

Abstract

Electron-based dissociation (ExD) produces uncluttered mass spectra of intact proteins while preserving labile post-translational modifications. However, technical challenges have limited this option to only a few high-end mass spectrometers. We have developed an efficient ExD cell that can be retrofitted in less than an hour into current LC/Q-TOF instruments. Supporting software has been developed to acquire, process, and annotate peptide and protein ExD fragmentation spectra. In addition to producing complementary fragmentation, ExD spectra enable many isobaric leucine/isoleucine and isoaspartate/aspartate pairs to be distinguished by side-chain fragmentation. The ExD cell preserves phosphorylation and glycosylation modifications. It also fragments longer peptides more efficiently to reveal signaling cross-talk between multiple post-translational modifications on the same protein chain and cleaves disulfide bonds in cystine knotted proteins and intact antibodies. The ability of the ExD cell to combine collisional activation with electron fragmentation enables more complete sequence coverage by disrupting intramolecular electrostatic interactions that can hold fragments of large peptides and proteins together. These enhanced capabilities made possible by the ExD cell expand the size of peptides and proteins that can be analyzed as well as the analytical certainty of characterizing their post-translational modifications.
Keywords: electron capture dissociation, electron transfer dissociation, proteomics, proteoform, isoaspartate
Introduction
Mass spectrometry is now indispensable for characterizing protein composition, structure, and function, but a surprisingly small fraction of the potentially available information is interpretable from current instruments. While major investments by manufacturers have improved the sensitivity, speed, and resolution of mass analyzers, the remaining challenge for improvement is the fragmentation of peptides and proteins. Currently, the majority of commercial tandem mass spectrometers employ collision-induced dissociation (CID) to analyze proteins. However, CID is a blunt tool, where collisions with gas molecules dispense vibrational energy throughout a peptide and thereby break weak bonds first.1 This creates three major difficulties. First, CID often induces the neutral loss of water and ammonia from side chains and unpredictable internal cleavages that clutter the resulting spectra. Second, CID loses information about fragile post-translational modifications such as phosphorylation, glycation, or acylation. Third, CID is generally restricted to modest sized peptides because longer peptides either disperse the collisional energy without fragmentation or break to yield many internal fragments. These three considerations make protein characterization for both basic research and the Biopharma industry laborious and notoriously incomplete.
Electron capture dissociation (ECD) and electron transfer dissociation (ETD) are known to overcome all three limitations imposed by CID. The fragmentation induced by electrons is primarily localized to the main chain N–Cα bond producing c and z ion fragments from the cleaved bond to yield exceptionally clean spectra even for large proteins.2,3 Sequencing of entire proteins becomes possible because the spectra are less congested and because ECD becomes more efficient with more highly charged proteins. In addition, labile PTMs such as phosphorylation and glycation are better preserved.4,5 Because CID and ECD spectra provide complementary information, the rate of misidentification can also be greatly diminished by using both types of fragmentation.6,7
The fundamental technical obstacle for efficient peptide fragmentation by ECD is the difficulty of keeping a large number of self-repulsing low-energy electrons in a small region overlapping with peptide or protein ions. We have overcome this difficulty by using shaped magnetic fields to confine high densities of low energy electrons to the ion flight path in an ExD cell.8−12 In the Agilent 6500 LC/Q-TOF family, we have now integrated the operation of the ExD cell with the instrument software to collect electron-based fragmentation spectra as easily as CID data. Additional software has been developed to process these spectra into the flexible mzML format annotated as fragmented by ECD. This hardware/software integration makes the multiple advantages of electron-based fragmentation readily available in a widely used, affordable family of instruments.
Materials and Methods
The ExD cell mounts to the front of a shortened collision cell for installation in Agilent Q-TOF instruments (Supplemental Figure 1). Hardware installation takes about an hour to complete in an existing Q-TOF and preserves resolution, sensitivity, and CID functionality. The ExD cell itself is a 30 mm long cylinder containing two high-temperature Sm2Co17 magnets and four electrostatic lenses disposed symmetrically around a central resistively heated electron-emitting filament (Figure 1). A total of eight DC voltages are supplied by an intelligent electronics controller to the elements in the ExD cell. When directed by the instrument acquisition software, the controller can quickly alter these DC voltages to perform ECD of an isolated precursor in MS2 or to transmit ions without fragmentation in MS1. The software also provides automated routines to optimize the operation of the ExD cell for each of these modes.
Figure 1.

Schematic illustrating how the magnetic field (left) and electrostatic field (right) in the ExD cell function together to trap low energy electrons (yellow) to promote efficient ECD. Electrons are emitted from the hot rhenium filament (red dots in the center) and are pulled by the electrostatic field into the central cavity of the ECD cell. The magnetic field confines electrons radially, while negative potentials on lenses 2 and 6 prevent electrons from escaping.
ECD Operation
Thermal electrons are produced by a hot filament loop located in the middle of the ExD cell, encircling the ion beam. The filament is made of rhenium wire and biased to a lower direct current (DC) potential relative to a surrounding titanium filament holder. The more positive DC potential placed on the holder pulls electrons from the filament toward the two electromagnetostatic lenses. The high-temperature magnets have been carefully designed to create a magnetic field that confines electrons radially and focuses the electrons toward the central axis (Figure 1). Both magnets are set to a positive bias relative to the filament that pulls electrons away from the filament and its holder symmetrically.
To prevent electrons from escaping the cell, the pair of electrostatic lenses just outside of the magnets are negatively biased relative to the filament (Figure 1). As a result, electrons become trapped within the magnets axially by the electrostatic field and confined radially by the magnetic field. Electrons fill the central axis of the cell until limited by charge repulsion between the confined electrons. As a consequence, movement of electrons along the axis slows to become near zero. These low-energy electrons are captured efficiently by peptide and protein cations.13 The radially restricted electron cloud helps to pull protein and peptide cations to the central axis and to traverse through the cell more efficiently. The electric potentials on the two outermost electrostatic lenses are optimized to guide ions in and out of the ExD cell. The outer potentials may need to be adjusted differently whether working with intact proteins versus small molecules.
Transmission without ECD
Making the filament DC bias approximately equal to the other elements in the ExD cell prevents electrons from leaving the filament. Thus, simply changing the DC biases on the lens elements quickly switches ECD cell operation to nearly pure transmission mode. The potentials of all the other lens elements were adjusted accordingly to facilitate ion transmission without the negative charge of trapped electrons along the cell’s central axis. By switching the applied DC potentials as appropriate for ECD or transmission, ECD fragmentation may be turned on when the instrument is set for MS2 and turned off to transmit intact analyte ions in MS1 mode.
Sample Preparation and Analysis
The reported spectra were acquired on an Agilent 6545XT AdvanceBio LC/Q-TOF equipped with an e-MSion ExD cell for ECD fragmentation. Ionization used the standard Jet Stream and an Agilent 1290 Infinity II HPLC system for chromatographic separations. Details of the sample preparation are described in Supporting Information.
Data Processing
ExD Viewer provides a suite of utilities to process and visualize ECD, CID, and EID data. In a typical workflow, ExD Viewer reads files in the Agilent.d format; provides options for baseline subtraction, centroiding, and summing MS2 spectra from the same precursor; and exports these processed spectra to mzML format. For bottom-up experiments, ExD Viewer can serve as a node in a KNIME workflow,14 which is a general data science project that includes the openMS functions supporting mass spectrometry. The output can be fed directly to a program such as MSGF+15 or Mascot for identification and scoring of peptides found in a digest (Figure 2). Because many third-party resources may not provide options for working with ECD fragmentation, ExD Viewer can also annotate ECD spectra as ETD, such that c and z ions are identified as appropriate.
Figure 2.
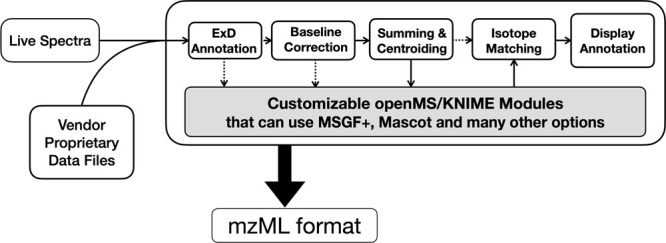
The generalized schema for analyzing electron-based fragmentation and CID data using ExD Viewer. In addition to processing data, the program can sum spectra from the same precursor to improve signal-to-noise and apply other processing using open-source openMS routines and export data as the standard mzML format readable by MSGF+ and many other open source programs.
ExD Viewer displays the fit of known to predicted isotopic clusters of fragments to the observed spectrum. For bottom-up experiments, ExD Viewer displays peptides identified by MSGF+ or Mascot and output in the standard.mzID format.
Results
Substance P is an 11 amino acid biopeptide used by Roman Zubarev in his early ECD experiments and has become our standard peptide for the routine optimization of the ExD cell. In the Agilent Q-TOF, the ExD cell typically yields 3–6% of the 2+ precursor into eight predominantly c ion fragments plus the charge-reduced precursor that effectively span the sequence (Figure 3A). Substance P also contains two prolines, whose cyclic structure prevents dissociation after electron capture. Only the z9 ion is present, most likely because it is the first z ion to cleave after lysine. Shorter C-terminal fragments have likely been neutralized by electron capture. Figure 3B shows the fragmentation of substance P (2+) collected under identical conditions as for 3A with the calculated optimal CID energy of 22 V. Approximately 60% of the precursor was fragmented under these conditions, and the resulting fragments provide near complete coverage from both the N- and C- ends with b and y ions as well as several a ions.
Figure 3.

Comparison of ECD with CID of Substance P. (A) ECD spectrum. (B) Corresponding CID spectrum with 22 V of activation and collected under identical conditions as the ECD spectra. In each case, 10 spectra collected at 20 Hz were averaged to reduce stochastic baseline noise. The multiple small peaks in the CID spectrum remained unchanged with longer signal averaging.
However, a majority of the precursor resulted in the loss of an ammonia and the C-terminal methionine to yield predominantly the b102+ precursor. Notably, the other CID peaks are comparable in intensity to the ECD peak, because ECD has yielded fewer but more informative fragments with more uniform intensity compared with CID. The entire CID m/z range contains fragments coming from neutral losses, side-chain fragmentation, and internal cleavages. Increasing CID energy only slightly increases the intensity of b and y ions but produces far more spectral congestion (Supplemental Figure 2A). The ECD spectrum in Figure 3A has 200 peaks greater than a noise threshold of 10 counts, while the CID spectrum at 22 V produced 750 peaks. Increasing the CID energy by another 3 V resulted in 1340 peaks. Supplemental Figure 2B shows that ECD can be performed simultaneously with CID. ECD may be turned on in a few milliseconds, allowing for complementary ECD and CID fragmentation data sets to be collected from a single chromatographic peak.
A significant advantage of the ExD cell is that the same or very similar operating profiles can work for both peptides and proteins, whereas CID energy needs to be carefully optimized for the size and charge of peptides. Furthermore, ECD efficiency increases rapidly with charge state. Hence, ECD fragmentation works even better with large peptides and intact proteins. This is shown with 6+ ubiquitin, a small protein with 76 amino acids and a mass of 8564.8 Da, as a standard to gauge the pattern and efficiency of fragmentation. We chose to use the 6+ charge state rather than the more easily fragmented 8+ to 13+ charge states because it is known to remain tightly folded during electrospray ionization16 and is a better model for larger proteins in a native-like conformation. The ExD cell produced c- and z-type fragments representing 95% sequence coverage for the 6+ charge state of ubiquitin (Figure 4). As expected, no ECD fragments were observed at the three prolines, whose cyclic chain maintains the covalent linkage after electron fragmentation of the N–Cα bond. Notably, side-chain fragmentation produced multiple w ions, which were observed for seven of the nine leucine residues and one of the five isoleucine residues in ubiquitin (Supplemental Figure 3). The side chain of leucine is more likely to undergo side-chain fragmentation because it loses an isopropyl secondary radical, whereas isoleucine loses less stable ethyl or methyl primary radicals.17 The mass difference between the w ion and the corresponding z ion allows these isobaric amino acids to be unambiguously assigned.18 Approximately 16% of the human proteome consists of leucine or isoleucine, which presents a significant source of ambiguity in peptide identification.19
Figure 4.

Effect of added collision energy on 5+ and 6+ ubiquitin fragmentation in the ExD cell. The fragmentation maps are produced with the ExD Viewer; c and z ions are shown as blue dots, b and y ions as green dots and finally a and x ions as purple dots (see Figure 2). Fragmentation coverage is shown for the following: (A) charge state 6+ without CID energy, (B) charge state 6+ with 10 V CID energy, (C) charge state 5+ without added collision energy, (D) charge state 5+ with 10 V added CID energy, (E) charge state 5+ with 30 V added collision energy. The observed fragments were manually curated by comparison to their isotopic distribution as shown in Figure 7 below. The purple shading indicates gaps in coverage between 2 or more amino acid residues.
Electron-based fragmentation is a highly localized process, allowing noncovalent interactions to persist after bond cleavage. When this occurs, the fragments produced by cleavage of backbone covalent bonds may be held together by noncovalent interactions. In this case, the ions will appear to be intact but charge reduced (often called “EC-no-D”). Internal vibrations which disrupt the noncovalent interactions and yield more efficient fragmentation may be introduced either in the source or downstream from the ExD cell, after cleavage has occurred. As shown in Figure 4a, we can achieve excellent coverage of 6+ ubiquitin without application of additional collisional energy, presumably because of sufficient activation in the source. Addition of 10 V collision energy improves coverage significantly however. With no additional postcleavage collisional activation, the more compact and challenging 5+ ubiquitin showed apparent cleavage of only 19 of 75 possible cleavages or 25% coverage, almost exclusively at the amino and carboxy termini (Figure 4C). Addition of 30 V collisional activation in the collision cell greatly improved the sequence coverage to 87% (Figure 4D). This capability is similar to EThcD,20 or with irradiative ion postactivation as reported.21−23
The addition of collision energy increases the number of b and y ions, including fragmenation on the N-terminal side of proline residues. To better illustrate the rich set of fragment ions that can be produced to support the identification of sequence fragments, we have illustrated the ions as color-coded dots in Figure 4. In addition to c and z ions, numerous a ions were observed, which can be produced from c ions by the loss of the amino-formyl moiety (H3N–C=O).13,24 Side-chain fragments are indicated as tilde marks above or below the one letter code for each amino acid for w ions and below for the less commonly observed d ions, respectively. This choice of displaying the fragmentation map of ubiquitin with ExD Viewer illustrates the multiple cleavages between each residue.
Isoaspartate
Asparagine and aspartate are both susceptible to spontaneous transformation to isoaspartate through a succinimide intermediate.25 This can affect the stability of biotherapeutics and is a major modification for long-lived proteins in vivo with pathological implications.26 While the transformation of asparagine to aspartate and isoaspartate results in an observable mass difference, aspartate and isoaspartate themselves are isobaric and cannot be distinguished by mass alone. Furthermore, isoaspartate cannot be distinguished from aspartate by CID because the main chain b and y fragments are also isobaric.27 In contrast, electron-based fragmentation breaks isoaspartate to give c + 57 Da and z −57 Da fragments that uniquely identify isoaspartate.
The synthetic peptide ECDisoDDELIGHTFLK was used to illustrate the distinctive fragmentation of isoaspartate (isoD) as well as side-chain fragmentation of other amino acids. The peptide eluted as a single chromatographic peak in a standard C18 reversed-phase HPLC run and subjected to MS2 of both the 2+ and 3+ precursors using ECD fragmentation. Both precursors gave the z-57 Da fragment characteristic of isoaspartate (Figure 5), which results from radical migration from the corresponding z ion to induce the loss of acetate. Aspartate was also distinguishable from isoaspartate by the loss of CO2.27,28 Side-chain cleavages yielded six w ions, which allowed leucine to be distinguished from isoleucine.
Figure 5.

Isoaspartate and side-chain fragmentation of a doubly charged synthetic peptide. The fragmentation coverage is also illustrated for the 3+ charge state measured under the same conditions.
ExD Fragmentation of Phosphopeptides
Previously, we have shown that the ExD cell can improve dissociation and sequence coverage for 2+ phosphopeptides over traditional ECD on an FTICR instrument,29 most likely because the ExD cell can better disrupt ionic interactions after electron capture without a measurable loss of phosphate groups from tyrosine, serine, or threonine.30 We used the ExD cell to confirm phosphorylation site localization as part of the HUPO Phosphopeptide Challenge.31 For this challenge, a set of peptides with up to three phosphate groups, along with their nonphosphorylated analogues, was provided to researchers to assess and promote the ability to accurately identify and quantify phosphopeptides. The location of the phosphate group within the phosphopeptides by MS2 methods was an important element of the challenge. Along with the neat mixture of phosphopeptides, the same set of peptides was provided in a peptide background consisting of a tryptic digest of yeast proteins in order to evaluate methods for phosphopeptide enrichment.
For the first HUPO phosphopeptide sample, HPLC-separated peptides were initially identified by CID.32 All 89 nonphosphopeptides on the list provided by the Challenge organizers were successfully identified. MS2 spectra were manually validated for each phosphopeptide.
The same acquisition strategy was used with the enriched HUPO Phosphopeptide-Yeast Digest sample; 287 distinct peptides were identified, of which 264 were phosphopeptides. The overall selectivity of the enrichment was approximately 92%. Moreover, 93 out of 94 phosphopeptides spiked into the yeast digest were identified from the enriched sample.
The fragmentation of the peptide VVEAVNSDSDSEFGIPK with two phosphorylation sites is illustrated in Figure 6. This sequence has three potential serine phosphorylation sites. The sample contained four of these peptides, differing only in phosphorylation. All four peptides were well-separated using the C18 column with the 90 min gradient as identified by CID. However, this left the challenge of identifying the specific phosphorylation sites for the mono and diphosphorylated peptides. Hence, a second targeted HPLC experiment was run to localize the specific phosphosites. The spectrum shows both phosphorylation sites with no loss of the phospho-moiety. Many cleavage sites exhibited golden triplets of a, b, and c ions from the amino terminus and golden pairs of y and z ions from the C-terminus.
Figure 6.

ECD fragmentation of a serine peptide with two phosphorylation sites. Multiple fragments between amino acid residues produce golden triplets and doublets to confirm assignments. To improve signal-to-noise, ExDProcess was used to sum four adjacent MS2 scans with the same precursor isolation window. This spectrum identifies the phosphorylation sites as Ser7 and Ser9, in which Ser11 is unmodified.
Lys-C Analysis of N-Linked Glycopeptides
Antibody characterization by mass spectrometry poses a number of difficult problems because of their size, their complex quaternary structure held by multiple disulfide bonds, and the number and lability of possible post-translational modifications. Many of these problems have been overcome, and mass spectrometry plays an important role in research and production of antibodies, a large and growing segment of the therapeutic drug market. However, complete analysis of antibodies is still a laborious process and is hindered by inadequate methods, particularly for studies of labile PTMs such as N-linked glycans, which play a crucial role in determining the stability of antibodies. Electron-based fragmentation is known to preserve N-linked glycans, which are particularly susceptible to loss during CID.5,33
Because ExD is more effective with more highly charged peptides, we investigated the cleavage of Infliximab into larger fragments using the endoproteinase Lys-C. The coverage map with Lys C is shown in Supplemental Figure 4. The basic software workflow shown in Figure 2 with the MSGF+ option was used to process the HPLC separated middle-down digest of the antibody Infliximab and characterize the N-glycosylated peptide.
ExD Viewer allows fragments to be verified through matching the theoretical isotopic distribution to the observed isotopic pattern. Clicking on the dot corresponding to a fragment displays a segment of the spectrum containing the ion of interest, with superposition of the theoretical isotopic distribution matched to the centroids of the observed spectra. As an example, an Infliximab peptide containing the N-glycan is shown in Figure 7, along with spectra for the three c ions bridging the N-glycan. In addition to unambiguously localizing the N-glycan, this spectrum also has intense peaks from diagnostic w ions, such as w4 ion from the side-chain fragmentation of leucine z4. As expected, no ECD c and z fragments are observed on the N-terminal side of proline, but the missing information was provided by b and y type fragments.
Figure 7.

The ECD spectrum of an N-glycopeptide from Infliximab using Lys C digestion. Within ExD Viewer, clicking on any fragment ion results in expansion of the spectrum to show the fit of the fragment to its theoretical isotope pattern.
Insulin Disulfide Analysis
Localization of disulfide bonds in proteins remains a significant challenge for mass spectrometry. Disulfide bonds are important for stabilizing the structure of proteins, especially extracellular proteins such as antibodies. The most prevalent proteomic methods entail reduction and alkylation of cysteine residues, which allows for dissociation within disulfide-protected regions but makes localization of the disulfide bonds impossible. ECD is well-known to efficiently cleave disulfide bonds13 and offers a way to analyze disulfide-linked sequences without off-line preparation or loss of information regarding the location of the disulfide bond.
We used insulin to demonstrate that this is now possible on widely available Q-TOF instruments. Insulin is a 5.7 kDa dimer consisting of two chains linked by two disulfide bonds. One of the chains has an additional intramolecular disulfide bond. The data were analyzed by considering each chain separately and each interchain disulfide bond separately. The mass of the other chain was considered as a modification of a cysteine in the case of a noncleaved disulfide bond, and all other cysteines were treated as dehydrocysteine. Cleavage of the peptide chains between the cystine ring provides evidence that a disulfide bond as well as the peptide chain has been broken. Both c and z ions found between the disulfide bonds are clear evidence that both the inter- and intrachain bonds have been broken. Interestingly, the intramolecular disulfide of the insulin chain a was only cleaved if the adjacent intermolecular disulfide was also cleaved. Figure 8 shows a composite of the peptide cleavage maps for chain a and b.
Figure 8.

Coverage map of bovine insulin. Fragmentation maps searched with each of the two interchain disulfides cleaved by ECD (indicated as an x between two cysteines).
Figure 9 shows that an interchain disulfide cleavage was observable from the intact antibody Infliximab, which allowed the light chain to be separated from the intact antibody.34 The mass of the light chain was readily deconvoluted to yield an average mass of 23 434.5 Da. To improve the signal-to-noise, the mass spectrum was collected in MS1 with the quadrupole acting as a low mass cutoff filter set to 2,300 m/z. This allowed for fragments from multiple charge states to be collected, which is useful for working with denatured proteins with multiple charge states in a relatively high mass range.
Figure 9.

ECD induced light chain cleavage from intact Infliximab. The light chain appears as a series of 11 charge states indicated with arrows. The mass of the light chain after deconvolution is shown in the inset.
Discussion
Proteomics has broadened its focus from inferring identities of tryptic peptides by shotgun approaches toward the more complete characterization of larger peptides and proteoforms with mapping and quantifying of PTMs. Arguably, the major limitation holding back the full characterization of biological macromolecules is the difficulty of producing fragmentation spectra to fully characterize proteoforms by CID alone. The ExD cell is a cost-effective alternative that effectively fragments peptides while preserving labile modifications such as phosphorylation and N-glycation.29,35 Additional advantages provided by the ExD cell include the side-chain fragmentation of leucine, isoleucine, and other amino acids as well as identifying isoaspartate isomerization and related protein aging artifacts. Applying ExD as an orthogonal method to collision-induced fragmentation of peptides can give higher confidence by producing golden pairs and triplets of fragment types observed at each residue.6,7 The sequence coverage maps shown in Figure 4 for ubiquitin and Figure 8 for insulin illustrate as many as five distinct main-chain fragment types that can be identified between each amino acid residue. While the N-terminus of proline is not effectively cleaved by ECD alone, it cleaves preferentially by CID (Figures 4, 7, and 8). Thus, the combination of ECD plus CID gives greater analytical certainty for characterizing proteoforms.
The ExD cell also substantially increases the size of peptides and proteins that are not readily fragmented by CID alone. Due to electrostatic attraction of electrons to positively charged peptides, the efficiency of electron capture increases with the square of protein’s charge.2,36−39 Because the energy from electron capture is locally focused on specific bonds, fragmentation is largely independent of the size of the peptide or protein.40 In contrast, activation by CID involves gradual heating of peptide chain, which limits the size of peptides that can be analyzed. Increasing CID energy further results in extensive internal cleavages and the extensive loss of PTMs.
The combination of ETD with HCD is well-known to improve sequence coverage.20 Similarly, modest CID activation with ExD greatly increases the coverage obtained in the interior of the protein as shown in Figure 4. The additional coverage was predominately from ECD fragments, supporting a role of CID in improving dissociation of fragments held together by noncovalent interactions. Additional ion activation helps improve the coverage for entire proteins up to the size of intact antibodies35,41 and even larger protein complexes.12,42
The ability of the ExD cell to sequence larger domains in proteins will help reveal the potential crosstalk of multiple post translation modifications on the same protein, which are frequently lost with bottom-up workflows. The amount of CID energy applied with ExD will need to be selected to maximize sequence coverage while minimizing the loss of labile PTMs. Fortunately, loss of phospho-moieties is minor even for doubly charged phosphopeptides held together by ionic interactions.29
Electron-based fragmentation is known to favor disulfide cleavage,13 allowing for the mapping of disulfide bonds in cystine cross-linked proteins as shown with insulin (Figure 8). The disulfide linking the heavy and light chains in antibodies is also cleaved by the ExD cell (Figure 9).
One of the major challenges we encountered from incorporating the ExD cell to existing mass spectrometers was the need to integrate the operation of ExD cell with data acquisition and then to annotate the resulting data to make data-analysis programs aware that electron fragmentation was used. Agilent provided direct access to its instrument control software, which allowed the coordination of the ExD electronics to transmit analytes without fragmentation during MS1 scans and rapidly turn on ECD with selectable levels of CID activation. To support subsequent data analysis, we developed a conversion utility to annotate MS2 spectra as fragmented with either ETD or ECD in mzML files to be processed by third party programs. In addition, ExD Viewer allows similar MS2 spectra to be averaged together, which is particularly valuable for analyzing larger proteins that require longer acquisition times to resolve the complex isotopic clusters.
The ExD Viewer suit integrates the rich tool set provided with the KNIME/openMS project.43 Over 180 tools to support mass spectrometry have been developed by the openMS consortium over the past 20 years.14,44 These have more recently been integrated into the larger KNIME data science package, which was originally developed for the pharmaceutical industry.45 KNIME provides a large collection of data science tools to complete complex workflows for data processing, analysis, and integration. Figure 2 illustrates a basic workflow using MSGF+ that was created by KNIME’s graphical user interface through connecting tools. By simply dragging in other tools, the workflow using MSGF+ could have been changed to use Mascot. Furthermore, the output from MSGF+ or Mascot could be further passed to Percolator to calculate false discovery rates.46 Far more complex work flows including batch processing can be readily created in minutes, saved locally, or shared through user-accessible online libraries.45,47 To help interpret the results, we have been developing the ExD Viewer tool to examine the fragmentation for each peptide or protein sequence coverage map and visually validate the results.
In summary, the retrofit of the ExD cell into Agilent LC/Q-TOF instruments greatly expands their analytical capabilities to probe larger peptides and proteins and offers new opportunities for improving macromolecular analysis. This includes capillary zone electrophoresis and capillary isoelectric separations of proteins.48 The ExD cell effectively fragments proteins on the fly without trapping and thus is compatible with ion mobility separations on Agilent’s 6560 IMS Q-TOF49 or Mobilion’s new SLIM front end.43
One exciting new application is to use increasing energy to induce collision induced unfolding (CIU) of proteins to observe the progressive unfolding of proteins by their increased drift time through the ion mobility separation. This unfolding of protein complexes substantially increases the coverage of proteins as large as alcohol dehydrogenase by the ExD cell50 and can provide additional insights into the structure of proteins.51 The ExD cell can readily produce more energetic electrons by increasing the voltage difference between the filament bias and the filament holder. These more energetic electrons open more channels for fragmentation that have been shown to further improve sequence coverage in macromolecules.52,53 We anticipate that the additional flexibility provided by the ExD cell will continue to expand the potential for existing mass spectrometers to analyze macromolecules more completely.
Acknowledgments
This work has been supported by the following SBIR awards GM122131 and GM123855.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jasms.0c00482.
The ExD/collision cell in relation to an Agilent Q-TOF, ECD vs 25 V CID spectra of substance P(2+), annotated ECD spectrum of top-down ubiquitin (6+), coverage of a Lys-C digest of the antibody Infliximab, and experimental methods for the mass spectrometer of the various peptides and proteins (PDF)
The authors declare the following competing financial interest(s): J.S.B., Y.V., and V.V. are founders of e-MSion, Inc and may have a perceived conflict of interest with respect to their positions at Oregon State University.
Supplementary Material
References
- Brodbelt J. S. Ion Activation Methods for Peptides and Proteins. Anal. Chem. 2016, 88, 30–51. 10.1021/acs.analchem.5b04563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zubarev R. A.; Horn D. M.; Fridriksson E. K.; Kelleher N. L.; Kruger N. A.; Lewis M. A.; Carpenter B. K.; McLafferty F. W. Electron capture dissociation for structural characterization of multiply charged protein cations. Anal. Chem. 2000, 72, 563–573. 10.1021/ac990811p. [DOI] [PubMed] [Google Scholar]
- Macias L. A.; Santos I. C.; Brodbelt J. S. Ion Activation Methods for Peptides and Proteins. Anal. Chem. 2020, 92, 227–251. 10.1021/acs.analchem.9b04859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stensballe A.; Jensen O. N.; Olsen J. V.; Haselmann K. F.; Zubarev R. A. Electron capture dissociation of singly and multiply phosphorylated peptides. Rapid Commun. Mass Spectrom. 2000, 14, 1793–1800. . [DOI] [PubMed] [Google Scholar]
- Khatri K.; Pu Y.; Klein J. A.; Wei J.; Costello C. E.; Lin C.; Zaia J. Comparison of Collisional and Electron-Based Dissociation Modes for Middle-Down Analysis of Multiply Glycosylated Peptides. J. Am. Soc. Mass Spectrom. 2018, 29, 1075–1085. 10.1007/s13361-018-1909-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savitski M. M.; Nielsen M. L.; Kjeldsen F.; Zubarev R. A. Proteomics-grade de novo sequencing approach. J. Proteome Res. 2005, 4, 2348–2354. 10.1021/pr050288x. [DOI] [PubMed] [Google Scholar]
- Zubarev R. A.; Zubarev A. R.; Savitski M. M. Electron capture/transfer versus collisionally activated/induced dissociations: solo or duet?. J. Am. Soc. Mass Spectrom. 2008, 19, 753–761. 10.1016/j.jasms.2008.03.007. [DOI] [PubMed] [Google Scholar]
- Voinov V. G.; Beckman J. S.; Deinzer M. L.; Barofsky D. F. Electron-capture dissociation (ECD), collision-induced dissociation (CID) and ECD/CID in a linear radio-frequency-free magnetic cell. Rapid Commun. Mass Spectrom. 2009, 23, 3028–3030. 10.1002/rcm.4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voinov V. G.; Bennett S. E.; Barofsky D. F. Electron-induced dissociation of peptides in a triple quadrupole mass spectrometer retrofitted with an electromagnetostatic cell. J. Am. Soc. Mass Spectrom. 2015, 26, 752–761. 10.1007/s13361-014-1074-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voinov V. G.; Deinzer M. L.; Beckman J. S.; Barofsky D. F. Electron capture, collision-induced, and electron capture-collision induced dissociation in Q-TOF. J. Am. Soc. Mass Spectrom. 2011, 22, 607–611. 10.1007/s13361-010-0072-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voinov V. G.; Hoffman P. D.; Bennett S. E.; Beckman J. S.; Barofsky D. F. Electron Capture Dissociation of Sodium-Adducted Peptides on a Modified Quadrupole/Time-of-Flight Mass Spectrometer. J. Am. Soc. Mass Spectrom. 2015, 26, 2096–2104. 10.1007/s13361-015-1230-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams J. P.; Morrison L. J.; Brown J. M.; Beckman J. S.; Voinov V. G.; Lermyte F. Top-Down Characterization of Denatured Proteins and Native Protein Complexes Using Electron Capture Dissociation Implemented within a Modified Ion Mobility-Mass Spectrometer. Anal. Chem. 2020, 92, 3674–3681. 10.1021/acs.analchem.9b04763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zubarev R. A.; Kruger N. A.; Fridriksson E. K.; Lewis M. A.; Horn D. M.; Carpenter B. K.; McLafferty F. W. Electron capture dissociation of gaseous multiply-charged proteins is favored at disulfide bonds and other sites of high hydrogen atom affinity. J. Am. Chem. Soc. 1999, 121, 2857–2862. 10.1021/ja981948k. [DOI] [Google Scholar]
- Rost H. L.; Sachsenberg T.; Aiche S.; Bielow C.; Weisser H.; Aicheler F.; Andreotti S.; Ehrlich H. C.; Gutenbrunner P.; Kenar E.; Liang X.; Nahnsen S.; Nilse L.; Pfeuffer J.; Rosenberger G.; Rurik M.; Schmitt U.; Veit J.; Walzer M.; Wojnar D.; Wolski W. E.; Schilling O.; Choudhary J. S.; Malmstrom L.; Aebersold R.; Reinert K.; Kohlbacher O. OpenMS: a flexible open-source software platform for mass spectrometry data analysis. Nat. Methods 2016, 13, 741–748. 10.1038/nmeth.3959. [DOI] [PubMed] [Google Scholar]
- Kim S.; Pevzner P. A. MS-GF+ makes progress towards a universal database search tool for proteomics. Nat. Commun. 2014, 5, 5277. 10.1038/ncomms6277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breuker K.; Oh H.; Horn D. M.; Cerda B. A.; McLafferty F. W. Detailed unfolding and folding of gaseous ubiquitin ions characterized by electron capture dissociation. J. Am. Chem. Soc. 2002, 124, 6407–6420. 10.1021/ja012267j. [DOI] [PubMed] [Google Scholar]
- Cooper H. J.; Hudgins R. R.; Hakansson K.; Marshall A. G. Characterization of amino acid side chain losses in electron capture dissociation. J. Am. Soc. Mass Spectrom. 2002, 13, 241–249. 10.1016/S1044-0305(01)00357-9. [DOI] [PubMed] [Google Scholar]
- Kelleher N. L.; Zubarev R. A.; Bush K.; Furie B.; Furie B. C.; McLafferty F. W.; Walsh C. T. Localization of labile posttranslational modifications by electron capture dissociation: the case of gamma-carboxyglutamic acid. Anal. Chem. 1999, 71, 4250–4253. 10.1021/ac990684x. [DOI] [PubMed] [Google Scholar]
- Savitski M. M.; Nielsen M. L.; Zubarev R. A. Side-chain losses in electron capture dissociation to improve peptide identification. Anal. Chem. 2007, 79, 2296–2302. 10.1021/ac0619332. [DOI] [PubMed] [Google Scholar]
- Frese C. K.; Altelaar A. F.; van den Toorn H.; Nolting D.; Griep-Raming J.; Heck A. J.; Mohammed S. Toward full peptide sequence coverage by dual fragmentation combining electron-transfer and higher-energy collision dissociation tandem mass spectrometry. Anal. Chem. 2012, 84, 9668–9673. 10.1021/ac3025366. [DOI] [PubMed] [Google Scholar]
- Riley N. M.; Westphall M. S.; Coon J. J. Activated Ion Electron Transfer Dissociation for Improved Fragmentation of Intact Proteins. Anal. Chem. 2015, 87, 7109–7116. 10.1021/acs.analchem.5b00881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley N. M.; Westphall M. S.; Coon J. J. Activated Ion-Electron Transfer Dissociation Enables Comprehensive Top-Down Protein Fragmentation. J. Proteome Res. 2017, 16, 2653–2659. 10.1021/acs.jproteome.7b00249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley N. M.; Westphall M. S.; Coon J. J. Sequencing Larger Intact Proteins (30–70 kDa) with Activated Ion Electron Transfer Dissociation. J. Am. Soc. Mass Spectrom. 2018, 29, 140–149. 10.1007/s13361-017-1808-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper H. J.; Hakansson K.; Marshall A. G. The role of electron capture dissociation in biomolecular analysis. Mass Spectrom. Rev. 2005, 24, 201–222. 10.1002/mas.20014. [DOI] [PubMed] [Google Scholar]
- Stephenson R. C.; Clarke S. Succinimide formation from aspartyl and asparaginyl peptides as a model for the spontaneous degradation of proteins. J. Biol. Chem. 1989, 264, 6164–6170. 10.1016/S0021-9258(18)83327-0. [DOI] [PubMed] [Google Scholar]
- Warmack R. A.; Shawa H.; Liu K.; Lopez K.; Loo J. A.; Horwitz J.; Clarke S. G. The l-isoaspartate modification within protein fragments in the aging lens can promote protein aggregation. J. Biol. Chem. 2019, 294, 12203–12219. 10.1074/jbc.RA119.009052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H.; Zubarev R. A. Mass spectrometric analysis of asparagine deamidation and aspartate isomerization in polypeptides. Electrophoresis 2010, 31, 1764–1772. 10.1002/elps.201000027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han H.; Xia Y.; McLuckey S. A. Ion trap collisional activation of c and z* ions formed via gas-phase ion/ion electron-transfer dissociation. J. Proteome Res. 2007, 6, 3062–3069. 10.1021/pr070177t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voinov V. G.; Bennett S. E.; Beckman J. S.; Barofsky D. F. ECD of Tyrosine Phosphorylation in a Triple Quadrupole Mass Spectrometer with a Radio-Frequency-Free Electromagnetostatic Cell. J. Am. Soc. Mass Spectrom. 2014, 25, 1730–1738. 10.1007/s13361-014-0956-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palumbo A. M.; Smith S. A.; Kalcic C. L.; Dantus M.; Stemmer P. M.; Reid G. E. Tandem mass spectrometry strategies for phosphoproteome analysis. Mass Spectrom. Rev. 2011, 30, 600–625. 10.1002/mas.20310. [DOI] [PubMed] [Google Scholar]
- Hoopmann M. R.; Kusebauch U.; Palmblad M.; Bandeira N.; Shteynberg D. D.; He L.; Xia B.; Stoychev S. H.; Omenn G. S.; Weintraub S. T.; Moritz R. L. Insights from the First Phosphopeptide Challenge of the MS Resource Pillar of the HUPO Human Proteome Project. J. Proteome Res. 2020, 19, 4754. 10.1021/acs.jproteome.0c00648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voinov V. G., Beckman J. S., Wu S., Newton K., Wu L., Hsiao J. J.: A Novel, Automated, and Highly Selective Phosphopeptide Enrichment for Phosphopeptide Identification and Phosphosite Localization. https://www.agilent.com/cs/library/applications/application-phosphopeptide-enrichment-5994-1235en-agilent.pdf. (2019).
- Mirgorodskaya E.; Roepstorff P.; Zubarev R. A. Localization of O-glycosylation sites in peptides by electron capture dissociation in a Fourier transform mass spectrometer. Anal. Chem. 1999, 71, 4431–4436. 10.1021/ac990578v. [DOI] [PubMed] [Google Scholar]
- Shaw J. B.; Liu W.; Vasil’ev Y. V.; Bracken C. C.; Malhan N.; Guthals A.; Beckman J. S.; Voinov V. G. Direct Determination of Antibody Chain Pairing by Top-down and Middle-down Mass Spectrometry Using Electron Capture Dissociation and Ultraviolet Photodissociation. Anal. Chem. 2020, 92, 766–773. 10.1021/acs.analchem.9b03129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fort K. L.; Cramer C. N.; Voinov V. G.; Vasil’ev Y. V.; Lopez N. I.; Beckman J. S.; Heck A. J. R. Exploring ECD on a Benchtop Q Exactive Orbitrap Mass Spectrometer. J. Proteome Res. 2018, 17, 926–933. 10.1021/acs.jproteome.7b00622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zubarev R. A. Reactions of polypeptide ions with electrons in the gas phase. Mass Spectrom. Rev. 2003, 22, 57–77. 10.1002/mas.10042. [DOI] [PubMed] [Google Scholar]
- Zubarev R. A. Electron-capture dissociation tandem mass spectrometry. Curr. Opin. Biotechnol. 2004, 15, 12–16. 10.1016/j.copbio.2003.12.002. [DOI] [PubMed] [Google Scholar]
- Zubarev R. A.; Haselmann K. F.; Budnik B. A.; Kjeldsen F.; Jensen F. Towards an understanding of the mechanism of electron-capture dissociation: a historical perspective and modern ideas. Eur. J. Mass Spectrom. 2002, 8, 337–349. 10.1255/ejms.517. [DOI] [Google Scholar]
- Zubarev R. A.; Kelleher N. L.; McLafferty F. W. Electron capture dissociation of multiply charged protein cations. A nonergodic process. J. Am. Chem. Soc. 1998, 120, 3265–3266. 10.1021/ja973478k. [DOI] [Google Scholar]
- Zhurov K. O.; Fornelli L.; Wodrich M. D.; Laskay U. A.; Tsybin Y. O. Principles of electron capture and transfer dissociation mass spectrometry applied to peptide and protein structure analysis. Chem. Soc. Rev. 2013, 42, 5014–5030. 10.1039/c3cs35477f. [DOI] [PubMed] [Google Scholar]
- Shaw J. B.; Malhan N.; Vasil’ev Y. V.; Lopez N. I.; Makarov A.; Beckman J. S.; Voinov V. G. Sequencing Grade Tandem Mass Spectrometry for Top-Down Proteomics Using Hybrid Electron Capture Dissociation Methods in a Benchtop Orbitrap Mass Spectrometer. Anal. Chem. 2018, 90, 10819–10827. 10.1021/acs.analchem.8b01901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vimer S.; Ben-Nissan G.; Morgenstern D.; Kumar-Deshmukh F.; Polkinghorn C.; Quintyn R. S.; Vasil’ev Y. V.; Beckman J. S.; Elad N.; Wysocki V. H.; Sharon M. Comparative Structural Analysis of 20S Proteasome Ortholog Protein Complexes by Native Mass Spectrometry. ACS Cent. Sci. 2020, 6, 573–588. 10.1021/acscentsci.0c00080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arndt J. R.; Wormwood Moser K. L.; Van Aken G.; Doyle R. M.; Talamantes T.; DeBord D.; Maxon L.; Stafford G.; Fjeldsted J.; Miller B.; Sherman M.: High resolution ion mobility-enabled peptide mapping for high-throughput critical quality monitoring. J. Am. Soc. Mass Spectrom. 2021, in press. 10.1021/jasms.0c00434 [DOI] [PubMed] [Google Scholar]
- Sturm M.; Bertsch A.; Gropl C.; Hildebrandt A.; Hussong R.; Lange E.; Pfeifer N.; Schulz-Trieglaff O.; Zerck A.; Reinert K.; Kohlbacher O. OpenMS - an open-source software framework for mass spectrometry. BMC Bioinf. 2008, 9, 163. 10.1186/1471-2105-9-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeuffer J.; Sachsenberg T.; Alka O.; Walzer M.; Fillbrunn A.; Nilse L.; Schilling O.; Reinert K.; Kohlbacher O. OpenMS - A platform for reproducible analysis of mass spectrometry data. J. Biotechnol. 2017, 261, 142–148. 10.1016/j.jbiotec.2017.05.016. [DOI] [PubMed] [Google Scholar]
- Brosch M.; Yu L.; Hubbard T.; Choudhary J. Accurate and sensitive peptide identification with Mascot Percolator. J. Proteome Res. 2009, 8, 3176–3181. 10.1021/pr800982s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rurik M.; Alka O.; Aicheler F.; Kohlbacher O. Metabolomics Data Processing Using OpenMS. Methods Mol. Biol. 2020, 2104, 49–60. 10.1007/978-1-0716-0239-3_4. [DOI] [PubMed] [Google Scholar]
- Shen X.; Xu T.; Hakkila B.; Hare M.; Wang Q.; Wang Q.; Beckman J.; Sun L.. Capillary zone electrophoresis-electron-capture collision-induced dissociation on a quadrupole time-of-flight mass spectrometer for top-down characterization of intact proteins. J. Am. Soc. Mass Spectrom. 2021, in press. 10.1021/jasms.0c00484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaskin R.; Voinov V.; Newton K.; Kurulugama R.; Stafford G.; Beckman J.; Barofsky D.; Costello C.. Ion Mobility Quadrupole Time-of-Flight Mass Spectrometer Modified for Electron Capture Dissociation of Glycans, Glycoconjugates, Peptides, and Proteins. Proceedings of the 64th ASMS Conference on Mass Spectrometry and Allied Topics, San Antonio, TX, June 5–9, 2016.
- Gadkari V. V.; Ramirez C. R.; Vallejo D. D.; Kurulugama R. T.; Fjeldsted J. C.; Ruotolo B. T. Enhanced Collision Induced Unfolding and Electron Capture Dissociation of Native-like Protein Ions. Anal. Chem. 2020, 92, 15489. 10.1021/acs.analchem.0c03372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M.; Liu W.; Shaw J. B. Charge Movement and Structural Changes in the Gas-Phase Unfolding of Multimeric Protein Complexes Captured by Native Top-Down Mass Spectrometry. Anal. Chem. 2020, 92, 1788–1795. 10.1021/acs.analchem.9b03469. [DOI] [PubMed] [Google Scholar]
- Li H.; Sheng Y.; McGee W.; Cammarata M.; Holden D.; Loo J. A. Structural Characterization of Native Proteins and Protein Complexes by Electron Ionization Dissociation-Mass Spectrometry. Anal. Chem. 2017, 89, 2731–2738. 10.1021/acs.analchem.6b02377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenaidee M. A.; Lantz C.; Perkins T.; Jung W.; Loo R. R. O.; Loo J. A. Internal Fragments Generated by Electron Ionization Dissociation Enhance Protein Top-Down Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2020, 31, 1896–1902. 10.1021/jasms.0c00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.