

Abstract
Geldanamycin (GDM) has been modified by different type neutral/acidic/basic substituents (1–7) and by quinuclidine motif (8), transformed into ammonium salts (9–13) at C(17). These compounds have been characterised by spectroscopic and x-ray methods. Derivative 8 shows better potency than GDM in MCF-7, MDA-MB-231, A549 and HeLa (IC50s = 0.09–1.06 µM). Transformation of 8 into salts 9–13 reduces toxicity (by 11-fold) at attractive potency, e.g. MCF-7 cell line (IC50∼2 µM). Our studies show that higher water solubility contributes to lower toxicity of salts than GDM in healthy CCD39Lu and HDF cells. The use of 13 mixtures with potentiators PEI and DOX enhanced anticancer effects from IC50∼2 µM to IC50∼0.5 µM in SKBR-3, SKOV-3, and PC-3 cancer cells, relative to 13. Docking studies showed that complexes between quinuclidine-bearing 8–13 and Hsp90 are stabilised by extra hydrophobic interactions between the C(17)-arms and K58 or Y61 of Hsp90.
Keywords: Ansamycins, benzoquinones, chaperone Hsp90, anticancer, SAR
Introduction
Geldanamycin (GDM, Figure 1), a natural ansa-macrolide produced by Streptomyces hygroscopicus, has two unique structural features, namely a rigid benzoquinone ring and an aliphatic ansa-bridge that are linked together to form a characteristic basket-like structure.1 Similar to other ansa-macrolides with benzene or benzoquinone core, it shows potent activity in different cancer cell lines.2–4 Unfortunately, the significant toxicity of GDM impedes its future medical applications. This toxicity is believed to originate from 1,4-Michael conjugate addition/aromatisation/oxidation cascade with glutathione, yielding a C(19)-adduct.5–7 In search for more active and less toxic benzoquinone and non-benzoquinone ansamycins as well as those with an expanded macrocyclic system, GDM was modified at the C(17), C(19) positions and at the methoxy, urethane or hydroxyl groups.2,8–10 The most promising candidates among the C(17)-derivatives of GDM are amine derivatives 17-DMAG and 17-AAG that are currently under consideration or are in various phases of clinical trials.2,11–13 Non-quinone analogues of GDM (reblastatin analogues, Bioteca, Cambridge, UK) or those containing halogens, saccharides, phenol groups at the core, were obtained by sequences of mutasynthetic, semi-synthetic, and total synthetic approaches. These analogues showed attractive activities against the cancer cell lines, such as human breast adenocarcinoma (SKBR-3, MCF-7, and MDA-MB-231), ovarian adenocarcinoma (SKOV-3), lung adenocarcinoma (A-549), and prostate adenocarcinoma (PC-3).2,4,14–20 Moreover, modifications at the ansa-macrolide correlated to the preparation of conjugates with biotin, foliate, or within corporated triazole bridge, that gained molecular probe features of improved biocompatibility.21–24 Overall, the ansa-bridge modifications of GDM led to a less effective anticancer potency than those performed at the rigid benzene or benzoquinone cores.25–33 This can be attributed to a restriction of the ansa-bridge flexibility that is necessary for binding the GDM analogues to their molecular target, i.e. heat shock proteins Hsp90. Hsp90 shows an ATPase activity, which is required to fold proteins in cells in order to achieve their full functionality.2,34–36 Crystal structures of GDM and 17-DMAG complexed with NBD of Hsp90 showed that the ligand conformation significantly differs from that of unbound molecule in solutions.27,28 Interestingly, modifications of GDM at C(17) and C(19) positions sometimes resulted in antiviral potency against herpes (HSV), hepatitis B and C (HCV, HBV) or HIV-1.37–39 Additionally, for GDM amine analogues where the C(17) substituent was bridged with the neighbouring quinone group at C(18), despite their good water solubility (e.g. guanidine-like derivatives), it was noticed that the bulkiness of the incorporated alkyl or aryl substituent was important for anticancer potency.40 Ge et al. obtained reduced (hydroquinone) and highly water-soluble GDM derivatives with allylamine or N,N-dimethylethylamine in the form of salts that are considered in cancer therapy in phase I clinical trials.41
Figure 1.
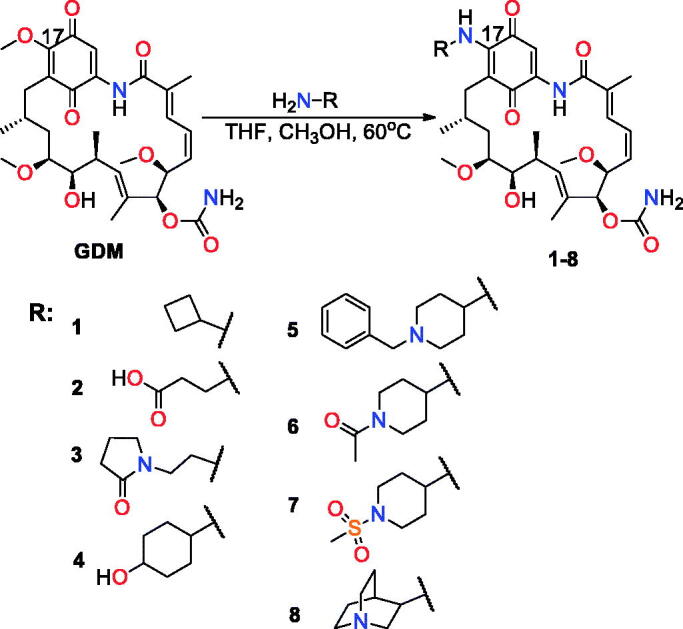
Geldanamycin (GDM) and its C(17)- analogues.
Recently, we have reported synthesis and anticancer activity of a series of C(17)-GDM derivatives and we found that rigidity of the C(17)-arm incorporating carbonyl group and lipophilicity are essential factors influencing an efficient binding of GDM analogues to Hsp90 and their anticancer potency.7 Here, we report the anticancer activity studies of the GDM amine-quinuclidine analogue 8 with C(17)-arm of basic, rigid, and bulky nature. The data have been determined for nine cancerous (MDA-MB-231, MCF-7, HeLa, HepG2, SKBR-3, SKOV-3, PC-3, U-87, and A-549) and two healthy (human lung fibroblasts CCD39Lu, human dermal fibroblasts HDF) cell lines. The activities of 8 are compared with the GDM derivatives 1–7 of acidic, basic, and neutral nature (Figure 1)7,23,40. Taking into account the fact that improved water solubility may lead to better bioavailability, derivative 8 was transformed into quaternary N-alkylammonium salts 9–13 of different N-tail structure (Figure 2). Moreover, GDM quinuclidine salts were also studied in mixtures with potentiators towards their anticancer potency and in order to obtain information about their toxicity.
Figure 2.
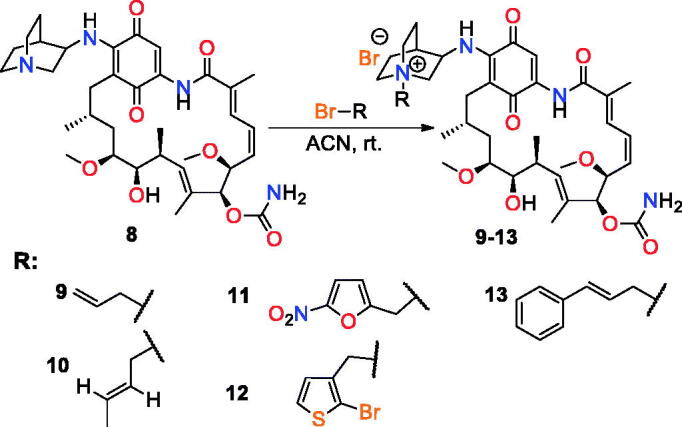
Quaternisation of the quinuclidine N atom within 8 leading to analogues 9–13.
Results and discussion
Synthesis of GDM analogues
GDM derivatives 1–8 were obtained according to the earlier reported protocol7 and their structures were confirmed by 1H, 13 C NMR, and 2D NMR, FT-IR, and ESI MS methods (Supplementary Material). Derivatives 1–3 formed single crystals that allowed to determine their structures by X-ray analysis (Table 1S, Figures 53S–59S, Supplemental Material). Comparison of the 1H and 13 C NMR spectra of 8 and its N-alkylammonium salts (9–13) with those of 1–7 showed only minor differences in the position of the proton and the carbon atom resonances assigned to the quinone and the ansa-bridge parts (Figure 60S; Supplemental Material). The NOESY contacts for GDM analogues indicated trans-configuration of the lactam group with the lactam proton directed into the macrocyclic cavity, as shown in Figure 60S. As found for 8 and 9–13 salts, the absolute value of the H(29)-C(29)-N(17)-H(17) torsion angle is in the range of 120–130° (3JN(17)H-H(29)∼6.5–8 Hz,) and the N-alkyl substituent of nitrogen N + (30) is oriented away from the quinone core (Figure 60S). The nitrogen atom N(17), linking the GDM and quinuclidine parts, seems to be present in a distorted sp2 hybridisation, as suggested by DFT calculations (Figure 60S). The above data indicated similar conformations of GDM derivatives in its “free form” in solution and solid, with only small changes in orientation of the C(17)-amine part relative to the quinone core.
Anticancer potency and toxicity in normal cells of GDM analogues
Quinuclidine derivative 8 and its quaternary N-alkylammonium salts (9–13) were initially tested in MDA-MB-231, MCF-7, HeLa, and HepG2 and healthy CCD39LU cell lines, and compared with biological test results of typical GDM C(17)-derivatives (1–7) (Table 1). Although GDM belongs to the known and active anticancer agents, its potency against breast cancer cells (MCF-7) is the lowest among the studied analogues (Table 1, IC50 = 3.51 µM). A comparison of GDM activity and its analogues in the MCF-7 indicates that derivatives 1, 3, and 8 are markedly more potent than GDM, which is reflected by their low IC50∼0.1 µM values. Furthermore, 1, 3, and 8 show higher SI indexes (∼77, 75, and 10) than GDM for the MCF-7 cell line. Derivatives 5–7 were also more potent than GDM in the MCF-7 cell line (IC50∼1 µM), with still favourable SIs∼10. Lipophilicity of the most promising analogues (1, 3, and 5–8) is relatively good (clogP = 0.7–3.4) and comparable with that of GDM (clogP∼2). Compound 2, bearing a terminal acidic group, showed the lowest activity in the MCF-7 cell line at a less favourable SI compared to GDM. Taking into account the binding models of GDM and its derivatives with Hsp90, one should expect that the carboxylic group might improve the binding strength as a result of the interaction with a positively charged K587,27,28. However, it is likely that in solution a flexible C(17) arm of 2 takes part in competitive intramolecular interactions (see Supplemental Material, Figure 51S), preventing the expected stabilising interaction with K58 of Hsp90. The existence of the H-bonded structures of 2 in solution and its relatively low lipophilicity (clogP∼0.4, Table 1), might explain the lower anticancer activity of 2 compared to the analogues 1, 3, and 8. In addition to low anticancer potency, compound 2 exhibits also very low toxicity when compared with GDM (IC50 (CCD39Lu) = 20.8 µM, Table 1). As for other biological data for 1–8, the quinuclidine analogue 8 shows the most beneficial potency in MDA-MB-231 (IC50 = 0.14 µM) among all ansamycins studied. The activity of 8 relative to GDM was improved, not only in MDA-MB-231 but also in other studied cancer cell lines (Table 1). When compared with 8, GDM was only more active in HepG2 cells. Lower activity than that of GDM was also observed for all other studied derivatives (1–7) and salts (9–13) in the HepG2 cancer cell line. The most promising quinuclidine analogue 8 exhibited relatively high toxicity in healthy cells CCD39Lu (IC50 = 0.87 µM) and had only low SI indexes (SIs < 1, Table 1).
Table 1.
The IC50 values [(µM) ± SD], selectivity indexes [SICCD39Lu] as well as clogP and solubility parameters for GDM and its analogues 1–8 and salts (9–13) obtained in MDA-MB-231, MCF-7, HeLa, HepG2, and CCD39Lu (1st panel) cell lines.
| Cmpd. | MDA-MB-231 | MCF-7 | HeLa | HepG2 | CCD39Lu | clogP | Solubility [mg/mL] |
|---|---|---|---|---|---|---|---|
| GDM | 1.04 ± 0.63 [SI 8.17] |
3.51 ± 0.21 [SI 2.42] |
1.42 ± 0.10 [SI 5.99] |
0.74 ± 0.32 [SI 11.48] |
8.50 ± 0.25 | 0.43/ 2.15* |
– |
| 1 | 2.45 ± 0.78 [SI > 4.08] |
0.13 ± 0.01 [SI > 76.92] |
1.67 ± 0.54 [SI > 5.99] |
1.75 ± 0.04 [SI > 5.71] |
>10 | 2.24 | – |
| 2 | 4.27 ± 1.54 [SI 4.87] |
7.62 ± 2.35 [SI 2.73] |
3.48 ± 0.51 [SI 5.98] |
3.04 ± 0.47 [SI 6.84] |
20.80 ± 5.14 | 0.43 | 1.47 |
| 3 | 1.84 ± 0.33 [SI 4.08] |
0.10 ± 0.21 [SI 75.00] |
1.25 ± 0.22 [SI 6.00] |
1.31 ± 0.54 [SI 5.73] |
7.50 ± 0.55 | 0.67 | 0.05 |
| 4 | >10 [SI > 1] |
>10 [SI > 1] |
>10 [SI > 1] |
>10 [SI > 1] |
>10 | 1.87 | 0.04 |
| 5 | 2.28 ± 0.54 [SI 6.71] |
1.84 ± 0.57 [SI 8.32] |
1.56 ± 0.14 [SI 9.81] |
1.63 ± 0.49 [SI 9.39] |
15.30 ± 0.12 | 3.40 | – |
| 6 | 3.16 ± 0.89 [SI > 3.16] |
0.61 ± 0.14 [SI > 16.39] |
4.75 ± 0.35 [SI > 2.11] |
3.38 ± 0.13 [SI > 2.96] |
>10 | 1.36 | – |
| 7 | 2.36 ± 0.35 [SI 5.34] |
0.96 ± 0.51 [SI 13.13] |
1.61 ± 0.61 [SI 7.83] |
1.68 ± 0.32 [SI 7.50] |
12.60 ± 0.21 | 1.38 | – |
| 8 | 0.14 ± 0.01 [SI 6.21] |
0.09 ± 0.01 [SI 9.67] |
1.06 ± 0.26 [SI 0.82] |
1.25 ± 0.22 [SI 0.70] |
0.87 ± 0.22 | 2.23 | 0.15 |
| 9 | 4.65 ± 2.65 [SI > 2.15] |
2.34 ± 0.56 [SI > 4.27] |
9.25 ± 0.98 [SI > 1.08] |
>10 [SI > 1] |
>10 | −1.10 | 11.9 |
| 10 | 4.64 ± 1.51 [SI > 2.16] |
2.51 ± 1.03 [SI > 3.98] |
7.43 ± 3.21 [SI > 1.35] |
>10 [SI > 1] |
>10 | −0.85 | 8.09 |
| 11 | 3.93 ± 1.60 [SI > 2.54] |
4.67 ± 2.00 [SI > 2.14] |
8.91 ± 3.19 [SI > 1.12] |
8.86 ± 0.17 [SI > 1.13] |
>10 | −0.81 | 2.30 |
| 12 | 5.01 ± 2.15 [SI > 2.00] |
2.65 ± 1.09 [SI > 3.77] |
9.12 ± 1.81 [SI > 1.10] |
>10 [SI > 1] |
>10 | 0.37 | 4.50 |
| 13 | 4.66 ± 2.49 [SI > 2.15] |
2.31 ± 0.97 [SI > 4.33] |
8.14 ± 0.64 [SI > 1.23] |
>10 [SI > 1] |
>10 | 0.61 | 5.03 |
clogP calculated by Molinspiration42.
Selectivity index [SIHDF] = IC50 normal cell line CCD39Lu/IC50 respective cancer cell line.
GDM: geldanamycin.
Considering the above results, we decided to further test compound 8 using the 2nd panel of cancer cell lines: SKBR-3, SKOV-3, PC-3, U-87, and A-549 and healthy cells HDF and to compare biological data with those of exemplary derivative 1 (Table 2). Our studies demonstrated that 8 is very active in the 2nd panel of cancer cell lines and its activity is even slightly better than that of GDM in the A-549 cell line (IC50 = 0.99 µM). The potency of 8 in SKBR-3, SKOV-3, PC-3, and U-87 cancer cell lines was lower or comparable to GDM (Table 2). In contrast with the results obtained in normal CCD39LU cell line, the toxicity of 8 in healthy cells HDF, is significantly lower than that for GDM (Table 2). The potency of low-cytotoxic 1 (IC50(HDF) = 30 µM; IC50(CCD39Lu) = > 10 µM), especially in the 2nd panel of SKBR-3, SKOV-3, PC-3, U-87, and A-549 cancer cell lines, was relatively low (IC50∼7.5 µM). Taking into regard the fact that another analogue of low cytotoxicity, 2 (IC50(CCD39Lu) = 20.8 µM), shows the best water solubility among 1–8 derivatives (Table 1), we decided to transform the most active quinuclidine analogue 8 into its better water-soluble N-alkylammonium salts to investigate the influence of a polar structure of this type on anticancer activity vs. toxicity. Biological tests of the salts 9–13 in the 1st panel of cancer cell lines (Table 1) revealed that similar to 8, they were generally more active than GDM in the MCF-7 cells (except 11, Table 1). A comparison of IC50 values between 8 and 9–13, indicated higher activity of the former. Interestingly, the transformation of 8 into its salts 9–13 was beneficial regarding low toxicities of 9–12 in normal cell line CCD39Lu (Table 1). Among quaternary salts, analogue 13 with a bulky N-cinnamyl quinuclidine moiety showed the best activity in MCF-7 with IC50 = 2.31 µM and with twice the value of SI index (SI = 4.33) when compared to GDM. Very good results in MCF-7 cells were also obtained for salts 9 (IC50 = 2.34 µM) and 10 (IC50 = 2.51 µM), which, like 13, had no heteroatoms in the tail attached to the quinuclidine unit (Table 1). Other biological data for the 1st panel of cell lines showed that quaternisation of quinuclidine nitrogen in 8 significantly decreased potency in MDA-MB-231, HeLa and HepG2 cancer cell lines while toxicity was reduced in normal CCD39Lu cells (Table 1). Anticancer studies of salts 9–13 in the 2nd panel of cell lines (Table 2) revealed that the most active analogue is compound 10 with no heteroatoms in the N-quinuclidine tail. Its IC50 values were oscillating around 1.7 µM (1.29 − 1.94 µM). The highest potency of 10 was registered in the A-549 cancer cell line (IC50 = 1.29 µM). Although a comparison of anticancer activities of GDM, 8 and 10 (Table 2) showed the lowest potency for 10, its activity remains attractive in the 2nd panel of cancer cells, at lower cytotoxicity than that of GDM in HDF and CCD39Lu cell lines. Salts 9 and 13, with substituents of similar nature as 10, also showed good anticancer activities with IC50∼2–3 µM and with a markedly reduced toxicity in HDF (IC50∼5.29 and 3.81 µM, Table 2). To conclude, considering the biological results for 9–13 obtained in the 1st and the 2nd panel of cancer cell lines, when compared to the activity data of GDM and 8, the most promising among the reported salts is salt 13 as it shows the best balance between the anticancer potency and toxicity in normal cells. It is relatively low toxic in both normal cell lines HDF (IC50 = 3.81 µM) and CCD39LU (IC50 > 10 µM) at still attractive level of anticancer potency IC50∼2 µM in MCF-7, SKBR-3, SKOV-3, PC-3, U-87, and A-549 cell lines.
Table 2.
Anticancer activities [IC50 (µM) ± SD] of GDM and its 1 and 8 analogues and salts (9–13) in SKBR-3, SKOV-3, PC-3, U-87, A-549 cells (2nd panel), and toxicity in human dermal fibroblasts (HDF) cell line [IC50 (µM) ± SD] together with selectivity indexes [SIHDF].
| Cmpd. | SKBR-3 | SKOV-3 | PC-3 | U-87 | A-549 | HDF |
|---|---|---|---|---|---|---|
| GDM | 0.87 ± 0.17 [SI 2.45] |
0.94 ± 0.09 [SI 2.27] |
0.73 ± 0.01 [SI 2.92] |
0.81 ± 0.12 [SI 2.62] |
0.99 ± 0.01 [SI 2.15] |
2.13 ± 0.11 |
| 1 | 7.44 ± 0.09 [SI 4.03] |
8.04 ± 0.24 [SI 3.73] |
7.02 ± 0.06 [SI 4.28] |
7.32 ± 0.49 [SI 4.10] |
9.01 ± 0.63 [SI 3.33] |
30.02 ± 1.03 |
| 8 | 1.49 ± 0.29 [SI 2.16] |
1.16 ± 0.92 [SI 2.78] |
1.47 ± 0.31 [SI 2.19] |
1.08 ± 0.16 [SI 3.43] |
0.94 ± 0.03 [SI 3.43] |
3.22 ± 0.18 |
| 9 | 3.12 ± 0.01 [SI 1.70] |
3.86 ± 0.05 [SI 1.37] |
3.01 ± 0.11 [SI 1.76] |
3.69 ± 0.08 [SI 1.43] |
3.09 ± 0.01 [SI 1.71] |
5.29 ± 0.17 |
| 10 | 1.94 ± 0.52 [SI 1.53] |
1.61 ± 0.13 [SI 1.84] |
1.88 ± 0.02 [SI 1.57] |
1.84 ± 0.11 [SI 1.61] |
1.29 ± 0.07 [SI 2.29] |
2.96 ± 0.36 |
| 11 | 6.49 ± 0.07 [SI 1.22] |
6.04 ± 0.38 [SI 1.31] |
6.22 ± 0.13 [SI 1.27] |
6.06 ± 0.58 [SI 1.31] |
6.83 ± 0.32 [SI 1.16] |
7.91 ± 0.91 |
| 12 | 8.49 ± 0.36 [SI 1.64] |
8.02 ± 0.17 [SI 1.73] |
7.94 ± 0.03 [SI 1.75] |
8.58 ± 0.19 [SI 1.62] |
8.94 ± 0.27 [SI 1.56] |
13.91 ± 0.42 |
| 13 | 2.02 ± 0.11 [SI 1.89] |
2.17 ± 0.06 [SI 1.76] |
2.05 ± 0.09 [SI 1.86] |
2.26 ± 0.15 [SI 1.67] |
2.06 ± 0.01 [SI 1.85] |
3.81 ± 0.11 |
Selectivity index [SIHDF] = IC50 normal cell line HDF/IC50 respective cancer cell line.
Binding mode of quinuclidine analogues of GDM and 9–13 salts with Hsp90
According to the earlier reports,7,26–28,32,43 we assumed that docking of 8, 10, and 13 into the target, i.e. ATP-binding pocket of Hsp90, requires trans/cis isomerisation of the lactam group and drastic conformational changes within the ansa-bridge owing to atropisomerisation process, i.e. flipping of the ansa-bridge from one side of the benzoquinone ring to the other (Figure 62S, Supplemental Material). DFT calculated energy barrier is 36.97 kcal/mol for the total atropisomerisation process of 10 requiring trans-cis lactam isomerisation crucial for binding with NBD of Hsp90 (Figures 3(b) and Figure 62S; Supplemental Material). This value is higher than that estimated from earlier studies of GDM (E = 16–21 kcal/mol).44,45 Structural comparison between complexes of Hsp90 with GDM and 8, showed that the mutual arrangements of the ansa-bridges and quinone cores relative to the Hsp90 key amino acids are similar in both cases.7,27 Atropisomerisation of active GDM analogues from the trans-lactam form (dominating in solution) into the cis-lactam one (bound with Hsp90) is evoked by the mutual induced fitting of human Hsp90 and the GDM derivative. The 8-Hsp90 complex is stabilised by the formation of intermolecular H-bonds between the ansamycin and F138, D54, D93, K112, and T184 of Hsp90, whereas, e.g. M98 and K58 are involved in hydrophobic interactions of the ligand in the ATP-binding pocket (Figure 3). Thus, the above binding mode of 8 is slightly improved relative to that known for GDM by an extra hydrophobic stabilisation of quinuclidine basket of 8 with K58, contributing also to improved anticancer potency of 8 relative to GDM. Comparison between calculated binding models of the most active derivative 8 and salts 10 and 13 of decreased cytotoxicity shows for the latter ones also possibility of hydrophobic stabilisation with K58 of Hsp90 (Figure 3). Comparison of binding energies of 10 (ΔH°f(10) = − 39 kcal/mol) or 8 (ΔH°f(8) = − 47 kcal/mol) with Hsp90 indicates the privileged formation of 8-Hsp90 complex. This result is in line with higher anticancer potency of higher lipophilic 8 (clogP = 2.23; = 0.15 mg/mL, Table 1) when compared to better water-soluble 10 (clogP = −0.85; = 8.09 mg/mL, Table 1). Compound 13, owing to the presence of its lengthy and bulky quinuclidine arm at C(17), has two alternative binding modes to Hsp90 (Figure 3(c,d)). The binding mode I of 13, is analogous to that of 8 and 10 (the interaction with K58, Figure 3(c)). The binding mode II of 13 to Hsp90 is realised via a conformational change within the quinuclidine basket, where the N+-cinnamyl tail is involved in an intermolecular C-H…π interaction with Y61 of Hsp90 (Figure 3(d)). A more favourable binding mode II to Hsp90 is excluded for 8 and 10 due to shorter quinuclidine arms at C(17). Overall, the higher anticancer activity of 8 compared to 10 and 13 can be explained by a higher energetic profit of binding 8 to Hsp90 (ΔH°f = − 47 kcal/mol) than for 10 (ΔH°f = − 39 kcal/mol) and 13 (ΔH°f = − 37 kcal/mol). Changes in lipophilicity (clogP) do not explain the differences in the anticancer effect of the tested salts (9–13). In turn, the better water solubility of salts 9, 10, and 13 ( > 5 mg/mL) seems to be in line with their better anticancer effects than 11 ( = 2.30 mg/mL) and 12 ( = 4.5 mg/mL), especially in the 2nd panel of tested cancer cell lines.
Figure 3.

Binding models revealing interactions between N-binding domain (NBD) of Hsp90 (PDB 3Q5J[43]) and new ansamyc in derivatives: 8/ΔH°f = −47 kcal/mol/(a), 10/ΔH°f = −39 kcal/mol/(b) and 13 in binding mode I /ΔH°f = −32 kcal/mol/(c), and 13 in binding mode II /ΔH°f = −37 kcal/mol/(d), compared to binding mode of macbecin II (PDB2VW535, pink), and optimised by MOG-PM6 method (Scigress F.J. 2.6, EU 3.1.9);46 amino acids of Hsp90 ATP-binding pocket are marked by yellow; intermolecular interactions (H-bonds) are marked by dots.
Anticancer tests of the most active quinuclidine analogues with potentiators
It is known that the effectiveness and potency of various antibiotics can be improved when they are used in cocktails with adjuvants and potentiators allowing, e.g. effective transportation of a drug into the target site of action.47–49 In order to evaluate the influence of the addition of doxorubicin (DOX; Figure 4) or branched polyethylenediamine – (PEI, polyethylenimine; Figure 4) on the activity of our lead compounds 8 and 13, tests were performed in SKBR-3, PC-3, SKOV-3, and HDF cells for their 1:1 equimolar mixtures (Table 3).
Figure 4.
Structures of tested selected potentiators of quinuclidine analogues of GDM.
Table 3.
Anticancer activities [IC50 (µM) ± SD] of 8 and 13 and their equimolar 1:1 mixtures with DOX and PEI in SKBR-3, SKOV-3, and PC-3, compared with toxicity determined in normal human dermal fibroblasts (HDF) cell line [IC50 (µM) ± SD] and selectivity indexes [SIHDF].
| Cmpd. | SKBR-3 | SKOV-3 | PC-3 | HDF |
|---|---|---|---|---|
| 8 | 1.49 ± 0.29 [SI 2.16] |
1.16 ± 0.92 [SI 2.78] |
1.47 ± 0.31 [SI 2.19] |
3.22 ± 0.18 |
| 8 + DOX | 1.06 ± 0.08 [SI 2.42] |
1.74 ± 0.02 [SI 1.48] |
1.71 ± 0.11 [SI 1.50] |
2.57 ± 0.07 |
| 8 + PEI | 2.61 ± 0.11 [SI 1.53] |
2.04 ± 0.01 [SI 1.96] |
2.68 ± 0.03 [SI 1.49] |
3.99 ± 0.22 |
| 13 | 2.02 ± 0.11 [SI 1.89] |
2.17 ± 0.06 [SI 1.76] |
2.05 ± 0.09 [SI 1.86] |
3.81 ± 0.11 |
| 13 + DOX | 1.33 ± 0.21 [SI 2.40] |
1.49 ± 0.16 [SI 2.14] |
1.09 ± 0.01 [SI 2.93] |
3.19 ± 0.31 |
| 13 + PEI | 0.55 ± 0.02 [SI 1.93] |
0.62 ± 0.05 [SI 1.71] |
0.59 ± 0.01 [SI 1.80] |
1.06 ± 0.06 |
| DOX | 0.72 ± 0.04 [SI 2.11] |
0.74 ± 0.06 [SI 2.05] |
0.68 ± 0.01 [SI 2.24] |
1.52 ± 0.03 |
| PEI | 0.53 ± 0.05 [SI 1.55] |
0.69 ± 0.01 [SI 1.19] |
0.51 ± 0.02 [SI 1.61] |
0.82 ± 0.11 |
Selectivity index [SIHDF] = IC50 normal cell line HDF/IC50 respective cancer cell line.
As shown in Table 3, DOX reveals a comparable activity at IC50∼0.7 µM towards all studied cancer cell lines at enhanced toxicity in the HDF normal cell line, relative to those of GDM (Table 2). Compounds 8 and 13 are of lower potency in studied cancer cell lines, although they simultaneously exhibit reduced toxic effects in HDF cell line, when referred to DOX and PEI. With the addition of PEI to 8 and 13 opposite changes in the potency were noted. The equimolar mixture 13 + PEI showed significantly improved potency relative to that of 13 (IC50(13+PEI)∼0.6 µM vs. IC50(13)∼2 µM). Interestingly 13 + PEI mixture showed even slightly higher anticancer potency (IC50 = 0.62 µM) than PEI itself in SKOV-3 cells. Unfortunately, with the increased potency of mixture 13 + PEI, its toxicity also showed a 3-fold increase in HDF cells in comparison with 13. In turn, the combined use of 8 with PEI resulted in decreased anticancer activity and cytotoxicity, both relative to 8 and PEI. The use of 8+DOX and 13+DOX mixtures improved potency in SKBR-3 cells relative to 8 and 13, respectively. For the 13+DOX mixture, the analogous result was also obtained in SKOV-3 and PC-3, in contrast to 8+DOX. The markedly enhanced anticancer activities of 13+DOX in studied cancer cell lines relative to that of 13, is accompanied by nearly preserved toxicity in HDF cells (IC50 level∼3 µM). Thus, our studies showed that the use of better water-soluble salt 13 than compound 8, in an equimolar mixture with PEI and DOX, contribute to markedly improved anticancer activity in SKBR-3, SKOV-3, and PC-3 (IC50∼3 µM), however, at the expense of increased toxicity in normal cells, when referred to 13. Furthermore, tests with 13+DOX mixture showed some available compromise between relatively preserved cytotoxicity (at the level IC50∼3 µM) and enhanced anticancer potency to IC50∼1 µM, especially in the PC-3 cell line (SI∼3).
Conclusions
The quinuclidine analogue 8 of GDM and its N-alkylammonium salts 9–13 have been synthesised and tested in nine cancer (MDA-MB-231, MCF-7, HeLa, HepG2, SKBR-3, SKOV-3, PC-3, U-87, and A-549) and two normal (CCD39Lu and HDF) cell lines. The biological activities of these compounds were compared with those of GDM and C(17)-analogues 1–7. Structural studies of 1–13 using 1D and 2D NMR and x-ray crystallography methods revealed nearly identical conformation of the ansa-bridge, the trans configuration of the lactam group and similar arrangement of the C(17) substituents relative to the quinone core in solution and in solid. The MTT assay revealed that 8 is the most active derivative among studied C(17)-analogues of GDM in the 1st panel of MDA-MB-231, MCF-7, HeLa and HepG2 cancer cell lines at low IC50s∼0.1–1 µM. The anticancer activities of 8 in these cancer cell lines were markedly higher than GDM, except for the HepG2 cell line. A higher potency of 8 than GDM was noted also in the A-549 cell line of the 2nd panel. Unfortunately, alongside the attractive potency of 8, higher and lower toxicities than GDM were noted in CCD39Lu and HDF normal cell lines, respectively. Studies in MCF-7 cancer cell line revealed improved activities of N-alkylammonium salts 9–13 when compared to GDM (e.g. IC50(13) = 2.31 µM vs. IC50(GDM) = 3.51 µM), at together reduced toxic effects in HDF and CCD39Lu normal cell lines (IC50 even > 10 µM). Our studies showed that the limited toxicity of 9–13 salts can be linked with their low clogP and improved water solubility relative to GDM (almost water-insoluble). In studied cancer cell lines, among 9–13 salts the most potent were those without heteroatoms in the attached tail at the nitrogen of C(17)-quinuclidine basket. Molecular docking of the most potent salts 8, 10, and 13 indicated the intermolecular hydrophobic stabilisation of the quaternary C(17)-quinuclidine arm with K58 or with Y61 of Hsp90. A more beneficial binding energy of the 8-Hsp90 complex than that for 10 or 13 complexes explains a higher anticancer activity of 8 than 10 and 13. Our studies also showed that quaternisation of the nitrogen within C(17)-quinuclidine containing arm can be a useful strategy in decreasing the toxicity of GDM analogues in normal cells, at a simultaneously improved or preserved anticancer activity in MCF-7 and A-549 cancer cell lines, respectively (e.g. compound 10). The use of 13 with potentiator PEI leads to improved or comparable anticancer activities relative to those of the salt and PEI, respectively. However, this beneficial anticancer effect was observed at the expense of increased toxicity in normal cells, when referred to 13.
Supplementary Material
Funding Statement
This work was supported by the Polish National Science Centre (NCN) under Grant Opus 13 no. UMO-2017/25/B/ST5/00291.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- 1.Baksh A, Kepplinger B, Isah HA, et al. Production of 17-O-demethyl-geldanamycin, a cytotoxic ansamycin polyketide, by streptomyces hygroscopicus DEM20745. Nat Prod Res 2017;31:1895–900. [DOI] [PubMed] [Google Scholar]
- 2.Franke J, Eichner S, Zeilinger C, Kirschning A.. Targeting heat-shock-protein 90 (Hsp90) by natural products: geldanamycin, a show case in cancer therapy. Nat Prod Rep 2013;30:1299–323. [DOI] [PubMed] [Google Scholar]
- 3.Fukuyo Y, Hunt CR, Horikoshi N.. Geldanamycin and its anti-cancer activities. Cancer Lett 2010;290:24–35. [DOI] [PubMed] [Google Scholar]
- 4.Hermane J, Eichner S, Mancuso L, et al. New geldanamycin derivatives with anti Hsp properties by mutasynthesis. Org Biomol Chem 2019;17:5269–78. [DOI] [PubMed] [Google Scholar]
- 5.Guo W, Reigan P, Siegel D, Ross D.. Enzymatic reduction and glutathione conjugation of benzoquinone ansamycin heat shock protein 90 inhibitors: relevance for toxicity and mechanism of action. Drug Metab Dispos 2008;36:2050–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cysyk RL, Parker RJ, Barchi JJ, et al. Reaction of geldanamycin and C17-substituted analogues with glutathione: product identifications and pharmacological implications. Chem Res Toxicol 2006;19:376–81. [DOI] [PubMed] [Google Scholar]
- 7.Skrzypczak N, Pyta K, Ruszkowski P, et al. Synthesis, structure and anticancer activity of new geldanamycin amine analogs containing C(17)- or C(20)- flexible and rigid arms as well as closed or open ansa-bridges. Eur J Med Chem 2020;202:112624. [DOI] [PubMed] [Google Scholar]
- 8.Kitson RRA, Moody CJ.. An improved route to 19-substituted geldanamycins as novel Hsp90 inhibitors-potential therapeutics in cancer and neurodegeneration. Chem Commun (Camb) 2013;49:8441–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li Z, Jia L, Wang J, et al. Discovery of diamine-linked 17-aroylamido-17-demethoxygeldanamycins as potent Hsp90 inhibitors. Eur J Med Chem 2014;87:346–63. [DOI] [PubMed] [Google Scholar]
- 10.Eichner S, Eichner T, Floss HG, et al. Broad substrate specificity of the amide synthase in S. hygroscopicus-new 20-membered macrolactones derived from geldanamycin. J Am Chem Soc 2012;134:1673–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mellatyar H, Talaei S, Pilehvar-Soltanahmadi Y, et al. Targeted cancer therapy through 17-DMAG as an Hsp90 inhibitor: overview and current State of the art. Biomed Pharmacother 2018;102:608–17. [DOI] [PubMed] [Google Scholar]
- 12.Talaei S, Mellatyar H, Asadi A, et al. Spotlight on 17-AAG as an Hsp90 inhibitor for molecular targeted cancer treatment. Chem Biol Drug Design 2019;93:760–86. [DOI] [PubMed] [Google Scholar]
- 13.Glaze ER, Lambert AL, Smith AC, et al. Preclinical toxicity of a geldanamycin analog, 17-(dimethylaminoethylamino)-17-demethoxygeldanamycin (17-DMAG), in rats and dogs: potential clinical relevance. Cancer Chemother Pharmacol 2005;56:637–47. [DOI] [PubMed] [Google Scholar]
- 14.Wu CZ, Jang JH, Woo M, et al. Enzymatic glycosylation of nonbenzoquinone geldanamycin analogs via bacillus UDP-glycosyltransferase. Appl Environ Microbiol 2012;78:7680–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hermane J, Bułyszko I, Eichner S, et al. New, non-Quinone fluorogeldanamycin derivatives strongly inhibit Hsp90. ChemBioChem 2015;16:302–11. [DOI] [PubMed] [Google Scholar]
- 16.Mohammadi-Ostad-Kalayeh S, Stahl F, Scheper T, et al. Heat shock proteins revisited: using a mutasynthetically generated reblastatin library to compare the inhibition of human and Leishmania Hsp90s. ChemBioChem 2018;19:562–74. [DOI] [PubMed] [Google Scholar]
- 17.Kirschning A, Hahn F.. Merging chemical synthesis and biosynthesis: a new chapter in the total synthesis of natural products and natural product libraries. Angew Chem Int Ed Engl 2012;51:4012–22. [DOI] [PubMed] [Google Scholar]
- 18.Bułyszko I, Dräger G, Klenge A, Kirschning A.. Evaluation of the synthetic potential of an AHBA knockout mutant of the rifamycin producer Amycolatopsis Mediterranei. Chemistry 2015;21:19231–42. [DOI] [PubMed] [Google Scholar]
- 19.Eichner S, Knobloch T, Floss HG, et al. The interplay between mutasynthesis and semisynthesis: generation and evaluation of an ansamitocin library. Angew Chem Int Ed Engl 2012;51:752–7. [DOI] [PubMed] [Google Scholar]
- 20.Mancuso L, Jürjens G, Hermane J, et al. Bioreduction of aryl azides during mutasynthesis of new ansamitocins. Org Lett 2013;15:4442–5. [DOI] [PubMed] [Google Scholar]
- 21.Harmrolfs K, Mancuso L, Drung B, et al. Preparation of New Alkyne-Modified Ansamitocins by Mutasynthesis. Beilstein J Org Chem 2014;10:535–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schax E, Walter JG, Märzhäuser H, et al. Microarray-based screening of heat shock protein inhibitors. J Biotechnol 2014;180:1–9. [DOI] [PubMed] [Google Scholar]
- 23.Tian ZQ, Liu Y, Zhang D, et al. Synthesis and biological activities of novel 17-aminogeldanamycin derivatives. Bioorg Med Chem 2004;12:5317–29. [DOI] [PubMed] [Google Scholar]
- 24.Greish K, Ray A, Bauer H, et al. Anticancer and antiangiogenic activity of HPMA copolymer-aminohexylgeldanamycin-RGDfK conjugates for prostate cancer therapy. J Control Release 2011;151:263–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li L, Wang L, You QD, Xu XL.. Heat shock protein 90 inhibitors: an update on achievements, challenges, and future directions. J Med Chem 2020;63:1798–822. [DOI] [PubMed] [Google Scholar]
- 26.Raman S, Singh M, Tatu U, Suguna K.. First structural view of a peptide interacting with the nucleotide binding domain of heat shock protein 90. Sci Rep 2015;5:17015–0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stebbins CE, Russo AA, Schneider C, et al. Crystal structure of an Hsp90-geldanamycin complex: targeting of a protein chaperone by an antitumor agent. Cell 1997;89:239–50. [DOI] [PubMed] [Google Scholar]
- 28.Jez JM, Chen JCH, Rastelli G, et al. Crystal structure and molecular modeling of 17-DMAG in complex with human Hsp90. Chem Biol 2003;10:361–8. [DOI] [PubMed] [Google Scholar]
- 29.Rastelli G, Tian ZQ, Wang Z, et al. Structure-based design of 7-carbamate analogs of geldanamycin. Bioorg Med Chem Lett 2005;15:5016–21. [DOI] [PubMed] [Google Scholar]
- 30.Immormino RM, Metzger LE, Reardon PN, et al. Different poses for ligand and chaperone in inhibitor-bound Hsp90 and GRP94: implications for paralog-specific drug design. J Mol Biol 2009;388:1033–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sawarkar R, Roy N, Rao S, et al. Heat shock protein 90 regulates development in dictyostelium discoideum. J Mol Biol 2008;383:24–35. [DOI] [PubMed] [Google Scholar]
- 32.Zhang MQ, Gaisser S, Nur-E-Alam M, et al. Optimizing natural products by biosynthetic engineering: discovery of nonquinone Hsp90 inhibitors †. J Med Chem 2008;51:5494–7. [DOI] [PubMed] [Google Scholar]
- 33.Wernimont AK, Tempel W, Lin YH, et al. Crystal structure of the amino-terminal domain of HSP90 from leishmania major, LMJF33.0312:M1-K213 in the presence of 17-DMAP-geldanamycin. Available from: 10.2210/pdb3q5j/pdb [DOI] [Google Scholar]
- 34.Bhat R, Tummalapalli SR, Rotella DP.. Progress in the discovery and development of heat shock protein 90 (Hsp90) inhibitors. J Med Chem 2014;57:8718–28. [DOI] [PubMed] [Google Scholar]
- 35.Massey AJ. ATPases as drug targets: insights from heat shock proteins 70 and 90. J Med Chem 2010;53:7280–6. [DOI] [PubMed] [Google Scholar]
- 36.Kitson RRA, Moody CJ.. Learning from nature: advances in geldanamycin- and radicicol-based inhibitors of Hsp90. J Org Chem 2013;78:5117–41. [DOI] [PubMed] [Google Scholar]
- 37.Shan G, Peng Z, Li Y, et al. A novel class of geldanamycin derivatives as HCV replication inhibitors targeting on Hsp90: synthesis, structure-activity relationships and anti-HCV activity in GS4.3 replicon cells. J Antibiot (Tokyo) 2011;64:177–82. [DOI] [PubMed] [Google Scholar]
- 38.Li YP, Shan GZ, Peng ZG, et al. Synthesis and biological evaluation of heat-shock protein 90 inhibitors: geldanamycin derivatives with broad antiviral activities. Antivir Chem Chemother 2010;20:259–68. [DOI] [PubMed] [Google Scholar]
- 39.Anderson I, Low JS, Weston S, et al. Heat shock protein 90 controls HIV-1 reactivation from latency. Proc Natl Acad Sci USA 2014;111:E1528–E1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schnur RC, Corman ML, Gallaschun RJ, et al. Inhibition of the oncogene product P185erbB-2 in vitro and in vivo by geldanamycin and dihydrogeldanamycin derivatives. J Med Chem 1995;38:3806–12. [DOI] [PubMed] [Google Scholar]
- 41.Ge J, Normant E, Porter JR, et al. Design, synthesis, and biological evaluation of hydroquinone derivatives of 17-amino-17-demethoxygeldanamycin as potent, water-soluble inhibitors of Hsp90. J Med Chem 2006;49:4606–15. [DOI] [PubMed] [Google Scholar]
- 42.Molinspiration V2018.10. Available from: https://www.Molinspiration.Com/
- 43.Bank, RPD. RCSB PDB - 3Q5J: crystal structure of the amino-terminal domain of HSP90 from Leishmania major, LMJF33.0312: M1-K213 in the presence of 17-DMAP-geldanamycin Available from: https://www.rcsb.org/structure/3Q5J [accessed 7 May 2020].
- 44.Thepchatri P, Eliseo T, Cicero DO, et al. Relationship among ligand conformations in solution, in the solid state, and at the Hsp90 binding site: geldanamycin and radicicol. J Am Chem Soc 2007;129:3127–34. [DOI] [PubMed] [Google Scholar]
- 45.Lee YS, Marcu MG, Neckers L.. Quantum chemical calculations and mutational analysis suggest heat shock protein 90 catalyzes trans-cis isomerization of Geldanamycin. Chem Biol 2004;11:991–8. [DOI] [PubMed] [Google Scholar]
- 46.Scigress package FJ 2.6/EU 3.1.9./2008–2019; Japan: Fujitsu. [Google Scholar]
- 47.Douafer H, Andrieu V, Phanstiel O, Brunel JM.. Antibiotic adjuvants: make antibiotics great again!. J Med Chem 2019;62:8665–81. [DOI] [PubMed] [Google Scholar]
- 48.Liu TY, Hussein WM, Giddam AK, et al. Polyacrylate-based delivery system for self-adjuvanting anticancer peptide vaccine. J Med Chem 2015;58:888–96. [DOI] [PubMed] [Google Scholar]
- 49.Domalaon R, Idowu T, Zhanel GG, Schweizer F.. Antibiotic hybrids: the next generation of agents and adjuvants against gram-negative pathogens? Clin Microbiol Rev 2018;31:e00077–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.