

Abstract
Wilson and Jungner’s recommendations for population-based screening have been used to guide decisions regarding candidate disease inclusion in newborn screening programs for the past 50 years. The advent of genomic-based technologies, including next-generation sequencing and its potential application to newborn screening, along with a changing landscape in terms of modern clinical practice and ethical, social, and legal considerations has led to a call for review of these criteria. Inborn errors of immunity (IEI) are a heterogeneous group of more than 450 genetically determined disorders of immunity, which are associated with significant morbidity and mortality, particularly where diagnosis and treatment are delayed. We argue that in addition to screening for severe combined immunodeficiency disease, which has already been initiated in several countries, other clinically significant IEI should be screened for at birth. Because of disease heterogeneity and identifiable genetic targets, a next-generation sequencing-based screening approach would be most suitable. A combination of worldwide experience and technological advances has improved our ability to diagnose and effectively treat patients with IEI. Considering IEI in the context of updated recommendations for population-based screening supports their potential inclusion as disease targets in newborn screening programs.
Keywords: Inborn errors of immunity, newborn screening, next-generation sequencing, severe combined immunodeficiency
The objective of newborn screening programs is to identify presymptomatic neonates affected by serious disorders, for which treatment is available and where early intervention reduces morbidity and mortality. Wilson and Jungner’s1 recommendations for population-based screening have guided decisions regarding candidate disease inclusion in newborn screening programs for the past 50 years. However, a changing landscape of clinical practice, newer technologies including genomic-based methodologies, and expanding views on the ethical, legal, and social implications of population-based screening practices call for review of these historic criteria.2–5
Inborn errors of immunity (IEI) are a heterogeneous group of more than 450 genetically determined disorders of immunity, which are associated with significant morbidity and mortality, particularly where diagnosis and treatment are delayed. Screening for severe combined immunodeficiency disease (SCID) has already been initiated in several countries6; however, we argue that other clinically significant IEI should also be screened for at birth, given that early identification and intervention can potentially improve patient outcomes. An increased international experience in managing patients with IEI and technological advances leading to development of new and improved therapeutics have translated into better clinical outcomes and quality of life for affected individuals. This alone warrants consideration of inclusion of these diseases in newborn screening programs. Given extensive disease heterogeneity, and the presence of identifiable genetic targets, we recommend that a next-generation sequencing (NGS)-based screening approach be considered. Proof of concept for this approach has been published for IEI specifically,7 and as a basis for screening for other inborn errors.5,8 Here, we consider IEI in the context of updated recommendations for population-based screening, and the argument for their inclusion as disease targets in newborn screening programs.
2020 VISION: A MODERN-DAY REVIEW OF THE WILSON AND JUNGNER CRITERIA
In their seminal article, “Principles and practice of screening for disease,” Wilson and Jungner9 outlined a series of recommendations for population-based disease screening. Since their publication in 1968, these guidelines have been a major point of reference and have informed decisions regarding inclusion of new disease candidates for population-based screening, including newborn screening programs. Since that time, there has been significant progress in our understanding of disease pathophysiology. Concurrently, technological advances have resulted in improved laboratory testing capabilities and the development of new and improved therapeutic options for a range of diseases. Given these developments, and the evolution of newer screening methodologies including genomic techniques, there has been a call for review and refinement of the classical Wilson and Jungner criteria (Table I). It has been suggested that these criteria be updated and brought into alignment with contemporary practices, including additional considerations that are relevant in the current era.2–4 A series of updated criteria have been proposed to aid decision making regarding selection of new candidate conditions for inclusion in screening programs, in the context of genetic screening and of broader considerations that have an increasingly prominent role, including ethical, social, and legal issues, logistics, and quality assurance.3,4 Dobrow et al2 published a series of consolidated screening principles, based on a process of systematic review and Delphi consensus (Table II), expanding on the classical Wilson and Jungner criteria. In addition, others have proposed additional important elements for consideration, including reducing/eliminating the “diagnostic odyssey” for patients and their families,3 provision of robust prescreening counseling, informed consent and the ability to “opt-out,”3,4 and integration of genetic counseling services for identified carriers.3 Importantly, program outcomes should be defined at the outset and be equitable in terms of access, and overall benefits of screening should outweigh any harms.4
TABLE I.
Original Wilson and Jungner criteria for population-based screening
| The condition should be an important public health concern |
| There should be a treatment for the condition |
| Facilities for diagnosis and treatment should be available |
| There should be a latent stage of the disease |
| There should be a test or examination for the condition |
| The test should be acceptable to the population |
| The natural history of the disease should be adequately understood |
| There should be an agreed policy on who to treat |
| The total cost of finding a case should be economically balanced in relationship to medical expenditure as a whole |
| Case finding should be a continuous process, not a “once and for all” project |
Reference: Wilson and Jungner.1
TABLE II.
Consolidated principles for screening (adapted from Dobrow et al2)
| Domain | Consolidated screening principles |
|---|---|
| Disease/condition principles | Epidemiology of disease/condition |
| • Adequately understood | |
| • Seen as important health problem (high/increasing incidence/prevalence, associated with significant morbidity/mortality) | |
| Natural history of disease/condition | |
| • Adequately understood | |
| • Well-defined | |
| • A detectable preclinical phase exists | |
| Target population for screening | |
| • Clearly defined (eg, age group) | |
| • Identifiable | |
| • Able to be reached | |
| Test/intervention principles | Screening test performance characteristics |
| • Appropriate for the purpose | |
| • All key components of the test are accurate (sensitivity, specificity, positive predictive value), reliable/reproducible | |
| • Acceptable to the target population | |
| • Possible to perform/administer safely, affordably, and efficiently | |
| Interpretation of screening test results | |
| • Clearly interpretable and determinate (with known distribution of test values and well-defined/agreed-upon cutoff points) to enable identification of screened individuals who require/do not require diagnostic testing and other postscreening care | |
| Postscreening test options | |
| • An agreed-upon course of action for screened individuals with positive screening test results (involving diagnostic testing, treatment/intervention, and follow-up) that will modify the natural history/clinical pathway for the disease/condition | |
| • Available, accessible, and acceptable to those affected | |
| • Results in improved outcomes (eg, improved quality of life, improved function, decreased mortality) | |
| • The testing burden on individuals is understood and acceptable | |
| • Minimal effects of false-positive and false-negative test results | |
| Program/system principles | Screening program infrastructure |
| • Adequate existing infrastructure (eg, financial, human health and information technology resources, facilities, equipment, test technology) or a clear plan to develop new infrastructure | |
| • Appropriate for the setting, to allow for timely access to all components of the testing program (recruitment, testing, information access, diagnosis, referral, treatment, follow-up, patient education and support, staff training, program management and evaluation) | |
| Screening program coordination and integration | |
| • All components of the screening program are coordinated and where possible integrated with the broader health care system (including formal system to inform, counsel, refer, and manage treatment of screened individuals) | |
| • Optimize continuity of care and ensure no screened individual is neglected | |
| Screening program acceptability and ethics | |
| • All components of the screening program are clinically, socially, and ethically acceptable to screened individuals, health professionals, and society | |
| • Effective methods for provision of informed choice to screened individuals, promoting autonomy and protecting their rights | |
| Screening program benefits and harms | |
| • Expected range and magnitude of benefits (eg, improved quality of life, improved function, and decreased mortality) and harms (eg, overdiagnosis and overtreatment) for screened individuals and society are clearly defined and acceptable | |
| • Supported by existing, high-quality scientific evidence (or addressed by ongoing studies), indicating the overall benefit or the program outweighs potential harms | |
| Economic evaluation of screening program | |
| • An economic evaluation (eg, cost-effectiveness, cost-benefit, and cost-utility analyses) is performed/planned from a health system or societal perspective | |
| • Assessment of full costs and effects of implementing, operating, and sustaining the screening program | |
| • Considering opportunity costs and effect of allocating resources to other nonscreening alternatives (eg, primary prevention, improved treatment, and other clinical services) for managing the disease or condition | |
| Screening program quality and performance management | |
| • The screening program has clear goals/objectives that explicitly link to program planning, monitoring, evaluating, and reporting activities | |
| • Dedicated information systems and funding | |
| • Ensure ongoing quality control and achievement of performance targets | |
Reference: Dobrow et al.2
NEWBORN SCREENING FOR IEI
IEI are a heterogeneous group of congenital defects of immunity, the hallmarks of which are severe, recurrent, or unusual infections, immune dysregulation, and other clinical features. These disorders are classified by the International Union of the Immunological Societies according to the predominant immunologic defect10 (Table III). IEI were previously thought to be rare entities; however, their overall prevalence is estimated to be at least 1:1000 individuals.11–13 Although some forms of IEI were described in the first decades of the 20th century,14–16 the disorders now known as Kostmann syndrome17,18 and Bruton’s agammaglobulinemia19,20 were the first disorders to be formally recognized as IEI in the 1950s. Since this time, and particularly over the past several years with the advent of improved genomic testing capabilities including NGS, there has been a rapid increase in the number of genetically determined forms of IEI, with more than 450 different monogenic causes described to date.10
TABLE III.
IUIS classification of inborn errors of immunity (2019)
| (1) | Immunodeficiencies affecting cellular and humoral immunity |
| 1. T− B+ severe combined immune deficiency | |
| 2. T−B− severe combined immune deficiency | |
| 3. Combined immunodeficiency (CID), generally less profound than SCID | |
| (2) | CIDs with associated or syndromic features |
| 1. Immunodeficiency with congenital thrombocytopenia | |
| 2. DNA repair defects other than those listed in (1) | |
| 3. Thymic defects with additional congenital anomalies | |
| 4. Immuno-osseous dysplasias | |
| 5. Hyper-IgE syndromes | |
| 6. Defects of vitamin B12 and folate metabolism | |
| 7. Anhidrotic ectodermodysplasia with immunodeficiency | |
| 8. Calcium channel defects | |
| 9. Other defects | |
| (3) | Predominantly antibody deficiencies |
| 1. Severe reduction in all serum immunoglobulin isotypes with profoundly decreased or absent B cells, agammaglobulinemia | |
| 2. Severe reduction in at least 2 serum immunoglobulin isotypes with normal or low number of B cells, CVID phenotype | |
| 3. Severe reduction in serum IgG and IgA with normal/elevated IgM and normal numbers of B cells, hyper-IgM | |
| 4. Isotype, light chain, or functional deficiencies with generally normal numbers of B cells | |
| (4) | Diseases of immune dysregulation |
| 1. Familial hemophagocytic lymphohistiocytosis (FHL) syndromes | |
| 2. FHL syndromes with hypopigmentation | |
| 3. Regulatory T-cell defects | |
| 4. Autoimmunity with or without lymphoproliferation | |
| 5. Immune dysregulation with colitis | |
| 6. Autoimmmune lymphoproliferative syndrome (Canale-Smith syndrome) | |
| 7. Susceptibility to EBV and lymphoproliferative conditions | |
| (5) | Congenital defects of phagocyte number or function |
| 1. Congenital neutropenias | |
| 2. Defects of motility | |
| 3. Defects of respiratory burst | |
| 4. Other nonlymphoid defects | |
| (6) | Defects of intrinsic and innate immunity |
| 1. Mendelian susceptibility to mycobacterial diseases | |
| 2. Epidermodysplasia verruciformis (human papillomavirus) | |
| 3. Predisposition to severe viral infection | |
| 4. Herpes simplex encephalitis | |
| 5. Predisposition to invasive fungal disease | |
| 6. Predisposition to mucocutaneous candidiasis | |
| 7. TLR signaling pathway deficiency with bacterial susceptibility | |
| 8. Other IEI related to nonhematopoeitic tissues | |
| 9. Other IEI related to leukocytes | |
| (7) | Autoinflammatory disorders |
| 1. Type I interferonopathies | |
| 2. Defects affecting the inflammasome | |
| 3. Non-inflammasome-related conditions | |
| (8) | Complement deficiencies |
| (9) | Bone marrow failure |
| (10) | Phenocopies of IEI |
| 1. Associated with somatic mutations | |
| 2. Associated with autoantibodies | |
IUIS, International Union of the Immunological Societies.
Reference: Tangye et al.10
One particularly significant form of IEI is SCID, which typically presents in early infancy and is uniformly fatal without allogeneic hematopoietic stem cell transplantation (HSCT) or other curative therapies. Evidence suggests that outcomes are significantly improved if HSCT is performed before the age of 3.5 months21 and before the infant has amassed a significant infectious burden and other disease complications.22,23 The only realistic way to achieve this is to identify affected infants in the asymptomatic phase, soon after birth. As such, SCID was identified as a candidate condition for newborn screening. Newborn screening programs for SCID have been successfully implemented in many countries around the world, using a DNA-based technique to quantify surrogate markers of T-cell generation (T-cell receptor excision circles [TRECs]), alone or in combination with their B-cell counterparts (kappa-deleting recombination excision circles [KRECs]).6 However, these methodologies will only identify those forms of IEI manifested by absent or low T and/or B cells, which constitute only a small subset of the described monogenetic causes of IEI.
As a candidate disease for inclusion in population-based screening programs, SCID fulfills Wilson and Jungner’s 10 criteria. There has been some debate as to whether screening for other forms of IEI, which could also be detected by a newborn screening test, is justified on the basis of these criteria.6 Our group and others have investigated methods by which to expand current screening capabilities to detect a greater number of distinct forms of IEI, using Guthrie card analyses.6 These proof-of-concept studies have included identification of granulocyte and complement disorders using protein-based methods,24–26 and screening for familial hemophagocytic lymphohistiocytosis (HLH) due to UNC13D inversion mutations using genetic copy number variant analysis.27 Given the significant genetic and phenotypic heterogeneity of IEI, screening for each of these diseases would require different testing modalities, and this is not practical from a logistic or economic perspective in the context of a newborn screening program. In recent years, NGS-based technologies have become more readily available, less expensive, and associated with faster turnaround times. NGS approaches have been suggested as an option for newborn screening for IEI and other conditions arising from inborn errors, enabling up-front, parallel sequencing of hundreds of disease-associated genes.5,7,28 This approach has already undergone preliminary evaluation in a small series evaluating up-front whole-exome sequencing (WES) of key immunodeficiency-associated genes as a newborn screening strategy.7 Although there were several limitations of this study, including the small cohort size, 1 case of IEI was identified in a cohort of 1349 screened newborns; however, further clinical information was not provided.
NGS-based approaches have also been evaluated for other conditions currently included in newborn screening programs, including inborn errors of metabolism (IEM). A recent study compared a WES-based screening approach to standard tandem mass spectrometry (MS/MS) in a large cohort of patients with IEM, finding the former approach to be less sensitive and specific, but helpful as an adjunct to biochemical screening.8 Further evaluation of an up-front WES approach for newborn screening is also under way as part of the National Institutes of Health INSIGHT program,29 and results from this and other evaluations of NGS-based newborn screening for different disorders are awaited.
THE DIAGNOSIS OF PRIMARY IMMUNODEFICIENCY DISEASES IS FREQUENTLY DELAYED, RESULTING IN SIGNIFICANT MORBIDITY AND MORTALITY
In the case of SCID, it has been demonstrated that patient outcomes are significantly improved with early diagnosis and treatment with HSCT.21 In the absence of early identification through either family history (an affected sibling or other family member) or early identification through a newborn screening program, this objective can rarely be achieved. It is well recognized that diagnosis of IEI is frequently delayed.12,13,30 For example, in the case of X-linked agammaglobulinemia, which is not identified in screening programs using a TREC-only approach, symptom onset and diagnosis would be anticipated between age 6 and 12 months, related to the waning of maternal antibody levels. However, in our small Swedish series, the mean age of diagnosis was 4 years.31 A larger, more recent international case series reported a diagnostic delay of more than 24 months after symptom onset in 34% of patients, and extended beyond 36 months in some cases.32 Unfortunately, delayed diagnosis remains a significant problem for many patients with IEI, owing to various factors, including phenotypic heterogeneity, which can lead to a “diagnostic odyssey,” and a lack of awareness and recognition of warning signs of IEI. Delayed diagnosis and commencement of appropriate treatment portends worse patient outcomes in terms of morbidity (often potentially avoidable complications have developed by the time a diagnosis is made) and mortality.
MANAGEMENT OF IEI
Over time, technological advances have led to a better understanding of the immunopathology underlying IEI, and worldwide experience in managing patients with different forms of these disorders has increased considerably. Together, this has translated into new and improved prophylactic and therapeutic options, leading to reduced morbidity and mortality, and improved quality of life for affected patients. Given the heterogeneity of different forms of IEI in terms of underlying pathophysiology, phenotype, and severity, treatment must be tailored both to the individual condition and to the individual patient. Broad management options for IEI are outlined in Fig 1. These span more empirical preventive and supportive measures such as antimicrobial prophylaxis and immunoglobulin replacement, to therapies targeting underlying immunologic defects or pathways. Curative management requires correction of the inborn genetic error underlying the disease, with current options including allogeneic HSCT or gene therapy (GT) for selected conditions. Clinical management can be instituted for most IEI, even if this simply involves measures such as recommending vaccination or monitoring for disease complications (Fig 2). The management options outlined above can be used individually or in combination, sequentially or concurrently (Fig 2). Further details are available in Table E1 in this article’s Online Repository at www.jacionline.org. This supports the argument for identification of individuals with IEI as early as possible, through newborn screening programs, to facilitate early treatment and reduce disease-associated complications that could otherwise be avoided.
FIG 1.
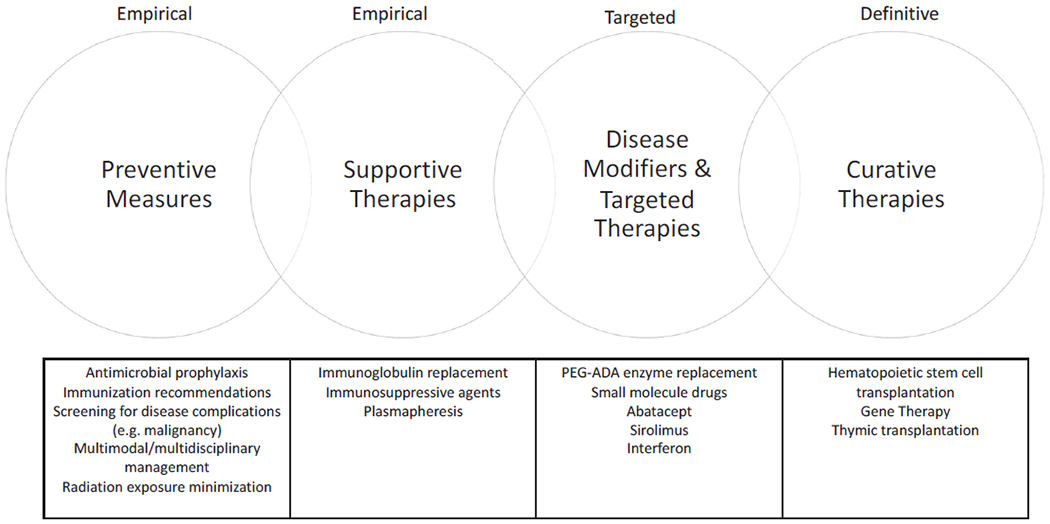
Treatment options for IEI. Management options for patients with IEI include preventive and supportive measures, targeted and disease-modifying therapies, and definitive or curative therapies. Patient management frequently involves 1 or more of these treatment strategies, concurrently or sequentially.
FIG 2.
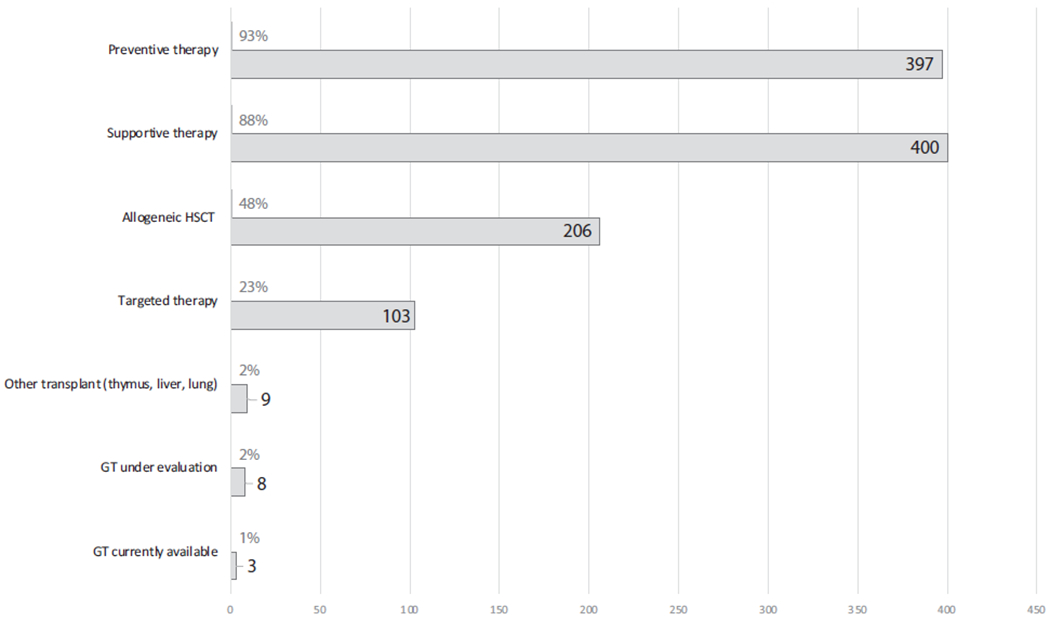
The proportion of monogenic IEI amenable to different clinical management strategies (n = 452). A clinical management strategy can be adopted for every IEI, and for most of these conditions, several treatment strategies exist. These are variably instituted, depending on disease severity, specific disease complications, and other individual patient factors. Further details regarding therapies for each monogenic IEI are available in Table E1.
Many forms of IEI can be managed with preventative and supportive measures
Diagnosis of an IEI enables a tailored management plan to be established, based on prevention and screening for expected complications, including those related to infection, immune dysregulation, or increased susceptibility to malignancy. Preventive measures such as antimicrobial prophylaxis, immunization, screening for malignancy, and avoiding exposure to ionizing radiation may be required in specific circumstances. Supportive therapies, such as immunoglobulin replacement in conditions with associated antibody deficiency, and immunosuppression for management of IEI-associated autoimmunity can be instituted. These approaches are somewhat empirical, and do not augment the underlying disease process but are efficacious in many patients with IEI, either alone or in conjunction with other therapies.
Specific forms of IEI can be managed with targeted or disease-modifying therapies
A greater understanding of immunologic mechanisms in health and disease, coupled with new drug development, has led to further options for the provision of targeted therapy for specific forms of IEI.33 These newer agents, including enzyme replacement therapy, biologic therapies, and small-molecule drugs (including cytokines, cytokine inhibitors, and agents inhibiting signal transduction molecules) target specific immunologic pathways and ameliorate defective immune responses at the molecular level. This contrasts with traditional agents such as corticosteroids and other immunosuppressants, which are nonspecific, have several off-target effects, and potentially more significant adverse effects. Specific examples are discussed below.
Enzyme replacement therapy.
The definitive treatment for adenosine deaminase (ADA)-deficiency SCID is either HSCT or GT; however, pegylated ADA enzyme replacement therapy is also available for the management of this condition.34–37 Enzyme replacement therapy is effective in achieving adequate detoxification and restoration of immunologic function, and can be used as an interim, bridging measure until curative therapy can be instituted.37
Cytokine therapies.
IFN-γ therapy has been used as an adjunctive measure in the treatment of chronic granulomatous disease to increase granulocyte superoxide production and enhance microbial killing.38 In addition, IFN-γ is effective in the treatment of Mendelian susceptibility to mycobacterial diseases due to defects in the IFN-γ-IL12 pathway.39
Anticytokine therapies.
The applications of anticytokine therapies for IEI are broad. For example, IL-1β antagonists (such as anakinra and canakinumab) are effective in the management of several autoinflammatory diseases, and anti-TNF agents such as etanercept may also be effective in some conditions.40 Emapalumab, an mAh against IFN-γ, has shown efficacy in patients with HLH, achieving disease control in a large proportion of patients who could then be successfully cured with allogeneic HSCT.41 Anti-IL-1β antagonists have also been used successfully as an adjunctive therapy for patients with HLH, along with other anticytokine monoclonal agents including anti-IL-6 (tocilizumab).42
Janus-activating kinase/signal transducer and activator of transcription inhibitors (Jakinibs).
Jakinibs are small molecules that inhibit the Janus-activating kinase (JAK)/signal transducer and activator of transcription (STAT) pathway, and include ruxolitinib, baracitinib (predominant JAK1/JAK2 blockade), and tofacitinib (predominant JAK1/JAK3 blockade).33 They have been demonstrated to be safe and efficacious in treating disease complications arising due to gain-of-function mutations in STAT143,44 and STAT343,45,46 by abrogating abnormal pathway activation.
Mammalian target of rapamycin inhibitors.
Mammalian target of rapamycin inhibitors such as sirolimus and everolimus have been used successfully in a range of conditions associated with immune dysregulation, including immunodeficiency, polyendocrinopathy, enteropathy, X-linked syndrome, and immunodeficiency, polyendocrinopathy, endocrinopathy, X-linked—like disease,47,48 activated phosphoinositide 3-kinase δ syndrome,49 and autoimmune lymphoproliferative syndrome.50
Selective PI3K inhibitors.
In addition to mammalian target of rapamycin inhibition for activated phosphoinositide 3-kinase δ syndrome, 2 selective inhibitors of PI3K have been developed and are currently under evaluation: leniolisib (oral inhibitor of the p110δ subunit of PI3Kδ) and nemiralisib (inhaled inhibitor of PI3Kδ).33
Cytotoxic T-lymphocyte-associated protein 4-immunoglobulin fusion proteins.
The cytotoxic T-lymphocyte-associated protein 4-immunoglobulin fusion protein abatacept has been used for the management of cytotoxic T-lymphocyte-associated protein 4 haploinsufficiency and LPS-responsive and beige-like anchor protein deficiency.51,52
Anti-CD20 m Abs.
Anti-CD20 agents (such as rituximab) are used to manage autoimmune and lymphoproliferative complications in several forms of IEI.33
Plerixafor.
Warts-hypogammaglobulinemia-immunodeficiency-myelokathexis syndrome is an IEI resulting from CXCR4 gene mutations that cause retention of mature neutrophils in the bone marrow and various degrees of pancytopenia. Plerixafor, a CXCR4 antagonist, has shown efficacy in these patients by improving blood cell counts and reducing the rate of infections.53
HSCT and GT are curative treatment options for many forms of IEI
Allogeneic HSCT and GT restore normal immunologic function by correcting the genetic defect underlying the immunodeficiency disease, through engraftment and differentiation of normal donor stem cells, or reinfusion of autologous stem cells following in vitro genetic correction, respectively. Given the inherent risks involved, HSCT is reserved for the more severe forms of IEI, which are correctable using this procedure, are not amenable to other definitive medical therapies, and where the potential benefits outweigh the risks of transplant. In some conditions, such as SCID, HSCT (or GT, if available) is an absolute indication. For other forms of IEI, HSCT is strongly recommended (eg, HLH), and for others, it is not routinely recommended because of the presence of other therapeutic options (eg, X-linked agammaglobulinemia, which can generally be well managed with immunoglobulin replacement therapy). The repertoire of “transplantable” conditions has increased over time, with international experience demonstrating that this is an effective, curative treatment options for both children and adults with different forms of IEI.54,55
GT was first attempted in the 1990s for the management of ADA deficiency and X-linked SCID; however, it was fraught with many challenges, including development of hematological malignancies in patients with X-linked SCID, Wiskott-Aldrich syndrome, X-linked chronic granulomatous disease, and now also in patients with ADA-SCID. Such serious adverse events reflected insertional mutagenesis due to integration of retroviral vectors within or near oncogenes (reviewed in Cavazzana et al56). Subsequently, there have been significant advancements in this field. In particular, no cases of leukemia have been observed in patients who have received GT based on self-inactivating retroviral and lentiviral vectors. Currently, GT is considered a very effective therapy for patients with ADA deficiency, particularly in the context of a lack of a suitably matched related donor.36,57 GT is currently under evaluation for various other immunodeficiencies, including X-linked SCID, X-linked chronic granulomatous disease, and Wiskott-Aldrich syndrome.58 In particular, excellent results have been recently reported in patients with X-linked SCID, with reconstitution of both T- and B-cell responses if low-dose chemotherapy is used in the preparative regimen, facilitating engraftment of gene-modified stem cells.59,60 It is likely that the number of conditions for which GT is available will continue to grow over time, offering further curative options for a wider range of patients. However, unless corporate social responsibilities are assumed by pharmaceutical companies in partnership with local governments, financial considerations may represent an important obstacle to broad international use of GT in the treatment of IEI, adding to ethical concerns about equitable access to cure for these disorders.61
Thymic transplantation.
Thymic transplantation has been developed for the management of patients with complete DiGeorge syndrome. Although T-cell—depleted HSCT is one treatment option for this condition, due to the complete athymia, only postthymic T-cell engraftment occurs with overall poor immune reconstitution.62 Thymic transplant has been demonstrated to be a favorable alternative approach, leading to successful T-cell reconstitution.63
The updated Wilson and Jungner criteria and the case for newborn screening for IEI using NGS-based screening
As for any screening program, a newborn screening program for IEI using NGS requires a robust clinical-pathological pipeline, an acceptable turnaround time, and favorable test performance characteristics. Furthermore, it must be subject to rigorous quality assurance and be economically feasible.28
Testing methodologies and strategies should be carefully selected.
Applying NGS technologies to newborn screening is a novel approach that enables concurrent testing, using one platform, for potentially hundreds of clinically actionable diseases5 and thus would be the ideal testing strategy for monogenic IEI. In terms of selecting the ideal NGS-based technique to apply to newborn screening for IEI, both WES and whole-genome sequencing (WGS) are available. Proof of concept for a WGS-based approach has been published for IEI7 and would be our favored approach given the ability to simultaneously sequence both intronic and exonic regions, thereby capturing pathogenic variants in intronic regions that would otherwise be missed by WES. This is particularly important for diseases in which pathogenic intronic genetic mutations are common, such as GATA2 deficiency.64 However, the use of WGS for newborn screening of IEI also comes with significant limitations. In particular, assessing the potential deleterious effects of novel variants may prove challenging, especially (but not only) for intronic and regulatory region variants.65–67
Turnaround time must be acceptable.
NGS-based technologies have evolved to yield more rapid results. Clinically actionable NGS results may now become available within 26 to 48 hours of sampling, as demonstrated in neonatal intensive care settings.68 It is anticipated that further technological advances and improved bioinformatic pipelines will also result in further decreases in turnaround times for routine workflow. However, although the test itself may be run and reported within 48 hours, in reality it may take several days or even weeks to process this information and analyze any variants present. Clinical review and second-tier testing will also be required in many cases; hence, the time between sampling and final diagnosis may be protracted.
Test performance characteristics must be favorable, and testing must be quality assured2.
Adhikari et al8 recently evaluated WES data in infants with various IEM identified by MS/MS, including 805 affected individuals and 385 who returned false-positive results on traditional screening. They compared the 2 methodologies as up-front screening tools, and found an overall lower sensitivity and specificity of WES-based screening (88% and 98.4%, respectively) compared with MS/MS (99% and 99.8%, respectively). This suggested that standalone WES was not superior to current methodology as the primary screening test for this group of disorders. WES did, however, have utility as an adjunctive test to resolve abnormal MS/MS results.8 Before instituting NGS-based mainstream newborn screening practice, it will be important to fully assess the testing characteristics for each condition and each gene. It is well described in the IEI literature that despite a clear clinical phenotype, no monogenic cause is identified in a proportion of affected patients, despite using the most up-to-date NGS technology and pipelines. These would be classified as “false-negative” reports. Sensitivity and specificity of NGS-based approaches will vary widely depending on a number of factors, including the number and type of genes screened, technological issues relating to read depth, assessment for copy number variants, deletions and single nucleotide polymorphisms, the specifics of the bioinformatic pipelines used, patient ethnicity and reference databases used, and variant calling strategies. Diagnostic accuracy may be improved by using various strategies, for example, applying different read length to genes with high homology to improve variant calling.69 As aforementioned, WGS-based screening would circumvent many of the shortfalls of WES, but it has its own limitations. Rigorous quality assurance activities are required to ensure appropriate test performance,2 including participation in internal and external quality control programs. Furthermore, and importantly, current genomic databases have poor representation of populations other than Northern Europeans. For variant calling to be robust and applicable to patients of other ethnicities, a larger number of individuals belonging to the same ethnic group should be included in genomic databases.
Health economic data must be evaluated2.
Health economic analyses are an essential consideration in newborn screening programs,2 and formal, detailed evaluation of the cost-benefit ratio of NGS-based newborn screening for IEI would need to be performed before adopting this approach. With the cost of NGS technologies continuing to decrease over time, this is likely to become more cost-effective and therefore represents a realistic option for future population-based screening. However, the cost of such a program goes well beyond the cost of the assay itself, and wider considerations of the economic impact of second-and third-tier testing, clinical follow-up, and treatment must also be considered. It is possible that identification of some genetic variants in asymptomatic infants would require long-term clinical follow-up, which will also carry an associated expense and also potentially be an extra burden on the health care system. This can be balanced by considering potential health savings earned through the benefits of early diagnosis—reduced acute health care service utilization, including hospital and intensive care unit admissions and averting expensive therapies through effective preventative measures and early definitive therapies. Formal health economic evaluations are required to assess the cost-benefit profile of screening for IEI using NGS, and this potentially needs to occur on a disease-specific basis.
NGS as a newborn screening strategy should interrogate genes where mutations give rise to actionable conditions, and take into account disease prevalence and penetrance5.
It has been established that the incidence of IEI is at least 1:1000 individuals11–13 although this is likely to be even higher due to underdiagnosis. Lists of genes included in newborn screening programs should be updated regularly, in alignment with new gene discovery, and only those genes with a clear disease phenotype should be included. We would not advocate for screening genes with no associated or well-described clinical phenotype. Irrespective of the approach used for molecular diagnosis of IEI, predicting the clinical phenotype may prove problematic, especially for disorders characterized by incomplete penetrance (such as cytotoxic T-lymphocyte-associated protein 4 deficiency and activated phosphoinositide 3-kinase δ syndrome)49,51 and for those genes where distinct clinical phenotypes are associated with gain-of-function, loss-of-function, and dominant-negative alleles (such as STAT1, STAT3, and CARD11).46,70–73 Hence, identifying variants in some genes may warrant a “watch and wait” approach rather than active treatment. However, even in this case, knowledge of a potential disease-causing variant provides an opportunity for clinical monitoring for disease manifestations, with the option of initiating early treatment if required at a later stage, therefore preventing diagnostic delay. In the first instance, we would advocate screening for those IEI where potentially curative therapies (allogeneic HSCT, GT) are available, including SCID, selected combined immunodeficiency diseases, and chronic granulomatous disease (Table E1). Further progression to screen for other clinically significant forms of IEI where supportive, targeted, or preventative measures can be instituted could then be considered. The selection of these diseases requires consideration of factors such as expected genotype-phenotype correlation, penetrance, and local population prevalence. It is anticipated that increasing worldwide experience in the diagnosis and management of these IEI will further guide selection of future candidate genes.
Management of variants of unknown significance.
Variants of unknown significance (VUS) are frequent findings in clinical practice and will also be encountered in NGS-based screening programs for IEI. First, it will be important to construct robust reference genome data for different ethnic groups to facilitate interpretation of sequencing results. To this purpose, it will be essential that for each ethnic group an appropriate number of subjects are included in genomic databases. Evaluation of VUS will potentially be even more challenging in presymptomatic infants who are yet to develop a clinical disease phenotype and will likely require second-line testing with immunologic assays and functional studies. Further genetic studies, including parental or familial studies, may also be required. Deciding on clinically actionable VUS may also present a further challenge. The time and costs associated with investigation of infants with VUS (which may be multiple) may be considerable, and should be factored into cost-benefit analyses, although it is anticipated that further advances in technology and experience will potentially lead to a more streamlined approach to the investigation of VUS in the future. One approach to help overcome this might be to run parallel screening tests: for example, a VUS in a SCID-associated gene in an infant with low TREC levels at birth is more likely to be clinically significant. It remains to be seen whether NGS-based screening will constitute standalone testing in the future, or continue to coexist with other assays (such as TREC/KREC screening for IEI, and MS/MS for IEM and other conditions).
There should be a latent stage of the disease, and treatment should be available3,4.
An asymptomatic period is a feature of all forms of IEI, including SCID. Delayed diagnosis of IEI is a frequent occurrence, and patients frequently experience a “diagnostic odyssey.” Treatment is delayed, and potentially preventable complications have often already developed by the time of diagnosis. This emphasizes the need for early identification of affected patients, with the newborn period an opportune time to identify serious, treatable disease. As we have demonstrated, a management plan can be instituted for almost every form of IEI, ranging from clinical surveillance and vaccination recommendations, through to HSCT and GT, further justifying their inclusion in newborn screening programs.
The screening test should be acceptable to the population, and ethical, legal, and social implications should be considered2
The proposed updated screening criteria appropriately have a strong focus on social, legal, and ethical considerations.2 This includes attention to obtaining informed consent,2 following detailed pretest counseling.3 The overall acceptability of genetic-based screening to the wider population requires further exploration, because there may be concerns in the community regarding proceeding with genetic testing, particularly if plans for management of biological material, data storage, and future access and use are not explicit. Concerns have been raised regarding the potential implications that genetic data may have on interaction with other agencies, for example, access to insurance. In the case that a genetic variant is identified in an infant, further testing may be required to clarify this finding, including parental and wider familial testing, which may also have significant social implications.
At present, most newborn screening programs operate on an “opt-out” basis, where testing occurs unless parents do not wish to proceed. Given the potential implications of genetic testing, clear information, careful pretest counseling, and obtaining fully informed consent would be required. Community concerns regarding implications of genetic testing may have an impact on uptake of newborn screening, and this requires evaluation before implementation.
Another social and ethical consideration that has arisen through current TREC-based screening for SCID is the identification of nontarget conditions, which do not have an available curative treatment option. Many of these conditions are ultimately lethal, regardless of early diagnosis or institution of supportive therapies, including severe forms of dyskeratosis congenita and ataxia telangiectasia (AT). AT is a syndrome arising from recessive mutations in the ATM gene, the product of which is ubiquitously expressed in a range of tissues and results in multisystem symptoms. Patients with AT present with variable immunodeficiency associated with the condition, but have severe, progressive neurological symptoms and significantly reduced life expectancy. No effective curative treatment exists at the current time. Newborns with AT frequently return an abnormal screening test due to their associated lymphopenia; hence, their underlying condition is diagnosed at birth, rather than as small toddlers after onset of neurological symptoms. Debate arose regarding ethical principles, and parental preferences, regarding identification of an early, presymptomatic diagnosis for which no curative treatment exists. This has been examined in a Dutch study, whose authors interviewed families of healthy newborns screened for SCID, and asked whether they would prefer an early (neonatal) diagnosis of AT, versus a late diagnosis.74 Eighty-two percent of families responded indicating they would prefer an early diagnosis, citing avoidance of diagnostic uncertainty and commencement of early management strategies.75 From the clinical perspective, AT identified in screened newborns enabled earlier identification and treatment for immunologic abnormalities, and also has the potential to facilitate earlier, coordinated approaches to multidisciplinary management.76 In addition to IEI, there are several lethal conditions that may be detected on newborn screening tests for which no treatment is available, and the benefits of very early identification of these infants in the presymptomatic phase also needs to be considered in the context of ethical and social factors. Ongoing discussion regarding these ethical and social issues is recommended and requires engagement of a range of representatives from different disciplines: medicine, law, philosophy, government, and importantly, families with affected children and the general community.
As highlighted, there are several potential ethical, social, and legal implications of adopting this approach that require careful, comprehensive discussion before implementation.
The roadmap to NGS-based screening for IEI
NGS-based screening technology represents a future direction for newborn screening programs, and as we have highlighted in the context of screening for IEI, there are many potential benefits to this approach. There are, however, several limitations, challenges, and important considerations that must be addressed before routinely adopting NGS-based newborn screening. Many of these questions remain unanswered and must be further evaluated and clarified in rigorous, prospective studies, which assess the entire screening process including ethical, legal, and social considerations, before implementation. Whether this technology will be used as standalone primary screening strategy, or as an adjunct to current methodologies, and whether this will differ for different conditions and different genes remains to be seen.
Conclusions
There have been significant advances in technology and clinical experience since publication of the original 1968 Wilson and Jungner criteria for population-based screening. In the current era of genomic medicine and the potential to harness NGS-based technologies for the purposes of newborn screening, updated criteria must be considered when selecting new disease candidates. IEI are a group of serious, often life-threatening conditions, where diagnosis and treatment are frequently delayed. An NGS-based newborn screening approach is ideal for the identification of newborns with monogenic forms of IEI, which are heterogeneous, but easily identifiable using a platform that enables concurrent screening for hundreds of individual diseases. There are several factors that must be considered and evaluated in detail before adopting this approach. These include testing strategies and characteristics, and economic, ethical, legal, and social considerations. Given the incidence of IEI, new and improved therapeutic options, and the possibility to institute a management plan for almost every affected patient, identification of affected individuals early in life through newborn screening programs is warranted.
Supplementary Material
Acknowledgments
L.D.N. is supported by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health (grant no. 1 ZIA AI001222-02).
Abbreviations used
- ADA
Adenosine deaminase
- AT
Ataxia telangiectasia
- GT
Gene therapy
- HLH
Hemophagocytic lymphohistiocytosis
- HSCT
Hematopoeitc stem cell transplantation
- IEI
Inborn errors of immunity
- IEM
Inborn errors of metabolism
- JAK
Janus-activating kinase
- LRBA
LPS-responsive and beige-like anchor protein
- MS/MS
Tandem mass spectrometry
- mTOR
Mammalian target of rapamycin
- NGS
Next-generation sequencing
- SCID
Severe combined immunodeficiency
- STAT
Signal transducer and activator of transcription
- TREC
T-cell receptor excision circle
- WES
Whole-exome sequencing
- VUS
Variant of unknown significance
- WGS
Whole-genome sequencing
GLOSSARY
- ALLOGENEIC HEMATOPOIETIC STEM CELL TRANSPLANTATION
Transplantation of hematopoietic stem cells from a healthy donor into a recipient.
- DIAGNOSTIC ODYSSEY
The difficult journey to diagnosis that is often encountered by patients with rare diseases. There is often a delay of several years between onset of symptoms and achieving a final diagnosis, which also frequently results in delayed treatment and development of potentially avoidable complications.
- ENZYME REPLACEMENT THERAPY
A form of treatment whereby an enzyme that is not produced endogenously as a result of an inborn error is administered to an affected patient as replacement therapy.
- GUTHRIE CARD ANALYSES
A procedure whereby blood is collected from a heel prick sample from a newborn, which is then applied to filter paper and tested to screen for a range of diseases such as inborn errors of metabolism and immunity and cystic fibrosis.
- INBORN ERRORS OF IMMUNITY
A group of heterogeneous, genetically determined disorders affecting the development and/or function of 1 or more components of the immune system. To be considered primary, the disorder must not be secondary in nature, such as being a result of other disease, drug treatment, or environmental exposure.
- KAPPA-DELETING RECOMBINATION EXCISION CIRCLES
Small, circular DNA segments that are produced during B-lymphocyte development in the bone marrow, which can be quantified and are surrogate markers for B-cell development.
- NEXT-GENERATION SEQUENCING (NGS)
A group of DNA sequencing techniques involving fragmentation of the genome, enabling rapid, massive, parallel sequencing of millions of nucleotide sequences within the entire genome.
- SEVERE COMBINED IMMUNODEFICIENCY DISEASE (SCID)
A severe, early-onset inborn error of immunity manifested by an abnormal number and/or function of T cells, with variably affected B and natural killer cells depending on the underlying genetic defect.
- T-CELL RECEPTOR EXCISION CIRCLES (TRECs)
Small, circular DNA segments that are produced during T-cell receptor rearrangement in naive T cells, which can be quantified and are surrogate markers of recent thymic emigrants.
- WHOLE-EXOME SEQUENCING
A next-generation technique in which all the protein-coding regions (exons) in a genome are sequenced.
Footnotes
Disclosure of potential conflict of interest: L. Hammarström is on the advisory board of ImmunoIVD. ImmunoIVD manufactures commercial kits for newborn screening for inborn errors of immunity using currently available technologies (T-cell receptor excision circle and kappa-deleting recombination excision circle quantitation). ImmunoIVD does not currently provide commercial assays for newborn screening using next-generation sequencing. In addition, ImmunoIVD does not have any future plans to establish such testing. The rest of the authors declare that they have no relevant conflicts of interest.
REFERENCES
- 1.Wilson J, Jungner G. The principles and practice of screening for disease. Geneva: World Health Organization; 1968. [Google Scholar]
- 2.Dobrow MJ, Hagens V, Chafe R, Sullivan T, Rabeneck L. Consolidated principles for screening based on a systematic review and consensus process. CMAJ 2018;190:E422–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Petros M Revisiting the Wilson-Jungner criteria: how can supplemental criteria guide public health in the era of genetic screening? Genet Med 2012;14:129–34. [DOI] [PubMed] [Google Scholar]
- 4.Andermann A, Blancquaert I, Beauchamp S, Déry V. Revisiting Wilson and Jungner in the genomic age: a review of screening criteria over the past 40 years. Bull World Health Organ 2008;86:317–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hendricks-Sturrup RM, Lu CY. When should genomic and exome sequencing be implemented in newborns? A call for an update to newborn screening guidelines. Genet Med 2020;22:809–10. [DOI] [PubMed] [Google Scholar]
- 6.King JR, Hammarström L. Newborn screening for primary immunodeficiency diseases: history, current and future practice. J Clin Immunol 2018;38:56–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pavey AR, Bodian DL, Vilboux T, Khromykh A, Hauser NS, Huddleston K, et al. Utilization of genomic sequencing for population screening of immunodeficiencies in the newborn. Genet Med 2017;19:1367–75. [DOI] [PubMed] [Google Scholar]
- 8.Adhikari AN, Gallagher RC, Wang Y, Currier RJ, Amatuni G, Bassaganyas L, et al. The role of exome sequencing in newborn screening for inborn errors of metabolism. Nat Med 2020;26:1392–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wilson JM, Jungner YG. Principles and practice of mass screening for disease [in Spanish]. Bol Oficina Sanit Panam 1968;65:281–393. [PubMed] [Google Scholar]
- 10.Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, et al. Human Inborn Errors of Immunity: 2019 Update on the Classification from in the US Immunodeficiency Network (USIDNet) Registry. J Clin Immunol 2020;40:1124–31.32880085 [Google Scholar]
- 11.Zhang Q, Frange P, Blanche S, Casanova JL. Pathogenesis of infections in HIV-infected individuals: insights from primary immunodeficiencies. Curr Opin Immunol 2017;48:122–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boyle JM, Buckley RH. Population prevalence of diagnosed primary immunodeficiency diseases in the United States. J Clin Immunol 2007;27:497–502. [DOI] [PubMed] [Google Scholar]
- 13.Kobrynski L, Powell RW, Bowen S. Prevalence and morbidity of primary immunodeficiency diseases, United States 2001-2007. J Clin Immunol 2014;34: 954–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Familiärer Wiskott A., angeborener Morbus Werlhofii. Monatsschr Kinderheilkd 1937;68:212–6. [Google Scholar]
- 15.Lutz W A propos de l’Epidermodysplasie verruciforme. Dermatology 1946;92: 30–43. [PubMed] [Google Scholar]
- 16.Glanzmann E, Riniker P. Essential lymphocytophthisis: new clinical aspect of infant pathology. Ann Paediatr 1950;175:1–32. [PubMed] [Google Scholar]
- 17.Kostmann R Hereditär reticulos: en ny systemsjukdom? Läkartidningen 1950;47:2861–8. [Google Scholar]
- 18.Kostmann R Infantile genetic agranulocytosis. A new recessive lethal disease in man. Acta Paediatr 1956;45:1–76. [PubMed] [Google Scholar]
- 19.Bruton OC. Agammaglobulinemia. Pediatrics 1952;9:722–8. [PubMed] [Google Scholar]
- 20.Vetrie D, Vorechovsky I, Sideras P, Holland J, Davies A, Flinter F, et al. The gene involved in X-linked agammaglobulinaemia is a member of the src family of protein-tyrosine kinases. Nature 1993;361:226–33. [DOI] [PubMed] [Google Scholar]
- 21.Brown L, Xu-Bayford J, Allwood Z, Slatter M, Cant A, Davies EG, et al. Neonatal diagnosis of severe combined immunodeficiency leads to significantly improved survival outcome: the case for newborn screening. Blood 2011;117:3243–6. [DOI] [PubMed] [Google Scholar]
- 22.Pai SY, Logan BR, Griffith LM, Buckley RH, Parrott RE, Dvorak CC, et al. Transplantation outcomes for severe combined immunodeficiency, 2000-2009. N Engl J Med 2014;371:434–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haddad E, Logan BR, Griffith LM, Buckley RH, Parrott RE, Prockop SE, et al. SCID genotype and 6-month posttransplant CD4 count predict survival and immune recovery. Blood 2018;132:1737–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dezfouli M, Bergström S, Skattum L, Abolhassani H, Neiman M, Torabi-Rahvar M, et al. Newborn screening for presymptomatic diagnosis of complement and phagocyte deficiencies. Front Immunol 2020;11:455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hamsten C, Skattum L, Truedsson L, von Döbeln U, Uhlén M, Schwenk JM, et al. Heat differentiated complement factor profiling. J Proteomics 2015;126:155–62. [DOI] [PubMed] [Google Scholar]
- 26.Janzi M, Sjöberg R, Wan J, Fischler B, von Döbeln U, Isaac L, et al. Screening for C3 deficiency in newborns using microarrays. PLoS One 2009;4:e5321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Borte S, Meeths M, Liebscher I, Krist K, Nordenskjöld M, Hammarström L, et al. Combined newborn screening for familial hemophagocytic lymphohistiocytosis and severe T- and B-cell immunodeficiencies. J Allergy Clin Immunol 2014;134:226–8. [DOI] [PubMed] [Google Scholar]
- 28.King J, Ludvigsson J, Hammarstroöm L. Newborn screening for primary immunodeficiency diseases: the past, the present and the future. Int J Neonat Screening 2017;3:19. [Google Scholar]
- 29.Berg JS, Agrawal PB, Bailey DB, Beggs AH, Brenner SE, Brower AM, et al. Newborn sequencing in genomic medicine and public health. Pediatrics 2017;139:e20162252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bazregari S, Azizi G, Tavakol M, Asgardoon MH, Kiaee F, Tavakolinia N, et al. Evaluation of infectious and non-infectious complications in patients with primary immunodeficiency. Cent Eur J Immunol 2017;42:336–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.King J, Borte S, Brodszki N, von Döbeln U, Smith CIE, Hammarström L. Kappa-deleting recombination excision circle levels remain low or undetectable throughout life in patients with X-linked agammaglobulinemia. Pediatr Allergy Immunol 2018;29:453–6. [DOI] [PubMed] [Google Scholar]
- 32.El-Sayed ZA, Abramova I, Aldave JC, Al-Herz W, Bezrodnik L, Boukari R, et al. X-linked agammaglobulinemia (XLA): phenotype, diagnosis, and therapeutic challenges around the world. World Allergy Organ J 2019;12:100018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bucciol G, Meyts I. Recent advances in primary immunodeficiency: from molecular diagnosis to treatment. F1000Res 2020;9:F1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hershfield MS, Buckley RH, Greenberg ML, Melton AL, Schiff R, Hatem C, et al. Treatment of adenosine deaminase deficiency with polyethylene glycol-modified adenosine deaminase. N Engl J Med 1987;316:589–96. [DOI] [PubMed] [Google Scholar]
- 35.Murguia-Favela L, Min W, Loves R, Leon-Ponte M, Grunebaum E. Comparison of elapegademase and pegademase in ADA-deficient patients and mice. Clin Exp Immunol 2020;200:176–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kohn DB, Hershfield MS, Puck JM, Aiuti A, Blincoe A, Gaspar HB, et al. Consensus approach for the management of severe combined immune deficiency caused by adenosine deaminase deficiency. J Allergy Clin Immunol 2019;143:852–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kuo CY, Garabedian E, Puck J, Cowan MJ, Sullivan KE, Buckley RH, et al. Adenosine Deaminase (ADA)-Deficient Severe Combined Immune Deficiency (SCID) in the US Immunodeficiency Network (USIDNet) Registry. J Clin Immunol 2020;40:1124–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marciano BE, Wesley R, De Carlo ES, Anderson VL, Barnhart LA, Darnell D, et al. Long-term interferon-gamma therapy for patients with chronic granulomatous disease. Clin Infect Dis 2004;39:692–9. [DOI] [PubMed] [Google Scholar]
- 39.Bustamante J, Boisson-Dupuis S, Abel L, Casanova JL. Mendelian susceptibility to mycobacterial disease: genetic, immunological, and clinical features of inborn errors of IFN-γ immunity. Semin Immunol 2014;26:454–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Almeida de Jesus A, Goldbach-Mansky R. Monogenic autoinflammatory diseases: concept and clinical manifestations. Clin Immunol 2013;147:155–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Locatelli F, Jordan MB, Allen C, Cesaro S, Rizzari C, Rao A, et al. Emapalumab in children with primary hemophagocytic lymphohistiocytosis. N Engl J Med 2020;382:1811–22. [DOI] [PubMed] [Google Scholar]
- 42.La Rosée P, Horne A, Hines M, von Bahr Greenwood T, Machowicz R, Berliner N, et al. Recommendations for the management of hemophagocytic lymphohistiocytosis in adults. Blood 2019;133:2465–77. [DOI] [PubMed] [Google Scholar]
- 43.Forbes LR, Vogel TP, Cooper MA, Castro-Wagner J, Schussler E, Weinacht KG, et al. Jakinibs for the treatment of immune dysregulation in patients with gain-of-function signal transducer and activator of transcription 1 (STAT1) or STAT3 mutations. J Allergy Clin Immunol 2018;142:1665–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Higgins E, Al Shehri T, McAleer MA, Conlon N, Feighery C, Lilic D, et al. Use of ruxolitinib to successfully treat chronic mucocutaneous candidiasis caused by gain-of-function signal transducer and activator of transcription 1 (STAT1) mutation. J Allergy Clin Immunol 2015;135:551–3. [DOI] [PubMed] [Google Scholar]
- 45.Fabre A, Marchal S, Barlogis V, Mari B, Barbry P, Rohrlich PS, et al. Clinical aspects of STAT3 gain-of-function germline mutations: a systematic review. J Allergy Clin Immunol Pract 2019;7:1958–69.e9. [DOI] [PubMed] [Google Scholar]
- 46.Milner JD, Vogel TP, Forbes L, Ma CA, Stray-Pedersen A, Niemela JE, et al. Early-onset lymphoproliferation and autoimmunity caused by germline STAT3 gain-of-function mutations. Blood 2015;125:591–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bindl L, Torgerson T, Perroni L, Youssef N, Ochs HD, Goulet O, et al. Successful use of the new immune-suppressor sirolimus in IPEX (immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome). J Pediatr 2005;147:256–9. [DOI] [PubMed] [Google Scholar]
- 48.Yong PL, Russo P, Sullivan KE. Use of sirolimus in IPEX and IPEX-like children. J Clin Immunol 2008;28:581–7. [DOI] [PubMed] [Google Scholar]
- 49.Maccari ME, Abolhassani H, Aghamohammadi A, Aiuti A, Aleinikova O, Bangs C, et al. Disease evolution and response to rapamycin in activated phosphoinositide 3-kinase δ syndrome: the European Society for Immunodeficiencies-Activated Phosphoinositide 3-Kinase δ Syndrome Registry. Front Immunol 2018;9:543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bride KL, Vincent T, Smith-Whitley K, Lambert MP, Bleesing JJ, Seif AE, et al. Sirolimus is effective in relapsed/refractory autoimmune cytopenias: results of a prospective multi-institutional trial. Blood 2016;127:17–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schwab C, Gabrysch A, Olbrich P, Patino V, Warnatz K, Wolff D, et al. Phenotype, penetrance, and treatment of 133 cytotoxic T-lymphocyte antigen 4-insufficient subjects. J Allergy Clin Immunol 2018;142:1932–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lo B, Zhang K, Lu W, Zheng L, Zhang Q, Kanellopoulou C, et al. AUTOIMMUNE DISEASE. Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science 2015;349:436–40. [DOI] [PubMed] [Google Scholar]
- 53.McDermott DH, Pastrana DV, Calvo KR, Pittaluga S, Velez D, Cho E, et al. Plerixa for for the treatment of WHIM syndrome. N Engl J Med 2019;380:163–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Freeman AF. Hematopoietic stem cell transplantation in primary immunodeficiencies beyond severe combined immunodeficiency. J Pediatric Infect Dis Soc 2018;7:S79–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fox TA, Chakraverty R, Burns S, Carpenter B, Thomson K, Lowe D, et al. Successful outcome following allogeneic hematopoietic stem cell transplantation in adults with primary immunodeficiency. Blood 2018;131:917–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cavazzana M, Six E, Lagresle-Peyrou C, André-Schmutz I, Hacein-Bey-Abina S. Gene therapy for X-linked severe combined immunodeficiency: where do we stand? Hum Gene Ther 2016;27:108–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cicalese MP, Ferrua F, Castagnaro L, Rolfe K, De Boever E, Reinhardt RR, et al. Gene therapy for adenosine deaminase deficiency: a comprehensive evaluation of short- and medium-term safety. Mol Ther 2018;26:917–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang ZY, Thrasher AJ, Zhang F. Gene therapy and genome editing for primary immunodeficiency diseases. Genes Dis 2020;7:38–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mamcarz E, Zhou S, Lockey T, Abdelsamed H, Cross SJ, Kang G, et al. Lentiviral gene therapy combined with low-dose busulfan in infants with SCID-X1. N Engl J Med 2019;380:1525–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.De Ravin SS, Wu X, Moir S, Anaya-O’Brien S, Kwatemaa N, Littel P, et al. Lentiviral hematopoietic stem cell gene therapy for X-linked severe combined immunodeficiency. Sci Transl Med 2016;8:335ra57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fischer A, Dewatripont M, Goldman M. Benefit Corporation: a path to affordable gene therapies? Nat Med 2019;25:1813–4. [DOI] [PubMed] [Google Scholar]
- 62.Janda A, Sedlacek P, Hönig M, Friedrich W, Champagne M, Matsumoto T, et al. Multicenter survey on the outcome of transplantation of hematopoietic cells in patients with the complete form of DiGeorge anomaly. Blood 2010;116:2229–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Davies EG, Cheung M, Gilmour K, Maimaris J, Curry J, Furmanski A, et al. Thymus transplantation for complete DiGeorge syndrome: European experience. J Allergy Clin Immunol 2017;140:1660–70.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hsu AP, Johnson KD, Falcone EL, Sanalkumar R, Sanchez L, Hickstein DD, et al. GATA2 haploinsufficiency caused by mutations in a conserved intronic element leads to MonoMAC syndrome. Blood 2013;121:3830-7.S1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Khourieh J, Rao G, Habib T, Avery DT, Lefevre-Utile A, Chandesris MO, et al. A deep intronic splice mutation of STAT3 underlies hyper IgE syndrome by negative dominance. Proc Natl Acad Sci U S A 2019;116:16463–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Starokadomskyy P, Gemelli T, Rios JJ, Xing C, Wang RC, Li H, et al. DNA polymerase-α regulates the activation of type I interferons through cytosolic RNA:DNA synthesis. Nat Immunol 2016;17:495–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Boisson B, Honda Y, Ajiro M, Bustamante J, Bendavid M, Gennery AR, et al. Rescue of recurrent deep intronic mutation underlying cell type-dependent quantitative NEMO deficiency. J Clin Invest 2019;129:583–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Farnaes L, Hildreth A, Sweeney NM, Clark MM, Chowdhury S, Nahas S, et al. Rapid whole-genome sequencing decreases infant morbidity and cost of hospitalization. NPJ Genom Med 2018;3:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Trier C, Fournous G, Strand JM, Stray-Pedersen A, Pettersen RD, Rowe AD. Next-generation sequencing of newborn screening genes: the accuracy of short-read mapping. NPJ Genom Med 2020;5:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Holland SM, DeLeo FR, Elloumi HZ, Hsu AP, Uzel G, Brodsky N, et al. STAT3 mutations in the hyper-IgE syndrome. N Engl J Med 2007;357:1608–19. [DOI] [PubMed] [Google Scholar]
- 71.Liu L, Okada S, Kong XF, Kreins AY, Cypowyj S, Abhyankar A, et al. Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J Exp Med 2011;208:1635–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dupuis S, Jouanguy E, Al-Hajjar S, Fieschi C, Al-Mohsen IZ, Al-Jumaah S, et al. Impaired response to interferon-alpha/beta and lethal viral disease in human STAT1 deficiency. Nat Genet 2003;33:388–91. [DOI] [PubMed] [Google Scholar]
- 73.Lu HY, Bauman BM, Arjunaraja S, Dorjbal B, Milner JD, Snow AL, et al. The CBM-opathies—a rapidly expanding spectrum of human inborn errors of immunity caused by mutations in the CARD11-BCL10-MALT1 complex. Front Immunol 2018;9:2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Blom M, Schoenaker MHD, Hulst M, de Vries MC, Weemaes CMR, Willemsen M, et al. Dilemma of reporting incidental findings in newborn screening programs for SCID: parents’ perspective on ataxia telangiectasia. Front Immunol 2019;10:2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Blom M, Pico-Knijnenburg I, Sijne-van Veen M, Boelen A, Bredius RGM, van der Burg M, et al. An evaluation of the TREC assay with regard to the integration of SCID screening into the Dutch newborn screening program. Clin Immunol 2017;180:106–10. [DOI] [PubMed] [Google Scholar]
- 76.Mandola AB, Reid B, Sirror R, Brager R, Dent P, Chakroborty P, et al. Ataxia telangiectasia diagnosed on newborn sreening—case cohort of 5 years’ experience. Front Immunol 2019;10:2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.