CYP2E1 in non-alcoholic fatty liver diseases. Exp. Mol. Pathol 99, 677–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, and Huber W (2010). Differential expression analysis for sequence count data. Genome Biol. 11, R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arab JP, Karpen SJ, Dawson PA, Arrese M, and Trauner M (2017). Bile acids and nonalcoholic fatty liver disease: Molecular insights and therapeutic perspectives. Hepatology 65, 350–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asrani SK, Devarbhavi H, Eaton J, and Kamath PS (2019). Burden of liver diseases in the world. J. Hepatol 70, 151–171. [DOI] [PubMed] [Google Scholar]
- Atilano-Roque A, Roda G, Fogueri U, Kiser JJ, and Joy MS (2016). Effect of Disease Pathologies on Transporter Expression and Function. J. Clin. Pharmacol 56 (Suppl 7), S205–S221. [DOI] [PubMed] [Google Scholar]
- Bala S, Csak T, Saha B, Zatsiorsky J, Kodys K, Catalano D, Satishchandran A, and Szabo G (2016). The pro-inflammatory effects of miR-155 promote liver fibrosis and alcohol-induced steatohepatitis. J. Hepatol 64, 1378–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bale SS, Golberg I, Jindal R, McCarty WJ, Luitje M, Hegde M, Bhushan A, Usta OB, and Yarmush ML (2015). Long-term coculture strategies for primary hepatocytes and liver sinusoidal endothelial cells. Tissue Eng. Part C Methods 21, 413–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedogni G, Nobili V, and Tiribelli C (2014). Epidemiology of fatty liver: an update. World J. Gastroenterol 20, 9050–9054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedict M, and Zhang X (2017). Non-alcoholic fatty liver disease: An expanded review. World J. Hepatol 9, 715–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertola A (2018). Rodent models of fatty liver diseases. Liver Res. 2, 3–13. [Google Scholar]
- Bird GL, Sheron N, Goka AK, Alexander GJ, and Williams RS (1990). Increased plasma tumor necrosis factor in severe alcoholic hepatitis. Ann. Intern. Med 112, 917–920. [DOI] [PubMed] [Google Scholar]
- Bischel LL, Lee S-H, and Beebe DJ (2012). A practical method for patterning lumens through ECM hydrogels via viscous finger patterning. J. Lab. Autom 17, 96–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishehsari F, Magno E, Swanson G, Desai V, Voigt RM, Forsyth CB, and Keshavarzian A (2017). Alcohol and Gut-Derived Inflammation. Alcohol Res. 38, 163–171. [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breher-Esch S, Sahini N, Trincone A, Wallstab C, and Borlak J (2018). Genomics of lipid-laden human hepatocyte cultures enables drug target screening for the treatment of non-alcoholic fatty liver disease. BMC Med. Genomics 11 (1), 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunt EM (2017). Nonalcoholic fatty liver disease and the ongoing role of liver biopsy evaluation. Hepatol. Commun 1, 370–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caro AA, and Cederbaum AI (2004). Oxidative stress, toxicology, and pharmacology of CYP2E1. Annu. Rev. Pharmacol. Toxicol 44, 27–42. [DOI] [PubMed] [Google Scholar]
- Castera L, Friedrich-Rust M, and Loomba R (2019). Noninvasive Assessment of Liver Disease in Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology 156, 1264–1281.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chacko KR, and Reinus J (2016). Spectrum of Alcoholic Liver Disease. Clin. Liver Dis 20, 419–427. [DOI] [PubMed] [Google Scholar]
- Chai J, Cai SY, Liu X, Lian W, Chen S, Zhang L, Feng X, Cheng Y, He X, He Y, et al. (2015). Canalicular membrane MRP2/ABCC2 internalization is determined by Ezrin Thr567 phosphorylation in human obstructive cholestasis. J. Hepatol 63, 1440–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang JYL (2017). Targeting bile acids and lipotoxicity for NASH treatment. Hepatol. Commun 1, 1002–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang JYL (2018). The gut’s feeling on bile acid signaling in NAFLD. Hepatobiliary Surg. Nutr 7, 151–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung KW, Han NI, Choi SW, Ahn BM, Yoon SK, Nam SW, Lee YS, Han JY, and Sun HS (2002). Increased microfilaments in hepatocytes and biliary ductular cells in cholestatic liver diseases. J. Korean Med. Sci 17, 795–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen JI, and Nagy LE (2011). Pathogenesis of alcoholic liver disease: interactions between parenchymal and non-parenchymal cells. J. Dig. Dis 12, 3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabb DW, Im GY, Szabo G, Mellinger JL, and Lucey MR (2020). Diagnosis and Treatment of Alcohol-Associated Liver Diseases: 2019 Practice Guidance From the American Association for the Study of Liver Diseases. Hepatology 71, 306–333. [DOI] [PubMed] [Google Scholar]
- de Chaumont F, Dallongeville S, Chenouard N, Hervé N, Pop S, Provoost T, Meas-Yedid V, Pankajakshan P, Lecomte T, Le Montagner Y, et al. (2012). Icy: an open bioimage informatics platform for extended reproducible research. Nat. Methods 9, 690–696. [DOI] [PubMed] [Google Scholar]
- Deharde D, Schneider C, Hiller T, Fischer N, Kegel V, Lübberstedt M, Freyer N, Hengstler JG, Andersson TB, Seehofer D, et al. (2016). Bile canaliculi formation and biliary transport in 3D sandwich-cultured hepatocytes in dependence of the extracellular matrix composition. Arch. Toxicol 90, 2497–2511. [DOI] [PubMed] [Google Scholar]
- Denechaud PD, Lopez-Mejia IC, Giralt A, Lai Q, Blanchet E, Delacuisine B, Nicolay BN, Dyson NJ, Bonner C, Pattou F, et al. (2016). E2F1 mediates sustained lipogenesis and contributes to hepatic steatosis. J. Clin. Invest 126, 137–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyson JK, Anstee QM, and McPherson S (2014). Non-alcoholic fatty liver disease: a practical approach to diagnosis and staging. Frontline Gastroenterol. 5, 211–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgammal A, Eves D, Albaghli A, Kane D, Durcan R, Storey D, and Gilligan P (2015). The Blood Alcohol Concentration Testing Emergency Room Investigation Analysis Study: A 1-Year Review of Blood Alcohol Concentration Testing in an Emergency Department. Adv. Emerg. Med 2015 (1). [Google Scholar]
- Farrell GC, van Rooyen D, Gan L, and Chitturi S (2012). NASH is an inflammatory disorder: Pathogenic, prognostic and therapeutic implications. Gut Liver 6, 149–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fickert P, Zollner G, Fuchsbichler A, Stumptner C, Weiglein AH, Lammert F, Marschall HU, Tsybrovskyy O, Zatloukal K, Denk H, and Trauner M (2002). Ursodeoxycholic acid aggravates bile infarcts in bile duct-ligated and Mdr2 knockout mice via disruption of cholangioles. Gastroenterology 123, 1238–1251. [DOI] [PubMed] [Google Scholar]
- Gentric G, Maillet V, Paradis V, Couton D, L’Hermitte A, Panasyuk G, Fromenty B, Celton-Morizur S, and Desdouets C (2015). Oxidative stress promotes pathologic polyploidization in nonalcoholic fatty liver disease. J. Clin. Invest 125, 981–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waller-Evans H, Hue C, Fearnside J, Rothwell AR, Lockstone HE, Caldérari S, Wilder SP, Cazier JB, Scott J, and Gauguier D (2013). Nutri-genomics of high fat diet induced obesity in mice suggests relationships between susceptibility to fatty liver disease and the proteasome. PLoS ONE 8 (12), e82825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wannamethee SG, Perry IJ, and Gerald Shaper A (1999). Nonfasting serum glucose and insulin concentrations and the risk of stroke. Stroke 30, 1780–1786. [DOI] [PubMed] [Google Scholar]
- Hines IN, and Wheeler MD (2004). Recent advances in alcoholic liver disease III. Role of the innate immune response in alcoholic hepatitis. Am. J. Physiol. Gastrointest. Liver Physiol 287, G310–G314. [DOI] [PubMed] [Google Scholar]
- Hotta K, Yoneda M, Hyogo H, Ochi H, Mizusawa S, Ueno T, Chayama K, Nakajima A, Nakao K, and Sekine A (2010). Association of the rs738409 polymorphism in PNPLA3 with liver damage and the development of nonalcoholic fatty liver disease. BMC Med. Genet 11, 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iravani F, Hosseini N, and Mojarrad M (2018). Role of MicroRNAs in Pathophysiology of Non-alcoholic Fatty Liver Disease and Non-alcoholic Steatohepatitis. Middle East J. Dig. Dis 10, 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang K-J, Otieno MA, Ronxhi J, Lim H-K, Ewart L, Kodella KR, Petropolis DB, Kulkarni G, Rubins JE, Conegliano D, et al. (2019). Reproducing human and cross-species drug toxicities using a Liver-Chip. Sci. Transl. Med 11 (517), eaax5516. [DOI] [PubMed] [Google Scholar]
- Jansen PLM, Ghallab A, Vartak N, Reif R, Schaap FG, Hampe J, and Hengstler JG (2017). The ascending pathophysiology of cholestatic liver disease. Hepatology 65, 722–738. [DOI] [PubMed] [Google Scholar]
- Kakehashi A, Stefanov VE, Ishii N, Okuno T, Fujii H, Kawai K, Kawada N, and Wanibuchi H (2017). Proteome characteristics of non-alcoholic steatohepatitis liver tissue and associated hepatocellular carcinomas. Int. J. Mol. Sci 18 (2), 434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang M, Kim J, An H-T, and Ko J (2017). Human leucine zipper protein promotes hepatic steatosis via induction of apolipoprotein A-IV. FASEB J. 31, 2548–2561. [DOI] [PubMed] [Google Scholar]
- Kasendra M, Tovaglieri A, Sontheimer-Phelps A, Jalili-Firoozinezhad S, Bein A, Chalkiadaki A, Scholl W, Zhang C, Rickner H, Richmond CA, et al. (2018). Development of a primary human Small Intestine-on-a-Chip using biopsy-derived organoids. Sci. Rep 8, 2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klassen LW, Thiele GM, Duryee MJ, Schaffert CS, DeVeney AL, Hunter CD, Olinga P, and Tuma DJ (2008). An in vitro method of alcoholic liver injury using precision-cut liver slices from rats. Biochem. Pharmacol 76, 426–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuttippurathu L, Juskeviciute E, Dippold RP, Hoek JB, and Vadigepalli R (2016). A novel comparative pattern analysis approach identifies chronic alcohol mediated dysregulation of transcriptomic dynamics during liver regeneration. BMC Genomics 17 (260). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Zhang Y, Pluchon P, Robens J, Herr K, Mercade M, Thiery J-P, Yu H, and Viasnoff V (2016). Extracellular matrix scaffolding guides lumen elongation by inducing anisotropic intercellular mechanical tension. Nat. Cell Biol 18, 311–318. [DOI] [PubMed] [Google Scholar]
- Lan Y, Yan N, and Wang W (2016). Application of PDMS pervaporation membranes filled with tree bark biochar for ethanol/water separation. RSC Adv. 6 (53), 47637–47645. [Google Scholar]
- Li H, Toth E, and Cherrington NJ (2018). Alcohol Metabolism in the Progression of Human Nonalcoholic Steatohepatitis. Toxicol. Sci 164, 428–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang JQ, Teoh N, Xu L, Pok S, Li X, Chu ESH, Chiu J, Dong L, Arfianti E, Haigh WG, et al. (2018). Dietary cholesterol promotes steatohepatitis related hepatocellular carcinoma through dysregulated metabolism and calcium signaling. Nat. Commun 9 (1), 4490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber CS (2004). Alcoholic fatty liver: its pathogenesis and mechanism of progression to inflammation and fibrosis. Alcohol 34, 9–19. [DOI] [PubMed] [Google Scholar]
- Liu C, Bonaccurso E, and Butt HJ (2008). Evaporation of sessile water/ethanol drops in a controlled environment. Phys. Chem. Chem. Phys 10, 7150–7157. [DOI] [PubMed] [Google Scholar]
- Liu Y, Cheng F, Luo YX, Hu P, Ren H, and Peng ML (2017). [The role of cytochrome P450 in nonalcoholic fatty liver induced by high-fat diet: a gene expression profile analysis]. Zhonghua Gan Zang Bing Za Zhi 25, 285–290. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magdaleno F, Blajszczak C, and Nieto N (2017). Dietary cholesterol promotes steatohepatitis related hepatocellular carcinoma through dysregulated metabolism and calcium signaling. Biomolecules 7 (1), 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuin C, Goodman A, Chernyshev V, Kamentsky L, Cimini BA, Karhohs KW, Doan M, Ding L, Rafelski SM, Thirstrup D, et al. (2018). CellProfiler 3.0: Next-generation image processing for biology. PLoS Biol. 16, e2005970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon KV, Gores GJ, and Shah VH (2001). Pathogenesis, diagnosis, and treatment of alcoholic liver disease. Mayo Clin. Proc 76, 1021–1029. [DOI] [PubMed] [Google Scholar]
- Meroni M, Longo M, Rametta R, and Dongiovanni P (2018). Genetic and Epigenetic Modifiers of Alcoholic Liver Disease. Int. J. Mol. Sci 19 (12), 3857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer K, Ostrenko O, Bourantas G, Morales-Navarrete H, Porat-Shliom N, Segovia-Miranda F, Nonaka H, Ghaemi A, Verbavatz J-M, Brusch L, et al. (2017). A Predictive 3D Multi-Scale Model of Biliary Fluid Dynamics in the Liver Lobule. Cell Syst. 4, 277–290.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min HK, Kapoor A, Fuchs M, Mirshahi F, Zhou H, Maher J, Kellum J, Warnick R, Contos MJ, and Sanyal AJ (2012). Increased hepatic synthesis and dysregulation of cholesterol metabolism is associated with the severity of nonalcoholic fatty liver disease. Cell Metab. 15, 665–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakakariya M, Ono M, Amano N, Moriwaki T, Maeda K, and Sugiyama Y (2012). In vivo biliary clearance should be predicted by intrinsic biliary clearance in sandwich-cultured hepatocytes. Drug Metab. Dispos 40, 602–609. [DOI] [PubMed] [Google Scholar]
- Natarajan SK, Stringham BA, Mohr AM, Wehrkamp CJ, Lu S, Phillippi MA, Harrison-Findik D, and Mott JL (2017). FoxO3 increases miR-34a to cause palmitate-induced cholangiocyte lipoapoptosis. J. Lipid Res 58, 866–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaou N, Gathercole LL, Marchand L, Althari S, Dempster NJ, Green CJ, van de Bunt M, McNeil C, Arvaniti A, Hughes BA, et al. (2019). AKR1D1 is a novel regulator of metabolic phenotype in human hepatocytes and is dysregulated in non-alcoholic fatty liver disease. Metabolism 99, 67–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura Y, Adachi N, and Koyama H (2007). Human Mus81 and FANCB independently contribute to repair of DNA damage during replication. Genes Cells 12, 1111–1122. [DOI] [PubMed] [Google Scholar]
- Ochs CJ, Kasuya J, Pavesi A, and Kamm RD (2014). Oxygen levels in thermoplastic microfluidic devices during cell culture. Lab Chip 14, 459–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orman ES, Odena G, and Bataller R (2013). Alcoholic liver disease: pathogenesis, management, and novel targets for therapy. J. Gastroenterol. Hepatol 28 (Suppl 1), 77–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owada Y, Tamura T, Tanoi T, Ozawa Y, Shimizu Y, Hisakura K, Matsuzaka T, Shimano H, Nakano N, Sakashita S, et al. (2018). Novel non-alcoholic steatohepatitis model with histopathological and insulin-resistant features. Pathol. Int 68, 12–22. [DOI] [PubMed] [Google Scholar]
- Petersen MC, Vatner DF, and Shulman GI (2017). Regulation of hepatic glucose metabolism in health and disease. Nat. Rev. Endocrinol 13, 572–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirola CJ, and Sookoian S (2018). Multiomics biomarkers for the prediction of nonalcoholic fatty liver disease severity. World J. Gastroenterol 24, 1601–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popper H (1981). Cholestasis: the future of a past and present riddle. Hepatology 1 (2), 187–191. [DOI] [PubMed] [Google Scholar]
- Qiu B, Wang Y, Fan S, Liu J, Jian S, Qin Y, Xiao Z, Tang X, and Wang W (2019). Ethanol mass transfer during pervaporation with PDMS membrane based on solution-diffusion model considering concentration polarization. Separ. Purif. Tech 220 (1), 276–282. [Google Scholar]
- Rehm J, Mathers C, Popova S, Thavorncharoensap M, Teerawattananon Y, and Patra J (2009). Global burden of disease and injury and economic cost attributable to alcohol use and alcohol-use disorders. Lancet 373, 2223–2233. [DOI] [PubMed] [Google Scholar]
- Reif R, Karlsson J, Günther G, Beattie L, Wrangborg D, Hammad S, Begher-Tibbe B, Vartak A, Melega S, Kaye PM, et al. (2015). Bile canalicular dynamics in hepatocyte sandwich cultures. Arch. Toxicol 89, 1861–1870. [DOI] [PubMed] [Google Scholar]
- Remmler J, Schneider C, Treuner-Kaueroff T, Bartels M, Seehofer D, Scholz M, Berg T, and Kaiser T (2018). Increased Level of Interleukin 6 Associates With Increased 90-Day and 1-Year Mortality in Patients With End-Stage Liver Disease. Clin. Gastroenterol. Hepatol 16, 730–737. [DOI] [PubMed] [Google Scholar]
- Ruttkay-Nedecky B, Nejdl L, Gumulec J, Zitka O, Masarik M, Eckschlager T, Stiborova M, Adam V, and Kizek R (2013). The role of metallothionein in oxidative stress. Int. J. Mol. Sci 14, 6044–6066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santhekadur PK, Kumar DP, and Sanyal AJ (2018). Preclinical models of non-alcoholic fatty liver disease. J. Hepatol 68, 230–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaglioni F, Ciccia S, Marino M, Bedogni G, and Bellentani S (2011). ASH and NASH. Dig. Dis 29, 202–210. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt-Arras D, and Rose-John S (2016). IL-6 pathway in the liver: From physiopathology to therapy. J. Hepatol 64, 1403–1415. [DOI] [PubMed] [Google Scholar]
- Segovia-Miranda F, Morales-Navarrete H, Kücken M, Moser V, Seifert S, Repnik U, Rost F, Brosch M, Hendricks A, Hinz S, et al. (2019). Three-dimensional spatially resolved geometrical and functional models of human liver tissue reveal new aspects of NAFLD progression. Nat. Med 25, 1885–1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seitz HK, and Stickel F (2006). Risk factors and mechanisms of hepatocarcinogenesis with special emphasis on alcohol and oxidative stress. Biol. Chem 387, 349–360. [DOI] [PubMed] [Google Scholar]
- Si M, and Lang J (2018). The roles of metallothioneins in carcinogenesis. J. Hematol. Oncol 11, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singal AK, Bataller R, Ahn J, Kamath PS, and Shah VH (2018). ACG clinical guideline: Alcoholic liver disease. Am. J. Gastroenterol 113, 175–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smalling RL, Delker DA, Zhang Y, Nieto N, Mcguiness MS, Liu S, Friedman SL, Hagedorn CH, and Wang L (2013). Am. J. Physiol. Liver Physiol 305, G364–G374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stättermayer AF, Rutter K, Beinhardt S, Wrba F, Scherzer TM, Strasser M, Hofer H, Steindl-Munda P, Trauner M, and Ferenci P (2014). Role of FDFT1 polymorphism for fibrosis progression in patients with chronic hepatitis C. Liver Int. 34, 388–395. [DOI] [PubMed] [Google Scholar]
- Steger C (1998). IEEE Trans. Pattern Anal. Mach. Intell 20, 113–125. [DOI] [PubMed] [Google Scholar]
- Swift B, Pfeifer ND, and Brouwer KLR (2010). Sandwich-cultured hepatocytes: an in vitro model to evaluate hepatobiliary transporter-based drug interactions and hepatotoxicity. Drug Metab. Rev 42, 446–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo G, and Bala S (2010). Alcoholic liver disease and the gut-liver axis. World J. Gastroenterol 16, 1321–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y, and Fukusato T (2014). Histopathology of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J. Gastroenterol 20, 15539–15548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurman RG (1998). Am. J. Physiol. Liver Physiol 275, G605–G611. [DOI] [PubMed] [Google Scholar]
- Tommasi S, and Besaratinia A (2019). Dna hydroxymethylation at the interface of the environment and nonalcoholic fatty liver disease. Int. J. Environ. Res. Public Health 16, E2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topper JN, and Gimbrone MA Jr. (1999). Blood flow and vascular gene expression: fluid shear stress as a modulator of endothelial phenotype. Mol. Med. Today 5, 40–46. [DOI] [PubMed] [Google Scholar]
- Toshikuni N, Tsutsumi M, and Arisawa T (2014). Clinical differences between alcoholic liver disease and nonalcoholic fatty liver disease. World J. Gastroenterol 20, 8393–8406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukamoto H (2015). Metabolic reprogramming and cell fate regulation in alcoholic liver disease. Pancreatology 15 (4, Suppl), S61–S65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukamoto H, Machida K, Dynnyk A, and Mkrtchyan H (2009). “Second hit” models of alcoholic liver disease. Semin. Liver Dis 29, 178–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tung BY, and Carithers RL Jr. (1999). Cholestasis and alcoholic liver disease. Clin. Liver Dis 3, 585–601. [DOI] [PubMed] [Google Scholar]
- Urbaschek R, McCuskey RS, Rudi V, Becker KP, Stickel F, Urbaschek B, and Seitz HK (2001). Endotoxin, endotoxin-neutralizing-capacity, sCD14, sICAM-1, and cytokines in patients with various degrees of alcoholic liver disease. Alcohol. Clin. Exp. Res 25, 261–268. [PubMed] [Google Scholar]
- Wong T, Dang K, Ladhani S, Singal AK, and Wong RJ (2019). Prevalence of Alcoholic Fatty Liver Disease Among Adults in the United States, 2001–2016. JAMA 321, 1723–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Zhang XB, and Zheng ZM (2013). Suppression of KIF14 expression inhibits hepatocellular carcinoma progression and predicts favorable outcome. Cancer Sci. 104, 552–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye P, Liu J, Xu W, Liu D, Ding X, Le S, Zhang H, Chen S, Chen M, and Xia J (2019). Dual-Specificity Phosphatase 26 Protects Against Nonalcoholic Fatty Liver Disease in Mice Through Transforming Growth Factor Beta-Activated Kinase 1 Suppression. Hepatology 69, 1946–1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Nguyen LH, Zhou K, Tu HC, Sehgal A, Nassour I, Li L, Gopal P, Goodman J, Singal AG, et al. (2018). Knockdown of Anillin Actin Binding Protein Blocks Cytokinesis in Hepatocytes and Reduces Liver Tumor Development in Mice Without Affecting Regeneration. Gastroenterology 154, 1421–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, and Zhong W (2017). Targeting the gut barrier for the treatment of alcoholic liver disease. Liver Res. 1, 197–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-seq data generated during this study are available at the NCBI Gene Expression Omnibus (GEO) database repository (GEO: GSE175396).