

Abstract
Platelet dense granules form using mechanisms shared by melanosomes in melanocytes and by subsets of lysosomes in more generalized cells. Consequently, disorders of platelet dense granules can reveal how organelles form and move within cells. Models for the study of new vesicle formation include isolated δ-storage pool deficiency, combined αδ-storage pool deficiency, Hermansky-Pudlak syndrome (HPS), Chediak-Higashi syndrome, Griscelli syndrome, thrombocytopenia absent radii syndrome, and Wiskott-Aldrich syndrome. The molecular bases of dense granule deficiency are known for the seven subtypes of HPS, as well as for Chediak-Higashi syndrome, Griscelli syndrome, and Wiskott-Aldrich syndrome. The gene products involved in these disorders help elucidate the generalized process of the formation of vesicles from extant membranes such as the Golgi.
Keywords: Platelet dense granules, storage pool deficiency, Hermansky-Pudlak syndrome, lysosome-related organelles, intracellular vesicle formation
This review concentrates on the dense bodies of platelets, or δ-granules, paying special attention to relevant human disorders. For some such disorders (e.g., Hermansky-Pudlak syndrome [HPS]), the molecular bases have been determined, whereas other dense-granule deficiency states, such as isolated δ-storage pool deficiency (SPD), are distinguished only by their clinical and histological characteristics. Still other diseases, such as Chediak-Higashi syndrome (CHS), Griscelli syndrome (GS), and thrombocytopenia absent radii (TAR) syndrome, may not lack dense bodies entirely, but manifest evidence for reduced number, size, or function of δ-granules. We first describe the dense granule itself and then discuss its disorders in humans.
THE DENSE GRANULE
Contents and Function
Normal human platelets contain three to eight dense granules, each measuring 200 to 300 nm in diameter.1 Some dense granules possess a clear halo encircling a calcium-containing core that is electron dense on un-stained whole mounts2 (Fig. 1). Dense granules are also highly osmophilic, appearing dark on transmission electron microscopy.3 They contain serotonin, adenosine diphosphate (ADP), adenosine triphosphate (ATP), and pyrophosphate.3,4 Serotonin is actively taken up from the blood by a plasma membrane transporter and accumulates in the slightly acidic (pH 6.1) dense granule. These vesicles contain approximately two thirds of the ATP and ADP in platelets at a molar ratio of 2:3, much less than the ratio in plasma (8:1).3 Dense granule membranes house the lysosomal membrane proteins LAMP2 and CD63 (granulophysin or LAMP3), but not LAMP1.5,6 They also contain P-selectin and the integrin αIIbβ3, which are constituents of the α-granule and plasma membrane.
Figure 1.
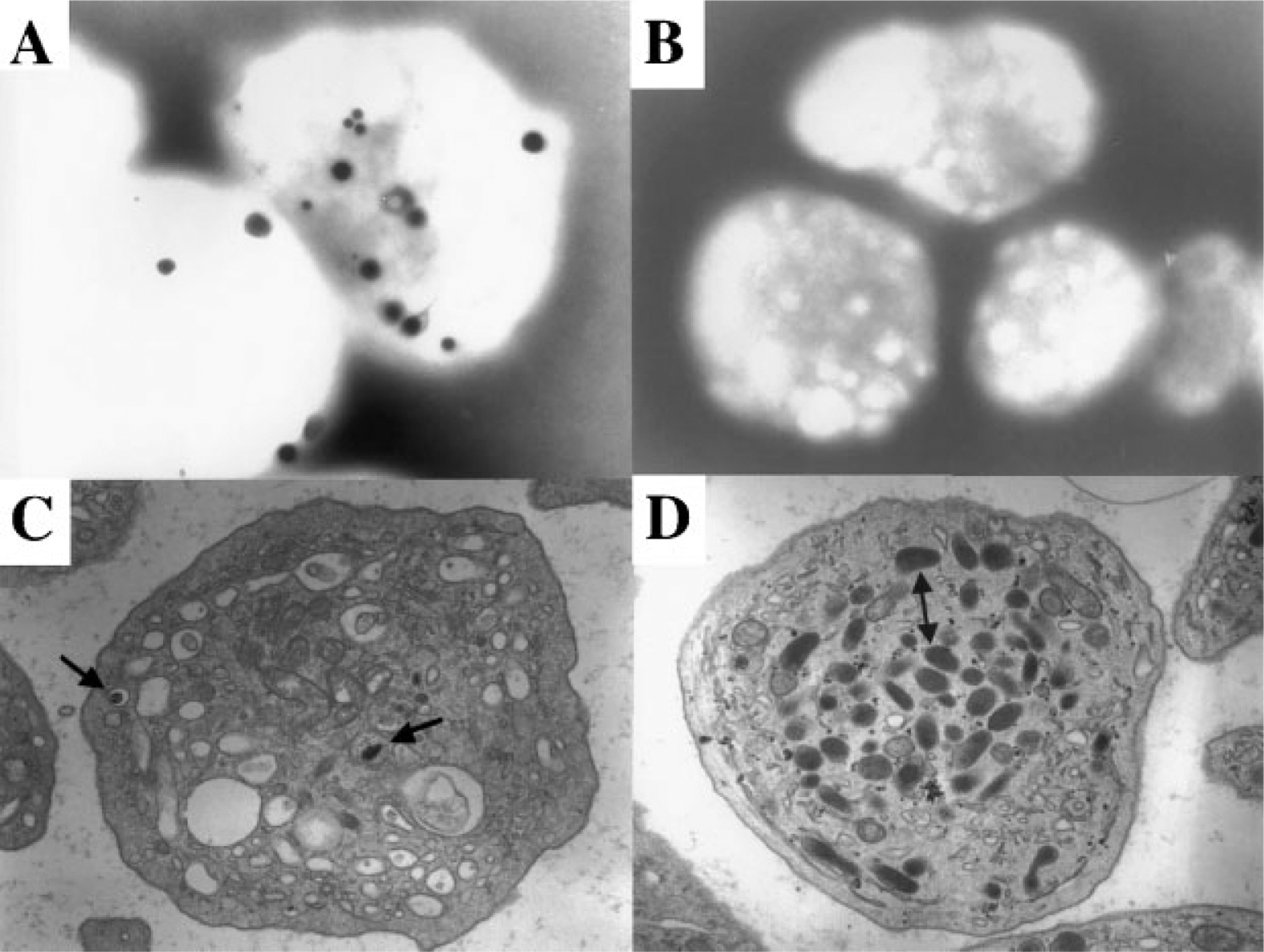
Electron microscopy of platelets showing presence and absence of dense bodies. (A) Whole-mount electron microscopy showing dark, electron-dense granules in the platelets of a 45-year-old woman with albinism but not Hermansky-Pudlak syndrome (HPS). (B) Whole-mount electron microscopy showing absence of dense bodies in the platelets of a 34-year-old man with HPS-1. (C) Transmission electron microscopy showing dense bodies (arrows) in the platelet of a patient with gray platelet syndrome. Note absence of α-granules. (D) Transmission electron microscopy showing absence of dense bodies in a platelet from a 30-year-old woman with HPS-1. Double arrow points to α-granules. (Photographs courtesy of Dr. James G. White, University of Minnesota.)
Upon activation of a platelet, its dense granule membranes fuse with the plasma membrane via the mechanism of soluble N-ethymaleimide-sensitive factor attachment protein receptor (SNARE) complex formation.7 SNARE proteins are inherent components of both the granule membrane and the plasma membrane that recognize each other and form a complex that facilitates docking of the granule to the plasma membrane. This docking does not require ATP,8 but it does require priming by other proteins, including N-ethylmaleimide-sensitive factor and synaptosome-associated proteins (SNAPs), specifically SNAP-23, to form a fusion complex.7
With exocytosis, the dense granule’s contents are disgorged, and its ADP activates and recruits other platelets.3 Serotonin causes vasoconstriction and assists in the binding of adhesive proteins to their platelet receptors.3
Biogenesis
Like α-granules, dense granules originate from the Golgi complex9,10 and are transported to the periphery of megakaryocytes via microtubules and actin filaments, which effect proplatelet formation and elongation. Along this path, a dense granule engages with a multivesicular body (MVB), the key storage and sorting compartment involved in the biogenesis of both α- and dense granules.10,11 There exist different types of MVBs. Type 1 MVBs have many internal vesicles that contain only the α-granule proteins β-thromboglobulin and von Willebrand factor (vWF). Type 2 MVBs have internal vesicles that contain CD63 and also have electron-dense material containing β-thromboglobulin and vWF.11
Young megakaryocytes, with an immature demarcating membrane system, have multivesicular bodies but few α- and dense granules.11,12 As megakaryocytes enlarge, MVBs become fewer, the number of α- and dense granules increases, the demarcating membrane system develops, and the granules are transported into the developing proplatelets.11,12
Storage Pool Deficiencies
The term SPD describes a heterogeneous group of congenital bleeding disorders caused by deficiency of granule-bound substances in platelets.1,13,14SPD platelets have decreased amounts of ADP, serotonin, calcium, and pyrophosphate.15 The discovery of platelets with isolated a-granule deficiency (α-SPD or gray platelet syndrome)16 or deficiency of both α- and dense granules (combined αδ-SPD),13 expanded the definition of SPD. Currently, δ-SPD defines patients with dense granule defects13 that are either acquired (as in myeloproliferative or rheumatologic disorders14) or congenital. We will deal here only with inherited δ-SPDs.
ISOLATED δ-SPD
δ-SPD results in easy bruising, mucous membrane bleeding, and excessive postoperative and postpartum hemorrhage,1,13,14,17 which can be exacerbated by aspirin or other antiplatelet agents. Typically, platelet counts are normal14,17 and the bleeding time is prolonged. The primary aggregation response is normal, but the secondary aggregation wave is absent or muted. In one study, 23% of 106 patients with storage pool deficiency (based on decreased content of adenine nucleotides and serotonin) had completely normal platelet aggregation results.14
Diagnosis of δ-SPD requires measurement of dense granule constituents and/or electron microscopy to demonstrate the absence of dense granule-limiting membranes and contents.13 Adenine nucleotides are reduced, with an increased ratio of ATP to ADP.13,15 Platelet serotonin is variably reduced13,15 and lysosomal enzymes are normal.13,18 Whole-mount electron microscopy recognizes calcium,18 but it cannot differentiate the absence of a dense granule from the absence of its calcium. Both full and empty δ-granules take up the fluorescent dye quinacrine (mepacrine) which can be used to quantitate dense granules by fluorescent microscopy.19,20
The causative gene, basic defect, and mode of inheritance for δ-SPD remain unknown, although evidence for autosomal dominant inheritance has been presented.1,13,17,19 Congenital δ-SPD appears to be underdiagnosed. Nieuwenhuis et al14 reviewed 390 patients with a bleeding tendency; among the 145 with a prolonged bleeding time and normal platelet counts, 27 (18%) had congenital δ-SPD. δ-SPD patients have a propensity to develop myeloproliferative disorders.21
Patients with δ-SPD should be instructed to avoid aspirin and other antiplatelet agents. Platelet transfusions effectively treat hemorrhage in patients with δ-SPD. Desmopressin normalizes the bleeding time in some patients.22 Oral and topical antifibrinolytic agents such as tranexamic acid and ε-amino caproic acid are useful for treating epistaxis and oropharyngeal bleeding.
COMBINED αδ-SPD
Patients with αδ-SPD have diminution of dense granules and a variable deficiency of α-granules and their constituents.13,17 The α-granules may be entirely absent from some platelets but their number may be close to normal in others.13 αδ-SPD can present in mosaic form, with some platelets having normal and some having decreased α- and dense granules.23
Patients with αδ-SPD appear healthy except for their bleeding tendency, which resembles that of δ-SPD and manifests with a prolonged bleeding time.13,17 Platelet counts are normal but platelet aggregation studies may or may not be normal. Diagnosis requires measurement of dense and α-granule constituents and/or electron microscopy. Lysosomal enzymes of αδ-SPD platelets are normal.
The frequency of αδ-SPD is not known, but the disorder appears to be less common than isolated δ-SPD.13,17 Among 43 patients with prolonged bleeding times, seven had αδ-SPD.17 The mode of inheritance appears to be autosomal dominant. The basic defect remains unknown, and no causative gene has been identified.
The gunmetal mouse has reduced α- and dense granules as well as disorganized megakaryocyte internal membranes and impaired retention of α-granule proteins. This mouse has mutations in RabggtA and defective activity of geranylgeranyl transferase, an enzyme that attaches lipid geranylgeraniol groups to certain rab proteins.24 Rabs are small guanosine triphos-phatases (GTPases), which split GTP to provide energy for membrane fusion events and for attachment to cytoskeletons. Human αδ-SPD may result from mutations in RabggtA.
HERMANSKY-PUDLAK SYNDROME
The bulk of our knowledge concerning dense granule deficiency disorders derives from studies of HPS. This disease, named after two Czechoslovakian pathologists who described the disorder in 1959,25 consists of oculocutaneous albinism and a platelet SPD. Much of our understanding of HPS derives from the study of a large isolate in northwest Puerto Rico, where ~450 individuals have HPS-1, due to a founder effect.26,27 However, there are now seven known HPS-causing genes, and seven subtypes of HPS. All are autosomal recessive, and no clinical findings have been reported in heterozygotes. There exist at least 14 HPS mice, and each human subtype is associated with a murine model, leaving seven mice scurrying to find a human counterpart. There exist mutants of Drosophila with pigment granule defects due to impaired HPS-related genes, and yeast with defects in vesicle proteins for sorting that affect vesicle formation and trafficking.28,29
Clinical Findings
All HPS patients have oculocutaneous albinism, with variable hypopigmentation of hair, skin, and irides.30,31 In certain HPS subtypes, the hypopigmentation is virtually indiscernible (Fig. 2). Ophthalmic abnormalities are always apparent, however.27,32 Most patients have congenital nystagmus, and visual acuities range from 20/50 to 20/400. Iris transillumination indicates a finite decrease in iris pigment, and retinal fundi are pale in a scattered pattern. Visual evoked potentials reveal abnormal decussation of optic nerve fibers.
Figure 2.
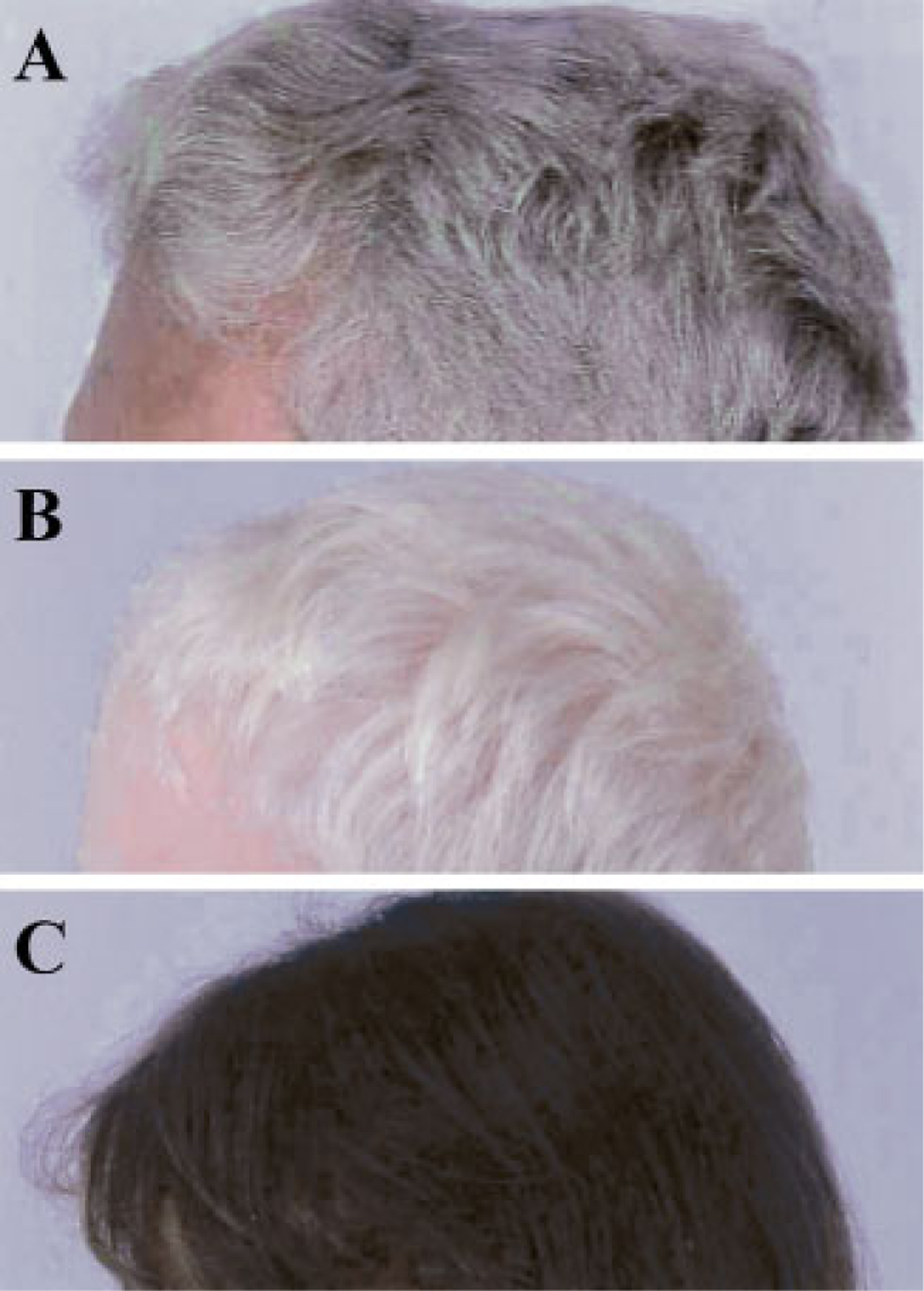
Hair of three HPS patients, all of Puerto Rican ancestry. (A) Typical gray-brown hair of a 54-year-old man with HPS-1. (B) Completely white hair of a 4-year-old boy with HPS-1. (C) Nearly normal, dark hair of a 5-year-old girl with HPS-3.
By definition, all HPS patients lack platelet dense bodies,2 apparently due to a defect in organelle development. Platelet aggregation studies show absence of the secondary wave in response to ADP and epinephrine and an impaired response to collagen.15 The bleeding time is usually prolonged, and mucus membrane bleeding, bruising, epistaxis, and metromenorrhagia are common.27 Many patients have received whole blood or platelet transfusions, but death from bleeding complications virtually never occurs.
Some patients with some types of HPS develop pulmonary fibrosis, which is generally fatal between the fourth and sixth decades.27,33 This may have an inflammatory component34 and begins as mild restrictive lung disease. Approximately 15% of all patients have granulomatous colitis,27,35 which may involve any portion of the alimentary tract, but largely affects the colon. The colitis resembles Crohn’s disease in histology and response to treatment. A lipid-protein complex of unknown etiology called ceroid lipofuscin has been found in the cells and tissues of many HPS patients, largely Puerto Ricans with HPS-1.27
The diagnosis of HPS relies on the finding of oculocutaneous albinism and absent platelet dense bodies on whole-mount electron microscopy.3,28 Transmission electron microscopy sometimes provides equivocal results. The definition of HPS may expand as we discover defects in the number or size of dense bodies rather than their complete absence.
Treatment of HPS is limited.27,29 The albinism requires skin protection, and visual aids are critically important. Bleeding can be controlled topically with thrombin and Gelfoam, and intravenous 1-desamino-8-d-arginine vasopressin is used prophylactically for procedures such as tooth extractions or biopsies. Birth control pills regulate menstrual bleeding. For large bleeds or surgeries, platelet or red blood cell transfusions may be required.
HPS Subtypes
The amount of information available for any HPS subtype reflects the number of affected individuals (Table 1). HPS-1 represents the most severe form of the disease. HPS-1 occurs in 1 of every 1800 northwest Puerto Rican citizens and has variable dermatologic36 and ophthalmologic findings, bleeding complications, colitis, and pulmonary involvement, which might respond to therapy with the investigational drug pirfenidone.37 HPS-1 patients of Japanese, Swiss, and northern European descent manifest typical phenotypes.
Table 1.
Vital Information about HPS Genes
| HPS Subtype | Gene | Chromosome | Size | Mouse | No. of Patients | Comments | |||
|---|---|---|---|---|---|---|---|---|---|
| gDNA (kb) | mRNA (kb)* | Exons | Protein (kd)* | ||||||
| 1 | HPS-1 | 10q23.1–13.3 | 31.5 | 3.7 | 20 | 79.3 | pale ear | 450–500 | NW PR isolate |
| 2 | AP3B1 | 5q14.1 | 292.3 | 4.2 | 27 | 140 | pearl | 4 | |
| 3 | HPS-3 | 3q24 | 43.9 | 4.4 | 17 | 113.7 | cocoa | 23 | Central PR isolate |
| 4 | HPS-4 | 22cen-q12.3 | 32.3 | 4.5 | 14 | 76.9 | light ear | 14 | Interacts with HPS-1 |
| 5 | HPS-5 | 11p14 | 43.4 | 4.8 | 23 | 127.4 | ruby eye-2 | 3 | |
| 6 | HPS-6 | 10q24.32 | 2.7 | 2.7 | 1 | 83.0 | ruby eye | 3 | |
| 7 | HPS-7 | 6p22.3 | 140.2 | 1.5 | 10 | 39.5 | sandy | 1 | |
mRNA and protein size of the longest transcript forms.
HPS, Hermansky-Pudlak syndrome; PR, Puerto Rico.
HPS-2 affects only four known patients who had neutropenia and increased childhood infections, oculocutaneous albinism, and bleeding.38–40 None of the patients has pulmonary fibrosis or colitis but all are too young to inform us whether this subtype is associated with lung disease.
HPS-3 occurs in an isolated region of central Puerto Rico, as well as among Ashkenazi Jews and others.41 Approximately 25 HPS-3 patients are known. The hypopigmentation is extremely mild, so that occasional patients have been diagnosed with ocular (rather than oculocutaneous) albinism. Ophthalmic involvement is also attenuated, along with the bleeding tendency. Colitis does occur in HPS-3, but pulmonary fibrosis probably does not.
HPS-4 affects ~15 individuals and resembles HPS-1, complete with fatal pulmonary fibrosis.42,43 HPS-5 has been reported in one person, a 3-year old Turkish boy with a bleeding time of greater than 7 minutes.44 We know of four additional patients, none with pulmonary fibrosis; two are ~50 years of age. HPS-6 has been reported in one patient, a 39-year-old Belgian woman with no pulmonary or gastrointestinal symptoms.44 We have diagnosed two additional patients, but they are too young for us to determine if pulmonary involvement will occur. One case of HPS-7 has been described in a 48-year-old Portuguese woman with albinism and a bleeding time of 13 minutes.45 She had dyspnea on exertion but normal pulmonary function tests.
Molecular Defects
The HPS1 gene is 31.5-kb in length, 2103 bp of which comprise its 20 exons (Table 1).26 The murine gene, hps1, is mutated in the pale ear mouse.27 The most frequent HPS1 mutation is a 16-bp duplication in exon 15 common in northwest Puerto Rico.26,27 A polymerase chain reaction (PCR)-based assay is available from GeneDx, Gaithersburg, MD. There exist at least 13 other HPS1 mutations, including deletions, insertions, nonsense, and splice site mutations.26,46 No genotype or phenotype correlation has appeared in HPS-1. The HPS1 gene product is a 700-amino acid protein with a molecular weight of 79.3 kd. It is not modified by glycosylation and has no homology to known proteins, although it shares a stretch of amino acids (DKF(L/V)KNRG) with the LYST protein of CHS.
The defective gene in HPS-2 is called AP3B147 (Table 1), has 27 exons within 292.3 kb of genomic DNA, and transcribes a message of 4.2 kb. Homologous genes are mutated in the pearl mouse and in a Drosophila mutant called ruby. Six AP3B1 mutations have been found in four affected humans, including an in-frame deletion, a missense, and two nonsense mutations.38,39 The patient with two nonsense mutations is more severely affected than the brothers with the in-frame mutations. AP3B1 codes for the β3A subunit of AP3, a heterotetrameric complex responsible for vesicle formation from the trans-Golgi network.38–40,48
The HPS3 gene consists of 3921 bp and 17 exons41 (Table 1). Its mRNA is 4.4 kb, and the murine counterpart is mutated in the cocoa mouse. A founder mutation arose in central Puerto Rico in ~1880 to 1890 and consists of a 3904-bp deletion.41 At least six other HPS3 mutations have been described, including IVS5 + 1G > C, a splicing mutation found among the Ashkenazi Jews.49 The HPS3 gene product has 1004 amino acids with a molecular weight of 113.7 kd, a clathrin binding site, no transmembrane regions, and no known function.
The HPS4 gene consists of 14 exons and spans 43.9 kb (Table 1). Several transcripts exist; the major one is 4.5 kb in size. The corresponding mouse mutant is light ear.42 At least nine different HPS4 mutations exist in 15 humans, including insertions, deletions, nonsense, and missense mutations.42,43 No genotype or phenotype correlation has been demonstrated. The function of the HPS4 product is unknown, but it appears to interact with the HPS1 gene product.50
The HPS5 gene spans 43.4 kb of DNA and codes for a 4.8-kb transcript with 23 exons producing a 127.4-kd protein (Table 1). The mouse model is ruby eye-2, which has 81% sequence homology with the human gene. Only one mutation has been reported. The HPS5 gene product has no transmembrane domains or signal sequences, and its function is unknown.44
The HPS6 gene consists of a single exon of 2.7 kb, the product of which is an 83.0-kd protein of unknown function. The murine model is ruby eye.44
The gene causing HPS-7, DTNBP1, consists of 10 exons spanning 140 kb of DNA. The human transcript is 1.5 kb in size, and murine mutations produce the sandy mouse.45 One human mutation, 307C > T (Q103X), has been identified. The human gene product, called dysbindin, has a molecular mass of 39.5 kd. Dysbindin is a component of the biogenesis of lysosome-related organelles complex-1 (BLOC-1; see the following discussion). It also binds to dystrobrevins, which are components of a dystrophin-associated protein complex in muscle and other cells.45
Cell Biology and Gene Function
Megakaryocytes have not been cultured from HPS patients. However, the results of studies of lysosomes in fibroblasts and melanosomes in melanocytes can be applied to dense bodies in platelets. Lysosomes undergo biogenesis in a manner similar to that of lysosome-related organelles (LROs), which include melanosomes and platelet dense bodies; they are formed from the trans-Golgi network and endosomes by processes involving vesicle sorting, budding, targeting, transport, maturation, and fusion.
AP3 functions in LRO biogenesis.47 In HPS-2 fibroblasts (lacking the β3A subunit of AP3), trafficking of lysosomal membrane proteins LAMP1 and LAMP3 through the plasma membrane is enhanced, suggesting that the plasma membrane provides a default pathway for these proteins that operates when normal AP3 function is blocked.39,47 In fact, the LAMP3 distribution in fibroblasts of the different HPS subtypes corresponds with the severity of the clinical phenotype. In HPS-2 melanocytes, tyrosinase expression is reduced and limited to multivesicular bodies in the perinuclear region, suggesting that tyrosinase trafficking is mediated by AP3.51 We infer that the platelets of HPS-2 patients also exhibit defective sorting or trafficking of dense granule proteins. Granulophysin/CD63 and LAMP2 are examples of proteins enriched in the dense body membrane and carrying an AP3 tyrosine sorting signal.52
In addition to AP3B1, all HPS-causing genes encode novel proteins with no recognizable homology to other proteins. HPS-1 is largely cytoplasmic; a portion is membrane bound.53 Some HPS proteins interact with each other in oligomeric complexes called BLOCs. For example, the HPS-1 and HPS-4 proteins interact in BLOC-3,50 which explains the similar phenotype of patients with HPS1 or HPS4 defects. Morphologic studies of HPS-1 melanocytes showed similar membranous complexes containing membrane-bound chambers, unpigmented and pigmented melanosomes, irregular deposits of tyrosinase, and granular or amorphous material—presumably the products of missorted platelet dense granule components.54 Missorting of tyrosine-related protein-1 and tyrosinase was also demonstrated by transfecting normal melanocytes with antisense HPS1 cDNA.55
The HPS-5 and HPS-6 proteins interact in BLOC-2.44 HPS-5 and HPS-6 patients show a similar, mild clinical phenotype. Their fibroblasts show decreased amounts of LAMP1 and LAMP3 reaching peripheral lysosomes, but no accumulation of LAMPs in the perinuclear area (Fig. 3; Huizing, unpublished results).
Figure 3.
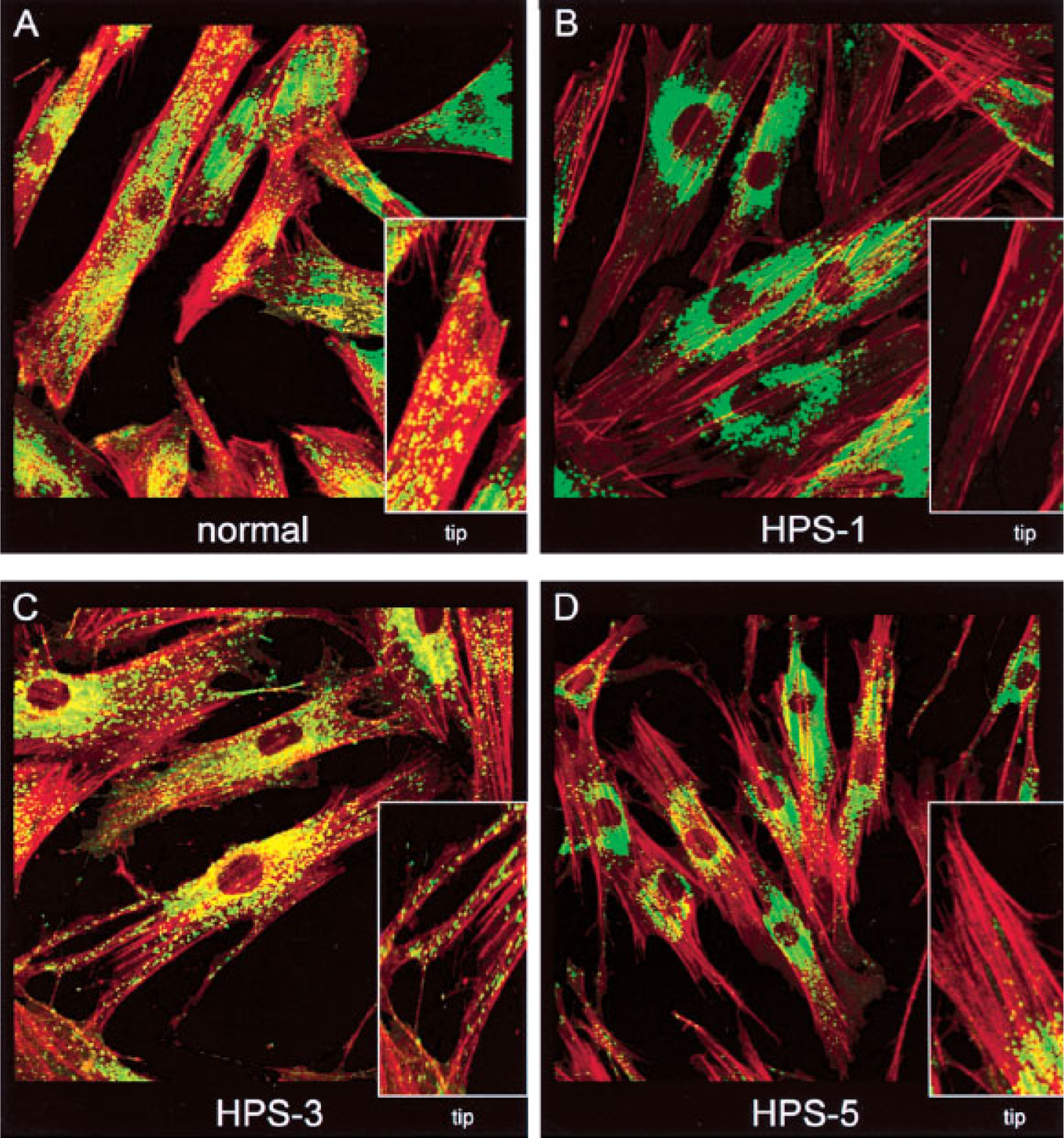
Intracellular distribution of LAMP3 and F-actin in normal, Hermansky-Pudlak syndrome (HPS)-1, HPS-3, and HPS-5 fibroblasts. Fibroblasts were fixed on coverslips and stained with mouse monoclonal antibodies to LAMP3 (as described in Huizing et al41) and F-actin (Phalloidin, Molecular Probes, Eugene, OR). F-actin staining (red) was employed to mark the outline of the cells, in particular the dendritic tips. Insets show isolated dendritic tips. (A) Normal fibroblasts show a punctate LAMP3 (green) distribution throughout the cell, including at the ends of the dendritic tips. (B) HPS-1 fibroblasts show a clumped accumulation of LAMP3 in the perinuclear area. (C) HPS-3 fibroblasts appear to have a nearly normal LAMP3 distribution, with LAMP3 reaching into the dendritic tips. (D) HPS-5 cells have minimal LAMP3 transport into the tips, but LAMP3 does not accumulate in clumps in the perinuclear area, as in HPS-1. (Photographs produced by Heidi Dorward.)
The HPS7 protein is a homologue of the murine sandy gene.45 The sandy protein is part of BLOC-1, which also contains the proteins coded for by the pallid, muted, and cappuccino murine HPS genes.45,56,57 HPS7, the only component of BLOC-1 to cause HPS in humans, is also called dysbindin, a β-dystrobrevin-binding protein. Dystrobrevins are components of the dystrophin-associated protein complex (DPC) in both muscle and nonmuscle cells.58 The specific functions of HPS7 in BLOC-1 and of the DPC in fibroblasts, melanocytes, and platelets remain to be determined.
CHEDIAK-HIGASHI SYNDROME
CHS is a rare autosomal recessive disorder characterized by defective platelet dense granules, oculocutaneous albinism, immune deficiency, and progressive neurological dysfunction.59,60 Many cell types in CHS patients contain giant cytoplasmic inclusions, which are enlarged vesicles.59
Clinical Findings
Most CHS patients present in early childhood with severe bacterial infections and succumb to an accelerated phase characterized by hemophagocytic lymphohistiocytosis due to uncontrolled T-cell activation.59 Approximately 10 to 15% of patients exhibit a milder immune phenotype, survive into adulthood, and develop a progressive neurological syndrome in the third to fifth decades, manifesting ataxia, gait problems, weakness and sensory deficits.59,60 Rarely, patients who present in childhood with severe infections and a moderate pigment defect escape progression to the accelerated phase.60 The beige mouse is the murine counterpart of CHS patients.
In CHS, platelet counts are normal prior to development of the accelerated phase.59,61 However, a dense-granule defect causes bleeding and a prolonged bleeding time. CHS patients have an impaired secondary wave of platelet aggregation, with an increased ATP-to-ADP ratio, decreased platelet serotonin, and decreased platelet calcium.62 Some humans63 and mink64 with CHS have a normal number and shape of dense bodies, but other reports indicate absent or markedly reduced dense bodies in CHS platelets from humans65 as well as mice66 and cattle.67 Beige mice showed mildly decreased mepacrine-containing granules.
CHS patients can manifest complete oculocutaneous albinism or apparently normal pigmentation.59 Hair, eyebrows, and eyelashes may have a silvery-gray metallic sheen. Pigment in the hair shaft is distributed in homogeneous small clumps.59,60
CHS patients have defective cytotoxic T-cell and natural killer-cell function, decreased chemotaxis of macrophages and polymorphonuclear leukocytes, and delayed intracellular killing.59 Antigen presentation, which requires appropriate vesicular trafficking, is also defective. Phagocytic activity is normal but fusion of phagosomes with lysosomes is delayed.
Defective cytotoxic T-cell function results in an inability to turn off activated lymphocytes, especially after Epstein-Barr virus infection.59 As a result, most patients develop an accelerated phase of nonmalignant lymphohistiocytic infiltration, cytokine overproduction, and multiorgan damage. Response to chemotherapy is poor. Bone marrow transplantation cures the immune deficiency and related lymphohistiocytosis,59 but does not prevent the progressive neurological defect.
The LYST Gene and Protein Function
CHS results from mutations in the lysosomal trafficking regulator (LYST) gene located on chromosome 1q42.1–42.2, which encodes a large cytoplasmic protein of 3801 amino acids (425 kd) and has unknown function.68 In some patients, no LYST mutations could be identified, suggesting the existence of other CHS-causing genes.60
Several proteins interact with LYST, including the SNARE complex protein HRS, signaling protein 14-3-3, and casein kinase II.69 LYST may act as an adaptor that brings into close proximity proteins that mediate intracellular membrane fusion reactions. LYST contains a beige and Chediak domain (BEACH), a series of WD-40 repeats, ARM motifs, and HEAT repeats.29 These motifs play roles in vesicle transport and fusion.
GRISCELLI SYNDROME
GS is a rare autosomal recessive disorder characterized by abnormal pigmentation, immunodeficiency, and development of the accelerated phase.70 GS is caused by mutations in the RAB27A gene, a small GTPase involved in vesicular transport and organelle dynamics.71 GS patients have no obvious bleeding prior to the accelerated phase, and whether they have a defect in platelet dense granules has not been verified. The ashen mouse, a model of GS with mutated RAB27A, exhibits a reduced number of platelet dense granules,72 although this finding varied depending on the genetic background.73 GS patients have hypopigmented hair with a silvery-gray sheen and pigment clumps that are larger and less homogeneously distributed than in CHS.70 GS cells also lack the giant cytoplasmic granules of CHS. In GS, defective granule exocytosis of T cells results in susceptibility to viral infections and eventually an accelerated phase, similar to CHS.
THROMBOCYTOPENIA ABSENT RADII SYNDROME
TAR is a developmental disorder characterized by thrombocytopenia and bilateral absence of the radii in the presence of thumbs.74 Platelet counts are extremely low (15 to 30 × 109/L) in infancy, but increase with age and may improve to almost normal by adulthood. The thrombocytopenia becomes symptomatic within the first 4 months of life in more than 90% of cases.74 Some patients have cardiac and genitourinary involvement. TAR could be either autosomal recessive or dominant.
In one TAR patient, the secondary wave of platelet aggregation in response to ADP and epinephrine was absent, and aggregation in response to collagen was poor.75
WISKOTT-ALDRICH SYNDROME
Wiskott-Aldrich syndrome (WAS) is an X-linked recessive disease of microthrombocytopenia, immunodeficiency, and eczema.76 X-linked thrombocytopenia (XLT) a milder phenotype without immune deficiency, and WAS are both caused by mutations in the WASP gene.77,78 The WASp protein is exclusively expressed in the cytoplasm of hematopoietic cells. It regulates signal-mediated actin cytoskeleton rearrangement by interacting with phosphatidylinositol (4,5)-bisphosphate (PIP2) and Rho-like GTPases (specifically, CDC42) and by stimulating the ARP2/3 actin-nucleating complex.79
Bleeding manifestations in WAS range from minor petechiae to serious gastrointestinal or intracranial hemorrhage. Among 154 WAS patients in one study, 30% had life-threatening bleeding before diagnosis.76 The number of serious bleeding events was similar in all platelet count subgroups, suggesting a qualitative platelet defect.
Several WAS patients have a marked reduction in dense granules, adenine nucleotide storage pools, and platelet aggregation.80–82 A reduction in α-granules and mitochondria has also been reported in WAS platelets.81 Patients with XLT also showed a reduced number of dense granules, a-granules and mitochondria.82 Some carrier mothers have a poor secondary wave of aggregation in response to epinephrine.80
CONCLUSION
As occurs repeatedly in medicine, investigation of a disease state leads to elucidation of a normal function that was previously a mystery. For the collection of disorders involving defective δ-granules, that function consists of intracellular vesicle formation from extant membranes. The analysis of this function, however, remains in its infancy, in part because a molecular etiology has been discovered for only a fraction of the pertinent diseases. In the future, our understanding of vesicle formation will emanate from knowledge of the genes and proteins involved in a gamut of δ-storage pool disorders, only a portion of which are currently recognized.
Already, δ-SPD states have taught us a good deal about the cell biology of vesicle formation and trafficking. The HPS diseases tell us that melanosomes in melanocytes and dense bodies in platelets share some elements of their biogenesis. These diseases also reveal that proper intracellular vesicle formation is required for regulation of localized inflammatory responses; when disturbed, the result is granulomatous colitis or pulmonary fibrosis. HPS-2 disease has also emphasized the importance of the β3A subunit of AP3 in the formation of neutrophils via a specific protein, neutrophil elastase.83 Studies of CHS have uncovered the function of intracellular vesicles in fighting bacterial infections, and investigations into GS indicate the importance of the distal movement of vesicles within cells in controlling T cell activation.84 With the exception of the role of AP3 in vesicle formation, however, we do not know how the specific molecular defects of δ-SPDs cause their pathology.
Another issue also remains incompletely understood. If HPS genes are ubiquitously expressed, why is the phenotype limited to pigment dilution, bleeding, and sometimes pulmonary and gastrointestinal involvement? One possible explanation is that the formation of melanosomes and dense bodies requires a single, specific pathway, whereas vesicle formation in more generic cells employs redundant mechanisms.
The future remains filled with potential for investigations into δ-SPDs. In all likelihood, subsets of lysosomes will be described based on markers that define them as counterparts of dense bodies or melanosomes, and the behavior of these subsets will be chronicled. Megakaryocytes will be cultured from patients with all varieties of δ-SPD, and the genesis of dense bodies, as well as platelets themselves, will be followed. Immuno-cytochemistry and videoimaging will be applied to the study of normal and mutant megakaryocytes in culture. Given that scores of different proteins work together to accomplish the complex task of new vesicle formation, new diseases involving defects in these proteins will undoubtedly be discovered, and the causative genes will be numbered among those required for vesicle formation in general. Dysfunction will reveal function.
ACKNOWLEDGMENTS
We appreciate the advice and assistance of Dr. Robert Kleta. Dr. James G. White provided the platelet electron micrographs, and Heidi Dorward provided the cell photographs.
REFERENCES
- 1.Israels SJ, McNicol A, Robertson C, Gerrard GM. Platelet storage pool deficiency: diagnosis in patients with prolonged bleeding times and normal platelet aggregation. Br J Haematol 1990;75:118–121 [DOI] [PubMed] [Google Scholar]
- 2.Witkop CJ, Krumwiede M, Sedano H, White JG. Reliability of absent platelet dense bodies as a diagnostic criterion for Hermansky-Pudlak syndrome. Am J Hematol 1987;26:305–311 [DOI] [PubMed] [Google Scholar]
- 3.McNicol A, Israels S. Platelet dense granules: structure, function and implications for haemostasis. Thromb Res 1999; 95:1–18 [DOI] [PubMed] [Google Scholar]
- 4.Holmsen H, Weiss HJ. Secretable storage pools in platelets. Annu Rev Med 1979;30:119–134 [DOI] [PubMed] [Google Scholar]
- 5.Nishibori M, Cham B, McNicol A, et al. The protein CD63 in platelet dense granules is deficient in a patient with Hermansky-Pudlak syndrome, and appears identical to granulophysin. J Clin Invest 1993;91:1775–1782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Israels SJ, McMillan EM, Robertson C, Singhory S, McNicol A. The lysosomal granule membrane protein, LAMP-2, is also present in platelet dense granule membranes. Thromb Haemost 1996;75:623–629 [PubMed] [Google Scholar]
- 7.Polgar J, Lane WS, Chung SH, Houng AK, Reed GL. Phosphorylation of SNAP-23 in activated human platelets. J Biol Chem 2003;278:44369–44376 [DOI] [PubMed] [Google Scholar]
- 8.Morimoto T, Ogihara S. ATP is required in platelet serotonin exocytosis for protein phosphorylation and priming of secretory vesicles docked on the plasma membrane. J Cell Sci 1996;109:113–118 [DOI] [PubMed] [Google Scholar]
- 9.Jones OP. Origin of megakaryocyte granules from Golgi vesicles. Anat Rec 1960;138:105–114 [DOI] [PubMed] [Google Scholar]
- 10.Youssefian T, Cramer EM. Megakaryocyte dense granule components are sorted in multivesicular bodies. Blood 2000; 95:4004–4007 [PubMed] [Google Scholar]
- 11.Heijnen HFG, Debili N, Vainchencker W, et al. Multivesicular bodies are an intermediate stage in the formation of platelet α-granules. Blood 1998;91:2313–2325 [PubMed] [Google Scholar]
- 12.King SM, Reed GL. Development of platelet secretory granules. Semin Cell Dev Biol 2002;13:293–302 [DOI] [PubMed] [Google Scholar]
- 13.Weiss HJ, Witte LD, Kaplan KL, et al. Heterogeneity in storage pool deficiency: studies on granule-bound substances in 18 patients including variants deficient in alpha-granules, platelet factor 4, beta-thromboglobulin and platelet-derived growth factor. Blood 1979;54:1296–1319 [PubMed] [Google Scholar]
- 14.Nieuwenhuis K, Akkerman JWN, Sixma JJ. Patients with a prolonged bleeding time and normal aggregation tests may have storage pool deficiency:studies on one hundred six patients. Blood 1987;70:620–623 [PubMed] [Google Scholar]
- 15.Holmsen H, Weiss HJ. Further evidence for a deficient storage pool of adenine nucleotides in platelets from some patients with thrombopathia-“Storage pool deficiency”. Blood 1972;39:197–209 [PubMed] [Google Scholar]
- 16.Raccuglia G Gray platelet syndrome, a variety of qualitative platelet disorder. Am J Med 1971;51:818–828 [DOI] [PubMed] [Google Scholar]
- 17.Pujol-Moix N, Hernandez A, Escolar G, et al. Platelet ultrastructural morphology for diagnosis of partial δ-storage pool disease in patients with mild platelet dysfunction and/or thrombocytopenia of unknown origin. A study of 24 cases. Haematologica 2000;85:619–626 [PubMed] [Google Scholar]
- 18.White JG. Inherited abnormalities of the platelet membrane and secretory granules. Hum Pathol 1987;18:123–139 [DOI] [PubMed] [Google Scholar]
- 19.Weiss HJ, Lages B, Vicic W, Tsung LJ, White JG. Heterogeneous abnormalities of platelet dense granule ultrastructure in 20 patients with congenital storage pool deficiency. Br J Haematol 1993;83:282–295 [DOI] [PubMed] [Google Scholar]
- 20.Lorez HP, DaPrada M, Rendu F, Pletscher A. Mepacrine, a tool for investigating the 5-hydroxytryptainineorganells of blood platelets by fluorescence microscopy. J Clin Med 1977; 89:200–206 [PubMed] [Google Scholar]
- 21.Gerrard JM, Israels ED, Bishop AJ, et al. Inherited platelet-storage pool deficiency associated with a high incidence of acute myeloid leukemia. Br J Haematol 1991;79:246–255 [DOI] [PubMed] [Google Scholar]
- 22.Nieuwenhuis HK, Sixma JJ. 1-Desamino-8-D-arginine vasopressin (Desmopressin) shortens the bleeding time in storage pool deficiency. Ann Intern Med 1988;108: 65–67 [DOI] [PubMed] [Google Scholar]
- 23.Biddle DA, Neto TG, Nguyen ND. Platelet storage pool deficiency of α and δ granules. Arch Pathol Lab Med 2001;125:1125–1126 [DOI] [PubMed] [Google Scholar]
- 24.Detter J, Zhang Q, Mules E, et al. Rab geranylgeranyl transferase alpha mutation in the gunmetal mouse reduces Rab prenylation and platelet synthesis. Proc Natl Acad Sci USA 2000;97:4144–4149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hermansky F, Pudlak P. Albinism associated with hemorrhagic diathesis and unusual pigmented reticular cells in the bone marrow: Report of two cases with histochemical studies. Blood 1959;14:162–169 [PubMed] [Google Scholar]
- 26.Oh J, Bailin T, Fukai K, et al. Positional cloning of a gene for Hermanksky-Pudlak syndrome, a disorder of cytoplasmic organelles. Nat Genet 1996;14:300–306 [DOI] [PubMed] [Google Scholar]
- 27.Gahl WA, Brantly M, Kaiser-Kupfer MI, et al. Genetic defects and clinical characteristics of patients with a form of oculocutaneous albinism (Hermansky-Pudlak syndrome). N Engl J Med 1998;338:1258–1264 [DOI] [PubMed] [Google Scholar]
- 28.Huizing M, Anikster Y, Gahl WA. Hermansky-Pudlak syndrome and related disorders of organelle formation. Traffic 2000;1:823–835 [DOI] [PubMed] [Google Scholar]
- 29.Huizing M, Anikster Y, Gahl WA. Hermansky-Pudlak syndrome and Chediak-Higashi syndrome: Disorders of vesicle formation and trafficking. Thromb Haemost 2001; 86:233–245 [PubMed] [Google Scholar]
- 30.Gahl WA. Hermansky-Pudlak Syndrome. In: Gene Reviews at Gene Tests: Medical Genetics Information Resource (database online). Copyright, University of Washington, Seattle. 1977–2003. Available at: www.genetests.org. Accessed January 2003 [Google Scholar]
- 31.King RA, Hearing VJ, Creel DJ, Oetting WS. Albinism. In: Scriver CR, Beaudet AL, Sly WS, Valle DL, eds. The Metabolic and Molecular Bases of Inherited Disease. 8th ed, Vol 4. New York: McGraw-Hill; 1995:5587–5628 [Google Scholar]
- 32.Iwata F, Reed GF, Caruso RC, et al. Correlation of visual acuity and ocular pigmentation with the 16-bp duplication in the HPS-1 gene of Hermansky-Pudlak syndrome, a form of albinism. Ophthalmology 2000;107:783–789 [DOI] [PubMed] [Google Scholar]
- 33.Brantly M, Avila NA, Shotelersuk V, et al. Pulmonary function and high-resolution CT findings in patients with an inherited form of pulmonary fibrosis, Hermansky-Pudlak syndrome due to mutations in HPS-1. Chest 2000;117:129–136 [DOI] [PubMed] [Google Scholar]
- 34.Harmon KR, Witkop CJ, White JG, et al. Pathogenesis of pulmonary fibrosis: platelet derived growth factor precedes structural alterations in the Hermansky-Pudlak syndrome. J Lab Clin Med 1994;123:617–627 [PubMed] [Google Scholar]
- 35.Schinella RA, Greco MA, Cobert BL, Denmark LW, Cox RP. Hermansky-Pudlak syndrome with granulomatous colitis. Ann Intern Med 1980;92:20–23 [DOI] [PubMed] [Google Scholar]
- 36.Toro J, Turner M, Gahl WA. Dermatologic manifestations of Hermansky-Pudlak syndrome in patients with and without a 16-base pair duplication in the HPS-1 gene. Arch Dermatol 1999;135:774–780 [DOI] [PubMed] [Google Scholar]
- 37.Gahl WA, Brantly M, Troendle J, et al. Effect of pirfenidone on the pulmonary fibrosis of Hermansky-Pudlak syndrome. Mol Genet Metab 2002;76:234–242 [DOI] [PubMed] [Google Scholar]
- 38.Shotelersuk V, Dell’Angelica EC, Hartnell L, Bonifacino JS, Gahl WA. A new variant of Hermansky-Pudlak syndrome due to mutations in a gene responsible for vesicle formation. Am J Med 2000;108:423–427 [DOI] [PubMed] [Google Scholar]
- 39.Huizing M, Scher CD, Strovel E, et al. Nonsense mutations in ADTB3A cause complete deficiency of the β3A subunit of adaptor complex-3 and severe Hermansky-Pudlak syndrome Type 2. Pediatr Res 2002;51:150–158 [DOI] [PubMed] [Google Scholar]
- 40.Clark RH, Stinchcombe JC, Day A, et al. Adaptor protein 3-dependent microtubule-mediated movement of lytic granules to the immunological synapse. Nat Immuno 2003;4:1111–1120 [DOI] [PubMed] [Google Scholar]
- 41.Anikster Y, Huizing M, White J, et al. Mutation of a new gene causes a unique form of Hermansky-Pudlak syndrome in a genetic isolate of central Puerto Rico. Nat Genet 2001;28: 376–380 [DOI] [PubMed] [Google Scholar]
- 42.Suzuki T, Li W, Zhang Q, et al. Hermansky-Pudlak syndrome is caused by mutations in HPS4, the human homologue of the mouse light ear gene. Nat Genet 2002;30:321–324 [DOI] [PubMed] [Google Scholar]
- 43.Anderson PD, Huizing M, Claassen DA, White J, Gahl WA. Hermansky-Pudlak syndrome type 4 (HPS-4): clinical and molecular characteristics. Hum Genet 2003;113:10–17 [DOI] [PubMed] [Google Scholar]
- 44.Zhang Q, Zhao B, Li W, et al. Ru2 and Ru encode mouse orthologs of the genes mutated in human Hermansky-Pudlak syndrome types 5 and 6. Nat Genet 2003;33:145–153 [DOI] [PubMed] [Google Scholar]
- 45.Li W, Zhang Q, Oiso N, et al. Hermansky-Pudlak syndrome type 7 (HPS-7) results from mutant dysbindin, a member of the biogenesis of lysosome-related organelles complex 1 (BLOC-1). Nat Genet 2003;35:84–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hermos CR, Huizing M, Kaiser-Kupfer MI, Gahl WA. Hermansky-Pudlak syndrome type 1: gene organization, new mutations, and clinical/molecular review of non-Puerto Rican cases. Hum Mutat 2002;20:482. [DOI] [PubMed] [Google Scholar]
- 47.Dell’Angelica EC, Shotelersuk V, Aguilar RC, Gahl WA, Bonifacino JS. Altered trafficking of lysosomal proteins in Hermansky-Pudlak syndrome due to mutations in the β3A subunit of the AP-3 adaptor. Mol Cell 1999;3:11–21 [DOI] [PubMed] [Google Scholar]
- 48.Huizing M, Gahl WA. Disorders of vesicles of lysosomal lineage: the Hermansky-Pudlak syndromes. Curr Mol Med 2002;2:451–467 [DOI] [PubMed] [Google Scholar]
- 49.Huizing M, Anikster Y, Fitzpatrick DL, et al. Hermansky-Pudlak syndrome type 3 in Ashkenazi Jews and other non-Puerto Rican patients with hypopigmentation and platelet storage pool deficiency. Am J Hum Genet 2001;69:1022–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nazarian R, Falcon-Perez JM, Dell’Angelica EC. Biogenesis of lysosome-related organelles complex 3 (BLOC-3): a complex containing the Hermansky-Pudlak syndrome (HPS) proteins HPS1 and HPS4. Proc Natl Acad Sci USA 2003; 100:8770–8775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huizing M, Sarangarajan R, Strovel E, et al. AP-3 mediates tyrosinase but not TRP-1 trafficking in human melanocytes. Mol Biol Cell 2001;12:2075–2085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Israels SJ, Gerrard JM, Jacques YV, et al. Platelet dense granule membranes contain both granulophysin and p-selectin (GM-140). Blood 1992;80:143–152 [PubMed] [Google Scholar]
- 53.Dell’Angelica EC, Aguilar RC, Wolins N, et al. Molecular characterization of the protein encoded by the Hermansky-Pudlak syndrome type 1 gene. J Biol Chem 2000;275:1300–1306 [DOI] [PubMed] [Google Scholar]
- 54.Boissy RE, Zhao Y, Gahl WA. Altered protein localization in melanocytes from Hermansky-Pudlak syndrome: support for the role of the HPS gene product in intracellular trafficking. Lab Invest 1998;78:1037–1048 [PubMed] [Google Scholar]
- 55.Sarangarajan R, Budev A, Zhao Y, Gahl WA, Boissy RE. Abnormal translocation of tyrosinase and tyrosinase-related protein 1 in cutaneous melanocytes of Hermansky-Pudlak Syndrome and in melanoma cells transfected with anti-sense HPS1 cDNA. J Invest Dermatol 2001;117:641–646 [DOI] [PubMed] [Google Scholar]
- 56.Ciciotte SL, Gwynn B, Moriyama K, et al. Cappuccino, a mouse model of Hermansky-Pudlak syndrome, encodes a novel protein that is part of the pallidin-muted complex (BLOC-1). Blood 2003;101:4402–4407 [DOI] [PubMed] [Google Scholar]
- 57.Falcon-Perez JM, Starcevic M, Gautam R, Dell’Angelica EC. BLOC-1, a novel complex containing the pallidin and muted proteins involved in the biogenesis of melanosomes and platelet-dense granules. J Biol Chem 2002;277:28191–2819 [DOI] [PubMed] [Google Scholar]
- 58.Benson MA, Newey SE, Martin-Rendon E, Hawkes R, Blake DJ. Dysbindin, a novel coiled-coil-containing protein that interacts with the dystrobrevins in muscle and brain. J Biol Chem 2001;276:24232–24241 [DOI] [PubMed] [Google Scholar]
- 59.Introne W, Boissy RE, Gahl WA. Clinical, molecular and cell biological aspects of Chediak-Higashi Syndrome. Mol Genet Metab 1999;68:283–303 [DOI] [PubMed] [Google Scholar]
- 60.Karim MA, Suzuki K, Fukai K, et al. Apparent genotype-phenotype correlation in childhood, adolescent, and adult Chediak-Higashi syndrome. Am J Med Genet 2002;108: 16–22 [PubMed] [Google Scholar]
- 61.Apitz-Castro R, Cruz MR, Ledezma E, et al. The storage pool deficiency in platelets from humans with the Chediak-Higashi syndrome: study of six patients. Br J Haematol 1985;59:471–483 [DOI] [PubMed] [Google Scholar]
- 62.Buchanan GR, Handin RI. Platelet function in the Chediak-Higashi syndrome. Blood 1976;47:941–948 [PubMed] [Google Scholar]
- 63.Boxer GJ, Holmsen H, Robkin L, et al. Abnormal platelet function in Chediak-Higashi Syndrome. Br J Haematol 1977; 35:521–533 [DOI] [PubMed] [Google Scholar]
- 64.Davis WC, Spicer SS, Greene WB, Padgett GA. Ultrastructure of cells in bone marrow and peripheral blood of normal mink and mink with the homologue of the Chediak-Higashi trait in humans. Am J Pathol 1971;63:411–424 [PMC free article] [PubMed] [Google Scholar]
- 65.Rendu F, Breton-Gorius J, Lebret M, ei al. Evidence that abnormal platelet function in human Chediak-Higashi Syndrome is the result of a lack of dense bodies. Am J Pathol 1983;111:307–314 [PMC free article] [PubMed] [Google Scholar]
- 66.Lorez HP, Da Prada M. Fluorescence microscopical study of 5-hydroxytryptamine storage organelles in mepacrine-incubated blood platelets of beige mice (Chediak-Higashi syndrome). Experientia 1978;34:663–664 [DOI] [PubMed] [Google Scholar]
- 67.Menard M, Meyers KM. Storage pool deficiency in cattle with the Chediak-Higashi syndrome result from an absence of dense granule precursors in their megakaryocytes. Blood 1988;72:1726–1734 [PubMed] [Google Scholar]
- 68.Nagle DL, Karim MA, Woolf EA, et al. Identification and mutation analysis of the complete gene for Chediak-Higashi syndrome. Nat Genet 1996;14:307–311 [DOI] [PubMed] [Google Scholar]
- 69.Tchernev VT, Mansfield TA, Giot L, et al. The Chediak-Higashi protein interacts with SNARE complex and signal transduction proteins. Mol Med 2002;8:56–64 [PMC free article] [PubMed] [Google Scholar]
- 70.Sanal O, Ersoy F, Tezcan I, et al. Griscelli Disease: Genotype-phenotype correlation in an array of clinical heterogeneity. J Clin Immunol 2002;22:237–243 [DOI] [PubMed] [Google Scholar]
- 71.Menasche G, Pastural E, Feldmann J, et al. Mutations in RAB27A cause Griscelli syndrome associated with haemophagocytic syndrome. Nat Genet 2000;25:173–176 [DOI] [PubMed] [Google Scholar]
- 72.Wilson SM, Yip R, Swing DA, et al. A mutation in Rab27a causes the vesicle transport defects observed in ashen mice. Proc Natl Acad Sci USA 2000;97:7933–7938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Novak EK, Gautam R, Reddington M, et al. The regulation of platelet-dense granules by Rab27a in the ashen mouse, a model of Hermansky-Pudlak and Griscelli syndromes, is granule specific and dependent on genetic background. Blood 2002;100:128–135 [DOI] [PubMed] [Google Scholar]
- 74.Greenhalgh KL, Howell RT, Bottani A, et al. Thrombocytopenia-absent radius syndrome: a clinical genetic study. J Med Genet 2002;39:876–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Day JH, Holmsen H. Platelet adenine nucleotide “Storage pool deficiency” in thrombocytopenic absent radii syndrome. JAMA 1972;221:1053–1054 [PubMed] [Google Scholar]
- 76.Sullivan KE, Mullen CA, Blaese RM, Winkelstein JA. A multiinstitutional survey of the Wiskott-Aldrich syndrome. J Pediatr 1994;125:876–885 [DOI] [PubMed] [Google Scholar]
- 77.Derry J, Ochs HD, Francke U. Isolation of a novel gene mutated in Wiskott-Aldrich syndrome. Cell 1994;78:635–644 [DOI] [PubMed] [Google Scholar]
- 78.Villa A, Notarangelo L, Macchi P, et al. X-linked thrombocytopenia and Wiskott-Aldrich syndrome are allelic diseases with mutations in the WASP gene. Nat Genet 1995;9:414–417 [DOI] [PubMed] [Google Scholar]
- 79.Badour K, Zhang J, Siminovitch KA. The Wiskott-Aldrich syndrome protein: forging the link between actin and cell activation. Immunol Rev 2003;192:98–112 [DOI] [PubMed] [Google Scholar]
- 80.Baldini MG. The nature of the platelet defect in the Wiskott-Aldrich syndrome. Ann N Y Acad Sci 1972;201: 437–444 [DOI] [PubMed] [Google Scholar]
- 81.Grottum KA, Hovig T, Holmsen H, et al. Wiskott-Aldrich syndrome: qualitative platelet defects and short platelet survival. Br J Haematol 1969;17:373–388 [DOI] [PubMed] [Google Scholar]
- 82.Stormorken H, Hellum B, Egeland T, Abrahamsen TG, Hovig T. X-linked thrombocytopenia and thrombocyte-pathia: attenuated Wiscott-Aldrich Syndrome. Thromb Haemost 1991;65:300–305 [PubMed] [Google Scholar]
- 83.Benson KF, Li FQ, Person RE, et al. Mutations associated with neutropenia in dogs and humans disrupt intracellular transport of neutrophil elastase. Nat Genet 2003;35:90–96 [DOI] [PubMed] [Google Scholar]
- 84.Trambas CM, Griffiths GM. Delivering the kiss of death. Nat Immunol 2003;4:399–403 [DOI] [PubMed] [Google Scholar]