

Abstract
Holoprosencephaly (HPE) is a dramatic human brain malformation sequence with an extreme variable phenotypic spectrum and genetic heterogeneity, variable degree of severity and unknown etiology, in many cases. HPE is classified into syndromic, chromosomal, and non-syndromic, non-chromosomal. The most cases of HPE are syndromic. We present an atypical case of syndromic alobar HPE associated with digynic triploidy fetus, prenatally diagnosed, early at 18 weeks of gestation, by ultrasound (US) and complex genetic investigations. The US examination was performed with a specialized US machine, General Electric Voluson E10 OLED BT18, using two-dimensional (2D) scanning, three-dimensional (3D) image reconstruction, four-dimensional (4D) spatiotemporal image methodology and the highest power Doppler US technology. A detailed US examination of the fetus revealed several major abnormalities of the fetal head and severe facial malformations. Based on the antenatal US findings, the fetus was diagnosed with alobar HPE. After a careful examination and genetic counseling, additional cytogenetic investigations and molecular genetic analyses were performed, which revealed an abnormal number of 69 chromosomes, digynic triploidy (69,XXY). Two days later, the parents choose to interrupt the current gestation because of major fetal malformations. The pathological examination of the embryo reaffirmed the antenatal diagnostics.
Keywords: holoprosencephaly, syndromic, triploidy, ultrasound, prenatal diagnosis
⧉ Introduction
Holoprosencephaly (HPE), a major congenital abnormality in brain development, relates to a variety of disturbance deriving from inadequate separation of the embryonic forebrain, during incipient stages of embryogenesis, into separate hemispheres [1,2].
HPE is classified into syndromic, chromosomal, and non-syndromic, non-chromosomal. The most cases of HPE are syndromic. Syndromic HPE is a rare developmental malformation, affecting one in 20 000 to one in 40 000 newborns [3,4]. Both sexes are equally affected [3]. The most usual chromosomal syndromes include aneuploidies, like trisomies 13, 16, 18, 21, monosomy 13q syndrome (long arm 13 deletion syndrome), monosomy 18p syndrome (short arm 18 deletion syndrome), and less often, Young–Madders syndrome and triploidy syndrome [5].
Four types of non-syndromic, non-chromosomal HPE exists, based on the nature of the brain malformations and severity of the defect. In descending order of severity, the four categories are: alobar HPE type, semilobar HPE variety, lobar HPE form and middle interhemispheric (MIH) fusion [6,7,8].
The alobar HPE form, a very rare and lethal complex human malformation [9], is the most serious expression of the defect, consisting in a large monoventricle, thalamic fusion and agenesis of the callosal commissure, absent cerebral falx, olfactory bulbs and optic visual tracts [2, 10]. The affected fetus is most often stillborn, or dies, in the neonatal period, or in the first half year of life [3, 11,12].
The etiology of HPE is highly polymorphic, and remains being explained [13,14]. In the etiology of non-syndromic, non-chromosomal HPE, to date, seven genes have been positively implicated: Sonic hedgehog (SHH) (142945), Zic family member 2 (ZIC2) (609637), SIX homeobox 3 (SIX3) (157170), transforming growth factor-beta (TGF-β)-induced factor homeobox 1 (TGIF) (142946), patched (PTCH) (610828), glioma-associated oncogene family zinc finger 2 (GLI2) (610829) and teratocarcinoma-derived growth factor 1 (TDGF1) [15].
Regarding the etiology of syndromic, chromosomal HPE, the most common chromosomal abnormality is trisomy 13, which is reported in 24–45% of newborns with this trisomy [16].
Aim
The specific aim of the study is to present a very rare association of two major conditions: fetal alobar HPE and digynic triploidy (69,XXY), early prenatally diagnosed by detailed fetal anatomy scan and specific genetic testing. The ultimate aim of research is to reveal the importance of early prenatal diagnosis in the prevention of severe major disorders incompatible with life.
⧉ Case presentation
A 34-year-old pregnant woman, primipara, with a history of overweight, presented to the Division of Maternal Fetal Medicine of a private medical center, from Bucharest, Romania, in February 2018, at 17 weeks of gestation, because of an abnormal fetal ultrasound (US) study. The present pregnancy is the first gestation of a young European, non-consanguineous family, from the urban area.
In the first phase, the pregnant woman was examined by a perinatologist. The US examination was performed with a specialized US machine, General Electric Voluson E10 OLED BT18 US Medical System (Austria), using two-dimensional (2D) scanning, three-dimensional (3D) image reconstruction, four-dimensional (4D) spatiotemporal image methodology and the highest power Doppler US technology. According to protocol standard techniques, US scans were performed transabdominally with a 3.5 MHz curvilinear transducer, to determine longitudinal frames of reference for biometric parameters of growing and development of the fetus.
The US investigation showed a unique viable in utero pregnancy, with a medium fetal age in accordance with 18 weeks and four days. Fetal age was determined according to the last menstrual period (LMP) and validated by an estimate based on echographic measurements of the fetus. Sonographic fetal weight, 249±36 g, was estimated based on Hadlock’s growth equation.
A detailed US examination of the fetus revealed several major abnormalities of the fetal head and severe facial malformations.
At the level of the fetal head, the supratentorial brain was replaced by cerebrospinal fluid, a thin brain mantle (about 1–2 mm) and a fused thalami (Figure 1). The corpus callosum, falx cerebri, cavum septum pellucidum and third ventricle were not visualized. Also, no optic tracts and olfactory bulbs were present.
Figure 1.
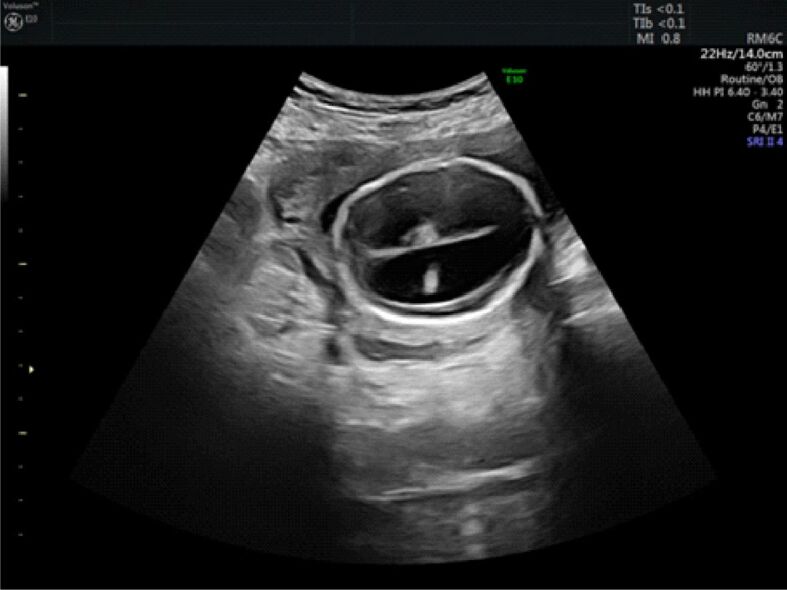
Ultrasound image of the fetal head showing hydrocephalus, fused thalami, the choroid plexus that appear to float freely in the ventricular cavity and thin brain mantle (about 1 mm)
The facial structures were dysmorphic with frontal bosses, nasal anomalies (depressed nasal bridge), and severe ocular and orbital abnormalities, such as hypotelorism, hyperplasia of the supraorbital portion of the frontal bones and the posterior protrusion of the orbit into the middle cranial fossa (Figure 2).
Figure 2.
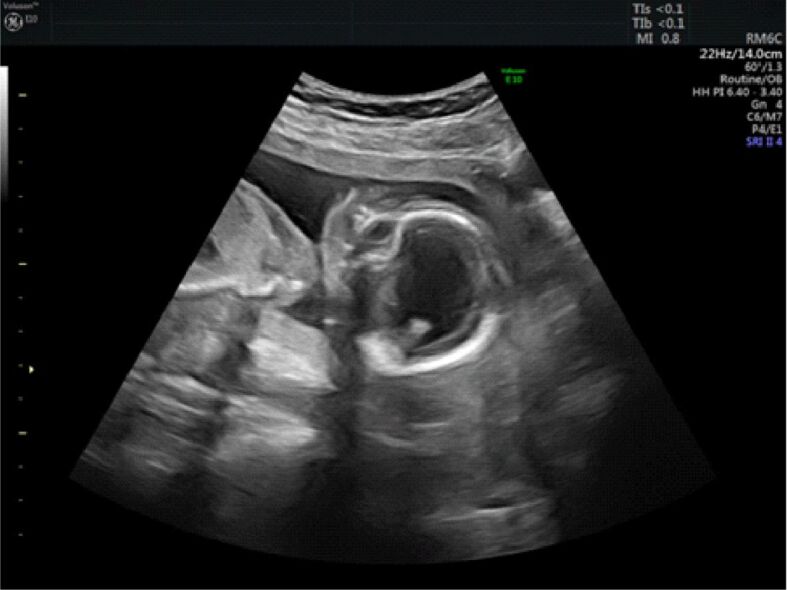
Ultrasound image of the fetal head showing large ventricle, the absence of the corpus callosum, choroid plexus that appear to float freely in the ventricular cavity (posterior) and thin brain mantle (about 2 mm), prominent forehead, depressed nasal bridge and orbital abnormalities
The fetal US head biometry measurements report the following remarkable abnormal values of the standard and additional parameters: head biparietal diameter (BPD): 4.64 cm (high value), occipitofrontal head diameter (OFD): 6.05 cm (high value), head circumference (HC): 16.89 cm (high value), transverse cerebellar diameter (Cereb) (Hill): 1.99 cm (normal range), interocular distance (IOD): 1.27 cm, and binocular distance (BOD): 2.8 cm (low value) (Figures 3,4,5,6).
Figure 3.
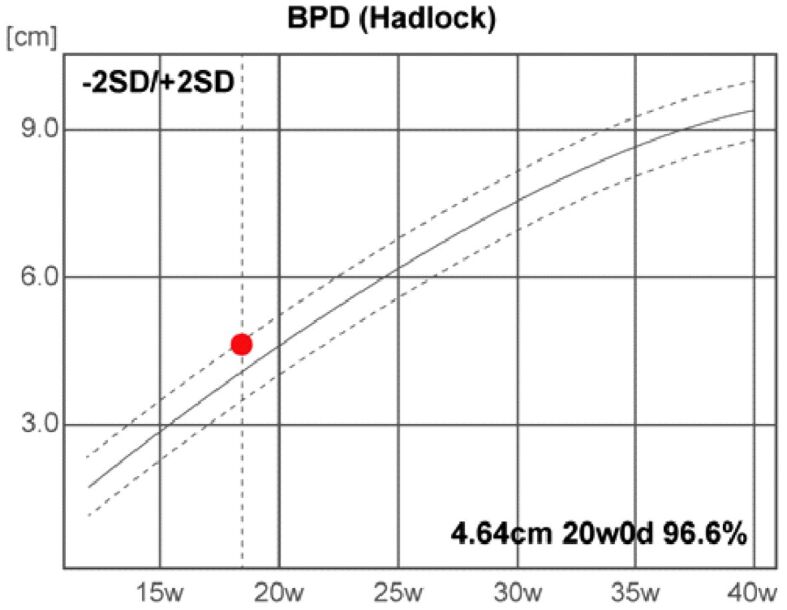
Head biparietal diameter (BPD). d: Days; SD: Standard deviation; w: Weeks
Figure 4.
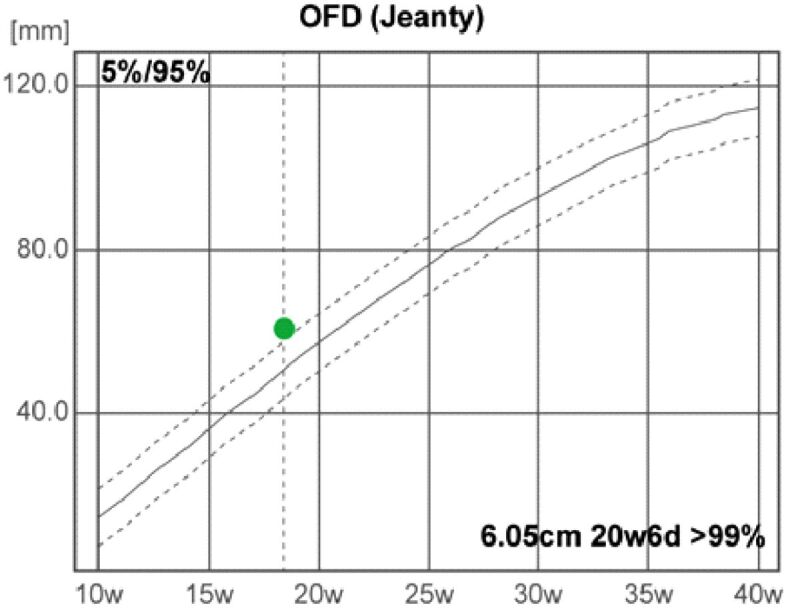
Occipitofrontal diameter (OFD). d: Days; w: Weeks
Figure 5.
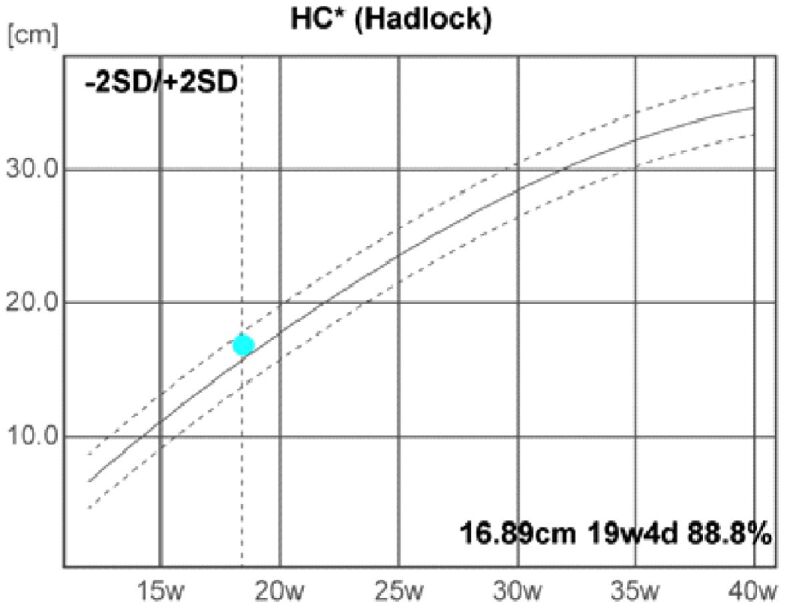
Head circumference (HC). d: Days; SD: Standard deviation; w: Weeks
Figure 6.
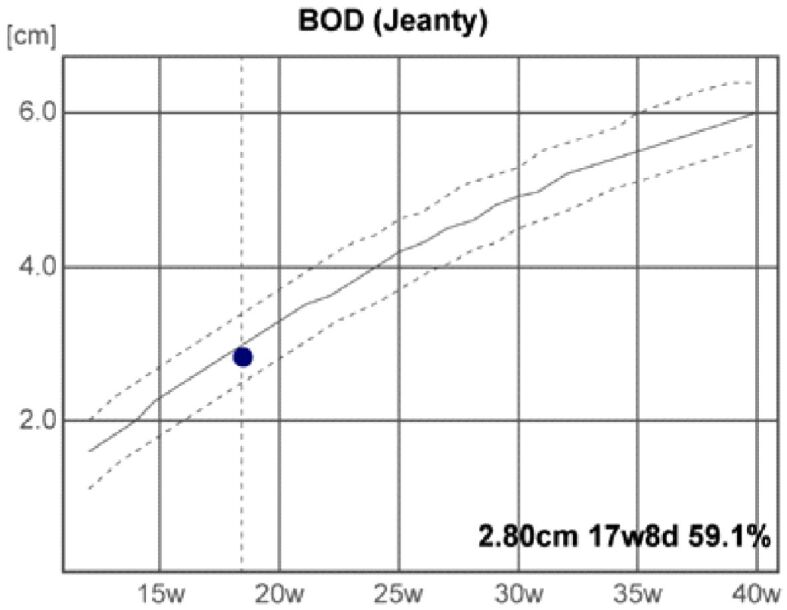
Binocular distance (BOD). d: Days; w: Weeks
The other biometric parameters of fetal head structures examined as part of the fetal morphology US examination, recorded the following significant values: anterior horn of the lateral ventricle (Va): 15.24 mm, posterior horn of the lateral ventricle (Vp): 17.93 mm, cisterna magna diameter (CM): 2.79 mm, hemisphere diameter (HEM): 20.59 mm, fetal nasal bone length (NBL): 6.26 mm and neck fold (NF): 2.26 mm.
The following measurements of additional structures examined in fetal anomaly US screening, were also performed: transthoracic diameter (TTD), anterior-posterior fetal thigh diameter (APTD), abdominal circumference (AC), anterior-posterior abdominal diameter (APAD), transverse abdominal diameter (TAD), femur length (FL) and humerus length (HL). Detailed data corresponding to the fetal US biometry are provided and illustrated in tabulations (Figures 7 and 8).
Figure 7.
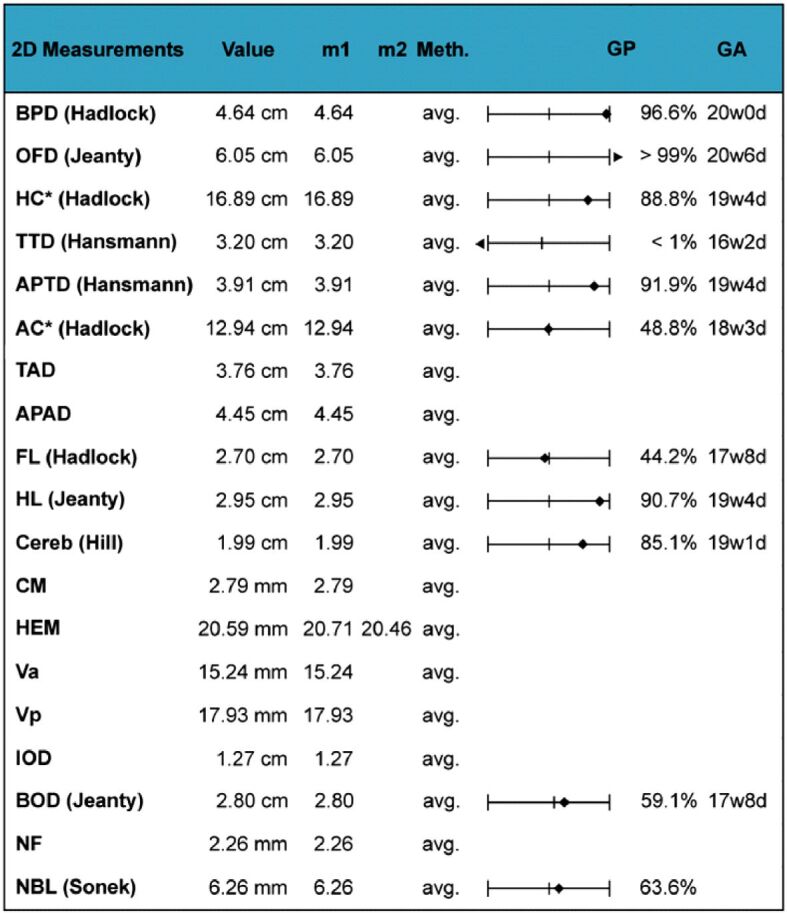
Ultrasound fetal biometry interpretation. 2D: Two-dimensional; AC: Abdominal circumference; APAD: Anterior-posterior abdominal diameter; APTD: Anterior-posterior fetal thigh diameter; avg.: Average; BOD: Binocular distance; BPD: Biparietal diameter; Cereb: Transverse cerebellar diameter; CM: Cisterna magna diameter; d: Days; FL: Femur length; GA: Gestational age; GP: Gestational pregnancy; HC: Head circumference; HEM: Hemisphere diameter; HL: Humerus length; IOD: Interocular distance; m1: Measurement 1; m2: Measurement 2; Meth.: Method; NBL: Nasal bone length; NF: Neck fold; OFD: Occipitofrontal diameter; TAD: Transverse abdominal diameter; TTD: Transthoracic diameter; Va: Anterior horn of the lateral ventricle; Vp: Posterior horn of the lateral ventricle; w: Weeks
Figure 8.
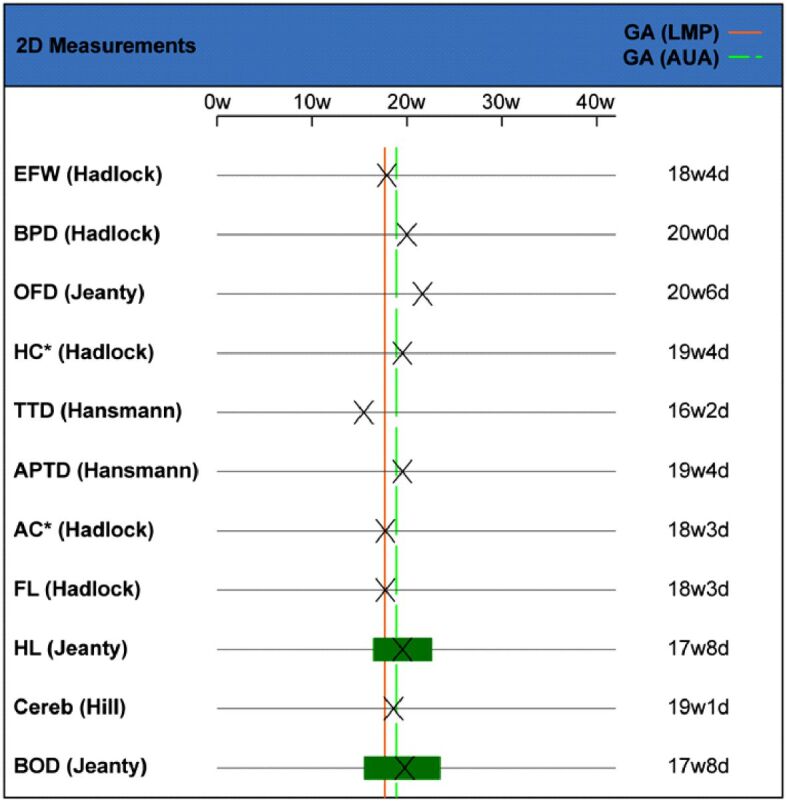
Ultrasound fetal biometry: correlation between 2D measurements and GA. 2D: Two-dimensional; AC: Abdominal circumference; APTD: Anterior-posterior fetal thigh diameter; AUA: Actual ultrasound age; BOD: Binocular distance; BPD: Biparietal diameter; Cereb: Transverse cerebellar diameter; d: Days; EFW: Estimated fetal weight; FL: Femur length; GA: Gestational age; HC: Head circumference; HL: Humerus length; LMP: Last menstrual period; OFD: Occipitofrontal diameter; TTD: Transthoracic diameter; w: Weeks
Based on the fetal biometric determinations, we calculated the following parameters, which provided the following results: cephalic index (CI) ratio: 77% (normal range), FL/AC ratio: 20% (inferior limit of the normal range), FL/BPD ratio: 60% (very low value), FL/HC ratio (Hadlock): 0.15 (low value), HC/AC ratio (Campbell): 1.30 (high value), Va/HEM ratio (Hansmann): 0.74 (high value), Vp/HEM ratio (Nicolaides): 0.87 (high value) (Figures 9 and 10; Table 1).
Figure 9.
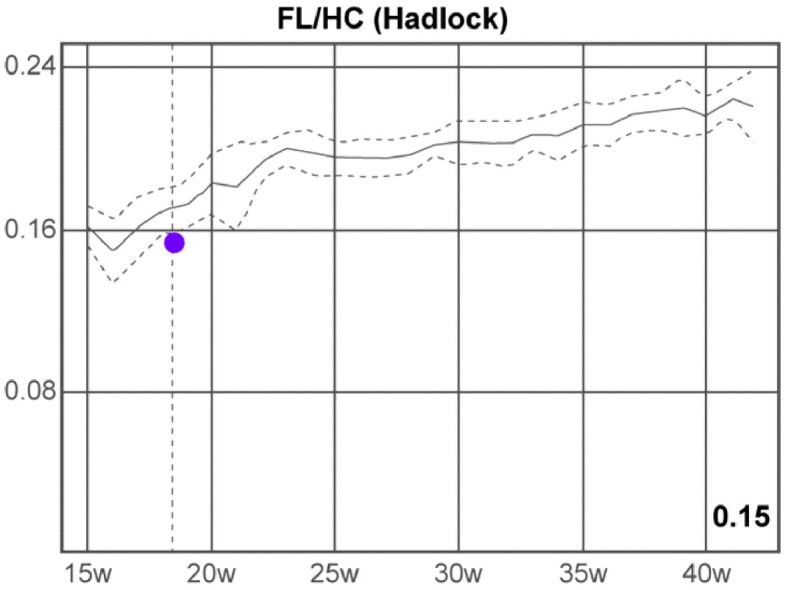
Femur length/head circumference (FL/HC) ratio (Hadlock). w: Weeks
Figure 10.
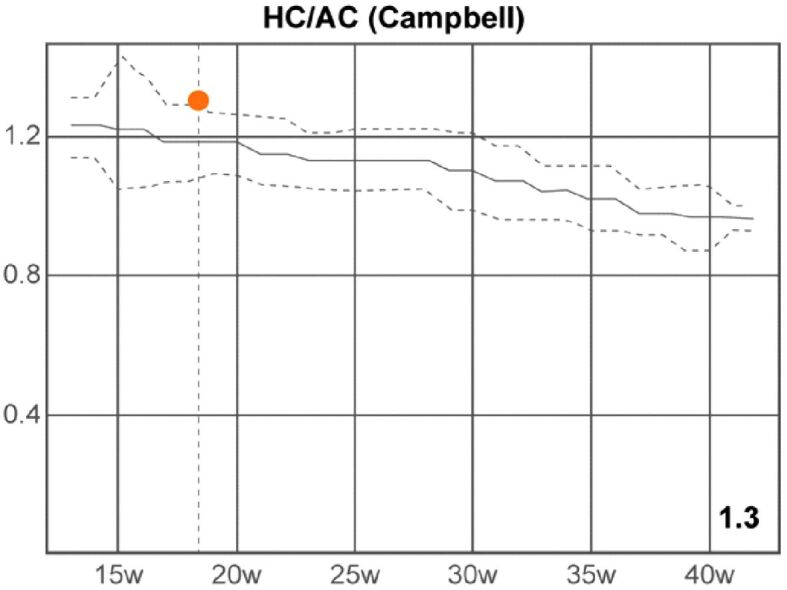
Head circumference/abdominal circumference (HC/AC) ratio (Campbell). w: Weeks
Table 1.
Obstetrics report: 2D calculations
|
2D calculations |
Value |
Normal range |
|
CI (BPD/OFD ratio) |
77% |
70–86% |
|
FL/AC ratio |
20% |
20–24% |
|
FL/BPD ratio |
60% ⇓ ⇓ |
GA: OOR |
|
FL/HC ratio (Hadlock) |
0.15 ⇓ |
0.16–0.18 |
|
HC/AC ratio (Campbell) |
1.30 ⇑ |
1.08–1.28 |
|
Va/HEM ratio (Hansmann) |
0.74 ⇑ |
0.37–0.59 |
2D: Two-dimensional; AC: Abdominal circumference; BPD: Biparietal diameter; CI: Cephalic index; FL: Femur length; GA: Gestational age; HC: Head circumference; HEM: Hemisphere; OFD: Occipitofrontal head diameter; OOR: Out of range; Va: Anterior horn of the lateral ventricle
We also performed Doppler velocimetry of fetal middle cerebral artery (MCA) and umbilical artery (UA), analyzing the following parameters: pulsatility index (PI), resistivity index (RI), peak systolic velocity (PSV), end-diastolic velocity (EDV), systolic/diastolic (S/D) ratio, time-averaged peak velocity (TAmax) and fetal heart rate (HR). Detailed results of the parameters of Doppler examination are summarized in Table 2.
Table 2.
Obstetrics report: Doppler measurements
|
Doppler measurements |
Value |
m1 |
|
|
Right MCA |
|
|
PSV [cm/s] |
27.59 |
27.59 |
|
EDV [cm/s] |
2.80 |
2.80 |
|
TAmax [cm/s] |
12.48 |
12.48 |
|
MDV [cm/s] |
2.80 |
2.80 |
|
RI |
0.90 |
0.90 |
|
PI |
1.99 |
1.99 |
|
S/D ratio |
9.85 |
9.85 |
|
HR [bpm] |
146 |
146 |
|
|
UA |
|
|
PSV [cm/s] |
17.15 |
-14.68 |
|
EDV [cm/s] |
11.93 |
-11.93 |
|
TAmax [cm/s] |
13.46 |
-13.46 |
|
MDV [cm/s] |
11.60 |
-11.60 |
|
RI |
0.47 |
0.19 |
|
PI |
0.74 |
0.20 |
|
S/D ratio |
2.65 |
1.23 |
|
HR [bpm] |
441 |
441 |
bpm: Beats per minute; EDV: End-diastolic velocity; HR: Heart rate; m1: Measurement 1; MCA: Middle cerebral artery; MDV: Minimum diastolic velocity; PI: Pulsatility index; PS: Peak systolic velocity; RI: Resistivity index; S/D: Systolic/diastolic; TAmax: Time-averaged peak velocity; UA: Umbilical artery
Also, additional cytogenetic investigations and molecular genetic analysis were performed to establish the cause of HPE. Amniocentesis was done to obtain a small quantity of amniotic fluid. From the sample of amniotic liquid, two techniques were applied, both of them providing the same result: 69,XXY triploidy.
The identification of heteroploidies of the autosomal chromosomes 13, 18, 21 and sex chromosomes X, Y was made by multiplex quantitative fluorescence polymerase chain reaction (QF–PCR) targeting 26 markers, by direct deoxyribonucleic acid (DNA) amplification, followed by fluorescent capillary electrophoresis of amplified DNA fragments.
QF–PCR uses specially selected DNA markers (M), named short tandem repeats (STRs). STRs were amplified by PCR using fluorescently labeled primers. The number of copies of each STR marker corresponds with the number of copies of the chromosomes analyzed.
The PCR product were subjected to separation by fluorescent capillary electrophoresis using the genetic analyzer, and the results was analyzed with the GeneMapper™ software.
Each marker generates a specific profile of fluorescent signals (peaks), profile that was analyzed quantitatively by calculating the peak height size and peak areas of these signals.
QF–PCR results for rapid detection of chromosomal aneuploids have revealed 69,XXY triploidy, and the biallelic model with 1:2 ratio (paternally allele to maternally allele ratio) indicates a maternally source of the triploid syndrome (Table 3). The method has an accuracy of 99.8%.
Table 3.
Details of the molecular markers: name of the marker, cytogenetic localization, fluorescent label, and the range of the allele resulted from the PCR tests
|
Laboratory results | ||
|
Locus |
Alleles |
Ratio |
|
Marker 21-1 (locus 21q21.1) |
141 |
– |
|
Marker 21-2 (locus 21q21.3) |
183, 187 |
1:2 |
|
Marker 21-3 (locus 21q21.1) |
243, 260 |
1:2 |
|
Marker 21-4 (locus 21q22.3) |
290, 298 |
2:1 |
|
Marker 21-5 (locus 21q21.3) |
372, 384, 396 |
1:1:1 |
|
Marker 21-6 (locus 21q22.13) |
462, 468, 470 |
1:1:1 |
|
Marker 18-1 (locus 18p12.3) |
213 |
– |
|
Marker 18-2 (locus 18p12.3) |
320, 324 |
2:1 |
|
Marker 18-3 (locus 18q22.1) |
387, 391, 403 |
1:1:1 |
|
Marker 18-4 (locus 18q11.32) |
369, 373, 384 |
1:1:1 |
|
Marker 18-5 (locus 18q11.31) |
461, 476 |
1:2 |
|
Marker 13-1 (locus 13q21.1) |
123, 131 |
2:1 |
|
Marker 13-2 (locus 13q12.12) |
254, 269, 278 |
1:1:1 |
|
Marker 13-3 (locus 13q21.32-q21.33) |
403, 406 |
1:2 |
|
Marker 13-4 (locus 13q13.1) |
432, 455, 463 |
1:1:1 |
|
Marker 13-5 (locus 13q13.3) |
454, 462, 470 |
1:1:1 |
|
Marker X/Y-1 (locus Xp22.2/Yp11.2) |
103, 109 |
2:1 |
|
Marker X/Y-2 (locus Xp22.11/Yp11.31) |
165 |
– |
|
Marker X/Y-3 (locus Xq21.31/Yp11.31) |
195 |
– |
|
Marker X/Y-4 (locus Xp22.33/Yp11.32) |
242, 246 |
2:1 |
|
Marker X-1 (locus Xq26.2) |
147 |
– |
|
Marker X-2 (locus Xq26.2-q26.3) |
276, 291 |
1:1 |
|
Marker X-3 (locus Xq27.1-q27.2) |
323, 339 |
1:1 |
|
Marker Y-1 (locus Yp11.31) |
240 |
– |
|
Marker control (locus 3p24.2/Xq21.1) |
132, 136 |
3:2 |
|
Marker control (locus 7q34/Xq13) |
183, 203 |
3:2 |
PCR: Polymerase chain reaction
Also, amniotic cells were cultured according with the standard cytogenetic methods, in two independent cultures (two flasks). Mitotic metaphases were prepared following a standard protocol and the chromosomes were banded by Giemsa staining [R-bands by heating using Giemsa (RHG) technique, banding score >4). Cytogenetic analysis was performed by two independent observers using CaryoSystems image analysis software. The karyotyping nomenclature followed the guidelines provided by International System for Human Cytogenomic Nomenclature (ISCN, 2016).
In our case, 20 metaphases were analyzed, of which 10 were karyotyped. Prenatal cytogenetic analysis revealed the presence of an extra haploid set of chromosomes in each cell, resulting an abnormal number of 69 chromosomes (69,XXY triploidy) (Figure 11). The sensitivity of the method is 999‰.
Figure 11.
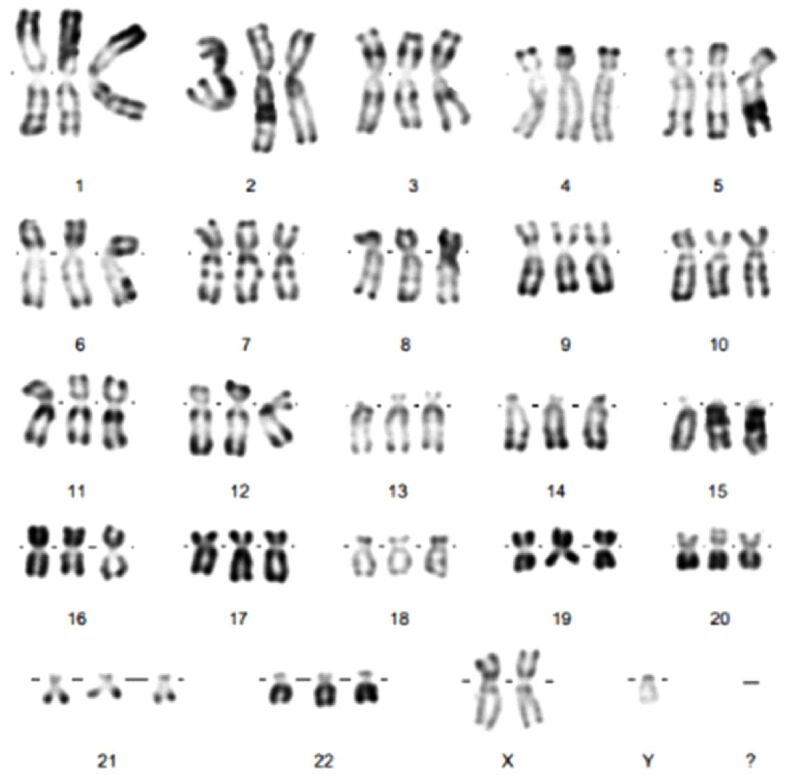
Fetal karyotype: 69,XXY. Specimen type: Amniotic fluid. Giemsa staining (RHG technique). RHG: R-bands by heating using Giemsa
After a careful US examination and genetic counseling, based on the antenatal US findings, the fetus was diagnosed with syndromic alobar HPE.
As a result, the couple was briefed regarding to the phenotypic characteristics of HPE and the gravity of cerebrofacial anomalies, which is why, two days later, the parents chose therapeutic abortion for medical reasons, finally anatomopathologically confirmed.
Currently, the mother is well, and the couple has decided to perform additional genetic investigations to prevent the appearance of a new affected fetus.
⧉ Discussions
Triploidy, a rare chromosomal numeric abnormality, generated by the existence of a supplementary haploid chromosome set, is incompatible with postnatal life [17]. The three possible chromosome complement are: 69,XXX; 69,XXY; and 69,XYY [18]. The prevalence of fetal triploidy is estimate to one per 100 of human conceptuses. Because the most of fetuses with triploidy are spontaneously aborted in the first trimester of pregnancy, at the beginning of the second trimester of pregnancy, the prevalence of triploidy is about one in each 5000 gestations [19,20,21,22].
There are diverse mechanisms that may produce triploidy: diandric triploidy, when the supplementary haploid chromosome set has paternal origin, or digynic triploidy, when the supplementary haploid chromosome set has maternal origin [19]. Digynic triploidies origin result from fertilization of a primary oocyte, a diploid oocyte or retention of a polar body [22]. The substrate of human triploidy is disputed [23,24]. About 75–86% of cases are diandric triploidy, generally resulting from the fecundation of a female haploid reproductive cell by two male haploid gametes (dispermy) [21, 25]. Triploidy of maternal origin is more frequently found in the fetal period, probably because of the longer survival in comparison with triploidy of paternal origin [26]. The mechanisms underlying digynic triploidy are still uncertain [27]. Triploidy is associated with intrauterine growth restriction, craniofacial dysmorphisms with fetal central nervous system anomalies (in 50% of the cases), multiple malformations (such as congenital heart defects, renal and respiratory defects, gastrointestinal tract anomalies, genitourinary and skeletal malformations) and placental abnormalities (molar placental changes), leading inevitably to spontaneous abortion or neonatal death, being one of the most common causes of miscarriages in the first trimester of pregnancy [21,28,29,30,31,32 ]. The association of triploidy with HPE is very rare [17, 33]. HPE is a developmental disorder, a dramatic central nervous system malformation, resulting from the improper diverticulation and segmentation of the prosencephalon, with an extremely heterogeneous etiology [34]. There are a few theories citing the mechanical causes, environmental, and genetic factors, infectious disease, but the definitive cause of HPE remains unknown, in many cases [35,36]. Up to now, seven genes have been incriminated in the etiology of HPE. A molecular genetic testing can be carried out by DNA sequencing and allele specific quantification for the major genes SHH, ZIC2, SIX3 and TGIF. Unfortunately, in over 70% of situations, the molecular genetic basis of HPE remains undiscovered [15]. For families with no identifiable etiology, HPE can be antenatal diagnosed by US examination performed by an expert with competence in ultrasonography and certification in maternal and fetal medicine [37,38].
As a result of contemporary progress in the evolution and development of super-resolution fetal ultrasonography, prenatal diagnosis of congenital malformations like HPE is today possible. The US machines provide exceptional images that permit an accurate diagnosis of cephalic as well as extracephalic abnormalities [39]. Fetal US findings of alobar HPE, the most serious form of HPE, can be early diagnosed in the second trimester of pregnancy. At the obstetric sonography examination, these structural anomalies include absence of median structures, a monoventricular cavity, fused thalami, and facial malformations [40,41]. In the present case, using 2D, 3D and 4D imaging technology, identification of earlier fused thalami at 18 weeks of pregnancy was achievable. Furthermore, missing of median structures echosignal and third ventricle was observed. The accurate sonographic images were representative of alobar HPE [42]. Also, after the US scan, following a comprehensive consultation with a specialist in obstetrics and fetology expert, it was determined to complete supplementary genetic analysis, respectively amniocentesis and QF–PCR, as well as fetal karyotype [43,44,45]. QF–PCR is a rapid prenatal test that detects the presence of numerical aberrations of chromosomes 13, 18, 21, X, Y, which are the most common viable aneuploids, by amplification of repeated sequences at chromosome-specific polymorphic loci. In our case, the primers used have been developed and manufactured in accordance with accredited quality systems International Organization for Standardization (ISO) 13485: 2012, by Lloyd’s Register Quality Assurance Ltd. [46].
During the investigations, the couple were sustained by a multidisciplinary unit with vast experience in the fetal US diagnosis and antenatal genetic evaluation [47,48,49].
HPE is an essential perinatally healthcare problem because of the gravity of cerebrofacial anomalies spectrum and its unsatisfactory prognosis [50,51]. Survival ratio differs in each type of HPE, but generally, lethality corresponds with the severeness of the cerebral malformation and, by expansion, gravity of the craniofacial malformations [13]. Prenatal US investigation, complex genetic testing and couple’s genetic counseling was essential in the prenatal diagnosis and management of the case [52,53]. Finally, the fetal autopsy provided important data for parental genetic counseling, and management of future pregnancies [54].
⧉ Conclusions
The strength of this study is in the elaborate prenatal US findings analysis in a unique case of fetal syndromic HPE present in a fetus with digynic triploidy. This study provided an illustration of the variety of the structural anomalies that appeared in association with triploidy. The findings identified by prenatal US examination underline the supremacy of US imaging technology and the importance of prenatal genetic tests, including amniocentesis, cytogenetic tests, and molecular genetic analysis, whenever abnormalities and growth disturbance are identified, because these are frequently present in fetuses with chromosomal aberrations diseases. Prenatal US examination was decisive in the early prenatal diagnosis, at 18 weeks of gestation, and optimized management of the malformed fetus with an extremely severe type of syndromic HPE associated with digynic triploidy. This new research demonstrated once again the potentiality of prenatal US scan and the power of genetic evaluation in the prevention of fetal morbidity and mortality by extremely severe genetic and structural malformations.
Conflict of interest
The authors declare that they have no conflict of interests.
References
- 1.Nyberg DA, Mack LA, Bronstein A, Hirsch J, Pagon RA. Holoprosencephaly: prenatal sonographic diagnosis. AJR Am J Roentgenol. 1987;149(5):1051–1058. doi: 10.2214/ajr.149.5.1051. [DOI] [PubMed] [Google Scholar]
- 2.Măluţan AM, Dudea M, Ciortea R, Mureşan M, Bucuri CE, Mihu C, Mihu D. Cyclopia and proboscis - the extreme end of holoprosencephaly. Rom J Morphol Embryol. 2017;58(4):1555–1559. [PubMed] [Google Scholar]
- 3.Bendavid C, Rochard L, Dubourg C, Seguin J, Gicquel I, Pasquier L, Vigneron J, Laquerrière A, Marcorelles P, Jeanne-Pasquier C, Rouleau C, Jaillard S, Mosser J, Odent S, David V. Array-CGH analysis indicates a high prevalence of genomic rearrangements in holoprosencephaly: an updated map of candidate loci. Hum Mutat. 2009;30(8):1175–1182. doi: 10.1002/humu.21016. [DOI] [PubMed] [Google Scholar]
- 4.Golden JA. Holoprosencephaly. In: Squire LR, editor. Encyclopedia of Neuroscience. Academic Press; 2009. pp. 1181–1187. [Google Scholar]
- 5.Kruszka P, Muenke M. Syndromes associated with holoprosencephaly. Am J Med Genet C Semin Med Genet. 2018;178(2):229–237. doi: 10.1002/ajmg.c.31620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tripathi AK, Agrawal D, Sedain G. Hydrocephalic holoprosencephaly: an oxymoron? Insights into etiology and management. J Pediatr Neurosci. 2009;4(1):41–43. doi: 10.4103/1817-1745.49108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Simon EM, Hevner RF, Pinter JD, Clegg NJ, Delgado M, Kinsman SL, Hahn JS, Barkovich AJ. The middle interhemispheric variant of holoprosencephaly. AJNR Am J Neuroradiol. AJNR Am J Neuroradiol. 2002;2002;2323(1)(4):151–156. 742–742. [PMC free article] [PubMed] [Google Scholar]
- 8.Ritivoiu M, Brezan F, Codreanu I, Stamate M, Anca I. Ultrasound diagnosis in two cases of severe craniofacial anomalies. Med Ultrason. 2013;15(4):330–332. doi: 10.11152/mu.2013.2066.154.mrit2. [DOI] [PubMed] [Google Scholar]
- 9.Salama GS, Kaabneh MA, Al-Raqad MK, Al-Abdallah IM, Shakkoury AG, Halaseh RA. Cyclopia: a rare condition with unusual presentation – a case report. Clin Med Insights Pediatr. 2015;9:19–23. doi: 10.4137/CMPed.S21107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ionescu CA, Calin D, Navolan D, Matei A, Dimitriu M, Herghelegiu C, Ples L. Alobar holoprosencephaly associated with a rare chromosomal abnormality: case report and literature review. Medicine (Baltimore) 2018;97(29):e11521–e11521. doi: 10.1097/MD.0000000000011521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Manea AM, Cioboata D, Stroescu R, Boia M, Motoc M. Biochemical study of cerebrospinal liquid composition on a lot of newborns with hydrocephaly. Rev Chim (Bucharest) 2019;70(2):495–497. [Google Scholar]
- 12.Saunders ES, Shortland D, Dunn PM. What is the incidence of holoprosencephaly. J Med Genet. 1984;21(1):21–26. doi: 10.1136/jmg.21.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Raam MS, Solomon BD, Muenke M. Holoprosencephaly: a guide to diagnosis and clinical management. Indian Pediatr. 2011;48(6):457–466. doi: 10.1007/s13312-011-0078-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paulussen ADC, Schrander-Stumpel CT, Tserpelis DCJ, Spee MK, Stegmann APA, Mancini GM, Brooks AS, Collée M, Maat-Kievit A, Simon MEH, van Bever Y, Stolte-Dijkstra I, Kerstjens-Frederikse WS, Herkert JC, van Essen AJ, Lichtenbelt KD, van Haeringen A, Kwee ML, Lachmeijer AMA, Tan-Sindhunata GMB, van Maarle MC, Arens YHJM, Smeets EEJGL, de Die-Smulders CE, Engelen JJM, Smeets HJ, Herbergs J. The unfolding clinical spectrum of holoprosencephaly due to mutations in SHH, ZIC2, SIX3 and TGIF genes. Eur J Hum Genet. 2010;18(9):999–1005. doi: 10.1038/ejhg.2010.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dubourg C, Bendavid C, Pasquier L, Henry C, Odent S, David V. Holoprosencephaly. Orphanet J Rare Dis. 2007;2:8–8. doi: 10.1186/1750-1172-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Costa AD, Schultz R, Rosemberg S. Alobar holoprosencephaly and trisomy 13 (Patau syndrome) Autops Case Rep. 2013;3(2):5–10. doi: 10.4322/acr.2013.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Toufaily MH, Roberts DJ, Westgate MN, Holmes LB. Triploidy: variation of phenotype. Am J Clin Pathol. 2016;145(1):86–95. doi: 10.1093/ajcp/aqv012. [DOI] [PubMed] [Google Scholar]
- 18.McWeeney DT, Munné S, Miller RC, Cekleniak NA, Contag SA, Wax JR, Polzin WJ, Watson WJ. Pregnancy complicated by triploidy: a comparison of the three karyotypes. Am J Perinatol. 2009;26(9):641–645. doi: 10.1055/s-0029-1220794. [DOI] [PubMed] [Google Scholar]
- 19.Chuang TY, Chang SY, Chen CP, Lin MH, Chen CY, Chen SW, Chern SR, Lee CC, Town DD, Wang W. Digynic triploidy in a fetus presenting with semilobar holoprosencephaly. Taiwan J Obstet Gynecol. 2018;57(6):881–884. doi: 10.1016/j.tjog.2018.11.001. [DOI] [PubMed] [Google Scholar]
- 20.Engelbrechtsen L, Brøndum-Nielsen K, Ekelund C, Tabor A, Skibsted L, Danish Fetal Medicine Study Group Detection of triploidy at 11–14 weeks’ gestation: a cohort study of 198 000 pregnant women. Ultrasound Obstet Gynecol. 2013;42(5):530–535. doi: 10.1002/uog.12460. [DOI] [PubMed] [Google Scholar]
- 21.Zalel Y, Shapiro I, Weissmann-Brenner A, Berkenstadt M, Leibovitz Z, Bronshtein M. Prenatal sonographic features of triploidy at 12–16 weeks. Prenat Diagn. 2016;36(7):650–655. doi: 10.1002/pd.4834. [DOI] [PubMed] [Google Scholar]
- 22.Kolarski M, Ahmetovic B, Beres M, Topic R, Nikic V, Kavecan I, Sabic S. Genetic counseling and prenatal diagnosis of triploidy during the second trimester of pregnancy. Med Arch. 2017;71(2):144–147. doi: 10.5455/medarh.2017.71.144-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zaragoza MV, Surti U, Redline RW, Millie E, Chakravarti A, Hassold TJ. Parental origin and phenotype of triploidy in spontaneous abortions: predominance of diandry and association with the partial hydatidiform mole. Am J Hum Genet. 2000;66(6):1807–1820. doi: 10.1086/302951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Masset H, Tšuiko O, Vermeesch JR. Genome-wide abnormalities in embryos: origins and clinical consequences. Prenat Diagn. 2021;41(5):554–563. doi: 10.1002/pd.5895. [DOI] [PubMed] [Google Scholar]
- 25.McFadden DE, Jiang R, Langlois S, Robinson WP. Dispermy – origin of diandric triploidy: brief communication. Hum Reprod. 2002;17(12):3037–3038. doi: 10.1093/humrep/17.12.3037. [DOI] [PubMed] [Google Scholar]
- 26.Doshi N, Surti U, Szulman AE. Morphologic anomalies in triploid liveborn fetuses. Hum Pathol. 1983;14(8):716–723. doi: 10.1016/s0046-8177(83)80145-2. [DOI] [PubMed] [Google Scholar]
- 27.Brancati F, Mingarelli R, Dallapiccola B. Recurrent triploidy of maternal origin. Eur J Hum Genet. 2003;11(12):972–974. doi: 10.1038/sj.ejhg.5201076. [DOI] [PubMed] [Google Scholar]
- 28.Benacerraf BR. Intrauterine growth retardation in the first trimester associated with triploidy. J Ultrasound Med. 1988;7(3):153–154. doi: 10.7863/jum.1988.7.3.153. [DOI] [PubMed] [Google Scholar]
- 29.Ludwig K, Pizzi M, Fassan M, Daolio C, Margiotti K, Consoli F, Salmaso R, Rugge M. Double trouble" or an amplification of the triploidy phenotype. Fetal Pediatr Pathol. 2013;31(1):60–65. doi: 10.3109/15513815.2012.671444. [DOI] [PubMed] [Google Scholar]
- 30.McFadden DE, Pantzar JT. Placental pathology of triploidy. Hum Pathol. 1996;27(10):1018–1020. doi: 10.1016/s0046-8177(96)90277-4. [DOI] [PubMed] [Google Scholar]
- 31.Gassner R, Metzenbauer M, Hafner E, Vallazza U, Philipp K. Triploidy in a twin pregnancy: small placenta volume as an early sonographical marker. Prenat Diagn. 2003;23(1):16–20. doi: 10.1002/pd.506. [DOI] [PubMed] [Google Scholar]
- 32.Lindor NM, Ney JA, Gaffey TA, Jenkins RB, Thibodeau SN, Dewald GW. A genetic review of complete and partial hydatidiform moles and nonmolar triploidy. Mayo Clin Proc. 1992;67(8):791–799. doi: 10.1016/s0025-6196(12)60805-2. [DOI] [PubMed] [Google Scholar]
- 33.Bekdache GN, Begam M, Al Safi W, Mirghani H. Prenatal diagnosis of triploidy associated with holoprosencephaly: a case report and review of the literature. Am J Perinatol. 2009;26(7):479–483. doi: 10.1055/s-0029-1214248. [DOI] [PubMed] [Google Scholar]
- 34.Geng X, Oliver G. Pathogenesis of holoprosencephaly. J Clin Invest. 2009;119(6):1403–1413. doi: 10.1172/JCI38937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Callahan J, Harmon C, Aleshire J, Hickey B, Jones B. Alobar holoprosencephaly with cebocephaly. J Diagn Med Sonogr. 2017;33(1):39–42. [Google Scholar]
- 36.Raman R, Mukunda Jagadesh G. Antenatal diagnosis of alobar holoprosencephaly. Case Rep Radiol. 2014;2014:724671–724671. doi: 10.1155/2014/724671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Children’s Hospital Los Angeles. Diagnostic Testing for Maternal-Fetal Medicine. Available online: https://www.chla.org/diagnostic-testing-maternal-fetal-medicine[ Accesed 29.04.2020]
- 38.Tekendo-Ngongang C, Muenke M, Kruszka P. Holopros-encephaly overview 2000 [updated 2020 Mar 5] In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, editors. GeneReviews® [Internet] Seattle WA: University of Washington; 1993–2020. [Google Scholar]
- 39.Wenghoefer M, Ettema AM, Sina F, Geipel A, Kuijpers-Jagtman AM, Hansmann H, Borstlap WA, Bergé S. Prenatal ultrasound diagnosis in 51 cases of holoprosencephaly: craniofacial anatomy, associated malformations, and genetics. Cleft Palate Craniofac J. 2010;47(1):15–21. doi: 10.1597/08-036.1. [DOI] [PubMed] [Google Scholar]
- 40.Chaudhari HD, Thakkar G, Darji P, Khokhani P. Prenatal ultrasound diagnosis of holoprosencephaly and associated anomalies. BMJ Case Rep. 2012;2012:bcr0320126129–bcr0320126129. doi: 10.1136/bcr-03-2012-6129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.El-Dessouky SH, Aboulghar MM, Gaafar HM, Abdella RM, Sharaf MF, Ateya MI, Elarab AE, Zidan WH, Helal RM, Aboelsaud SM, Eid MM, Abdel-Salam GMH. Prenatal ultrasound findings of holoprosencephaly spectrum: unusual associations. Prenat Diagn. 2020;40(5):565–576. doi: 10.1002/pd.5649. [DOI] [PubMed] [Google Scholar]
- 42.Achiron R, Achiron A, Lipitz S, Mashiach S, Goldman B. Holoprosencephaly: alobar. TheFetus.net. 1994 https://sonoworld.com/Client/Fetus/page.aspx?id=115 [Google Scholar]
- 43.Atef SH, Hafez SS, Mahmoud NH, Helmy SM. Prenatal diagnosis of fetal aneuploidies using QF-PCR: the Egyptian study. J Prenat Med. 2011;5(4):83–89. [PMC free article] [PubMed] [Google Scholar]
- 44.Huo P, Luo Q, Li J, Jiao B, Rong L, Zhang J, Wu X. High accuracy of quantitative fluorescence polymerase chain reaction combined with non-invasive prenatal testing for mid-pregnancy diagnosis of common fetal aneuploidies: a single-center experience in China. Exp Ther Med. 2019;18(1):711–721. doi: 10.3892/etm.2019.7625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Badenas C, Rodríguez-Revenga L, Morales C, Mediano C, Plaja A, Pérez-Iribarne MM, Soler A, Clusellas N, Borrell A, Sánchez MÁ, Miró E, Sánchez A, Milà M, Jiménez W. Assessment of QF-PCR as the first approach in prenatal diagnosis. J Mol Diagn. 2010;12(6):828–834. doi: 10.2353/jmoldx.2010.090224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ochshorn Y, Shira AB, Jonish A, Lessing JB, Pauzner D, Ezer O, Evans M, Orr-Urtreger A, Yaron Y. Quantitative fluorescence polymerase chain reaction (QF-PCR) for rapid prenatal diagnosis of aneuploidy. Am J Obstet Gynecol. 2004;191(6 Suppl):S44–S44. [Google Scholar]
- 47.Ko H, Chang TY, Lussier EC, Olisova K, Sung CY, Chen PK, Li WC, Yang TY, Wang RX. Multidisciplinary team approach to the prenatal management of orofacial clefts: a single center cohort study in Taiwan. Sci Rep. 2020;10(1):13916–13916. doi: 10.1038/s41598-020-70906-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bijma HH, van der Heide A, Wildschut HIJ. Decision-making after ultrasound diagnosis of fetal abnormality. Reprod Health Matters. 2008;16(31 Suppl):82–89. doi: 10.1016/S0968-8080(08)31372-X. [DOI] [PubMed] [Google Scholar]
- 49.Albu CC, Albu DF, Albu SD, Patrascu A, Musat AR, Goganau AM. Early prenatal diagnosis of an extremely rare association of Down syndrome and transposition of the great vessels. Rev Chim (Bucharest) 2019;70(7):2574–2578. [Google Scholar]
- 50.Zaręba K, Makara-Studzińska M, Ciebiera M, Gierus J, Jakiel G. Role of social and informational support while deciding on pregnancy termination for medical reasons. Int J Environ Res Public Health. 2018;15(12):2854–2854. doi: 10.3390/ijerph15122854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yi L, Liu Z, Deng C, Li X, Wang K, Deng K, Mu Y, Zhu J, Li Q, Wang Y, Dai L. Epidemiological characteristics of holoprosencephaly in China, 2007–2014: a retrospective study based on the national birth defects surveillance system. PLoS One. 2019;14(6):e0217835–e0217835. doi: 10.1371/journal.pone.0217835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Albu CC, Stancu IG, Grigore LG, Albu DF, Albu ŞD, Pătraşcu A, Gogănău AM. Impact of genetic testing and family health history of cystic fibrosis in the early prenatal diagnosis and prevention of a new case of genetic disorder. Rom J Morphol Embryol. 2019;60(2):667–671. [PubMed] [Google Scholar]
- 53.Albu CC, Albu DF, Muşat AR, Stancu IG, Albu ŞD, Pătraşcu A, Gogănău AM. The crucial role of SRY gene in the determination of human genetic sex: 46,XX disorder of sex development. Rom J Morphol Embryol. 2019;60(4):1311–1316. [PubMed] [Google Scholar]
- 54.Venkataswamy C, Gurusamy U, Lakshmi SV. Second-trimester fetal autopsy: a morphological study with prenatal USG correlations and clinical implications. J Lab Physicians. 2018;10(3):338–345. doi: 10.4103/JLP.JLP_134_17. [DOI] [PMC free article] [PubMed] [Google Scholar]