

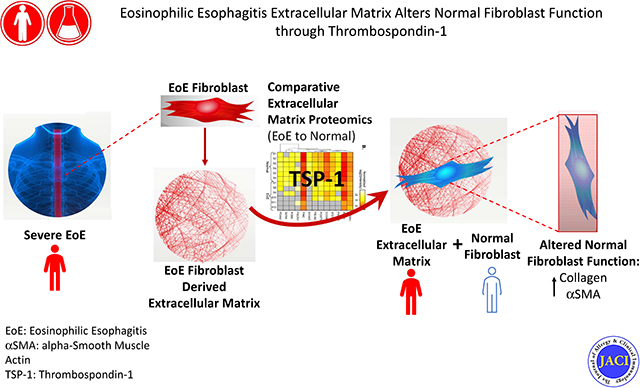
Abstract
Background.
Eosinophilic esophagitis (EoE) is a chronic Th2 disorder complicated by tissue fibrosis and loss of esophageal luminal patency. The fibrostenotic esophagus does not respond well to therapy but pro-fibrotic therapeutic targets are largely unclear.
Objective.
To utilize proteomics and primary cell as a novel approach to determine relevant pro-fibrotic factors.
Methods.
We utilized primary esophageal EoE and normal fibroblasts, their derivative extracellular matrixes (ECMs), an approach of fibroblast culture on autologous versus opposing ECM, and proteomics to elucidate EoE ECM proteins that dysregulate cellular function.
Results.
We cultured esophageal fibroblasts from normal or severe EoE esophagi on autologous versus opposing extracellular matrix (ECM). The EoE ECM proteome shifted normal esophageal fibroblast protein expression. Proteomic analysis demonstrated that thrombospondin-1 (TSP-1) is detected only in the EoE ECM, is central in the EoE ECM protein-protein interactome, is significantly elevated in active EoE biopsies, and induces fibroblast collagen I production.
Conclusion.
EoE fibroblasts secrete a unique ECM proteome that reflects their in vivo state and induces collagen I and α-smooth muscle actin protein expression from normal fibroblasts. TSP-1 is a previously unappreciated pro-fibrotic molecule in EoE.
Clinical Implications.
The novel approach of culturing tissue fibroblasts on autologous versus opposing ECM has clinical utility for identifying previously unappreciated disease-specific druggable ECM targets in EoE and, potentially other, fibrotic disorders.
Keywords: Eosinophil, Esophagus, Fibrosis, Interactome, Remodeling, Thrombospondin-1
Capsule summary
Using proteomics analyses of diseased and normal esophageal fibroblasts cultured on autologous or opposing derivative extracellular matrixes, thrombospondin-1 was discovered and validated as a pathogenic mediator of fibrosis in EoE.
Graphical Abstract
Introduction
Eosinophilic esophagitis (EoE) is an increasingly prevalent chronic allergic disease that leads to esophageal rigidity and lost luminal patency.1–3 EoE is instigated by food and environmental antigens with resultant Th2 inflammation and tissue remodeling comprised of basal cell hyperplasia, epithelial barrier breakdown, esophageal fibrosis and myofibroblast activation.1, 4, 5 Patients with a fibrostenotic esophagus often require repeated esophageal dilations and their disease is commonly resistant to therapeutic interventions such as topical corticosteroids.6 EoE almost uniformly progresses to a strictured esophagus when untreated or unresponsive to therapies.2, 3
Wound healing is a key feature of Th2 diseases but the molecular drivers that promote progression from physiologic to pathologic remodeling are still under investigation.7 A paucity of human tissue samples and adequate surrogate markers for fibrosis create a clinical challenge. While physiologic esophageal remodeling reverses with successful anti-inflammatory therapies such as topical glucocorticoids or dietary antigen elimination diets, pathologic remodeling is unremitting and can be progressive despite therapy but the risk factors for progression are unclear.7 The EoE esophagus demonstrates increased fibrosis, angiogenesis, and smooth muscle hypertrophy and can serve as a model system for fibrotic diseases.7 Once remodeling occurs, the physical and chemical environment of the diseased extracellular matrix (ECM) is sufficient to drive fibrosis and smooth muscle cell hypertrophy via mechanotransductive signals.8, 9 Transforming growth factor beta-1 (TGFβ1) and tumor necrosis factor (TNFα) are important mediators of EoE remodeling.10, 11 However, unique EoE extracellular matrix (ECM) components which could be new therapeutic targets have not been evaluated using an unbiased approach.
Proteomic analysis has created a new framework for understanding human protein dysregulation during disease states. The ECM modulates disease severity, activity and complications in many contexts including development and cancer,12 but the ECM is difficult to study in human EoE due to the paucity of tissue, leaving its constituents largely enigmatic. While endogenous tissue samples are small and unlikely to be amenable to a proteomic approach in EoE, cultured primary cells are a more abundant resource for probing the human ECM proteome. A clearer understanding of the most pathogenic and distinct proteins in a diseased ECM would facilitate our assessment and management of fibrotic diseases. Currently, gauging EoE severity is challenging and limited to assessing fibrosis in small tissue biopsies, which often do not contain adequate subepithelial tissue, or to using research tools such as endoscopic functional lumen imaging planimetry (EndoFLIP).13–15 To date, it has been unclear if EoE and normal esophageal fibroblasts retain cellular memory of their in vivo functional characteristics and if they can serve as a source that reveals disease relevant ECM molecules. If this were the case, then novel approaches to study the pro-fibrotic ECM become attainable.
Herein we conduct an analysis of the ECM proteome using esophageal fibroblasts from severe EoE patients and normal controls. We use a methodologic approach of culturing diseased and normal fibroblasts on non-autologous and autologous matrix to understand the sufficiency of the diseased ECM to alter normal fibroblast function. Our approaches reveal the capacity of the diseased ECM to change normal fibroblast function and elucidates the multi-functional cell-cell and cell-ECM interaction molecule, thrombospondin-1 (TSP-1) in active EoE. This approach could be valuable for determining specific therapeutic targets in fibrotic diseases.
Methods
See Supplemental Methods for Detailed Descriptions
Disease definition
Active EoE was defined as greater than or equal to 15 eosinophils per high power field (hpf) in subjects with a poor responsive to standard therapy (Table S1). Written informed consent / assent was obtained from all parents and patients older than 7 years under the University of California, San Diego / Rady Children’s Hospital, San Diego IRB approved protocol 181690. Utilization of organ donor esophagi is not considered human subjects research and studies are completed under the University of California, San Diego approved IRB exempt protocol 130835. Healthy esophageal tissue from donors was provided by the Arkansas Regional Organ Recovery Agency.
ECM decellularization and fibroblast culture
Primary fibroblasts were isolated from healthy donor esophagi or EoE esophageal biopsies using collagenase 10 and cultured to confluency in 10%FBS/DMEM media (Gibco). Cells were washed (PBS, Sigma-Aldrich) and ECM was decellularized with 20mM NH4OH. Fibroblasts were placed on autologous or non-autologous decellularized ECM for 7–12 days. All samples for comparator experiments were passage matched and collected and analyzed simultaneously. Experiments were repeated in biological replicates at 3 different timepoints using fibroblasts from 5 EoE and 5 normal subjects. For in vitro studies, fibroblasts were treated with human TSP-1 (R&D systems) for 24 hours and with the TGFβ receptor I inhibitor SB431542 (10uM, Tocris) for the final 48 hours of culture. See supplemental methods for details.
LC-MS-MS
Trypsin-digested peptides were analyzed by ultra-high-pressure liquid chromatography (UPLC) coupled with tandem mass spectroscopy (LC-MS/MS) using nanospray ionization and Orbitrap fusion Lumos hybrid mass spectrometer (Thermo) interfaced with nano-scale reversed-phase UPLC (Thermo Dionex UltiMate™ 3000 RSLC nano System) using a 25 cm, 75-micron ID glass capillary packed with 1.7-μm C18 (130) BEH™ beads (Waters corporation). Fibroblasts from 3–5 different EoE and normal donors run in replicates were utilized.
Proteomic data analysis
The normalized label-free quantification (LFQ) intensity in the log2 scale were used for the differential expression analysis in this study.
Protein-protein interaction (PPI) network analysis
The Search Tool for the Retrieval of Interacting Genes (STRING) database is a widely used database that integrates direct and indirect protein-protein interactions (PPI).16 The 24 differentially expressed genes identified were uploaded, and interactions with at least medium confidence (interaction score > 0.4) were selected.
Functional Annotation and Enrichment Analysis
Database for annotation, visualization and integrated discovery (DAVID) Bioinformatics Resource was used to carry out functional annotation and enrichment analysis with functional annotation clustering.17 Similar annotation contents were grouped into clusters representing similar biological functions. DAVID employed Fisher’s exact method (modified EASE score) to test whether a gene set is enriched by the input genes. To decide the degree of enrichment, an appropriate background set (the reference background) of genes must be set up as a baseline to perform the comparison in the enrichment analysis. In this study, we used the 3,131 proteins identified in the proteomic data as the reference background, and the 7 differentially expressed proteins (DEPs) identified as the query proteins.18 The functional annotation clustering was done with default parameters. Classification stringency was set as medium. The raw p values were used in functional annotation.
Gene set enrichment analysis (GSEA)
GSEA was performed using the Broad Institute GSEA software. A ranked list of genes was made using the ratio of protein intensity from differential gene expression analysis to run through the GSEA pre-ranked protocol. GSEA-pre-rank analysis was processed to detect significant secretory pathways: Trafficking, Golgi glycosylation, COPI/COPII, Protein folding, ERAD, GPI biosynthesis, ER glycosylation, Dolichol pathway, ER calcium homeostasis, Unfolded Protein Response, Translocation, Signal Peptide, Secreted Protein, and Membrane Protein for the ranked list of genes.19, 20 The criteria for considering a secretory pathway as significant is p<0.05. The leading edge analysis allows for the GSEA to determine which subsets (referred to as the leading edge subset) of genes contributed the most to the enrichment signal of a given gene set’s leading edge or core enrichment. The leading edge analysis is determined from the enrichment score (ES), which is defined as the maximum deviation from zero.
PCR analysis
Quantitative PCR was performed as previously described.10
Immunoblotting
Fibroblasts or biopsies were lysed in RIPA buffer with protease inhibitors and run for immunoblotting.10
Immunohistochemistry
Paraffin embedded specimens were deparaffinized and processed as previously described.10 All images were analyzed using ImageJ software under identical light microscope and imaging settings. TSP1 clone ab1823 (Abcam) and CD47 (Supplemental figures) clone HPA044659 (Sigma Aldrich) were used for immunochemistry.
Statistics
Proteomic analysis
We performed differential expression analysis on each protein in the proteomic data using a two-sample t-test. The normalized log2(LFQ) of each protein were used as the final log2(LFQ) for calling the differential expression. Proteins with FDR-adjusted P<.05 and absolute fold change >1.5 were considered as differentially expressed proteins (DEPs). For protein-protein interaction (PPI) network analysis (STRING) and functional annotation and enrichment analysis (DAVID), a threshold of 1.5-fold change is widely used for identifying differentially expressed proteins in the proteomics analysis.21, 22
Immunoblot and immunohistochemical samples were compared using a Wilcoxon signed rank test for paired samples, a Mann-Whitney U test for unpaired samples, or a repeated measures one way ANOVA with multiple comparisons for >2 sample comparisons (for TSP-1 and inhibitor treatments). Two tailed P<.05 was considered significant.
Results
EoE fibroblast derived matrix can alter normal fibroblasts
To understand if the ECM derived from diseased esophageal EoE fibroblasts could functionally alter normal fibroblasts, we isolated and utilized esophageal fibroblasts from 5 children with severe, active, therapy refractory EoE and 5 normal organ donors (clinical characteristics in Table S1). Passage matched esophageal fibroblasts were grown to confluency, the ECM was decellularized, and EoE or normal fibroblasts were cultured on autologous or non-autologous ECM (Figure 1A). Transcriptional analysis for collagen I, fibronectin, and α-smooth muscle actin (αSMA)as representative markers of fibrotic and myofibroblast gene expression, respectively, demonstrated no significant alterations following culture on opposing, as compared to autologous, ECM (Figure 1B).
Figure 1. EoE ECM alters normal fibroblast protein expression of collagen I and αSMA.

(A) Experimental design of fibroblast-ECM interaction experiments. (B) qPCR of fibrotic and myofibroblast genes (n=4 different normal and n=4 different EoE). Each dot: mean of subject replicates. Lines and bars: mean and SEM. (C) Representative immunoblot of fibroblasts cultured on autologous or non-autologous decellularized matrix. NL: normal, EoE: eosinophilic esophagitis. (D) Normalized quantitation of replicate experiments from 5 different normal and 5 different EoE subjects. Dots: mean of subject replicates.
ECM from fibrotic diseases can alter ribosome bound transcripts.23 Thus, we hypothesized that the ECM from EoE esophageal fibroblasts would alter protein expression from normal esophageal fibroblasts despite the absence of mRNA changes. We cultured EoE and normal esophageal fibroblasts on autologous or non-autologous ECM and analyzed protein expression for collagen I, fibronectin and αSMA via immunoblotting. Collagen I was significantly and consistently increased in normal esophageal fibroblasts following culture on an EoE fibroblast derived ECM (p<0.05, n=5 EoE and 5 normal different patient fibroblasts) (Figure 1C, D- left panel). αSMA also was consistently and significantly increased in normal fibroblasts cultured on EoE, as compared to autologous, ECM (p<0.05) (Figure 1C, D, left panel). In contrast, fibronectin expression was not altered in normal fibroblasts cultured on EoE ECM (Figure 1C, D- left panel). Normal ECM was not consistently able to change the expression of collagen I, αSMA, or fibronectin in EoE fibroblasts cultured on normal ECM (Figure 1C, D- right panel). These data demonstrate that the EoE ECM alters protein expression in normal esophageal fibroblasts. Further, these data support the hypothesis that EoE ECM proteins or physical properties can increase the fibrotic and myofibroblast potential of normal esophageal fibroblasts.
Normal cells cultured on EoE matrix have altered secretory signals on proteomic analysis
To better understand the global differences induced in normal fibroblasts by EoE ECM, we analyzed the proteomic profiles of normal esophageal fibroblasts cultured on autologous versus EoE ECM. Tandem mass spectrometry (UPLC-MS-MS) with nanoscale reversed-phase tandem mass tag labeling revealed 7 differentially expressed proteins (DEPs) (Figure 2A), all of which were downregulated. DAVID analysis demonstrated that these DEPs were enriched (enrichment score for cluster=1.12) for proteins with signal peptides (p=3.53e-02) and that were predicted to be located in the ECM (p=4.96e-02) (Table S2).
Figure 2. Proteomic analysis of Normal fibroblasts on autologous versus EoE ECM.

(A) Fibroblasts cultured on autologous or opposing (EoE) ECM (n=2 normal, n=3 EoE). (Left panel) Volcano plot of the differentially expressed proteins (DEPs) in normal fibroblasts cultured on EoE ECM (NE) versus normal fibroblasts cultured on autologous ECM (C=Control). Significant differential expression: indicated by red and blue dashed lines and FDR adjusted p-value < 0.05 (black dashed line) (Left panel). (Right panel) Heat map of the down-regulated proteins. (B) US-MS-MS of normal and EoE ECM. (Left panel) Volcano plot of dysregulated proteins in EoE versus normal decellularized matrix. (Right panel) Heat map of dysregulated proteins in EoE versus normal decellularized matrix. (C) Protein-protein interaction network (STRING) of EoE DEPs. (D) Molecular functions enriched in the ECM DEPs.
Given the enrichment for signal peptide containing proteins among the DEPs in normal fibroblasts cultured EoE ECM, we conducted a protein set enrichment analysis to investigate changes in secretory pathways and products.19, 20 Our results demonstrated that the EoE ECM caused downregulation in four specific secretory pathways (trafficking, GPI biosynthesis, proteins with signal peptide and membrane proteins). These analyses support the ability of the EoE ECM proteome to globally alter normal fibroblast protein secretion (Table S3).
TSP-1 is a central member of the EoE ECM proteome
Given the ability of the EoE ECM to alter normal fibroblast protein expression and secretion, we sought to decipher candidate proteins in the EoE ECM proteome that might be responsible for the altered function of normal fibroblasts cultured on EoE, as compared with autologous, ECM. UPLC-MS-MS proteomics and analysis was used to compare the decellularized ECM from EoE (n=3) and normal (n=3) fibroblasts. There were 24 DEPs in total when comparing the EoE and normal ECM proteome (Table 1 and Tables S4, S5). Thirteen DEPs were present in both EoE and normal ECMs. Eleven DEPs were uniquely expressed in either the EoE (3) or normal (8) ECM (Table 1). The EoE ECM proteome had more down-regulated (12) than up-regulated (1) DEPs (Figure 2B). The 3 proteins unique to the EoE ECM proteome were thrombospondin-1 (TSP-1), antithrombin 3 (ANT3), and bromodomain containing protein 9 (BRD9) (p=0.63 to 1.5e-90) (Table 1). A protein-protein interaction (PPI) network analysis completed using the Search Tool for the Retrieval of Interacting Genes (STRING) database24 revealed that 16 of the DEPs formed an ECM interactome (Figure 2C, Table S6) (observed versus expected edges=4 fold, enrichment score p=3.05e-10) with TSP-1 as a central member. Consistent with the concept that TSP-1 was functioning as a pivotal member of the EoE ECM proteome, a gene ontology (GO) analysis revealed TSP-1’s involvement in each of the 9 binding related functions (Figure 2D, Table S7).
Table 1.
Differentially expressed proteins in the ECM proteome from EoE vs. Normal fibroblasts.
| Categorya | Protein | Proteinb | Protein Intensity |
Z-test on the log2(Ratio) |
|||||
|---|---|---|---|---|---|---|---|---|---|
| Normal | EoE | Ratio | log2(Ratio)c | z score | t statistic | p-value | |||
| P01008 | ANT3 | 0.00 | 2.00 | 5.32 | 2.41 | 59.95 | −59.70 | 1.50E–90 | |
| EoE (+), | Q9H8M2 | BRD9 | 0.00 | 2.00 | 5.32 | 2.41 | 59.95 | −59.70 | 1.50E–90 |
| Normal (−) | P07996 | TSP1 | 0.38 | 1.62 | 4.32 | 2.11 | 48.40 | −48.20 | 6.29E–80 |
| P04908 | H2A1B | 2.00 | 0.00 | 0.00 | −Inf | – | – | – | |
| P13674 | P4HA1 | 2.00 | 0.00 | 0.00 | −Inf | – | – | – | |
| P15311 | EZR1 | 2.00 | 0.00 | 0.00 | −Inf | – | – | – | |
| EoE (−), | P17844 | DDX5 | 2.00 | 0.00 | 0.00 | −Inf | – | – | – |
| Normal (+) | P55010 | IF5 | 2.00 | 0.00 | 0.00 | −Inf | – | – | – |
| Q04828 | AK1C1 | 2.00 | 0.00 | 0.00 | −Inf | – | – | – | |
| Q05707 | COEA1 | 2.00 | 0.00 | 0.00 | −Inf | – | – | – | |
| Q9HCB6 | SPON1 | 2.00 | 0.00 | 0.00 | −Inf | – | – | – | |
There are two categories of proteins: (1) the proteins significantly expressed in the EoE but not or very low expression in normal ECM denoted in red=‘EoE (+), Normal (−)’ and (2) proteins significantly expressed in normal but not in the EoE ECM denoted in blue= ‘EoE (−), Normal (+)’.
For the protein in ‘EoE (−), Normal (+)’, z-test cannot be performed due to 0 expression
The font colors for genes represent the three categories match those in Figure 2C.
ANT3: antithrombin 3; BRD9: bromodomain containing protein 9; TSP1: thrombospondin-1
TSP-1 is expressed in active EoE and induces collagen I in esophageal fibroblasts
TSP-1 is a functionally complex, TGFβ1-induced glycoprotein that, itself, activates TGFβ1, mediates cell-cell adhesion, cell-matrix adhesion, cell motility, collagen fibrillogenesis, and angiogenesis.25–28 Each of these functions are potentially relevant to EoE remodeling. We used EoE and normal biopsies to validate that our in vitro fibroblast ECM reflected the in vivo pathogenic state. EoE esophageal fibroblasts had higher TSP-1 levels intracellularly and in their supernatants compared to normal fibroblasts (Figure 3A). TSP-1 was higher in active than inactive EoE biopsies on immunoblotting (Figure 3B). In esophageal biopsies, TSP-1 was localized in both the epithelium and subepithelial lamina propria of active EoE patients. Active EoE biopsies demonstrated significantly higher numbers of TSP-1 expressing cells in the subepithelial lamina propria on immunohistochemical analysis (Figure 3C, Supplemental Table 1). TSP-1 was found both intra- and extracellularly in active EoE biopsies, consistent with its secretion and reports of TSP-1 retention in the ECM as insoluble punctate droplets.29 TSP-1 has multiple binding partners, including CD47. Quantification of CD47 expressing cells demonstrated that active EoE biopsies had higher numbers of CD47+ cells as compared with normal esophagi (Supplemental Figure 1). Together, these data support that proteomic analysis of esophageal fibroblasts cultured on EoE versus normal ECM can reveal disease associated molecules such as TSP-1 in vivo.
Figure 3. Thrombospondin-1 is elevated in active EoE.

(A) Representative immunoblots of EoE and normal esophageal fibroblasts extracts and supernatants probed for TSP-1 with quantification of protein normalized to β-tubulin for cell extracts (Dots: different subject fibroblasts). (B) Representative immunoblot and quantitation of active and inactive EoE biopsies probed for TSP-1. Dots: different patients. Lines and bars: mean and SEM. (C) Normal and EoE esophageal biopsies stained and quantitated for TSP-1 localization. Dots: average TSP-1 positive cells in 3–5 hpfs in the lamina propria of different subjects. Lines and bars: mean and SEM.
We hypothesized that treatment of esophageal fibroblasts with TSP-1 might mimic some of the changes induced in normal fibroblasts by the EoE ECM. Normal fibroblasts treated with recombinant human TSP-1 demonstrated a dose-dependent increase in collagen I expression (Figure 4A). The same trend was seen in EoE fibroblasts but was less robust (Supplemental Figure 2), suggesting that EoE fibroblasts had become accustomed to TSP-1 exposure. In contrast, neither fibronectin nor αSMA were altered by TSP-1 treatment (Figure 4A). These data demonstrate that TSP-1 can modify collagen I production from esophageal fibroblasts and thereby partially phenocopy the effects of the EoE ECM proteome on normal esophageal fibroblast protein expression.
Figure 4. Thrombospondin-1 increases collagen I, is induced by TGFβ1 and collagen induction by EoE ECM is blocked by TGFβ-receptor I inhibition.

(A) Representative immunoblot and quantitation of normal fibroblasts treated with TSP-1. Dots: biological replicates. (B) qPCR of TGFβ1 treated normal fibroblasts. Dots: biological replicates. (C) TSP-1 protein quantitation of normal fibroblasts treated with TGFβ1. Dots: biological replicates. (D) Collagen I immunoblot quantitation of TGFβ receptor I inhibitor (SB431542) treated normal fibroblasts cultured on EoE ECM. Bars: mean and SEM (n=6 biological replicates).
TGFβ1 expressing cells are increased in EoE. In order to understand if TGFβ1 could be involved in the induction of TSP-1, we treated esophageal fibroblasts with TGFβ1 and found significant increases in TSP-1 transcript and protein (Figure 4B, C). EoE ECM-induced collagen I induction in normal fibroblasts was blocked when the TGFβ1 receptor I was chemically inhibited (Figure 4D). Together, these data demonstrate that TSP-1 in the EoE ECM induces collagen I and that this effect is blocked when signals, such as TGFβ1, that increase TSP-1 are downregulated.
Discussion
EoE is an antigen driven Th2 disorder of increasing prevalence in children and adults that manifests clinically with symptoms of esophageal dysfunction and progresses to esophageal rigidity and strictures when untreated or unresponsive to therapy.1, 2,6 A physically rigid matrix propagates and exacerbates inflammation-induced structural cell dysfunction, thereby contributing to esophageal dysmotility and loss of luminal patency.8, 9 The remodeled, fibrostenotic esophagus is difficult to treat and therapeutic failure or incomplete response is common.6, 30 For those patients with the most severe disease and the highest number of complications, therapeutic options are limited. Our understanding of the master regulators in the EoE ECM that drive and/or predict disease complications and progression also is limited. This paucity of knowledge leaves a significant gap for disease course prediction and challenges the development of impactful and novel therapeutic interventions, a common issue for many fibrosing diseases. Herein, we utilized high sensitivity proteomics to analyze active EoE and normal esophageal fibroblasts and their derivative ECMs to understand salient molecules in the EoE ECM interactome. Further, our data provides a manner in which to understand other fibrosing diseases.
We describe several novel findings. First, we demonstrate that the EoE esophageal fibroblast derived ECM is distinct from that of a normal fibroblast. Secondly, we show that the unique EoE ECM proteome dysregulates normal esophageal fibroblast protein expression. Thirdly, we found elevated TSP-1 in the EoE ECM proteome and validated its increased expression in vivo during active EoE. Fourthly, we demonstrate the ability of TSP-1 to induce collagen I production from normal fibroblasts and show that this is likely dependent on TGFβ1. Together these data support our hypothesis that high sensitivity proteomic analysis using esophageal structural cells in vitro can elucidate meaningful disease relevant molecules in vivo in EoE and distinguish new disease mechanisms that could form the basis for clinically relevant interventions.
TSP-1 is a complex and multifunctional protein.25, 26, 29 It can be a central regulator of the ECM proteome since it binds to many ECM constituents including collagen I, heparin, and fibrinogen, to cellular receptors including CD47 and 36, and to growth factors and proteases such as MMP2, TGFβ1, and urokinase plasminogen activator, all of which have demonstrated elevation in active EoE.4,31 Our current data and that in publicly available databases supports increased levels of TSP-1 receptors such as CD47 and CD36 in active EoE.32 In addition, TSP-1 can activate TGFβ1.25 Indeed, multiple groups, including ours, have shown that TGFβ1 elevation and dysregulation are disease driving and/or modifying in EoE.10, 33 Since TGFβ1 is able to induce TSP-1 expression from esophageal fibroblasts and TSP-1 can activate latent TGFβ1, the diseased ECM likely creates a feed forward loop for EoE disease severity. Future studies to dissect this mechanism are of interest. Given that other proteins in addition to TSP-1 were dysregulated in the ECM proteome, understanding their roles in EoE is also of interest. Since pathologic remodeling, unlike the physiologic process of wound healing, seems not to be adequately downregulated by standard therapies, these types of positive loops are likely to represent relevant disease mechanisms.
ECM is a complex structure containing large molecules such as collagen that are needed in the proper proportions for fibrillogenesis and cross-linking. When ECM constituents are imbalanced, there are significant ramifications on ECM elasticity, cell migration, activation of cytokines, and structural cell function. Indeed, altered collagen cross linking has been recently shown in EoE pathogenesis.11 It is interesting that the EoE esophagus becomes rigid over time but that a genetic predisposition to increased flexibility such as that seen in Ehler’s Danlos, Marfan’s and benign hypermobility syndromes also associate with EoE.34, 35 Our proteomic analysis demonstrated alterations in multiple binding pathways in the ECM including heparin. These findings imply that the perturbations in the balance of ECM components may underlie EoE pathogenesis.
Our data also has implications for disease management. Fibroblasts can be isolated and cultured for approximately 8 passages from a single biopsy specimen from the pediatric esophagus. Our current finding of TSP-1 from EoE fibroblasts that was validated in esophageal biopsy specimens implies that some degree of functional memory is retained by the diseased esophageal fibroblasts in vitro. Harnessing this observation could be a powerful tool for EoE clinical care since distinctly dysregulated ECMs could be a reflection clinical disease severity. Further, understanding the differences between the normal and dysregulated ECMs derived from esophageal fibroblasts may characterize factors in the ECM that could protect from or drive disease complications. Gauging EoE severity is challenging and currently functional remodeling is assessed by EndoFLIP and fibrosis is estimated on subepithelial tissue.7, 13, 14, 36 However, neither of these tools is universally available due the paucity of deep tissue in most biopsies and the limited number of centers performing EndoFLIP. Our findings support the concept that EoE fibroblasts, their derivative ECM, and the interaction between EoE ECM and normal fibroblasts may perform as a new tool for predicting and assessing disease severity. It is important to note that our normal fibroblasts are obtained from organ donor esophagi and therefore likely represent a more normal state than that seen in “normal” patients undergoing esophageal biopsy since these people have clinical complaints that warrant an endoscopic evaluation. Whether our approach has generalizability will require larger studies using higher sample numbers and patients with varying disease severities and different diseases such as GERD.
We acknowledge that our study has weaknesses. There number of patients whose fibroblasts were utilized was small. However, severe EoE is relatively rare in children and we believe the best starting point for our type of study is to compare those patients with most severe disease to those who have a normal esophagus. Our normal donors were older than the EoE subjects but current experiences do not demonstrate clear differences between adult and pediatric EoE disease mechanisms in vitro or in vivo.1,8 In addition, all ECMs have an insoluble fraction that cannot be assessed. We were surprised to see down-regulation of some proteins that have been previously been demonstrated to be up-regulated in the esophagus of EoE patients, such as periostin.37 It is possible that molecules such as periostin become trapped in the insoluble fraction when performing proteomic studies. Alternatively, it is possible that periostin is not made primarily by fibroblasts but rather is being induced in other cells such as epithelial cells in EoE.37
In conclusion, we have utilized a high sensitivity proteomic approach to distinguish the EoE and normal ECM proteomes, demonstrated that a single factor, TSP-1, determined by proteomic analysis, is elevated in vivo and that TSP-1 alters the function of normal fibroblasts. This type of approach can help us understand patient phenotypes and detect novel therapeutic targets in fibrotic diseases.
Supplementary Material
Acknowledgments
Sources of Funding: This work was supported by grants from the NIH/NIAID/NIDDK R01AI092135 (SA), K24AI135034 (SA). Additional funding from R01DK114457 (SA), the Novo Nordisk Foundation provided to the Center for Bio sustainability at the Technical University of Denmark, NNF10CC1016517 (A.W.T.C.), and R35 GM119850 (N.E.L).
Abbreviations
- BRD9
Bromodomain containing protein 9
- DAVID
Database for annotation, visualization and integrated discovery
- DEP
Differentially expressed protein
- ECM
Extracellular matrix
- EndoFLIP
Endoscopic functional lumen imaging probe
- EoE
Eosinophilic esophagitis
- ES
Enrichment score
- GO
Gene ontology
- GSEA
Gene set enrichment analysis
- Hpf
high power field
- LFQ
Label-free quantification
- PAI-1
Plasminogen activator inhibitor-1
- PPI
Protein-protein interaction
- STRING
Search Tool for the Retrieval of Interacting Genes
- TGFβ1
Transforming growth factor-beta-1
- TNFα
Tumor necrosis factor-alpha
- TSP-1
Thrombospondin-1
- UPLC-MS/MS
Ultra high-pressure liquid chromatography-tandem mass spectrometry
- αSMA
alpha-Smooth muscle actin
Footnotes
Conflict of Interest: SSA and RD are co-inventors of oral viscous budesonide patented by the University of California, San Diego and licensed by Takeda. S.S.A. has consulting agreements with AstraZeneca, Astellas, AImmune, and Gossamer Bio. The other authors declare no relevant conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an article that has undergone enhancements after acceptance, such as the addition of a cover page and metadata, and formatting for readability, but it is not yet the definitive version of record. This version will undergo additional copyediting, typesetting and review before it is published in its final form, but we are providing this version to give early visibility of the article. Please note that, during the production process, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dellon ES, Liacouras CA, Molina-Infante J, Furuta GT, Spergel JM, Zevit N, et al. Updated international consensus diagnostic criteria for eosinophilic esophagitis: Proceedings of the AGREE conference. Gastroenterology 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dellon ES, Kim HP, Sperry SL, Rybnicek DA, Woosley JT, Shaheen NJ. A phenotypic analysis shows that eosinophilic esophagitis is a progressive fibrostenotic disease. Gastrointest Endosc 2014; 79:577–85 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schoepfer AM, Safroneeva E, Bussmann C, Kuchen T, Portmann S, Simon HU, et al. Delay in diagnosis of eosinophilic esophagitis increases risk for stricture formation in a time-dependent manner. Gastroenterology 2013; 145:1230–6 e1–2. [DOI] [PubMed] [Google Scholar]
- 4.Azouz NP, Ynga-Durand MA, Caldwell JM, Jain A, Rochman M, Fischesser DM, et al. The antiprotease SPINK7 serves as an inhibitory checkpoint for esophageal epithelial inflammatory responses. Sci Transl Med 2018; 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aceves SS, Newbury RO, Dohil R, Bastian JF, Broide DH. Esophageal remodeling in pediatric eosinophilic esophagitis. J Allergy Clin Immunol 2007; 119:206–12. [DOI] [PubMed] [Google Scholar]
- 6.Eluri S, Runge TM, Cotton CC, Burk CM, Wolf WA, Woosley JT, et al. The extremely narrow-caliber esophagus is a treatment-resistant subphenotype of eosinophilic esophagitis. Gastrointest Endosc 2016; 83:1142–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nhu QM, Aceves SS. Tissue Remodeling in Chronic Eosinophilic Esophageal Inflammation: Parallels in Asthma and Therapeutic Perspectives. Front Med (Lausanne) 2017; 4:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Muir AB, Dods K, Henry SJ, Benitez AJ, Lee D, Whelan KA, et al. Eosinophilic Esophagitis-Associated Chemical and Mechanical Microenvironment Shapes Esophageal Fibroblast Behavior. J Pediatr Gastroenterol Nutr 2016; 63:200–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tkachenko E, Rawson R, La E, Doherty TA, Baum R, Cavagnero K, et al. Rigid substrate induces esophageal smooth muscle hypertrophy and eosinophilic esophagitis fibrotic gene expression. J Allergy Clin Immunol 2016; 137:1270–2 e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rawson R, Yang T, Newbury RO, Aquino M, Doshi A, Bell B, et al. TGF-beta1-induced PAI-1 contributes to a profibrotic network in patients with eosinophilic esophagitis. J Allergy Clin Immunol 2016; 138:791–800 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kasagi Y, Dods K, Wang JX, Chandramouleeswaran PM, Benitez AJ, Gambanga F, et al. Fibrostenotic eosinophilic esophagitis might reflect epithelial lysyl oxidase induction by fibroblast-derived TNF-alpha. J Allergy Clin Immunol 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Walker C, Mojares E, Del Rio Hernandez A. Role of Extracellular Matrix in Development and Cancer Progression. Int J Mol Sci 2018; 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carlson DA, Hirano I, Zalewski A, Gonsalves N, Lin Z, Pandolfino JE. Improvement in Esophageal Distensibility in Response to Medical and Diet Therapy in Eosinophilic Esophagitis. Clin Transl Gastroenterol 2017; 8:e119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Menard-Katcher C, Benitez AJ, Pan Z, Ahmed FN, Wilkins BJ, Capocelli KE, et al. Influence of Age and Eosinophilic Esophagitis on Esophageal Distensibility in a Pediatric Cohort. Am J Gastroenterol 2017; 112:1466–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hassan M, Aceves S, Dohil R, Gharibans A, Newbury R, Proudfoot J, et al. Esophageal Compliance Quantifies Epithelial Remodeling in Pediatric Patients With Eosinophilic Esophagitis. J Pediatr Gastroenterol Nutr 2019; 68:559–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Banerjee I, Carrion K, Serrano R, Dyo J, Sasik R, Lund S, et al. Cyclic stretch of embryonic cardiomyocytes increases proliferation, growth, and expression while repressing Tgf-beta signaling. J Mol Cell Cardiol 2015; 79:133–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang JK, Carlin DE, Yu MK, Zhang W, Kreisberg JF, Tamayo P, et al. Systematic Evaluation of Molecular Networks for Discovery of Disease Genes. Cell Syst 2018; 6:484–95 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009; 4:44–57. [DOI] [PubMed] [Google Scholar]
- 19.Feizi A, Gatto F, Uhlen M, Nielsen J. Human protein secretory pathway genes are expressed in a tissue-specific pattern to match processing demands of the secretome. NPJ Syst Biol Appl 2017; 3:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Proteomics. Tissue-based map of the human proteome. Science 2015; 347:1260419. [DOI] [PubMed] [Google Scholar]
- 21.Dunn J, Ferluga S, Sharma V, Futschik M, Hilton DA, Adams CL, et al. Proteomic analysis discovers the differential expression of novel proteins and phosphoproteins in meningioma including NEK9, HK2 and SET and deregulation of RNA metabolism. EBioMedicine 2019; 40:77–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen C, Hao X, Geng Z, Wang Z. ITRAQ-based quantitative proteomic analysis of MG63 in response to HIF-1alpha inducers. J Proteomics 2020; 211:103558. [DOI] [PubMed] [Google Scholar]
- 23.Parker MW, Rossi D, Peterson M, Smith K, Sikstrom K, White ES, et al. Fibrotic extracellular matrix activates a profibrotic positive feedback loop. J Clin Invest 2014; 124:1622–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res 2015; 43:D447–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Belotti D, Capelli C, Resovi A, Introna M, Taraboletti G. Thrombospondin-1 promotes mesenchymal stromal cell functions via TGFbeta and in cooperation with PDGF. Matrix Biol 2016; 55:106–16. [DOI] [PubMed] [Google Scholar]
- 26.Resovi A, Pinessi D, Chiorino G, Taraboletti G. Current understanding of the thrombospondin-1 interactome. Matrix Biol 2014; 37:83–91. [DOI] [PubMed] [Google Scholar]
- 27.Murphy-Ullrich JE. Thrombospondin 1 and Its Diverse Roles as a Regulator of Extracellular Matrix in Fibrotic Disease. J Histochem Cytochem 2019; 67:683–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Min-DeBartolo J, Schlerman F, Akare S, Wang J, McMahon J, Zhan Y, et al. Thrombospondin-I is a critical modulator in non-alcoholic steatohepatitis (NASH). PLoS One 2019; 14:e0226854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adams JC, Bentley AA, Kvansakul M, Hatherley D, Hohenester E. Extracellular matrix retention of thrombospondin 1 is controlled by its conserved C-terminal region. J Cell Sci 2008; 121:784–95. [DOI] [PubMed] [Google Scholar]
- 30.Eluri S, Runge TM, Hansen J, Kochar B, Reed CC, Robey BS, et al. Diminishing Effectiveness of Long-Term Maintenance Topical Steroid Therapy in PPI Non-Responsive Eosinophilic Esophagitis. Clin Transl Gastroenterol 2017; 8:e97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beppu L, Yang T, Luk M, Newbury RO, Palmquist J, Dohil R, et al. MMPs-2 and −14 are Elevated in Eosinophilic Esophagitis and Reduced Following Topical Corticosteroid Therapy. J Pediatr Gastroenterol Nutr 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sherrill JD, Kc K, Wu D, Djukic Z, Caldwell JM, Stucke EM, et al. Desmoglein-1 regulates esophageal epithelial barrier function and immune responses in eosinophilic esophagitis. Mucosal Immunol 2014; 7:718–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Duong LD, Rawson R, Bezryadina A, Manresa MC, Newbury RO, Dohil R, et al. TGFbeta1 single-nucleotide polymorphism C-509T alters mucosal cell function in pediatric eosinophilic esophagitis. Mucosal Immunol 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abonia JP, Wen T, Stucke EM, Grotjan T, Griffith MS, Kemme KA, et al. High prevalence of eosinophilic esophagitis in patients with inherited connective tissue disorders. J Allergy Clin Immunol 2013; 132:378–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Frischmeyer-Guerrerio PA, Guerrerio AL, Oswald G, Chichester K, Myers L, Halushka MK, et al. TGFbeta receptor mutations impose a strong predisposition for human allergic disease. Sci Transl Med 2013; 5:195ra94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kwiatek MA, Hirano I, Kahrilas PJ, Rothe J, Luger D, Pandolfino JE. Mechanical properties of the esophagus in eosinophilic esophagitis. Gastroenterology 2011; 140:82–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blanchard C, Mingler MK, McBride M, Putnam PE, Collins MH, Chang G, et al. Periostin facilitates eosinophil tissue infiltration in allergic lung and esophageal responses. Mucosal Immunol 2008; 1:289–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.