

Abstract
We describe severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)-specific T cell responses, soluble markers of inflammation, and antibody levels and neutralization capacity longitudinally in 70 individuals with PCR-confirmed SARS-CoV-2 infection. Participants represent a spectrum of illness and recovery, including some with persistent viral shedding in saliva and many experiencing post-acute sequelae of SARS-CoV-2 infection (PASC). T cell responses remain stable for up to 9 months. Whereas the magnitude of early CD4+ T cell immune responses correlates with severity of initial infection, pre-existing lung disease is independently associated with higher long-term SARS-CoV-2-specific CD8+ T cell responses. Among participants with PASC 4 months following coronavirus disease 2019 (COVID-19) symptom onset, we observe a lower frequency of CD8+ T cells expressing CD107a, a marker of degranulation, in response to Nucleocapsid (N) peptide pool stimulation, and a more rapid decline in the frequency of N-specific interferon-γ-producing CD8+ T cells. Neutralizing antibody levels strongly correlate with SARS-CoV-2-specific CD4+ T cell responses.
Keywords: SARS-CoV-2, COVID-19, T cell, immunity, post-acute sequelae of SARS-CoV-2 infection, PASC, long COVID
Graphical abstract
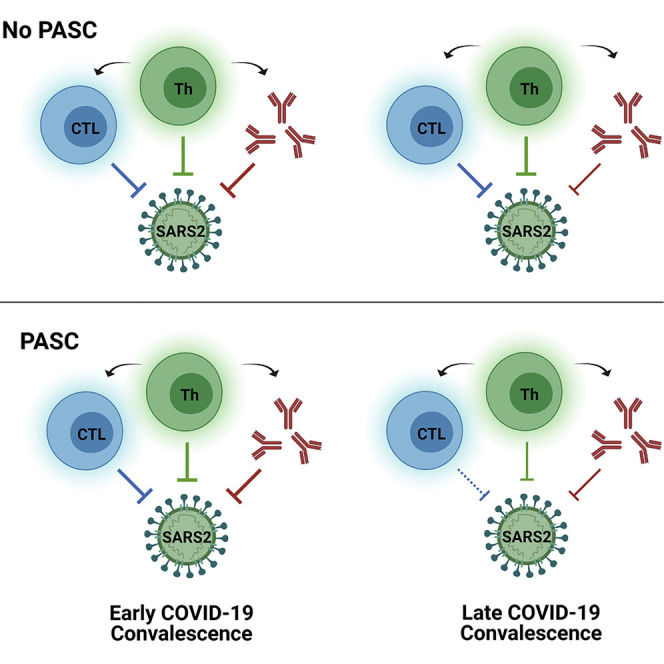
CD4+ and CD8+ T cell responses following natural infection with COVID-19 are stable over 8 months. Individuals with PASC demonstrate a lower frequency of CD8+ T cells expressing CD107a, a marker of degranulation, and a more rapid decline in the frequency of N-specific interferon-γ-producing CD8+ T cells.
Introduction
Most people generate detectable and durable severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)-specific CD4+ and CD8+ T cell responses following natural infection (Braun et al., 2020; Breton et al., 2021; Dan et al., 2021; Grifoni et al., 2020; Peng et al., 2020; Rydyznski Moderbacher et al., 2020; Sekine et al., 2020; Zhou et al., 2020). However, current understanding of the factors associated with the magnitude and long-term persistence of the cellular immune response and its relationship to clinical outcomes, humoral responses, and soluble markers of inflammation remain limited. Regardless of the development of long-term immunity, a significant proportion of people recovering from coronavirus disease 2019 (COVID-19) develop post-acute sequelae of SARS-CoV-2 infection (PASC; also known as “long COVID”) and persistent symptoms that can be prolonged and interfere with activities of daily life (Nalbandian et al., 2021). As a result, there is currently intense interest in understanding whether potentially important immunologic differences exist among groups experiencing rapid versus prolonged COVID-19 recovery, but data from this latter group are lacking (Carfì et al., 2020; Datta et al., 2020; Drew et al., 2020; Hellmuth et al., 2021; Huang et al., 2021; Peluso et al., 2021; Tenforde et al., 2020). In addition, few studies have investigated inflammatory responses in carefully curated PASC cohorts or in those with prolonged viral RNA shedding.
To address these issues, we measured SARS-CoV-2-specific T cell responses, soluble markers of inflammation, antibody levels and neutralization capacity, and viral RNA in saliva longitudinally up to 8.9 months following infection in a diverse group of 70 individuals with PCR-confirmed SARS-CoV-2 infection with varying degrees of initial disease severity and PASC in northern California enrolled in the Long-Term Impact of Infection with Novel Coronavirus (LIINC) cohort (Peluso et al., 2021). We demonstrate that, whereas the magnitude of the early CD4+ T cell immune response is determined by the severity of initial infection (participants requiring hospitalization or intensive care), pre-existing lung disease was significantly associated with higher long-term SARS-CoV2-specific CD8+ T cell responses, independent of initial disease severity or age. By contrast, participants with PASC 4 months following the initial infection had lower CD8+ T cell responses over time. Neutralizing antibody (NAb) levels were strongly correlated with SARS-CoV-2-specific CD4+, but not CD8+, T cell responses.
Results
Characterization of a clinically diverse COVID-19 cohort over 8 months of recovery
In order to evaluate adaptive immune and inflammatory responses over a range of COVID-19 presentations, we selected 70 cohort participants that represented a wide range of initial disease presentations, from those with no or mild symptoms to those requiring hospitalization or treatment in an intensive care unit (ICU). The first study time point (T1) occurred a median 53 days after symptom onset (interquartile range [IQR] 38–64.5). We prioritized inclusion of participants enrolled during early recovery (within 40 days following onset of symptoms) and those with samples available at later time points after symptom onset in order to include participants that developed PASC. Peripheral blood mononuclear cells (PMBCs), plasma, serum, and saliva were collected longitudinally between 1 and 8 months after symptom onset. One participant had antibody and inflammatory marker data but insufficient cell viability for T cell analyses.
Overall, 48.6% of participants were female, 25.7% identified as Latino or Latina (Latinx), and 55.7% identified as white (non-Latinx), as shown in Table 1 . In addition, 25.7% of participants were hospitalized and 14.3% reported receiving ICU care. A significantly higher proportion of males than females was hospitalized (38.9 versus 11.8%; p = 0.02), and all participants who received ICU care were male (p < 0.01). A significantly higher proportion of hospitalized participants (61.1%) identified as Latinx (p < 0.001). The majority of participants did not have underlying medical comorbidities, but 18.6% were previously diagnosed with lung disease (e.g., asthma, chronic obstructive pulmonary disease), 12.9% with hypertension, and 8.6% had diabetes mellitus (DM). Given the large number of individuals enrolled prior to widespread availability of COVID-19-specific therapies, only 4.3% reported having received remdesivir, 11.4% received hydroxychloroquine with or without azithromycin, and one participant each reported receiving systemic corticosteroids and convalescent plasma.
Table 1.
Participant demographics, comorbidities, clinical presentations, and saliva SARS-CoV2 PCR positivity at the first study visit
| All participants | Sex | Hospitalized | Saliva PCR+ at time of first sample collectiona | ||||
|---|---|---|---|---|---|---|---|
| Female | Male | Yes | No | Yes | No | ||
| n | 70b | 34 | 36 | 18 | 52 | 7 | 54c |
| Age (median, IQR) | 43 (36, 53) | 43 (36.8, 53) | 44.5 (36, 55.3) | 50.5 (40.3, 56.8) | 40 (36, 53) | 36 (37, 50) | 43 (35.5, 53.3) |
| Female, n (%)d | 34 (48.6) | – | – | 4 (22.2)∗ | 30 (57.7) | ||
| Race/ethnicity, n (%) | |||||||
| Latinx | 18 (25.7) | 6 (17.6) | 12 (33.3) | 11 (61.1)∗∗∗ | 7 (13.5) | 0 (0) | 13 (24.1) |
| White (non-Latinx) | 39 (55.7) | 23 (67.6) | 16 (44.4) | 3 (16.7) | 19 (67.9) | 6 (85.7) | 30 (55.6) |
| Black | 3 (4.3) | 3 (8.8) | 0 (0) | 1 (5.6) | 2 (7.1) | 0 (0) | 2 (3.7) |
| Asian | 10 (14.3) | 2 (5.9) | 8 (22.2) | 3 (16.7) | 2 (7.1) | 1 (14.3) | 9 (16.7) |
| ICU admission, n (%) | 10 (14.3) | 0 (0)∗∗ | 10 (100) | – | – | 1 (14.3) | 6 (11.1) |
| Underlying medical condition, n (%) | |||||||
| Lung disease | 13 (18.6) | 9 (26.5) | 4 (11.1) | 5 (27.8) | 8 (15.4) | 3 (42.9) | 10 (18.5) |
| Autoimmune disease | 3 (4.3) | 3 (8.8) | 0 (0) | 0 (0) | 3 (5.8) | 0 (0) | 2 (3.7) |
| Hypertension | 9 (12.9) | 6 (17.6) | 3 (8.3) | 4 (22.2) | 5 (9.6) | 0 (0) | 7 (13) |
| Cancer | 1 (1.4) | 1 (2.9) | 0 (0) | 0 (0) | 1 (1.9) | 0 (0) | 1 (1.9) |
| Diabetes mellitus | 6 (8.6) | 2 (5.9) | 4 (11.1) | 5 (27.8)∗∗ | 1 (1.9) | 0 (0) | 4 (7.4) |
| Symptoms during acute infection, n (%) | 68 (97.1) | 32 (94.1) | 36 (100) | 18 (100) | 50 (96.2) | 7 (100) | 53 (98.1) |
| Fever/chills | 56 (80) | 24 (70.6) | 32 (88.9) | 17 (94.4) | 39 (75) | 6 (85.7) | 43 (79.6) |
| Cough/SOB | 61 (87.1) | 28 (82.4) | 33 (91.7) | 17 (94.4) | 44 (84.6) | 4 (57.1)∗ | 50 (92.6) |
| Sore throat/runny nose | 46 (65.7) | 22 (64.7) | 24 (66.7) | 10 (55.6) | 36 (69.2) | 3 (42.9) | 37 (68.5) |
| Chest pain or palpitations | 7 (10) | 2 (5.9) | 5 (13.9) | 3 (16.7) | 4 (1.9) | 1 (14.3) | 4 (7.4) |
| Neurological/cognitive | 49 (70) | 26 (76.5) | 23 (63.9) | 13 (72.2) | 36 (69.2) | 5 (71.4) | 37 (68.5) |
| Fatigue | 63 (90) | 31 (91.2) | 32 (88.9) | 16 (88.9) | 47 (90.4) | 6 (85.7) | 50 (92.6) |
| Smell/taste changes | 52 (74.3) | 28 (82.4) | 24 (66.7) | 13 (72.2) | 39 (75) | 4 (57.1) | 43 (79.6) |
| Gastrointestinal symptoms | 43 (61.4) | 24 (70.6) | 19 (52.8) | 14 (77.8)∗ | 29 (55.8) | 3 (42.9) | 33 (61.1) |
| Musculoskeletal symptoms | 48 (68.6) | 34 (70.6) | 24 (66.7) | 14 (77.8) | 34 (65.4) | 3 (42.9) | 38 (70.4) |
p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001 by Fisher’s exact test (within column comparisons); SOB, shortness of breath.
No saliva sample was PCR+ 1 month following time of first collection.
One participant had antibody and inflammatory marker data, but insufficient cell viability for T cell analyses.
Sixty-one of 70 participants had saliva collected during the first sample collection time point.
Percentage of participants for the column N are shown throughout the table.
Saliva was collected on 61 participants. Seven (11.5%) had detectable SARS-CoV-2 RNA using both Nucleocapsid 1 (N1) and N2 qPCR probes at the first collection time point (Table 1). All cycle threshold (Ct) values were >35 for each assay reaction, and no viral RNA was detected at the subsequent collection time point a median of 80.5 days following acute illness (n = 54). The median time from symptom onset to T1 was significantly shorter for those with detectable saliva RNA than for those without PCR positivity (31 versus 57 days, p < 0.001; Table 2 ).
Table 2.
Persistent symptoms at the first study visit and 4 months following COVID-19 diagnosis
| All participants | Sex |
Hospitalized |
Saliva PCR+ at time of first sample collectiona |
||||
|---|---|---|---|---|---|---|---|
| Female | Male | Yes | No | Yes | No | ||
| n | 70a | 34 | 36 | 18 | 52 | 7 | 54b |
| Days from symptom onset to 1st sample (median, IQR) | 53 (38, 64.5) | 50 (37.8, 63.3) | 55 (41.3, 70) | 73 (51.8, 89.8) | 48 (37.3, 60.8) | 31 (29, 38)∗∗∗ | 57 (41.8, 66) |
| Persistent symptoms at time of first sample, n (%)c | 32 (45.8) | 18 (52.9) | 14 (38.9) | 9 (50) | 23 (44.2) | 1 (14.3) | 26 (48.1) |
| Fever/chills | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Cough/SOB | 15 (21.4) | 8 (23.6) | 7 (19.4) | 6 (33.3) | 9 (17.3) | 1 (14.3) | 11 (20.4) |
| Sore throat/runny nose | 3 (4.3) | 0 (0) | 3 (8.3) | 1 (5.6) | 2 (3.8) | 0 (0) | 3 (5.6) |
| Chest pain or palpitations | 2 (2.9) | 1 (2.94) | 1 (2.8) | 1 (5.6) | 1 (1.9) | 0 (0) | 2 (3/7) |
| Neurological | 14 (20) | 6 (17.6) | 8 (22.2) | 5 (27.8) | 9 (17.3) | 1 (14.3) | 10 (18.5) |
| Fatigue | 11 (15.7) | 7 (20.6) | 4 (11.1) | 4 (22.2) | 7 (13.5) | 0 (0) | 8 (14.8) |
| Smell/taste changes | 15 (21.4) | 9 (26.5) | 6 (16.7) | 6 (33.3) | 9 (17.3) | 0 (0) | 14 (25.9) |
| Gastrointestinal symptoms | 7 (10) | 5 (14.7) | 2 (5.6) | 3 (16.7) | 4 (7.7) | 0 (0) | 7 (13) |
| Musculoskeletal symptoms | 6 (8.6) | 4 (11.8) | 2 (5.6) | 3 (16.7) | 3 (5.8) | 0 (0) | 6 (11.1) |
| Days from symptom onset to T4 sample (median, IQR) | 125 (120, 133) | 127 (122, 132) | 123 (116, 134) | 125.5 (120.3, 134.5) | 125 (120, 125) | 115 (113.5, 124.5) | 123 (115.5, 130) |
| >3 Persistent symptoms at 4 months, n (%) | 20 (30.8) | 13 (39.4) | 7 (21.9) | 6 (37.5) | 14 (28.6) | 0 (0) | 17 (33.3) |
| 1–2 persistent symptoms at 4 months, n (%) | 15 (23.1) 65 | 7 (21.2) | 8 (25) | 3 (18.8) | 12 (24.5) | 1 (20) | 11 (21.6) |
| No persistent symptoms at 4 months, n (%) | 30 (46.2) | 13 (39.4) | 17 (53.1) | 7 (43.8) | 23 (46.9) | 4 (80) | 23 (45.1) |
SOB, shortness of breath.
p < 0.001 by Mann-Whitney U test.
No saliva sample was PCR+ 1 month following time of first collection (n = 54).
Saliva was available from 61 of 70 participants at the first sample collection time point.
Percentage of participants for the column N are shown throughout the table.
Clinical characteristics of acute infection and recovery
Ninety-seven percent of participants had COVID-19-related symptoms at the time of or immediately following their initial diagnosis, and 45.8% had persistence of at least one COVID-19-attributed symptom during the initial LIINC study visit, which was a median of 53 days since symptom onset (or initial time of initial clinical PCR positivity for asymptomatic individuals; Table 1). Interestingly, 53.8% had COVID-19-related symptoms approximately 4 months after initial illness (median 123 days [IQR 115–130.5] since initial symptom onset). Although not statistically significant, a higher proportion of females than males reported persistent symptoms at T1 (52.9 versus 38.9%) and 4-month (T4; 45.5 versus 27.3%) study visit (Table 2), despite lower hospitalization rates and similar time to study enrollment. Pulmonary symptoms, including cough and shortness of breath, fever, and loss or change in smell or taste, fatigue, and neurological symptoms (including headache, difficulties with concentration, attention, brain fog, neuropathies) were the most commonly experienced symptoms during acute infection (all reported in >70% of participants) and during the first study visit (Table 2). No significant relationship between persistent symptoms at any time point and RNA detection in saliva was observed.
SARS-CoV-2-specific T cell responses are stable over 8 months following initial infection
We applied two different methods to ascertain the frequency of SARS-CoV-2-specific CD4+ and CD8+ T cells in the peripheral blood after stimulation with Spike (S) or N peptide pools: the activation-induced marker (AIM) assay, which measures co-expression of OX40 and CD137 (4-1BB) on CD4+ T cells or co-expression of CD69 and CD137 on CD8+ T cells (Grifoni et al., 2020) (gating strategy shown in Figure S1), and an intracellular cytokine staining (ICS) assay to ascertain the frequencies of CD8+ and CD4+ T cells expressing interferon-γ (IFNγ), co-expressing tumor necrosis factor alpha (TNF-α) and IFNγ, and IFNγ cells expressing the degranulation marker CD107a. The percentage of CD8+ T cells identified in the ICS assay by the expression of CD107a and/or IFNγ that co-express Granzyme B was also measured (gating strategy shown in Figure S2). Overall, 192 unique sample time points across 69 participants ranging from 26 to 266 days (8.9 months) after onset of symptoms were tested and yielded interpretable results from either AIM or ICS assays. A large majority of participants had detectable S or N protein SARS-CoV-2-specific non-naive (memory) CD4+ (100%) and CD8+ (95.7%) T cell responses greater than the upper IQR of five historical blood-banked samples collected prior to November 2019 for at least one time point over the entire study period by either AIM or ICS assays (Table S1). An upper quartile cut-off was used given potential for noise in certain pre-COVID-19 samples that may have been due to prior coronavirus infection cross-reactivity as has previously been shown (Sekine et al., 2020). A median of 23.1 (IQR 10.3; 36.6)% of IFNγ+ CD8+ T cells (N and S specific) and 61.5 (IQR 44.4; 76.0)% of IFNγ+ CD4+ T cells (N and S specific) produced TNF-α across all time points sampled (p < 0.001).
The frequency of SARS-CoV-2 S- and N-specific memory CD4+ and CD8+ T cells (measured as AIM+, or ICS+: IFNγ+, IFNγ+/TNF-α+, or IFNγ−/CD107a+) for the most part did not significantly change over the sampling period (i.e., slopes were not different from zero in linear mixed effects models). The exception to this was a modest decrease in the frequency of CD4+ T cells expressing IFNγ+ in response to N peptide stimulation (−0.0065 log2 change in percentage per day; p = 0.011). These data demonstrate long-term stability of SARS-CoV-2-specific T cell responses in our cohort (Figure 1 ; Figure S3).
Figure 1.

Long-term durability of SARS-CoV-2-specific CD4 and CD8 cell responses
Gating strategy for identifying SARS-CoV-2-specific memory T cell responses in the activation-induced marker (AIM) (A) and intracellular cytokine staining (ICS) (B) assays. Percentage of AIM+ Nucleocapsid (N)-specific CD4+ (C) and CD8+ (D) T cell responses over time (Spike [S]-specific responses were similar and are shown in Figure S3; n = 68 participants). Percentage of IFNγ+ Nucleocapsid (N)- or S-specific CD4 (E and F) and CD8 (G and H) T cell responses over time (n = 64 participants). Points and connecting lines represent raw data for each individual. Solid line and shaded region represent the median model prediction and 95% prediction interval, respectively, from linear mixed effects modeling including individual effects. Dashed lines represent assay limits of detection. T mem, T memory cells (i.e., excluding naive CCR7+CD45RA+ T cells). Longitudinal responses as measured by IFNγ+/TNF-α dual expression and IFNγ+/CD107a expressing lymphocytes are show in Figure S3.
Overall, a majority of SARS-CoV-2-specific CD8+ T cells that expressed either IFNγ or CD107a at the early and late memory cross-sectional analysis time points expressed Granzyme B (median 66.7% and 77.3% for N- and S-responsive cells at T1 and 55.6% and 67.1% at T4 within both IFNγ+ and IFNγ−CD107a+ cells). No significant differences between T1 and T4 within N- and S-specific T cells were observed (all p > 0.68).
Differences in SARS-CoV-2-specific T cell responses across demographic and disease factors
We next asked whether the frequency of SARS-CoV-2-specific CD4+ and CD8+ T cells (AIM+, IFNγ+, IFNγ+/TNF-α+, and IFNγ−/CD107a) were impacted by various clinical and demographic factors. We first used cross-sectional analyses at the T1 and T4 time points. We then performed longitudinal analyses using linear mixed effects modeling over all time points, with days from onset of symptoms and individual factors (i.e., sex, race/ethnicity, age, COVID-19-related hospitalization, prior ICU care, and the presence of persistent COVID-19-related symptoms) as model parameters. Longitudinal models incorporated random variability for each participant.
We observed differences in the frequency of SARS-CoV-2-specific memory CD4+ and/or CD8+ T cell responses according to several demographic and clinical parameters in both cross-sectional time points (T1 and T4) and longitudinal linear mixed effects models. We highlight key differences in IFNγ-expressing cell populations identified by the ICS assay in Figure 2 (differences in polyfunctional responses as determined by co-expression of IFNγ and TNF-α followed a similar pattern and are shown in Figure S4). Participants hospitalized during acute infection had significantly higher frequencies of N- and S-specific IFNγ+ CD4+ T cells at T1 and S-specific IFNγ+ CD4+ T cells at T4 than non-hospitalized participants (Figure 2A). We also observed higher N and S IFNγ+ CD4+ responses in hospitalized participants using mixed effects models across all longitudinal data points through the last collection time point at study month 8 (p = 0.025 and 0.068; Figure 2B). The subset of hospitalized participants who were admitted to the ICU had significantly higher frequencies of N-specific IFNγ+ CD4+ T cells at both T1 and T4 than participants that did not require ICU care, regardless of hospitalization (Figure 2C). The N-specific IFNγ+ CD4+ T cell responses were also significantly higher for participants receiving ICU care across time in linear effects modeling (p = 0.024).
Figure 2.

Relationships between SARS-CoV-2-specific T cell responses and participant demographic and clinical factors
(A) Frequency of SARS-CoV-2-specific CD4+ and CD8+ T cells as measured by the ICS assay in study participants who were (A) hospitalized versus not hospitalized at early (left, median 53 days after onset of symptoms) versus late (right, median 123 days from onset of symptoms) cross-sectional analysis time points.
(B) Longitudinal frequency of IFNγ+ memory T cells in hospitalized versus non-hospitalized participants.
(C) Frequency of SARS-CoV-2-specific T cells as measured by the ICS assay in study participants who required ICU care.
(D) Frequency of SARS-CoV-2-specific T cells as measured by the ICS assay in study participants <50 versus ≥50 years of age at early and late cross-sectional analysis time points.
(E and F) Cross-sectional (E) and longitudinal (F) frequency of SARS-CoV-2-specific CD8+ T cells in study participants who had diagnosed pulmonary disease prior to infection.
(G and H) Frequency of SARS-CoV-2-specific CD8+ T cells among participants identifying as (G) white (non-Latinx), Latinx, Asian, or black, and as (H) male or female. All data points are shown as individual points.
For (A), (C), (D), (E), (G), and (H), dots represent individual participant values, and bars and lines in cross-sectional data represent median values and interquartile ranges: ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001 by non-parametric Kruskal-Wallis tests with Dunn adjustment for analyses incorporating comparisons across more than two variables (T1 ICS IFNγ N CD8 and CD4 data available for 61 participants, T1 IFNγ ICS S CD8 and CD4 n = 60, T4 IFNγ N and S CD4 and CD8 n = 46.) For (B) and (F), solid line and shaded region represent the median model prediction (including individual effects) and 50% prediction interval, respectively. mem T, T memory cells. Responses by demographic and clinical factors as measured by IFNγ+/TNF-α dual expression and AIM assay are show in Figure S4 and Figure S5, respectively.
Although we did not observe differences between participants <50 or ≥50 years of age at T1, those aged ≥50 years had significantly higher percentages of N- and S-specific IFNγ+ CD4+ T cells at T4 (Figure 2D). Interestingly, participants with a pre-existing history of pulmonary disease had significantly higher percentages of S-specific IFNγ+ CD4+ T cells and S-specific IFNγ+ CD8+ T cells at both T1 and T4 (Figure 2E). We observed significantly higher N and S IFNγ+ CD8+ (p = 0.018, 0.016; Figure 2F) and S IFNγ+ CD4+ (p = 0.024) T cell responses in participants with prior pulmonary disease in mixed effects models across time through study month 8.
A significantly higher percentage of S-specific IFNγ+ CD8+ T cells was observed in Asian than white (non-Latinx) participants (Figure 2G) at T1 only, but there were no differences between Latinx and white (non-Latinx) participants at T1 or T4 by the ICS assay. By contrast, we observed a significantly higher percentage of S AIM+ CD4+ T cells in Latinx versus white (not-Latinx) participants at T1 (p = 0.025), but not at T4 (p = 0.97). Linear mixed effects modeling also revealed a higher overall percentage of N AIM+ CD4+ T cells (p = 0.022) and S AIM+ CD8+ T cells (p = 0.003) in Latinx versus white (non-Latinx) participants across time. Despite higher overall levels, the percentage of S AIM+ CD8+ T cells declined in the Latinx, but not in the white non-Latinx population (p = 0.006).
We did not find significant differences in the frequency of SARS-CoV-2-specific CD4+ and CD8+ T cells by ICS assay between males and females by ICS (Figure 2H), but did identify a modestly lower percentage of N AIM+ CD4 T cells in females (p = 0.04) at T1. However, there were no significant differences in CD4+ or CD8+ T cell responses between participants with and without hospitalization, ICU care, prior pulmonary disease, and age <50 years by AIM assay in cross-sectional analyses at the T1 or T4 time points (Figure S5).
There were no significant differences in CD4+ or CD8+ T cell responses by AIM or ICS assays between participants with and without positive saliva PCR detection at T1 (Figure 3 A).
Figure 3.

Relationships between SARS-CoV-2-specific T cell responses, persistent viral shedding, and PASC
(A) The percentage of SARS-CoV-2-specific CD4+ and CD8+ T cells at T1 as measured by the AIM and ICS assays in study participants with detectable SARS-CoV-2 RNA in saliva at T1.
(B) SARS-CoV-2-specific T cell frequency at T1 in study participants with any COVID-19-related symptoms at T1.
(C and D) SARS-CoV-2-specific T cell frequency at T1 (C) and T4 (D) in study participants with post-acute sequelae of SARS-CoV-2 infection (PASC; persistent symptoms) at T4.
(E) Longitudinal frequency of N-specific IFNγ+ and IFNγ−/CD107a+ memory (mem) CD8+ T cells in those with PASC. For (A)–(D), dots represent individual participant values, and bars and lines in cross-sectional data represent median values and interquartile ranges: ∗p < 0.05 by non-parametric Kruskal-Wallis tests (T1 N and S AIM data available for 67 participants, ICS IFNγ N CD8 and CD4 n = 61, T1 ICS IFNγ S CD8 and CD4 n = 60, T4 IFNγ N and S CD4 and CD8 n = 46.) For (E), solid line and shaded region represent the median model prediction (including individual effects) and 50% prediction interval, respectively (n = 61 participants: 32 with PASC, 29 without PASC). T mem, T memory cells.
SARS-CoV-2-specific T cell responses in participants with persistent symptoms
There were no differences in T cell responses at T1 between participants with and without persistent symptoms at this initial time point by any assay (Figure 3B), nor were there any significant differences observed in T cell responses at T1 in participants who developed PASC at T4 (Figure 3C). However, at T4, N and S CD8+ T cell responses (IFNγ+ and IFNγ−/CD107a+, but not AIM+) trended lower in participants with PASC, with the decreased frequency of IFNγ−/CD107a+ N-specific CD8+ T cells reaching statistical significance (p = 0.044; Figure 3D). In order to determine whether the lower frequency of N-specific CD8+ T cells in individuals with PASC was driven by individuals with more symptoms, we performed cross-sectional analyses using two PASC groups: those with one to two symptoms at T4 and those with three or more symptoms. Interestingly, we identified a significantly lower percentage of IFNγ−/CD107a+ cells in those with 1–2 symptoms (p = 0.017). In mixed effects modeling, the frequency of N-specific IFNγ+ CD8+ T cells was overall higher (p = 0.02), but declined for those experiencing PASC at T4 in comparison with those not experiencing PASC (Figure 3E; p = 0.0007).
Interactions and independent associations between T cell responses and demographic and clinical factors
In order to determine the relationships between significant covariates identified in univariate cross-sectional analyses as above (COVID-19-related hospitalization, pre-existing lung disease, age, and persistent symptoms at T4), we performed linear regression modeling using log2-transformed data for T cell responses at T1 and T4 and performed tests of interactions between covariates. Differences in N- and S-specific IFNγ+ CD4+ T cell responses at T1 and S-specific IFNγ+ CD4+ T cell response at T4 between hospitalized and non-hospitalized participants remained significant at T1 when adjusted for age (all p < 0.042). Furthermore, increased percentages of N- and S-specific IFNγ+ CD8+ T cells at the first time point and N-specific IFNγ+ CD8+ T cells at T4 observed in participants with pre-existing lung disease were significant in regression models adjusted for age (all p < 0.042). Similarly, all increases in N- and S-specific IFNγ+ CD8+ T cell responses observed in those with pre-existing lung disease in univariate analyses remained significant when adjusted for prior hospitalization (all p < 0.024).
We identified significant interactions between hospitalization and age for S-specific IFNγ+ CD4+ T cells at T4 (p = 0.007), demonstrating that with more advanced age, there were lower percentages of IFNγ-producing CD4+ T cells in hospitalized participants even though the percentage of IFNγ+ CD4+ T cells were generally higher for age ≥50 and hospitalization in univariate analyses described above. Interestingly, participants with a prior history of lung disease and COVID-19-related hospitalization had lower N-specific IFNγ+ CD8+ T cells at T4 (p = 0.024), despite higher levels observed in those with a pre-existing lung disease only.
SARS-CoV-2 specific antibody responses and neutralization capacity
We quantified immunoglobulin G (IgG) antibody titers to full-length SARS-CoV-2 N, S, and N fragment (N.361) and the S receptor binding domain (RBD) proteins using an in-house Luminex assay. We also tested NAb responses using pseudoviruses expressing the S protein in the presence of autologous sera from approximately 4 months post-symptom onset (median 125 days [IQR 120–133]) across the entire study population (n = 66; 4 participants did not have NAb data available). Consistent with data that we and others (Chen et al., 2020a, 2020b; Gaebler et al., 2021; Gudbjartsson et al., 2020; Kowitdamrong et al., 2020; Lei et al., 2020; Long et al., 2020; Naaber et al., 2020; Seow et al., 2020; Zhao et al., 2020) have previously demonstrated, N-, S-, and RBD-specific antibody levels remained stable (Figure 4 A; Figure S6). However, antibody neutralization declined. Higher antibody titers were observed at T1 and T4 in those who were hospitalized (including the subgroup requiring ICU level care; data not shown) and those who identified as Latinx, and at T4 only among participants ≥50 years of age (N-specific responses only for the latter) (all p < 0.035; Figures 4B–4D). No significant differences were observed in T1 antibody levels between participants with and without persistent saliva SARS-CoV-2 RNA detection or symptoms at T1. In addition, there were no significant differences between early and late (T1 and T4) antibody levels and those with PASC at T4 (Figure 4E).
Figure 4.

SARS-CoV-2-specific antibody levels and neutralization responses
Longitudinal antibody response (relative light unit [RU]) as measured using the Luminex IgG assay across the N, N fragment (N.361), S, and receptor binding domain (RBD), along with antibody-neutralizing capacity (infectious dose, 50% [ID50] of S pseudovirus in presence of participant serum) for all participants over time. Differences at T1 and T4 in the levels of N-, S-, and RBD-specific antibody responses for participants grouped by (B) hospitalization during acute infection, (C) Latinx versus white-non Latinx race/ethnicity, (D) age, and (E) PASC (symptoms at T4). ID50 values from neutralization assays in cross-sectional neutralization antibody (NAb) analyses of participants grouped by (F) sex, (G) hospitalization during acute infection, and (H) PASC. Solid line and shaded region represent the median model prediction and 95% prediction interval, respectively, from linear mixed effects modeling including individual effects. Dashed lines represent assay limits of detection. Dots represent individual participant values, and solid bars and lines represent median values and interquartile ranges (T1 antibody data available on 66 participants and 50 at T4). ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001 by non-parametric analyses. Linear mixed effects modeling of NAb responses by various clinical and demographic factors are shown in Figure S6.
In linear regression modeling of antibody responses to determine the independent relationships between covariates of interest, we observed that increased N and RBD antibody levels observed in univariate analyses at T1 in hospitalized participants remained statistically significant when adjusted for Latinx race/ethnicity (p = 0.02, 0.05). However, the increases in antibody levels observed in Latinx participants were not significant in adjusted analyses, suggesting that differences in antibody responses are primarily driven by initial disease severity, rather than by demographic factors.
NAb responses were modestly lower in female participants at T1 time point (p = 0.013), but there were no significant differences observed at the last cross-sectional analysis time point (Figure 4F). As expected, NAb responses were significantly higher in participants that were hospitalized at T1 and T4 (Figure 4G). There were no significant differences in neutralization infectious dose, 50% (ID50) between those with and without symptoms at T1 or between those with and without PASC at T4 at either T1 or T4 (Figure 4H). In longitudinal analyses, N-, N.361-, S-, and RBD-specific antibody levels were higher across all time points for hospitalized participants (p = 0.024, 0.012, 0.014, and 0.043, respectively).
Soluble markers of inflammation
We measured longitudinal circulating levels of interleukin-6 (IL-6), IL-10, IP-10, D-Dimer, soluble cluster of differentiation 14 (sCD14), and sCD163 in a random subset of 57 individuals through study month 4 (Figure S7). There were no significant changes in the levels of soluble markers over time, with the exception of a modest decline in sCD14 (p = 0.006). No significant differences were observed between demographic and clinical factor groups or between those with and without PASC in cross-sectional analysis at T1 and T4 (Figures 5A and 5B). Interferon gamma-induced protein 10 (IP-10) levels were modestly higher in the hospitalized participants across time points (p = 0.028).
Figure 5.

Soluble markers of inflammation in participants with and without PASC and correlations with antibody and T cell responses
(A and B) Differences in the levels of IL-6, IL-10, IP-10, D-Dimer, sCD14, and sCD163 at (A) T1 and (B) T4 in participants with and without PASC (T1 cytokine data available on 57 participants and 40 at T4).
(C) Spearman correlation matrix heatmap (r values) between weighted average antibody and T cell response across all time points.
(D) Individual correlation plots between NAb weighted average ID50 and the percentage of CD4+ T cells co-expressing IFNγ+ in response to N and S peptide pool stimulation across all time points (n = 61).
(E) Spearman correlation matrix heatmap incorporating soluble markers of inflammation and T cell and antibody responses. All data points are shown. Linear mixed effects modeling of markers of inflammation are shown in Figure S7.
Relationships between inflammatory markers, humoral immunity, and virus-specific T cell responses
In order to explore relationships between T cell responses and antibody levels and soluble inflammatory markers, we generated Spearman correlation matrices. Given the variation in time from initial symptoms to sample collection times and the number of sample time points for each participant, correlation analyses were performed using weighted averages for all T cell, antibody, and cytokine data across all time points. N-, N.361-, S-, and RBD-specific antibody levels were significantly correlated with N- and S-specific CD4+ T cell responses (as measured by expression IFNγ as well as dual expression of IFNγ and TNF-α), but much less so with CD8+ T cell responses (Figure 5C). NAb responses were most strongly correlated with S- and N-specific CD4+ T cell IFNγ+ and IFNγ+/TNF-α+ ICS results and also correlated with S- and N-specific AIM responses (Figure 5D).
Spearman correlation analysis was performed for the soluble markers of inflammation and T cell and antibody responses using weighted averages. Overall, there were modest positive associations between IL-1, IL-10, and sCD14 levels with the frequency of CD8+ T cell responses as measured by ICS and negative association with D-Dimer levels with CD8+ T cell ICS responses, but many of these associations did not reach statistical significance (Figure 3E;). We did observe a negative correlation between IL-10 levels and RBD-specific antibody responses; however. IL-6, IL-10, and IP-10 were strongly positively correlated with each other, but not with D-Dimer, sCD14, or sCD163.
Discussion
In this analysis, we demonstrate several important findings related to immune responses in individuals recovering from SARS-CoV-2 infection. First, we observed that clinical factors related to medical history and acute infection were associated with variability in T cell responses over the long term. Second, individuals experiencing PASC demonstrated subtle differences in immune responses compared with those without persistent symptoms, suggesting that further immunophenotyping of such individuals could provide clues to the biological mechanism(s) that underlie this poorly understood condition that is of growing clinical concern. Finally, we identified strong relationships between cellular and humoral immune responses that could have implications for understanding the durability of immunity to natural infection and/or vaccination.
Data from this well-characterized, diverse cohort build upon recent studies demonstrating that adaptive immune responses are relatively stable over 8 months following infection with SARS-CoV-2 (Braun et al., 2020; Breton et al., 2021; Dan et al., 2021; Grifoni et al., 2020; Le Bert et al., 2021; Peng et al., 2020; Rydyznski Moderbacher et al., 2020; Tan et al., 2021; Yao et al., 2021) by identifying important differences in CD4+ and CD8+ T cell responses between various groups across early and later convalescent time points, including those with and without PASC.
We identified certain characteristics of acute illness that may determine long-term immunologic outcomes. For example, those requiring hospitalization or intensive care during acute infection had higher levels of memory CD4+ T cell responses during recovery. Whereas the higher percentage of SARS-CoV-2-specific CD4+ T cell responses initially observed in hospitalized participants converged with those in participants who were not hospitalized approximately 4 months following acute infection similar to one prior report (Dan et al., 2021), CD4+ T cell responses remained elevated in those who had required ICU level care. We identified persistently higher virus-specific CD8+ T cell responses in the subset of participants with pre-existing pulmonary disease. These differences remained significant after adjusting for prior hospitalization and age. In addition, we observed an interesting interaction in that those with pre-existing lung disease had “lower” SARS-CoV-2-specific CD8+ T cell responses if hospitalized. These results suggest that the relationships between various demographic and clinical factors are complex and will require large, diverse, and well-curated cohorts to more fully understand relationships between long-term immunity and different components of the immune response.
PASC has recently been identified as a major public health concern, and there is now intense interest in understanding the cause and effects of this condition (Carfì et al., 2020; Datta et al., 2020; Hellmuth et al., 2021; Huang et al., 2021; Nalbandian et al., 2021; Tenforde et al., 2020). Given the high prevalence of persistent symptoms among the study population, we had an opportunity to study immune responses in individuals with or without PASC and identified several potentially important patterns. Although the severity of initial infection was similar between individuals with and without PASC, we identified lower and more rapidly waning month 4 N-specific CD8+ T cell responses (IFNγ−/CD107a+ and IFNγ+) in those with PASC. We cannot distinguish whether the lower frequency of degranulating virus-specific CD8+ T cells in individuals with PASC represents decreased functional capacity of these cells or dysfunction of the immune response, perhaps in response to persistent antigen stimulation, or another process. Few participants in our study experienced persistent shedding of virus in saliva by PCR, which has shown to be as sensitive as testing nasopharyngeal swabs (Sakanashi et al., 2021). Importantly, we observed no association between viral shedding, PASC symptoms, or immune responses, suggesting that if persistent viral stimulation is associated with PASC, it is likely to be present in deeper tissues (Gaebler et al., 2021; de Melo et al., 2021) and not readily detectable at superficial sites. PASC is likely to be a multifactorial process, and larger, well-powered studies of PASC will be needed to provide further mechanistic understanding of this important group.
We observed a strong correlation between antibody-neutralizing capacity and virus-specific CD4+ responses across ICS and AIM assays. We opted to use weighted averages for these analyses to represent a measure of the immune response over the entire study interval. These results are consistent with those of other studies that have shown an association between the magnitude of SARS-CoV-2-specific CD4+ T cells and antibody levels within 2 months (Grifoni et al., 2020; Rydyznski Moderbacher et al., 2020). However, longer term analyses have not identified such correlations beyond 8 months following infection (Chia et al., 2021; Dan et al., 2021). Nonetheless, these data suggest the potential importance of follicular helper T cell responses in lymph nodes during the first 4–8 months of recovery, and further tissue-based analyses will be important to more clearly define the coordinated adaptive and humoral immune response in diverse populations. Interestingly, antibody neutralization appeared to wane, suggesting that there may be a dissociation between binding antibody levels and function over time; this warrants further study.
Strengths of this study include the diversity of the cohort, both in racial/ethnic diversity reflective of groups disproportionately affected by the pandemic in California (Chamie et al., 2020; Richardson et al., 2020) as well as the even distribution of individuals according to disease severity followed longitudinally over 8 months from initial symptoms and a high proportion of individuals experiencing persistent symptoms. There are several notable limitations. First, the time frame of sampling was limited to the recovery phase of COVID-19, and it is possible that there are clinically important biological correlates that could have been identified had samples from the infectious period been available. Second, while there is intense interest in understanding PASC, there remains no consensus case definition for this condition. Nonetheless, we observed potentially important differences in T cell responses in those with and without PASC using detailed longitudinal phenotyping to carefully define persistent symptoms in the cohort. And third, this study involved a large number of analyses and comparator groups using assays with inherent intra- and inter-participant variation and a high degree of collinearity between study factors, making it difficult to make specific biological or causal inferences. As a result, we used a targeted approach to focus analyses on primary endpoints of interest, and we acknowledge our results are hypothesis generating and need to be confirmed in future studies and/or in other cohorts.
In this study, we observed important patterns across assays measuring adaptive and humoral immune responses for various clinical factors such as initial clinical severity defined by hospitalization or ICU care, pre-existing pulmonary disease, and PASC. These data suggest that apart from severity of initial COVID-19 illness, pre-existing medical conditions may have important influence over the longitudinal adaptive immune responses and that immune responses may be linked with the development of post-acute sequelae.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD45RA (BV421) Clone HI100 | Biolegend | Cat# 304130 |
| CD14 (V500) Clone M5E2 | BD | Cat# 561391 |
| CD19 (V500) Clone HIB19 | BD | Cat# 561121 |
| Live/Dead (Aqua) | Invitrogen | Cat# L34966 |
| CD8 (BV650) Clone RPA-T8 | BioLegend | Cat#301042 |
| CD4 (BV605) Clone RPA-T4 | BD | Cat# 562659 |
| CCR7 (FITC) Clone G043H7 | Biolegend | Cat# 353216 |
| CD69 (PE) Clone FN50 | BD | Cat# 555531 |
| OX40 (PE-Cy7) Clone Ber-ACT35 | Biolegend | Cat# 350012 |
| CD137 (APC) Clone 4B4-1 | Biolegend | Cat# 309810 |
| CD3 (AF700) Clone OKT3 | Biolegend | Cat# 317340 |
| CD40L (PE-Dazzle) Clone 24-31 | Biolegend | Cat# 310840 |
| IL-2 (BV421) Clone MQ1-17H12 | Biolegend | Cat# 500328 |
| CD14 (BV510) Clone M5E2 | Biolegend | Cat# 301842 |
| CD19 (BV510) Clone HIB19 | Biolegend | Cat# 302242 |
| TCR γ/Δ (BV510) Clone B1 | Biolegend | Cat# 331220 |
| CD27 (BV570) Clone O323 | Biolegend | Cat# 302825 |
| CD4 (BV650) Clone OKT4 | Biolegend | Cat# 317436 |
| CD8 (BV711) Clone SK1 | Biolegend | Cat# 344733 |
| CD45RA (BV785) Clone HI100 | Biolegend | Cat# 304140 |
| Granzyme B (FTIC) Clone Ber-ACT35 | Biolegend | Cat# 350012 |
| Interferon-γ (PE-CF594) Clone BG11 | Biolegend | Cat# 515403 |
| CD3 (PE-Cy5.5) Clone SK7 | eBioscience | Cat# 35-0036-42 |
| CD107a (APC) Clone H4A3 | Biolegend | Cat# 328620 |
| TNFα (AF700) Clone 6402 | R&D Systems | Cat# IC9677N |
| R-PE-conjugated AffiniPure F(ab’)2 Fragment Goat Anti Human IgG, Fcy Fragment Specific | Jackson Immunoresearch | Cat# 109-116-098 |
| Chemicals, peptides, and recombinant proteins | ||
| PepTivator SARS-CoV-2 Prot_S | Miltenyi Biotec | 130-126-700 |
| PepTivator SARS-CoV-2 Prot_N | Miltenyi Biotec | 130-126-698 |
| CD3/CD28 Dynabeads | GIBCO | 11131D |
| Phytohemagglutinin | Sigma-Aldrich | L1668-5MG |
| SepMate Tubes | 85460 | |
| Spike Protein | Pilarowski et al., 2021; Amanat et al., 2020 | N/A |
| N Protein | Pilarowski et al., 2021; Amanat et al., 2020 | N/A |
| RBD | Pilarowski et al., 2021; Amanat et al., 2020 | N/A |
| N Protein (Fragment N361) | Pilarowski et al., 2021; Amanat et al., 2020 | N/A |
| Critical commercial assays | ||
| PhenoSense SARS-CoV-2 neutralizing antibody | LabCorp | N/A |
| Human IL-6 ELISA Kit | Millipore Sigma-Aldrich | RAB0306-1KT |
| Human IL-10 ELISA Kit | Millipore Sigma-Aldrich | RAB0244-1KT |
| Human IP-10 ELISA Kit | Invitrogen | KAC2361 |
| Quantikine ELISA Human CD14 Immunoassay | R&D Systems | DC140 |
| Human CD163 (M130) ELISA Kit | ThermoFisher Scientific/Invitrogen | EHCD163 |
| Human D-Dimer ELISA Kit | ThermoFisher Scientific/Invitrogen | EHDDIMER |
| QIAamp Viral RNA Kit | QIAGEN | Cat. No. / ID: 52904 |
| 2019-nCoV RUO Kit (N1/N2 qPCR) | Integrated DNA Technologies | Cat# 10006713 |
| MagPlex Microspheres | Luminex | Cat # MC10018, MC10014, MC10026 MC10055, MC10013, MC10044 |
| Software and algorithms | ||
| Prism (version 9.1) | GraphPad Software | https://www.graphpad.com |
| R | The R Project | https://www.r-project.org/ |
| SPSS Statistics | IBM Software | https://www.ibm.com/products/spss-statistics |
| XPONENT Software | Luminex | https://www.luminexcorp.com/xponent/#overview |
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Dr. Timothy J. Henrich (timothy.henrich@ucsf.edu)
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
LIINC Study
Participants were volunteers in the University of California, San Francisco-based Long-term Impact of Infection with Novel Coronavirus (LIINC) study. LIINC is an observational cohort that enrolls individuals with SARS-CoV-2 infection documented by clinical nucleic acid amplification testing who have recovered from the acute phase of infection. Volunteers are recruited by clinician- or self-referral. They are eligible to enroll between 14 and 90 days after onset of COVID-19 symptoms and are offered monthly visits until 4 months after illness onset; they are then seen every 4 months thereafter. At each study visit, participants undergo a detailed clinical interview that includes demographic information, COVID-19 diagnosis, illness, and treatment history, assessment of medical comorbidities and concomitant medications, and evaluation of ongoing symptoms and quality of life. At the first visit, participants were asked to assess their level of disability related to the worst point in their acute illness according to 3 measures on a 3-point scale: mobility, ability to perform self-care, and ability to perform routine work and household obligations. Biospecimens are collected and stored.
For the current study, we selected LIINC participants who had at least two time points available for analysis. We excluded participants with HIV infection. We randomly selected participants that experienced low initial disease severity (defined as 4 or fewer points on the symptom severity scale without hospitalization), moderate severity (5-7 points without hospitalization), and highly severe disease (greater than 7 points and/or hospitalized) in order to have a sample population representing a wide spectrum of initial disease severity. We also assessed whether participants had persistent symptoms that they attributed to COVID-19. We defined a persistent symptom as a symptom that was noted to be newly present during acute infection that remained present at follow-up. The characteristics of the participants, including gender and age, are displayed in Table 1. The median age was 43 years (IQR 36-53 years) and the cohort was 48.6% female.
Ethics Statement
All participants sign a written informed consent and the study was approved by the University of California, San Francisco Institutional Review Board (IRB# 20-30479).
Method details
PBMC Isolation
Whole blood was collected from EDTA tubes (COVID-19 participants) or from Buffy coats from healthy unexposed, anonymous donors collected and stored prior to November, 2019 in the San Francisco, Bay Area. Whole blood was centrifuged for 10 min at 1600 rpm to separate plasma. Plasma was then removed and stored at −80°C. Peripheral blood mononuclear cells (PBMCs) were isolated by density-gradient Ficol-Paque (GE Healthcare, Chicago, IL) using SepMate tubes (StemCell Technologies, Cambridge, MA). PBMC were cryopreserved in heat inactivated fetal bovine serum (Phoenix Scientific, Bangkok, Thailand) containing 10% DMSO (Sigma-Aldrich) and stored in liquid nitrogen.
Saliva SARS-CoV-2 PCR Testing
We collected unexpectorated saliva during early visit time points for RNA PCR quantification and detection. RNA was isolated from 150 μl of saliva using the QIAmp Viral Kit (QIAGEN) and tested using the CDC SARS-CoV-1 (2019-nVoV) qPCR probe assay kit targeting N1 and N2 (Integrated DNA Technologies) including human RPP30 and SARS-CoV-2 positive control as per manufacturer protocols. PCR was performed in quadruplicate for both N1 and N2 assays.
Peptide Pools
SARS-CoV-2 specific T cell peptide pools were purchased from Miltenyi Biotec (PepTivator SARS-CoV-2 Prot_S and PepTivator SARS-CoV-2 Prot_N) and resuspended in DMSO. SARS-CoV-2 S and N are pools of lyophilized peptides, consisting of 15 amino acids length with 11 amino acids overlap, covering the immunodominant sequence domains of the Spike (“S”) or Nucleocapsid (“N”) proteins of SARS-CoV-2.
Activated Induced Cell Marker Assay
Cryopreserved PBMC were thawed in 10 mL of complete RPMI (GIBCO) containing 10% fetal bovine serum and stimulated in complete RPMI containing 10% human AB serum (Sigma-Aldrich). Cells were then cultured for 24 hours in 96-wells U bottom plates at 1x106 PBMC per well in the presence of either SARS-CoV-2 peptide pools (1 ug/ml), 10ug/ml phytohemagglutinin (PHA, Sigma-Aldrich) as positive control, or equimolar DMSO as negative control as previously described (Grifoni et al., 2020). All samples were analyzed on a BD LSR-II analyzer and analyzed with FlowJo X software. A complete list of antibodies are listed in the Key resources table. Whenever possible, longitudinal samples from individual participants were included in the same assay to minimize potential batch testing effects.
Intracellular Cytokine Staining Assay
We implemented an in-house ICS assay as previously described with minor modifications to be consistent with other published SARS-CoV-2-specific assays (Henrich et al., 2017; Morley et al., 2019; Scully et al., 2018). Briefly, cells were rested overnight before stimulation. Cells were cultured for 8 hours at 37°C in 96-wells U bottom plates at 1x106 PBMC per well in presence of either SARS-CoV-2 peptides [1 μg/ml], CD3/CD28 beads (GIBCO Dynabeads) as positive control, or equimolar DMSO as negative control. All conditions were in the presence of Golgi-plug containing brefeldin A (eBioscience), monensin (eBioscience), anti-CD28 (Biolegend, Clone CD28.2), and CD107a. After an 8-hour incubation, plates were put at 4°C overnight. The following day, cells were washed and surfaced stained for 20 min at room temp in the dark. Following surface staining, cells were washed twice with PBS and then fixed/permeabilized (BD Cytofix/Cytoperm) for 45 min at 4°C in the dark. Cells were then washed twice with fix/perm wash buffer (BD Perm/Wash) and stained with intracellular antibodies for 45min at 4°C in the dark. A complete list of antibodies are listed in the Key resources table. All samples were analyzed on a BD LSR-II analyzer and analyzed with FlowJo X software. Whenever possible, longitudinal samples from individual participants were included in the same assay to minimize potential batch testing effects.
Soluble Markers of Inflammation
Six different ELISA kits were used to detect the following markers, respectively: IL-6, IL-10, IP-10, D-Dimer, CD163, and CD14. Each kit was developed by a manufactured brand and standard protocol and dilutions were used in the preparation and analysis of each plate as follows: IL-6 ELISAs were prepared and conducted using the Millipore Sigma-Aldrich Human IL-10 ELISA Kit (Product Number RAB0306). The highest standard concentration used was 1000 pg/mL, decreasing concentration at a 3-fold dilution. Samples were run in duplicate without further dilution. IL-10 ELISAs were prepared and conducted using the Millipore Sigma-Aldrich Human IL-10 ELISA Kit. The highest standard concentration used was 150 pg/mL, decreasing concentration at a 2-fold dilution. Samples were run in duplicate without further dilution. D-Dimer ELISAs were conducted using the ThermoFisher Scientific Invitrogen Human D-Dimer ELISA Kit. The highest standard concentration used was 60 pg/mL, decreasing concentration at a 3-fold dilution. Samples were run in duplicate at a 250,000-500,000-fold dilution. IP-10 ELISAs were conducted using the Invitrogen Human IP-10 ELISA Kit. The highest standard concentration used was 500 pg/mL, decreasing concentration at a 2-fold dilution. Samples were run in duplicate without further dilution. CD163 ELISAs were prepared and conducted using the ThermoFisher Scientific Invitrogen Human CD163 (M130) ELISA Kit. The highest standard used was 8000 pg/mL, decreasing at a serial dilution of 40%. Samples were run in duplicate at a 50-fold dilution. CD14 ELISAs were prepared and conducted using the RnD Systems Quantikine ELISA Human CD14 Immunoassay. The highest concentration used was 8000 pg/mL, decreasing concentration at a 2-fold dilution. Samples were run in duplicate at a 200-250-fold dilution. Colorimetric changes were detected and quantified using the Spectramax M3 Plate Reader.
SARS-CoV-2 Antibody Testing
Serum was tested for antibodies at UCSF using an in-house multiplex microsphere assay (Luminex platform) to detect IgG against SARS-CoV-2 Spike, receptor binding domain (RBD), and two preparations of the N protein (on full length and one fragment). We used a published protocol with modifications (Amanat et al., 2020; Pilarowski et al., 2021; Wu et al., 2020). Plasma samples were diluted to 1:100 in blocking buffer A (1xPBS, 0.05% Tween, 0.5% bovine serum albumin (BSA), 0.02% sodium azide). Antigen concentrations used for bead coupling were as follows: S, 4 ug/mL; RBD, 2 ug/mL; and N, 3 ug/mL. Concentration values were calculated from the Luminex median fluorescent intensity (MFI) using a plate-specific standard curve from serial dilutions of a pool of positive control samples (https://github.com/EPPIcenter/flexfit). A cutoff for positivity was established for each antigen as the maximum concentration value observed across 114 pre-pandemic SARS-CoV-2 negative control samples tested on the platform.
PhenoSense SARS CoV-2 nAb Assay
The measurement of nAb activity using the PhenoSense SARS CoV-2 nAb Assay (Monogram Biosciences, South San Francisco, CA) is performed by generating HIV-1 pseudovirions that express the SARS CoV-2 Spike protein. The pseudovirus is prepared by co-transfecting HEK293 producer cells with an HIV-1 genomic vector that contains a firefly luciferase reporter gene together with a SARS CoV-2 Spike protein expression vector. Neutralizing antibody activity is measured by assessing the inhibition of luciferase activity in HEK293 target cells expressing the ACE2 receptor and TMPRSS2 protease following pre-incubation of the pseudovirions with serial dilutions of the serum specimen.
Data are displayed by plotting the percent inhibition [% Inhibition = 100% – ((RLU(Pseudovirus+Sample+Cells)) ÷ (RLU(Pseudovirus+Diluent+Cells)) x 100%)] of luciferase activity expressed as relative light units (RLU) versus the log10 reciprocal of the serum/plasma dilution. Neutralizing antibody titers are reported as the reciprocal of the serum dilution conferring 50% inhibition (ID50) of pseudovirus infection. To insure that the measured nAb activity is SARS CoV-2 nAb specific, each test specimen is also assessed using a non-specific pseudovirus (specificity control) that expresses a non-reactive envelope protein of an unrelated virus (e.g., avian influenza virus H10N7).
Quantification and statistical analysis
Flow cytometric, antibody and cytokine data were generated blinded to participant information. Comparisons of M0 values across comparator groups incorporated non-parametric Mann-Whitney or Kruskal-Wallis test with Dunn correction for multiple comparisons using Prism v. 8 (GraphPad Software). Adjusted P values a reported in analyses involving multiple comparisons. Fisher’s exact test when any N < 5 was used to compare tabular data. Spearman Rank Correlation analysis was used to compare T cell, antibody and soluble markers of inflammation (Prism). Whereas using all time points from all participants may lead to oversampling of some individuals, for correlations we used calculated weighted averages, representing the outcome of interest across all time points for each individual. Linear regression modeling was performed using SPSS v. 27 (IBM) including covariates of interest identified in the univariate analyses. For longitudinal analyses, linear mixed effects modeling was performed for each immunologic outcome (log transformed) in R (version 4.0) using lme4 package (version 1.1) with time and individual factors (e.g., Age, Sex, Ethnicity, Hospitalization, ICU admission, Symptoms at the first study time point and the month 4 time point, prior history of pulmonary disease) as predictors, and random effects based on participant. Sensitivity analyses were performed excluding month 8 data (when available) to rule out assay batch effects.
Acknowledgments
We are grateful to the LIINC study participants and to the clinical staff who provided care to these individuals during their acute illness period and recovery. We acknowledge LIINC study team members Tamara Abualhsan, Mireya Arreguin, Jennifer Bautista, Monika Deswal, Heather Hartig, Marian Kerbleski, Lynn Ngo, Fatima Ticas, and Meghann Williams for their contributions to the program. We are grateful to Khamal Anglin, Grace Bronstone, Jessica Chen, Michelle Davidson, Kevin Donohue, Peyton Ellis, Sarah Goldberg, Scott Lu, Jonathan Massachi, Sujata Mathur, Irum Mehdi, Victoria Wong Murray, Enrique Martinez Ortiz, Jesus Pineda-Ramirez, Mariela Romero, Paulina Rugart, Hannah Sans, Joshua Shak, Jaqueline Tavs, and Jacob Weiss for assistance with data entry and validation. We thank Shane Crotty and Alessandro Sette for providing experimental details and advice regarding the AIM assay. We thank the UCSF Core Immunology Laboratory for processing some samples. We are grateful to the UCSF AIDS Specimen Bank for specimen processing and for managing the LIINC biospecimen repository. This work was supported by the National Institute of Allergy and Infectious Diseases (NIH/NIAID) 3R01AI141003-03S1 (to T.J.H.); NIH/NIAID R01AI158013 (to M.G. and M.S.); and the Zuckerberg San Francisco Hospital Department of Medicine and Division of HIV, Infectious Diseases, and Global Medicine. This work is also supported by a Merck Investigator Studies Program grant (to T.J.H.). M.J.P. is supported on NIH T32 AI60530-12 and by the UCSF Resource Allocation Program. S.T. is supported by the Schmidt Science Fellow, in partnership with the Rhodes Trust. I.R.B. and S.T. acknowledge research funding from the MIDAS Coordination Center COVID-19 Urgent Grant Program (MIDASNI2020-5). A.N.D. is supported by NIH R01 AI141003 and R01 HD068174 and the UCSF Resource Allocation Program. The flow cytometry core is supported by the UCSF/Gladstone Institute of Virology & Immunology CFAR (P30 AI027763).
Author contributions
M.J.P., A.N.D., F.T.A., R.L.R., and T.J.H. designed the study, which was supported through funding to M.A.S., M.G., J.D.K., J.N.M., S.G.D., and T.J.H. M.J.P., L.T., R.H., V.T., Y.H., and E.A.F. collected clinical data and biospecimens. L.T., C.C.N., N.S.I., S.E.M., J.D., C.T., J.H., K.T., and O.J. performed PBMC and plasma isolation and storage. Specimens were analyzed by L.T., C.C.N., N.S.I., S.E.M., J.D., J.H., K.T., and O.J. in the laboratories of B.G. and T.J.H. L.T., T.W., and C.J.P. performed and interpreted the neutralization assays. M.J.P., A.N.D., S.T., F.T.A., R.L.R., and T.J.H. performed and/or interpreted the statistical analyses. M.J.P., A.N.D., R.L.R., and T.J.H. drafted the initial manuscript. All authors edited, reviewed, and approved the final manuscript.
Declaration of interests
L.T., T.W., and C.J.P. are employees of Monogram Biosciences, a division of LabCorp. T.J.H. reports grants from Merck, Gilead Biosciences, and Bristol-Myers Squibb outside the submitted work.
Published: July 26, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109518.
Supplemental information
Data and code availability
-
•
De-identified data reported in this paper will be shared by the lead contact upon request.
-
•
All original code is available in this paper’s supplemental information.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Amanat F., Stadlbauer D., Strohmeier S., Nguyen T.H.O., Chromikova V., McMahon M., Jiang K., Arunkumar G.A., Jurczyszak D., Polanco J., et al. A serological assay to detect SARS-CoV-2 seroconversion in humans. Nat. Med. 2020;26:1033–1036. doi: 10.1038/s41591-020-0913-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun J., Loyal L., Frentsch M., Wendisch D., Georg P., Kurth F., Hippenstiel S., Dingeldey M., Kruse B., Fauchere F., et al. SARS-CoV-2-reactive T cells in healthy donors and patients with COVID-19. Nature. 2020;587:270–274. doi: 10.1038/s41586-020-2598-9. [DOI] [PubMed] [Google Scholar]
- Breton G., Mendoza P., Hägglöf T., Oliveira T.Y., Schaefer-Babajew D., Gaebler C., Turroja M., Hurley A., Caskey M., Nussenzweig M.C. Persistent cellular immunity to SARS-CoV-2 infection. J. Exp. Med. 2021;218:e20202515. doi: 10.1084/jem.20202515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carfì A., Bernabei R., Landi F., Gemelli Against COVID-19 Post-Acute Care Study Group Persistent Symptoms in Patients After Acute COVID-19. JAMA. 2020;324:603–605. doi: 10.1001/jama.2020.12603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamie G., Marquez C., Crawford E., Peng J., Petersen M., Schwab D., Schwab J., Martinez J., Jones D., Black D., et al. Community Transmission of Severe Acute Respiratory Syndrome Coronavirus 2 Disproportionately Affects the Latinx Population During Shelter-in-Place in San Francisco. Clin. Infect. Dis. 2020;2020:ciaa1234. doi: 10.1093/cid/ciaa1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Pan Z., Yue S., Yu F., Zhang J., Yang Y., Li R., Liu B., Yang X., Gao L., et al. Disease severity dictates SARS-CoV-2-specific neutralizing antibody responses in COVID-19. Signal Transduct. Target. Ther. 2020;5:180. doi: 10.1038/s41392-020-00301-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Zuiani A., Fischinger S., Mullur J., Atyeo C., Travers M., Lelis F.J.N., Pullen K.M., Martin H., Tong P., et al. Quick COVID-19 Healers Sustain Anti-SARS-CoV-2 Antibody Production. Cell. 2020;183:1496–1507.e16. doi: 10.1016/j.cell.2020.10.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chia W.N., Zhu F., Ong S.W.X., Young B.E., Fong S.-W., Le Bert N., Tan C.W., Tiu C., Zhang J., Tan S.Y., et al. Dynamics of SARS-CoV-2 neutralising antibody responses and duration of immunity: a longitudinal study. Lancet Microbe. 2021;2:e240–e249. doi: 10.1016/S2666-5247(21)00025-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dan J.M., Mateus J., Kato Y., Hastie K.M., Yu E.D., Faliti C.E., Grifoni A., Ramirez S.I., Haupt S., Frazier A., et al. Immunological memory to SARS-CoV-2 assessed for up to 8 months after infection. Science. 2021;371:eabf4063. doi: 10.1126/science.abf4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta S.D., Talwar A., Lee J.T. A Proposed Framework and Timeline of the Spectrum of Disease Due to SARS-CoV-2 Infection: Illness Beyond Acute Infection and Public Health Implications. JAMA. 2020;324:2251–2252. doi: 10.1001/jama.2020.22717. [DOI] [PubMed] [Google Scholar]
- de Melo G.D., Lazarini F., Levallois S., Hautefort C., Michel V., Larrous F., Verillaud B., Aparicio C., Wagner S., Gheusi G., et al. COVID-19-related anosmia is associated with viral persistence and inflammation in human olfactory epithelium and brain infection in hamsters. Sci. Transl. Med. 2021;13:eabf8396. doi: 10.1126/scitranslmed.abf8396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew D.A., Nguyen L.H., Steves C.J., Menni C., Freydin M., Varsavsky T., Sudre C.H., Cardoso M.J., Ourselin S., Wolf J., et al. Rapid implementation of mobile technology for real-time epidemiology of COVID-19. Science. 2020;368:1362–1367. doi: 10.1126/science.abc0473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaebler C., Wang Z., Lorenzi J.C.C., Muecksch F., Finkin S., Tokuyama M., Cho A., Jankovic M., Schaefer-Babajew D., Oliveira T.Y., et al. Evolution of antibody immunity to SARS-CoV-2. Nature. 2021;591:639–644. doi: 10.1038/s41586-021-03207-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grifoni A., Weiskopf D., Ramirez S.I., Mateus J., Dan J.M., Moderbacher C.R., Rawlings S.A., Sutherland A., Premkumar L., Jadi R.S., et al. Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans with COVID-19 Disease and Unexposed Individuals. Cell. 2020;181:1489–1501.e15. doi: 10.1016/j.cell.2020.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudbjartsson D.F., Norddahl G.L., Melsted P., Gunnarsdottir K., Holm H., Eythorsson E., Arnthorsson A.O., Helgason D., Bjarnadottir K., Ingvarsson R.F., et al. Humoral Immune Response to SARS-CoV-2 in Iceland. N. Engl. J. Med. 2020;383:1724–1734. doi: 10.1056/NEJMoa2026116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellmuth J., Barnett T.A., Asken B.M., Kelly J.D., Torres L., Stephens M.L., Greenhouse B., Martin J.N., Chow F.C., Deeks S.G., et al. Persistent COVID-19-associated neurocognitive symptoms in non-hospitalized patients. J. Neurovirol. 2021;27:191–195. doi: 10.1007/s13365-021-00954-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henrich T.J., Hatano H., Bacon O., Hogan L.E., Rutishauser R., Hill A., Kearney M.F., Anderson E.M., Buchbinder S.P., Cohen S.E., et al. HIV-1 persistence following extremely early initiation of antiretroviral therapy (ART) during acute HIV-1 infection: An observational study. PLoS Med. 2017;14:e1002417. doi: 10.1371/journal.pmed.1002417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C., Huang L., Wang Y., Li X., Ren L., Gu X., Kang L., Guo L., Liu M., Zhou X., et al. 6-month consequences of COVID-19 in patients discharged from hospital: a cohort study. Lancet. 2021;397:220–232. doi: 10.1016/S0140-6736(20)32656-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowitdamrong E., Puthanakit T., Jantarabenjakul W., Prompetchara E., Suchartlikitwong P., Putcharoen O., Hirankarn N. Antibody responses to SARS-CoV-2 in patients with differing severities of coronavirus disease 2019. PLoS ONE. 2020;15:e0240502. doi: 10.1371/journal.pone.0240502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Bert N., Clapham H.E., Tan A.T., Chia W.N., Tham C.Y.L., Lim J.M., Kunasegaran K., Tan L.W.L., Dutertre C.-A., Shankar N., et al. Highly functional virus-specific cellular immune response in asymptomatic SARS-CoV-2 infection. J. Exp. Med. 2021;218:e20202617. doi: 10.1084/jem.20202617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei Q., Li Y., Hou H.-Y., Wang F., Ouyang Z.-Q., Zhang Y., Lai D.-Y., Banga Ndzouboukou J.-L., Xu Z.-W., Zhang B., et al. Antibody dynamics to SARS-CoV-2 in asymptomatic COVID-19 infections. Allergy. 2020;76:551–561. doi: 10.1111/all.14622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long Q.-X., Tang X.-J., Shi Q.-L., Li Q., Deng H.-J., Yuan J., Hu J.-L., Xu W., Zhang Y., Lv F.-J., et al. Clinical and immunological assessment of asymptomatic SARS-CoV-2 infections. Nat. Med. 2020;26:1200–1204. doi: 10.1038/s41591-020-0965-6. [DOI] [PubMed] [Google Scholar]
- Morley D., Lambert J.S., Hogan L.E., De Gascun C., Redmond N., Rutishauser R.L., Thanh C., Gibson E.A., Hobbs K., Bakkour S., et al. Rapid development of HIV elite control in a patient with acute infection. BMC Infect. Dis. 2019;19:815. doi: 10.1186/s12879-019-4374-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naaber P., Hunt K., Pesukova J., Haljasmägi L., Rumm P., Peterson P., Hololejenko J., Eero I., Jõgi P., Toompere K., Sepp E. Evaluation of SARS-CoV-2 IgG antibody response in PCR positive patients: Comparison of nine tests in relation to clinical data. PLoS ONE. 2020;15:e0237548. doi: 10.1371/journal.pone.0237548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalbandian A., Sehgal K., Gupta A., Madhavan M.V., McGroder C., Stevens J.S., Cook J.R., Nordvig A.S., Shalev D., Sehrawat T.S., et al. Post-acute COVID-19 syndrome. Nat. Med. 2021;27:601–615. doi: 10.1038/s41591-021-01283-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peluso M.J., Kelly J.D., Lu S., Goldberg S.A., Davidson M.C., Mathur S., Durstenfeld M.S., Spinelli M.A., Hoh R., Tai V., et al. Rapid implementation of a cohort for the study of post-acute sequelae of SARS-CoV-2 infection/COVID-19. medRxiv. 2021 [Google Scholar]
- Peng Y., Mentzer A.J., Liu G., Yao X., Yin Z., Dong D., Dejnirattisai W., Rostron T., Supasa P., Liu C., et al. Broad and strong memory CD4+ and CD8+ T cells induced by SARS-CoV-2 in UK convalescent individuals following COVID-19. Nat. Immunol. 2020;21:1336–1345. doi: 10.1038/s41590-020-0782-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilarowski G., Lebel P., Sunshine S., Liu J., Crawford E., Marquez C., Rubio L., Chamie G., Martinez J., Peng J., et al. Performance Characteristics of a Rapid Severe Acute Respiratory Syndrome Coronavirus 2 Antigen Detection Assay at a Public Plaza Testing Site in San Francisco. J. Infect. Dis. 2021;223:1139–1144. doi: 10.1093/infdis/jiaa802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson S., Hirsch J.S., Narasimhan M., Crawford J.M., McGinn T., Davidson K.W., Barnaby D.P., Becker L.B., Chelico J.D., Cohen S.L., et al. Presenting Characteristics, Comorbidities, and Outcomes Among 5700 Patients Hospitalized With COVID-19 in the New York City Area. JAMA. 2020;323:2052–2059. doi: 10.1001/jama.2020.6775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rydyznski Moderbacher C., Ramirez S.I., Dan J.M., Grifoni A., Hastie K.M., Weiskopf D., Belanger S., Abbott R.K., Kim C., Choi J., et al. Antigen-Specific Adaptive Immunity to SARS-CoV-2 in Acute COVID-19 and Associations with Age and Disease Severity. Cell. 2020;183:996–1012.e19. doi: 10.1016/j.cell.2020.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakanashi D., Asai N., Nakamura A., Miyazaki N., Kawamoto Y., Ohno T., Yamada A., Koita I., Suematsu H., Hagihara M., et al. Comparative evaluation of nasopharyngeal swab and saliva specimens for the molecular detection of SARS-CoV-2 RNA in Japanese patients with COVID-19. J. Infect. Chemother. 2021;27:126–129. doi: 10.1016/j.jiac.2020.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scully E.P., Rutishauser R.L., Simoneau C.R., Delagrèverie H., Euler Z., Thanh C., Li J.Z., Hartig H., Bakkour S., Busch M., et al. Inconsistent HIV reservoir dynamics and immune responses following anti-PD-1 therapy in cancer patients with HIV infection. Ann. Oncol. 2018;29:2141–2142. doi: 10.1093/annonc/mdy259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekine T., Perez-Potti A., Rivera-Ballesteros O., Strålin K., Gorin J.-B., Olsson A., Llewellyn-Lacey S., Kamal H., Bogdanovic G., Muschiol S., et al. Robust T Cell Immunity in Convalescent Individuals with Asymptomatic or Mild COVID-19. Cell. 2020;183:158–168.e14. doi: 10.1016/j.cell.2020.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seow J., Graham C., Merrick B., Acors S., Pickering S., Steel K.J.A., Hemmings O., O’Byrne A., Kouphou N., Galao R.P., et al. Longitudinal observation and decline of neutralizing antibody responses in the three months following SARS-CoV-2 infection in humans. Nat. Microbiol. 2020;5:1598–1607. doi: 10.1038/s41564-020-00813-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan A.T., Linster M., Tan C.W., Le Bert N., Chia W.N., Kunasegaran K., Zhuang Y., Tham C.Y.L., Chia A., Smith G.J.D., et al. Early induction of functional SARS-CoV-2-specific T cells associates with rapid viral clearance and mild disease in COVID-19 patients. Cell Rep. 2021;34:108728. doi: 10.1016/j.celrep.2021.108728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenforde M.W., Kim S.S., Lindsell C.J., Billig Rose E., Shapiro N.I., Files D.C., Gibbs K.W., Erickson H.L., Steingrub J.S., Smithline H.A., et al. Symptom Duration and Risk Factors for Delayed Return to Usual Health Among Outpatients with COVID-19 in a Multistate Health Care Systems Network - United States, March-June 2020. MMWR Morb. Mortal. Wkly. Rep. 2020;69:993–998. doi: 10.15585/mmwr.mm6930e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L., Hall T., Ssewanyana I., Oulton T., Patterson C., Vasileva H., Singh S., Affara M., Mwesigwa J., Correa S., et al. Optimisation and standardisation of a multiplex immunoassay of diverse Plasmodium falciparum antigens to assess changes in malaria transmission using sero-epidemiology. Wellcome Open Res. 2020;4:26. doi: 10.12688/wellcomeopenres.14950.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao L., Wang G.-L., Shen Y., Wang Z.-Y., Zhan B.-D., Duan L.-J., Lu B., Shi C., Gao Y.-M., Peng H.-H., et al. Persistence of Antibody and Cellular Immune Responses in COVID-19 patients over Nine Months after Infection. J. Infect. Dis. 2021;2021:jiab255. doi: 10.1093/infdis/jiab255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J., Yuan Q., Wang H., Liu W., Liao X., Su Y., Wang X., Yuan J., Li T., Li J., et al. Antibody Responses to SARS-CoV-2 in Patients With Novel Coronavirus Disease 2019. Clin. Infect. Dis. 2020;71:2027–2034. doi: 10.1093/cid/ciaa344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R., To K.K.-W., Wong Y.-C., Liu L., Zhou B., Li X., Huang H., Mo Y., Luk T.-Y., Lau T.T.-K., et al. Acute SARS-CoV-2 Infection Impairs Dendritic Cell and T Cell Responses. Immunity. 2020;53:864–877.e5. doi: 10.1016/j.immuni.2020.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
De-identified data reported in this paper will be shared by the lead contact upon request.
-
•
All original code is available in this paper’s supplemental information.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.