

Neuronal cell death is the main hallmark of Parkinson’s disease (PD). It is an irreversible process promoted by neurotoxins and/or genetic mutations. Different types of cell death have been associated with PD. The mechanisms by which neurons decide to specific type of cell death remain elusive. However, it is well known that cell death can be either programmed or not. Apoptosis is a programmed cell death that involves the release of cytochrome C from damaged mitochondria to cytosol and the activation of caspases leading to nuclear condensation. Necrosis is a caspase-independent cell death characterized by a gain in cell volume, rupture of plasma membrane and leak of cell contents, inflammation, and affects neighbouring cells. It was classified as a non-programmed cell death, but there are types of necrotic death triggered by a protein activation cascade, including necroptosis.
Necrostatin-1 (Nec-1) inhibits necroptosis: Nec-1 inhibits necroptosis, a programmed necrotic cell death, which is regulated by subsequent activation of and interaction with the receptor-interacting protein (RIP) 1, RIP3 and the mixed lineage kinase domain-like (MLKL) protein. The MLKL protein is crucial for the rupture of the plasma membrane and the release of cell contents, and it triggers inflammation, oxidative stress and cell death (Alegre-Cortés et al., 2020). Nec-1 has been shown to be neuroprotective in different cells (Ito et al., 2017) and animal models of neurodegenerative diseases (Iannielli et al., 2018), such as PD. Although Nec-1 has also been related to the inhibition of nonnecroptotic cell death (Ito et al., 2017), it is indeed an inhibitor of RIP1 kinase activity. Nec-1 has analogs, such as Nec-1s (the stable variant) that seems to be more efficient in inhibiting the activity of this serine/threonine protein kinase (Iannielli et al., 2018). The activation of necroptosis is generally related to the tumor necrosis factor (TNF) receptor pathway (Remijsen et al., 2014), but other studies have demonstrated that the strong generation of reactive oxygen species (ROS; Zhang et al., 2017) and neurotoxins (Lin et al., 2020) can lead to the cascade activation of the RIP1 and RIP3 proteins (Figure 1). In this regard, necroptosis has been detected in neuronal cells exposed to neurotoxins [6-hydroxydopamine (Oñate et al., 2020), rotenone (Alegre-Cortés et al., 2020), and 1-methyl-4-phenylpyridinium iodide], in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced mouse models (Lin et al., 2020), and in postmortem brain samples (Oñate et al., 2020) but not in fibroblasts from patients with PD. However, when cells from genetic and sporadic PD patients were exposed to the neurotoxin rotenone, they displayed an increase in MLKL phosphorylation (Alegre-Cortés et al., 2020). There is no doubt that necroptosis is activated in PD models; however, this activation could be a priori related to the sensitivity of the model. Despite the inhibition of necroptosis by Nec-1 (Alegre-Cortés et al., 2020) or Nec-1s (Oñate et al., 2020), other cell death processes are still activated in certain experimental disease models. Most likely, the inhibition of necroptosis activates a switch between cell death pathways, which depends on Nec-1 dose-responses and disease models. Given that multiple pathways of cell death (necroptosis, apoptosis, and autophagy) are simultaneously involved in rotenone toxicity, Nec-1 inhibits rotenone-induced necroptosis but does not reverse the inhibition of mitochondrial complex I and seems to potentiate apoptosis and/or necrosis. Consistently, knockout of RIP3 or MLKL did not attenuate neuronal apoptosis in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced mice (Lin et al., 2020). Therefore, we surmised that neuroprotection with Nec-1 was partial because of the sustained mitochondrial ROS generation, the presence of nuclear condensation, and the detection of propidium iodide-positive cells. However, Goodall et al. (2016) suggested that the autophagy machinery could control this switch. In fact, they described a strong interaction between necrosome components and proteins of the autophagosome formation. Autophagy-related gene (ATG) protein such as sequestosome 1 (SQSTM1) promotes necrosome/necroptosis activation upon TNF-related apoptosis inducing ligand treatment. The knockdown of sqstm1 as well as Nec-1 abrogates this interaction and promotes apoptosis.
Figure 1.
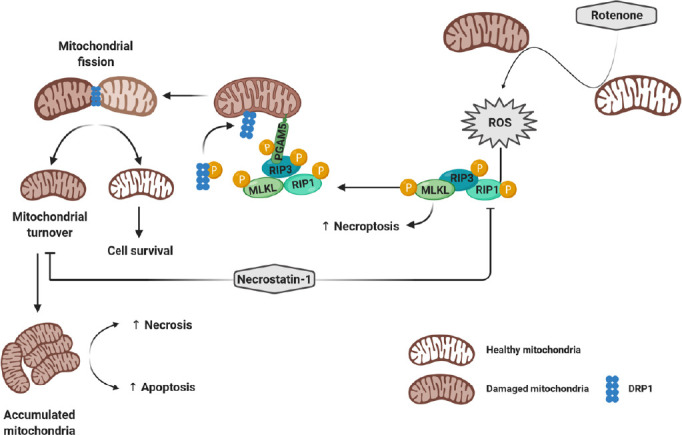
Necrostatin-1 downregulates rotenone-activated necroptosis and mitophagy.
Through the inhibition of mitochondrial complex I, rotenone triggers ROS generation, which leads to the activation of the necrosome complex (RIP1-RIP3-MLKL) and necroptosis. Meanwhile, RIP3 activates the serine/threonine phosphatase protein PGAM5. The latter recruits into mitochondria and dephosphorylates DRP1 to promote the mitochondrial fission and turnover. However, necrostatin-1 inhibits both rotenone-induced necroptosis and mitophagy and enhances necrosis and apoptotic cell death. DRP1: Dynamin-related protein 1; MLKL: mixed lineage kinase domain-like protein; PGAM5: phosphoglycerate mutase 5; RIP: receptor-interacting protein; ROS: reactive oxygen species.
Necrostatin-1 downregulates mitophagy: Autophagy is a dynamic cellular mechanism that engulfs the cytosolic cargo, into a double membrane structure called autophagosome, to be later degraded in autophagolysosomes. In most neurodegenerative diseases, autophagy is impaired, which affects the turnover of misfolded proteins and damaged organelles. This impairment, which can be due to either exacerbation or inhibition of this catabolic process, can sensitize cells to death. Optimal autophagy induction generally maintains cell viability and metabolism. This catabolic process can be triggered by different stress stimuli to promote cell protection and survival. Although rotenone is an inhibitor of mitochondrial complex I and an efficient ROS generator, it induces mitophagy and necroptosis in neuronal cells (Alegre-Cortés et al., 2020). Consequently, the inhibition of necroptosis by Nec-1 promotes an increase of mitochondrial mass and further facilitates apoptosis and necrotic cell death in PD patients and rotenone-exposed cells. The inhibitory effect of Nec-1 on autophagy has been reported in 6-hydroxydopamine-treated neurons (Wu et al., 2015). Additionally, RIP1 knockdown upregulated autophagy, while Nec-1 was shown to downregulate autophagy (Yonekawa et al., 2015). This suppression of autophagy is RIP1 kinase-independent. Therefore we think that autophagy inhibition could provoke the accumulation of damaged mitochondria in rotenone-exposed cells. Remarkably, Nec-1 prevented mitophagy in human fibroblasts and SH-SY5Y cells even in the presence of carbonyl cyanide 3-chlorophenylhydrazone (CCCP; Alegre-Cortés et al., 2020). Thus, the inhibition of mitophagy by Nec-1 could occur through the RIP1 protein as an adapter and not as a kinase. Consequently, RIP1 kinase inhibition does not alleviate rotenone-induced mitochondrial damage and fails to reverse mitochondrial dysfunction and mitochondrial ROS generation (Figure 1). Previous studies have associated the activation of necroptosis with dynamin-related protein 1 (DRP1)-dependent mitochondrial fission. Upon TNF treatment, RIP3 phosphorylates a mitochondrial phosphatase protein, phosphoglycerate mutase 5 (PGAM5), which, in turn, leads to the dephosphorylation (serine 637) and activation of DRP1 (Remijsen et al 2014). Mitochondrial fission promotes the clearance of damaged mitochondria via mitophagy and protects the healthy mitochondria. However, it remains unclear whether DRP1 promotes the removal of damaged mitochondria or potentiates TNF-induced necroptosis. We hypothesized that the implication of DRP1 might precede MLKL activation, even though Remijsen et al. (2014) reported that mitophagy did not contribute to necroptosis. We have not tested this hypothesis in PD models but did observe that the mitochondrial mass increased, the lysosomal size was enlarged and Nec-1 prevented CCCP-induced mitophagy (Alegre-Cortés et al., 2020). In fact, CCCP induces mitophagy in SH-SY5Y cells via the increased expression of the PGAM5, DRP1 and PTEN-induced putative kinase/Parkin proteins. The loss of PGAM5 or DRP1 reduces autophagosome formation, prevents CCCP-induced mitophagy, and enhances CCCP-induced apoptosis (Park et al., 2018). We think that the use of Nec-1 affects not only the RIP1-RIP3-MLKL but also the RIP1-PGAM5-DRP1 axis, preventing mitochondrial fission and subsequent mitochondrial turnover (Figure 1). By inhibiting PINK1/Parkin-mediated mitophagy, Nec-1 affects mitochondrial morphology and mitochondrial clearance, which could enhance the effect of any parkinsonian toxin. It would be interesting and crucial to combine Nec-1 with other drugs regulating or modulating autophagy/mitophagy and oxidative stress.
We thank FUNDESALUD for helpful assistance.
This work was supported by grants from Junta de Extremadura, Spain, No. IB18048 (to EAC), the ONCE Foundation (to GMC), the Isabel Gemio Foundation (to SMSYD), the Instituto de Salud Carlos III, CIBERNED, No. CB06/05/004 (to JMF), and partially supported by the “Fondo Europeo de Desarrollo Regional” (FEDER) from the European Union.
Additional file 1: Open peer review report 1 (79.3KB, pdf) .
Footnotes
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewer: Abraam M. Yakoub, Stanford University, USA.
C-Editors: Zhao M, Wang L; T-Editor: Jia Y
References
- 1.Alegre-Cortés E, Muriel-González A, Canales-Cortés S, Uribe-Carretero E, Martínez-Chacón G, Aiastui A, López de Munain A, Niso-Santano M, Gonzalez-Polo RA, Fuentes JM, Yakhine-Diop SMS. Toxicity of necrostatin-1 in Parkinson’s disease models. Antioxidants (Basel) 2020;9:524. doi: 10.3390/antiox9060524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goodall ML, Fitzwalter BE, Zahedi S, Wu M, Rodriguez D, Mulcahy-Levy JM, Green DR, Morgan M, Cramer SD, Thorburn A. The autophagy machinery controls cell death switching between apoptosis and necroptosis. Dev Cell. 2016;37:337–349. doi: 10.1016/j.devcel.2016.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Iannielli A, Bido S, Folladori L, Segnali A, Cancellieri C, Maresca A, Massimino L, Rubio A, Morabito G, Caporali L, Tagliavini F, Musumeci O, Gregato G, Bezard E, Carelli V, Tiranti V, Broccoli V. Pharmacological inhibition of necroptosis protects from dopaminergic neuronal cell death in Parkinson’s disease models. Cell Rep. 2018;22:2066–2079. doi: 10.1016/j.celrep.2018.01.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ito K, Eguchi Y, Imagawa Y, Akai S, Mochizuki H, Tsujimoto Y. MPP+ induces necrostatin-1- and ferrostatin-1-sensitive necrotic death of neuronal SH-SY5Y cells. Cell Death Discov. 2017;3:17013. doi: 10.1038/cddiscovery.2017.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lin QS, Chen P, Wang WX, Lin CC, Zhou Y, Yu LH, Lin YX, Xu YF, Kang DZ. RIP1/RIP3/MLKL mediates dopaminergic neuron necroptosis in a mouse model of Parkinson disease. Lab Invest. 2020;100:503–511. doi: 10.1038/s41374-019-0319-5. [DOI] [PubMed] [Google Scholar]
- 6.Oñate M, Catenaccio A, Salvadores N, Saquel C, Martinez A, Moreno-Gonzalez I, Gamez N, Soto P, Soto C, Hetz C, Court FA. The necroptosis machinery mediates axonal degeneration in a model of Parkinson disease. Cell Death Differ. 2020;27:1169–1185. doi: 10.1038/s41418-019-0408-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Park YS, Choi SE, Koh HC. PGAM5 regulates PINK1/Parkin-mediated mitophagy via DRP1 in CCCP-induced mitochondrial dysfunction. Toxicol Lett. 2018;284:120–128. doi: 10.1016/j.toxlet.2017.12.004. [DOI] [PubMed] [Google Scholar]
- 8.Remijsen Q, Goossens V, Grootjans S, Van den Haute C, Vanlangenakker N, Dondelinger Y, Roelandt R, Bruggeman I, Goncalves A, Bertrand MJ, Baekelandt V, Takahashi N, Berghe TV, Vandenabeele P. Depletion of RIPK3 or MLKL blocks TNF-driven necroptosis and switches towards a delayed RIPK1 kinase-dependent apoptosis. Cell Death Dis. 2014;5:e1004. doi: 10.1038/cddis.2013.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu JR, Wang J, Zhou SK, Yang L, Yin JL, Cao JP, Cheng YB. Necrostatin-1 protection of dopaminergic neurons. Neural Regen Res. 2015;10:1120–1124. doi: 10.4103/1673-5374.160108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yonekawa T, Gamez G, Kim J, Tan AC, Thorburn J, Gump J, Thorburn A, Morgan MJ. RIP1 negatively regulates basal autophagic flux through TFEB to control sensitivity to apoptosis. EMBO Rep. 2015;16:700–708. doi: 10.15252/embr.201439496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Y, Su SS, Zhao S, Yang Z, Zhong CQ, Chen X, Cai Q, Yang ZH, Huang D, Wu R, Han J. RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat Commun. 2017;8:14329. doi: 10.1038/ncomms14329. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.