

Abstract
The engagement of a T cell with an antigen-presenting cell (APC) or activating surface results in the formation within the T cell of several distinct actin and actomyosin networks. These networks reside largely within a narrow zone immediately under the T cell’s plasma membrane at its site of contact with the APC or activating surface, i.e., at the immunological synapse. Here we review the origin, organization, dynamics, and function of these synapse-associated actin and actomyosin networks. Importantly, recent insights into the nature of these actin-based cytoskeletal structures were made possible in several cases by advances in light microscopy.
Keywords: T cell, actin, myosin, immunological synapse, lytic granule, formin
INTRODUCTION
Upon contact with an antigen-presenting cell (APC), the portion of the T cell’s cortex in contact with the APC undergoes a dramatic increase in the assembly of several actin and actomyosin networks (1–10). The dynamics of these unique networks drive in cooperative fashion the centripetal transport and positioning of proteins within the T cell’s plasma membrane. The culmination of these events is the formation of a mature immunological synapse (IS) characterized by three distinct zones within the T cell: the central supramolecular activation cluster (cSMAC) and two surrounding radial symmetric rings, the peripheral SMAC (pSMAC) and the distal SMAC (dSMAC) (11, 12) (Figure 1a). This review focuses on the creation, organization, dynamics, and function of the actin and actomyosin networks that span these three zones. Toward that end, we begin with a review of the basic principles of actin assembly and myosin biochemistry. We then explain how T cells employ these basic principles to create distinct actin and actomyosin networks at the immune synapse. Finally, we describe how these distinct networks drive the maturation of the IS, the adhesion of the T cell to the APC, and one major T cell effector function—the polarized secretion of lytic granules (LGs) by cytotoxic T cells (CTLs).
Figure 1.
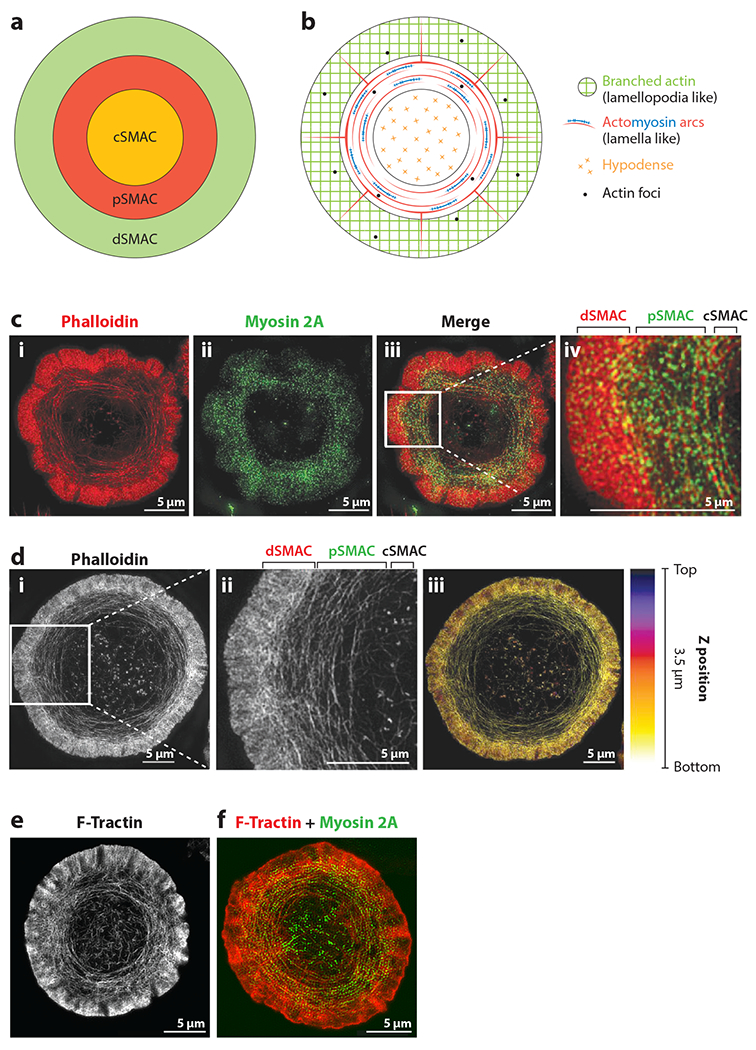
The four actin networks at the T cell IS. (a) The three distinct functional zones within the mature IS. (b) The spatial relationships between these three IS zones and the four actin networks at the IS. (c) A SIM image at the bottom of an activated Jurkat T cell that was stained for F-actin using phalloidin (i, red) and myosin 2A (ii, green). Subpanel iii shows the overlaid image, while the magnified inset in subpanel iv shows in greater detail the lamellipodia-like branched actin network spanning the dSMAC, the lamella-like actomyosin arc network spanning the pSMAC, and the actin hypodense network in the cSMAC. The green doublets that are concentrated in the pSMAC region in subpanel iv are individual myosin 2 bipolar filaments (reproduced from Reference 48). Note that the colors used in the drawings in panels a and b are not meant to match the colors in the micrographs in panels c, d, and e. (d) A 3D SIM image of an activated Jurkat T cell stained for F-actin using phalloidin (i). The inset in subpanel ii shows in greater detail the three networks noted in panel c. Color coding the 3D projection of this cell according to Z position (iii) shows that these actin networks are largely confined to the plane of the IS (reproduced from Reference 48). These networks can be considered, therefore, as 2D structures (which makes live TIRF-SIM possible) (see also 122). That said, the branched actin network in the dSMAC can sometimes lift off the surface, as shown by Yi et al. (47) and, more recently, Fritzsche et al. (50). (e) A still image from a TIRF-SIM movie of an activated Jurkat T cell expressing the indirect F-actin reporter F-Tractin tagged with GFP (adapted from Reference 48; see also Supplemental Video 1). (f) A still image from a two-color TIRF-SIM movie of an activated Jurkat T cell expressing Td-Tomato-tagged F-Tractin (red) and GFP-tagged myosin 2A (green) (adapted from Reference 48; see also Supplemental Video 2). Abbreviations: cSMAC, central SMAC; dSMAC, distal SMAC; GFP, green fluorescent protein; IS, immunological synapse; pSMAC, peripheral SMAC; SIM, structured illumination microscopy; SMAC, supramolecular activation cluster; TIRF, total internal reflection.
Our review is centered around four distinct actin networks that coexist at the IS: the lamellipodia-like branched actin network at the periphery of the IS (the dSMAC), the lamella-like actomyosin arc network in the medial portion of the IS (the pSMAC), the hypodense actin network at the center of the IS (the cSMAC), and actin foci in the dSMAC and pSMAC (Figure 1b). For each network, we describe what it looks like, where it comes from, and what it does. More specifically, we describe the signals that stimulate its formation, the nucleation-promoting factors (NPFs) and nucleation machines that create it, the specifics of its architecture and dynamics, the signature molecules it contains, and current understanding of its functions. Most of this information has come from imaging T cells on activating surfaces (13). While this approach places the IS in an imaging plane that is ideal for high-resolution and superresolution microscopy, it is not entirely physiological. We close, therefore, with a discussion of where the field must go to visualize these actin networks in truly physiological settings.
BASIC PRINCIPLES OF ACTIN ASSEMBLY
Although cells typically contain copious amounts of F-actin at steady state, these filaments do not arise spontaneously, as spontaneous filament nucleation and elongation are strongly suppressed in cells by two mechanisms (14–16). First, small actin seeds like dimers and trimers that form spontaneously are extremely unstable and very rapidly dissociate back to monomers rather than elongate into filaments. Second, most of the actin monomer in cells is bound to profilin. While the profilin:actin complex works perfectly as a substrate for actin filament elongation (with profilin being released immediately upon addition), it is completely incapable of undergoing spontaneous nucleation. What all this means is that actin filaments do not form on their own. Rather, they are made only when the cell decides to make them through the action of specific nucleating machines. This is the first major principle of actin filament assembly in cells.
Cells make actin filaments using two actin filament nucleation machines: the Arp2/3 complex and the formins (17–21). Amazingly, these two nucleation machines create networks of dramatically different architecture. The seven-member Arp2/3 complex creates branched actin networks by first binding to the side of an existing filament (the mother filament) and then nucleating a branch (the daughter filament) that grows off the side of the mother at a 70° angle (22). The rapid capping of these branched filaments by Capping Protein results in a dendritic actin network that exhibits sufficient tensile strength to push the cell edge forward. The quintessential Arp2/3-generated actin structure is the lamellipodium, a flat, sheet-like extension found at the leading edge of mesenchymal cells. Formins, on the other hand, create linear actin networks by nucleating single filaments and then elongating them while simultaneously holding on to their growing end (23, 24). Filament nucleation and elongation involve cooperation between the formin’s two FH2 domains, which rock back and forth on the growing filament end to allow in alternating fashion the addition of a profilin:actin complex to each strand of the actin filament. Assembly is accelerated by the formin’s FH1 domain, which feeds profilin:actin complexes to the FH2 domains. Common cellular structures whose formation involves formin-dependent nucleation/assembly include filopodia, diverse types of stress fibers that occupy the lamella of mesenchymal cells, and the contractile ring of dividing cells.
Importantly, the Arp2/3 complex and formins (at least Dia and FMNL1, the two major formins expressed in T cells) (25) are by themselves largely inactive. For Arp2/3, its ability to robustly nucleate a branch off the side of a mother filament requires its transient interaction with an NPF. The two major NPFs for Arp2/3 are WASp and WAVE (14, 18, 19). Even more importantly, WASp and WAVE are intrinsically inactive. In the case of WASp, it undergoes autoinhibitory intramolecular folding in the cytoplasm, which blocks its ability to activate the Arp2/3 complex (26, 27). Several signals at the plasma membrane, most notably Cdc42-GTP, the polyphosphoinositide PIP2, and the adaptor protein Nck, synergize to simultaneously recruit, unfold, and activate WASp, allowing it to then promote Arp2/3-dependent branched actin nucleation immediately under the membrane. In the case of WAVE, it too is held inactive in the cytoplasm, but in this case through an interaction with four other proteins that, together with WAVE, constitute the WAVE Regulatory Complex (WRC) (28, 29). Binding of the WRC to both Rac1-GTP and the polyphosphoinositide PIP3 in the plasma membrane alters the conformation of the WRC such that WAVE can now promote Arp2/3-dependent nucleation. Finally, the formins Dia and FMNL1 are also intrinsically inactive because they undergo intramolecular folding in the cytoplasm, which blocks their ability to nucleate and elongate actin filaments (20). Both formins are simultaneously unfolded and activated for linear filament assembly by binding to RhoA-GTP and PIP2 in the plasma membrane.
Several major principles of actin filament assembly are embedded in the regulatory and assembly mechanisms just described. First, actin filament nucleation and assembly occur almost exclusively at the interface between the plasma membrane and the cytoplasm (or between intracellular membranes and the cytoplasm). For branched networks, this involves the continued recruitment of fresh Arp2/3 molecules to the leading edge of the growing network, where they are then activated by a membrane-bound NPF to promote further branching. For linear networks, this means the same membrane-associated formin that initiated the assembly of a filament remains on the end of that filament, where it then drives the addition of monomers while simultaneously holding on to both the growing end and the membrane (this process is often referred to as insertional polymerization). Simply stated, all assembly action occurs in a very narrow zone just under the plasma membrane (or just outside of an organelle membrane). Second, a common set of “make-actin” signals, most notably active Rho family GTPases and polyphosphoinositides, drive actin assembly (specific adaptors, which are not covered here except for Nck for WASp and WRC for WAVE, also play important roles). Finally, by controlling the signals at membranes that drive the recruitment and activation of formins and the NPFs for Arp2/3, cells can exert almost total control over when and where actin filaments are made.
A final principle of actin assembly that is particularly relevant to recent studies in T cells is that the amount of actin monomer available at steady state for filament creation is limiting, such that the Arp2/3 complex and formins are competing for it. This central paradigm was demonstrated initially in budding yeast, where inhibition of Arp2/3 was shown to result in a very large increase in the amount of formin-dependent actin structures (and vice versa using formin inhibition) (30). Subsequent studies extended this observation to vertebrate cells, and showed that the greater effect is seen on formin-dependent structures when Arp2/3 is inhibited, as Arp2/3 is consuming the majority of monomer at steady state (31–33). A major implication of this concept is that for actin assembly to continue, fresh actin monomer must be continuously generated by actin filament disassembly. The major player driving actin filament disassembly is the F-actin-severing protein cofilin (34). It makes sense, therefore, that the signals that drive Arp2/3-dependent assembly downstream of WAVE (the major consumer of monomer in most cells, including T cells) also drive cofilin activation (35). Without cofilin-driven filament turnover, filament assembly would rapidly slow as Arp2/3 quickly becomes starved for monomer. Of note, this principle means that blocking actin filament disassembly with jasplakinolide also leads to the rapid diminution of assembly as the monomer pool rapidly shrinks.
BASIC PRINCIPLES OF MYOSIN 2 FUNCTION
Nonmuscle myosin 2 s (denoted here simply as myosin 2), of which there are three in vertebrates (myosin 2A, myosin 2B, and myosin 2C), are the major actin-based contractile machine in all nonmuscle cell types, including T cells (which express primarily myosin 2A) (36–38). Myosin 2s are similar structurally and functionally to muscle myosins and, like muscle myosins, perform their cellular duties in the form of bipolar filaments (albeit much smaller filaments containing only ~30 myosins, consistent with the tight working environment within nonmuscle cells). When the motor domains at each end of these ~300-nm-long myosin 2 bipolar filaments engage actin filaments of opposing orientation, they drive the sliding of these opposing actin filaments past each other, resulting in contraction (just as in the muscle sarcomere). Common cellular functions powered by myosin 2 bipolar-filament-dependent actin filament sliding are the closure of the contractile ring during cytokinesis and the generation of tension on focal adhesions by myosin 2–rich stress fibers. Notably, myosin 2 filaments drive motility primarily in cooperation with linear, formin-generated actin structures.
Cells regulate where and when myosin 2–based contractions occur by regulating where and when myosin 2 monomers are assembled into bipolar filaments through the action of multiple regulatory light chain (RLC) kinases (37, 38). RLC phosphorylation activates myosin 2s by converting them from folded monomers, which are mechanically silent and incapable of assembling into filaments, into extended monomers, which readily assemble into filaments and are capable of walking on F-actin. One major RLC kinase is the Rho-associated coiled-coil kinase (commonly referred to as ROCK), which is activated by Rho-GTP and appears to play a leading role in the assembly and maintenance of large-scale actomyosin structures in the lamella of mesenchymal cells. Of note, myosin 2s are very unlikely to act as vesicle motors. As “low-duty ratio” motors, myosin 2s act as ensembles to drive the contraction of actin networks, not as unitary, processive, cargo-binding, “high-duty ratio” motors like myosin V (39).
Myosin 2 function in cells can be blocked conditionally and rapidly using the cell-permeant, small-molecule inhibitor blebbistatin, which selectively inhibits all myosin 2 isoforms by stabilizing their motor domains in a weak actin-binding state, effectively dissociating them from actin (40). Of note, the original version of blebbistatin rapidly degrades when illuminated with blue light [e.g., when imaging green fluorescent protein (GFP)] (41). This causes it not only to lose activity but also to generate free radicals that are toxic to cells. A newer version, para-nitroblebbistatin, does not have this issue (42).
THE FOUR ACTIN NETWORKS AT THE T CELL IMMUNOLOGICAL SYNAPSE
Most of what we know about the formation, organization, and dynamics of actin and actomyosin networks at the IS has come from imaging T cells engaged with activating surfaces (coated glass, planar lipid bilayers), which position this cortical structure in the ideal imaging plane. Within minutes of contact with an activating surface, two major actin networks, one less conspicuous actin structure, and a region containing much less actin (albeit still functionally important) are evident at the maturing IS (Figures 1b, 2a). Below we review the source, organization, and dynamics of these four actin networks. In the following sections, we review the contributions they make to T cell function.
Figure 2.
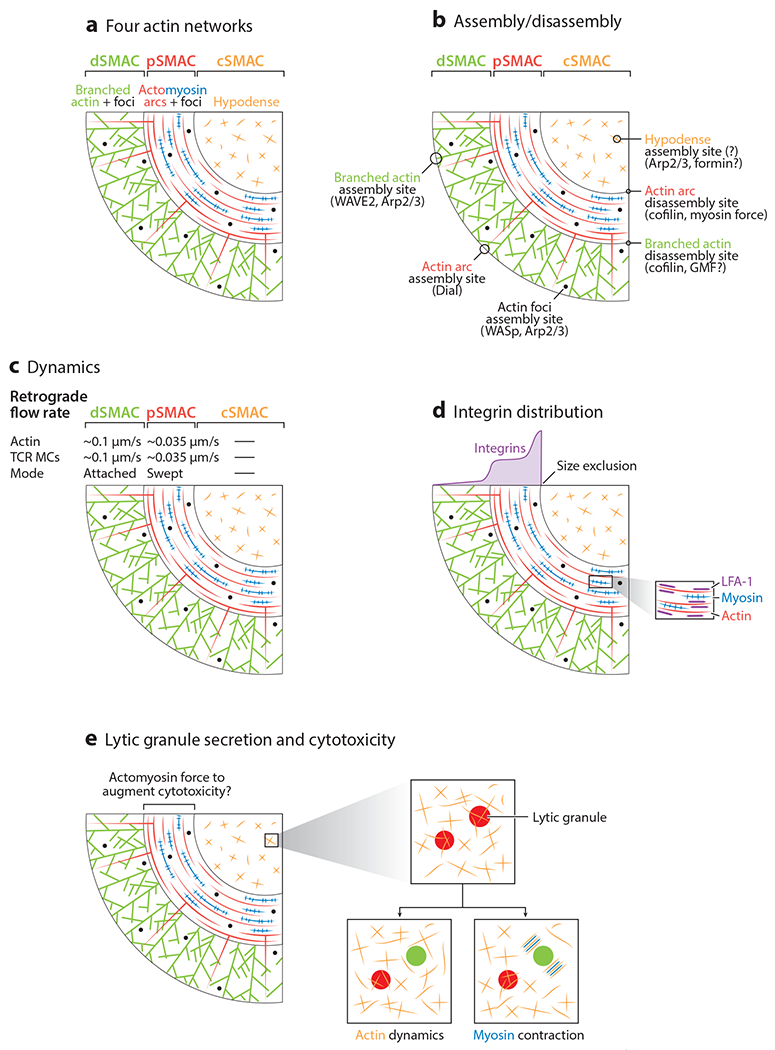
Key characteristics of the four actin networks. (a) An expanded view of the four actin networks at the IS. It should be noted that while the boundary between the branched actin network and actomyosin arc network, as well as the boundary between the actomyosin network and the actin hypodense center of the IS, can be both sharp and symmetric (see, for example Figure 1c–f), they can be more graded in some cells. (b) Assembly sites for the four actin networks, along with the nucleation-promoting factors and nucleation machines that drive their formation. Also shown are the two major sites of actin filament disassembly, along with key players in the disassembly process. Many other actin regulators play important roles in creating, organizing, and disassembling these actin networks. Some of these important proteins are HS1, IQGAP, ezrin/moesin, coronin 1A, L-plastin, CARMIL, and GMF (123–131). Due to space limitations, we could not address the roles of these proteins in depth. (c) Dynamics of the four actin networks at the IS. Note that while the actin in the cSMAC is static in terms of directional flow, it is dynamic on a nanoscale. (d) Distribution of integrins (purple) in the mature synapse, which accumulate across the actomyosin-arc-rich pSMAC and, most dramatically, at the pSMAC/cSMAC boundary (because the LFA-1:ICAM pairs are size-excluded from the cSMAC). The expanded view shows that the actin arcs (red) are decorated with open, active LFA-1 (purple) as well as with myosin 2 bipolar filaments (blue) (see 48). (e) Activity-dependent, nanoscale, actin filament dynamics and myosin 2 contractility within the cSMAC promote lytic granule secretion by increasing the size of pores in the fine actin meshwork that normally restricts granule access to the plasma membrane fusion machinery (the red and green circles indicate restricted and unrestricted granules, respectively; the view is from just the APC side of the T cell plasma membrane looking into the T cell). The actomyosin arc network in the pSMAC is the likely source of the T cell–based force that augments cytotoxicity by straining the target cell plasma membrane. Abbreviations: APC, antigen-presenting cell; cSMAC, central SMAC; dSMAC, distal SMAC; IS, immunological synapse; MC, microcluster; pSMAC, peripheral SMAC; SMAC, supramolecular activation cluster; TCR, T cell receptor.
THE BRANCHED ACTIN NETWORK IN THE dSMAC
Far and away the most obvious actin network at the IS is the bright actin ring that forms at the periphery of the IS (i.e., the dSMAC portion of the IS) shortly after contact with an activating surface (43–53) (Figures 1c–f,2a; see also Supplemental Video 1). This ring, which can be seen in both Jurkat T cells and primary T cells without very sophisticated microscopy (Figure 1c), is composed almost entirely of a branched actin network created by the Arp2/3 complex located all along the outer edge of the IS (Figure 2b). The assembly of this network, which is analogous to the lamellipodium of a crawling mesenchymal cell, serves initially to drive the spreading of the T cell across the activating surface. Once the cell is fully spread, continued assembly of this network drives a radially symmetric inward flow of F-actin commonly referred to as retrograde flow, which moves inward at ~0.1 μm/s (47,48, 52) (Figure 2c). This network occupies just the dSMAC portion of the IS, as it is largely disassembled at the dSMAC/pSMAC boundary, based on imaging (it disappears rather abruptly at this boundary; see, for example, Figure 1c–f, Supplemental Video 1) and on the rapid appearance of a bright actin ring at this boundary when actin filament disassembly is blocked using jasplakinolide (47) (Figure 2b). Of note, seminal work employing fluorescence speckle microscopy revealed a similarly abrupt disassembly of the lamellipodium in mesenchymal cells (54).
This branched network can be imaged dynamically using GFP-actin because the Arp2/3 complex efficiently incorporates modified actins into filaments. Importantly, the assembly of this network can be rapidly, potently, and conditionally blocked using the membrane-permeant, small-molecule inhibitor of the Arp2/3 complex, CK-666 (55). Finally, the core machinery driving the assembly of this branched actin network is as follows. First, activation of the lipid kinase PI3K downstream of T cell receptor (TCR) engagement leads to the conversion of PIP2 to PIP3 in the plasma membrane (56). PIP3 then recruits the Rac GEF DOCK2, leading to elevated levels of active Rac-GTP in the membrane (57, 58). These two “make-actin” signals then recruit and activate WRC complexes containing WAVE2 at the plasma membrane (29, 56, 59). Finally, active WAVE2 promotes branched nucleation by the Arp2/3 complex just under the plasma membrane at the outer edge of the IS to drive cell spreading and then retrograde actin flow (59, 60) (Figure 3a).
Figure 3.
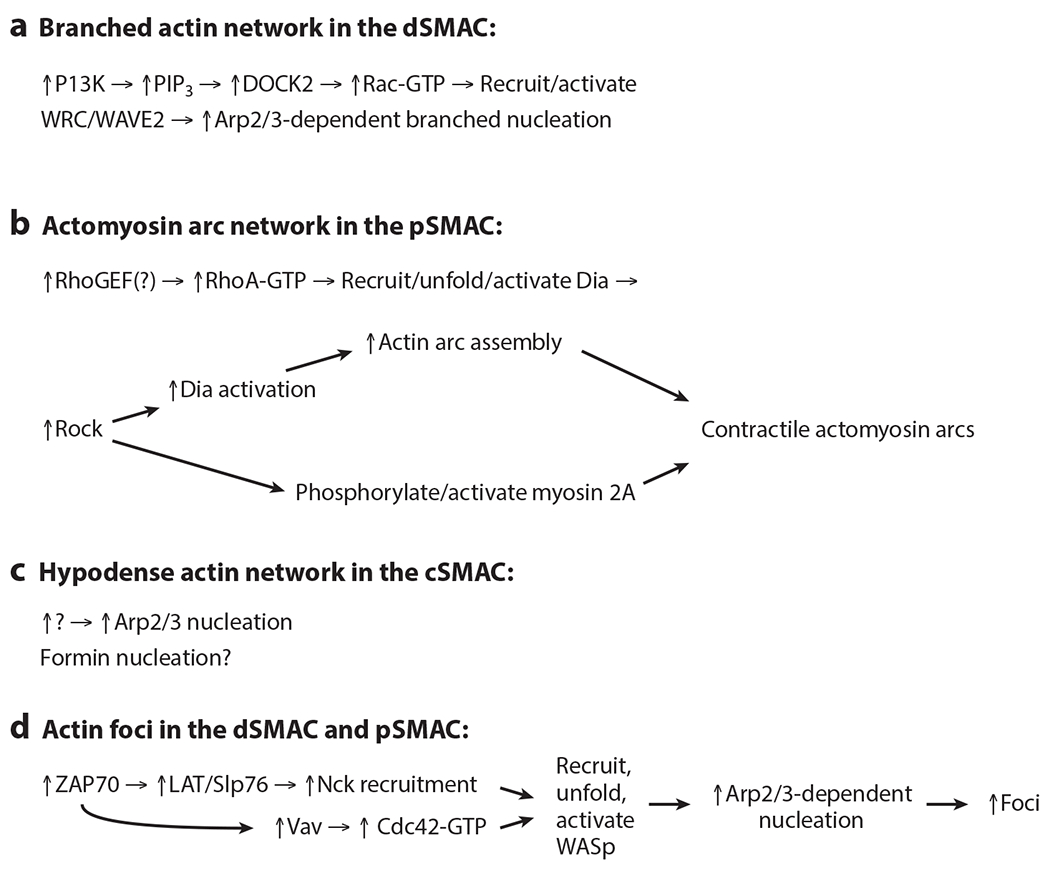
Origins of the four actin networks. “Make-actin” signals drive the formation of the four actin networks at the IS. While the generation of these signals is in every case downstream of the engagement of the TCR (and its coreceptors), we do not attempt to include TCR-proximal signaling pathways in this figure. Abbreviations: IS, immunological synapse; TCR, T cell receptor.
THE ACTOMYOSIN ARC NETWORK IN THE pSMAC
The second major actin network at the IS is the myosin 2–rich actin arc network that occupies the pSMAC portion of the IS (47–52) (Figures 1c–f,Figure 2a; see also Supplemental Videos 1, 2). The actin making up this network, which is analogous to actomyosin networks in the lamella of mesenchymal cells, is created by the formin Dia1 functioning at the plasma membrane at the IS outer edge (48) (Figure 2b). There Dia1 generates linear actin filaments that dive down through the branched actin network in the dSMAC and then bend upon exiting the dSMAC to give rise to concentric actin arcs in the pSMAC (48) (Figure 2b). These linear filaments have the topology of filaments generated by membrane-bound formin, which can either protrude outward to form filopodia or dive down into the cell, as is the case at the IS. Arc assembly is rapidly, potently, and conditionally inhibited using the pan-formin inhibitor SMIFH2 (61), as well as by knockdown of Dia1 (48). Importantly, the arcs are heavily decorated with bipolar filaments of myosin 2A (Figures 1c,f and Figure 2a; Supplemental Video 2) and so are thought to undergo a telescoping contraction as they move inward, much as occurs in the contractile ring of a dividing cell (48, 52, 62). The average inward speed of this radial symmetric, contracting actomyosin network is ~0.035 μm/s, or about one-third the rate of polymerization-driven retrograde actin flow in the dSMAC (47, 48) (Figure 2c). Myosin 2 is required to organize the arcs into concentric structures, as inhibition of myosin 2 using blebbistatin results in nonconcentric, highly disorganized arcs (48). Therefore, one can either block the formation of the arcs with SMIFH2 or disrupt their concentric organization with blebbistatin if one wishes to inhibit the function of this network. Finally, this actomyosin arc network occupies just the pSMAC portion of the IS, as it is largely disassembled at the pSMAC/cSMAC boundary based on imaging (it disappears rather abruptly at this boundary; see, for example, Figure 1c–f; Supplemental Videos 1, 2), and on the rapid appearance of a bright actin ring at this boundary when actin filament disassembly is blocked using jasplakinolide (47) (Figure 2b).
While actin arcs are visible by phalloidin staining in untransfected cells (Figure 1c,d) or in live cells using an indirect reporter for F-actin like F-Tractin (Figure 1e,f; Supplemental Videos 1, 2), they are not visible using GFP-actin (44–46) because, unlike the Arp2/3 complex, most if not all formins do not incorporate modified actins efficiently (63). Indeed, the fact that the arcs label very poorly with GFP-actin is a clear indication that they are formin-generated structures. Because most investigators have historically used GFP-actin to follow IS actin dynamics, the existence of the arcs was missed for many years (although strong myosin 2 staining at the pSMAC had been seen). That said, their existence in the pSMAC, where the T cell accumulates its integrin LFA-1 to drive adhesion to the APC (see below), should not be surprising given that the lamella of mesenchymal cells is dominated by linear, formin-generated, myosin 2-decorated actin structures that colocalize with integrin-based adhesions.
The pSMAC actin arc network is considerably more challenging to image than the branched network in the dSMAC, as it is much dimmer. Phalloidin-stained or F-Tractin-labeled arcs can be seen using high-resolution confocal imaging [e.g., spinning disc confocal microscopy with a high-numerical-aperture objective (47)] or, more recently, superresolution confocal microscopy using Zeiss Airyscan (unpublished observation). That said, their organization and dynamics are best imaged using a combination of total internal reflection (TIRF) illumination, which provides superior signal-to-noise ratio (and can be used because these structures are very close to the ventral plasma membrane), and structured illumination, which provides ~100-nm X/Y resolution, allowing one to image individual arcs (48) (Figure 1d–f; Supplemental Videos 1, 2).
Actin arc formation increases dramatically following the addition of the Arp2/3 inhibitor CK-666, most likely because of the increase in actin monomer made available to Dia (48). Moreover, the Dia-generated linear filaments embedded within the dSMAC that give rise to the arcs transform upon CK-666 addition into surface spikes as the surrounding branched network disappears (25, 48). Consistent with the fact that the arcs are eliminated by the knockdown of Dia, the tips of these CK-666-induced spikes stain strongly for endogenous Dia (48). Finally, both spike formation and the increase in arc content caused by CK-666 addition are blocked by the simultaneous addition of SMIFH2 (48).
Two self-organizing properties propel the formation of the radially symmetric contractile structure composing the pSMAC. First, the filaments used to create the arcs all have their barbed ends at the plasma membrane because they are formin generated (Figure 2b). Consequently, these filaments present their pointed ends upon entering the pSMAC, just as filaments do upon entering the bare zone of the sarcomere. Second, stochastic variations in the direction the linear filaments bend as they exit the dSMAC and enter the pSMAC (Figure 2a) should ensure that bipolar myosin 2 filaments are routinely engaged with actin filaments of opposing orientation, a prerequisite for the myosin 2–dependent contraction and inward movement of the arcs (48).
The recruitment of Dia1 to the plasma membrane is most likely driven by its binding to both RhoA-GTP and PIP2 (Figure 3b). Recruitment also serves to unfold and activate Dial by disrupting the interaction between the two domains that serve to stabilize its folded, inactive conformation (the DID and DAD domains) (20). The signals that drive the loading of RhoA with GTP in T cells have not been defined. Finally, the phosphorylation of Dial’s DAD domain by the kinase ROCK may also help to unfold and activate it (20) (Figure 3b). This would fit nicely with the fact that myosin 2, which is required to organize and contract the actin structures created by Dial, is strongly activated by the ROCK-dependent phosphorylation of its RLC and the ROCK-dependent inhibition of the RLC phosphatase PP2A (36–38) (Figure 3b).
Finally, while this actomyosin network was initially identified in Jurkat T cells (47, 48), it is also present in synapses formed on activating surfaces by primary T cells (49). Indeed, the arc network formed by primary T cells occupies an even greater fraction of total IS area than that formed by Jurkat T cells. Moreover, the localization of endogenous myosin 2 on the arcs of primary T cells often appears almost sarcomeric in nature (48).
THE HYPODENSE F-ACTIN NETWORK IN THE cSMAC
The relatively abrupt, large-scale disassembly of the actomyosin arcs at the inner aspect of the pSMAC yields a cSMAC with greatly diminished F-actin density (43–52) (Figures 1c–f Figure 2a; Supplemental Videos 1, 2). Indeed, the cSMAC is commonly referred to as actin-poor, actin hypodense, or even actin depleted based on typical confocal images of the cSMAC in mature synapses generated on activating surfaces. While the cSMAC is clearly actin poor compared to the major actin networks in the dSMAC and pSMAC, high-resolution confocal images often reveal small amounts of F-actin distributed across it (47, 52). More importantly, imaging this cSMAC actin in mature synapses made by natural killer (NK) and T cells on activating surfaces using several super-resolution imaging modalities [most notably stimulated emission depletion (STED) microscopy and TIRF–structured illumination microscopy (SIM)] has revealed a fine isotropic network of filaments that pervades the cSMAC and that comprises small actin foci embedded within both straight and branched filaments/fibers (64, 65). While this network is largely static in terms of directional flow (Figure 2c), it is dynamic on a nanoscale (66). Experiments using CK-666 indicate that a significant fraction of this fine actin network is created by the Arp2/3 complex, although the specific site of nucleation and the NPF involved remain unknown (66) (Figures 2b, 3c). As for the linear filaments, experiments using SMIFH2 to test a possible contribution from formins were inconclusive (66). That said, the substantial increase in linear filament content in the cSMAC following CK-666 addition seen in this study argues that at least one formin is contributing (66), although the specific formin involved, the signals that recruit/activate it, and its exact site of nucleation remain unknown (Figures 2b, 3c). One distinct possibility is that the linear actin structures in the normal cSMAC are simply remnants of the formin-generated actin arcs that span the pSMAC. Finally, simultaneous imaging of F-actin and LG secretion at the cSMAC is consistent with granule release triggering an increase in actin assembly at the cSMAC that is preceded by a rise in plasma membrane PIP2 content, and that serves to restrict further LG secretion and promote serial killing (67). The specific nucleator, NPF, and signal (other than PIP2) driving this actin assembly have not been identified.
ACTIN FOCI IN THE dSMAC AND pSMAC
The last of the four actin networks at the T cell IS are small actin foci found throughout the dSMAC and pSMAC in synapses made by primary T cells on activating surfaces (68) (Figure 2a). These structures, which represent a minor fraction of total synaptic F-actin, are created by the Arp2/3 complex in conjunction with the NPF WASp (Figure 2b). Consistently, they are greatly diminished in T cells that lack WASp (68). Moreover, they are the only IS actin structure that is diminished in T cells lacking WASp because the Arp2/3 complex uses WAVE2 rather than WASp to create the branched network in the dSMAC, and the arc network in the pSMAC is formin generated (Figures 2b, 3). In other words, the appearance and organization of actin and actomyosin at synapses formed by WASp null T cells are essentially normal (2, 4, 59, 69). Approximately 35% of TCR microclusters colocalize with actin foci, although what distinguishes these TCR microclusters from the remainder that lack foci is unclear (68). Foci present at the periphery of the IS move inward at the same rate as actin retrograde flow in the dSMAC (~0.1 μm/s) and disappear by the time they reach the pSMAC/cSMAC boundary (Figure 2a,c). Finally, many actin foci are not associated with TCR microclusters. This fact, together with their general appearance (i.e., actin dots), their dependence on WASp and Arp2/3 for formation, and the fact that their formation also requires HS1 (the hematopoietic version of cortactin, which serves to stabilize actin branches), makes them quite similar to podosomes (70). Staining with a podosome-specific marker like TKS4 would help resolve this ambiguity.
The pathway leading to foci formation is the pathway most often cited in reviews regarding actin assembly at the IS, in which proximal signaling downstream of TCR engagement leads to the recruitment of the adaptor protein Slp76 to LAT clusters at the plasma membrane (71, 72) (Figure 3d). Slp76 then recruits Vav, which serves as a guanine nucleotide exchange factor (GEF) for Cdc42, the specific Rho GTPase that recruits, unfolds, and activates WASp (73, 74). Finally, Slp76 also recruits the adaptor protein Nck, which also helps bring WASp to the membrane. Interestingly, Jurkat T cells apparently do not make actin foci (68). This might be because Jurkats lack the lipid phosphatase PTEN, which generates PIP2 from PIP3. PTEN loss should result, therefore, in reduced PIP2 levels in the plasma membrane, which in turn would impair the recruitment of WASp and the formation of foci. Perhaps the expression of PTEN in Jurkats, or the partial suppression of PI3K activity, might augment foci formation in Jurkats by shifting the balance between PIP2 and PIP3 in favor of PIP2. Of note, formin inhibition in Jurkats not only blocks arc formation but also results in the appearance of numerous actin foci at the IS, possibly downstream of increased Arp2/3-dependent nucleation (48). The precise relationship between these actin foci and those reported by Kumari et al. (68) has not been explored, however.
FUNCTION OF ACTIN AND ACTOMYOSIN STRUCTURES AT THE T CELL IMMUNOLOGICAL SYNAPSE
Actin and actomyosin structures at the IS must accomplish at least four major tasks: (a) promote the assembly of TCR microclusters to drive TCR signaling, (b) power the centripetal transport of TCR microclusters to drive IS maturation and microcluster fate, (c) promote the activation of integrins and their positioning to drive T cell–APC attachment, and (d) control/facilitate major T cell effector functions like polarized secretion. We next discuss the contributions made by the four actin networks described above to these four major tasks.
ASSEMBLY OF TCR MICROCLUSTERS AND SUBSEQUENT SIGNALING
TCR microclusters assemble largely within the branched actin network in the dSMAC but can further enlarge as they move inward across the actin arc network in the pSMAC (46–48, 52). Global disassembly of F-actin using latrunculin dramatically inhibits TCR microcluster formation and, therefore, TCR-dependent signaling and T cell activation (6–8). TCR microclusters signal robustly while transiting the dSMAC and pSMAC, but they stop signaling upon reaching the cSMAC because they become dissociated from key adaptor molecules (75). Consistently, slowing inward actin flow by increasing adhesion strength, which in primary T cells will retard inward actin flow, results in increased signaling because it lengthens the residence time of TCR microclusters in the dSMAC and pSMAC (76, although see 77). Conversely, halting the polymerization-dependent retrograde flow of the branched network in the dSMAC using the Arp2/3 inhibitor CK-666, an inhibitor of PI3K to block PIP3 synthesis, or via knockdown/knockout of DOCK2, WAVE2, or WRC components impairs many aspects of TCR signaling (47, 52, 56, 59, 60). These defects include the phosphorylation and activation of proximal signaling molecules like ZAP70 and LAT, as well as more downstream components like PLCƴ1 activation and subsequent calcium signaling. Similar signaling defects are seen after halting the myosin 2–dependent inward flow of the actomyosin arc network in the pSMAC using the myosin inhibitor blebbistatin or the formin inhibitor SMIFH2 (48, 62). Consistent with the defect in calcium signaling exhibited by WASp null T cells (6), the WASp-dependent actin foci that associate with TCR microclusters, and that are the only obvious WASp-dependent actin structure at the IS, also appear to contribute to PLCƴ1 recruitment, activation, and subsequent calcium flux (68).
CENTRIPETAL TRANSPORT OF TCR MICROCLUSTERS
Over a period of ~10 min following attachment to an activating surface, TCR microclusters form within the outer dSMAC ring and are transported inward to accumulate at the cSMAC. Importantly, their centripetal transport can be visualized in an ideal imaging plane using planar lipid bilayers (PLBs) containing freely diffusing, labeled molecules to engage and activate the TCR [anti-CD3 in the case of Jurkats, peptide–major histocompatibility complex (pMHC) in the case of primary T cells harboring a clonal TCR]. Moreover, the organization and dynamics of the four IS actin networks described above can be visualized simultaneously using a dynamic, indirect reporter for F-actin-like F-Tractin. Studies employing these approaches have provided very strong evidence that the centripetal transport of TCR microclusters is driven largely if not entirely by the inward flow of the two major actin networks composing the dSMAC and pSMAC (47, 48, 52; although see 78). First, the rates of centripetal TCR microcluster transport across the IS measured using PLBs, ~0.1 μm/s across the dSMAC and ~0.035 μm/s across the pSMAC, match exactly the rates of retrograde actin flow in the dSMAC and inward actomyosin arc flow in the pSMAC, respectively (47,48, 52) (Figure 2c). Second, blocking the assembly of the branched actin network in the dSMAC using cytochalasin D (a blocker of actin polymerization), or blocking the assembly/organization of the actomyosin arc network in the pSMAC using SMIFH2 or blebbistatin, inhibits TCR microcluster transport across their respective regions (47, 48, 52, 62). Finally, blocking both networks simultaneously completely halts both the inward flow of actin and the inward movement of TCR microclusters (47). In summary, these studies argue strongly that the inward flow of the branched actin network in the dSMAC, which is driven by continuous Arp2/3-dependent polymerization at the outer edge of the IS, followed by the inward flow of the actin arcs in the pSMAC, which is driven by their myosin 2–dependent contraction, together drive the centripetal transport of TCR microclusters to the cSMAC. Stated more simply, the cSMAC is at the end of an actin- and actomyosin-driven conveyor belt that propels inward TCR microcluster transport. Importantly, the actin monomer required to continuously feed this escalator is generated constantly by the ongoing disassembly of the branched and arc networks at the inner aspects of the dSMAC and pSMAC, respectively (Figure 2b).
How exactly are TCR microclusters moved inward by these actin and actomyosin structures? Two recent papers have provided insight into this central question. In the first paper, Murugesan et al. (48) showed using 3D SIM imaging of fixed cells that microclusters typically reside in between the actomyosin arcs that populate the pSMAC. This distribution is consistent with the idea that these contractile arcs drive inward TCR microcluster movement by sweeping the microclusters inward (Figure 2c). Importantly, high-speed, live-cell, TIRF-SIM imaging of actin arcs and TCR microclusters on PLBs revealed exactly this sweeping phenomenon (48). This and other data obtained in Jurkat and primary T cells (49) argue that these telescoping actomyosin arcs drive inward TCR microcluster transport across the pSMAC via the frictional coupling mechanism proposed by Jay Groves and colleagues, where the collision of moving cortical actin structures with microclusters simply pushes them inward, like a broom pushing dirt (79–81). While this mechanism does not completely exclude direct physical interaction between the arcs and TCR microclusters, such interactions, if they occur, would likely be very transient.
Regarding TCR microcluster transport across the dSMAC portion of the IS, recent work from the Rosen lab combining in vitro reconstitution with live-cell imaging has provided evidence that TCR microclusters within the branched dSMAC physically associate with this network as it flows inward by means of basic regions in WASp and its adaptor Nck that bind to F-actin (82) (Figure 2c). Importantly, as TCR microclusters exit the dSMAC, they lose Nck (and presumably WASp), and therefore their direct connection to F-actin. At this point, the microclusters are picked up and moved inward across the pSMAC by the sweeping action of the actomyosin arcs described by Murugesan et al. (48) (Figure 2c). In support of this model, overexpression in Jurkats of a chimeric adaptor that prevents the normal uncoupling of TCR microclusters from F-actin at the dSMAC/pSMAC boundary caused microclusters to now undergo a more circumferential inward path in the pSMAC, presumably because they are now bound to the telescoping actomyosin arcs (82). Together, these data argue that WASp and Nck act as a clutch to connect TCR microclusters to actin and that changes in TCR microcluster composition at the dSMAC/pSMAC boundary cause the mode by which microclusters are transported inward to also change, from an actin-attached mode in the dSMAC to an actin-detached, sweeping mode in the pSMAC (Figure 2c). These results (82), together with those of Murugesan et al. (48), also explain how these two actin networks manage to move TCR microclusters in a relatively straight path to the cSMAC despite differing in the way they engage the microclusters, and in their overall orientation relative to the cell edge (primarily perpendicular for the dSMAC network and parallel for the arc pSMAC network) (Figure 2a). Implicit in this model is the idea that changes in actin architecture and organization may exert control over TCR microcluster composition, and that altering the position of the dSMAC/cSMAC boundary should alter the timing of such compositional changes. Also implicit in this model, given that it is WASp centric, is that the TCR microcluster–associated, WASp-dependent actin foci described by Kumari et al. (68) may play a role in coupling TCR microclusters to the branched actin network in the dSMAC. Consistent with this idea, recent work has implicated a complex of DOCK8, WIP, and WASp in forming actin foci and linking TCRs to the actin cytoskeleton (83).
ACTIVATING AND POSITIONING INTEGRINS
Attachment of the T cell to an APC is driven largely by the binding of the T cell’s major integrin LFA-1 to the integrin ICAM-1 on the surface of the APC (8, 84, 85). In mature synapses, LFA-1 is found primarily in the actomyosin-rich pSMAC, with the highest density being at the pSMAC/cSMAC boundary (47) (Figure 2d). This organization creates a circular adhesive gasket around the cSMAC in T cell:APC conjugates. The creation of this gasket, when coupled with the repositioning of the T cell’s centrosome to the plasma membrane at the cSMAC, which positions the T cell’s secretory machinery in the direction of the target cell, serves to focus and limit the T cell’s effector functions to the bound APC (86, 87).
As with TCR microclusters, the inward transport of LFA-1 clusters at the IS can be followed using PLBs (in this case containing labeled ICAM-1) (46, 47, 52). While the inward transport of integrin clusters in these images is obvious, how their rate compares with the rates of inward actin flow in the dSMAC and pSMAC is difficult to ascertain owing to difficulties in tracking individual integrin clusters. Nonetheless, the inward movement of integrin clusters clearly depends on the inward flow of the branched actin and the actomyosin arcs (46–48, 52). LFA-1:ICAM1 pairs are excluded from the cSMAC because of the size of their extracellular domains, which are larger than the TCR:pMHC extracellular domains congregated at the cSMAC (Figure 2d).
The tight association of LFA-1 with ICAM-1 requires that LFA-1 assume its high-affinity, extended-open conformation (84, 85). The conversion of LFA-1 from its low-affinity, bent-closed conformation to its high-affinity, extended-open conformation involves several steps. First, TCR activation leads, in a process known as inside-out signaling, to the recruitment of the Rap1-RIAM complex to the cytoplasmic tail of LFA-1 (88, 89). There, the complex promotes the recruitment, unfolding, and activation of the actin-associated, LFA-1-binding protein talin, which serves to coax LFA-1 from its bent-closed conformation to its second low-affinity state, the extended-closed conformation. Finally, a force perpendicular to the plasma membrane is applied to the cytoplasmic tail of LFA-1’s β1 subunit to convert LFA-1 from its low-affinity, extended-closed conformation into its high-affinity, extended-open conformation (84, 85, 90–92). This force is typically provided by a cortical flow of actin under the plasma membrane that is connected to LFA-1’s β1 tail through talin. For T cells, this actin flow arises downstream of TCR ligation in the form of the two major actin networks composing the dSMAC and pSMAC.
In mesenchymal cells, myosin-dependent pulling forces on cortical actin also contribute significantly to integrin activation, as well as to the maturation and maintenance of extracellular-matrix-bound focal adhesions (93, 94). Indeed, myosin inhibition in these cells leads to a rapid disassembly of their focal adhesions. By contrast, a recent study in T cells using an antibody that specifically recognizes LFA-1’s extended-open conformation, together with inhibitors of either actin flow or myosin function, argued that actin retrograde flow is almost entirely responsible for LFA-1 activation, i.e., that myosin contractility makes only a minor contribution (85). That said, the stability of T cell:APC conjugates is greatly reduced by myosin inhibition, as well as by blocking the formation of actomyosin arcs in the pSMAC using SMIFH2 (48, 62). Moreover, these actomyosin arcs are highly decorated with open-extended LFA-1 (48, 49) (Figure 2d). These results, together with the fact that LFA-1:ICAM1 pairs accumulate in the actomyosin-arc-rich pSMAC and the fact that the pSMAC associates much more tightly with activating surfaces than the dSMAC (47, 50), argue that myosin contractility must also contribute to LFA-1-dependent adhesion in T cells.
One conceivable way to reconcile this apparent discrepancy can be found in the literature on integrin activation and focal adhesion maturation in mesenchymal cells (93, 94). Focal adhesions typically begin as small structures known as focal contacts that form just inside the leading edge. These focal contacts then mature into larger focal adhesions as the cell advances. Importantly, the force associated with actin retrograde flow alone appears sufficient to activate enough integrins to form and maintain focal contacts (95). The conversion of these nascent adhesions into mature focal adhesions, on the other hand, requires forces generated by the myosin 2–based contraction of actin structures that are attached to focal adhesions, such as ventral stress fibers. By analogy, actin flow in the dSMAC may be sufficient to activate a large fraction of LFA-1, but the force created by the myosin 2–dependent contraction of the actomyosin arcs in the pSMAC may be required for the maturation and maintenance of the adhesive structures connecting the T cell to the APC (Figure 2d). Central to this idea is the fact that several key proteins in adhesions (e.g., talin and vinculin) are mechanosensitive and require large forces to be fully active (96, 97). This activation typically involves the strain-dependent unfolding of the protein, leading to the exposure of cryptic sites for protein-protein interactions that drive adhesion maturation. In mesenchymal cells these activation steps (e.g., the opening of talin to allow vinculin binding) are very strongly promoted by myosin 2–dependent force generation. Something similar may be going on in the pSMAC. That said, adhesion maturation in T cells, at least as typically defined in mesenchymal cells (usually an increase in the size of the adhesion and its content of clutch components and regulatory molecules), has not been demonstrated (although see 77). This may have a lot to do with imaging on PLBs, because they offer very little resistance to the inward movement of LFA-1:ICAM1 pairs (except perhaps at the pSMAC/cSMAC boundary—see below). This is important because the activating force exerted on LFA-1’s β1 cytoplasmic tail by cortical actin flow within the T cell will increase significantly if the ICAM-1 to which LFA-1 is bound is not free to move. This has been shown directly in T cells by measuring their content of extended-open LFA-1 when bound to APCs in which the mobility of ICAM-1 in the APC’s membrane was reduced (98). It may well be that under these conditions, adhesion maturation within the T cell occurs, and that the forces exerted on these adhesions by the actomyosin arcs in the pSMAC help drive this maturation.
Finally, integrin pairs may be excluded from the cSMAC not only because they are prevented from entering it by the size of their extracellular domains (99) but also because the actomyosin arcs pulling on LFA-1 to sustain inside-out signaling disassemble at the pSMAC/cSMAC boundary (Figure 2d). This should result in the loss of myosin-based tension on LFA-1 at this boundary, leading to LFA-1:ICAM-1 uncoupling (like what occurs at focal adhesions in mesenchymal cells following blebbistatin addition). On the flip side of this equation, an increase in myosin-based contractile tension within the arcs that probably occurs when their inward movement is thwarted by their attachment to integrin-based adhesion complexes that cannot proceed into the cSMAC may promote rapid arc disassembly at this boundary by cofilin (Figure 2b), as cofilin-dependent severing is thought to be potentiated by strain placed on actin by myosin contractility (100).
CONTROLLING AND FACILITATING LYTIC GRANULE SECRETION
Secretion of LGs at the cSMAC portion of the IS by CTLs usually occurs after the establishment in the pSMAC of an adhesive gasket around the cSMAC, and after the repositioning of the CTL’s centrosome and attached interphase microtubule array to the plasma membrane at the cSMAC. This latter event serves to point the T cell’s microtubule-based secretory machinery in the direction of the bound target cell, while both events help limit killing to the target cell (86, 87). As discussed above, imaging T cells and their close brethren NK cells on activating surfaces using two superresolution imaging modalities (STED and SIM) has revealed a fine isotropic actin meshwork that pervades the mature cSMAC (64, 65) (Figures 1, Figure 2a). Moreover, this meshwork exhibits an average pore size about half the diameter of a LG, consistent with it serving as a barrier to LG secretion (64–66). Importantly, T cell/NK cell activation leads to the appearance of larger pores in the meshwork that would allow a LG to pass. Moreover, the point in time following activation that these larger pores appear is the same point in time that LG secretion increases (~5 min for T cells, ~30 min for NK cells) (64–66). Equally important, studies using jasplakinolide to freeze actin dynamics, and blebbistatin to inhibit myosin 2, argue that nanoscale-level actin filament dynamics and myosin 2–based contractions within this fine actin meshwork drive the activation-dependent increases in meshwork pore size that lead to LG secretion (66) (Figure 2e).
It is useful to consider these results in the broader context of cortical F-actin and secretion. Traditionally, cortical actin has been thought of as a barrier to vesicle secretion, such that cells must clear it first before vesicles can dock and fuse (101). Indeed, the thick mat of cortical F-actin that pervades the initial site of contact between the T cell and the APC seen recently using lattice light sheet (LLS) microscopy would almost certainly block secretion at the immature synapse (102). Moreover, those same images show that this thick mat of actin largely disappears as the synapse matures. What probably remains at the center of the mature synapse, however, is the fine actin meshwork seen by superresolution imaging of T cells on activating surfaces (64, 65). Nanoscale-level actin filament/fiber rearrangements within this fine actin meshwork caused by actin dynamics and myosin contractility would then serve to regulate LG secretion (66). Taken together, these observations call for a more nuanced appreciation for the role of F-actin at the lytic synapse, just like what has evolved in the field of neuroendocrine cell secretion (103). Of note, it seems likely that this hypodense cSMAC actin network serves purposes beyond just regulating secretion, such as supporting the internalization (104) and/or ESCRT-dependent shedding of TCR microclusters (105, 106).
Finally, exciting recent work from a collaboration between the Huse and Kam labs has provided evidence that actomyosin-based forces exerted by the T cell on the target cell increase the efficiency of target cell killing by increasing the efficiency with which perforin forms pores in the target cell membrane (107). Several key observations led to this discovery. First, DOCK-2 knockdown T cells were shown to form tiny synapses (consistent with a block in the DOCK-2-dependent formation of the branched actin network composing the dSMAC), to generate reduced force (measured using a micropipette pulling assay and micropillar deflection assay), and to kill poorly. Conversely, PTEN knockdown T cells were shown to make larger synapses (consistent with an enhancement of DOCK-2-dependent branched actin network assembly downstream of elevated PIP3 levels), to generate more force, and to kill much more effectively. Most importantly, DOCK-2 and PTEN knockdown cells (as well as myosin 2 knockdown T cells, which phenocopied DOCK-2 knockdown cells) exhibited no defects in calcium flux, centrosome repositioning, or LG release (as measured by the expression of LAMP1 on their surface). This result argued that the difference in cytotoxic function between these cells cannot be explained by a difference in the release of lytic components, and it raised the possibility that force exerted by the T cell on the target cell might promote in some way the ability of lytic components to kill the target. Much of the remainder of the paper provided evidence that increasing strain within the target cell membrane increases the speed and magnitude of perforin pore formation (shown using both purified perforin and perforin delivered by the T cell). This would of course augment the entry of granzymes into the target cell, thereby potentiating target cell killing. Together, these data argue that forces created by the T cell’s actomyosin cytoskeleton at the IS help drive cytotoxicity by increasing tension within the target cell’s plasma membrane, thereby promoting perforin pore formation (107).
A last point worth considering given the focus of this review is where exactly in the T cell synapse these forces are generated. Relevant to this question, Basu et al. (107) imaged T cells on micropillars (to mark sites of force generation) expressing pHluorin-LAMP1 (to mark sites of exocytosis) to provide spatial correlation between force generation and LG release. Importantly, secretion typically appeared at hotspots of micropillar defection/force generation, and quantitation supported this correlation. They also quantified where within the synapse these coupled micropillar defection events and degranulation events were most likely to occur. These events were seen most often within an annular region about halfway between the center of the IS and the outer edge. This is roughly where, in principle, the myosin-rich actin arc network composing the pSMAC should be. That would make a lot of sense given that this network is almost certainly the major contractile structure in T cells, as well as the major site of adhesion to the target cell (which is required for transcellular force transmission) (Figure 2e). Moreover, the actin-hypodense cSMAC is unlikely to generate much force. Despite making good sense (108,109), drawing these conclusions should be done with caution because the organization of actin and myosin in micropillar-engaged T cells, where the activating molecules are presented as point sources and are not mobile, probably does not exactly mirror their organization on PLBs (i.e., radial symmetric in organization and inward flow). What is needed, therefore, is a way to correlate in space and time force generation and LG exocytosis using an activating surface that allows normal actomyosin organization and dynamics. Perhaps this could be accomplished by modifying traction force microscopy to include imaging LG release (110, 111).
IMAGING ACTIN AND MYOSIN IN T CELL:ANTIGEN-PRESENTING CELL CONJUGATES
Much of what is stated above about actin and myosin at the T cell IS was learned by imaging T cells using activating surfaces. The strength of this approach comes from the fact that essentially all imaging modalities, including superresolution imaging modalities like SIM, provide much greater resolution in the XY plane (the plane of the IS on an activating surface) than in the Z plane (the plane of the IS in a typical T cell:APC conjugate). But even PLBs (which are much more physiological than glass surfaces coated with activating molecules, because the molecules can move and reorganize when presented in the bilayer) are far from physiological. For example, because PLBs provide little resistance to the movement of activating molecules, efforts to study phenomena like TCR mechanotransduction and adhesion maturation are somewhat problematic (112–114). That said, the information gained from this ideal imaging plane should be extrapolatable to the more physiological context of T cell:APC conjugates in vitro (and to the most physiological context of conjugates in living tissue), just as the information gained from studying focal adhesions in 2D (where they are large and relatively stable) has turned out to be extrapolatable to focal adhesions in more physiological 3D environments (where they are much smaller and more dynamic) (115).
What is clearly needed to push the field forward is the ability to visualize the actin and actomyosin structures described above, along with the dynamics of TCR microclusters and integrin clusters, in live T cell:APC conjugates at the superresolution level. One way to approach this is to improve the imaging of conjugates in which the IS is aligned in the Z plane (the typical orientation for conjugates). The imaging modality best suited for this is LLS (116) in SIM mode, as it provides nearly isotropic resolution (~ 150 nm in X, ~ 180 nm in Y, and ~250 in Z). Running LLS in SIM mode is important, because running it in dither mode provides only diffraction-limited images, which most likely cannot resolve fine structures in conjugates like the actomyosin arcs (102). LLS in SIM mode is relatively slow, however, so it might not allow live visualization of IS formation, as does dither mode. One way to improve this situation might be to place the T cell on top of a very flat APC to create a conjugate in which the IS is in the XY plane, much as has been done recently to image B cell–APC interactions (117). This approach would take advantage of the slightly higher resolution LLS-SIM provides in XY than in Z, and it would also be amenable to other high-resolution imaging modalities like instant SIM (ISIM) and Airyscan.
The best live-cell images of F-actin within T cells engaged with an APC to date were obtained using LLS in diffraction-limited dither mode (102). While these images showed with remarkable clarity the dynamics of the branched actin network in the dSMAC, including its contribution to a flow of F-actin up the sides of the APC-bound T cell (analogous to rearward-flowing dorsal ruffles seen in mesenchymal cells), none of the other three actin networks described above were visible. These latter networks, all of which are much fainter actin structures than the dSMAC, would presumably become visible using one or more of the cell set-up and superresolution imaging modalities described above.
Challenges other than those associated with imaging also abound. Many of these arise from the fact that the IS is unlikely to remain flat in conjugates. Instead, it will undulate, possess interdigitations, and exhibit much less radial symmetry (118–120). As a result, it is unlikely that the organization of actin and myosin at synapses formed in conjugates will be as well defined and symmetric as that of actin and myosin in synapses formed on activating surfaces. Nevertheless, the hope is that the organization and function of actin and actomyosin gleaned from studying T cells on activating surfaces will be extrapolatable to conjugates, albeit after considerable averaging and analysis of conjugate images. Finally, there are challenges associated with the fact that T cells often function in ways not in exact agreement with common themes permeating the literature. For example, T cells can kill without forming well-defined synapses and without centrosome repositioning (121). How actin and myosin participate in these noncanonical synapses and secretory events will also be important to address.
Supplementary Material
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Cannon JL, Burkhardt JK. 2002. The regulation of actin remodeling during T-cell-APC conjugate formation. Immunol. Rev 186(1):90–99 [DOI] [PubMed] [Google Scholar]
- 2.Huang Y, Burkhardt JK. 2007. T-cell-receptor-dependent actin regulatory mechanisms. J. Cell Sci 120(5):723–30 [DOI] [PubMed] [Google Scholar]
- 3.Billadeau DD, Nolz JC, Gomez TS. 2007. Regulation of T-cell activation by the cytoskeleton. Nat. Rev. Immunol 7(2):131–43 [DOI] [PubMed] [Google Scholar]
- 4.Burkhardt JK, Carrizosa E, Shaffer MH. 2008. The actin cytoskeleton in T cell activation. Annu. Rev. Immunol 26(1):233–59 [DOI] [PubMed] [Google Scholar]
- 5.Hammer JA, Burkhardt JK. 2013. Controversy and consensus regarding myosin II function at the immunological synapse. Curr. Opin. Immunol 25(3):300–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burkhardt JK. 2013. Cytoskeletal function in the immune system. Immunol. Rev 256(1):5–9 [DOI] [PubMed] [Google Scholar]
- 7.Kumari S, Curado S,Mayya V, Dustin ML. 2014.T cell antigen receptor activation and actin cytoskeleton remodeling. Biochim. Biophys. Acta Biomembr 1838(2):546–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Le Floc’h A, Huse M. 2015. Molecular mechanisms and functional implications of polarized actin remodeling at the T cell immunological synapse. Cell. Mol. Life Sci 72(3):537–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jankowska KI, Burkhardt JK. 2017. Analyzing actin dynamics at the immunological synapse. Methods Mol. Biol 1584:7–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ritter AT, Angus KL, Griffiths GM. 2013. The role of the cytoskeleton at the immunological synapse. Immunol. Rev 256(1):107–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dustin ML, Baldari CT. 2017. The immune synapse: past, present, and future. Methods Mol. Biol 1584:1–5 [DOI] [PubMed] [Google Scholar]
- 12.Xie J, Tato CM, Davis MM. 2013. How the immune system talks to itself: the varied role of synapses. Immunol. Rev 251(1):65–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dustin ML, Starr T, Varma R, Thomas VK. 2007. Supported planar bilayers for study of the immunological synapse. Curr. Protoc. Immunol 76(1):18.13.1–35 [DOI] [PubMed] [Google Scholar]
- 14.Welch MD, Mullins RD. 2002. Cellular control of actin nucleation. Annu. Rev. Cell Dev. Biol 18(1):247– 88 [DOI] [PubMed] [Google Scholar]
- 15.Pollard TD, Borisy GG. 2003. Cellular motility driven by assembly and disassembly of actin filaments. Cell 112(4):453–65 [DOI] [PubMed] [Google Scholar]
- 16.Pollard TD. 2016. What we know and do not know about actin. In The Actin Cytoskeleton: Handbook of Experimental Pharmacology, ed. Jockusch BM, pp. 331–47. Cham, Switz.: Springer; [DOI] [PubMed] [Google Scholar]
- 17.Kovar DR. 2006. Molecular details of formin-mediated actin assembly. Curr. Opin. Cell Biol 18(1):11–17 [DOI] [PubMed] [Google Scholar]
- 18.Chhabra ES, Higgs HN. 2007. The many faces of actin: matching assembly factors with cellular structures. Nat. Cell Biol 9(10):1110–21 [DOI] [PubMed] [Google Scholar]
- 19.Pollard TD. 2007. Regulation of actin filament assembly by Arp2/3 complex and formins. Annu. Rev. Biophys. Biomol. Struct 36(1):451–77 [DOI] [PubMed] [Google Scholar]
- 20.Kühn S, Geyer M. 2014. Formins as effector proteins of Rho GTPases. Small GTPases 5(3):e983876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chesarone MA, Goode BL. 2009. Actin nucleation and elongation factors: mechanisms and interplay. Curr. Opin. Cell Biol 21(1):28–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Swaney KF, Li R. 2016. Function and regulation of the Arp2/3 complex during cell migration in diverse environments. Curr. Opin. Cell Biol 42:63–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goode BL, Eck MJ. 2007. Mechanism and function of formins in the control of actin assembly. Annu. Rev. Biochem 76:593–627 [DOI] [PubMed] [Google Scholar]
- 24.Breitsprecher D, Goode BL. 2013. Formins at a glance. J. Cell Sci 126(1):1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gomez TS, Kumar K, Medeiros RB, Shimizu Y, Leibson PJ, Billadeau DD. 2007. Formins regulate the actin-related protein 2/3 complex-independent polarization of the centrosome to the immunological synapse. Immunity 26(2):177–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matalon O, Reicher B, Barda-Saad M. 2013. Wiskott-Aldrich syndrome protein—dynamic regulation of actin homeostasis: from activation through function and signal termination in T lymphocytes. Immunol. Rev 256(1):10–29 [DOI] [PubMed] [Google Scholar]
- 27.Alekhina O, Burstein E, Billadeau DD. 2017. Cellular functions of WASP family proteins at a glance. J. Cell Sci 130(14):2235–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen Z, Borek D, Padrick SB, Gomez TS, Metlagel Z, et al. 2010. Structure and control of the actin regulatory WAVE complex. Nature 468(7323):533–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen B, Chou H-T, Brautigam CA, Xing W, Yang S, et al. 2017. Rac1 GTPase activates the WAVE regulatory complex through two distinct binding sites. eLife 6:1–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burke TA, Christensen JR, Barone E, Suarez C, Sirotkin V, Kovar DR. 2014. Homeostatic actin cytoskeleton networks are regulated by assembly factor competition for monomers. Curr. Biol 24(5):579–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fritzsche M, Erlenkämper C, Moeendarbary E, Charras G, Kruse K. 2016. Actin kinetics shapes cortical network structure and mechanics. Sci. Adv 2(4):e1501337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rotty JD, Wu C, Haynes EM, Suarez C, Winkelman JD, et al. 2015. Profilin-1 serves as a gatekeeper for actin assembly by Arp2/3-dependent and -independent pathways. Dev. Cell 32(1):54–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lomakin AJ, Lee K, Han SJ, Bui DA, Davidson M, et al. 2015. Competition for actin between two distinct F-actin networks defines a bistable switch for cell polarization. Nat. Cell Biol 17(11):1435–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kanellos G, Frame MC. 2016. Cellular functions of the ADF/cofilin family at a glance. J. Cell Sci 129(17):3211–18 [DOI] [PubMed] [Google Scholar]
- 35.Roybal KT, Buck TE, Ruan X, Cho BH, Clark DJ, et al. 2016. Computational spatiotemporal analysis identifies WAVE2 and cofilin as joint regulators of costimulation-mediated T cell actin dynamics. Sci. Signal 9(424):rs3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vicente-Manzanares M, Ma X, Adelstein RS, Horwitz AR. 2009. Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat. Rev. Mol. Cell Biol 10(11):778–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Beach JR, Hammer JA. 2015. Myosin II isoform co-assembly and differential regulation in mammalian systems. Exp. Cell Res 334(1):2–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shutova MS, Svitkina TM. 2018. Mammalian nonmuscle myosin II comes in three flavors. Biochem. Biophys. Res. Commun 506:394–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hammer JA, Sellers JR. 2012. Walking to work: roles for class V myosins as cargo transporters. Nat. Rev. Mol. Cell Biol 13(1):13–26 [DOI] [PubMed] [Google Scholar]
- 40.Sakamoto T, Limouze J, Combs CA, Straight AF, Sellers JR. 2005. Blebbistatin, a myosin II inhibitor, is photoinactivated by blue light. Biochemistry 44(2):584–88 [DOI] [PubMed] [Google Scholar]
- 41.Straight AF, Cheung A, Limouze J, Chen I, Westwood NJ, et al. 2003. Dissecting temporal and spatial control of cytokinesis with a myosin II inhibitor. Science 299(5613):1743–47 [DOI] [PubMed] [Google Scholar]
- 42.Képiró M, Várkuti BH, Végner L, Vörös G, Hegyi G, et al. 2014. para-Nitroblebbistatin, the non-cytotoxic and photostable myosin II inhibitor. Angew. Chemie Int. Ed 53(31):8211–15 [DOI] [PubMed] [Google Scholar]
- 43.Parsey MV, Lewis GK. 1993. Actin polymerization and pseudopod reorganization accompany anti-CD3-induced growth arrest in Jurkat T cells. J. Immunol 151(4):1881–93 [PubMed] [Google Scholar]
- 44.Bunnell SC, Kapoor V, Trible RP, Zhang W, Samelson LE. 2001. Dynamic actin polymerization drives T cell receptor-induced spreading. Immunity 14(3):315–29 [DOI] [PubMed] [Google Scholar]
- 45.Bunnell SC, Hong DI, Kardon JR, Yamazaki T, McGlade CJ, et al. 2002. T cell receptor ligation induces the formation of dynamically regulated signaling assemblies. J. Cell Biol 158(7):1263–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kaizuka Y, Douglass AD, Varma R, Dustin ML, Vale RD. 2007. Mechanisms for segregating T cell receptor and adhesion molecules during immunological synapse formation in Jurkat T cells. PNAS 104(51):20296–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yi J, Wu XS, Crites T, Hammer JA. 2012. Actin retrograde flow and actomyosin II arc contraction drive receptor cluster dynamics at the immunological synapse in Jurkat T cells. Mol. Biol. Cell 23(5):834–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Murugesan S, Hong J, Yi J, Li D, Beach JR, et al. 2016. Formin-generated actomyosin arcs propel T cell receptor microcluster movement at the immune synapse. J. Cell Biol 215(3):383–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hong J, Murugesan S, Betzig E, Hammer JA. 2017. Contractile actomyosin arcs promote the activation of primary mouse T cells in a ligand-dependent manner. PLOS ONE 12(8):e0183174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fritzsche M, Fernandes RA, Chang VT, Colin-York H, Clausen MP, et al. 2017. Cytoskeletal actin dynamics shape a ramifying actin network underpinning immunological synapse formation. Sci. Adv 3(6):e1603032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ashdown GW, Burn GL, Williamson DJ, Pandžić E, Peters R, et al. 2017. Live-cell super-resolution reveals F-actin and plasma membrane dynamics at the T cell synapse. Biophys. J 112(8):1703–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Babich A, Li S, O’Connor RS,Milone MC, Freedman BD, Burkhardt JK. 2012. F-actin polymerization and retrograde flow drive sustained PLCγ1 signaling during T cell activation. J. Cell Biol 197(6):775–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yu Y, Fay NC, Smoligovets AA, Wu H-J, Groves JT. 2012. Myosin IIA modulates T cell receptor transport and CasL phosphorylation during early immunological synapse formation. PLOS ONE 7(2):e30704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ponti A, Machacek M, Gupton SL, Waterman-Storer CM, Danuser G. 2004. Two distinct actin networks drive the protrusion of migrating cells. Science 305(5691):1782–86 [DOI] [PubMed] [Google Scholar]
- 55.Hetrick B, Han MS, Helgeson LA, Nolen BJ. 2013. Small molecules CK-666 and CK-869 inhibit actin-related protein 2/3 complex by blocking an activating conformational change. Chem. Biol 20(5):701– 12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Le Floc’h A, Tanaka Y, Bantilan NS, Voisinne G, Altan-Bonnet G, et al. 2013. Annular PIP3 accumulation controls actin architecture and modulates cytotoxicity at the immunological synapse. J. Exp. Med 210(12):2721–37. Correction. 2013. J. Exp. Med. 210(12):2721–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sanui T, Inayoshi A, Noda M, Iwata E, Oike M, et al. 2003. DOCK2 is essential for antigen-induced translocation of TCR and lipid rafts, but not PKC-θ and LFA-1, in T cells. Immunity 19(1):119–29 [DOI] [PubMed] [Google Scholar]
- 58.Nishikimi A, Kukimoto-Niino M, Yokoyama S, Fukui Y. 2013. Immune regulatory functions of DOCK family proteins in health and disease. Exp. Cell Res 319(15):2343–49 [DOI] [PubMed] [Google Scholar]
- 59.Nolz JC, Gomez TS, Zhu P, Li S, Medeiros RB, et al. 2006. The WAVE2 complex regulates actin cytoskeletal reorganization and CRAC-mediated calcium entry during T cell activation. Curr. Biol 16(1):24–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zipfel PA, Bunnell SC, Witherow DS, Gu JJ, Chislock EM, et al. 2006. Role for the Abi/Wave protein complex in T cell receptor-mediated proliferation and cytoskeletal remodeling. Curr. Biol 16(1):35–46 [DOI] [PubMed] [Google Scholar]
- 61.Rizvi SA, Neidt EM, Cui J, Feiger Z, Skau CT, et al. 2009. Identification and characterization of a small molecule inhibitor of formin-mediated actin assembly. Chem. Biol 16(11):1158–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ilani T, Vasiliver-Shamis G, Vardhana S, Bretscher A, Dustin ML. 2009. T cell antigen receptor signaling and immunological synapse stability require myosin IIA. Nat. Immunol 10(5):531–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen Q, Nag S, Pollard TD. 2012. Formins filter modified actin subunits during processive elongation. J. Struct. Biol 177(1):32–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rak GD,Mace EM, Banerjee PP, Svitkina T, Orange JS. 2011. Natural killer cell lytic granule secretion occurs through a pervasive actin network at the immune synapse. PLOS Biol 9(9):e1001151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Brown ACN, Oddos S, Dobbie IM, Alakoskela J-M, Parton RM, et al. 2011. Remodelling of cortical actin where lytic granules dock at natural killer cell immune synapses revealed by super-resolution microscopy. PLOS Biol. 9(9):e1001152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Carisey AF, Mace EM, Saeed MB, Davis DM, Orange JS. 2018. Nanoscale dynamism of actin enables secretory function in cytolytic cells. Curr. Biol 28(4):489–502.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ritter AT, Kapnick SM, Murugesan S, Schwartzberg PL, Griffiths GM, Lippincott-Schwartz J. 2017. Cortical actin recovery at the immunological synapse leads to termination of lytic granule secretion in cytotoxic T lymphocytes. PNAS 114(32):E6585–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kumari S, Depoil D,Martinelli R,Judokusumo E, Carmona G, et al. 2015. Actin foci facilitate activation of the phospholipase C-γ in primary T lymphocytes via the WASP pathway. eLife 4(4):1–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cannon JL, Burkhardt JK. 2004. Differential roles for Wiskott-aldrich syndrome protein in immune synapse formation and IL-2 production. J. Immunol 173(3):1658–62 [DOI] [PubMed] [Google Scholar]
- 70.Eddy RJ, Weidmann MD, Sharma VP, Condeelis JS. 2017. Tumor cell invadopodia: invasive protrusions that orchestrate metastasis. Trends Cell Biol. 27(8):595–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fuller CL, Braciale VL, Samelson LE. 2003. All roads lead to actin: the intimate relationship between TCR signaling and the cytoskeleton. Immunol. Rev 191(1):220–36 [DOI] [PubMed] [Google Scholar]
- 72.Courtney AH, Lo W-L, Weiss A. 2018. TCR signaling: mechanisms of initiation and propagation. Trends Biochem. Sci 43(2):108–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tybulewicz VL. 2005. Vav-family proteins in T-cell signalling. Curr. Opin. Immunol 17(3):267–74 [DOI] [PubMed] [Google Scholar]
- 74.Razidlo GL, Schroeder B, Chen J, Billadeau DD, McNiven MA. 2014. Vav1 as a central regulator of invadopodia assembly. Curr Biol. 24(1):86–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Varma R, Campi G, Yokosuka T, Saito T, Dustin ML. 2006. T cell receptor-proximal signals are sustained in peripheral microclusters and terminated in the central supramolecular activation cluster. Immunity 25(1):117–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nguyen K, Sylvain NR, Bunnell SC. 2008. T cell costimulation via the integrin VLA-4 inhibits the actin-dependent centralization of signaling microclusters containing the adaptor SLP-76. Immunity 28(6):810–21 [DOI] [PubMed] [Google Scholar]
- 77.Jankowska KI, Williamson EK, Roy NH, Blumenthal D, Chandra V, et al. 2018. Integrins modulate T cell receptor signaling by constraining actin flow at the immunological synapse. Front. Immunol 9:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hashimoto-Tane A, Yokosuka T, Sakata-Sogawa K, Sakuma M, Ishihara C, et al. 2011. Dynein-driven transport of T cell receptor microclusters regulates immune synapse formation and T cell activation. Immunity 34(6):919–31 [DOI] [PubMed] [Google Scholar]
- 79.DeMond AL, Mossman KD, Starr T, Dustin ML, Groves JT. 2008. T cell receptor microcluster transport through molecular mazes reveals mechanism of translocation. Biophys. J 94(8):3286–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yu C,Wu H-J, Kaizuka Y,Vale RD, Groves JT. 2010. Altered actin centripetal retrograde flow in physically restricted immunological synapses. PLOS ONE 5(7):e11878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Smoligovets AA, Smith AW, Wu H-J, Petit RS, Groves JT. 2012. Characterization of dynamic actin associations with T-cell receptor microclusters in primary T cells. J. Cell Sci 125(3):735–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ditlev JA, Vega AR, Köster DV, Su X, Lakoduk A, et al. 2018. A composition-dependent molecular clutch between T cell signaling clusters and actin. bioRxiv 316414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Janssen E, Tohme M, Hedayat M, Leick M, Kumari S, et al. 2016. A DOCK8-WIP-WASp complex links T cell receptors to the actin cytoskeleton. J. Clin. Investig 126(10):3837–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Springer TA,Dustin ML. 2012. Integrin inside-out signaling and the immunological synapse. Curr. Opin. Cell Biol 24(1):107–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Comrie WA, Burkhardt JK. 2016. Action and traction: cytoskeletal control of receptor triggering at the immunological synapse. Front. Immunol 7:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Stinchcombe JC, Majorovits E, Bossi G, Fuller S, Griffiths GM. 2006. Centrosome polarization delivers secretory granules to the immunological synapse. Nature 443(7110):462–65 [DOI] [PubMed] [Google Scholar]
- 87.de la Roche M, Asano Y, Griffiths GM. 2016. Origins of the cytolytic synapse. Nat. Rev. Immunol 16(7):421–32 [DOI] [PubMed] [Google Scholar]
- 88.Raaijmakers JH, Bos JL. 2009. Specificity in Ras and Rap signaling. J. Biol. Chem 284(17):10995–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lagarrigue F, Kim C, Ginsberg MH. 2016. The Rap1-RIAM-talin axis of integrin activation and blood cell function. Blood 128(4):479–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Comrie WA, Babich A, Burkhardt JK. 2015. F-actin flow drives affinity maturation and spatial organization of LFA-1 at the immunological synapse. J. Cell Biol 208(4):475–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nolz JC, Medeiros RB, Mitchell JS, Zhu P, Freedman BD,et al. 2007. WAVE2 regulates high-affinity integrin binding by recruiting vinculin and talin to the immunological synapse. Mol. Cell. Biol 27(17):5986– 6000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nolz JC,Nacusi LP, Segovis CM, Medeiros RB, Mitchell JS, et al. 2008. The WAVE2 complex regulates T cell receptor signaling to integrins via Abl- and CrkL-C3G-mediated activation of Rap1. J. Cell Biol 182(6):1231–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gardel ML, Schneider IC, Aratyn-Schaus Y, Waterman CM. 2010. Mechanical integration of actin and adhesion dynamics in cell migration. Annu. Rev. Cell Dev. Biol 26:315–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Case LB, Waterman CM. 2015. Integration of actin dynamics and cell adhesion by a three-dimensional, mechanosensitive molecular clutch. Nat. Cell Biol 17(8):955–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Galbraith CG, Yamada KM, Sheetz MP. 2002. The relationship between force and focal complex development. J. Cell Biol 159(4):695–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Klapholz B, Brown NH. 2017. Talin—the master of integrin adhesions. J. Cell Sci 130(15):2435–46 [DOI] [PubMed] [Google Scholar]
- 97.Sun Z, Guo SS, Fässler R. 2016. Integrin-mediated mechanotransduction. J. Cell Biol 215(4):445–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Comrie WA, Li S, Boyle S, Burkhardt JK. 2015. The dendritic cell cytoskeleton promotes T cell adhesion and activation by constraining ICAM-1 mobility. J. Cell Biol 208(4):457–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hartman NC, Nye JA, Groves JT. 2009. Cluster size regulates protein sorting in the immunological synapse. PNAS 106(31):12729–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wilson CA, Tsuchida MA, Allen GM, Barnhart EL, Applegate KT, et al. 2010. Myosin II contributes to cell-scale actin network treadmilling through network disassembly. Nature 465(7296):373–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gutiérrez LM, Villanueva J. 2018. The role of F-actin in the transport and secretion of chromaffin granules: an historic perspective. Pflügers Arch. Eur. J. Physiol 470(1):181–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ritter AT, Asano Y, Stinchcombe JC, Dieckmann NMG, Chen B-C, et al. 2015. Actin depletion initiates events leading to granule secretion at the immunological synapse. Immunity 42(5):864–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hammer JA. 2018. Immunology: Is actin at the lytic synapse a friend or a foe? Curr. Biol 28(4):R155–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Čemerski S, Das J, Giurisato E, Markiewicz MA, Allen PM, et al. 2008. The balance between T cell receptor signaling and degradation at the center of the immunological synapse is determined by antigen quality. Immunity 29(3):414–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Choudhuri K, Llodrá J, Roth EW, Tsai J, Gordo S, et al. 2014. Polarized release of T-cell-receptor-enriched microvesicles at the immunological synapse. Nature 507(7490):118–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Dustin ML, Choudhuri K. 2016. Signaling and polarized communication across the T cell immunological synapse. Annu. Rev. Cell Dev. Biol 32:303–25 [DOI] [PubMed] [Google Scholar]
- 107.Basu R, Whitlock BM, Husson J, Le Floc’h A, Jin W, et al. 2016. Cytotoxic T cells use mechanical force to potentiate target cell killing. Cell 165(1):100–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Basu R, Huse M. 2017. Mechanical communication at the immunological synapse. Trends Cell Biol. 27(4):241–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Huse M 2017. Mechanical forces in the immune system. Nat. Rev. Immunol 17(11):679–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hui KL, Balagopalan L, Samelson LE, Upadhyaya A.2015. Cytoskeletal forces during signaling activation in Jurkat T-cells. Mol. Biol. Cell 26(4):685–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Upadhyaya A 2017. Mechanosensing in the immune response. Semin. Cell Dev. Biol 71:137–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Chen W, Zhu C. 2013. Mechanical regulation of T-cell functions. Immunol. Rev 256(1):160–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Depoil D, Dustin ML. 2014. Force and affinity in ligand discrimination by the TCR. Trends Immunol. 35(12):597–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Santos LC, Blair DA, Kumari S, Cammer M, Iskratsch T, et al. 2016. Actin polymerization-dependent activation of Cas-L promotes immunological synapse stability. Immunol. Cell Biol 94(10):981–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kubow KE, Horwitz AR. 2011. Reducing background fluorescence reveals adhesions in 3D matrices. Nat. Cell Biol 13(1):5–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chen B, Legant WR, Wang K, Shao L, Milkie DE, et al. 2014. Lattice light-sheet microscopy: imaging molecules to embryos at high spatiotemporal resolution. Science 346(6208):1257998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wang JC, Bolger-Munro M, Gold MR. 2018. Imaging the interactions between B cells and antigen-presenting cells. Methods Mol. Biol 1707:131–61 [DOI] [PubMed] [Google Scholar]
- 118.Singleton K, Parvaze N, Dama KR, Chen KS, Jennings P, et al. 2006. A large T cell invagination with CD2 enrichment resets receptor engagement in the immunological synapse. J. Immunol 177(7):4402–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Roybal KT,Mace EM, Mantell JM, Verkade P, Orange JS, Wülfing C. 2015. Early signaling in primary T cells activated by antigen presenting cells is associated with a deep and transient lamellal actin network. PLOS ONE 10(8):e0133299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Roybal KT, Mace EM, Clark DJ, Leard AD, Herman A, et al. 2015. Modest interference with actin dynamics in primary T cell activation by antigen presenting cells preferentially affects lamellal signaling. PLOS ONE 10(8):e0133231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bertrand F, Muller S, Roh K-H, Laurent C, Dupre L, Valitutti S. 2013. An initial and rapid step of lytic granule secretion precedes microtubule organizing center polarization at the cytotoxic T lymphocyte/target cell synapse. PNAS 110(15):6073–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Clausen MP, Colin-York H, Schneider F, Eggeling C, Fritzsche M. 2017. Dissecting the actin cortex density and membrane-cortex distance in living cells by super-resolution microscopy. J. Phys. D. Appl. Phys 50(6):64002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Gomez TS, McCarney SD, Carrizosa E, Labno CM, Comiskey EO, et al. 2006. HS1 functions as an essential actin-regulatory adaptor protein at the immune synapse. Immunity 24(6):741–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Gorman JA, Babich A, Dick CJ, Schoon RA, Koenig A, et al. 2012. The cytoskeletal adaptor protein IQGAP1 regulates TCR-mediated signaling and filamentous actin dynamics. J. Immunol 188(12):6135– 44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Liang Y, Cucchetti M, Roncagalli R, Yokosuka T, Malzac A, et al. 2013. The lymphoid lineage-specific actin-uncapping protein Rltpr is essential for costimulation via CD28 and the development of regulatory T cells. Nat. Immunol 14(8):858–66 [DOI] [PubMed] [Google Scholar]
- 126.Roncagalli R, Cucchetti M,Jarmuzynski N, Grégoire C, Bergot E, et al. 2016. The scaffolding function of the RLTPR protein explains its essential role for CD28 co-stimulation in mouse and human T cells. J. Exp. Med 213(11):2437–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Schober T, Magg T, Laschinger M, Rohlfs M, Linhares ND, et al. 2017. A human immunodeficiency syndrome caused by mutations in CARMIL2. Nat. Commun 8:14209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Goode BL, Sweeney MO, Eskin JA. 2018. GMF as an actin network remodeling factor. Trends Cell Biol. 28:749–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Morley SC.2013. The actin-bundling protein L-plastin supportsT-cell motility and activation. Immunol. Rev 256(1):48–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Mace EM, Orange JS. 2014. Lytic immune synapse function requires filamentous actin deconstruction by Coronin 1A. PNAS 111(18):6708–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Shaffer MH, Dupree RS, Zhu P, Saotome I, Schmidt RF, et al. 2009. Ezrin and moesin function together to promote T cell activation. J. Immunol 182(2):1021–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.