

Abstract
Sporadic late-onset Alzheimer’s disease (SLOAD) and familial early-onset Alzheimer’s disease (FEOAD) associated with dominant mutations in APP, PSEN1 and PSEN2, are thought to represent a spectrum of the same disorder based on near identical behavioral and histopathological features. Hence, FEOAD transgenic mouse models have been used in past decades as a surrogate to study SLOAD pathogenic mechanisms and as the gold standard to validate drugs used in clinical trials. Unfortunately, such research has yielded little output in terms of therapeutics targeting the disease’s development and progression. In this short review, we interrogate the widely accepted view of one, dimorphic disease through the prism of the Bmi1+/– mouse model and the distinct chromatin signatures observed between SLOAD and FEOAD brains.
Keywords: aging, Alzheimer's disease, BMI1, epigenetics, familial, late-onset, sporadic
Overview of Alzheimer’s Disease
Alzheimer’s disease (AD) is the most common form of dementia in the world affecting up to 35 million people worldwide (Cacace et al., 2016). AD cases can be categorized according to the age of onset and the presence of a family history of the disease or lack thereof. Early-onset AD (EOAD) designates cases that manifest before 65 years old whereas AD at and after 65 years old is considered late-onset AD (LOAD). Familial AD (FAD) indicates a positive family history while sporadic AD (SAD) indicates no familial history of the disease. Differentiating familial and sporadic cases can prove difficult, especially in large cohorts, when family history is missing or incomplete. Nonetheless, LOAD is by far the most predominant form of AD, accounting for 90–99% of all AD patients, depending on the estimate (Bird, 2008; Cacace et al., 2016) (Figure 1). Of the total LOAD cases, according to one study, approximately 40% were familial LOAD (FLOAD) whereas the remaining 60% were sporadic LOAD (SLOAD) (Jarvik et al., 1993) (Figure 1). On the other hand, EOAD, representing only 1–10% of total AD cases, is comprised of up to 60% familial EOAD (FEOAD) cases and thus approximately 40% sporadic EOAD (SEOAD) cases (Bird, 2008; Cacace et al., 2016). Unfortunately, comprehensive statistics such as these remain very limited given that often studies employ only one axis (e.g. late vs. early onset). Moreover, family history may be unobtainable to researchers. Thus, even though these estimates are based on relatively small populations, they provide a useful overview of the disease. Further study of diverse AD populations would greatly improve our understanding of this challenging disorder.
Figure 1.
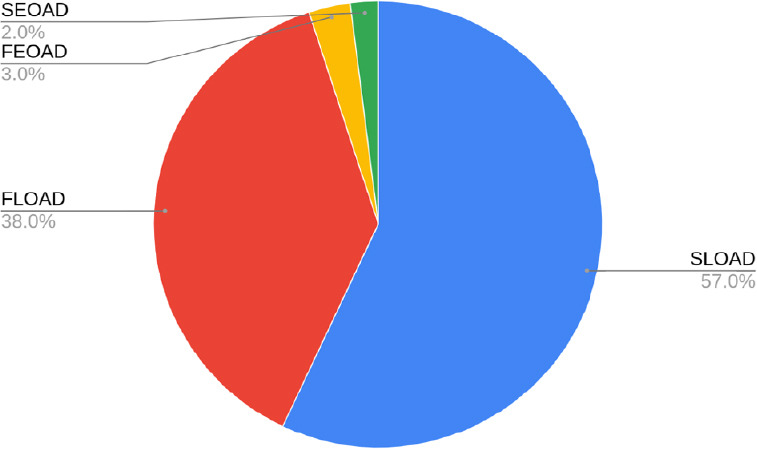
Breakdown of total AD cases.
Each percentage value is based on the total number of AD cases. From largest to smallest, SLOAD is 57%, FLOAD is 38%, FEOAD is 3% and SEOAD is 2%. For the purposes of this figure, LOAD is considered 95% of the total (Bird, 2008; Cacace et al., 2016), 60% of which is SLOAD and 40% is FLOAD (Jarvik et al., 1993). EOAD, here 5% of the total cases, is made up of 60% FEOAD and 40% SEOAD cases (Bird, 2008; Cacace et al., 2016). AD: Alzheimer’s disease; EOAD: early-onset Alzheimer’s disease; FEOAD: familial early-onset Alzheimer’s disease; FLOAD: familial late-onset Alzheimer’s disease; LOAD: late-onset Alzheimer’s disease; SEOAD: sporadic early-onset Alzheimer’s disease; SLOAD: sporadic late-onset Alzheimer’s disease.
LOAD and EOAD are neurodegenerative diseases affecting primarily the cortex and the hippocampus, and are generally considered the same disease, despite the different age of onset, with the amyloid beta peptide (Aβ42) considered as the central etiological factor (Nussbaum and Ellis, 2003). In all cases, patients’ episodic memory deteriorates as the pathologies progress, and so do the main cognitive functions, e.g. critical judgment, orientation, language, eventually leading to a loss of autonomy (Blennow et al., 2006). The pathological hallmarks comprise extracellular senile plaques formed by the aggregation of Aβ42, intracellular tangles of the hyperphosphorylated form of Tau protein, neuronal loss and synaptic degeneration (Blennow et al., 2006).
Genetics of Alzheimer’s Disease
FEOAD
The discovery of highly penetrant mutations in the amyloid beta precursor protein (APP), presenilin 1 (PSEN1) and presenilin 2 (PSEN2) genes has revealed important aspects of the mechanisms underlying FEOAD. However, these known mutations can only explain between 5% and 10% of FEOAD cases (Cacace et al., 2016). Some EOAD families also present a duplication of one allele of APP, resulting in three gene copies (Van Cauwenberghe et al., 2016). FEOAD-associated genes are all involved in the amyloid cascade: APP can either be cleaved by β-secretase (BACE1) in the amyloidogenic pathway or follow the non-amyloidogenic pathway and be processed by α-secretase. The products of both pathways are subsequently cleaved by γ-secretase. The former pathway leads to the production of the non-physiologic Aβ42 peptide, which is fibrillogenic and aggregation-prone, causing the formation of the characteristic extracellular plaques (Blennow et al., 2006). Therefore, according to the amyloid cascade hypothesis, an imbalance between the production and clearance of Aβ42 is the triggering cause of the disease (Yin and Wang, 2018). PSEN1 and PSEN2 are highly homologous genes which encode for the catalytic subunits of γ-secretase. Mutations in these loci therefore impair the enzymatic activity, the localization and the conformation of the complex, leading to an aberrant accumulation and further aggregation of Aβ42 (Escamilla-Ayala et al., 2020). The proteins have different biological roles, demonstrated by the different impacts they have once mutated; missense mutations in PSEN1 are responsible for the most serious form of FEOAD, with an onset as early as 25 years old. Whereas those occurring in PSEN2 might not have complete penetrance and are linked to older age of onset (Van Cauwenberghe et al., 2016).
SLOAD
While the modes of transmission of FEOAD are generally understood, those responsible for SLOAD are much less clear. Interestingly, mutations in the APP gene have been identified in small populations of cortical neurons from SAD patients, attributed to seemingly random somatic mutations (Lee et al., 2019). However, it is unclear what the prevalence of such somatic gene mosaicism is and whether it is sufficient to provoke AD. Thus, SLOAD is thought to be the result of complex interactions between both environmental and human genetic factors (Chouraki and Seshadri, 2014).
Apolipoprotein E (APOE) is the gene most robustly associated with the risk of developing LOAD, both familial and sporadic. APOE is the main apolipoprotein present in the brain, which is involved, amongst other things, in the transport of lipids and responsible for their internalization through specific binding to cell-surface lipoprotein receptors (Huang and Mahley, 2014). Three APOE allelic variants exist, which differ by nucleotides at two sites in the gene: E2 is considered protective (the rarest allele, ~8.5% worldwide frequency), E3 is considered neutral (the most common allele, ~78% worldwide) and E4 increases LOAD risk by 3-fold in carriers and 15-fold in E4 homozygotes (Corder et al., 1993; Saunders et al., 1993). In patients, these figures of allelic frequency are overturned, since E4 is the dominant allele both in LOAD, found in ~50% of the subjects affected, and in EOAD, carried by 44.31% of the patients enrolled in one study (Verghese et al, 2011; Jia et al., 2020). It should also be taken into account that the percentages here listed slightly vary between different ethnic groups. However, different studies evidenced a notable disparity concerning the effect of APOE4 on disease progression in EOAD vs. LOAD (van der Vlies et al., 2009; Jochemsen et al., 2012). LOAD APOE4-carriers experience more rapid disease progression than LOAD APOE4-negative individuals, whereas EOAD APOE4 carriers exhibit a slower disease progression than EOAD APOE4-negative individuals. The biological reasons behind this effect are yet to be elucidated and are more likely to be found in the interaction of the genotype with environmental cues and other factors, rather than in the genetic background solely. It is indeed well-known that APOE4 modulatory effect on the clinical AD features strongly correlates with the age of onset of the disease (van der Flier et al., 2011).
The mechanisms linking APOE to the etiology of AD are unclear, but it has been implicated in amyloid clearance and p-Tau pathology as well as neuroinflammation (Carter, 2005; Castellano et al., 2011; Dorey et al., 2014; Tai et al., 2015; Rebeck, 2017; Buckley et al., 2019; Fernandez et al., 2019). More recently, expression of APOE4 in cultured human neurons was shown to induce degeneration of GABAergic neurons, suggesting that APOE4 entails toxic gain-of-function activities (Wang et al., 2018). Conversely, the protective role of APOE2 has been explained with a weaker ability of this isoform to stimulate Aβ synthesis by neurons via the engagement of a MAPK signaling pathway; APOE4, APOE3, APOE2 respectively exhibit a decreased potency in doing so (Huang et al., 2017). Further, the protective genotype also correlates with specific morphological features in the brain of healthy heterozygous individuals, i.e. a wider entorhinal cortex in children (Shaw et al., 2007) and a thicker hippocampus in the adults (Fennema-Notestine et al. 2011), while the homozygotes exhibit even more prominent characteristics, such as a larger volume of the grey matter in the anterior cingulate and medial prefrontal areas and less severe tau tangles and Aβ plaques (Reiman et al., 2020).
Since the discovery of the correlation between APOE4 and AD decades ago, at least 20 new alleles of other genes have been reported to have an impact on the risk of developing the disease (Dong et al., 2017). Different strategies have been used to identify new hits, mainly GWAS, the candidate gene approach, statistical testing of SNPs, the study of the structural variants as a genetic marker and gene-environment interactions (Chouraki and Seshadri, 2014; Van Cauwenberghe et al., 2016). In the end, many of the variants described in these studies occur in loci either involved in the processing of APP, or in the modulation of Tau toxicity. Nevertheless, they are also part of broader biochemical processes, i.e. the immune response (e.g. CR1 (Lambert et al., 2009), TREM2 (Jonsson et al., 2013) and CD33 (Naj et al., 2011)), cholesterol and lipid metabolism (e.g. ABCA7 (Hollingworth et al., 2011), CLU (Harold et al., 2009)), apoptosis (e.g. HRK (Bis et al., 2012)), trafficking of endosomal vesicles (e.g. PICALM (Harold et al., 2009), SORL1 (Rogaeva et al., 2007), BIN1 (Harold et al., 2009, Chapuis et al., 2013)), and the regulation of the cytoskeleton (e.g. FERMT2 (Lambert et al., 2013)), expanding the number of dysregulated pathways in AD. However, each of these alleles only mildly modulates the risk of the disease (effect sizes with odds ratio ≤ 2.0), implying that APOE4 remains the most relevant LOAD susceptibility gene (Van Cauwenberghe et al., 2016). Nonetheless, there is no mutation that can be uniquely and directly associated with LOAD and it is difficult to distinguish between genetic causal mutations and biomarkers (Lutz et al., 2016). Concluding, none of the risk alleles described so far is necessary nor sufficient to explain the insurgence of the pathology, although models of combinatorial risk variants have been developed (Sims et al., 2020). For this reason, we advance an epigenetic framework underlying the development or progression of SLOAD based on seeming disparate epigenetic signatures between FEOAD and SLOAD.
Non-Genetic Alzheimer’s Disease Risk Factors
Among many other risk factors for AD, ageing is by far the most relevant (Hou et al., 2019). In 2019, 5.8 million Americans were living with AD, 81% of whom were aged 75 or older (Alzheimer’s Association, 2019). Meta-analysis studies showed that the incidence rate is 7 times higher in subjects aged 85–89 years compared to individuals from 70 to 74 years old (Petersen et al., 2001).
Other risk factors include family history and female sex. Having a relative who is affected by AD more than doubles the chances of developing the pathology (Alzheimer’s Association, 2019) and in every age group considered, the percentage of females affected is higher of that of the males (Viña and Lloret, 2010). Moreover, both cardiovascular diseases and risk factors of developing them are associated with AD (de Bruijn and Ikram, 2014; Reitz and Mayeux, 2014), as well as a number of environmental factors. For instance, viral infections, aluminum and other trace metals, and an unhealthy diet all have been demonstrated to increase the risk of AD (Grant et al., 2002; Armstrong, 2019). Similarly, lifestyle habits such as lack of sleep, high blood pressure and sedentary behavior affect AD risk (Reitz and Mayeux, 2014), while education and intellectual engagement in old age are protective factors (Sando et al., 2008; Stern, 2012).
Mouse Models of AD
FEOAD models
To date, there are 184 mouse models of AD according to Alzforum (Alzforum.org). However, because causal mutations associated with SLOAD are not known, there is a relative dearth of such models. Given the high penetrance of FEOAD-associated mutations, many AD models utilize one or more of them. In some cases, FEOAD-associated mutations are introduced in conjunction with SLOAD-associated risk genes however, assessments of AD-related pathologies remain incomplete or absent in these mouse strains. For thorough reviews of the current FEOAD-associated genetic mouse models, see Onos et al. (2016) and Esquerda-Canals et al. (2017).
Characterized in 2003, the 3xTg-AD mouse continues to be a popular preclinical AD model and can help illustrate the advantages and disadvantages of such FEOAD models. This strain contains three human transgenes coding for the mutant APPSwe, PS1m146V and TauP301L proteins (Oddo et al., 2003). The most notable AD-related pathologies are replicated in this mouse, with some important caveats. Firstly, Aβ plaques are detectable in the hippocampus at 6 months, with severity increasing with age; cortical Aβ plaques appear at 12 months and also exhibit an age-dependent progression (Oddo et al., 2003; Belfiore et al., 2019). In fact, Tau phosphorylation shows the same pattern of age- and region-specific appearance (Belfiore et al., 2019). Tau tangles have been shown at 12 months in the hippocampus particularly in pyramidal neurons of the CA1 region (Oddo et al., 2003). However, there are important considerations concerning the 3xTg-AD mouse tauopathy. Firstly, mutations of the MAPT gene are exceedingly rare in human cases of AD; in fact, they are more often associated with frontotemporal dementia (Strang et al., 2019). Secondly, Tau isolated from 3xTg-AD mouse brains fails to replicate important characteristics of Tau isolated from human AD brains. Notably, human AD-related Tau has the prion-like ability to transform normal Tau into the pathological, filamentous form both in vitro and in vivo, it often lacks the N-terminal domain and it is hyperphosphorylated to a much greater extent than 3xTg-AD mouse-derived Tau (Li et al., 2019). In terms of cognitive decline, the 3xTg-AD mouse presents cognitive impairment as early as 4 months which persists with age (Oddo et al., 2003). At last, the 3xTg-AD mouse model contains an aberrant gene copy number for APP, PSEN1 and MAPT, a condition that does not reflect AD genetics, even in those rare FEOAD patients carrying three wild type alleles of APP.
As we will highlight, recent studies elaborate a particular epigenetic profile of SLOAD in terms of markers of heterochromatin and DNA damage response machinery. In contrast to analyses of human SLOAD samples, the 3xTg-AD mouse shows increased levels of the heterochromatin mark H3K9me3 compared to non-transgenic mice, a trend that is consistent from young to old age (Walker et al., 2013). Lardenoije et al. (2019) review epigenetic aspects of multiple, prominent FEOAD mouse models even though the available data focus on DNA methylation, hydroxymethylation and the related genes. More broadly, divergent protein homology between humans and mice may have disparate effects on mouse models of FEOAD and SLOAD. For example, all three FEOAD-associated proteins have an amino acid sequence homology greater than 90% between mice and humans. However, LOAD risk-related proteins APOE and TREM2 exhibit approximately 70% and 50% sequence homology, respectively (Liao et al., 2015; Penney et al., 2020). Moreover, mouse ApoE is more amyloidogenic than any of the human isoforms (Fagan et al., 2002). For these reasons, AD therapeutics tested on FEOAD-associated genetic mouse models may have little to no efficacy in the case of SLOAD pathogenesis.
LOAD models
Recently reviewed in Zhang et al. (2020), several LOAD mouse models exist. In these various models, AD phenotypes are induced via metabolic dysregulation, traumatic brain injury, Adeno-associated virus 1 (AAV1) gene transduction, toxin exposure, perturbed metal ion homeostasis or aging. Of particular interest is the final category, aging, and the corresponding model, the senescence-accelerated mouse prone 8 (SAMP8) model. Although the exact cause of accelerated senescence has not yet been determined, this model may shed light on the relationship between aging and AD (Griñán-Ferré et al., 2018).
In many respects, the SAMP8 mouse exhibits important AD-associated pathologies including shortened lifespan, reviewed by Griñán-Ferré et al. (2018). To further go into details, age-dependent deposition of Aβ in the hippocampus is observed, but it does not culminate in Aβ plaques (Akiguchi et al., 2017). Similarly, there is an age-related increase in phosphorylated tau, yet neurofibrillary tangles in the brain do not seem to form (Akiguchi et al., 2017). To a limited extent, neurodegeneration in the aged SAMP8 mouse has been reported. In the basal forebrain, there seems to be a 20% reduction in the density of cholinergic neurons (Tooyama et al., 1997). The authors relate this to the memory deficits observed in these mice. Such cognitive phenotypes, based on the Morris Water Maze and Novel Object Recognition for example, are reported as early as 2 months (Akiguchi et al., 2017). As for chromatin modifications, an emphasis of the current review, there still remains much to be explored in the SAMP8 model. Firstly, in the hippocampus, SAMP8 mice show significant downregulation of HDAC3 and SIRT1, in addition to an upregulation of miRNAs that have been found to be dysregulated in the hippocampus of AD patients; these miRNAs are speculated to antagonize senescence-controlling genes (Cosín-Tomás et al., 2014). Nonetheless, reliable quantifications of important histone modifications (e.g. H3K9me3, H3K27me3, H4K20me3) remain largely absent. In an investigation of oxidative damage, SAMP8 mice exhibit notable nucleic acid oxidation, especially in brain tissue (Gan et al., 2012). This is evidenced by 8-oxo-2′-deoxyguanosine, a product of oxidative damage. Thus, further investigation of the DNA damage response (DDR) pathway (e.g. γH2AX, p-ATM, and p-ATR) is warranted to better characterize levels of DNA damage and response. Although the SAMP8 mouse model is promising in terms of many AD-associated pathologies, it has not been shown that its epigenetic signatures mirror human cases of SLOAD. As we will lay out, particular epigenetic anomalies seem to characterize SLOAD, thus a model lacking these features may not be adequate to explore early SLOAD pathogenic mechanisms or effective therapeutics.
Bmi1+/– mouse model of SLOAD
Recently, two reports using both in vitro and in vivo models of BMI1 deficiency revealed that they display notable SLOAD-associated phenotypes (Flamier et al., 2018; El Hajjar et al., 2019). BMI1 is an integral part of the Polycomb Repressive Complex 1 (PRC1), which maintains transcriptional repression at developmental and senescence-associated genes mainly through mono-ubiquitination of histone H2A at lysine 119 (Bhattacharya et al., 2015). The BMI1 protein exhibits a well-conserved sequence homology between mouse and man (Bhattacharya et al., 2015; Abdouh et al., 2016). Notably, the protein is also abundantly found at the repeat DNA sequence-rich heterochromatin, and BMI1 inactivation in primary human cells or in mice results in loss of heterochromatin, as shown using the H3K9me3, HP1, ATRx and DEK1 heterochromatin markers (Abdouh et al. 2016).
While Bmi1-deficient mice (Bmi1–/–) exhibit severe developmental growth defects, premature ageing features, cerebellar, cortical and retinal degeneration and die prematurely (van der Lugt et al., 1994; Chatoo et al., 2009; Barabino et al., 2016), Bmi1 hemi-deficient mice (Bmi1+/–) develop normally, retain fertility and best recapitulate SLOAD (El Hajjar et al., 2019) (Table 1). This begs the question, what advantages and disadvantages does this new model of SLOAD have over previously established models? Firstly, Bmi1+/– mice exhibit an approximate 50% reduction in Bmi1 protein in cortical tissue (El Hajjar et al., 2019). This mirrors the ~50% reduction in BMI1 expression that is reported in the human frontal cortex and hippocampus in SLOAD samples (Flamier et al., 2018). Notably, BMI1 reduction is not present in FEOAD or other neurodegenerative conditions (Flamier et al., 2018). What is striking is that reduced BMI1 expression is also observed in induced pluripotent stem cell (iPSC)-derived neurons from SLOAD patients (Flamier et al., 2018), suggesting an underlying mechanism that persists despite reprogramming from fibroblast to iPSC to neuron.
Table 1.
Comparative analysis of BMI1 expression and chromatin state between SLOAD and FEOAD
| Disease/model | Genetics | Age of onset | BMI1 expression | Heterochromatin | DDR | Sources |
|---|---|---|---|---|---|---|
| SLOAD | Polygenic | +++ | Reduced | Affected | +++ | Flamier et al. (2018); El Hajjar et al. (2019) |
| Bmi1+/– mice | Bmi1 deficiency | +++ | Reduced | Affected | +++ | Flamier et al. (2018); El Hajjar et al. (2019) |
| SAMP8 mice | Unknown, derived from AKR/J background | + | Unknown | Unknown | +++ | Gan et al. (2012); Akiguchi et al. (2017); Griñán-Ferré et al. (2018) |
| FEOAD | APP, PSEN1 or PSEN2 mutations | + | Normal | Normal | + | Flamier et al. (2018) |
| 3xTg-AD mice | PS1m146V, tauP301L, APPSwe | + | Normal | Unknown | Unknown | Walker et al. (2013); Flamier et al. (2018) |
SLOAD excludes SEOAD and FLOAD cases homozygous for APOE4. FEOAD designates cases with known pathogenic mutations. DDR: DNA damage response; FEOAD: familial early-onset Alzheimer’s disease; FLOAD: familial late-onset Alzheimer’s disease; SEOAD: sporadic early-onset Alzheimer’s disease; SLOAD: sporadic late-onset Alzheimer’s disease.
Thoroughly described in El Hajjar et al. (2019), Bmi1 hemi-deficient mice exhibit many of the important AD-associated phenotypes regarding both behavior and histopathology. Just as it is the case in human AD, the disease onset in Bmi1+/– mice is age-dependent, including extracellular amyloid accumulations (although rare), tauopathy, neurodegeneration and cognitive decline (El Hajjar et al., 2019). In this study, amyloid pathology was evidenced by immunoblotting for the C99 fragment, a product of pathogenic APP cleavage by β-secretase. In parallel with the accumulation of the amyloidogenic peptide, Bace1 levels are significantly increased. Although amyloid plaques were not observed upon cortical sectioning, amyloid reactivity was increased in the neuronal soma (El Hajjar et al., 2019). Yet, this can be expected given murine App is much less prone to aggregate into plaques than the human isoform (Bharadwaj, 2019). However, when Bmi1+/– mice were crossed with human APP transgenic mice, the resulting progeny displayed an even more advanced disease-related phenotypes, indicative of possible interaction between these two pathways (El Hajjar et al., 2019). Tauopathy in the Bmi1+/– mouse included p-Tau accumulation and large p-Tau deposits in the cortex, whereas p-Tau tangles were not observed. Neurodegeneration is evidenced by an approximate 20% reduction in NeuN-positive cortical neurons supported by an approximate 2-fold increase in apoptotic neurons in old Bmi1+/– mice compared to WT littermates (El Hajjar et al., 2019). In the hippocampus there is a marked reduction of neuronal density coupled with an increase in apoptosis. Finally, cognitive decline is characterized behaviorally by the Morris water maze probe test and at the cellular level by diminished LTP in the hippocampal CA1 region.
For the purposes of this review, the epigenetic profile of this model sets it apart from others. At an early age, 2–3 months-old in vivo and embryonic day 18.5 in vitro, Bmi1+/– neurons already exhibit loss of heterochromatin, and this before any indication that DNA damage is present (El Hajjar et al., 2019). In the aged Bmi1+/– mouse (15 months old), neuronal heterochromatin depletion persists, repetitive genomic elements are de-repressed and chromocenters appear smaller and more numerous (El Hajjar et al., 2019). Importantly, DDR proteins such as p-ATM, p-ATR and γH2AX accumulated preferentially at repetitive elements in cerebral cortex extracts, as shown using chromatin immuno-precipitation (ChIP) analysis, indicative of heterochromatic genome instability (El Hajjar et al., 2019).
Chromatin Signatures May Distinguish FEOAD from SLOAD
As we attempt to discern differences between FEOAD and SLOAD, there is mounting evidence that chromatin signatures may be a distinguishing factor. Already, global but modest heterochromatin reduction is a well-characterized phenomenon associated with normal cellular aging, reviewed in Kane (2019). However, severe heterochromatin depletion is observed in SLOAD frontal cortex sections compared to non-demented age-matched controls, which correlates with reduced BMI1 expression (El Hajjar et al., 2019). Hence, it is striking to note that despite severe brain neurodegeneration, brain samples from patients with FEOAD do not present reduced BMI1 expression or depletion of heterochromatin (Flamier et al., 2018; El Hajjar et al., 2019). Although, given the small sample size, independent confirmation of these results is essential, it seems that the aforementioned modest aging-related heterochromatin reduction cannot explain the marked difference observed between FEOAD and SLOAD cases.
Specifically, heterochromatin loss in SLOAD frontal cortical sections consists of significantly less H3K9me3 nuclear staining, in conjunction with smaller and more numerous chromocenters (El Hajjar et al., 2019). ChIP analysis revealed that in age-matched controls, BMI1 and H3K9me3 enrichment occurs at repetitive elements such as McBOX, SATIII and SATA, which is depleted in the context of SLOAD (El Hajjar et al., 2019). Interestingly, there is an accumulation of DDR proteins (i.e. p-ATM, p-ATR and p-CHK1) in SLOAD brains (frontal cortex) that is absent even in FEOAD samples (El Hajjar et al., 2019). Lastly, ChIP revealed that p-ATR and γH2AX were enriched at genomic repeats in AD brains (El Hajjar et al., 2019). Seemingly, de-repressed repetitive elements are more susceptible to DNA damage accumulation, an observation also shown in a stem cell model of aging (Zhang et al., 2015). As a consequence of accumulated DNA damage, the constitutively activated DDR pathway seems to enhance amyloid and tau pathologies in cortical neurons of Bmi1–/– mice (El Hajjar et al., 2019).
Heterochromatin loss following BMI1 deficiency may be attributable not only to BMI1’s direct chromatin maintenance functions, but additionally to its protective functions against oxidative damage. In post-mitotic neurons specifically, BMI1 represses the pro-oxidant activity of p53, namely p53’s repressive control over the transcription of antioxidant defense genes (Chatoo et al., 2009). BMI1 may also directly repress the expression of pro-oxidant genes (Liu et al., 2009). The resulting increase of pro-oxidant conditions following BMI1 deficiency may thus also drive heterochromatin loss, although this remains to be tested. As shown in the case of tauopathy in Drosophilia, heterochromatin loss can be driven by oxidative damage, culminating in neuronal apoptosis (Frost et al., 2014). The aberrant gene expression, which results from heterochromatin loss, is cited as a likely underlying mechanism of cell death. Ago3, a regulator of retrotransposon activity and a homolog of human PIWIL1, is one such gene that becomes deregulated following heterochromatin loss (Frost et al., 2014). In human AD brains, specifically hippocampal neurons that stained positively for phosphorylated tau, both chromatin relaxation (using the H3K9me2 antibody) and a corresponding dysregulated gene expression profile were observed (Frost et al., 2014). Going forward, it is important to bolster the data showing oxidative damage induces heterochromatin loss. For example, can ROS scavengers mitigate heterochromatin loss in BMI1-deficient neurons? Moreover, the identity of such de-repressed loci is paramount. Is oxidative damage-induced heterochromatin de-repression random, or are there particular features that render some loci more vulnerable?
Decreased H3K9me3 levels are not only an important indicator of de-repressed constitutive heterochromatin, they may also be implicated in diminished DNA damage repair capacity. H3K9me3 at double-strand DNA breaks (DSBs) is essential to activate TIP60 which goes on to activate ATM as part of the DDR pathway (Sun et al., 2009). However, a recent study in cancer cells showed that hypermethylation of H3K9 surrounding DSBs masked the H3K9me3 signal that is essential for homologous DNA repair. Mechanistically, H3K9 hypermethylation impaired the recruitment of TIP60 and ATM at DNA breaks (Sulkowski et al., 2020). Cited above, DNA damage accrues preferentially at de-repressed repetitive elements in SLOAD models, which may be indicative of increased susceptibility to DNA damage or of diminished DNA repair capacity. Of course, these two phenomena may be present simultaneously. In the context of SLOAD, it is possible that depletion of H3K9me3 at such loci results in an analogous disruption of the DDR pathway; however this is not yet shown. In summary, the Bmi1+/– mouse model appears to recapitulate new cardinal features of SLOAD that are not found in FEOAD brains or mouse models.
Future Directions
Going forward, it is important to gather more robust data supporting BMI1 deficiency and chromatin anomalies as a differentiating factor between FEOAD and SLOAD. These phenotypes have been highlighted here, albeit in studies of very small sample sizes. These observations have been revelatory because these pathologies precede other AD-associated phenotypes in the Bmi1+/– mouse model. Perhaps there are other chromatin or gene-expression anomalies that develop before amyloid and Tau pathologies in SLOAD. Moreover, it remains to be shown whether BMI1 deficiency is present or not in FEOAD iPSC-derived neurons, as is the case with SLOAD iPSC-derived neurons. Nonetheless, considering the current AD mouse models, the Bmi1+/– model has notable advantages. It presents chromatin anomalies from a very early age whereas more severe AD-associated pathologies develop much later (e.g. Aβ42 accumulation, tauopathy and neuronal loss among others), and it does not rely on overexpressed FEOAD-associated human genes which are suspected to induce many nonspecific anomalies. Delineating FEOAD and SLOAD from one another, if it is in fact the case, is thus essential for continued research and drug development. If we are indeed dealing with two, etiologically distinct disorders, they are likely to require two distinct treatments.
Footnotes
Conflicts of interest: AF and GB are co-founders and shareholders of StemAxonTM. The corporation was however not involved in this work.
Financial support: This work was supported by grants from the National Science and Engineering Research Council of Canada (NSERC) (to GB), Canadian Institutes of Health Research (CIHR) (to GB), Maisonneuve-Rosemont Hospital Foundation and Fondation de la Famille Pierre Theroux (to GB). RH is supported by a fellowship from the Maisonneuve-Rosemont Hospital Foundation. AF is supported by post-doctoral fellowship from the Jane Coffin Childs Fund.
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewer: Annamaria Cimini, University of L’Aquila, Italy.
Funding: This work was supported by grants from the National Science and Engineering Research Council of Canada (NSERC) (to GB), Canadian Institutes of Health Research (CIHR) (to GB), Maisonneuve-Rosemont Hospital Foundation and Fondation de la Famille Pierre Theroux (to GB). RH is supported by a fellowship from the Maisonneuve-Rosemont Hospital Foundation. AF is supported by post-doctoral fellowship from the Jane Coffin Childs Fund.
P-Reviewer: Cimini A; C-Editors: Zhao M, Qiu Y; T-Editor: Jia Y
References
- 1.Abdouh M, Hanna R, El Hajjar J, Flamier A, Bernier G. The polycomb repressive complex 1 protein BMI1 is required for constitutive heterochromatin formation and silencing in mammalian somatic cells. J Biol Chem. 2016;291:182–197. doi: 10.1074/jbc.M115.662403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Akiguchi I, Pallàs M, Budka H, Akiyama H, Ueno M, Han J, Yagi H, Nishikawa T, Chiba Y, Sugiyama H, Takahashi R, Unno K, Higuchi K, Hosokawa M. SAMP8 mice as a neuropathological model of accelerated brain aging and dementia: Toshio Takeda’s legacy and future directions. Neuropathology. 2017;37:293–305. doi: 10.1111/neup.12373. [DOI] [PubMed] [Google Scholar]
- 3.Alzforum Research Models: Alzheimer's Disease. 2020. [Accessed August 5, 2020]. Alzforum Available at: https://www.alzforum.org/research-models/alzheimers-disease .
- 4.Alzheimer's Association (2019) 2019 Alzheimer's disease facts and figures. Alzheimers Dement. 15:321–387. [Google Scholar]
- 5.Armstrong R. Risk factors for Alzheimer’s disease. Folia Neuropathol. 2019;57:87–105. doi: 10.5114/fn.2019.85929. [DOI] [PubMed] [Google Scholar]
- 6.Barabino A, Plamondon V, Abdouh M, Chatoo W, Flamier A, Hanna R, Zhou S, Motoyama N, Hébert M, Lavoie J, Bernier G. Loss of Bmi1 causes anomalies in retinal development and degeneration of cone photoreceptors. Development. 2016;143:1571–1584. doi: 10.1242/dev.125351. [DOI] [PubMed] [Google Scholar]
- 7.Belfiore R, Rodin A, Ferreir E, Velazquez R, Branca C, Caccamo A, Oddo S. Temporal and regional progression of Alzheimer’s disease-like pathology in 3xTg-AD mice. Aging cell. 2019;18:e12873. doi: 10.1111/acel.12873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bharadwaj P. Animal Models of Alzheimer’s Disease. In: Martins RN, Brennan CS, Fernando WMADB, Brennan MA, Fuller SJ, editors. Neurodegeneration and Alzheimer's Disease: The Role of Diabetes, Genetics, Hormones, and Lifestyle. Hoboken: John Wiley and Sons Limited; 2019. pp. 291–310. [Google Scholar]
- 9.Bhattacharya R, Banerjee Mustafi S, Street M, Dey A, Dwivedi SKD. Bmi-1: At the crossroads of physiological and pathological biology. Genes Dis. 2015;2:225–239. doi: 10.1016/j.gendis.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bird TD. Genetic aspects of Alzheimer disease. Genet Med. 2008;10:231–239. doi: 10.1097/GIM.0b013e31816b64dc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bis JC, DeCarli C, Smith AV, van der Lijn F, Crivello F, Fornage M, Debette S, Shulman JM, Schmidt H, Srikanth V, Schuur M, Yu L, Choi SH, Sigurdsson S, Verhaaren BF, DeStefano AL, Lambert JC, Jack CR, Jr, Struchalin M, Stankovich J, et al. Common variants at 12q14 and 12q24 are associated with hippocampal volume. Nat Genet. 2012;44:545–551. doi: 10.1038/ng.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blennow K, de Leon MJ, Zetterberg H. Alzheimer’s disease. Lancet. 2006;368:387–403. doi: 10.1016/S0140-6736(06)69113-7. [DOI] [PubMed] [Google Scholar]
- 13.Buckley RF, Mormino EC, Chhatwal J, Schultz AP, Rabin JS, Rentz DM, Acar D, Properzi MJ, Dumurgier J, Jacobs H, Gomez-Isla T, Johnson KA, Sperling RA, Hanseeuw BJ. Associations between baseline amyloid sex, and APOE on subsequent tau accumulation in cerebrospinal fluid. Neurobiol Aging. 2019;78:178–185. doi: 10.1016/j.neurobiolaging.2019.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cacace R, Sleegers K, Van Broeckhoven C. Molecular genetics of early-onset Alzheimer’s disease revisited. Alzheimers Dement. 2016;12:733–748. doi: 10.1016/j.jalz.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 15.Carter DB. The interaction of amyloid-beta with ApoE. In: Harris JR, Fahrenholz F, editors. Alzheimer's disease. Boston: Springer; 2005. pp. 255–272. [DOI] [PubMed] [Google Scholar]
- 16.Castellano JM, Kim J, Stewart FR, Jiang H, DeMattos RB, Patterson BW, Fagan AM, Morris JC, Mawuenyega KG, Cruchaga C, Goate AM, Bales KR, Paul SM, Bateman RJ, Holtzman DM. Human apoE isoforms differentially regulate brain amyloid- peptide clearance. Sci Transl Med. 2011;3:89ra57. doi: 10.1126/scitranslmed.3002156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chapuis J, Hansmannel F, Gistelinck M, Mounier A, Van Cauwenberghe C, Kolen KV, Geller F, Sottejeau Y, Harold D, Dourlen P, Grenier-Boley B, Kamatani Y, Delepine B, Demiautte F, Zelenika D, Zommer N, Hamdane M, Bellenguez C, Dartigues JF, Hauw JJ, et al. Increased expression of BIN1 mediates Alzheimer genetic risk by modulating tau pathology. Mol Psychiatry. 2013;18:1225–1234. doi: 10.1038/mp.2013.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chatoo W, Abdouh M, David J, Champagne M-P, Ferreira J, Rodier F, Bernier G. The polycomb group gene Bmi1 regulates antioxidant defenses in neurons by repressing p53 Pro-oxidant activity. J Neurosci. 2009;29:529–542. doi: 10.1523/JNEUROSCI.5303-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chouraki V, Seshadri S. Genetics of Alzheimer’s disease. Adv Genet. 2014;87:245–294. doi: 10.1016/B978-0-12-800149-3.00005-6. [DOI] [PubMed] [Google Scholar]
- 20.Corder E, Saunders A, Strittmatter W, Schmechel D, Gaskell P, Small G, Roses A, Haines J, Pericak-Vance M. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 21.Cosín-Tomás M, Alvarez-López MJ, Sanchez-Roige S, Lalanza JF, Bayod S, Sanfeliu C, Pallàs M, Escorihuela RM, Kaliman P. Epigenetic alterations in hippocampus of SAMP8 senescent mice and modulation by voluntary physical exercise. Front Aging Neurosci. 2014;6:51. doi: 10.3389/fnagi.2014.00051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Bruijn RF, Ikram MA. Cardiovascular risk factors and future risk of Alzheimer’s disease. BMC Med. 2014;12:130. doi: 10.1186/s12916-014-0130-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dong HK, Gim JA, Yeo SH, Kim HS. Integrated late onset Alzheimer’s disease (LOAD) susceptibility genes: cholesterol metabolism and trafficking perspectives. Gene. 2017;597:10–16. doi: 10.1016/j.gene.2016.10.022. [DOI] [PubMed] [Google Scholar]
- 24.Dorey E, Chang N, Liu QY, Yang Z, Zhang W. Apolipoprotein E amyloid-beta, and neuroinflammation in Alzheimer’s disease. Neurosci Bull. 2014;30:317–330. doi: 10.1007/s12264-013-1422-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.El Hajjar J, Chatoo W, Hanna R, Nkanza P, Tétreault N, Tse YC, Wong TP, Abdouh M, Bernier G. Heterochromatic genome instability and neurodegeneration sharing similarities with Alzheimer’s disease in old Bmi1+/− mice. Sci Rep. 2019;9:594. doi: 10.1038/s41598-018-37444-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Escamilla-Ayala A, Wouters R, Sannerud R, Annaert W. Contribution of the Presenilins in the cell biology structure and function of γ-secretase. Semin Cell Dev Biol. 2020;105:12–26. doi: 10.1016/j.semcdb.2020.02.005. [DOI] [PubMed] [Google Scholar]
- 27.Esquerda-Canals G, Montoliu-Gaya L, Güell-Bosch J, Villegas S. Mouse models of Alzheimer’s disease. J Alzheimers Dis. 2017;57:1171–1183. doi: 10.3233/JAD-170045. [DOI] [PubMed] [Google Scholar]
- 28.Fagan AM, Watson M, Parsadanian M, Bales KR, Paul SM, Holtzman DM. Human and murine ApoE markedly alters Aβ metabolism before and after plaque formation in a mouse model of Alzheimer’s disease. Neurobiol Dis. 2002;9:305–318. doi: 10.1006/nbdi.2002.0483. [DOI] [PubMed] [Google Scholar]
- 29.Fennema-Notestine C, Panizzon M, Thompson W, Chen C-H, Eyler L, Fischl B, Franz C, Grant M, Jak A, Jernigan T, Lyons M, Neale M, Seidman L, Tsuang M, Xian H, Dale A, Kremen W. Presence of ApoE ε4 allele associated with thinner frontal cortex in middle age. J Alzheimers Dis. 2011;26(Suppl 3):49–60. doi: 10.3233/JAD-2011-0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fernandez CG, Hamby ME, McReynolds ML, Ray WJ. The role of APOE4 in disrupting the homeostatic functions of astrocytes and microglia in aging and Alzheimer’s disease. Front Aging Neurosci. 2019;11:14. doi: 10.3389/fnagi.2019.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Flamier A, El Hajjar J, Adjaye J, Fernandes KJ, Abdouh M, Bernier G. Modeling late-onset sporadic Alzheimer’s disease through BMI1 deficiency. Cell Rep. 2018;23:2653–2666. doi: 10.1016/j.celrep.2018.04.097. [DOI] [PubMed] [Google Scholar]
- 32.Frost B, Hemberg M, Lewis J, Feany MB. Tau promotes neurodegeneration through global chromatin relaxation. Nat Neurosci. 2014;17:357–366. doi: 10.1038/nn.3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gan W, Nie B, Shi F, Xu XM, Qian JC, Takagi Y, Hayakawa H, Sekiguchi M, Cai JP. Age-dependent increases in the oxidative damage of DNA RNA, and their metabolites in normal and senescence-accelerated mice analyzed by LC-MS/MS: urinary 8-oxoguanosine as a novel biomarker of aging. Free Radic Biol Med. 2012;52:1700–1707. doi: 10.1016/j.freeradbiomed.2012.02.016. [DOI] [PubMed] [Google Scholar]
- 34.Grant WB, Campbell A, Itzhaki RF, Savory J. The significance of environmental factors in the etiology of Alzheimer’s disease. J Alzheimers Dis. 2002;4:179–189. doi: 10.3233/jad-2002-4308. [DOI] [PubMed] [Google Scholar]
- 35.Griñán-Ferré C, Corpas R, Puigoriol-Illamola D, Palomera-Ávalos V, Sanfeliu C, Pallàs M. Understanding epigenetics in the neurodegeneration of Alzheimer’s disease: SAMP8 mouse model. J Alzheimers Dis. 2018;62:943–963. doi: 10.3233/JAD-170664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Williams A, Jones N, Thomas C, Stretton A, Morgan AR, Lovestone S, Powell J, Proitsi P, Lupton MK, Brayne C, Rubinsztein DC, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet. 2009;41:1088–1093. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hollingworth P, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Williams A, Jones N, Thomas C, Stretton A, Morgan AR, Lovestone S, Powell J, Proitsi P, Lupton MK, Brayne C, Rubinsztein DC, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet. 2011;43:429–435. doi: 10.1038/ng.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hou Y, Dan X, Babbar M, Wei Y, Hasselbalch SG, Croteau DL, Bohr VA. Ageing as a risk factor for neurodegenerative disease. Nat Rev Neurol. 2019;15:565–581. doi: 10.1038/s41582-019-0244-7. [DOI] [PubMed] [Google Scholar]
- 39.Huang Y, Mahley RW. Apolipoprotein E: Structure and function in lipid metabolism neurobiology, and Alzheimer’s diseases. Neurobiol Dis. 2014;72:3–12. doi: 10.1016/j.nbd.2014.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang YWA, Zhou B, Wernig M, Südhof TC. ApoE2, ApoE3, and ApoE4 differentially stimulate APP transcription and Aβ secretion. Cell. 2017;168:427–441. doi: 10.1016/j.cell.2016.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jarvik G, Larson EB, Goddard K, Schellenberg GD, Wijsman EM. Influence of apolipoprotein E genotype on the transmission of Alzheimer disease in a community-based sample. Am J Hum Genet. 1996;58:191–200. [PMC free article] [PubMed] [Google Scholar]
- 42.Jia L, Xu H, Chen S, Wang X, Yang J, Gong M, Wei C, Tang Y, Qu Q, Chu L, Shen L, Zhou C, Wang Q, Zhao T, Zhou A, Li Y, Li F, Li Y, Jin H, Qin Q, JiaoH , Li Y, Zhang H, Lyu D, Shi Y, Song Y, Jia J. The APOE ε4 exerts differential effects on familial and other subtypes of Alzheimer’s disease. Alzheimers Dement. 2020 doi: 10.1002/alz.12153. doi: 10.1002/alz.12153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jochemsen HM, Muller M, van der Graaf Y, Geerlings MI. APOE ε4 differentially influences change in memory performance depending on age. The SMART-MR study. Neurobiol Aging. 2012;33:832.e15–22. doi: 10.1016/j.neurobiolaging.2011.07.016. [DOI] [PubMed] [Google Scholar]
- 44.Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, Bjornsson S, Huttenlocher J, Levey AI, Lah JJ, Rujescu D, Hampel H, Giegling I, Andreassen OA, Engedal K, Ulstein I, Djurovic S, Ibrahim-Verbaas C, Hofman A, Ikram MA, et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med. 2013;368:107–116. doi: 10.1056/NEJMoa1211103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kane AE, Sinclair DA. Epigenetic changes during aging and their reprogramming potential. Crit Rev Biochem Mol Biol. 2019;54:61–83. doi: 10.1080/10409238.2019.1570075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, Combarros O, Zelenika D, Bullido MJ, Tavernier B, Letenneur L, Bettens K, Berr C, Pasquier F, Fiévet N, Barberger-Gateau P, Engelborghs S, De Deyn P, Mateo I, Franck A, Helisalmi S, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet. 2009;41:1094–1099. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- 47.Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, DeStafano AL, Bis JC, Beecham GW, Grenier-Boley B, Russo G, Thorton-Wells TA, Jones N, Smith AV, Chouraki V, Thomas C, Ikram MA, Zelenika D, Vardarajan BN, Kamatani Y, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet. 2013;45:1452–1458. doi: 10.1038/ng.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lardenoije R, van den Hove D, Havermans M, van Casteren A, Le KX, Palmour R, Lemere CA, Rutten B. Age-related epigenetic changes in hippocampal subregions of four animal models of Alzheimer’s disease. Mol Cell Neurosci. 2018;86:1–15. doi: 10.1016/j.mcn.2017.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee MH, Siddoway B, Kaeser GE, Segota I, Rivera R, Romanow WJ, Liu CS, Park C, Kennedy G, Long T, Chun J. Publisher Correction: Somatic APP gene recombination in Alzheimer’s disease and normal neurons. Nature. 2019;566:E6. doi: 10.1038/s41586-019-0905-0. [DOI] [PubMed] [Google Scholar]
- 50.Li L, Jiang Y, Hu W, Tung YC, Dai C, Chu D, Gong CX, Iqbal K, Liu F. Pathological alterations of Tau in Alzheimer’s disease and 3xTg-AD mouse brains. Mol Neurobiol. 2019;56:6168–6183. doi: 10.1007/s12035-019-1507-4. [DOI] [PubMed] [Google Scholar]
- 51.Liao F, Zhang TJ, Jiang H, Lefton KB, Robinson GO, Vassar R, Sullivan PM, Holtzman DM. Murine versus human apolipoprotein E4: differential facilitation of and co-localization in cerebral amyloid angiopathy and amyloid plaques in APP transgenic mouse models. Acta Neuropathol Commun. 2015;3:70. doi: 10.1186/s40478-015-0250-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu J, Cao L, Chen J, Song S, Lee IH, Quijano C, Liu H, Keyvanfar K, Chen H, Cao LY, Ahn BH, Kumar NG, Rovira II, Xu XL, van Lohuizen M, Motoyama N, Deng CX, Finkel T. Bmi1 regulates mitochondrial function and the DNA damage response pathway. Nature. 2009;459:387–392. doi: 10.1038/nature08040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lutz MW, Crenshaw D, Welsh-Bohmer KA, Burns DK, Roses AD. New genetic approaches to AD: lessons from APOE-TOMM40 phylogenetics. Curr Neurol Neurosci Rep. 2016;16:48. doi: 10.1007/s11910-016-0643-8. [DOI] [PubMed] [Google Scholar]
- 54.Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, Gallins PJ, Buxbaum JD, Jarvik GP, Crane PK, Larson EB, Bird TD, Boeve BF, Graff-Radford NR, De Jager PL, Evans D, Schneider JA, Carrasquillo MM, Ertekin-Taner N, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet. 2011;43:436–441. doi: 10.1038/ng.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nussbaum RL, Ellis CE. Alzheimer’s disease and Parkinson’s disease. N Engl J Med. 2003;348:1356–1364. doi: 10.1056/NEJM2003ra020003. [DOI] [PubMed] [Google Scholar]
- 56.Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 57.Onos KD, Sukoff Rizzo SJ, Howell GR, Sasner M. Toward more predictive genetic mouse models of Alzheimer’s disease. Brain Res Bull. 2016;122:1–11. doi: 10.1016/j.brainresbull.2015.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Penney J, Ralvenius WT, Tsai LH. Modeling Alzheimer’s disease with iPSC-derived brain cells. Mol Psychiatry. 2020;25:148–167. doi: 10.1038/s41380-019-0468-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Petersen RC, Doody R, Kurz A, Mohs RC, Morris JC, Rabins PV, Ritchie K, Rossor M, Thal L, Winblad B. Current concepts in mild cognitive impairment. Arch Neurol. 2001;58:1985. doi: 10.1001/archneur.58.12.1985. [DOI] [PubMed] [Google Scholar]
- 60.Rebeck GW. The role of APOE on lipid homeostasis and inflammation in normal brains. J Lipid Res. 2017;58:1493–1499. doi: 10.1194/jlr.R075408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Reiman EM, Arboleda-Velasquez JF, Quiroz YT, Huentelman MJ, Beach TG, Caselli RJ, Chen Y, Su Y, Myers AJ, Hardy J, Paul Vonsattel J, Younkin SG, Bennett DA, De Jager PL, Larson EB, Crane PK, Keene CD, Kamboh MI, Kofler JK, Duque L, et al. Exceptionally low likelihood of Alzheimer’s dementia in APOE2 homozygotes from a 5,000-person neuropathological study. Nat Commun. 2020;11:667. doi: 10.1038/s41467-019-14279-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Reitz C, Mayeux R. Alzheimer disease: epidemiology diagnostic criteria, risk factors and biomarkers. Biochem Pharmacol. 2014;88:640–651. doi: 10.1016/j.bcp.2013.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rogaeva E, Meng Y, Lee JH, Gu Y, Kawarai T, Zou F, Katayama T, Baldwin CT, Cheng R, Hasegawa H, Chen F, Shibata N, Lunetta KL, Pardossi-Piquard R, Bohm C, Wakutani Y, Cupples LA, Cuenco KT, Green RC, Pinessi L, et al. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat Genet. 2007;39:168–177. doi: 10.1038/ng1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sando SB, Melquist S, Cannon A, Hutton M, Sletvold O, Saltvedt I, White LR, Lydersen S, Aasly J. Risk-reducing effect of education in Alzheimer’s disease. Int J Geriatr Psychiatry. 2008;23:1156–1162. doi: 10.1002/gps.2043. [DOI] [PubMed] [Google Scholar]
- 65.Saunders AM, Strittmatter WJ, Schmechel D, St. George-Hyslop PH, Pericak-Vance MA, Joo SH, Rosi BL, Gusella JF, Crapper-MacLachlan DR, Alberts MJ, Hulette C, Crain B, Goldgaber D, Roses AD. Association of apolipoprotein E allele 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology. 1993;43:1467–1467. doi: 10.1212/wnl.43.8.1467. [DOI] [PubMed] [Google Scholar]
- 66.Shaw P, Lerch JP, Pruessner JC, Taylor KN, Rose AB, Greenstein D, Clasen L, Evans A, Rapoport JL, Giedd JN. Cortical morphology in children and adolescents with different apolipoprotein E gene polymorphisms: an observational study. Lancet Neurol. 2007;6:494–500. doi: 10.1016/S1474-4422(07)70106-0. [DOI] [PubMed] [Google Scholar]
- 67.Sims R, Hill M, Williams J. The multiplex model of the genetics of Alzheimer’s disease. Nat Neurosci. 2020;23:311–322. doi: 10.1038/s41593-020-0599-5. [DOI] [PubMed] [Google Scholar]
- 68.Stern Y. Cognitive reserve in ageing and Alzheimer’s disease. Lancet Neurol. 2012;11:1006–1012. doi: 10.1016/S1474-4422(12)70191-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Strang KH, Golde TE, Giasson B I. MAPT mutations, tauopathy, and mechanisms of neurodegeneration. Lab Invest. 2019;99:912–928. doi: 10.1038/s41374-019-0197-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sulkowski PL, Oeck S, Dow J, Economos NG, Mirfakhraie L, Liu Y, Noronha K, Bao X, Li J, Shuch BM, King MC, Bindra RS, Glazer PM. Oncometabolites suppress DNA repair by disrupting local chromatin signalling. Nature. 2020;582:586–591. doi: 10.1038/s41586-020-2363-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sun Y, Jiang X, Xu Y, Ayrapetov MK, Moreau LA, Whetstine JR, Price BD. Histone H3 methylation links DNA damage detection to activation of the tumour suppressor Tip60. Nat Cell Biol. 2009;11:1376–1382. doi: 10.1038/ncb1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tai LM, Ghura S, Koster KP, Liakaite V, Maienschein-Cline M, Kanabar P, Collins N, Ben-Aissa M, Lei AZ, Bahroos N, Green SJ, Hendrickson B, Van Eldik LJ, LaDu MJ. APOE-modulated Aβ-induced neuroinflammation in Alzheimer’s disease: current landscape novel data, and future perspective. J Neurochem. 2015;133:465–488. doi: 10.1111/jnc.13072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tooyama I, Sasaki K, Oomura Y, Li AJ, Kimura H. Effect of acidic fibroblast growth factor on basal forebrain cholinergic neurons in senescence-accelerated mice. Exp Gerontol. 1997;32:171–179. doi: 10.1016/s0531-5565(96)00071-x. [DOI] [PubMed] [Google Scholar]
- 74.Van Cauwenberghe C, Van Broeckhoven C, Sleegers K. The genetic landscape of Alzheimer disease: clinical implications and perspectives. Genet Med. 2016;18:421–430. doi: 10.1038/gim.2015.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.van der Flier WM, Pijnenburg YA, Fox NC, Scheltens P. Early-onset versus late-onset Alzheimer’s disease: the case of the missing APOE ε4 allele. Lancet Neurol. 2011;10:280–288. doi: 10.1016/S1474-4422(10)70306-9. [DOI] [PubMed] [Google Scholar]
- 76.van der Lugt NM, Domen J, Linders K, van Roon M, Robanus-Maandag E, te Riele H, van der Valk M, Deschamps J, Sofroniew M, van Lohuizen M. Posterior transformation neurological abnormalities, and severe hematopoietic defects in mice with a targeted deletion of the bmi-1 proto-oncogene. Genes Dev. 1994;8:757–769. doi: 10.1101/gad.8.7.757. [DOI] [PubMed] [Google Scholar]
- 77.van der Vlies AE, Koedam ELGE, Pijnenburg YAL, Twisk JWR, Scheltens P, van der Flier VM. Most rapid cognitive decline in APOE epsilon4 negative Alzheimer’s disease with early onset. Psychol Med. 2009;39:1907–1911. doi: 10.1017/S0033291709005492. [DOI] [PubMed] [Google Scholar]
- 78.Verghese PB, Castellano JM, Holtzman DM. Roles of apolipoprotein E in Alzheimer’s disease and other neurological disorders. Lancet Neurol. 2011;23:1–7. doi: 10.1016/S1474-4422(10)70325-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Viña J, Lloret A. Why women have more Alzheimer’s disease than men: gender and mitochondrial toxicity of amyloid-beta peptide. J Alzheimers Dis 20 Suppl. 2010;2:S527–533. doi: 10.3233/JAD-2010-100501. [DOI] [PubMed] [Google Scholar]
- 80.Walker MP, LaFerl FM, Oddo SS, Brewer GJ. Reversible epigenetic histone modifications and Bdnf expression in neurons with aging and from a mouse model of Alzheimer’s disease. Age (Dordr) 2013;35:519–531. doi: 10.1007/s11357-011-9375-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang C, Najm R, Xu Q, Jeong D-E, Walker D, Balestra ME, Yoon SY, Yuan H, Li G, Miller ZA, Miller BL, Malloy MJ, Huang Y. Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector. Nat Med. 2018;24:647–657. doi: 10.1038/s41591-018-0004-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yin Y, Wang Z. ApoE and neurodegenerative diseases in aging. Adv Exp Med Biol. 2018;1086:77–92. doi: 10.1007/978-981-13-1117-8_5. [DOI] [PubMed] [Google Scholar]
- 83.Zhang L, Chen C, Mak MS, Lu J, Wu Z, Chen Q, Han Y, Li Y, Pi R. Advance of sporadic Alzheimer’s disease animal models. Med Res Rev. 2020;40:431–458. doi: 10.1002/med.21624. [DOI] [PubMed] [Google Scholar]
- 84.Zhang W, Li J, Suzuki K, Qu J, Wang P, Zhou J, Liu X, Ren R, Xu X, Ocampo A, Yuan T, Yang J, Li Y, Shi L, Guan D, Pan H, Duan S, Ding Z, Li M, Yi F, et al. A Werner syndrome stem cell model unveils heterochromatin alterations as a driver of human aging. Science. 2015;348:1160–1163. doi: 10.1126/science.aaa1356. [DOI] [PMC free article] [PubMed] [Google Scholar]