

Abstract
Two major obstacles that limit the widespread usage of polymeric nanocarriers include the complexity of formulation methods and their stability during storage. To address both of these issues, here we present morphologically complex nanocarriers in a hydratable powder form, which bypasses the need for expensive, harsh, and/or time-consuming nanocarrier fabrication techniques. The powders are composed of carbohydrates and self-assembling polymer amphiphiles having a low glass transition temperature. Hydration requires less than one minute and only involves the addition of aqueous media (water or saline) to rapidly obtain self-assembled micelles, worm-like micelles (i.e. filomicelles), or polymersomes from poly(ethylene glycol)-b-poly(propylene sulfide) (PEG-b-PPS) polymers. The formulated powders are highly stable, achieving hydration into monodisperse nanocarriers following >6 months of storage. Diverse drug cargoes were efficiently encapsulated during hydration, including hydrophobic small molecules for micellar morphologies, as well as individual and concurrent loading of both hydrophobic and hydrophilic molecules for vesicular morphologies. Hydrated polymersomes are shown to load hydrophilic biological macromolecules, and encapsulated enzymes retain bioactivity. Furthermore, we demonstrate that inclusion of lipid-anchored ligands in powder form permits the surface-display of targeting ligands and enhances target cell uptake, thereby extending this technology to targeted drug delivery applications. Our powder-based formulation strategy was extendable to commercially available polymer amphiphiles, including PEG-b-polystyrene and PEG-b-polycaprolactone. The formulated nanotechnologies described herein are highly modular, require minimal preparation, remain stable in ambient long-term storage (bypassing cold chain requirements), and will enable their use in medicine (human and veterinary), research, and commercial applications from cosmetics to agriculture.
We present a storage-stable carbohydrate-based powder technology that forms monodisperse polymeric nanocarriers of diverse morphology upon simple hydration with aqueous media. This platform loads a wide range of cargoes and is capable of targeted delivery.
Graphical Abstract
Introduction
The broad use of self-assembled nanocarriers such as liposomes, micelles and polymersomes in medical, research, and commercial applications is burdened by a large number of resources and skills needed to prepare stable, reliable formulations. Nanocarrier formulations are commonly prepared using emulsification1, cosolvent evaporation2, thin-film hydration3,4, or flash nanoprecipitation5-10 techniques. These methods utilize multi-step procedures, harsh conditions (e.g. organic solvents, sonication), and/or equipment, as well as significant allocations of time and/or financial resources. Aside from the more commonly recognized concerns, such as the exposure of biologic cargo (proteins, nucleic acids, etc.) to harsh organic solvents, the use of these methods to fabricate nanocarriers often requires a trained professional multiple days to prepare stable, monodisperse formulations. This is problematic, since endpoint users of nanocarriers usually do not have the training and/or time to prepare formulations for their intended application(s). Furthermore, many nanocarrier platforms are often formulated as aqueous suspensions and become unstable in storage over time, largely due to oxidation and hydrolysis concerns. These stability issues propagate to various problems ranging from agglomeration to cargo leakage, rendering stored formulations unusable shortly after the point of creation.
As compared to traditional aqueous suspensions, hydratable powders offer useful solutions to each of the aforementioned issues. The powder form is scalable, can be stored for prolonged time periods at room temperature, is compatible with sterilization via gamma irradiation, requires minimal time and skill from the endpoint user, and bypasses concerns involving the exposure of protein cargo to conditions capable of irreversible denaturation (and a corresponding loss of bioactivity). Powder forms of self-assembling lipid-based platforms, such as liposomes11-13 and niosomes14,15, have been developed by leveraging the nanocarrier-stabilizing effects of carbohydrates. However, formulations that employ analogous strategies have not been developed for polymeric self-assembling nanocarriers to date. Here, we present hydratable powders that successfully form polymeric self-assembled nanocarriers of diverse morphology. Polymeric nanocarriers bypass many of the disadvantages of their lipid-based counterparts, such as pro-liposomal powders16. For instance, lipid-based systems require multiple additional components (phospholipid mixtures, cholesterol, etc.) to optimize self-assembly and nanocarrier integrity, are confined to spherical morphologies, and suffer from both cargo leakage and hydrolysis in storage. These issues can be overcome, to some degree, by the lyophilization of formed nanocarriers (where compatible). However, lyophilized nanocarriers have their own set of disadvantages, including the choice of a suitable cryoprotectant, compromised nanostructure integrity, and cargo leakage issues after hydration.
Here, we develop a minimalist powdered form of hydratable polymeric nanocarriers that requires only the addition of a carbohydrate and subsequent desiccation to permit the facile storage and rapid preparation of delivery vehicles for diverse applications. These powders are created using a simple slurry method, consisting of self-assembling polymer(s) and carbohydrate in a volatile organic solvent that are mixed and then subjected to a solvent evaporation step. The successful formation of stable nanocarriers depends only on the ratio of self-assembling polymer amphiphile and carbohydrate additive. Furthermore, the morphological diversity of our hydratable polymeric systems has broad utility for medical and commercial applications, including differential loading and cargo release profiles, and provides a set of drug delivery vehicles with distinct circulation times as well as biodistributions at the organ- and cellular-levels3,17,18. Our formulation strategies simultaneously address various issues that are unique to certain morphologies of polymeric nanocarriers. For example, spherical micelle and cylindrical/filamentous micelle morphologies exhibit stability concerns in storage, resulting in nanocarrier disassembly, cargo loss, and/or cargo aggregation with time. By developing powdered forms, nanocarriers can be self-assembled at the time of use, eliminating issues arising from storage, handling, or hydration of already formed nanocarriers (e.g. lyophilized powders). Importantly, the ease of preparation for this methodology will extend the utility of polymeric nanocarriers to a broad community of scientists, engineers, and clinicians.
Results and Discussion
Powder formulation and characterization of nanocarrier self-assembly upon hydration
Poly(ethylene glycol)-b-poly(propylene sulfide) (PEG-b-PPS) diblock copolymers19 offer a versatile stimuli-responsive3,19-22 drug delivery platform that is capable of self-assembling into morphologically diverse nanocarriers, including micelle (MC), cylindrical micelle (filomicelle; FM), vesicular polymersome (PS) morphologies3,9,18,23. Furthermore, PEG-b-PPS nanocarriers are non-inflammatory24 and non-toxic in non-human primates25. Importantly, the low glass transition temperature of the PPS block (< −30 °C)26 allows sufficient chain flexibility for nanocarrier self-assembly at room temperature via a wide range of methods7,9,10. Our past studies demonstrate that PEG-b-PPS copolymers are amenable to self-assembly by thin film hydration. However, the formation of nanocarriers requires hydration periods of ≥24 h and vesicular PS nanocarriers require additional extrusion steps to improve monodispersity3,4,17,27. Furthermore, substantial material loss is a common issue with thin film hydration protocols due to retention of polymer on the vessel wall and incomplete hydration into aqueous media, which also makes the technique challenging to scale.
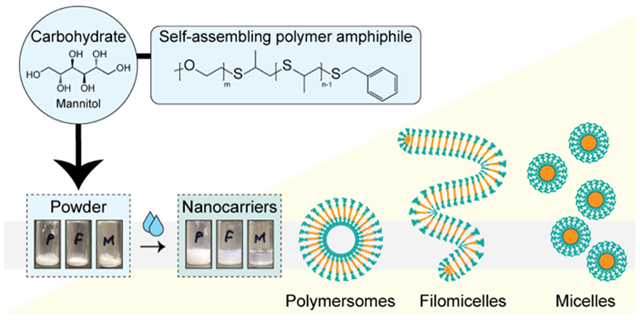
We hypothesized that suitable mixtures of polymer amphiphiles and carbohydrates would yield scalable powders upon desiccation that could rapidly and efficiently form stable nanocarriers upon the addition of aqueous media. Formulations were developed consisting of a 1:3 ratio (polymer:carbohydrate) of PEG-b-PPS polymer and mannitol (Fig. 1A). Mannitol is a sugar alcohol and is commonly employed as an excipient in the pharmaceutical industry28, including as a cryoprotectant for biomedical products29,30. Furthermore, Mannitol is widely used in the development of carbohydrate-based proliposome formulations11. To examine whether MC, FM, or PS morphologies favourably self-assemble upon hydration, we prepared powders with PEG-b-PPS diblock copolymers that differ in their hydrophilic weight fraction (Fig. 1A; Fig. S1; Table S1). Powders were hydrated in aqueous media (here, water). The simplicity and efficiency of the hydration procedure presented in Supplementary Movie S1.
Figure 1. Formation of morphologically diverse nanocarriers upon hydration.

(A) Schematic of formulation strategy and illustrative depiction of the resulting PEG-b-PPS nanocarrier morphologies. Powders consisting of PEG-b-PPS polymer and mannitol carbohydrate are prepared. Upon adding water and vortexing, the nanocarriers rapidly self-assemble into the expected morphology based on the hydrophilic (PEG) weight fraction (fPEG) range of the polymer. Abbreviations: polymersome (PS); filomicelle (FM); micelle (MC). (B) Transmission electron microscopy (TEM) of negatively stained hydrated nanocarriers (30,000X; scale bar = 200 nm). Orange arrows point to MC. The theoretical cargo loading compatibility is depicted below each micrograph and is experimentally demonstrated later in this manuscript.
Morphological analysis by transmission electron microscopy (TEM) of negatively stained specimens demonstrates spherical and cylindrical morphologies self-assemble successfully upon hydration (Fig. 1B). The diameter of PEG-b-PPS PS exceeded that of the MC, as expected23. FM had a cross-sectional diameter that was comparable to that of the MC, but a length exceeding 1 μm (Fig. 1B). This is in agreement with our past reports on these worm-like structures3,23.
Differences in nanocarrier size are also readily observable through transmittance measurements (Fig. S2). As expected, MC formulations are transparent, FM are translucent, and the vesicular PS are more opaque (Fig. S2). Results for spherical morphologies were corroborated by dynamic light scattering (DLS), where representative number average diameter and polydispersity index (PDI) values are presented (Table 1). These values are representative of ten separate batches of hydrated nanocarriers. Hydrated PEG-b-PPS MC and PS were monodisperse with an average diameter of 22.6 nm and 78.5 nm, respectively (Table 1). The vesicular morphology of PS was confirmed by small-angle x-ray scattering (SAXS) using synchrotron radiation (Fig. S3).
Table 1.
Summary of hydrated nanocarrier physicochemical properties, cargo loading, and ligand display.
| Nanocarrier | Cargo or Ligand |
Efficiency (%)* |
D (nm)† | PDI† | ζ (mV)†† |
|---|---|---|---|---|---|
| Micelles (MC) | None | N/A | 22.6 | 0.06 | −5.0 ± 0.9 |
| DiI MC | DiI | >95.0 | 26.5 | 0.07 | −2.2 ± 0.8 |
| MC +peptide | Targeting ligand | >95.0 | 26.7 | 0.08 | −0.3 ± 0.3 |
| Filomicelles (FM) | None | N/A | - | - | −7.5 ± 0.5 |
| DiI FM | DiI | >95.0 | - | - | −7.2 ± 0.2 |
| FM +peptide | Targeting ligand | >95.0 | - | - | −0.3 ± 0.2 |
| Polymersomes (PS) | None | N/A | 78.5 | 0.12 | −4.4 ± 0.1 |
| DiI PS | DiI | >95.0 | 75.0 | 0.12 | −1.1 ± 0.2 |
| PS +peptide | Targeting ligand | >95.0 | 72.3 | 0.16 | 9.8 ± 0.1 |
| AP PS | AP††† | 16.9 ± 0.2 | 82.9 | 0.12 | −3.0 ± 0.6 |
| Dex-TMR PS | Dex-TMR††† | 10.8 ± 0.5 | 73.9 | 0.09 | −0.5 ± 0.3 |
Efficiency (%) = loading efficiency for cargo; display efficiency for targeting ligands.
Number-average diameter and polydispersity index (PDI) determined by DLS. Note: FM (cylinders) are excluded from DLS analysis (Stokes-Einstein equation assumes spheres).
Zeta potential is reported as mean ± s.d. (n=3).
Hydrophilic cargo (only applicable to the vesicular PS morphology)
Regarding the importance of the 1:3 polymer:carbohydrate ratio for this formulation strategy, we note the most important consideration is that the carbohydrate sufficiently coats the bottom surface of the vessel used for powder preparation. This depends on the amount of polymer used and the dimensions of the vessel floor (bottom surface). For the amount of polymer used in this study (10 mg) and the glass vials used (inner diameter of 12 mm), this corresponded to a minimum of a 1:3 ratio (i.e. 10 mg polymer to 30 mg carbohydrate). With this polymer amount/mass fixed, the use of lower amounts of carbohydrate resulted in incomplete vial coatings and powders that were not hydratable by pipetting or mild agitation. We further note that increasing the carbohydrate amount further did not enhance nanocarrier self-assembly. We therefore proceeded with the 1:3 ratio since this was the minimum amount of carbohydrate that produced easy-to-hydrate powders while avoiding an excessive cost of the excipient.
We further assessed the storage properties of the powdered formulations and the hydrated nanocarrier suspensions, where relevant. Nanocarriers formed by hydrating powders for 1 month or 6 months were monodisperse, with physical properties similar to those of nanocarriers formed from freshly prepared powders (Table S2). This is particularly important for micellar nanocarriers, which often have stability issues in storage in suspension form. The resulting powders avoid these issues, since they exhibit high storage stability and can be hydrated on demand to prepare formulations with minimal effort.
Due to their higher stability compared to MC31, vesicular nanocarriers such as PS are more frequently stored in suspension form for longer periods of time. In our studies, hydrated suspensions of PS stored at room temperature for 8 months remained stable. The stored PS formulations were monodisperse with an average diameter of 68.9 nm (Table S3). PS were still readily observable after storage for 1 year at room temperature (Fig. S4).
For completeness, we note that nanocarrier self-assembly upon hydration was also permitted using different volatile organic solvents (Fig. S5) and carbohydrates (Fig. S6). Nanocarriers formed from powders that were prepared with alternative solvents (Table S4) and carbohydrates (Table S5) were monodisperse with an average diameter consistent with nanocarriers prepared from powders formed with mannitol and dichloromethane (Table 1).
Morphological and crystallographic characterization of polymer-carbohydrate powders
Employed as an excipient, the crystallinity of mannitol, as well as other carbohydrates, is known to influence its utility as a stabilizing agent for retaining the activity of lyophilized enzymes32-34, and in promoting the self-assembly of pro-liposomes11. To examine the structural properties and crystallinity of the polymer-carbohydrate powders in greater detail, we characterized each formulation using scanning electron microscopy (SEM) and powder x-ray diffraction (XRD) (Fig. 2). In all powder formulations, PEG-b-PPS polymers coated the crystalline mannitol carbohydrate. This is observable by SEM as a smooth layer that renders the uncoated carbohydrate to have a more amorphous and rough appearance (Fig. 2A). Similar observations were made for powders prepared with alternative carbohydrates, where a smooth polymer coating is visible by SEM (Fig. S7). This surface coating on a water-soluble carrier (carbohydrate) increases the surface area that is available for interaction with the aqueous media. Compared to thin film hydration, the carbohydrate thus allows hydration to proceed quickly and efficiently without leaving behind material on the vessel wall.
Figure 2. Powder characterization by scanning electron microscopy and X-ray diffraction.

(A) Scanning electron microscopy (SEM) micrographs of mannitol (M), mannitol + micelle polymer (M+MC), mannitol + filomicelle polymer (M+FM), and mannitol + polymersome polymer (M+PS) powders. Top panel = 300X magnification (scale bar = 100 μm). Bottom panel = 1500X magnification (scale bar = 30 μm). (B) Powder x-ray diffraction analysis of mannitol alone (control) and mannitol in complex with PEG-b-PPS polymers in powdered form.
XRD analysis further confirms our interpretations of SEM micrographs (Fig. 2B). Diffuse peaks of lower intensity are observed in polymer-coated mannitol samples compared to mannitol control (Fig. 2B). This peak broadening is observed due to the introduction of disorder that results from coating ordered mannitol crystals with polymer amphiphiles, which disrupts x-ray diffraction. These analyses demonstrate the powder formulations consist of self-assembling polymer amphiphiles that integrate stably with the carbohydrate additives.
Hydrated nanocarriers retain diverse hydrophobic payloads, are non-toxic, and are differentially endocytosed by immune cells.
Nanocarriers hold broad utility, including their usage in drug delivery and imaging for transporting payloads having low water solubility to specific tissues and cell types. We therefore prepared powder formulations containing hydrophobic cargos and assessed loading efficiency upon hydration. All nanocarriers loaded DiI hydrophobic dye with >95% efficiency (Table 1). Representative images of hydrated DiI-loaded nanocarrier formulations are presented in Fig. S8. The high loading efficiency of DiI is consistent with our previously reported work for PEG-b-PPS nanocarriers loading lipophilic tracers, prepared via thin-film hydration3,17, flash nanoprecipitation7,10,18, and cosolvent evaporation2,35. More modest loading efficiencies were found for curcumin (Table S6), which is explainable by its relatively lower hydrophobicity than DiI. Physicochemical analysis by DLS demonstrates cargo loading did not produce substantial changes to nanocarrier size, and the resulting nanocarrier suspensions were monodisperse (Table 1). Electrophoretic light scattering (ELS) analysis further demonstrates zeta potential was not altered by cargo loading (Table 1).
While cytotoxic nanocarrier systems are useful for cancer therapy, it is generally favorable for the nanocarrier to be biologically inert and non-toxic at relevant concentrations. Greater than 85% viability was observed for macrophages cultured in the presence of hydrated nanocarriers dosed by polymer concentration in the range of 0.25-1.0 mg/mL for 24 h (Fig. 3A). With exception to the 0.5 mg/mL MC treatment group, all viabilities exceeded 90% (Fig. 3A). For comparison, brief cellular exposure to 70% ethanol (toxicity control), decreased cell viability to below 30% (Fig. 3A). Comparing cell viability in the presence of nanocarriers (>85% mean viability in all cases) suggests all hydrated nanocarriers were non-toxic at the high concentrations administered. As an orthogonal assessment of toxicity, an MTT assay further demonstrate the nanocarriers to be non-toxic over 24 h. In these studies, the viability of cells treated with hydrated nanocarriers was significantly higher than that of cells treated with water-soluble carboplatin, a cytotoxic anti-cancer agent (Fig. S9).
Figure 3. Hydrated nanocarriers are non-toxic and demonstrate morphology-dependent uptake by macrophages.

(A, B) Analysis of macrophage (A) viability and (B) median fluorescence intensity (MFI) after incubation with nanocarriers at the specified polymer concentration for 2 h. For the viability studies in (A), PBS (white bar) and short duration exposure to 70% ethanol (grey bar) were included as controls. Significant differences in cell viability versus toxicity control (70% ethanol) were determined by ANOVA with Tukey’s multiple comparisons test. and a 5% significance level. ****p<0.0001. (C) Flow cytometric analysis of nanocarrier uptake after pre-treating cells with PBS (no inhibitor), 50 μM CytD (phagocytosis inhibitor), or 50 μM CPZ (clathrinmediated endocytosis inhibitor). Nanocarriers were administered at a 1.0 mg/mL polymer concentration for endocytosis inhibitor studies. For (B,C) significant differences were determined by two-way ANOVA with Tukey’s multiple comparisons test and a 5% significance level. Comparisons within concentration groupings: ****p<0.0001, ***p≤0.0005, **p<0.005, *p<0.05. For all analyses, cellular viability and nanocarrier uptake studies used a 2 h nanocarrier incubation period. Cells were cultured at 37 °C, 5% CO2. The mean ± s.e.m. (n=3) is displayed.
We next quantified differential cellular uptake by assessing the difference in the median fluorescence intensity (MFI) (Fig. 3B). For each hydrated DiI-loaded nanocarrier, the MFI generally increased in a concentration-dependent fashion (Fig. 3B). Furthermore, larger nanostructures, PS and FM, were taken up by macrophages to a lesser extent than the hydrated MC at similar concentrations, as expected due to differences in diffuse rate and nanoparticle count (Fig. 3B ). Cellular uptake studies performed after cellular pre-treatment confirmed our findings of the morphology-dependent extent of internalization. Both cytochalasin D (CytD)36, a general phagocytosis inhibitor, and chlorpromazine (CPZ)37, an inhibitor of clathrin-mediated endocytosis, decreased uptake of all structures (Fig. 3C). Collectively, these viability and cellular uptake studies demonstrate the hydrated polymeric nanocarriers are non-toxic to immune cells and are differentially internalized by cells to an extent that is size- and morphology-dependent. The resulting nanocarriers therefore fulfil basic requirements for the intracellular delivery of drugs, vaccines, and tracers/diagnostics.
Preparation of hydratable, morphologically diverse targeted drug delivery vehicles.
Targeted drug delivery vehicles consist of drug-loaded nanocarriers displaying a ligand that binds to a unique molecular feature that is enriched on the surface of one or more cell type(s) of interest. The goal of these vehicles is to increase the drug concentration at the site of action to improve efficacy, while minimizing side effects that are associated with off-target drug uptake. Lipid-anchored targeting ligands offer a modular approach for functionalizing amphiphilic nanocarriers for targeted drug delivery applications without the need for covalent modification of the polymer. Powdered formulations that consist of a self-assembling polymer amphiphile, lipid-anchored targeting ligand, and carbohydrate additive would offer a rapid and customizable platform for facile on-demand preparation of targeted drug delivery vehicles.
We prepared powders consisting of PEG-b-PPS polymer with weight fractions that self-assemble into MC, FM, or PS morphologies, mannitol, and a model lipid-anchored targeting peptide of the form palmitoleic acid-PEG6-peptide (Fig. 4A). Powders were prepared to include a linear peptide at a 5% molar ratio (peptide:polymer). Our past targeting studies demonstrate this 5% molar ratio yields an optimal ligand-mediated increase in uptake by the target cell type, whereas embedding ligand at higher molar ratios does not produce substantial increases in targeting enhancements27,35. After hydration, the nanocarriers successfully embedded the lipid-anchored targeting peptides at over >95% efficiency (Table 1).
Figure 4. Hydrated PEG-b-PPS nanocarriers can display lipid-anchored targeting peptides and targeting functionality.

(A) Powders were prepared to consist of PEG-b-PPS, mannitol, and lipid-anchored targeting peptide. Nanocarriers self-assembled upon hydration. MC (top), FM (middle), and PS (bottom) displaying a linear targeting peptide is displayed. Cryo-TEM micrographs were acquired at 10,000X. Scale bar = 100 nm. (B, C) Demonstration of targeted micellar delivery of hydrophobic cargo (B) and targeted vesicular delivery of biological cargo (C). MC and PS nanocarriers were prepared to displayed cyclic RGD peptides at 1% and 5% molar ratios, respectively. The fold change in uptake by primary Human Umbilical Vein Endothelial Cells (HUVECs) was quantified as the median fluorescence intensity (MFI) above untargeted control. The mean ± s.e.m. is displayed. Statistically significant differences in fold changes were determined between the two molar ratio groups using an unpaired t test and a 5% significance level. **p<0.01.
Cryo-TEM was performed to examine the morphology of the targeted drug delivery vehicles formed after hydration (Fig. 4A). For Cryo-TEM performed on PEG-b-PPS nanocarriers, the dense hydrophobic PPS core is the source of contrast, whereas the PEG corona is not readily visible due to its insufficient contrast. As demonstrated by representative Cryo-TEM micrographs, the expected morphologies were retained in the presence of peptide embedding (Fig. 4A). Aberrations in the hydrophobic PPS core due to the lipid anchor were not observed (Fig. 4A), and the morphologies did not deviate from the nanocarriers prepared in the absence of targeting peptide (Fig. 1B). DLS and ELS demonstrate the spherical structures to be monodisperse with a diameter comparable to that of nanocarriers prepared in the absence of lipid-anchored targeting ligand (Table 1).
We examined targeting functionality in vitro. The model cyclic RGD peptide was used for these studies, which exhibits greater stability/lower degradation than its linear counterparts38,39 and have been used in a large number of targeting applications40-42. RGD peptides bind to beta integrin receptors43-46, which are present on the surface of a variety of endothelial cell types47, including Human Umbilical Vein Endothelial Cells (HUVECs)48. For this analysis, micelle powders were prepared with DiI as a model hydrophobic cargo whereas PS powders were prepared with 70 kDa Dex-TMR as a model hydrophilic cargo. Lipid-anchored cyclic RGD peptides of the form, palmitoleic acid-PEG6-cyclic RGD, were included at 1% or 5% molar ratios (peptide:polymer) and formed stable nanostructures (Table S7). HUVECs were treated with hydrated formulations and peptide-mediated enhancements in cellular uptake were quantified versus untargeted control (Fig. 4B, C). For both cases, the display of cyclic RGD at the nanocarrier surface-enhanced uptake and the magnitude of the increase was significantly greater for nanocarriers displaying the peptide at a 5% molar ratio (Fig. 4B, C).
These results demonstrate the powder-based formulation strategy established herein is capable of forming morphologically diverse polymeric targeted drug delivery vehicles. The surface display of the model cyclic RGD peptide enhanced the micellar delivery of a hydrophobic tracer cargo and PS delivery of a biological tracer. The powdered form bypasses ligand stability issues common to stored liquid suspensions, which could otherwise diminish targeting performance with time. The modularity of these powders and the diversity of the hydrated targeted drug delivery vehicles enable their convenient preparation for a broad range of applications where precise control over nanocarrier-cellular interactions is required.
Hydrated vesicles load diverse hydrophilic cargo and preserve enzyme bioactivity
We next sought to examine the properties unique to the vesicular nanocarriers, such as facile encapsulation of water-soluble payloads. PS are capable of loading biologics, such as carbohydrates, nucleic acid, and protein cargo into their aqueous lumen. Alkaline phosphatase (AP) and 70 kDa dextran-tetramethylrhodamine (Dex-TMR) were chosen as two model hydrophilic cargo. Alkaline phosphatase is an enzyme that is capable of non-specific dephosphorylation, which we selected to assess retention of enzyme activity following our powder nanocarrier loading protocol49. Dex-TMR is a high molecular weight polysaccharide conjugated to TMR fluorophore, and is commonly employed as a hydrophilic tracer18.
The enzyme cargo was prepared in aqueous media, which was then used to hydrate powder consisting of PEG-b-PPS PS polymer and mannitol carbohydrate. Following hydration, AP and Dex-TMR loaded into PS at 16.9 ± 0.2% and 10.8 ± 0.5%, respectively (Table 1). These results are consistent with the observation that higher molecular weight hydrophilic cargo load into PS at greater efficiency (AP molecular weight > 100 kDa). Furthermore, hydrated PS formations are also capable of dual loading hydrophobic and hydrophilic cargo. The hydrated PS dual loaded Dex-TMR (hydrophilic cargo) and curcumin (hydrophobic cargo) with a loading efficiency of 16.3 ± 1.1% and 22.9 ± 0.4%, respectively (Table S6). The resulting PS bearing biological cargo are monodisperse, with physicochemical characteristics similar to that of PS prepared without cargo or with hydrophobic cargo (Table 1). TEM demonstrates the PS nanocarriers have the expected spherical morphology (Fig. 5A) and do not deviate substantially from unloaded structures (Fig. 1B).
Figure 5. Morphological characterization of biologic-loaded polymersomes and bioactivity assessment of enzymatic cargo.

(A) TEM of negatively stained PS loaded with Dex-TMR (left) or AP enzyme (right) following hydration. (B) Characterization of AP enzyme activity in unfiltered or SEC-filtered AP-loaded PS formulations following cargo release with 1% triton. BCIP/NBT substrate was administered and the activity of AP enzyme monitoring with time by measuring the absorbance of 630 nm light. (C) Cell viability of RAW 264.7 macrophages treated with polymersomes ([polymer] = 1.0 mg/mL) loaded with Dex-TMR hydrophilic tracer. Cells treated with PBS or 70% ethanol (EtOH) were included as controls. Significant differences in cell viability versus the EtOH treatment group were determined by ANOVA with Dunnett’s multiple comparisons test and a 5% significance level. (D) Median fluorescence intensity (MFI) of macrophages treated with PS ([polymer] = 1.0 mg/mL) encapsulating Dex-TMR. Where specified, cells were pre-treated with 50 μM CytD or 50 μM CPZ inhibitor prior to PS administration. Statistically significant differences in MFI were determined by ANOVA with Tukey’s multiple comparisons test and a 5% significance level. The mean ± s.e.m. (n=3) is presented in all cases. For all statistical tests, ****p<0.0001, *p<0.05.
AP-loaded PS were either left unpurified (a mixture of free, unloaded AP and AP loaded into PS) or were purified by size exclusion chromatography (SEC) using a Sepharose 6B column. The latter condition permits the examination of substrate turnover by the encapsulated enzymes, without confounding effects from product formation mediated by the diffuse enzyme that was not loaded into nanostructures. Filtered or unfiltered AP-loaded PS aliquots were treated with 1% triton to break the PS structure, and 5-bromo-4-chloro-3-indolyl phosphate (BCIP)/nitro blue tetrazolium (NBT) substrate was administered. AP retained bioactivity following encapsulation into PS (Fig. 5B). PS loaded with Dex-TMR hydrophilic tracer are non-toxic to macrophages (Fig. 5C) and are readily endocytosed by macrophages in vitro (Fig. 5D).
Our analyses demonstrate the PS powders permit the encapsulation of hydrophilic cargo with efficiency on par with most current hydrophilic encapsulation methods. However, we note that this loading efficiency is lower than that achieved by flash nanoprecipitation7,18. The results presented herein demonstrate that encapsulated enzymes retain bioactivity. Furthermore, PS bearing hydrophilic tracers are non-toxic and their cellular internalization is readily detectable.
Self-assembly of polymeric nanostructures upon hydration of commercially available polymers
We found the utility of the developed formulation strategy extends beyond the PEG-b-PPS platform, and is generally applicable to self-assembling polymer amphiphiles having low glass transition temperature. This includes polymer systems that are commercially available, such as PEG-b-polystyrene polymer that forms micelles based on its fPEG ratio (Fig. 6A; Table S8). As observed through SEM, PEG-b-polystyrene polymer coats mannitol carbohydrate to produce a more amorphous appearance of the formed powder (Fig. 6B). Further support for successful coating is provided by the XRD profile of the polymer-carbohydrate powder, where peak broadening is observed together with decreases in peak intensity (Fig. 6C).
Figure 6. Powdered formulations of a commercially available polymer amphiphile and characterization of resulting nanocarriers that self-assemble upon hydration.

(A) Illustration and image of powder formed from mannitol and commercially available PEG-b-polystyrene polymer. (B) SEM micrographs of powders (left = 300X magnification; right = 1500X magnification). (C) Powder XRD of mannitol (control; top) and mannitol coated with PEG-b-polystyrene (bottom). (D) TEM micrograph of hydrated PEG-b-polystyrene micelles (30,000X magnification; Scale bar = 200 nm). (E-G) Cellular viability and uptake studies performed with PEG-b-polystyrene nanocarriers and RAW 264.7 macrophages. (E) Viability of macrophages after 2 h treatment with PEG-b-polystyrene nanocarriers dosed at the specified polymer concentration. PBS- or 70% ethanol (EtOH)-treated cells were included as controls. Significant differences in cell viability versus the EtOH-treated cells were determined by ANOVA with Dunnett’s multiple comparisons test and a 5% significance level. (F) Median fluorescence intensity (MFI) of macrophages treated with PEG-b-polystyrene nanocarriers (0.25-1.0 mg/mL polymer concentration) for 2 h. (G) Cellular MFI after pre-treatment with CytD (50 mM) or CPZ (50 mM) endocytosis inhibitors prior to nanocarrier administration. For (F, G), significant differences in cellular uptake were determined by ANOVA with Tukey’s multiple comparisons test and a 5% significance level. The mean ± s.e.m. (n=3) is displayed in E-G.
MC self-assembled after hydrating PEG-b-polystyrene powders with aqueous media (Fig. 6D). These formulations were monodisperse, with an average diameter of 39.8 nm and zeta potential of −8.5 ± 0.8 (Table S8). PEG-b-polystyrene nanocarriers encapsulated hydrophobic small molecules at high efficiency (Table S9). DiI-loaded PEG-b-polystyrene nanocarriers were non-toxic to macrophages in vitro (Fig. 6E). Macrophages internalized these nanocarriers in a manner that was concentration-dependent (Fig. 6F). The cellular uptake of these nanocarriers is mostly abolished by pre-treating macrophages with endocytosis inhibitors prior to nanocarrier administration (Fig. 6G).
Powders were also prepared with PEG-b-polycaprolactone to examine the ability to produce hydratable vesicular nanocarriers from a commercially available polymer. PEG-b-polycaprolactone is expected to form PS based on its fPEG ratio. After hydration, PEG-b-polycaprolactone self-assembly required a heating step at 60 °C for 30 min after hydration with aqueous media to allow the formation of monodisperse vesicles (Table S8). However, this short timeline is still a convenient approach to fabricate PEG-b-polycaprolactone vesicles as compared to the thin-film hydration method that traditionally requires an overnight incubation at 60 °C to self-assemble monodisperse vesicular nanostructures50. We note that for this platform, the requirement of heat can thermally denature most protein biologics and could degrade certain heat-sensitive small molecule drugs. However, this strategy requires a much lower duration of heat exposure than thin film. Lastly, PEG-b-polycaprolactone powders successfully permit the encapsulation of both hydrophilic and hydrophobic cargo without comprising the structural integrity of the nanocarrier, which is consistent with successful vesicle formation (Table S9). Further investigations are needed to understand the relationship between the hydrophilic weight fraction and glass transition temperature of this polymer, as well as the requirement for an additional heating step for successful nanocarrier self-assembly upon hydration of the powder form.
Conclusions
We developed a carbohydrate-based powdered formulation strategy that is scalable and permits nanocarrier self-assembly from polymer amphiphiles after the addition of water or saline. The formulated powders are robust, accommodating a variety of carbohydrates and organic solvents. We further demonstrate this formulation strategy is extendable to unique polymer systems, such as PEG-b-PPS, as well as polymer amphiphiles that are commercially available. Importantly, powders incorporating polymer amphiphiles of distinct hydrophilic weight fractions self-assembled into the expected morphologies upon hydration, as demonstrated by the successful formation of MC, FM and PS. The hydration procedure was efficient, with no observable loss of polymer to the vessel, which is an additional benefit of this technique over thin film hydration. Hydrated micellar and vesicular nanocarriers load hydrophobic payloads with high efficiency. Specifically, the formulation strategy described herein may permit loading of hydrophobic compounds without further filtration steps (logP > 4), but we note that in some cases (e.g., hydrophobic compounds with lower logP values) the ratio of polymer to drug will require further optimization or filtration to remove any unencapsulated compound(s). Hydrated vesicular PS encapsulated diverse hydrophilic payloads without exposure to organic solvents and retained the bioactivity of enzyme cargo. The hydratable PS were also capable of dually loading hydrophobic and hydrophilic cargoes. Since hydrophilic cargoes typically load into self-assembled nanocarriers with lower efficiency than hydrophobic cargoes, we note that formulations incorporating hydrophilic payloads may need a filtration step as required for some biological applications where an unencapsulated payload fraction is undesirable. However, cases exist where nanocarriers bearing hydrophilic cargoes are administered in clinical settings without removal of unencapsulated fractions51. In all cases, the hydrated nanocarriers were non-toxic, and were internalized by cells at different extents that are consistent with the combination of nanocarrier size and shape. All nanocarrier morphologies were capable of displaying lipid-anchored targeting ligands upon hydration. As we have previously demonstrated that diverse ligands of this format can be easily tailored and incorporated into PEG-b-PPS nanocarriers for different targeting applications27,35,52-54, we expected these lipid anchored constructs to be a customizable and modular feature of this technology. Our studies with the model cyclic RGD peptide demonstrated the successful enhancement of MC, FM, and PS uptake by HUVECs.
Finally, the powders remain stable after long-term storage. Powders stored for a period of 6 months successfully form monodisperse nanocarriers following hydration. This feature bypasses numerous issues associated with the storage of nanocarrier suspensions, which is an issue that gained worldwide attention during the development of vaccines against COVID-1955. The powder formulations developed herein solve numerous issues surrounding the preparation and stability of polymeric nanocarriers of diverse morphology. These technologies have potential utility in a variety of drug delivery and commercial applications.
Supplementary Material
Acknowledgements
M.P.V. gratefully acknowledges support from the Ryan Fellowship, the International Institute for Nanotechnology at Northwestern University, and the Northwestern University Multidisciplinary Visual Sciences Training Program (T32 Fellowship funded by NEI Award 2T32EY025202-06). We acknowledge staff and instrumentation support from the Structural Biology Facility at Northwestern University, the Robert H Lurie Comprehensive Cancer Center of Northwestern University and NCI CCSG P30 CA060553. The Gatan K2 direct electron detector was purchased with funds provided by the Chicago Biomedical Consortium with support from the Searle Funds at The Chicago Community Trust. This work made use of the EPIC facility of Northwestern University’s NUANCE Center, which has received support from the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF ECCS-1542205); the MRSEC program (NSF DMR-1121262) at the Materials Research Center; the International Institute for Nanotechnology (IIN); the Keck Foundation; and the State of Illinois, through the IIN. This work made use of the IMSERC at Northwestern University, which has received support from the NSF (CHE-1048773); Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF ECCS-1542205); the State of Illinois and International Institute for Nanotechnology (IIN). This work was supported by the Northwestern University – Flow Cytometry Core Facility supported by Cancer Center Support Grant (NCI CA060553). SAXS experiments were performed at the DuPont-Northwestern-Dow Collaborative Access Team (DND-CAT) that is located at Sector 5 of the Advanced Photon Source (APS). DND-CAT is supported by Northwestern University, E.I. DuPont de Nemours & Co., and The Dow Chemical Company. This research was supported by the National Institutes of Health Director's New Innovator Award (grant no. 1DP2HL132390-01), National Institute of Allergy and Infectious Diseases (grants no. 5R21AI137932 and 1R01AI145345), and the National Science Foundation CAREER Award no. 1453576.
Footnotes
Conflicts of interest
The authors state that there are no conflicts to declare.
Notes and references
- 1.Matoori S, Bao Y, Schmidt A, Fischer EJ, Ochoa-Sanchez R, Tremblay M, Oliveira MM, Rose CF and Leroux J-C, Small, 2019, 15, e1902347. [DOI] [PubMed] [Google Scholar]
- 2.Stack T, Vahabikashi A, Johnson M and Scott E, J Biomed Mater Res A, 2018, 106, 1771–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Karabin NB, Allen S, Kwon H-K, Bobbala S, Firlar E, Shokuhfar T, Shull KR and Scott EA, Nature Communications, , DOI: 10.1038/s41467-018-03001-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yi S, Karabin NB, Zhu J, Bobbala S, Lyu H, Li S, Liu Y, Frey M, Vincent M and Scott EA, Front. Bioeng. Biotechnol, , DOI: 10.3389/fbioe.2020.00542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnson BK and Prud’homme RK, AIChE J, 2003, 49, 2264–2282. [Google Scholar]
- 6.Saad WS and Prud’homme RK, Nano Today, 2016, 11, 212–227. [Google Scholar]
- 7.Allen S, Osorio O, Liu Y-G and Scott E, J Control Release, 2017, 262, 91–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Allen SD, Bobbala S, Karabin NB, Modak M and Scott EA, ACS Appl Mater Interfaces, 2018, 10, 33857–33866. [DOI] [PubMed] [Google Scholar]
- 9.Bobbala S, Allen SD and Scott EA, Nanoscale, 2018, 10, 5078–5088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Allen S, Vincent M and Scott E, J Vis Exp, 2018, e57793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bobbala SKR and Veerareddy PR, J Liposome Res, 2012, 22, 285–294. [DOI] [PubMed] [Google Scholar]
- 12.Nekkanti V, Wang Z and Betageri GV, AAPS PharmSciTech, 2016, 17, 851–862. [DOI] [PubMed] [Google Scholar]
- 13.Janga KY, Jukanti R, Velpula A, Sunkavalli S, Bandari S, Kandadi P and Veerareddy PR, Eur J Pharm Biopharm, 2012, 80, 347–357. [DOI] [PubMed] [Google Scholar]
- 14.Veerareddy PR and Bobbala SKR, Drug Dev Ind Pharm, 2013, 39, 909–917. [DOI] [PubMed] [Google Scholar]
- 15.Gurrapu A, Jukanti R, Bobbala SR, Kanuganti S and Jeevana JB, Advanced Powder Technology, 2012, 23, 583–590. [Google Scholar]
- 16.Nekkanti V, Venkatesan N and Betageri GV, Curr Pharm Biotechnol, 2015, 16, 303–312. [DOI] [PubMed] [Google Scholar]
- 17.Yi S, Allen SD, Liu Y-G, Ouyang BZ, Li X, Augsornworawat P, Thorp EB and Scott EA, ACS Nano, 2016, 10, 11290–11303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vincent MP, Bobbala S, Karabin NB, Frey M, Liu Y, Navidzadeh JO, Stack T and Scott EA, Nat Commun, 2021, 12, 648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Napoli A, Valentini M, Tirelli N, Müller M and Hubbell JA, Nature Materials, 2004, 3, 183–189. [DOI] [PubMed] [Google Scholar]
- 20.Napoli A, Boerakker MJ, Tirelli N, Nolte RJM, Sommerdijk NAJM and Hubbell JA, Langmuir, 2004, 20, 3487–3491. [DOI] [PubMed] [Google Scholar]
- 21.Vasdekis AE, Scott EA, O’Neil CP, Psaltis D and Hubbell Jeffrey. A., ACS Nano, 2012, 6, 7850–7857. [DOI] [PubMed] [Google Scholar]
- 22.Bobbala S, Allen SD, Yi S, Vincent M, Frey M, Karabin NB and Scott EA, Nanoscale, 2020, 12, 5332–5340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karabin NB, Vincent MP, Allen SD, Bobbala S, Frey MA, Yi S, Yang Y and Scott EA, bioRxiv, 2020, 2020.09.02.280404. [Google Scholar]
- 24.Scott EA, Stano A, Gillard M, Maio-Liu AC, Swartz MA and Hubbell JA, Biomaterials, 2012, 33, 6211–6219. [DOI] [PubMed] [Google Scholar]
- 25.Allen SD, Liu Y-G, Bobbala S, Cai L, Hecker PI, Temel R and Scott EA, Nano Res., 2018, 11, 5689–5703. [Google Scholar]
- 26.Cerritelli S, O’Neil CP, Velluto D, Fontana A, Adrian M, Dubochet J and Hubbell JA, Langmuir, 2009, 25, 11328–11335. [DOI] [PubMed] [Google Scholar]
- 27.Yi S, Zhang X, Sangji MH, Liu Y, Allen SD, Xiao B, Bobbala S, Braverman CL, Cai L, Hecker PI, DeBerge M, Thorp EB, Temel RE, Stupp SI and Scott EA, Adv Funct Mater, 2019, 29, 1904399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ohrem HL, Schornick E, Kalivoda A and Ognibene R, Pharm Dev Technol, 2014, 19, 257–262. [DOI] [PubMed] [Google Scholar]
- 29.Lee MK, Kim MY, Kim S and Lee J, Journal of Pharmaceutical Sciences, 2009, 98, 4808–4817. [DOI] [PubMed] [Google Scholar]
- 30.Cui Y, Cui P, Chen B, Li S and Guan H, Drug Development and Industrial Pharmacy, 2017, 43, 519–530. [DOI] [PubMed] [Google Scholar]
- 31.Discher BM, Won Y-Y, Ege DS, Lee JC-M, Bates FS, Discher DE and Hammer DA, Science, 1999, 284, 1143–1146. [DOI] [PubMed] [Google Scholar]
- 32.Izutsu K, Yoshioka S and Terao T, Pharm Res, 1993, 10, 1232–1237. [DOI] [PubMed] [Google Scholar]
- 33.Izutsu K, Yoshioka S and Kojima S, Pharm Res, 1994, 11, 995–999. [DOI] [PubMed] [Google Scholar]
- 34.Izutsu K, Yoshioka S and Terao T, Chem Pharm Bull (Tokyo), 1994, 42, 5–8. [DOI] [PubMed] [Google Scholar]
- 35.Stack T, Vincent M, Vahabikashi A, Li G, Perkumas KM, Stamer WD, Johnson M and Scott E, Small, 2020, 2004205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jackman MR, Shurety W, Ellis JA and Luzio JP, Journal of Cell Science, 1994, 107, 2547–2556. [DOI] [PubMed] [Google Scholar]
- 37.Sasso L, Purdie L, Grabowska A, Jones AT and Alexander C, Journal of Interdisciplinary Nanomedicine, 2018, 3, 67–81. [Google Scholar]
- 38.Bogdanowich-Knipp SJ, Chakrabarti S, Williams TD, Dillman RK and Siahaan TJ, J Pept Res, 1999, 53, 530–541. [DOI] [PubMed] [Google Scholar]
- 39.Bogdanowich-Knipp SJ, Jois DS and Siahaan TJ, J Pept Res, 1999, 53, 523–529. [DOI] [PubMed] [Google Scholar]
- 40.Amin M, Badiee A and Jaafari MR, Int J Pharm, 2013, 458, 324–333. [DOI] [PubMed] [Google Scholar]
- 41.Dubey PK, Mishra V, Jain S, Mahor S and Vyas SP, J Drug Target, 2004, 12, 257–264. [DOI] [PubMed] [Google Scholar]
- 42.Song Z, Lin Y, Zhang X, Feng C, Lu Y, Gao Y and Dong C, Int J Nanomedicine, 2017, 12, 1941–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pierschbacher MD and Ruoslahti E, Nature, 1984, 309, 30–33. [DOI] [PubMed] [Google Scholar]
- 44.Kim JP, Zhang K, Kramer RH, Schall TJ and Woodley DT, J Invest Dermatol, 1992, 98, 764–770. [DOI] [PubMed] [Google Scholar]
- 45.Ruoslahti E and Pierschbacher MD, Cell, 1986, 44, 517–518. [DOI] [PubMed] [Google Scholar]
- 46.Yamada KM and Kennedy DW, J Cell Biochem, 1985, 28, 99–104. [DOI] [PubMed] [Google Scholar]
- 47.Zitzmann S, Ehemann V and Schwab M, Cancer Res, 2002, 62, 5139–5143. [PubMed] [Google Scholar]
- 48.Cressman S, Sun Y, Maxwell EJ, Fang N, Chen DDY and Cullis PR, Int J Pept Res Ther, 2009, 15, 49–59. [Google Scholar]
- 49.Holtz KM and Kantrowitz ER, FEBS Letters, 1999, 462, 7–11. [DOI] [PubMed] [Google Scholar]
- 50.Ahmed F and Discher DE, Journal of Controlled Release, 2004, 96, 37–53. [DOI] [PubMed] [Google Scholar]
- 51.James SF, Chahine EB, Sucher AJ and Hanna C, Ann Pharmacother, 2018, 52, 673–680. [DOI] [PubMed] [Google Scholar]
- 52.Nagaraj R, Stack T, Yi S, Mathew B, Shull KR, Scott EA, Mathew MT and Bijukumar DR, Nanomaterials, 2020, 10, 581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stack T, Liu Y, Frey M, Bobbala S, Vincent M and Scott E, Nanoscale Horiz., 2021, 6, 393–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vincent MP, Stack T, Vahabikashi A, Li G, Perkumas KM, Ren R, Gong H, Stamer WD, Johnson M and Scott EA, bioRxiv, 2021, 2021.05.19.444878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shin MD, Shukla S, Chung YH, Beiss V, Chan SK, Ortega-Rivera OA, Wirth DM, Chen A, Sack M, Pokorski JK and Steinmetz NF, Nat Nanotechnol, 2020, 15, 646–655. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.