

Abstract
The androgen receptor (AR) is critical in the progression of prostate cancer (PCa). Small molecule antagonists that bind to the ligand binding domain (LBD) of the AR have been successful in treating PCa. However, the structural basis by which the AR antagonists manifest their therapeutic efficacy remains unclear, due to the lack of detailed structural information of the AR bound to the antagonists. We have performed accelerated molecular dynamics (aMD) simulations of LBDs bound to a set of ligands including a natural substrate (dihydrotestosterone), an agonist (RU59063) and three antagonists (bicalutamide, enzalutamide and apalutamide) as well as in the absence of ligand (apo). We show that the binding of AR antagonists at the substrate binding pocket alter the dynamic fluctuations of H12, thereby disrupting the structural integrity of the agonistic conformation of AR. Two antagonists, enzalutamide and apalutamide, induce considerable structural changes to the agonist conformation of LBD, when bound close to H12 of AR LBD. When the antagonists bind to the pocket with different orientations having close contact with H11, no significant conformational changes were observed, suggesting the AR remains in the functionally activated (agonistic) state. The simulations on a drug resistance mutant F876L bound to enzalutamide demonstrated that the mutation stabilizes the agonistic conformation of AR LBD, which compromises the efficacy of the antagonists. Principal component analysis (PCA) of the structural fluctuations shows that the binding of enzalutamide and apalutamide induce conformational fluctuations in the AR, which are markedly different from those caused by the agonist as well as another antagonist, bicalutamide. These fluctuations could only be observed with the use of aMD.
Subject terms: Biophysics, Drug discovery, Chemistry
Introduction
Androgen Receptor (AR) is a ligand-activated transcription factor that regulates the expression of many target genes that are crucial for male sexual differentiation and development. The AR plays a critical role in the development and proliferation of prostate cancer (PCa)1,2. In the early phase of PCa, androgen ablation therapy or chemotherapy with antiandrogens are established as the standard therapies. However, castration-resistant prostate cancer (CRPC) occurs after a few years of the standard treatment, which becomes more aggressive and ultimately lethal3–5. CRPC is still predominantly dependent upon the AR signaling pathway for its progression. The cellular mechanisms to resist the existing therapies include the AR gene amplification, the AR mutations, the androgen-independent AR activation and the appearance of AR splice variants6. The second generation AR antagonists, enzalutamide (brand name: Xtandi) and apalutamide (brand name: Erleada) are approved for clinical use in CRPC, which show much improved therapeutic efficacy over the first generation antagonists4,7,8.
The AR, a subfamily of nuclear receptors (NRs), consists of two well-defined structural domains, a DNA binding domain and a C-terminal ligand binding domain (LBD); there is also a highly variable N-terminal domain. The LBD has been a major interest in the drug discovery efforts, where most ligands and cofactors bind to9. Upon binding of a ligand to the ligand binding pocket in the LBD, NRs undergo conformational changes that are associated with the activation function-2 (AF-2). Biochemical evidence suggests that upon binding of an agonist such as testosterone, the repositioning of H12 and the formation of hydrophobic cleft follow, that promote the interaction of the AR with the LXXLL motif of co-activators10,11. On the contrary, the binding of antagonists abolishes the recruitment of co-activators by preventing H12 from folding over into the space that facilitates AR interactions with co-activators. The structural details of several NR antagonists bound to the LBD of other NR have been reported. An estrogen receptor alpha (ERα) antagonist, raloxifene, induces repositioning of H12 by its bulky side chain and disrupts the overall surface topography of AF-212. Despite many attempts, the structural basis of the AR antagonism is not fully understood. There are over 90 x-ray crystal structures of the human AR LBD deposited in Protein Data Bank. However, all of the existing crystal structures represent functionally activated ARs either wild type bound to agonists or drug-resistant mutant ARs bound to antagonists (Table 1)13.
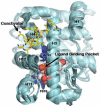
Table 1.
X–ray crystal structures of AR LBD from Protein Data Bank and their 3D structural alignment complexed with various ligands.
| PDB ID | AR type | Ligand | |
|---|---|---|---|
| 1XQ3 | Wild type | AR agonist (R1881)14 |
|
| 1T63 | Wild type | AR agonist (dihydrotestosterone; DHT)15 | |
| 2AX6 | T877A mutant | AR antagonist (hydroxyflutamide)16 | |
| 2AXA | Wild type | AR agonist (S1)16 | |
| 1Z95 | W741L mutant | AR antagonist (R-bicalutamide)17 | |
| 2OZ7 | T877A mutant | AR antagonist (cyproterone acetate)18 | |
| 2PIP | Wild type | AR agonist (DTH)/BF3 inhibitor (K10)19 | |
| 3V49 | Wild type | SARM (diarylhydantoins)20 | |
| 5JJM | Wild type | AR agonist (DHT)21 | |
| 5VO4 | Wild type | SARM (pyrrole-2-carbonitrile derivatives)22 | |
The key mutant residues (W741, T877 and F876) near the binding site for the ligands are represented as colored in blue. Image was created by using Visual Molecular Dynamics (VMD) v1.9.352.
Computational studies have elucidated the molecular basis of AR antagonism. Bisson et al. proposed that the sulfonyl group of R-bicalutamide pushes W741 from its activating position, thereby achieving the observed antagonism. They also showed that the decreased activity of hydroxyflutamide (HFT) on wild type AR is caused by the weaker interaction between HFT and Met895 compared to T877A mutant AR23. Zhou and co-workers employed replica exchange molecular dynamics (REMD) simulations to study the structural changes of the WT and the T877A mutant ARs bound to HFT. Conformations of the H12 sampled from the REMD simulations were shown to have a correlation with the agonistic/antagonistic activities of the ligand24. By performing MD simulations and quantum mechanics (QM) calculations, Osguthorpe and Hagler demonstrated that bicalutamide can access an additional binding pocket (B-site) of the AR LBD, which results in the instability of H1225. Based on 10 ns MD simulations, Balbas et al. showed F876L mutation in AR allows enzalutamide to act as an agonist26. Xu et al. analyzed the structural stability of AR upon binding of dihydrotestosterone (DHT) and co-activator (SRC), based on 20 ns MD simulations27. Liu et al. demonstrated that the ligand binding affects the AR–coactivator interactions as well as the allosteric regulation pathway from ligands to co-activator, based on MD simulations and free energy calculations28. By using homology modeling, Wang et al. constructed a putative antagonistic structure of the AR and proposed the role of H12 in the molecular function of the AR29. Duan et al. carried out microsecond unbiased MD simulations as well as bias-exchange metadynamics MD simulations to quantify the conformational characteristics of antagonist bound AR LBD30. Recently, Hu et al. summarized the recent advances in the discovery of AR antagonists using computer-aided drug design approaches31. The computational analyses comprehensively support the mechanistic hypothesis that the AR antagonists cause conformational changes to the AR LBD, which can be correlated with the antagonism of the AR. However, to our best knowledge, the influences of the second-generation antagonists such as enzalutamide and apalutamide to the conformational dynamics of the AR LBDs have not been elucidated in detail. Moreover, a comprehensive comparison of the conformational changes in AR LBDs bound to the agonists, the first- and the second-generation antagonists has not been carried out. In addition, a structure activity-relationship that is readily applicable to the discovery of novel AR antagonists is in dire need for the rapid screening of next-generation AR antagonists.
Herein, we examined the detailed structural changes of the AR LBDs caused by the binding of antagonists by means of computational modeling and molecular dynamics (MD) simulations of the various ligands bound to the AR LBD. We have studied conformational dynamics on the LBDs of the wild type and F876L mutant AR in the presence of the first and the second-generation antagonists, the agonists, and in the absence of ligands (apo) using accelerated MD simulations. We found that the binding of agonists stabilizes the AR LBDs by retaining the agonistic conformation, that are in agreement with the previous experimental and computational results. The simulations revealed that the wild type AR bound to antagonist showed distinctive conformational dynamics depending on the initial pose and the nature of the antagonists. We explored two distinct binding poses of the N-methylbenzamide moiety of enzalutamide in the AR. The results show that the structural changes of α-helix 12 (H12) only occurs when the N-methylbenzamide of enzalutamide binds proximal to the H12, but not when it directs toward α-helix 11 (H11). We also found that the F876L mutation of the AR stabilizes the agonistic conformation, regardless of the conformation of enzalutamide in the binding pocket. Binding of bicalutamide to the wild type AR destabilizes the H1–H3 loop significantly, unlike the case of enzalutamide. The comparison of two distinct conformational changes induced by bicalutamide and enzalutamide suggested two different mode of antagonistic actions, in spite of their structural similarities. The finding suggested a new mechanistic scenario for AR antagonism caused by the second-generation antagonists, where a large conformational change in H12 directly hampers the binding of the cofactors to AR LBDs. Finally, we performed principal component analysis (PCA) on the conformational dynamics of the AR in the presence of agonists, antagonists, and the absence of ligand. The analysis suggested that one can correlate the efficacy of each ligand with the conformational fluctuation of LBD sampled from aMD simulations.
Results and discussion
Structures of AR ligands in the ligand binding domain of AR
Figure 1 shows the chemical structures of the five AR ligands studied here. Each represents a specific class of ligands: dihydrotestosterone (DHT) is a natural agonist and RU59063 (RU5) is an agonist and the lead compound we used for the development of enzalutamide and apalutamide. R-Bicalutamide (BCA), enzalutamide (ENZ) and apalutamide (APL) are AR antagonists, approved for therapeutic application by the FDA. APL has a similar chemical structure to ENZ, but shows a greater efficacy and a higher therapeutic index with lower steady-state plasma levels than ENZ32. The chemical structures of the ligands can be divided into three structural units that determine the binding positions. The C3 ketone on the A-ring of DHT is essential for the AR binding by interacting with the key residues on H3, H5 and β3 of the AR LBD. A number of non-steroidal AR agonists or antagonists replace the A-ring of DHT with substituted aromatic rings like a 3-trifluoromethyl-4-cyanophenyl (Fig. 1) or a 3-chloro-4-cyanophenyl group. The central parts of the structures occupy the ligand binding pocket covered with H3, H4 and H11 and control the shape of the ligand. The left-most segment plays a key role to produce agonist or antagonist properties by stabilizing or disrupting H12 (vide infra).
Figure 1.

Representative AR agonists and antagonists. Two proposed binding conformations for enzalutamide in AR LBD. Images were created by using Chemdraw and VMD v1.9.352.
We initially carried out 250 ns conventional MD simulations with the bound AR ligands. The backbone root-mean-square deviation (RMSD) variation of the AR bound to ENZ was small (< 2 Å), and there were no notable changes from the starting agonistic conformation (Figure S1). As such, we applied accelerated MD simulations to observe large-scale conformational dynamics of the LBDs in apo and bound to the ligands of interest33. Accelerated MD (aMD) is a method to greatly enhance conformational sampling of biomolecules. Using aMD can facilitate conformational transitions of proteins over 500-fold than the conventional MD simulations. The aMD method has been successfully applied to study conformational dynamics of proteins and protein–ligand complexes34–36. We monitored the conformational changes of the AR LBD, particularly the pockets near H11 and H12 as well as the cofactor binding region near the ligand binding pocket during aMD simulations. To analyze further the structural fluctuations of the AR LBD simulated, principal component analyses (PCA) were employed to examine the relationships of conformations observed when agonists and antagonists bind to the AR37,38.
Table 2 summarizes the systems which have been simulated with aMD. Two PDB structures (2AXA and 2PIP) were chosen as initial structures for the simulations. Both of the PDB structures represent agonistic conformations of the wild type (WT) AR, which are co-crystallized with a synthetic agonist S116 and DHT19. The ligands were removed for the simulations of apo AR. We conducted constant temperature MD simulations of 250 ns duration and then commenced aMD simulations for 250 ns, starting from the last structure of the cMD simulations (see SI for details).
Table 2.
Model systems simulated with aMD in this study.
| PDBs:2AXA, 2PIP | Wild type AR | Mutant AR |
|---|---|---|
| apo | No ligand | No ligand |
| Agonist | DHT | – |
| RU59063a | – | |
| Antagonist | ENZa | (W876L) ENZa |
| APLa | – | |
| BCAa | (W741L) BCA |
aThe simulations were performed with the ligands in both conformation 1 and 2.
Accelerated MD simulations of AR in apo and AR bound to agonists
Figure 2 summarizes the aMD simulations of AR in apo. Figure 2a shows the backbone RMSDs of the simulated apo AR LBDs from the initial X-ray crystal structures. For both of the aMD trajectories initiated from the two PDB structures, the backbone RMSDs gradually increase to 3.0 Å after 250 ns. In the absence of ligands, both PDB structures undergo structural changes from the initial agonist-bound conformation. To examine residue specific atomistic fluctuations, root mean square fluctuations (RMSF) were calculated after superimposing all the trajectories to the initial conformation (Fig. 2b). We observed significant RMSF changes in a loop region H1–H3, H8–H9 and H9–H10. In particular, a series of movement of several amino acids (P682 to D695, colored in red in Fig. 2c) on H1–H3 loop was observed. Although the functional role of the loop is not well understood in the AR, the H1–H3 loop in glucocorticoid receptor (GR) serves as the interaction for a heat-shock protein (Hsp90) and the co-chaperone complexes and influences the hormone binding affinity39,40. Due to the conformational similarity of LBD to the other steroid receptors, we speculate that this loop in the AR may also affect interactions with the chaperon machineries.
Figure 2.

Conformational changes of apo AR during 250 ns aMD simulations. (a) The backbone RMSD. (b) RMSF analysis. (c) The comparison of the initial X-ray crystal structure of the AR (PDB ID: 2AXA) after removal of the ligand (S1) and the resulting structure after 250 ns aMD simulation. The regions showing RMSF > 2.5 Å are colored in red. Images were created by using VMD v1.9.3, plugin52 and Gnuplot v5.257.
The binding of a natural substrate (DHT) and a synthetic agonist (RU5) maintained the agonistic conformation after 250 ns of aMD simulations (Figs. 3 and S2). Upon binding to DHT, the RMSD changes in the AR did not exceed 3.0 Å from the initial conformations that represent agonistic conformation of the AR (Figs. 3a and S2B). The binding of RU5 retained the structural integrity of the AR LBD (Figs. 3b and S2C). Of note, two possible orientations of thiohydantoin group of RU5 were considered (conformation 1 and 2 in Fig. 3b). However, the RMSD from the initial agonistic conformation did not exceed 2.5 Å through most of the simulation period. These results demonstrate that the binding of agonists stabilize the AR LBD by holding the structural components that surround the ligand binding pocket, when compared with the structural changes that were observed from the AR in apo. In the following, we demonstrate that the AR LBD becomes unstable upon binding to the antagonists of AR.
Figure 3.

The wild type ARs bound to the agonists. (a) The snapshots of the initial structure (PDB ID: 2PIP) of the wild type ARs bound to DHT and the resulting structure after 250 ns aMD simulations. Initial and 250 ns simulated structures are colored in cyan and lime, respectively. (b) The wild type ARs bound to RU5s. Initial structures and 250 ns aMD simulated structures are colored in cyan and tan, respectively. Images were created by using VMD v1.9.352.
Binding of ENZ displaces H12 of the AR
We examined plausible binding modes of ENZ in the ligand binding pocket of AR, as high resolution structural information of the AR LBD bound to ENZ is not available. In the beginning, we explored viable conformers of ENZ by means of quantum mechanical calculations. As the head group (benzonitrile) and the core group (thiohydantoin) can rotate across the C–N bond connecting the two functional moieties, multiple energetic minimums were expected. Our quantum chemical calculations demonstrated that the two preferred conformers differ in Gibbs free energy by 1.5 kcal/mol (Fig. 4), suggesting that both of the conformers are accessible in vivo. The two conformers are termed as conformation 1 and conformation 2, respectively. Starting from the two conformers, we explored the possible conformations of ENZ in the binding pocket of AR LBD based on the molecular docking simulations and the top scored binding pose of ENZ was selected as a starting structure for MD simulations. For conformation 1, the N-methylbenzamide tail stretches toward H11 (Fig. 5b). For conformation 2, the tail of ENZ is placed in the space between H4 and H12 (Fig. 5c). The pose of the tail group in conformation 2 resembles the conformation of the tail group of an agonist (S1) that was co-crystallized with the AR LBD16 and that of BCA proposed by Osguthorpe and Hagler25. In the following, we demonstrate that the binding of ENZ in conformation 2 induces conformational changes in the surrounding regions of the binding pocket of AR LBD.
Figure 4.

Two lowest energy conformers of ENZ. (a) Conformer with the second lowest in energy (conformation 1) and (b) the lowest in energy conformation (conformation 2). The dihedral angles of the four highlighted atoms (C-C-N-C) are shown. Images were created by using CYLView v.1.0.56156.
Figure 5.

Conformational changes of the wild type AR bound to ENZ during 250 ns aMD simulations. (a) The backbone RMSDs of AR-ENZ complexes relative to the initial agonist conformation. The visualizations of initial and 250 ns simulated structures of the wild type AR bound to ENZ in (b) conformation 1 and (c) conformation 2. Images were created by using Gnuplot v5.257 and VMD v1.9.352.
Figure 5 summarizes aMD simulations of the wild type AR LBDs bound to the two distinct conformers of ENZ. When ENZ is bound in conformation 2 large conformational changes of the AR occur as shown in Fig. 5a,c. The backbone RMSDs from an initial structure (PDB ID: 2AXA) with ENZ in conformation 2 increased to > 3 Å after 10 ns and reached to 4.5 Å in 50 ns. From aMD simulations using another crystal structure of AR LBD (PDB code: 2PIP), we also found the backbone RMSD increased to 2.5 Å (Fig. 5a), when ENZ is bound in conformation 2. The RMSD changes of conformation 1 compared to the initial structure were not as dramatic as, the value remained < 2.5 Å throughout the simulations, except for 130 ns to 140 ns (Fig. 5a). The magnitude of RMSD change was comparable to those of DHT or RU5 bound to AR. The results suggested that conformation 2, where the tail group of ENZ orients toward H12, can disturb the structural stability of the agonistic conformation.
Ensemble averaging of the MD and the aMD simulations confirmed the statistical significance of the increased backbone RMSD in the AR LBD bound to ENZ in conformation 2. Figure S11 summarizes the distributions of backbone RMSDs, measured from the single 250 ns MD and aMD trajectories and the ensembles of 5-independent 250 ns MD and aMD trajectories. For the conventional MD simulations, the distributions were centered at 1–1.5 Å, suggesting minor conformational changes in the agonistic conformation, regardless of the number of independent MD trajectories considered. For the aMD simulations of AR LBDs bound to ENZ in conformation 2, we observed right-shift in the distribution maximum relative to that of ENZ in conformation 1. Of note, the right-shifts in the maximum observed from the ensemble-averaged distributions of ENZ in conformation 2 were less pronounced than those computed from single 250 ns aMD trajectories. The observation suggested that ensemble averaging of conformational changes based on multiple independent aMD simulations is necessary for prognostic aMD simulations, where one aims at predicting the efficacy of ligand prior to experimental tests.
Figure 5b,c compare the initial and the final structures of the wild type AR bound to ENZ, sampled from 250 ns aMD simulations (PDB code: 2AXA). From the aMD simulations with conformation 2, we observed significant structural differences arose from the repositioning of H12 and connecting loop between H11 and H12. The Cα RMSF over the time indicated that a number of residues, including H885 to M895 on H11–H12, loop over H12 and the residues (K912 to H917) in the end of C-terminus, showed significant fluctuations in the course of aMD simulations (Figures S3 and S4). The increment in RMSD and RMSF correlated with the tilt in H12 relative to H11. We measured the relative angle between H11 and H12 from the MD and the aMD trajectories of AR bound to ENZ (Figure S12). From the aMD simulations using 2AXA bound to ENZ in conformation 2, the angle decreased from 70° to 51° in 20 ns, that complies with the increment of backbone RMSD to > 3.0 Å in 10 ns. From the aMD simulations of ENZ in conformation 2 bound to another agonistic conformation of AR (PDB code: 2PIP), the angle began to decrease to 50° after 70 ns from the initiation of simulations, that coincided with the onset of increment in RMSD to > 3.0 Å.
Figure 6 highlights the regions in the AR LBD that show significant conformational changes upon binding of ENZ in conformation 2. In the initial agonistic structure, H12 is located in the close vicinity of the ligand binding pocket surrounding H4 and H11 (Cα distances of 7–11 Å from H4). During the aMD simulation, the N-methylbenzamide moiety of ENZ extends toward H12 that results in a large displacement of the helix from the initial position that covers the ligand binding pocket (Cα distances of 9–15 Å from H4). The bulky and hydrophobic side chains of H12 (M895, L898, V903, I906 and L907) facing the binding pocket were influenced by ENZ. Repositioning of H12 causes a dynamic movement of the H11–H12 loop and displacement of F-domain composed of β-sheet as well as the flexible loop. In short, the binding of ENZ in conformation 2 repositions H12 by repulsive interactions with the bulky and hydrophobic side chains on H12.
Figure 6.

Representative snapshots for the detailed binding mode of ENZ in conformation 2 in the AR LBD. The initial structures of ENZ and key residue are colored in cyan and the simulated structures are colored in yellow. Images were created by using VMD v1.9.352.
F876L stabilizes AR LBD bound to ENZ
Point mutations in the ligand binding pocket of AR can lead to the drug resistance by converting antagonists into agonists. T877A, W741L, F876L are the most prevalent drug resistant mutations for HFT (the active form of flutamide), BCA and ENZ, respectively41–43. We performed aMD simulations of the AR F876L mutant in the presence of ENZ in conformation 1 and 2 and in the absence of the ligands, as to understand the origin of drug resistance caused by the mutation. For all simulations, we observed RMSDs < 2.5 Å from the initial agonistic structure (Figure S5A). Unlike the wild type apo AR, F876L mutant in apo state was stable during 250 ns simulations without any significant conformational changes (Figure S5B). The binding of ENZ in conformation 1 to the mutant AR stabilizes the agonistic conformation, similarly to ENZ in conformation 1 (Figure S5C). Interestingly, the N-methylbenzamide tail of ENZ in conformation 2 moves into the space along H11 without disturbing H12 after 200 ns (Fig. 7a).
Figure 7.

Conformational dynamics of the F876L mutant AR bound to ENZ in conformation 2. (a) Comparison of the ligand binding pocket at 0 and 250 ns. Initial structures are colored in cyan and simulated structures with ENZ in conformation 2 are colored in blue. (b) Comparison of the ligand binding pocket of the wild type (yellow) and F876L mutant ARs (blue) bound to ENZ in conformation 2 after 250 ns aMD simulation. Images were created by using VMD v1.9.352.
An analysis of the ligand binding pocket of AR F876L suggested that the mutation reduces steric demand between H11, H12, and ligand. First we observed that, when averaged over 250 ns of constant temperature MD simulations, the volume of the ligand binding pocket of the F876L mutant was 101 ± 30 Å3, which is 36% larger than that of the wild type AR (74 ± 26 Å3), which were measured by POcket Volume MEasurer (POVME) program44. The increased volume may reduce the steric demand to accommodate the tail group of ENZ in conformation 2. In the F876L mutant, the gem-dimethyl of ENZ in conformation 2 is placed toward H3, H11 and the end of H6, resulting in a repositioning of benzamide tail of ENZ into H11 (Fig. 7b). By contrast, in the wild type AR the F876 residue precludes the gem-dimethyl of ENZ from approaching the space crowded with bulky side chains of M788, F705 and L709. These results demonstrate that the F876L mutant AR bound to ENZ is close to the agonistic conformation of the AR regardless of the binding of ENZ.
Binding of APL repositions H6 and H11 as well as H12
Figure 8 represents conformational changes of the wild type AR bound to APL in conformation 2 during 250 ns aMD simulation. The structural differences between APL and ENZ are only the pyridine ring in the head and cyclobutyl moiety instead of dimethyl group on the thiohydantoin core. We wondered how this subtle structural difference affects the conformation of the AR LBD. We initially explored the conformers of APL by carrying out QM calculation. Two conformers having distinct orientation between the pyridyl head group and the thiohydantoin group differ in Gibbs free energy by 1.9 kcal/mol (Figure S6), suggesting that the two conformers are viable in solution. Therefore, we considered the two conformations as the starting point at the simulations, as in the case of ENZ. Our aMD simulations showed that only APL in conformation 2 perturbs initial agonistic conformation by repositioning H12 and H11–H12 connecting loop in much the same way as ENZ (Figs. 8a,b and S7). H12 began to move away from the ligand binding pocket after 100 ns, resulting in RMSD from the initial conformation greater than 3 Å. More importantly, in addition to the repositioning of H12, we observed additional movement of H11 and H6 the wild type AR bound to APL (Fig. 8c). We correlated these structural changes to the presence of cyclobutyl moiety in the core group of APL. The Cα RMSF over the time indicated that the residues V692-C694 and N699-Q701 on H1–H3 loop, Y781-R787 on H6 and H885-M895 on H11–H12 loop and H12 were significantly fluctuated during aMD simulations (Figure S7). We note that no hydrogen bonding interactions between the nitrogen on pyridine and amino acids of the ligand binding pocket were observed during simulations, suggesting that replacement of phenyl ring of ENZ with pyridine does not significantly affect the structural changes of AR caused by APL. In short, we demonstrated that the tail group shared by ENZ and APL induces structural fluctuations in H12 and the introduction of cyclobutyl moiety at the thiohydantoin core can induce additional structural changes in H11, which may explain increased therapeutic efficacy of APL over ENZ.
Figure 8.

Conformational changes of the wild type AR bound to APL in conformation 2. (a) Visualizations of 0 and 250 ns aMD simulations. (b) The backbone RMSDs. (c) Comparison of the ligand binding pocket of the simulated AR bound to ENZ (yellow) and APL (magenta) in conformation 2 after 250 ns. Images were created by using VMD v1.9.352 and Gnuplot v5.257.
BCA induces conformational changes in the H1–H3 loop and the H3 of AR
Bicalutamide (BCA) is a nonsteroidal AR antagonist known as the predecessor of ENZ. BCA has 5–8 times lower affinity to the AR than ENZ despite the structural similarity of the head group. The AR inhibitory mechanisms of BCA is primarily the blocking the binding of androgens to AR45. Of note, ENZ impedes the nuclear translocation of AR, the binding of AR to DNA and the interactions of AR with the coactivators. Moreover, ENZ does not show agonistic effect on the wild type AR7.
Figure 9a shows two of the viable conformations of BCA. We explored the structures and energies of conformations with quantum mechanical calculations and confirmed that BCA conformation 2 is 5.6 kcal/mol lower in Gibbs free energy than conformation 1. Of note, S1, the parent compound of BCA, was co-crystallized with the AR with the 4-fluorobenzenesulfonyl group positioned toward H1216. These findings strongly suggested that BCA binds to the AR in a similar orientation to that of S1. As such, in the following MD simulations, we considered the conformation BCA in the binding pocket of AR, which resembles that of S1 (Figure S8A).
Figure 9.

Comparison of conformational changes of the wild type AR bound to BCA. (a) Two conformers of BCA with the lowest energy; Left: conformation 1 with the second lowest energy; Right: conformation 2 with the lowest in energy conformation. The dihedral angles of the four highlighted atoms (O-C-C-O) are shown. Images were created by using CYLView v.1.0.56156. (b) The backbone RMSDs of aMD simulations relative to the initial conformation. Gnuplot v5.257. (c) Comparison of the wild type ARs bound to BCA in conformation 2 at the initial (silver) and simulated structure after 400 ns (blue).Images were created by using VMD v1.9.352.
We compared the conformational dynamics of AR bound to BCA using a series of aMD simulations. We constructed models of AR bound to BCA using two of available x-ray structure of AR (PDB ID: 2AXA and 2PIP). Figure 9b shows the plot of backbone RMSDs of the aMD simulations. The RMSD fluctuations were less than 3 Å relative to the initial conformation even after 400 ns of aMD simulations, which was less pronounced than that of the AR bound to ENZ. Since the structural difference between S1 and BCA is minor (Fig. S8A), we thought that BCA may not induce large conformational changes relative to ENZ. An investigation on the sampled conformations revealed that there are small but notable conformational fluctuations in several loop regions in the AR bound to BCA (Fig. 9c). The comparison of the simulated conformation after 400 ns with the initial conformation, we realized that the loop connecting H1 and H3 (H1–H3 loop) moved outward and the part of H3 tilted toward the H11–H12, when compared to the AR bound to DHT or ENZ (Figure S8B). On the contrary, aMD simulations of a drug resistant mutant (W741L) bound to BCA showed that the H1–H3 and the H11–H12 loops are stable after 250 ns (Figure S9), suggesting the observed conformational changes in the WT AR bound BCA can be correlated with the action of the antagonist. The plot of RMSF indicates the conformational fluctuation of the residues on this region was significantly increased as well (Figure S8C). We found that H12 and the connecting loop also fluctuate in this system, but not as much as that of the AR bound to ENZ.
Based on our computational results, we concluded that the binding of BCA to the AR LBD inhibits the binding of androgens by inducing destabilization of H1–H3 loop and a tilt of H3 as well as nudging H12 outward, whereas ENZ and APL antagonize the AR by significantly displacing H12. The conformation of BCA is in a bent shape formed by chiral center bearing a methyl and a hydroxyl group, and a benzene sulfonyl moiety. By contrast, ENZ and APL have a thiohydantoin ring at the core that connects the benzyl (pyridyl) head and the benzamide tail group via two nitrogen atoms on the ring. The rigid core and extended tail of ENZ and APL enable the conformations of the ligands to maintain the linearity during the aMD simulations, which result in the repositioning of H12. From these observations, we speculate that the core of the two antagonists (ENZ and APL) can modulate conformational dynamics of the AR LBD, which can explain the improved efficacies of ENZ and APL to the wild type AR than BCA.
PCA reveals distinct conformational fluctuation of the AR by the binding of ligands
Figure 10 summarizes the principal component analysis of the aMD trajectories of AR LBDs, which was performed to quantify the observed conformational fluctuations. We computed the principal components using the agglomerated aMD trajectories of the four aMD trajectories; AR LBDs bound to DHT, ENZ, BCA, and in apo. Prior to the computations of principal components, the backbone trajectories were aligned to an X-ray structure of the AR (PDB code: 2AXA), so as to remove the translation-rotational motions of the protein. We then calculated the projection of each aMD trajectory to the computed PCs, as shown in Fig. 10a. WT AR bound to ENZ showed a notable fluctuation along the first principal component (PC1); the conformational fluctuations of AR in apo were captured by the second principal component (PC2); the fluctuations of AR bound to BCA were characterized by the third principal component (PC3).
Figure 10.

PCA of trajectories obtained from aMD simulations of the wild type ARs bound to the ligands. (a) Projections of sampled coordinates onto PC axes for the first three PCs. Each color indicates aMD trajectories of AR bound with ENZ in conformation 2 (red), BCA (brown), DHT (purple) and no ligand (apo, black). (b) Projections of aMD trajectories of the wild type ARs bound to ENZ in conformation 2 (red), BCA (brown), F876L mutant AR bound to ENZ in conformation 2 (green) and W741L mutant AR bound to BCA (cyan) (c) Visualizations of the three PCs. Arrows show the direction and relative magnitude of Cα motions along the corresponding PC. Colors of arrows represent the positive (blue) and the negative (red) directions along each PC eigenvector. Images were created by using Gnuplot v5.257 and VMD v1.9.352.
The projection of aMD trajectories of drug resistant mutants bound to antagonists confirmed that the mutations minimizes the conformational changes in the agonistic conformation of AR. Using the computed PCs based on the aMD simulations of WT AR, we projected the backbone trajectories of W741L bound to BCA and F876L bound to ENZ (Fig. 10b). The projection demonstrated that the drug resistant mutations reduce the characteristic structural fluctuations found from the WT AR bound to the antagonist.
The direction of the backbone dynamics in each PC is also informative. Figure 10c represents the direction of those fluctuations on the AR structures. In PC1, the positive motion (colored in blue) moves H12 and the connecting loop away from the ligand binding pocket formed by H3, H4, and H11, which strongly supports the proposition that the repositioning of H12 is one of the key antagonistic mechanisms. In PC2, the dominant fluctuations occur at the flexible H1–H3 and H8–H9 loops. These movements correspond to the folding and unfolding of the loops that can influence the binding of co-regulators and co-chaperone complexes. Interestingly, AR bound to DHT fluctuates along the positive direction of PC2, which indicates the flexible H1–H3 and H8–H9 loops moves closer to H3 and H4, suggesting an increased stability of the AR. In PC3, the negative motion moves the H1–H3 and the H11–H12 loops outward, which characterizes the structural fluctuations observed from the WT AR bound to BCA. When combined, our PCA analysis showed significant differences in the conformational dynamics of WT AR bound to antagonists, agonist, and in apo.
We demonstrate the utility of PCA as a measure of the structure–activity relationship (SAR) of a ligand of AR. We plot the projections of aMD trajectories of AR bound to an agonist (RU5) and an antagonist (APL), as shown in Fig. 11. In a previous section, we showed the AR bound to RU5 exhibit reduced conformational fluctuations than the ARs bound to antagonist. The projection on the computed PCs confirmed the binding of RU5 minimal structural changes in AR (Fig. 11a). On the contrary, the projection of aMD trajectories of AR bound to APL showed that the ligand acts as an antagonist, that exhibited large fluctuations along the PC1 and PC2 (Fig. 11b), which resembles that of AR bound to ENZ.
Figure 11.

Projection of aMD trajectories of the wild type ARs bound to (a) RU5 and (b) APL in conformation 2. Images were created by using Gnuplot v5.257 and Chemdraw.
Conclusion
We have explored the conformational dynamics of the AR bound to the agonists (DHT and RU5), the antagonists (BCA, ENZ and APL), and in the absence of the ligand (apo). We carried out > 250 ns aMD simulations of the ligand bound AR LBD complexes and the AR LBD in apo, initiated from available agonistic conformations of AR LBD. With the aMD simulations, we were able to expedite the conformational changes in the AR LBDs and to observe the influence of ligands on AR LBD that provides insights into how antagonists destabilize the AR LBD. A large and differential repositioning of H12 manifests the influence of ENZ and APL to the WT AR. The N-methylbenzamide tail of ENZ and APL is a salient structural element that induces repulsive interactions with the bulky and hydrophobic side chains of H12, leading to the destabilization of H12. The drug resistant mutations (F876L and W741L) suppress the influence of the antagonists by stabilizing the agonistic conformation of the AR. The simulations of BCA bound to AR show less pronounced repositioning of H12 but rather significant fluctuations in the H1–H3 loop. These distinct conformational changes caused by the first and the second generation AR antagonists were observed only from the aMD simulations, but not from conventional MD simulations. PCA results account for the conformational dynamics obtained from the aMD trajectories of the AR LBDs. These findings indicate that aMD simulations can guide future drug discovery of new-generation AR antagonists.
Among the intriguing questions to be addressed in the future, the detailed interactions between the selective ligands and the AR LBDs are paramount. The detailed orientation of the tail group of ENZ and APL in the binding pocket of AR LBD remains elusive. Our computational analyses suggested the N-methylbenzamide group of ENZ and APL oriented toward H12 causes displacement of the helix, but we were not able to exclude the other possible orientation to occur in vivo. Future computational analyses including the comparisons of the binding free energies of the two orientations will give decisive information about these effects. The applicability of the PCA that we have introduced as a measure for the structure–activity relationship (SAR) of the ligands of AR will need further validations, which are underway in our lab.
Methods
Preparation of the ligands and proteins for MD simulations
The x-ray crystal structures of the AR LBD (2AXA and 2PIP) were retrieved from Protein Data Bank for the simulation. 2AXA is the structure of synthetic agonist (S1) bound wild type AR and 2PIP is co-crystalized with DHT and BF3 inhibitor (K10) and both were used as a starting structure. For the mutant AR, the residue 876 was mutated from phenylalanine to leucine using mutagenesis wizard in PyMOL. The structure of bicalutamide (BCA) was built based on the co-crystalized S1 by replacing the ether linkage with the sulfonyl group and the nitro group with cyano group. The other ligand structures were virtually generated since no structures of the AR bound to antagonists have been determined. Each ligand was optimized with density functional theory (DFT) at the B3LYP/6-31G* using Gaussian 09 software46. The optimized ligands were docked to the each PDB structure using Autodock Vina that uses a scoring function combined knowledge-based potentials and empirical approach to rank ligands docked to protein structures. Protein receptors and ligands were prepared using default criteria from AutoDock Vina47. Receptors were treated as rigid, and the ligands were treated as a flexible molecule with active rotatable bonds. To perform molecular docking, all hydrogen atoms were added to each protein and ligand of which coordinate files were generated as PDBQT file by merging nonpolar hydrogen atoms of the AR and calculating Gasteiger charges. The search space for docking was determined based on the three dimensional dimension of the original ligand from the crystal structure. A grid box for binding site was set as 20, 24 and 20 in the three dimensions (x, y and z centered at 26.3, 2.4 and 6.5, respectively) using 1.0 Å grid spacing. The predicted binding affinity of an agonist (FHM) was − 10.6 kcal/mol when we used 2AXA as the agonistic conformation. In each docking experiment, the pose with best calculated binding affinity (i.e. − 0.4 kcal/mol for conformation 1 or − 0.8 kcal/mol for conformation 2 for ENZ) was selected for further MD simulations. The docking simulations of the other ligands were performed with same condition as described above.
MD simulations
Molecular mechanics parameters were constructed using the Amber14 software package48. The Amber FF99SB force field was used for the proteins49. The generalized Amber Force Field (GAFF) was used to parameterize the ligands (DHT, RU59063, bicalutamide (BCA), enzalutamide (ENZ) and apalutamide (APL)). Partial atomic charges on each ligand were determined by the quantum mechanical (QM) calculations based on the Merz-Singh-Kollman algorithm using Gaussian 09. Initially, each ligand is docked in the ligand binding pocket manually. Each protein and ligand complex was immersed in a rectangular solvation box filled TIP3P water molecules, with a 15 Å margin from the solvation boundary50. Monovalent counter ions (Na+ or Cl−) were added to achieve electrostatic neutrality of the systems. Each solvated system consists of approximately 48,000 atoms. The second step of the preparatory steps was to equilibrate the solvated system. The equilibration steps were initiated by energy minimizations and then equilibrating each solvated system at 300 K using the NVT ensemble for 2 ns. Berendsen thermostat was used to control the temperature at the desired value, and then the isobaric ensemble (NPT) was used for next 4 ns. Extended system algorithm was used to control the system pressure at 1 atm. The coordinates of the carbon atoms were restrained during the first 6 ns equilibration period. A 250 ns equilibration period followed, without any restraint of the atomic coordinates. Langevin thermostat with a small friction coefficient (5 ps−1) and a Monte Carlo barostat were used to keep the temperature at 300 K, and the pressure at 1 atm throughout the equilibration period. For the entire equilibration steps, the time integration step was 2 fs. The SHAKE algorithm was employed to restrain all hydrogens to heavy atoms bond distances. The particle mesh EWALD method accelerated the computation of long-range electrostatic interactions.
Accelerated MD simulations
Accelerated MD enhances the conformational sampling of the protein complex by adding a non-negative boost potential to the system. We initially carried out 250 ns MD simulations of the LBDs bound to the ligands. The backbone root-mean-square deviation (RMSD) from the initial conformation were < 2.0 Å (Figure S1). Moreover, we were not able to identify conformational changes that are specific to each ligand considered. The observations prompted us to utilize aMD algorithm that is known to facilitate conformational changes otherwise difficult to sample from MD simulations. The Hamiltonian of a simulated system () is modified in such a way to elevate the local minimum via the boost potential (),
where is the modified Hamiltonian. The boost potential is defined as following:
where and are the boosting potentials of the total energy and the dihedral energy, respectively, is the total potential energy, is the control parameter of the boosting potential of the total potential energy, is the dihedral energy, and is the control parameter of the boosting potential of the dihedral energy. Of note, either of the boosting potentials ( or ) is applied, when the total energy () or the dihedral energy () is lower than the threshold values: and . We chose as to ensure each atom of a solvated system experience 0.2 kcal/mol of boosting potential and as to elevate 0.7 kcal/mol of dihedral energy of per each amino acid51. The four control parameters of the aMDs were determined as following.
where is the number of atoms in a solvated system, is the number amino acids, and and are the total and the dihedral energies averaged over the initial 250 ns MD simulations.
Once MD simulations were finished, trajectories were analyzed using the AmberTools14 software package. The backbone root-mean square deviations (RMSDs) between the simulated aMD trajectories and the initial PDB structures and the root-mean square fluctuations (RMSFs) per residue over the time were computed using VMD and plugin52. We note that we have not attempted to construct potential energy profile of the conformational ensemble, as the original implementation of aMD methodology suffers from difficulty in reconstruction of the unbiased energy surface. In order to obtain proper conformational free energy surface of AR-antagonists systems, it is desirable to perform the other potential approaches such as Gaussian accelerated molecular dynamics (GaMD)53 for the future investigation.
Principal component analysis
To understand conformational dynamics of AR bound to agonists and antagonists, principal component analysis (PCA) was performed with trajectories extracted from aMD simulations. With PCA, one can identify essential coordinates that describe the global motion of proteins54. Specifically, the diagonalization of interatomic correlation matrix results in characteristic modes of conformation motions. Each mode represents an orthogonal direction of global conformational motion of a protein, with an eigenvalue that quantifies the relative significance of the motion. A few large eigenvalue modes are of specific interest, as they represent significant conformational changes during the simulations. To process the aMD trajectories for PCA, the 250 ns MD trajectories of the ARs bound to the agonists and antagonists and apo AR were reoriented and aligned to the initial x-ray structure of the AR. PCA were performed for Cα atoms on snapshots at 250 ps intervals. MDAnalysis software55 was used to compute the first three principal components.
Supplementary Information
Acknowledgements
We are grateful to the National Institute of General Medical Sciences, National Institutes of Health (GM-124480 to K.N.H.) for financial support of this research.
Author contributions
M.E.J. and K.N.H. conceived the project. H.J.G. and J.P. designed, performed and analyzed the simulations. J.P. performed quantum mechanical calculations. H.J.G. and J.P. wrote the manuscript. M.E.J. and K.N.H. contributed to the review of the whole manuscript. All authors discussed the results and reviewed the final manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Hyo Jin Gim and Jiyong Park.
Contributor Information
Michael E. Jung, Email: jung@chem.ucla.edu
K. N. Houk, Email: houk@chem.ucla.edu
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-021-94707-2.
References
- 1.Heinlein CA, Chang C. Androgen receptor in prostate cancer. Endocr. Rev. 2004;25:276–308. doi: 10.1210/er.2002-0032. [DOI] [PubMed] [Google Scholar]
- 2.Elshan NGRD, Rettig MB, Jung ME. Molecules targeting the androgen receptor (AR) signaling axis beyond the AR-Ligand binding domain. Med. Res. Rev. 2019;39:910–960. doi: 10.1002/med.21548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scher HI, Sawyers CL. Biology of progressive, castration-resistant prostate cancer: Directed therapies targeting the androgen-receptor signaling axis. J. Clin. Oncol. 2005;23:8253–8261. doi: 10.1200/JCO.2005.03.4777. [DOI] [PubMed] [Google Scholar]
- 4.Chen CD, et al. Molecular determinants of resistance to antiandrogen therapy. Nat. Med. 2004;10:33–39. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- 5.Karantanos T, Corn PG, Thompson TC. Prostate cancer progression after androgen deprivation therapy: Mechanisms of castrate resistance and novel therapeutic approaches. Oncogene. 2013;32:5501–5511. doi: 10.1038/onc.2013.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chandrasekar T, Yang JC, Gao AC, Evans CP. Mechanisms of resistance in castration-resistant prostate cancer (CRPC) Transl. Androl. Urol. 2015;4:365–380. doi: 10.3978/j.issn.2223-4683.2015.05.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tran C, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324:787–790. doi: 10.1126/science.1168175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rathkopf DE, et al. Phase I study of ARN-509, a novel antiandrogen, in the treatment of castration-resistant prostate cancer. J. Clin. Oncol. 2013;31:3525–3530. doi: 10.1200/JCO.2013.50.1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang P, Chandra V, Rastinejad F. Structural overview of the nuclear receptor superfamily: Insights into physiology and therapeutics. Annu. Rev. Physiol. 2010;72:247–272. doi: 10.1146/annurev-physiol-021909-135917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Darimont BD, et al. Structure and specificity of nuclear receptor-coactivator interactions. Genes Dev. 1998;12:3343–3356. doi: 10.1101/gad.12.21.3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bevan CL, Hoare S, Claessens F, Heery DM, Parker MG. The AF1 and AF2 domains of the androgen receptor interact with distinct regions of SRC1. Mol. Cell. Biol. 1999;19:8383–8392. doi: 10.1128/MCB.19.12.8383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brzozowski AM, et al. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature. 1997;389:753–758. doi: 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- 13.Matias PM, et al. Structural basis for the glucocorticoid response in a mutant human androgen receptor (AR(ccr)) derived from an androgen-independent prostate cancer. J. Med. Chem. 2002;45:1439–1446. doi: 10.1021/jm011072j. [DOI] [PubMed] [Google Scholar]
- 14.He B, et al. Structural basis for androgen receptor interdomain and coactivator interactions suggests a transition in nuclear receptor activation function dominance. Mol. Cell. 2004;16:425–438. doi: 10.1016/j.molcel.2004.09.036. [DOI] [PubMed] [Google Scholar]
- 15.Estébanez-Perpiñá E, et al. The molecular mechanisms of coactivator utilization in ligand-dependent transactivation by the androgen receptor. J. Biol. Chem. 2005;280:8060–8068. doi: 10.1074/jbc.M407046200. [DOI] [PubMed] [Google Scholar]
- 16.Bohl CE, Miller DD, Chen J, Bell CE, Dalton JT. Structural basis for accommodation of nonsteroidal ligands in the androgen receptor. J. Biol. Chem. 2005;280:37747–37754. doi: 10.1074/jbc.M507464200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bohl CE, Gao W, Miller DD, Bell CE, Dalton JT. Structural basis for antagonism and resistance of bicalutamide in prostate cancer. Proc. Natl. Acad. Sci. USA. 2005;102:6201–6206. doi: 10.1073/pnas.0500381102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bohl CE, Wu Z, Miller DD, Bell CE, Dalton JT. Crystal structure of the T877A human androgen receptor ligand-binding domain complexed to cyproterone acetate provides insight for ligand-induced conformational changes and structure-based drug design. J. Biol. Chem. 2007;282:13648–13655. doi: 10.1074/jbc.M611711200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Estébanez-Perpiñá E, et al. A surface on the androgen receptor that allosterically regulates coactivator binding. Proc. Natl. Acad. Sci. USA. 2007;104:16074–16079. doi: 10.1073/pnas.0708036104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nique F, et al. Discovery of diarylhydantoins as new selective androgen receptor modulators. J. Med. Chem. 2012;55:8225–8235. doi: 10.1021/jm300249m. [DOI] [PubMed] [Google Scholar]
- 21.Nadal M, et al. Structure of the homodimeric androgen receptor ligand-binding domain. Nat. Commun. 2017;8:14388. doi: 10.1038/ncomms14388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Unwalla R, et al. Structure-based approach to identify 5-[4-hydroxyphenyl]pyrrole-2-carbonitrile derivatives as potent and tissue selective androgen receptor modulators. J. Med. Chem. 2017;60:6451–6457. doi: 10.1021/acs.jmedchem.7b00373. [DOI] [PubMed] [Google Scholar]
- 23.Bisson WH, Abagyan R, Cavasotto CN. Molecular basis of agonicity and antagonicity in the androgen receptor studied by molecular dynamics simulations. J. Mol. Graph. Model. 2008;27:452–458. doi: 10.1016/j.jmgm.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 24.Zhou J, Liu B, Geng G, Wu JH. Study of the impact of the T877A mutation on ligand-induced helix-12 positioning of the androgen receptor resulted in design and synthesis of novel antiandrogens. Proteins. 2010;78:623–637. doi: 10.1002/prot.22592. [DOI] [PubMed] [Google Scholar]
- 25.Osguthorpe DJ, Hagler AT. Mechanism of androgen receptor antagonism by bicalutamide in the treatment of prostate cancer. Biochemistry. 2011;50:4105–4113. doi: 10.1021/bi102059z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Balbas MD, et al. Overcoming mutation-based resistance to antiandrogens with rational drug design. Elife. 2013;2:e00499. doi: 10.7554/eLife.00499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu X, et al. Dynamic communication between androgen and coactivator: Mutually induced conformational perturbations in androgen receptor ligand-binding domain. Proteins. 2011;79:1154–1171. doi: 10.1002/prot.22951. [DOI] [PubMed] [Google Scholar]
- 28.Liu N, et al. Molecular dynamics simulations revealed the regulation of ligands to the interactions between androgen receptor and its coactivator. J. Chem. Inf. Model. 2018;58:1652–1661. doi: 10.1021/acs.jcim.8b00283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang X, et al. Effects of HO-/MeO-PBDEs on androgen receptor: In vitro investigation and helix 12-involved MD simulation. Environ. Sci. Technol. 2013;47:11802–11809. doi: 10.1021/es4029364. [DOI] [PubMed] [Google Scholar]
- 30.Duan M, et al. Structural diversity of ligand-binding androgen receptors revealed by microsecond long molecular dynamics simulations and enhanced sampling. J. Chem. Theory Comput. 2016;12:4611–4619. doi: 10.1021/acs.jctc.6b00424. [DOI] [PubMed] [Google Scholar]
- 31.Hu X, et al. Advances in the computational development of androgen receptor antagonists. Drug Discov. Today. 2020;25:1453–1461. doi: 10.1016/j.drudis.2020.04.004. [DOI] [PubMed] [Google Scholar]
- 32.Clegg NJ, et al. ARN-509: A novel antiandrogen for prostate cancer treatment. Cancer Res. 2012;72:1494–1503. doi: 10.1158/0008-5472.CAN-11-3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hamelberg D, Mongan J, McCammon JA. Accelerated molecular dynamics: A promising and efficient simulation method for biomolecules. J. Chem. Phys. 2004;120:11919–11929. doi: 10.1063/1.1755656. [DOI] [PubMed] [Google Scholar]
- 34.Gasper PM, Fuglestad B, Komives EA, Markwick PR, McCammon JA. Allosteric networks in thrombin distinguish procoagulant vs. anticoagulant activities. Proc. Natl. Acad. Sci. USA. 2012;109:21216–21222. doi: 10.1073/pnas.1218414109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kappel K, Miao Y, McCammon JA. Accelerated molecular dynamics simulations of ligand binding to a muscarinic G-protein-coupled receptor. Q. Rev. Biophys. 2015;48:479–487. doi: 10.1017/S0033583515000153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miao Y, et al. Accelerated structure-based design of chemically diverse allosteric modulators of a muscarinic G protein-coupled receptor. Proc. Natl. Acad. Sci. USA. 2016;113:E5675–5684. doi: 10.1073/pnas.1612353113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Balsera M, Wriggers W, Oono Y, Schulten K. Principal component analysis and long time protein dynamics. J. Phys. Chem. 1996;100:2567–2572. doi: 10.1021/jp9536920. [DOI] [Google Scholar]
- 38.Hess B. Convergence of sampling in protein simulations. Phys. Rev. E. 2002;65:031910-1-10. doi: 10.1103/PhysRevE.65.031910. [DOI] [PubMed] [Google Scholar]
- 39.Cluning C, et al. The helix 1–3 loop in the glucocorticoid receptor LBD is a regulatory element for FKBP cochaperones. Mol. Endocrinol. 2013;27:1020–1035. doi: 10.1210/me.2012-1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fuller PJ, Smith BJ, Rogerson FM. Cortisol resistance in the New World revisited. Trends Endocrinol. Metab. 2004;15:296–299. doi: 10.1016/j.tem.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 41.Veldscholte J, et al. A mutation in the ligand binding domain of the androgen receptor of human LNCaP cells affects steroid binding characteristics and response to anti-androgens. Biochem. Biophys. Res. Commun. 1990;173:534–540. doi: 10.1016/S0006-291X(05)80067-1. [DOI] [PubMed] [Google Scholar]
- 42.Korpal M, et al. An F876L mutation in androgen receptor confers genetic and phenotypic resistance to MDV3100 (enzalutamide) Cancer Discov. 2013;3:1030–1043. doi: 10.1158/2159-8290.CD-13-0142. [DOI] [PubMed] [Google Scholar]
- 43.Hara T, et al. Novel mutations of androgen receptor: A possible mechanism of bicalutamide withdrawal syndrome. Cancer Res. 2003;63:149–153. [PubMed] [Google Scholar]
- 44.Durrant JD, Votapka L, Sørensen J, Amaro RE. POVME 2.0: An enhanced tool for determining pocket shape and volume characteristics. J. Chem. Theory Comput. 2014;10:5047–5056. doi: 10.1021/ct500381c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Masiello D, Cheng S, Bubley GJ, Lu ML, Balk SP. Bicalutamide functions as an androgen receptor antagonist by assembly of a transcriptionally inactive receptor. J. Biol. Chem. 2002;277:26321–26326. doi: 10.1074/jbc.M203310200. [DOI] [PubMed] [Google Scholar]
- 46.Frisch, M. J. et al. Gaussian 09 (Gaussian Inc., 2009). https://gaussian.com/glossary/g09/.
- 47.Trott O, Olson AJ. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Case, D. A. et al. AMBER 14 (University of California, 2014) http://ambermd.org/doc12/Amber14.pdf.
- 49.Lindorff-Larsen K, et al. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins. 2010;78:1950–1958. doi: 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jorgensen W, Chandrasekhar J, Madura J, Impey R, Klein M. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. doi: 10.1063/1.445869. [DOI] [Google Scholar]
- 51.Pierce LC, Salomon-Ferrer R, Augusto F de Oliveira C, McCammon JA, Walker RC. Routine access to millisecond time scale events with accelerated molecular dynamics. J. Chem. Theory Comput. 2012;8:2997–3002. doi: 10.1021/ct300284c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Humphrey W, Dalke A, Schulten KVMD. Visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 53.Miao Y, Feher VA, McCammon JA. Gaussian accelerated molecular dynamics: Unconstrained enhanced sampling and free energy calculation. J. Chem. Theory Comput. 2015;11:3584–3595. doi: 10.1021/acs.jctc.5b00436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.David CC, Jacobs DJ. Principal component analysis: A method for determining the essential dynamics of proteins. Methods Mol. Biol. 2014;1084:193–226. doi: 10.1007/978-1-62703-658-0_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Michaud-Agrawal N, Denning EJ, Woolf TB, Beckstein O. MDAnalysis: A toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 2011;32:2319–2327. doi: 10.1002/jcc.21787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Legault, C. Y., Université de Sherbrooke, CYLview, 1.0b. http://www.cylview.org (2009).
- 57.Williams, T. & Kelley, C. Gnuplot 5.2: An Interactive Plotting Program. http://gnuplot.info (2011).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.