

Abstract
Dementia is a clinical syndrome that affects approximately 47 million people worldwide and is characterized by progressive and irreversible decline of cognitive, behavioral and sesorimotor functions. Alzheimer’s disease (AD) accounts for approximately 60–80% of all cases of dementia, and neuropathologically is characterized by extracellular deposits of insoluble amyloid-β (Aβ) and intracellular aggregates of hyperphosphorylated tau. Significantly, although for a long time it was believed that the extracellular accumulation of Aβ was the culprit of the symptoms observed in these patients, more recent studies have shown that cognitive decline in people suffering this disease is associated with soluble Aβ-induced synaptic dysfunction instead of the formation of insoluble Aβ-containing extracellular plaques. These observations are translationally relevant because soluble Aβ-induced synaptic dysfunction is an early event in AD that precedes neuronal death, and thus is amenable to therapeutic interventions to prevent cognitive decline before the progression to irreversible brain damage. The plasminogen activating (PA) system is an enzymatic cascade that triggers the degradation of fibrin by catalyzing the conversion of plasminogen into plasmin via two serine proteinases: tissue-type plasminogen activator (tPA) and urokinase-type plasminogen activator (uPA). Experimental evidence reported over the last three decades has shown that tPA and uPA play a role in the pathogenesis of AD. However, these studies have focused on the ability of these plasminogen activators to trigger plasmin-induced cleavage of insoluble Aβ-containing extracellular plaques. In contrast, recent evidence indicates that activity-dependent release of uPA from the presynaptic terminal of cerebral cortical neurons protects the synapse from the deleterious effects of soluble Aβ via a mechanism that does not require plasmin generation or the cleavage of Aβ fibrils. Below we discuss the role of the PA system in the pathogenesis of AD and the translational relevance of data published to this date.
Keywords: Alzheimer's disease, amyloid precursor protein, amyloid β, neuroserpin, plasmin, plasminogen activating system, plasminogen activator inhibitor-1, synapse, tissue-type plasminogen activator, urokinase-type plasminogen activator
Introduction
Dementia is a complex process characterized by a progressive and irreversible decline in at least two neuropsychiatric or cognitive domains that cannot be explained by systemic conditions such as delirium, or non neurodegenerative or primary neuropsychiatric disorders (Elahi and Miller, 2017). The different forms of dementias are classified according to the accumulation of specific protein aggregates in neurons, glia and the extracellular compartment. In line with these observations, most of the dementias that are not caused by cerebrovascular disease predominantly fall into one of the following six neurodegenerative proteinopathies: amyloid-β (Aβ), microtubule associated protein tau, TAR DNA-binding protein 43, fused in sarcoma, α-synuclein, and prion protein (DeTure and Dickson, 2019; Robinson et al., 2020).
Alzheimer’s disease (AD) accounts by 60–80% of all dementias. It affects approximately 46.8 million people worldwide, and this number is expected to reach 131.5 million by 2050 (Long and Holtzman, 2019). It is a dual proteinopathy characterized by the extracellular deposition of Aβ 1–40 and 1–42 fibrils in neuritic plaques, and intracellular aggregates of hyperphosphorylated tau in neurofibrillary tangles. Aβ 1–40 and 1–42 are generated by the amyloidogenic process of the amyloid precursor protein (APP). More specifically, the proteolytic processing of APP by α-secretase on the cell membrane generates soluble APPα (Chen et al., 2017; DeTure and Dickson, 2019), which has been implicated in neuronal plasticity and synaptogenesis (Muller et al., 2017). However, those APP molecules that fail to be processed by α-secretase are endocytosed and cleaved by β-secretase 1 and γ-secretase to generate Aβ 1–40 and 1–42 peptides (Mamada et al., 2015; Wang et al., 2017; Zhao et al., 2020), which have a harmful effect on cell survival and synaptic structure and function (Collaborators, 2019). Importantly, recent studies have shown that the soluble non-fibrillar form of Aβ is more toxic to the synapse that the fibrillar form (Ferreira et al., 2015).
Search Strategy
For the present review, we searched the literature using keywords such as dementia, Alzheimer’s disease, plasminogen activation system, tissue-type plasminogen activator, urokinase-type plasminogen activator, and plasmin on PubMed and Google Scholar from their inception to 2020. In addition, we also used modifications of the above main keywords to thoroughly search the literature. The major inclusion criteria preferred the literature comprising biological bases of Alzheimer’s disease and the plasminogen activation system.
Synaptic Dysfunction in Alzheimer’s Disease
The long-time proposed idea that the extracellular accumulation of insoluble Aβ peptides is the culprit of the cognitive and behavioral deficits observed in AD patients (Price and Morris, 1999) has been challenged by several unsuccessful clinical trials aimed at curtailing or preventing the formation of Aβ-containing extracellular plaques (Makin, 2018). In line with these observations, neuropathological and clinical studies have shown that the development of cognitive deficits in AD patients correlates with abnormalities in synaptic structure and function better than the number of tangles and insoluble Aβ-containing plaques (Terry et al., 1991; Selkoe, 2002). This concept is of significant translational importance, because synaptic dysfunction is an early event in the pathogenesis of AD that precedes neuronal death (Forner et al., 2017; Mondragon-Rodriguez et al., 2020) and thus is amenable to therapeutic interventions to prevent its development.
The last two decades of research have significantly contributed to our understanding of early synaptic dysfunction in AD. A growing body of experimental evidence indicates that neuronal activity regulates Aβ production (Kamenetz et al., 2003; Tampellini, 2015), and that while at low concentrations soluble Aβ induces presynaptic facilitation, at high concentrations triggers post-synaptic depression (Palop and Mucke, 2010; Marsh and Alifragis, 2018). In line with these observations, it has been shown that by decreasing the abundance of glutamatergic receptors in the post-synaptic density, elevated soluble Aβ attenuates excitatory synaptic transmission, thus causing the collapse of glutamatergic dendritic spines (He et al., 2019; Rolland et al., 2020). Additionally, several reports have shown that soluble Aβ blocks α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid and N-methyl-D-aspartate receptor receptor function, impairs long term potentiation, enhances long term depression, and augments the excitotoxic effect of glutamate by blocking its reuptake from the synaptic cleft (Charkhkar et al., 2015; Jackson et al., 2019; Liu et al., 2019). The translational relevance of these observations is underscored by the fact that increased soluble Aβ in the brain of AD patients is linked to synaptic depression and disruption of neuronal network activity (Palop and Mucke, 2010; Charkhkar et al., 2015). Importantly, the harmful effects of Aβ on the synapse are potentially reversible inasmuch as they precede neuronal death.
The Plasminogen Activation System
Plasminogen is an ubiquitous zymogen produced mainly in the liver. It binds not only to receptors broadly distributed on the cell surface of prokaryotic and eukaryotic cells, but also to molecules such as laminin, fibronectin, fibrin and thrombospondin. The proteolytic cleavage of plasminogen by tissue-type plasminogen activator (tPA), urokinase-type plasminogen activator (uPA), and specific bacterial proteins, generates plasmin, a potent trypsin-like proteinase with wide substrate specificity (Svenningsen et al., 2017). Inasmuch as unrestrained plasmin-induced proteolytic activity is potentially hazardous, cells secrete plasminogen activator inhibitors (PAIs) to tightly control the activation of plasminogen into plasmin. PAIs are members of the serine proteinase inhibitor superfamily (serpins) that are abundantly present in most body tissues, including the brain (Lin et al., 2020). The serpin molecules harbor a peptide, known as reactive center loop, that acts as a pseudosubstrate for the target enzyme. Cleavage of the reactive center loop by the proteinase triggers a conformational change that moves the proteinase from the top to the bottom of the serpin molecule. Simultaneously, the reactive center loop inserts into the β-sheet A of the serpin, thus irreversibly inactivating the proteinase (Huntington et al., 2000).
PAI-1 is a 50 kDa serpin that binds to and inhibits tPA and uPA. Following its secretion, PAI-1 is transformed into a latent conformation unable to form complexes with plasminogen activators. However, its interaction with vitronectin and heparin stabilizes its active conformation and broaden its specificity towards thrombin. PAI-1 also binds to uPA bound to its receptor (uPAR), thereby inhibiting plasmin-mediated degradation of the extracellular matrix. Neuroserpin is another serpin that preferentially binds to and inhibits tPA (Lee et al., 2017), and is localized in neurons of regions of the brain where tPA is also found. Significantly, the release of neuroserpin has been reported to have a neuroprotective effect (Yepes et al., 2000; Wang et al., 2015; Yang et al., 2016; Li et al., 2017; Wu et al., 2017).
Despite the fact that for a long time it was believed that the only function of the PA system was to catalyze the generation of plasmin from the zymogen plasminogen in the intravascular space and on the cell surface, a substantial body of experimental evidence generated in the last 20 years has shown that tPA and uPA also play a central role in central nervous system (CNS) function and dysfunction, and that some of their functions are plasminogen-independent. Indeed, tPA and uPA activity have been detected in well defined areas of the brain (Vassalli et al., 1991; Sappino et al., 1993; Chevilley et al., 2015; Lenoir et al., 2019), where they play a plethora of roles that not always require plasmin generation. Accordingly, tPA is crucial for the development of synaptic plasticity (Seeds et al., 1995) and regulation of blood-brain barrier (BBB) permeability (Yepes, 2015, 2016), while uPA induces neurogenesis in the developing brain (Dent et al., 1993), and its release in the mature central nervous system triggers astrocytic activation (Diaz et al., 2017) and promotes axonal and synaptic recovery following different forms of injury (Merino et al., 2016). Importantly, both activators are packed into synaptic vesicles located in the presynaptic and post-synaptic terminals where they are released by calcium-dependent mechanisms (Jeanneret et al., 2018; Diaz et al., 2020). Furthermore, once in the synaptic cleft both activators act as modulators of synaptic function (Diaz et al., 2017, 2018; Jeanneret et al., 2018). As it will be discussed below, a substantial body of evidence indicates that both plasminogen activators and their inhibitors play a role in the pathogenesis of dementias.
The Plasminogen Activation System in the Pathogenesis of Dementia
Below we discuss data supporting a role for five components of the PA system in the pathogenesis of AD: plasmin, tissue-type plasminogen activator, PAI-1, neuroserpin and urokinase-type plasminogen activator.
Plasmin in the brain of AD patients
In vitro studies have demonstrated that plasmin cleaves insoluble Aβ fibrils (Tucker et al., 2000a, b) and induces neuroprotection by prompting the conversion of pro-brain derived neurotrophic factor into brain derived neurotrophic factor (Angelucci et al., 2019). Furthermore, plasmin expression has been found to be restricted to lipid rafts, where it has been described to trigger α-secretase-induced cleavage of Aβ (Mattei et al., 2020). Remarkably, it has been reported that plasmin’s expression and activity are reduced in AD brains (Ledesma et al., 2000), likely due to neuroserpin-induced decrease in tPA activity (Fabbro and Seeds, 2009). However, other studies have failed to reproduce these observations, and instead have shown that the abundance of plasminogen and plasmin are unchanged in the brain of AD patients (Barker et al., 2010). These investigators have suggested that the reported decrease in plasmin is actually due to disruption of lipid rafts caused by abnormal cholesterol metabolism in the neuronal membrane of these patients (Dotti et al., 2004).
tPA in the pathogenesis of AD
tPA is a 70 kDa protein secreted as a single-chain precursor that is cleaved by plasmin at Arg275-Ile276 to generate an active two-chain form. The molecule of tPA is assembled by an amino terminal region (fibronectin- or finger-like domain), an EGF-like domain, two kringles, and a serine protease region with the activity-site residues His322, Asp371 and Ser478. Despite the fact that in the mature brain tPA is detected in most neurons, its enzymatic activity is limited to well-defined areas, namely amygdala, hypothalamus, hippocampal complex and dentate gyrus (Sappino et al., 1993). This apparent discrepancy between the expression of tPA and its enzymatic activity suggests that the role of this plasminogen activator in the central nervous system is not limited to tissue remodeling and fibrinolysis. In line with these observations, subsequent studies have found that membrane depolarization induces the rapid release of tPA from cerebral cortical neurons (Echeverry et al., 2010), and that this plasminogen activator mediates the development of neuronal plasticity in in vitro and in vivo models of long-term potentiation (Qian et al., 1993), learning (Seeds et al., 1995, 2003), stress-induced anxiety (Pawlak et al., 2003), and visual cortex plasticity (Muller and Griesinger, 1998).
During fibrinolysis, tPA binding to fibrin aggregates increases its affinity for plasminogen, thus triggering its cleavage into plasmin. Remarkably, it has been found that insoluble Aβ aggregates also activate tPA (Kingston et al., 1995). Furthermore, tPA mRNA is increased in cerebral cortical neurons treated with insoluble Aβ and in the brain of mice with a transgene with the Aβ precursor. Along these observations, it has been proposed that the main role of tPA is to prompt plasmin-induced cleavage of extracellular insoluble Aβ-containing plaques (Tucker et al., 2000a). However, in apparent discrepancy with these reports, in vivo studies with mouse models of AD have revealed that chronic elevation of Aβ actually decreases tPA activity by enhancing the inhibitory effect of PAI-1, and that the intracerebral injection of Aβ causes neuronal degeneration in animals genetically deficient on either tPA or plasminogen, but not in their wild-type controls (Melchor et al., 2003). Importantly, in seemingly contradiction with a proposed protective role of tPA in the brain of AD patients, other studies haven shown that this proteinase actually mediates the neurotoxic effect of Aβ via its ability to activate ERK 1/2 (Medina et al., 2005).
PAI-1: the culprit of plasminogen activator system dysfunction in AD?
PAI-1 is detected in various cell types, and its synthesis is induced by a variety of growth factors, hormones and inflammatory cytokines. This SERPIN not only inactivates tPA and uPA, but also modulates uPA-triggered proteolysis on the cell surface, and cell adhesion and migration. In line with these observations, multiple studies have shown that genetic deficiency of PAI-1 is linked to decreased thrombosis, accelerated neointima formation, and reduced lung inflammation, atherosclerosis and cancer invasion (Irigoyen et al., 1999). PAI-1 expression is increased in a murine aging model [klotho mutant (kl/kl) (Takeshita et al., 2002)], in senescent cells (Mu and Higgins, 1995), in the plasma of healthy aging humans (Yamamoto et al., 2005), and in the cerebrospinal fluid (Sutton et al., 1994) and the brain of AD patients (Liu et al., 2011). It has been reported that genetic deletion of PAI-1 in the brain of a murine model of AD reduces the deposition of Aβ, purportedly by increasing tPA-induced plasmin-mediated cleavage of Aβ-containing plaques (Liu et al., 2011). Together, these data have led to propose a model in which increased PAI-1 activity in the brain of AD patients abrogates plasmin-triggered cleavage of Aβ by blocking tPA-catalyzed conversion of plasminogen into plasmin.
Familial encephalopathy with neuroserpin inclusion bodies
Neuroserpin is a member of the SERPIN gene family originally identified as an axonally secreted protein (Osterwalder et al., 1996) in neurons of the central and peripheral nervous system (Hastings et al., 1997). It is widely expressed during the late stages of development. In contrast, its expression in the mature brain its expression is restricted to highly plastic areas such as the cerebral cortex, hippocampus and amygdala (Krueger et al., 1997). Furthermore, neuroserpin is also found in the pancreas, heart, kidney and testis, as well as in the pituitary and adrenal glands (Hill et al., 2000). Neuroserpin inhibits tPA and to a lesser extent uPA and plasmin, but does not exhibit inhibitory activity against thrombin (Hastings et al., 1997). Because neuroserpin preferentially inhibits tPA, in contrast with PAI-1 that inhibits tPA and uPA, and given its preferential neuronal localization, it has been proposed that this serpin is the selective inhibitor of tPA in neurons (Hastings et al., 1997).
The “mousetrap-like” inhibitory mechanism of the serpins described above has the disadvantage that mutations that destabilize and keep open their β-sheet A allow the insertion of the reactive center loop of other serpins, thus initiating a polymerization process that causes the formation of intracellular inclusions (Lomas and Carrell, 2002). In line with these observations, Davis et al. (1999a) described a familial form of early onset dementia that neuropathologically is characterized by neuroserpin-containing inclusion bodies disseminated throughout the cerebral cortex. Further studies identified four mutations (Ser49Pro, Ser52Arg, His338Arg and Gly392Glu) that cause the polymerization of human neuroserpin that the is retained as intraneuronal inclusion bodies in the deeper layers of the cerebral cortex and substantia nigra that characterize this disease (Davis et al., 1999b, 2002).
This autosomal dominant entity, denominated familial encephalopathy with neuroserpin inclusion bodies, is characterized by an insidious onset of cognitive decline, with perseveration and impaired attention, concentration, learning and memory. Other clinical manifestations commonly observed in these patients are tremor, seizures, myoclonic seizures and dysarthria.
uPA protects the synapse from the harmful effects of Aβ
uPA is a serine proteinase that upon binding to its receptor (uPAR) generates plasmin on the cell surface and activates cell signaling pathways by plasminogen-dependent and -independent mechanisms (Mahmood et al., 2018). In the developing brain uPA is abundantly detected in neurons and oligodendrocytes (Dent et al., 1993), where it is believed to play a role in the formation of dendritic and axonal branches. In contrast, the expression of uPA and uPAR in the mature brain is circumscribed to neuronal extensions, growth cones and a subpopulation of astrocytes (Merino et al., 2016), where its role is less well understood. However, recent experimental evidence indicates that the expression of uPAR increases in growth cones after an axonal injury, and that uPA/uPAR binding induces axonal recovery following a mechanical injury in vitro and an ischemic stroke in vivo (Merino et al., 2016, 2018), and promotes both, the formation of new synaptic contacts and the repair of those damaged by an ischemic injury in vivo (Diaz et al., 2017, 2018). In the mature brain uPA is abundantly found in the II and V layers of the human and murine frontal cortex, and biochemical analyses reveal that most of this uPA is associated with synaptic vesicles in the presynaptic terminal. Additionally, concentrations of glutamate that induce synaptic plasticity trigger its release via a calcium-dependent mechanism (Diaz et al., 2020).
It has been reported that Aβ induces the expression of uPA mRNA (Tucker et al., 2000a; Davis et al., 2003), and that the main role of uPA in the AD brain is to induce plasmin-mediated cleavage of insoluble Aβ-containing extracellular plaques (Tucker et al., 2002). However, four lines of experimental evidence support a role for uPA independent of its ability to cleave Aβ fibrils. First, the expression of uPA but not of its receptor (uPAR) is decreased in the synapse of AD patients and 5XFAD mice [express human APP with the Swedish (KM670/671NL), Florida (I716V), and London (V717I) mutations together with a mutant presenilin 1 (M146L, L286V) under the control of the murine Thy-1 promoter]. Significantly, this is a cell – specific finding, as the abundance of uPA is unchanged in astrocytes, kidneys and aorta of 5XFAD mice (Diaz et al., 2020). Second, Aβ oligomers halt the transcription of uPA mRNA and decrease the expression of uPA in the synapse but not in astrocytes (Diaz et al., 2020). Third, treatment with recombinant uPA abrogates the harmful effect of soluble Aβ on presynaptic function and the frequency of excitatory post-synaptic currents. And fourth, treatment with uPA attenuates the deleterious effect of Aβ on the number of synaptic contacts, via its ability to induce the expression of neuronal cadherin. Significantly, none of these effects requires the generation of plasmin. These data indicate that uPA plays a role in the early phases of the pathogenesis of AD by a mechanism independent of plasmin-induced cleavage of Aβ-containing plaques. More specifically, the results discussed above suggest a model, summarized in Figure 1, in which uPA is a homeostatic regulator of Aβ function in the synapse. More specifically, under physiological conditions Aβ-induced depression of postsynaptic function (Palop and Mucke, 2010) is modulated by uPA. However, an excessive increase in Aβ levels leads to Aβ-induced halting of uPA mRNA translation and decreased synaptic expression of uPA. This sequence of events leaves unopposed the harmful effect of Aβ on synaptic structure and function, with the subsequent development of cognitive impairment observed in AD patients. If this model is correct, then it is plausible to postulate that increasing the expression of uPA in the synapse may be a potential therapeutic strategy to prevent cognitive decline in AD patients.
Figure 1.
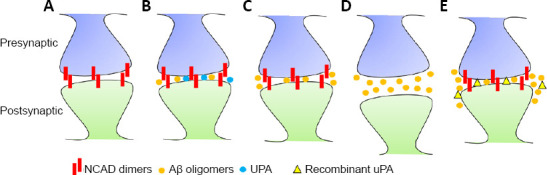
Model of the proposed homeostatic role of uPA on Aβ-induced synaptic damage.
Under physiological conditions NCAD dimers formation between pre- and post-synaptic terminals contribute to the formation of functional synaptic contacts (A). An increase in neuronal activity induces the release of both, Aβ and uPA in the synapse. Aβ increases presynaptic activity and depresses postsynaptic activity, and these effects are modulated by uPA (B). However, an abnormal increase in Aβ inhibits the translation of uPA (C), and this leaves unopposed the harmful effects of Aβ on NCAD dimer formation, with the subsequent detachment of pre- from post-synaptic terminals and ensuing collapse of synaptic structure and function (D). Treatment with recombinant uPA abrogates the harmful effect of Aβ on NCAD, thus preserving the structural and functional integrity of the synapse (E), and so preventing cognitive decline in AD. Aβ: Amyloid-β; NCAD: neuronal cadherin; uPA: urokinase-type plasminogen activator.
Footnotes
Conflicts of interest: None.
Financial support: This work was supported in part by National Institutes of Health Grant NS-NS091201 (to MY) and VA MERIT Award IO1BX003441 (to MY).
Copyright license agreement: The Copyright License Agreement has been signed by the author before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Funding: This work was supported in part by National Institutes of Health Grant NS-NS091201 (to MY) and VA MERIT Award IO1BX003441 (to MY).
C-Editors: Zhao M, Wang L; T-Editor: Jia Y
References
- 1.Angelucci F, Čechová K, Průša R, Hort J. Amyloid beta soluble forms and plasminogen activation system in Alzheimer’s disease: Consequences on extracellular maturation of brain-derived neurotrophic factor and therapeutic implications. CNS Neurosci Ther. 2019;25:303–313. doi: 10.1111/cns.13082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barker R, Love S, Kehoe PG. Plasminogen and plasmin in Alzheimer’s disease. Brain Res. 2010;1355:7–15. doi: 10.1016/j.brainres.2010.08.025. [DOI] [PubMed] [Google Scholar]
- 3.Charkhkar H, Meyyappan S, Matveeva E, Moll JR, McHail DG, Peixoto N, Cliff RO, Pancrazio JJ. Amyloid beta modulation of neuronal network activity in vitro. Brain Res. 2015;629:1–9. doi: 10.1016/j.brainres.2015.09.036. [DOI] [PubMed] [Google Scholar]
- 4.Chen GF, Xu TH, Yan Y, Zhou YR, Jiang Y, Melcher K, Xu HE. Amyloid beta: structure biology and structure-based therapeutic development. Acta Pharmacol Sin. 2017;38:1205–1235. doi: 10.1038/aps.2017.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chevilley A, Lesept F, Lenoir S, Ali C, Parcq J, Vivien D. Impacts of tissue-type plasminogen activator (tPA) on neuronal survival. Front Cell Neurosci. 2015;9:415. doi: 10.3389/fncel.2015.00415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davis J, Wagner MR, Zhang W, Xu F, Van Nostrand WE. Amyloid beta-protein stimulates the expression of urokinase-type plasminogen activator (uPA) and its receptor (uPAR) in human cerebrovascular smooth muscle cells. J Biol Chem. 2003;278:19054–19061. doi: 10.1074/jbc.M301398200. [DOI] [PubMed] [Google Scholar]
- 7.Davis RL, Shrimpton AE, Carrell RW, Lomas DA, Gerhard L, Baumann B, Lawrence DA, Yepes M, Kim TS, Ghetti B, Piccardo P, Takao M, Lacbawan F, Muenke M, Sifers RN, Bradshaw CB, Kent PF, Collins GH, Larocca D, Holohan PD. Association between conformational mutations in neuroserpin and onset and severity of dementia. Lancet. 2002;359:2242–2247. doi: 10.1016/S0140-6736(02)09293-0. [DOI] [PubMed] [Google Scholar]
- 8.Davis RL, Holohan PD, Shrimpton AE, Tatum AH, Daucher J, Collins GH, Todd R, Bradshaw C, Kent P, Feiglin D, Rosenbaum A, Yerby MS, Shaw CM, Lacbawan F, Lawrence DA. Familial encephalopathy with neuroserpin inclusion bodies. Am J Pathol. 1999a;155:1901–1913. doi: 10.1016/S0002-9440(10)65510-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davis RL, Shrimpton AE, Holohan PD, Bradshaw C, Feiglin D, Collins GH, Sonderegger P, Kinter J, Becker LM, Lacbawan F, Krasnewich D, Muenke M, Lawrence DA, Yerby MS, Shaw CM, Gooptu B, Elliott PR, Finch JT, Carrell RW, Lomas DA. Familial dementia caused by polymerization of mutant neuroserpin. Nature. 1999b;401:376–379. doi: 10.1038/43894. [DOI] [PubMed] [Google Scholar]
- 10.Dent MA, Sumi Y, Morris RJ, Seeley PJ. Urokinase-type plasminogen activator expression by neurons and oligodendrocytes during process outgrowth in developing rat brain. Eur J Neurosci. 1993;5:633–647. doi: 10.1111/j.1460-9568.1993.tb00529.x. [DOI] [PubMed] [Google Scholar]
- 11.DeTure MA, Dickson DW. The neuropathological diagnosis of Alzheimer’s disease. Mol Neurodegener. 2019;14:32. doi: 10.1186/s13024-019-0333-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Diaz A, Merino P, Manrique LG, Cheng L, Yepes M. Urokinase-type plasminogen activator (uPA) protects the tripartite synapse in the ischemic brain via ezrin-mediated formation of peripheral astrocytic processes. J Cereb Blood Flow Metab. 2018;39:2157–2171. doi: 10.1177/0271678X18783653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Diaz A, Merino P, Guo JD, Yepes MA, McCann P, Katta T, Tong EM, Torre E, Rangaraju S, Yepes M. Urokinase-type plasminogen activator protects cerebral cortical neurons from soluble Aβ-induced synaptic damage. J Neurosci. 2020;40:4251–4263. doi: 10.1523/JNEUROSCI.2804-19.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Diaz A, Merino P, Manrique LG, Ospina JP, Cheng L, Wu F, Jeanneret V, Yepes M. A cross talk between neuronal urokinase-type plasminogen activator (uPA) and astrocytic uPA receptor (uPAR) promotes astrocytic activation and synaptic recovery in the ischemic brain. J Neurosci. 2017;37:10310–10322. doi: 10.1523/JNEUROSCI.1630-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dotti CG, Galvan C, Ledesma MD. Plasmin deficiency in Alzheimer’s disease brains: causal or casual. Neurodegener Dis. 2004;1:205–212. doi: 10.1159/000080987. [DOI] [PubMed] [Google Scholar]
- 16.Echeverry R, Wu J, Haile WB, Guzman J, Yepes M. Tissue-type plasminogen activator is a neuroprotectant in the mouse hippocampus. J Clin Invest. 2010;120:2194–205. doi: 10.1172/JCI41722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elahi FM, Miller BL. A clinicopathological approach to the diagnosis of dementia. Nat Rev Neurol. 2017;13:457–476. doi: 10.1038/nrneurol.2017.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fabbro S, Seeds NW. Plasminogen activator activity is inhibited while neuroserpin is up-regulated in the Alzheimer disease brain. J Neurochem. 2009;109:303–315. doi: 10.1111/j.1471-4159.2009.05894.x. [DOI] [PubMed] [Google Scholar]
- 19.Ferreira ST, Lourenco MV, Oliveira MM, De Felice FG. Soluble amyloid-β oligomers as synaptotoxins leading to cognitive impairment in Alzheimer’s disease. Front Cell Neurosci. 2015;9:191. doi: 10.3389/fncel.2015.00191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Forner S, Baglietto-Vargas D, Martini AC, Trujillo-Estrada L, LaFerla FM. Synaptic impairment in Alzheimer’s disease: a dysregulated symphony. Trends Neurosci. 2017;40:347–357. doi: 10.1016/j.tins.2017.04.002. [DOI] [PubMed] [Google Scholar]
- 21.GBD 2016 Dementia Collaborators (2019) Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 18:88–106. doi: 10.1016/S1474-4422(18)30403-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hastings GA, Coleman TA, Haudenschild CC, Stefansson S, Smith EP, Barthlow R, Cherry S, Sandkvist M, Lawrence DA. Neuroserpin, a brain-associated inhibitor of tissue plasminogen activator is localized primarily in neurons. Implications for the regulation of motor learning and neuronal survival. J Biol Chem. 1997;272:33062–33067. doi: 10.1074/jbc.272.52.33062. [DOI] [PubMed] [Google Scholar]
- 23.He Y, Wei M, Wu Y, Qin H, Li W, Ma X, Cheng J, Ren J, Shen Y, Chen Z, Sun B, Huang FD, Shen Y, Zhou YD. Amyloid β oligomers suppress excitatory transmitter release via presynaptic depletion of phosphatidylinositol-4 5-bisphosphate. Nat Commun. 2019;10:1193. doi: 10.1038/s41467-019-09114-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hill RM, Parmar PK, Coates LC, Mezey E, Pearson JF, Birch NP. Neuroserpin is expressed in the pituitary and adrenal glands and induces the extension of neurite-like processes in AtT-20 cells. Biochem J. 2000;345(Pt 3):595–601. [PMC free article] [PubMed] [Google Scholar]
- 25.Huntington JA, Read RJ, Carrell RW. Structure of a serpin-protease complex shows inhibition by deformation. Nature. 2000;407:923–926. doi: 10.1038/35038119. [DOI] [PubMed] [Google Scholar]
- 26.Irigoyen JP, Muñoz-Cánoves P, Montero L, Koziczak M, Nagamine Y. The plasminogen activator system: biology and regulation. Cell Mol Life Sci. 1999;56:104–132. doi: 10.1007/PL00000615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jackson J, Jambrina E, Li J, Marston H, Menzies F, Phillips K, Gilmour G. Targeting the synapse in Alzheimer’s disease. Front Neurosci. 2019;13:735. doi: 10.3389/fnins.2019.00735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jeanneret V, Ospina JP, Diaz A, Manrique LG, Merino P, Gutierrez L, Torre E, Wu F, Cheng L, Yepes M. Tissue-type plasminogen activator protects the postsynaptic density in the ischemic brain. J Cereb Blood Flow Metab. 2018;38:1896–1910. doi: 10.1177/0271678X18764495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R. APP processing and synaptic function. Neuron. 2003;37:925–937. doi: 10.1016/s0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- 30.Kingston IB, Castro MJ, Anderson S. In vitro stimulation of tissue-type plasminogen activator by Alzheimer amyloid beta-peptide analogues. Nat Med. 1995;1:138–142. doi: 10.1038/nm0295-138. [DOI] [PubMed] [Google Scholar]
- 31.Krueger SR, Ghisu GP, Cinelli P, Gschwend TP, Osterwalder T, Wolfer DP, Sonderegger P. Expression of neuroserpin an inhibitor of tissue plasminogen activator, in the developing and adult nervous system of the mouse. J Neurosci. 1997;17:8984–8996. doi: 10.1523/JNEUROSCI.17-23-08984.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ledesma MD, Da Silva JS, Crassaerts K, Delacourte A, De Strooper B, Dotti CG. Brain plasmin enhances APP alpha-cleavage and Abeta degradation and is reduced in Alzheimer’s disease brains. EMBO Rep. 2000;1:530–535. doi: 10.1093/embo-reports/kvd107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee TW, Tsang VW, Loef EJ, Birch NP. Physiological and pathological functions of neuroserpin: Regulation of cellular responses through multiple mechanisms. Semin Cell Dev Biol. 2017;62:152–159. doi: 10.1016/j.semcdb.2016.09.007. [DOI] [PubMed] [Google Scholar]
- 34.Lenoir S, Varangot A, Lebouvier L, Galli T, Hommet Y, Vivien D. Post-synaptic release of the neuronal tissue-type plasminogen activator (tPA) Front Cell Neurosci. 2019;13:164. doi: 10.3389/fncel.2019.00164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li W, Asakawa T, Han S, Xiao B, Namba H, Lu C, Dong Q, Wang L. Neuroprotective effect of neuroserpin in non-tPA-induced intracerebral hemorrhage mouse models. BMC Neurol. 2017;17:196. doi: 10.1186/s12883-017-0976-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin H, Xu L, Yu S, Hong W, Huang M, Xu P. Therapeutics targeting the fibrinolytic system. Exp Mol Med. 2020;52:367–379. doi: 10.1038/s12276-020-0397-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu J, Chang L, Song Y, Li H, Wu Y. The role of NMDA receptors in Alzheimer’s disease. Front Neurosci. 2019;13:43. doi: 10.3389/fnins.2019.00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu RM, van Groen T, Katre A, Cao D, Kadisha I, Ballinger C, Wang L, Carroll SL, Li L. Knockout of plasminogen activator inhibitor 1 gene reduces amyloid beta peptide burden in a mouse model of Alzheimer’s disease. Neurobiol Aging. 2011;32:1079–1089. doi: 10.1016/j.neurobiolaging.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lomas DA, Carrell RW. Serpinopathies and the conformational dementias. Nat Rev Genet. 2002;3:759–768. doi: 10.1038/nrg907. [DOI] [PubMed] [Google Scholar]
- 40.Long JM, Holtzman DM. Alzheimer disease: an update on pathobiology and treatment strategies. Cell. 2019;179:312–339. doi: 10.1016/j.cell.2019.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mahmood N, Mihalcioiu C, Rabbani SA. Multifaceted role of the urokinase-type plasminogen activator (uPA) and its receptor (uPAR): diagnostic prognostic, and therapeutic applications. Front Oncol. 2018;8:24. doi: 10.3389/fonc.2018.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Makin S. The amyloid hypothesis on trial. Nature. 2018;559:S4–7. doi: 10.1038/d41586-018-05719-4. [DOI] [PubMed] [Google Scholar]
- 43.Mamada N, Tanokashira D, Hosaka A, Kametani F, Tamaoka A, Araki W. Amyloid β-protein oligomers upregulate the β-secretase BACE1, through a post-translational mechanism involving its altered subcellular distribution in neurons. Mol Brain. 2015;8:73. doi: 10.1186/s13041-015-0163-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marsh J, Alifragis P. Synaptic dysfunction in Alzheimer’s disease: the effects of amyloid beta on synaptic vesicle dynamics as a novel target for therapeutic intervention. Neural Regen Res. 2018;13:616–623. doi: 10.4103/1673-5374.230276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mattei V, Manganelli V, Martellucci S, Capozzi A, Mantuano E, Longo A, Ferri A, Garofalo T, Sorice M, Misasi R. A multimolecular signaling complex including PrPC and LRP1 is strictly dependent on lipid rafts and is essential for the function of tissue plasminogen activator. J Neurochem. 2020;152:468–481. doi: 10.1111/jnc.14891. [DOI] [PubMed] [Google Scholar]
- 46.Medina MG, Ledesma MD, Domínguez JE, Medina M, Zafra D, Alameda F, Dotti CG, Navarro P. Tissue plasminogen activator mediates amyloid-induced neurotoxicity via Erk1/2 activation. EMBO J. 2005;24:1706–1716. doi: 10.1038/sj.emboj.7600650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Melchor JP, Pawlak R, Strickland S. The tissue plasminogen activator-plasminogen proteolytic cascade accelerates amyloid-beta (Abeta) degradation and inhibits Abeta-induced neurodegeneration. J Neurosci. 2003;23:8867–8871. doi: 10.1523/JNEUROSCI.23-26-08867.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Merino P, Diaz A, Jeanneret V, Wu F, Torre E, Cheng L, Yepes M. Urokinase-type plasminogen activator (uPA) binding to the uPA receptor (uPAR) promotes axonal regeneration in the central nervous system. J Biol Chem. 2017;292:2741–2753. doi: 10.1074/jbc.M116.761650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Merino P, Diaz A, Manrique LG, Cheng L, Yepes M. Urokinase-type plasminogen activator (uPA) promotes ezrin-mediated reorganization of the synaptic cytoskeleton in the ischemic brain. J Biol Chem. 2018;293:9234–9247. doi: 10.1074/jbc.RA118.002534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mondragón-Rodríguez S, Salgado-Burgos H, Peña-Ortega F. Circuitry and synaptic dysfunction in Alzheimer’s disease: a new tau hypothesis. Neural Plast. 2020;2020:2960343. doi: 10.1155/2020/2960343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mu XC, Higgins PJ. Differential growth state-dependent regulation of plasminogen activator inhibitor type-1 expression in senescent IMR-90 human diploid fibroblasts. J Cell Physiol. 1995;165:647–657. doi: 10.1002/jcp.1041650324. [DOI] [PubMed] [Google Scholar]
- 52.Müller CM, Griesinger CB. Tissue plasminogen activator mediates reverse occlusion plasticity in visual cortex. Nat Neurosci. 1998;1:47–53. doi: 10.1038/248. [DOI] [PubMed] [Google Scholar]
- 53.Müller UC, Deller T, Korte M. Not just amyloid: physiological functions of the amyloid precursor protein family. Nat Rev Neurosci. 2017;18:281–298. doi: 10.1038/nrn.2017.29. [DOI] [PubMed] [Google Scholar]
- 54.Osterwalder T, Contartese J, Stoeckli ET, Kuhn TB, Sonderegger P. Neuroserpin, an axonally secreted serine protease inhibitor. EMBO J. 1996;15:2944–2953. [PMC free article] [PubMed] [Google Scholar]
- 55.Palop JJ, Mucke L. Amyloid-beta-induced neuronal dysfunction in Alzheimer’s disease: from synapses toward neural networks. Nat Neurosci. 2010;13:812–818. doi: 10.1038/nn.2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pawlak R, Magarinos AM, Melchor J, McEwen B, Strickland S. Tissue plasminogen activator in the amygdala is critical for stress-induced anxiety-like behavior. Nat Neurosci. 2003;6:168–174. doi: 10.1038/nn998. [DOI] [PubMed] [Google Scholar]
- 57.Price JL, Morris JC. Tangles and plaques in nondemented aging and “preclinical” Alzheimer’s disease. Ann Neurol. 1999;45:358–368. doi: 10.1002/1531-8249(199903)45:3<358::aid-ana12>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 58.Qian Z, Gilbert ME, Colicos MA, Kandel ER, Kuhl D. Tissue-plasminogen activator is induced as an immediate-early gene during seizure kindling and long-term potentiation. Nature. 1993;361:453–457. doi: 10.1038/361453a0. [DOI] [PubMed] [Google Scholar]
- 59.Robinson JL, Porta S, Garrett FG, Zhang P, Xie SX, Suh E, Van Deerlin VM, Abner EL, Jicha GA, Barber JM, Lee VM, Lee EB, Trojanowski JQ, Nelson PT. Limbic-predominant age-related TDP-43 encephalopathy differs from frontotemporal lobar degeneration. Brain. 2020;143:2844–2857. doi: 10.1093/brain/awaa219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rolland M, Powell R, Jacquier-Sarlin M, Boisseau S, Reynaud-Dulaurier R, Martinez-Hernandez J, André L, Borel E, Buisson A, Lanté F. Effect of Aβ oligomers on neuronal APP triggers a vicious cycle leading to the propagation of synaptic plasticity alterations to healthy neurons. J Neurosci. 2020;40:5161–5176. doi: 10.1523/JNEUROSCI.2501-19.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sappino AP, Madani R, Huarte J, Belin D, Kiss JZ, Wohlwend A, Vassalli JD. Extracellular proteolysis in the adult murine brain. J Clin Invest. 1993;92:679–685. doi: 10.1172/JCI116637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Seeds NW, Basham ME, Ferguson JE. Absence of tissue plasminogen activator gene or activity impairs mouse cerebellar motor learning. J Neurosci. 2003;23:7368–7375. doi: 10.1523/JNEUROSCI.23-19-07368.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Seeds NW, Williams BL, Bickford PC. Tissue plasminogen activator induction in Purkinje neurons after cerebellar motor learning. Science. 1995;270:1992–1994. doi: 10.1126/science.270.5244.1992. [DOI] [PubMed] [Google Scholar]
- 64.Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- 65.Sutton R, Keohane ME, VanderBerg SR, Gonias SL. Plasminogen activator inhibitor-1 in the cerebrospinal fluid as an index of neurological disease. Blood Coagul Fibrinolysis. 1994;5:167–171. doi: 10.1097/00001721-199404000-00002. [DOI] [PubMed] [Google Scholar]
- 66.Svenningsen P, Hinrichs GR, Zachar R, Ydegaard R, Jensen BL. Physiology and pathophysiology of the plasminogen system in the kidney. Pflugers Arch. 2017;469:1415–1423. doi: 10.1007/s00424-017-2014-y. [DOI] [PubMed] [Google Scholar]
- 67.Takeshita K, Yamamoto K, Ito M, Kondo T, Matsushita T, Hirai M, Kojima T, Nishimura M, Nabeshima Y, Loskutoff DJ, Saito H, Murohara T. Increased expression of plasminogen activator inhibitor-1 with fibrin deposition in a murine model of aging “Klotho” mouse. Semin Thromb Hemost. 2002;28:545–554. doi: 10.1055/s-2002-36699. [DOI] [PubMed] [Google Scholar]
- 68.Tampellini D. Synaptic activity and Alzheimer’s disease: a critical update. Front Neurosci. 2015;9:423. doi: 10.3389/fnins.2015.00423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 70.Tucker HM, Kihiko-Ehmann M, Estus S. Urokinase-type plasminogen activator inhibits amyloid-beta neurotoxicity and fibrillogenesis via plasminogen. J Neurosci Res. 2002;70:249–255. doi: 10.1002/jnr.10417. [DOI] [PubMed] [Google Scholar]
- 71.Tucker HM, Kihiko M, Caldwell JN, Wright S, Kawarabayashi T, Price D, Walker D, Scheff S, McGillis JP, Rydel RE, Estus S. The plasmin system is induced by and degrades amyloid-beta aggregates. J Neurosci. 2000a;20:3937–3946. doi: 10.1523/JNEUROSCI.20-11-03937.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tucker HM, Kihiko-Ehmann M, Wright S, Rydel RE, Estus S. Tissue plasminogen activator requires plasminogen to modulate amyloid-beta neurotoxicity and deposition. J Neurochem. 2000b;75:2172–2177. doi: 10.1046/j.1471-4159.2000.0752172.x. [DOI] [PubMed] [Google Scholar]
- 73.Vassalli JD, Sappino AP, Belin D. The plasminogen activator/plasmin system. J Clin Invest. 1991;88:1067–1072. doi: 10.1172/JCI115405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang L, Zhang Y, Asakawa T, Li W, Han S, Li Q, Xiao B, Namba H, Lu C, Dong Q. Neuroprotective effect of neuroserpin in oxygen-glucose deprivation- and reoxygenation-treated rat astrocytes in vitro. PLoS One. 2015;10:e0123932. doi: 10.1371/journal.pone.0123932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang X, Zhou X, Li G, Zhang Y, Wu Y, Song W. Modifications and trafficking of APP in the pathogenesis of Alzheimer’s disease. Front Mol Neurosci. 2017;10:294. doi: 10.3389/fnmol.2017.00294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wu W, Asakawa T, Yang Q, Zhao J, Lu L, Luo Y, Gong P, Han S, Li W, Namba H, Wang L. Effects of neuroserpin on clinical outcomes and inflammatory markers in Chinese patients with acute ischemic stroke. Neurol Res. 2017;39:862–868. doi: 10.1080/01616412.2017.1357780. [DOI] [PubMed] [Google Scholar]
- 77.Yamamoto K, Takeshita K, Kojima T, Takamatsu J, Saito H. Aging and plasminogen activator inhibitor-1 (PAI-1) regulation: implication in the pathogenesis of thrombotic disorders in the elderly. Cardiovasc Res. 2005;66:276–285. doi: 10.1016/j.cardiores.2004.11.013. [DOI] [PubMed] [Google Scholar]
- 78.Yang X, Asakawa T, Han S, Liu L, Li W, Wu W, Luo Y, Cao W, Cheng X, Xiao B, Namba H, Lu C, Dong Q, Wang L. Neuroserpin protects rat neurons and microglia-mediated inflammatory response against oxygen-glucose deprivation- and reoxygenation treatments in an in vitro study. Cell Physiol Biochem. 2016;38:1472–1482. doi: 10.1159/000443089. [DOI] [PubMed] [Google Scholar]
- 79.Yepes M. Tissue-type plasminogen activator is a neuroprotectant in the central nervous system. Front Cell Neurosci. 2015;9:304. doi: 10.3389/fncel.2015.00304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yepes M. Tissue-type plasminogen activator is a modulator of the synaptic vesicle cycle. Neural Regen Res. 2016;11:212–213. doi: 10.4103/1673-5374.177712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yepes M, Sandkvist M, Wong MK, Coleman TA, Smith E, Cohan SL, Lawrence DA. Neuroserpin reduces cerebral infarct volume and protects neurons from ischemia-induced apoptosis. Blood. 2000;96:569–576. [PubMed] [Google Scholar]
- 82.Zhao J, Liu X, Xia W, Zhang Y, Wang C. Targeting amyloidogenic processing of APP in Alzheimer’s disease. Front Mol Neurosci. 2020;13:137. doi: 10.3389/fnmol.2020.00137. [DOI] [PMC free article] [PubMed] [Google Scholar]