

Abstract
Heme nitric oxide/oxygen binding protein isolated from the obligate anaerobe Clostridium botulinum (Cb H-NOX) was previously reported to bind NO with a femtomolar KD (Nioche, P. et al. Science 2004, 306, 1550–1553). On the other hand, no oxyferrous Cb H-NOX was observed despite full conservation of the key residues that stabilize the oxyferrous complex in the H-NOX from Thermoanaerobacter tengcongensis (Tt H-NOX) (the same study). In this study, we re-measured the kinetics/affinities of Cb H-NOX for CO, NO, and O2. KD(CO) for the simple one-step equilibrium binding was 1.6 × 10−7 M. The KD(NO) of Cb H-NOX was 8.0 × 10−11 M for the first six-coordinate NO complex, and the previous femtomolar KD(NO) was actually an apparent KD for its multiple-step NO binding. An oxyferrous Cb H-NOX was clearly observed with a KD(O2) of 5.3 × 10−5 M, which is significantly higher than Tt H-NOX’s KD(O2) = 4.4 × 10−8 M. The gaseous ligand binding of Cb H-NOX provides another supportive example for the “sliding scale rule” hypothesis (Tsai, A.-L. et al. Antioxid. Redox Signal. 2012, 17, 1246–1263), and the presence of hydrogen bond donor Tyr139 in Cb H-NOX selectively enhanced its affinity for oxygen.
Graphical Abstract
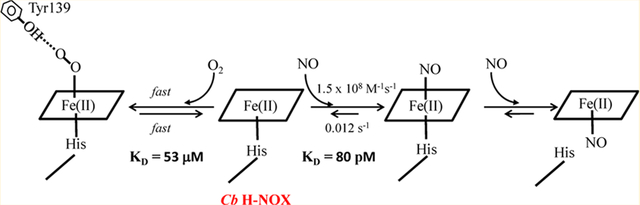
Heme nitric oxide/oxygen binding proteins (H-NOXs) are a family of bacterial hemoproteins, identified based on their significant sequence homology with the heme domain [β1(1–194)] of the only mammalian heme NO receptor soluble guanylyl cyclase (sGC).1,2 In bacterial genome, H-NOX proteins are either encoded in the same operon with putative histidine kinases or diguanylate cyclases, or are domains of methyl-accepting chemotaxis proteins (MCP). As sGC, all H-NOX proteins conspicuously bind nitric oxide (NO) and carbon monoxide (CO).3 On the other hand, H-NOXs isolated from different organisms exhibit a drastic difference in O2 binding.4 sGC and H-NOXs from facultative anaerobes, such as the one found in Vibrio cholera (Vc H-NOX),5 reacts with O2 but does not form a stable oxyferrous complex, while H-NOXs from obligate anaerobes, like the one found in Thermoanaerobacter tengcongensis (Tt H-NOX), bind O2 with strong affinities and form stable oxyferrous complexes. The binding of O2 to H-NOXs in obligate anaerobes may provide these organisms a mechanism to sense O2 for self-defense. In obligate anaerobe Clostridium botulinum (C. botulinum), genome screening identifies a two-domain protein with an N-terminal H-NOX domain (Cb H-NOX or CB-SONOHD) and a C-terminal MCP domain.6 Cb H-NOX has been reported to bind NO with a femtomolar dissociation constant, KD, much smaller than those of other H-NOXs; however, no oxyferrous Cb H-NOX was observed despite conservation of all the key residues which stabilize oxyferrous Tt H-NOX.6,7
All the H-NOXs and sGC possess a 5-coordinate (5c) heme with a proximal neutral histidine ligand (5c heme-His), as shown by structural determination6–10 or sequence homology comparison. Although the 5c histidine-ligated ferrous hemoproteins with different structural folds bind NO, CO, and O2 with different KD’s, covering a dynamic range up to 9 orders for each gaseous ligand, bioinformatics analysis reveals a simple “sliding scale rule” relationship between individual log KD sets of each hemoprotein.3,11 This hypothetic “sliding scale rule” states that for a 5c neutral histidine-ligated ferrous hemoprotein (or model compound), its log KD(NO), log KD(CO), and log KD(O2) fall on an approximately straight line in the plot of log KD’s versus ligand type, and the difference in the immediate protein environments around the heme cause a vertical parallel shift of such lines for each hemoprotein (or model compound) into a 9-order dispersion.3,11 The “sliding scale rule” has been examined against many hemoproteins, including sGC and H-NOX from Nostoc punctiforme (Ns H-NOX), and heme model compound, such as PPIX(1-MeIm).11,12 A recent example is Vc H-NOX, whose binding to the gaseous ligands is shown to also follow the “sliding scale rule”.5 Hemoproteins’ parallel pattern of log KD(NO)-log KD(CO)-log KD(O2) lines described in the “sliding scale rule” has been ascribed to three major factors, including the presence of a proximal neutral histidine ligand, direct distal steric hindrance, and proximal constraints for in-plane iron movement. This hypothetical rule provides an empirical tool to predict the affinities of a hemoprotein to any two of NO, CO, and O2 based on its measured affinity for the third one.3,11 On the other hand, deviations from the “sliding scale rule” are observed in some hemoproteins with a neutral histidine-ligated heme, and their log KD(NO)’s and/or log KD(O2)’s are significantly lower than the values predicted by parallel linear projection going through their observed log KD(CO)’s. These deviations are due to mechanisms which further stabilize the NO-heme and/or O2-heme complexes of these hemoproteins. The deviation of KD(NO)’s from the parallel linear pattern in some of these hemoproteins is attributed to the multiple steps in their NO bindings. In these hemoproteins, NO binding to heme first generates a 6-coordinate (6c) NO-heme-His complex, which later converts to a 5c NO-heme complex due to dissociation of the proximal histidine. Apparent affinity KD,apparent(NO), as the ratio between the off-rate of the final stable 5c NO-heme complex, koff(5c), and the on-rate of the initial 6c NO-heme-His complex, kon(6c), is much lower than the KD(NO) of the initial 6c NO-heme-His complex, KD(NO)(6c) = koff(6c)/kon(6c). A good example of multistep NO binding is found in sGC, whose KD,apparent(NO) is ~3 orders lower than its KD(NO)(6c), which agrees perfectly with the “sliding scale rule”.3 Enhancement of O2 affinity (lower KD(O2)) is usually observed in hemoproteins with a distal hydrogen-bond donor(s). For example, the presence of H64 in myoglobin and Y140 in Tt H-NOX results in KD(O2)’s much smaller than the predicted values based on linear extrapolation from their log KD(NO)-log KD(CO) lines.11 Cb H-NOX, with its reported extremely low KD(NO) but no detectable O2 binding, even with retaining of all key residues in stabilizing oxyferrous complex in Tt H-NOX,6 appears to be an outlier to the “sliding scale rule”.
In this study, we re-assessed the gaseous ligand binding of Cb H-NOX in an effort to clarify two important issues. First, the reported femtomolar KD(NO), 3.0 × 10−14 M, for Cb H-NOX is an apparent value;6 what is then the KD(NO)(6c) for Cb H-NOX? Second, is oxyferrous complex intermediate in Cb H-NOX observable? To define the KD(NO)(6c) of Cb H-NOX, we measured the kon(NO) and koff(NO) of its 6c NO-heme-His complex. This study demonstrates that logarithms of KD(NO)(6c) and KD(CO) in wt or Y139F Cb H-NOXs agree well with the “sliding scale rule”. Moreover, wild type Cb H-NOX is shown to bind O2 and form an oxyferrous complex, but its KD(O2) is three orders higher than that of Tt H-NOX. As a comparison, we also measured the kinetics and affinities of NO, CO, and O2 to wt Tt H-NOX; a complete set of parameters of gaseous ligand binding has so far not been available for Tt H-NOX. The affinities of wt Tt H-NOX for NO and CO also follow the “sliding scale rule”. Deviations of KD(O2)’s of Cb and Tt H-NOXs from the “sliding scale rule” are ascribed to the stabilizing hydrogen bonding of distal Y139 in Cb and Y140 in Tt H-NOX, respectively.
MATERIALS AND METHODS
Materials.
Carbon monoxide (CO) and nitric oxide (NO) gases were from Matheson-TriGas Inc. (Houston, TX). NO was prepurified by passing through a NaOH trap to remove nitrous and nitric acid contaminants. Sodium hydrosulfite (Na2S2O4), imidazole, heme, δ-aminolevulinic acid, isopropyl-1-thio-β-d-galactopyranoside, ampicillin, kanamycin, chloramphenicol, and egg lysozyme were from Sigma (St. Louis, MO). Restriction enzymes and other DNA modifying enzymes were purchased from New England BioLabs (Beverly, MA). Oligonucleotides were obtained from Integrated DNA Technologies (Coralville, IA). Reagents for DNA extraction and purification were from Qiagen (Valencia, CA). Immunoblotting reagents were from Bio-Rad (Hercules, CA). Plasmid vectors pET43.1a and pET28b, and Escherichia coli strains Rosetta 2(DE3)pLysS and Rosetta-gami B(DE3), and anti-HisTag monoclonal antibody were from Novagen (Madison, WI). TALON metal affinity resin was purchased from BD Biosciences Clontech (Palo Alto, CA). Other chemicals were all of reagent grade.
Construction of the Expression Vectors for H-NOXs.
The cDNA of wt Cb H-NOX from C. botulinum A strain (ATCC3502) was synthesized with the gene sequence optimized for E. coli codon usage, replacing several rare codons with high-frequency synonymous codons. The cDNA contained the codons for six histidines at the N-terminus. The cDNA was cloned into pBSK vector (Epoch LifeScience, Houston, TX) and amplified. The wt Cb H-NOX cDNA was then released by digesting with NcoI and XhoI, and subcloned into pET28b vector. The integrity of the resulting plasmid was confirmed by restriction digestion and DNA sequencing (Lone Star Labs, Houston, TX).
Y139F point mutation in Cb H-NOX was introduced using the QuikChange Site-directed Mutagenesis kit (Stratagene, La Jolla, CA). The mutagenic primer (base changes are underlined), 5′-CGCGGCATGTTTGATTTTTTTCTGGGCCTGCTG-3′, together with its complementary (antisense) strand, 5′-CAGCAGGCCCAGAAAAAAATCAAACATGCCGCG-3′, were used in a PfuUltra polymerase-initiated reaction, using wt Cb H-NOX in pET28b vector as the template. The DNA template of wt Cb H-NOX was then removed by digestion with DpnI, and the cDNA of Y139F Cb H-NOX was transformed into E. coli XL-10 competent cells. The mutation was verified by DNA sequencing.
The sequence of wt Tt H-NOX MB4 gene was first optimized for E. coli codon usage by replacing several rare codons with high-frequency synonymous codons. This optimized cDNA contained six histidine codons inserted upstream of the stop codon. The cDNA was amplified in pBSK vector and then subcloned into pET43.1a vector. The integrity of the resulting plasmid was confirmed by restriction digestion and DNA sequencing.
Y140F point mutation in Tt H-NOX was introduced using the QuikChange Site-directed Mutagenesis kit (Stratagene, La Jolla, CA). The mutagenic primer (base changes are underlined), 5′-CGCAAGATGTACGATTTCTTTCTGGGCCTGATC-3′, together with its complementary (antisense) strand, 5′-GATCAGGCCCAGAAAGAAATCGTACATCTTGCG-3′, were used in a PfuUltra polymerase-initiated reaction, using wt Tt H-NOX in pET43.1a vector as the template. The DNA template of wt Tt H-NOX was then removed by digestion with DpnI, and the cDNA of Y140F Tt H-NOX was transformed into E. coli XL-10 competent cells. The mutation was verified by DNA sequencing.
Expression and Purification of H-NOXs.
The wild type Cb and Tt H-NOXs and their point mutants were expressed and purified following the similar procedure as described previously5 and are summarized briefly as follows. The H-NOXs were overexpressed in E. coli Rosetta 2(DE3)pLysS strain (for wt and Y139F Cb H-NOXs) or Rosetta-gami B(DE3) strain (for wt and Y140F Tt H-NOXs). The cells were first grown at 37 °C in Terrific Broth medium containing chloramphenicol and kanamycin until A610 reached 0.8. Then heme, δ-aminolevulinic acid, and isopropyl-1-thio-β-d-galactopyranoside were added to the culture, and the cells were grown at 20 °C for another 48 h before harvest.
Cells harvested above were lysed by sonication in 100 mM potassium phosphate, pH 7.5 containing 100 mM NaCl, 10% (v/v) glycerol and egg lysozyme (3.0 mg/mL). Supernatant after centrifugation at 100,000g was loaded onto TALON affinity resin, and the H-NOXs were eluted with the same buffer (without lysozyme) containing 250 mM imidazole. The purities of the H-NOXs were typically ≥95%, checked with SDS-PAGE (data not shown).
Stopped-Flow Experiments.
The association and dissociation kinetics of NO, CO, and O2 to anaerobic ferrous H-NOXs were studied with an Applied Photophysics (Leather-head, UK) model SX-18MV stopped-flow instrument, and optically following the absorbance changes at different wavelengths with a monochromator or the whole spectral changes with a diode-array accessory. The sample handling unit was kept in an anaerobic chamber (COY Lab Products, Inc., Grass Lake, MI). Ferrous H-NOXs used in all the experiments were prepared by Na2S2O4 titration after five alternating cycles of vacuum and argon displacement in a tonometer. No extra Na2S2O4 existed in the final samples judged by the absence of the absorbance due to Na2S2O4. Rapid scan data was analyzed with the global analysis method using the Pro-Kineticist package by Applied Photophysics. Observed rate constants, kobs, of single wavelength data under pseudo-first order conditions were obtained by fitting the time courses of optical changes to the standard exponential function:
| (1) |
where Af, t, and a are the final optical absorbance, reaction time, and amplitude of optical change, respectively. Second-order association rate constant kon was derived from the slope in the secondary plots of kobs versus gaseous ligand concentration [L]:
| (2) |
The y-intercept corresponds to koff. However, in this study, koff constants for the NO and CO complexes are small, and fitting errors prevent extracting their accurate values based on the y-intercepts of the secondary plots. Dissociation constants were therefore determined using competition methods as explained in Results.
When measuring the formation rate of a 6c NO-heme-His complex in a H-NOX, the protein was mixed with an equal concentration of NO, and the kobs was obtained by fitting the time course to the equation of a second-order reaction:
| (3) |
where A0 is the initial optical absorbance and t is the reaction time. Second order rate constant in unit of M−1 s−1 was obtained by multiplying kobs with the difference extinction coefficient, Δε.
EPR Spectroscopy.
EPR spectra were recorded on a Bruker EMX spectrometer. Data analyses and spectral simulations were conducted using WinEPR and SimFonia programs furnished with the EMX system. EPR at 115 K was conducted using the following parameters: frequency, 9.29 GHz; modulation frequency, 100 kHz; modulation amplitude, 2.0 G; and time constant, 0.33 s.
EPR samples of NO complexes were prepared by injecting desired volumes of 2 mM NO stock into ferrous H-NOXs inside EPR tubes and the reactants were then mixed. The procedures were conducted inside the anaerobic chamber, and then the EPR tubes were transferred outside and quickly frozen in an ethanol/dry ice mixture. The times between NO injection and sample freezing were less than 1 min.
Computer Modeling.
Time courses of A422 during the reactions between 2.3 μM ferrous Cb H-NOX with varied levels of NO, 25 μM, 0.1 mM, 0.2 mM, 0.3 mM, 0.4 mM, 0.5 mM, 0.65 mM, 0.8 mM, and 1 mM were fitted simultaneously using SCoP Program (Simulation Resources Inc., Redlands, CA). The model used for the fitting was based on reactions I and II (Scheme 1, C ⇔ D ⇔ E). Parameters including the extinction coefficients for both 6c NO-heme-His and 5c NO-heme complexes and rate constant k2 were set to their experimentally measured values. Other kinetic parameters including rate constants k1, k−1, and k−2 and the extinction coefficient for the putative transient quaternary complex were allowed to float, but initial value for k−2 was ~0, and the ratio of k−1/k1 was equal to KD, determined by experiment. The global minimum of the error surface during the nonlinear regression for the multiple time courses was located by a “principal axis method” algorithm (PRAXIS), which is capable of random jumping out of local minima to find the global minimum.13 The goodness of fit was evaluated by “least squares” method, which was calculated as follows,
| (4) |
where E represents the error function, n is the number of data point in each experimental time course, and fk and yk are the fitted and experimental values at each time point, respectively. A good fit was indicated by its small value of E.
Scheme 1. Binding of Gaseous Ligands to Ferrous Cb H-NOXa.
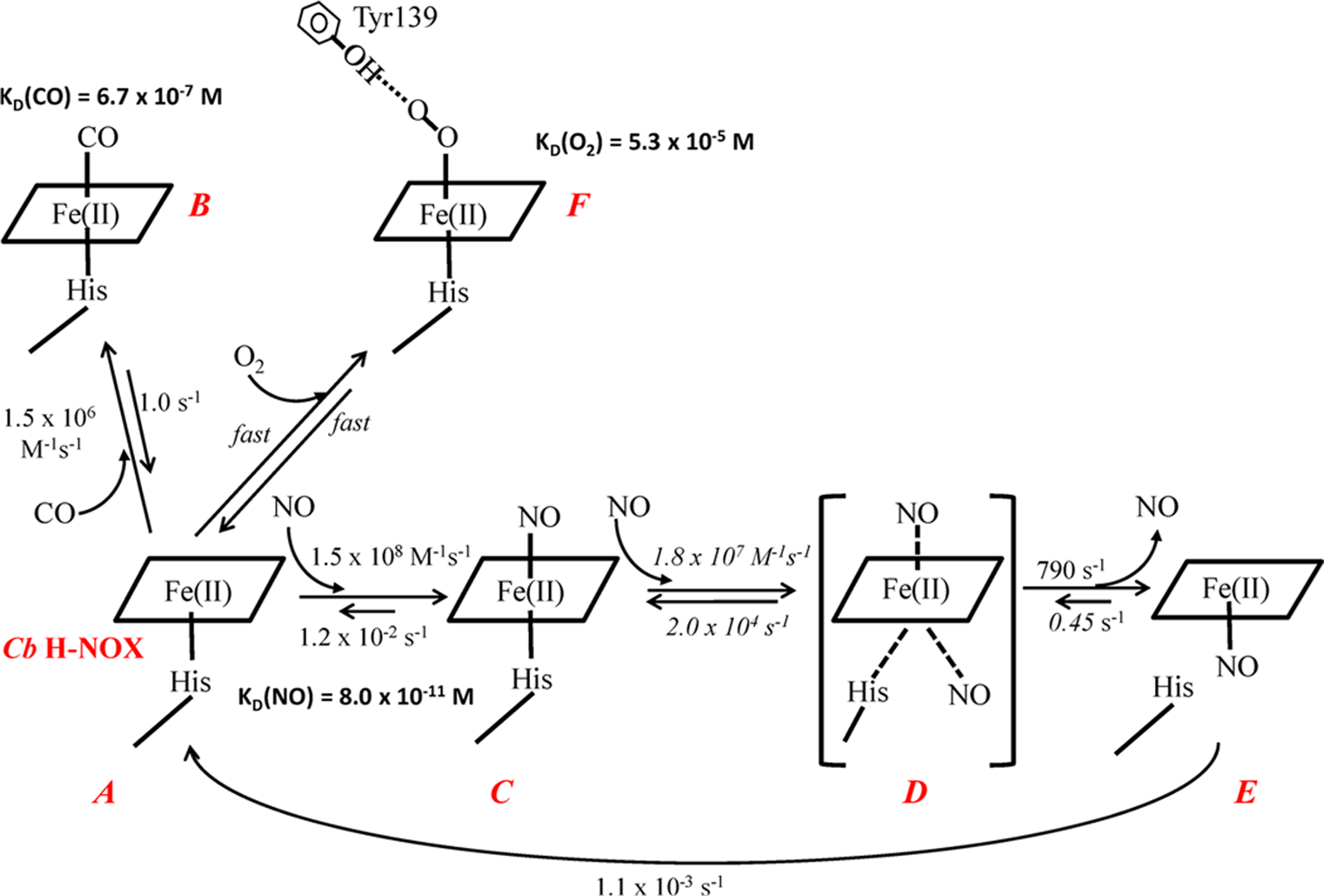
aEach heme species is labeled in red. The rate constants labeled in italic are estimated by computer modeling. Both association and dissociation of O2 to ferrous Cb H-NOX (A) are assumed to be fast, and the rate constants are not determined, although KD(O2) of Cb H-NOX was measured by stopped-flow titration method. KD(NO) is the affinity for 6c NO-heme-His complex. The putative hydrogen bonding from the hydroxyl group of Y139 to O2-heme is represented by a dashed line. Intermediate D, bracketed in parentheses, is a quaternary complex as proposed before32 and is not observed in experiment, for its extreme transient nature.
RESULTS
Kinetics of CO Binding to Ferrous Cb and Tt H-NOXs.
CO binding to Cb H-NOX was first characterized in this study since it was not reported before; and among NO, CO, and O2 ligands, the kinetics of CO binding to hemoproteins is the simplest. CO bound ferrous wt Cb H-NOX in a simple reversible one-step reaction (Scheme 1, A ⇔ B) which shifted the Soret peak from 431 to 424 nm with no spectral intermediate (Figure S1A). Observed rate constants, kobs, obtained by fitting the time courses at 424 nm exhibited a linear dependence on [CO] (Figure S1B) and the second-order association rate constant equals the slope, kon(CO) = 1.5 × 106 M−1 s−1 (Figure S1B, inset and Table 1). The dissociation rate constant, koff, was measured with the NO displacement method14 by reacting CO-heme complex with 1 mM NO (Figure S1C). The koff(CO) of wt Cb H-NOX thus determined was 1.0 s−1 (Table 1). The equilibrium dissociation constant, KD(CO) for wt Cb H-NOX, was calculated to be 6.7 × 10−7 M (Table 1).
Table 1.
Gas Ligand Binding Parameters: kon, koff, and KD of Cb and Tt H-NOXs, in Comparisons with Published Values
| O2 | CO | NO | |||||||
|---|---|---|---|---|---|---|---|---|---|
| H-NOX | kon (M−1 s−1) | koff (s−1) | KD (M) | kon (M−1 s−1) | koff (s−1) | KD (M) | kon (M−1 s−1) | koff (s−1) | KD (M) |
| wt Cb | ~2500b | 5.3 × 10−5 | 1.5 × 106 | 1.0 | 6.7 × 10−7 | 1.5 × 108 | 1.2 × 10−2 | 8.0 × 10−11 | |
| 1.1 × 10−3a | |||||||||
| 1.5 × 106d | ~ 108c | 3 × 10−6c | 3.0 × 10−14c | ||||||
| Y139F Cb | 3.4 × 106 | 1.1 | 3.3 × 10−7 | 4.3 × 107 | 5.0 × 10−3 | 1.2 × 10−10 | |||
| wt Tt | 4.3 × 107 | 1.9 | 4.4 × 10−8 | 3.3 × 106 | 0.5 | 1.6 × 10−7 | 1.5 × 108 | 3.4 × 10−3 | 2.3 × 10−11 |
| 1.36 × 107e | 1.2e | 8.97 × 10−8e | 2.18× 106g | 3.56g | 1.63 × 10−6 | 5.6 × 10−4 | |||
| 2.5 × 107f | 1.2f | 4.8 × 10−8f | 4.1 × 106g | ||||||
The CO binding kinetics to ferrous Y139F Cb H-NOX was similar to that of wt Cb H-NOX (Figure S2). Binding of CO to ferrous Y139F Cb H-NOX shifted the Soret peak from 432 to 424 nm in a single step reaction (Figure S2A). The kon(CO) of Y139F Cb H-NOX is slightly faster than that of wt Cb H-NOX, kon(CO) = 3.4 × 106 M−1 s−1 (Figure S2B, inset and Table 1). The dissociation rate constant of Y139F Cb H-NOX CO-heme complex, koff(CO) = 1.1 s−1 (Figure S2C and Table 1), is the same as that of wt Cb H-NOX CO-heme complex. The equilibrium constant of Y139F Cb H-NOX for CO, KD(CO) was calculated as 3.3 × 10−7 M (Table 1). Y139F Cb H-NOX binds CO with slightly higher affinity than wt Cb H-NOX.
The kinetics for CO binding to wt Tt H-NOX were similarly measured (data not shown), kon(CO) = 3.3 × 106 M−1 s−1 and koff(CO) = 0.5 s−1 (Table 1). The kon(CO) is quite similar to published values, either with laser photolysis or stopped-flow methods.15 On the other hand, koff(CO) measured in this study is about 7-fold lower than the published values.15 The reason for this discrepancy is unclear since similar NO displacement method was used in both studies.15 In this study, wt Tt H-NOX is measured to bind CO slightly more tightly than wt Cb H-NOX with a KD(CO) = 1.6 × 10−7 M (Table 1).
Binding of NO to Ferrous Cb and Tt H-NOXs.
To characterize the binding of NO to Cb H-NOX, a rapid-scan stopped-flow method was used to follow the optical changes during the reactions of ferrous Cb H-NOX with either stoichiometric (Figure 1A) or excess NO (10 equiv, Figure 1C). Ferrous wt Cb H-NOX bound stoichiometric NO promptly with most of the reaction lost in the dead time of the stopped-flow apparatus, indicating a very large kon(NO) (Figure 1A). The Soret peak shifted from 431 to 420 nm with isosbestic points at 424 and 455 nm (Figure 1A). The wavelength of the product indicates the formation of a typical 6c NO-heme-His complex (Figure 1A, inset, A → B). The conformation of the 6c NO-heme-His complex then began to change indicated by the decay of the Soret peak (Figure 1A, inset, B → C). In a longer reaction time, the Soret peak further decayed and shifted to 409 nm (Figure 1A), indicating breakage of Fe-His bond in the initial 6c NO-heme-His complex and conversion to a 5c NO-heme complex. When reacted with 10 equiv NO, a 6c NO-heme-His complex formed in the dead time of the stopped-flow apparatus as indicated by the first captured spectrum (1.28 ms after mixing) which exhibited an absorption maximum at 420 nm (Figure 1C). The observed optical change was a monophasic shift to 407 nm (Figure 1C), indicating conversion to a 5c NO-heme complex. However, the conversion was likely incomplete since a pure 5c NO-heme complex usually exhibits a Soret peak at wavelength ≤400 nm in many hemoproteins including H-NOXs and sGC.5,12,16,17
Figure 1.
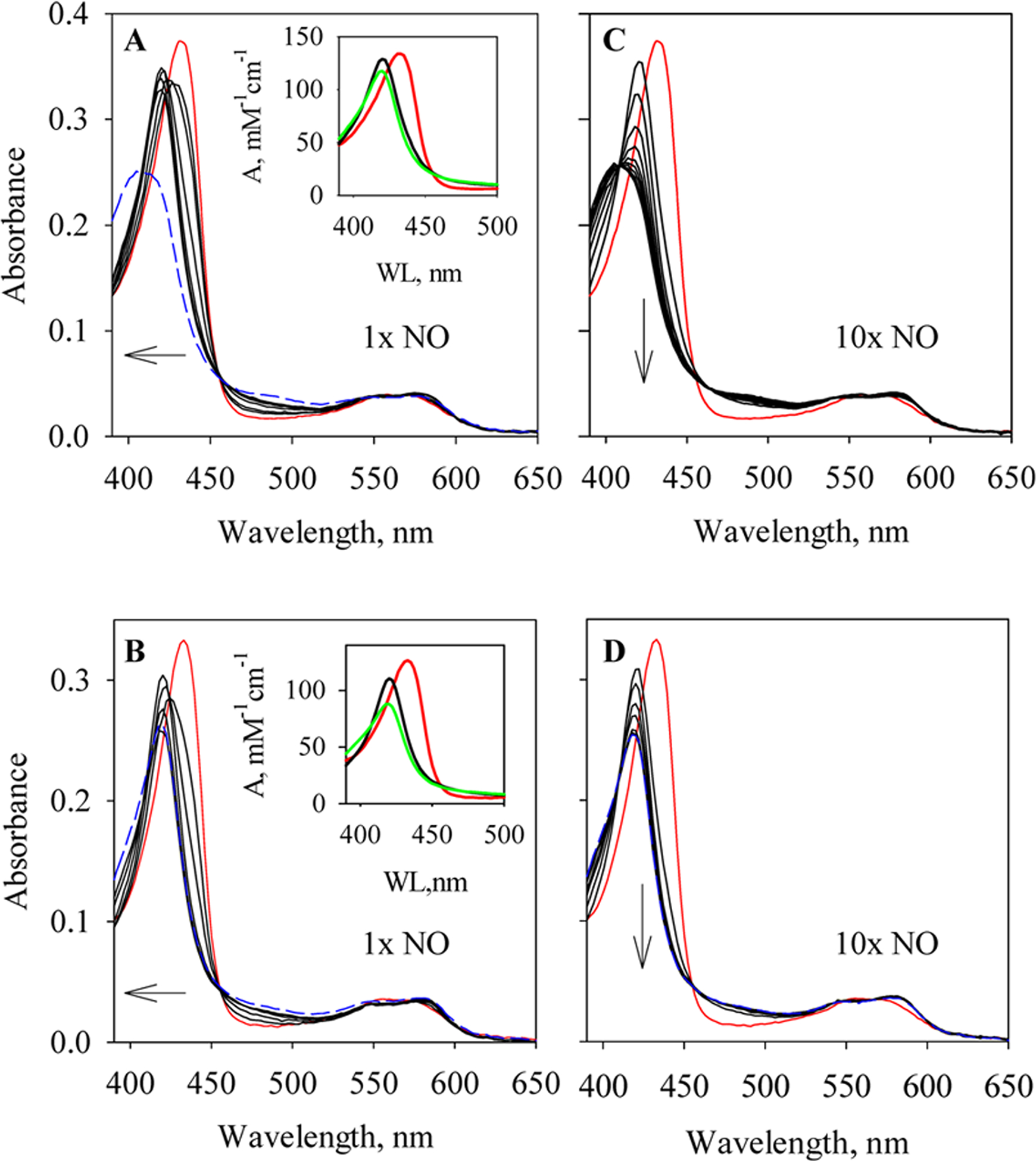
Rapid-scan of NO binding to wt and Y139F Cb H-NOXs. (A) Reaction of 3.3 μM wt Cb H-NOX with 1 equiv of NO monitored for 0.3 s (black). Spectra at selected time points, 1.28, 3.84, 11.5, 49.9, 152.3, and 254.7 ms, are presented. The spectrum of wt Cb H-NOX mixed with anaerobic buffer (red) represents the spectrum at 0 s of the reaction. The Soret peak shifts from 432 to 420 nm; and further shifts to 409 nm after 6 s (blue dashed). Inset. The Soret region of the two intermediates resolved from the 0.3 s reaction, B (black) and C (green), based on model A → B → C, are plotted together with that of ferrous wt Cb H-NOX (A, red). (B) Reaction of 3.4 μM Y139F Cb H-NOX with 1 equiv NO monitored for 0.3 s (black). Spectra at selected time points, 1.28, 3.84, 24.3, 126.7, and 254.7 ms, are presented. The spectrum of Y139F Cb H-NOX mixed with anaerobic buffer (red) represents the spectrum at 0 s of the reaction. The Soret peak shifts from 432 to 420 nm, and does not shift further up to 524 s (blue dashed). Inset. The Soret region of the two optically resolved intermediates from the 0.3 s reaction, B (black) and C (green), based on model A → B → C, are plotted together with that of ferrous Y139F Cb H-NOX (A, red). (C) Reaction of 3.3 μM wt Cb H-NOX with 10 equiv of NO monitored for 0.3 s (black). Spectra at selected time points, 1.28, 11.5, 24.3, 37.1, 49.9, 62.7, 75.5, 101.1, 152.3, 203.5, and 254.7 ms, are presented. Red line, same as in (A). The Soret peak shifts from 432 to 409 nm. (D) Reaction of 3.4 μM Y139F Cb H-NOX with 10 equiv NO monitored for 0.3 s (black). Spectra at selected time points, 1.28, 11.5, 24.3, 37.1, 75.5, 126.7, and 254.7 ms, are presented. Red line, same as in (B). The Soret peak shifts from 432 to 420 nm without any further shift after 524 s (blue dashed). The arrows indicate the directions of spectral changes.
The optical change during the reaction of ferrous Y139F Cb H-NOX with stoichiometric NO (Figure 1B) was similar to that of wt Cb H-NOX (Figure 1A), the Soret peak shifted from 432 to 420 nm with isosbestic points at 423 and 457 nm (Figure 1B), indicating the formation of a 6c NO-heme-His complex (Figure 1B, inset, A → B). Most of the optical change was lost in the dead time of the stopped-flow apparatus, indicating a very fast binding of NO to Y139F Cb H-NOX. The conformation of the 6c NO-heme-His complex then began to change, indicated by the decay of the Soret peak (Figure 1B, inset, B → C). However, different from the case in wt Cb H-NOX, the Soret peak further decayed slightly without further shift in wavelength even after much longer reaction time (Figure 1B), indicating that the initial 6c NO-heme-His complex in Y139F Cb H-NOX underwent certain conformational change(s) without dissociation of the proximal histidine ligand. In the reaction of Y139F Cb H-NOX with excess NO, a 6c NO-heme-His complex formed in the dead time of the stopped-flow apparatus, and the first spectrum captured at 1.28 ms exhibited an absorption peak at 420 nm (Figure 1D). However, in contrast to that in wt Cb H-NOX (Figure 1B), the Soret peak of the 6c NO-heme-His complex formed in Y139F Cb H-NOX with excess NO decayed monophasically without shift in wavelength (Figure 1D), indicating that certain conformational change(s) happened without dissociation of the proximal histidine ligand. In line with that observed previously, the Fe-His bond between heme and the proximal histidine in the 6c NO-heme-His complex of Cb H-NOX is stabilized by the Y139 to phenylalanine mutation.6
To determine the large formation rates of the 6c NO-heme-His complexes in wt and Y139F Cb H-NOXs, kon(NO), we measured the kinetics under true second-order conditions, using a [NO]:[Cb H-NOX] ratio of 1:1 as described before (Figure 2A,C).5,11 The time courses at 430 nm (wt Cb H-NOX) or 432 nm (Y139F Cb H-NOX) were fitted to a second-order, irreversible mechanism (eq 3, Figure 2A,C). The formation rate constants of 6c NO-heme-His complexes were obtained as kon(NO) = kobs × Δε and Δε430 nm = 37 mM−1 cm−1 for wt Cb H-NOX (Figure 1A), while Δε432 nm = 39.2 mM−1 cm−1 for Y139F Cb H-NOX (Figure 1C). The kon(NO) was 1.5 × 108 M−1 s−1 for wt Cb H-NOX (Table 1), similar to the value estimated previously for Cb H-NOX based on diffusion limitation assumption.6 Y139F Cb H-NOX had a slightly slower kon(NO), 4.3 × 107 M−1 s−1 (Table 1).
Figure 2.
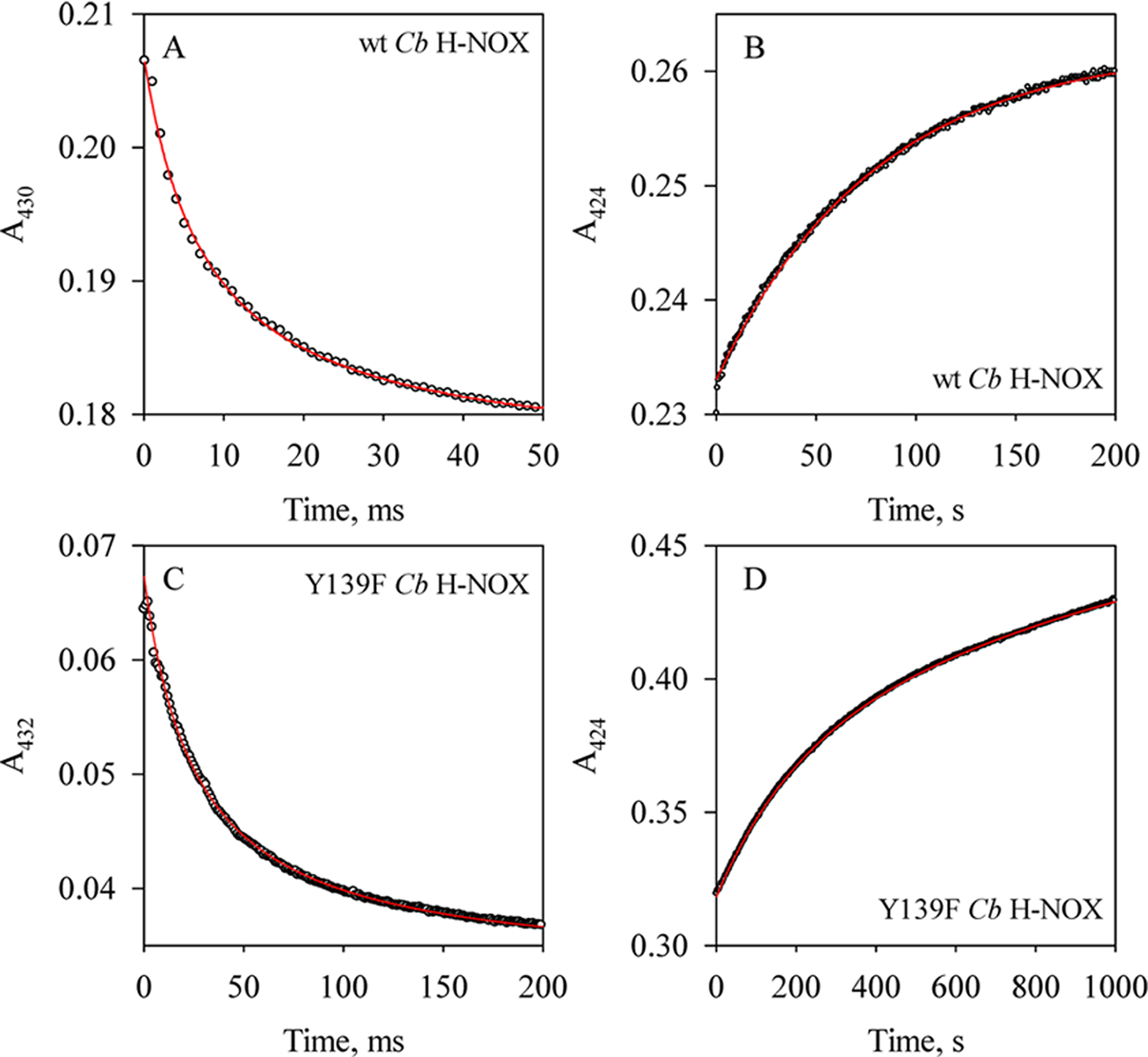
Kinetics of the formation/dissociation of 6c NO complexes of wt and Y139F Cb H-NOXs. (A) Time course of A430 during the reaction of 2.3 μM wt Cb H-NOX with 1 equiv of NO (circles) and its fit to the eq 3 (red line). (B) Time course of A424 during the sequential stopped-flow reaction, 2.3 μM wt Cb H-NOX was first reacted with 1 equiv of NO, aged for 100 ms and then reacted with 50 mM dithionite with 1 mM CO (circles), and fitting to equation (red line), where Amax is the maximal level of A424 and t is the reaction time. (C) Time course of A432 during the reaction of 0.5 μM Y139F Cb H-NOX with 1 equiv of NO (circles), and kobs obtained by fitting to eq 3 (red line). (D) Time course of A424 during the reaction of 3.0 μM Y139F Cb H-NOX with 1 equiv of NO then reacted with 50 mM dithionite with 1 mM CO (circles), and kobs obtained by fitting to the same equation as in (B) (red line).
The dissociation rate constants of the 6c NO-heme-His complexes in both wt and Y139F Cb H-NOXs, koff(NO), were measured using the sequential stopped-flow method.5,11 The 6c NO-heme-His complexes were generated by reacting either wt or Y139F Cb H-NOX with stoichiometric NO, aged for 100 ms, and then reacted with 0.5 mM CO in 25 mM Na2S2O4 to displace and consume the dissociated NO, respectively.5,11 Exponential increase in A424 was observed following the second mixing (Figure 2B,D), indicating the formation of CO-heme complex. Fitting the time courses at 424 nm to the single-exponential function (eq 1) yielded a dissociation rate constant koff(NO) of 0.012 s−1 for the 6c NO-heme-His in wt Cb H-NOX and a slightly slower rate of 5.0 × 10−3 s−1 for the same complex in Y139F Cb H-NOX (Table 1). The koff(NO) are much slower than kon(CO) of wt or Y139F Cb H-NOXs (Table 1), in line with the rate of NO displacement by CO being limited by the NO dissociation from the 6c NO-heme-His complexes. The KD(NO)(6c)’s, calculated as the ratio of koff(NO)/kon(NO), were 8.0 × 10−11 M and 1.2 × 10−10 M for wt and Y139F Cb H-NOXs, respectively (Table 1). The reaction to form 6c NO-heme-His complex in Cb H-NOX is represented in Scheme 1 (A ⇔ C).
In the presence of excess NO, the 6c NO-heme-His complex in wt Cb H-NOX converted to a 5c NO-heme complex (Scheme 1, E), and the kinetics of this process was studied under pseudo first-order conditions, following the time courses of A422 (Figure 3). Formation of 6c NO-heme-His (Scheme 1, C) complex was complete within the dead time of the stopped-flow apparatus, and the kobs’s are the rate constants for its further conversion to the 5c NO-heme complex (Figure S3). The kobs’s increased with [NO] and gradually leveled off at high [NO] (Figure 3, inset). This pattern of [NO]-dependence of kobs suggests (1) in the presence of excess NO, conversion to the 5c NO-heme complex presumably proceeds through a quaternary complex, NO-heme--NO(--His) (Scheme 1, D), formed by the reaction of 6c NO-heme-His complex with NO at the proximal side of heme (Scheme 1, C ⇔ D) and thus shows [NO]-dependence of kobs; (2) conversion from the hypothetic quaternary complex to the 5c NO-heme complex (Scheme 1, D ⇔ E) becomes rate-limiting at high [NO]. The formation of 5c NO-heme complex in wt Cb H-NOX with excess NO can be represented with reactions I and II:
| (I) |
| (II) |
At low [NO], the first step is rate-limiting and the kobs increases with [NO], while at high [NO], the second step becomes rate-limiting and the kobs levels off. The [NO]-dependence of kobs can be described using the following hyperbolic function:
| (5) |
Here the reassociation rate constant of NO with 5c NO-heme to form NO-heme--NO(--His) complex, k−2, is small and can be ignored. The k2 was estimated ~790 s−1 and KD ≈ 570 μM based on eq 5 (Figure 3, inset). Such a saturating behavior in the [NO]-dependence of 5c NO-heme formation kinetics was also observed for Vc H-NOX5 but not for sGC18 or Ns H-NOX.12 In fact the maximum formation rates for the 5c NO-heme complex are quite similar for Cb and Vc H-NOXs.5 A computer model was built based on reactions I and II to fit the time courses during the formation of the 5c NO-heme complex in Cb H-NOX. The values for k1 and k−1 obtained by simultaneous fitting to kinetic data obtained at nine different [NO] were 1.8 × 107 M−1 s−1 and 2.0 × 104 s−1, respectively (Figure S3). Although both k1 and k−1 were allowed to float during the fitting, KD = 1.1 mM of reaction I, calculated from the fitted values of k1 and k−1, agrees reasonably well with the experimentally measured value (Figure 3, inset). According to the mechanism, the hypothetic quaternary complex has the best chance accumulating enough for observation at high [NO]. However, at the highest [NO] available (1 mM), the Soret peak of the spectrum captured at the shortest reaction time (1.28 ms) was at 420 nm, the same as that observed at much lower [NO] and the subsequent spectral change exhibited a single one step shift to 407 nm (data not shown). Therefore, at the highest [NO], no spectral feature attributed to the predicted transient quaternary complex could be resolved in addition to the initial 6c and final 5c NO-heme complexes. The transient nature of the quaternary complex is due to the very large dissociation rate constant koff of the quaternary complex, 2 × 104 s−1 (Scheme 1, D → C). Moreover, the equilibrium constant, KD = koff/kon, between the 6c NO-heme-His complex and the quaternary complex NO-heme--NO(--His) complex, can be estimated to reach 1.1 at the highest [NO] available. This large KD led to the incomplete conversion from the 6c NO-heme-His complex to the 5c NO-heme complex (Scheme 1, C → E), although the conversion from the quaternary complex to the 5c complex is essentially irreversible (Scheme 1, D ⇔ E).
Figure 3.
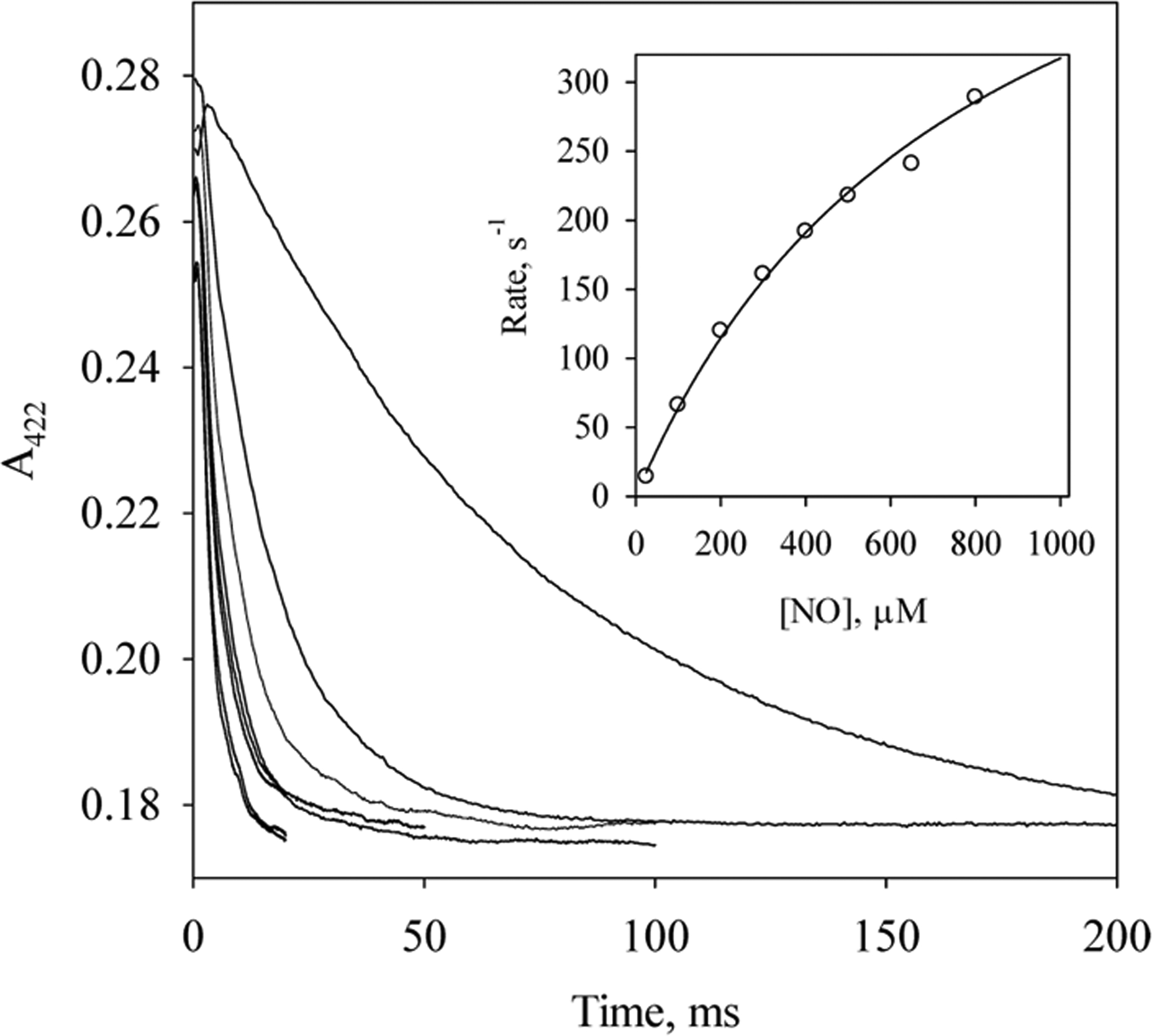
Saturation kinetics for the reaction of wt Cb H-NOX with excess NO. Time courses of A422 during the reactions of 2.3 μM Cb H-NOX with 25, 100, 200, 300, 400, 500, 650, 800, and 1000 μM NO (from top to bottom, respectively) at 24 °C. Inset. Dependence of observed rate constants, kobs (○) on [NO] and its fit to eq 5 (line).
The dissociation rate constants of the 5c NO-heme complex in wt Cb H-NOX, koff(5c) (Scheme 1, E → A), were measured by reacting the 5c NO-heme complex with 0.5 mM CO in 25 mM Na2S2O4 (data not shown). The 5c NO-heme exhibited a biphasic NO dissociation kinetics, with rates of 1.1 × 10−2 s−1 (45% total amplitude change) and 1.1 × 10−3 s−1 (55% total amplitude change) (Table 1). The rate of the fast phase of NO dissociation is essentially the same as the koff(NO) of its 6c NO-heme-His complex (Figure 2B) which did not fully transform to 5c NO-heme complex. The slow phase thus represents the NO dissociation from the 5c NO-heme complex in wt Cb H-NOX at a rate 10 times slower than from the corresponding 6c NO-heme-His complex (Table 1). The kinetic events during the NO binding to Cb H-NOX are summarized in Scheme 1 (A → E).
The association/dissociation rate constants for the 6c NO-heme-His complex in Tt H-NOX were measured using the same method as for Cb H-NOX, and we obtained kon(NO) = 1.5 × 108 M−1 s−1 and koff(NO) = 3.4 × 10−3 s−1 (Table 1). Although the koff(NO) of wt Tt H-NOX was measured before using similar method,19 it is 6-fold lower than that measured in this study. The reason for the discrepancy is not clear yet. Compared to the kinetic parameters of NO binding in Cb H-NOX, kon(NO) of the 6c NO-heme-His complex in wt Tt H-NOX is the same as in wt Cb H-NOX, but koff(NO) in wt Tt H-NOX is very similar to that in Y139F Cb H-NOX (Table 1). The KD(NO) of the 6c NO-heme-His complex in wt Tt H-NOX, 2.3 × 10−11 M, a little lower than that of the corresponding 6c NO-heme-His complex in Cb H-NOX (Table 1).
EPR of NO Complexes in Cb and Tt H-NOXs.
Spectroscopic studies indicate that distal tyrosine modulates the strength of the Fe-His bond in the 6c NO-heme-His complex in Cb H-NOX but not in Tt H-NOX.6,20 In this study, the NO-heme complexes formed in these H-NOXs were studied more in detail using EPR. In the presence of excess NO, the EPR spectrum of NO-heme complex in wt Cb H-NOX majorly exhibited features typical for a 5c NO-heme complex (Figure 4a), confirming breakage of the Fe–His bond in the initial 6c NO-heme-His complex, in line with the shift of absorption peak from 420 to 407 nm during the reaction of wt Cb H-NOX with excess NO (Figure 1C, and previously6). On the other hand, the small EPR feature at g = 1.978 indicated the existence of a minor portion of a 6c NO-heme-His complex (Figure 4a). The 6c NO-heme-His complex accounted for ~30% of total [spin] against a standard EPR spectrum of a pure 5c NO-heme complex (Figure 4c) obtained by subtracting 30% of a simulated 6c NO-heme-His EPR spectrum (Figure 4b) from spectrum 4a. EPR therefore demonstrates an incomplete conversion from the initial 6c NO-heme-His complex to 5c NO complex in wt Cb H-NOX, corroborating the observed Soret peak at 407 nm (Figure 1C) not that for a pure 5c NO-heme complex (≤400 nm). On the other hand, percentage of 6c and 5c NO complexes estimated by EPR varied from that estimated based on optical kinetic data (30% vs 45%) and the discrepancy could be due to the very low measurement temperature of EPR samples.
Figure 4.
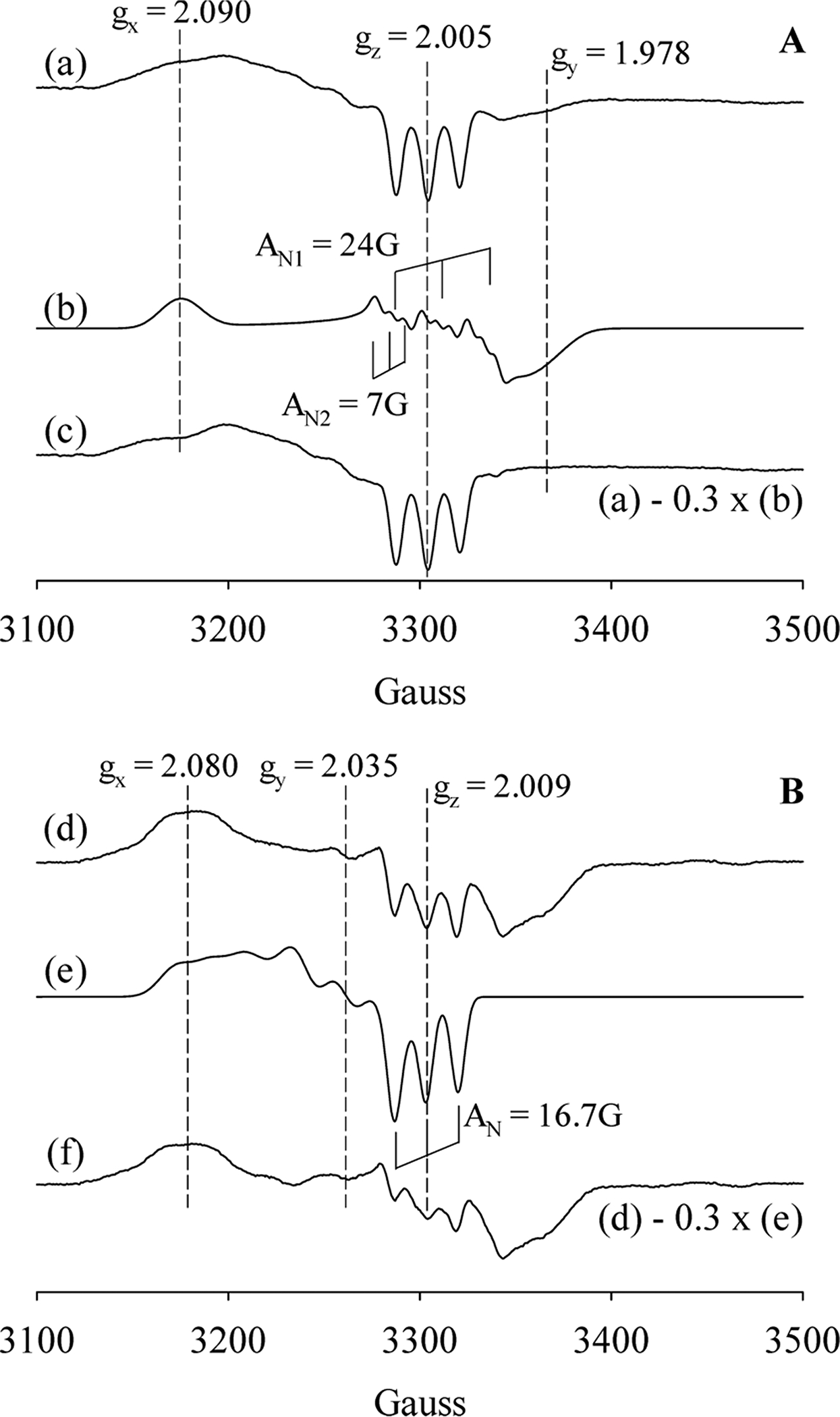
EPR of wt and Y139F Cb H-NOXs with excess NO. Panel A. (a) Reaction of 4.5 μM wt Cb H-NOX with 20 equiv of NO for 1 min, exhibiting EPR signatures of the majority a 5c NO-heme complex plus a minor portion of a 6c NO-heme-His complex. (b) EPR spectrum of a 6c NO-heme-His complex simulated using the following parameters: gx = 2.090, gy = 1.978, gz = 2.005; AN1: Ax = 6 G; Ay = 10 G and Az = 24.0 G; AN2: Ax = 6 G; Ay = 10 G and Az = 7 G. (c) Subtraction of 30% of (b) from (a) yields an EPR spectrum of a pure 5c NO-heme complex. Panel B. (d) Reaction of 6.0 μM Y139F Cb H-NOX reacted with 20 equiv NO for 1 min, exhibiting EPR signatures of the majority a 6c NO-heme-His complex with a minor portion of a 5c NO-heme complex. (e) EPR spectrum of a 5c NO-heme complex simulated using the following parameters: gx = 2.080, gy = 2.035, gz = 2.009; AN: Ax = 18 G; Ay = 20 G and Az = 16.7 G. (f) Subtraction of 30% of (e) from (d) yields an EPR spectrum of a pure 6c NO-heme-His complex.
In line with the optical data from this (Figure 1D) and previous studies,6 EPR data demonstrated the modulation effect of Y139F mutation on the stability of the Fe-His bond in the 6c NO-heme-His complex as significantly more EPR features due to a 6c NO-heme-His complex were observed in the reaction of Y139F Cb H-NOX with excess NO (Figure 4d). Some 6c NO-heme-His complex did convert to a 5c NO-heme complex in the 1 min reaction as indicated by the three-line hyperfine features observed at g = 2.009, which is typical of a 5c NO-heme complex (Figure 4d). About 30% 6c NO-heme-His was estimated to have converted, as indicated by the EPR of a nearly pure 6c NO-heme-His complex (Figure 4f) obtained by subtracting ~30% of a simulated 5c NO-heme EPR spectrum (Figure 4e) from spectrum 4d. Since optical data did not indicate significant conversion from 6c NO-heme-His complex to 5c NO-heme complex during the reaction of Y139F Cb H-NOX with excess NO (Figure 1D), the observed EPR features of a 5c NO-heme complex may be also introduced by the freezing of the EPR samples.
On the contrary to Cb H-NOX, optical data indicated that the corresponding Y140F mutation in Tt H-NOX does not change the coordination number of the NO complex, a 6c NO-heme-His complex is stable in both wt and Y140F Tt H-NOXs.20 With excess NO, both wt and Y140F Tt H-NOXs, EPR signatures typical of a 6c NO-heme-His complex were observed (Figure 5A,C), and the EPR spectra were nicely simulated as 6c NO-heme-His complexes (Figure 5B,D). Both sets of hyperfine splitting constants resulted from the NO nitrogen (AN1) and that from superhyperfine splitting from imidazole nitrogen (AN2) are similar: 24G/7G for wild type and 23.2G/6.7G for Y140F Tt H-NOX, respectively. Although the Fe–His bond stayed unbroken in both 6c NO-heme-His complexes, the EPR data demonstrated for the first time noticeably different lineshapes of the two 6c NO-heme-His complexes in wt and Y140F Tt H-NOXs (Figure 5). The most noticeable difference was the shift of gx from 2.090 to 2.069 of the 6c NO-heme-His (Figure 5A/B to 5C/D), indicating decreased heme rhombicity of the Y140F NO-heme-His complex. In conclusion, in Cb H-NOX, mutation of distal Y139 to phenylalanine stabilizes the Fe–His bond in its 6c NO-heme-His complex, but in Tt H-NOX, the same mutation Y140F alters the geometry of heme without an obvious effect on the stability of Fe–His bond. The association/dissociation kinetics of Y140F Tt H-NOX for NO and CO was also measured in the same way as above and is similar to that of wt Tt H-NOX (Table S1).
Figure 5.
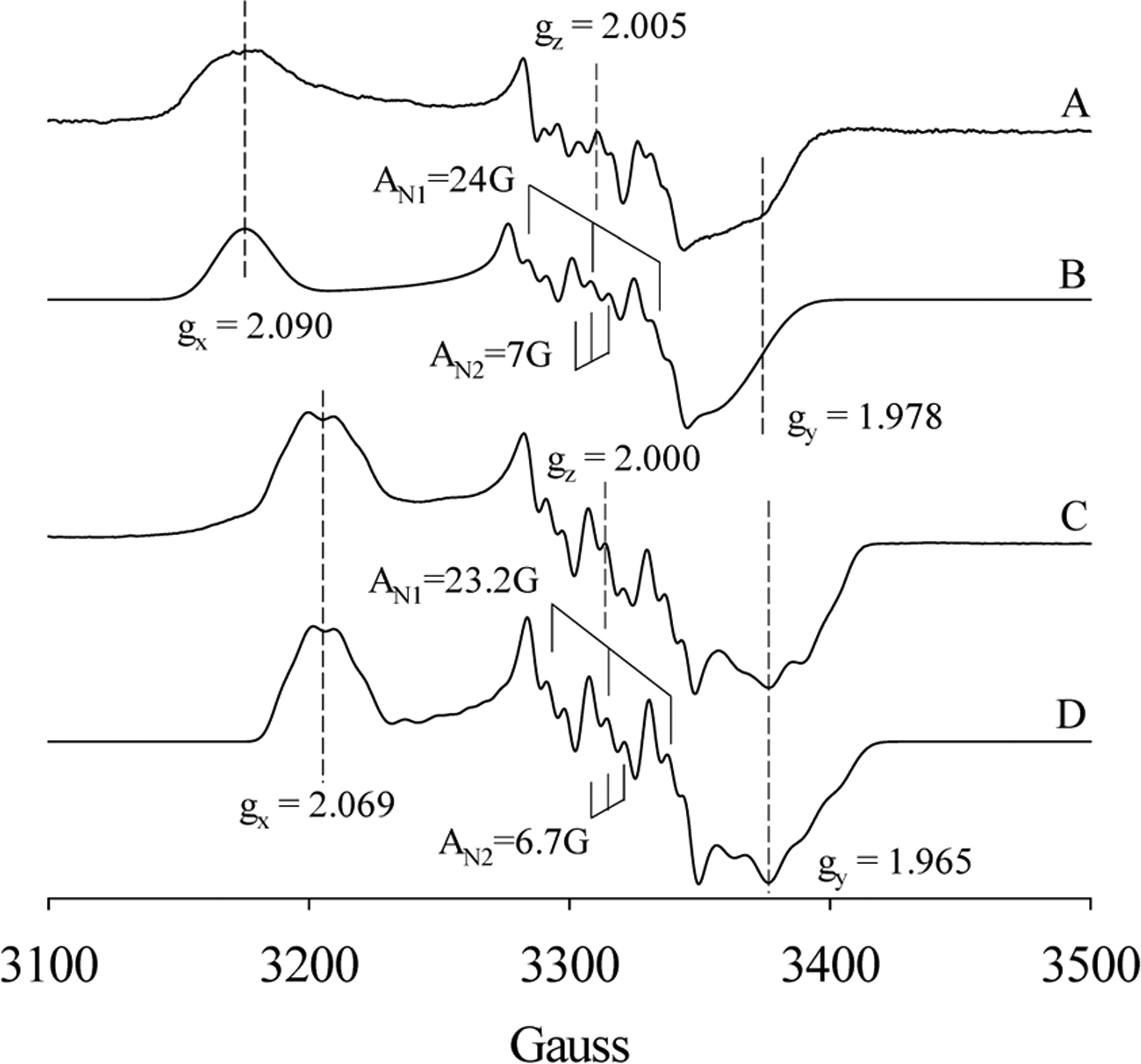
EPR of wt and Y140F Tt H-NOXs with excess NO. (A) NO-heme-His complex trapped after 13 μM wt Tt H-NOX reaction with 30 equiv of NO for 1 min. (B) Simulation of the EPR spectrum A using the following parameters: gx = 2.090, gy = 1.978, gz = 2.005; AN1: Ax = 6 G; Ay = 10 G and Az = 24.0 G; AN2: Ax = 6 G; Ay = 10 G and Az = 7 G. (C) NO-heme-His complex trapped after 95 μM Y140F Tt H-NOX reaction with 5 equiv of NO for 1 min. (D) Simulation of the EPR spectrum C using the following parameters: gx = 2.069, gy = 1.965, gz = 2.000; AN1: Ax = 10 G; Ay = 13 G, and Az = 22.2 G; AN2: Ax = 6 G; Ay = 13 G, and Az = 6.7 G.
Reaction of O2 with Ferrous Tt and wt and Y139F Cb H-NOXs.
Tt H-NOX has been shown to bind O2 tightly.21 In this study, the association/dissociation kinetics of O2 to Tt H-NOX was remeasured using another method and compared to the published values. The binding rate constant, kon(O2), was determined under true second-order reaction conditions, reacting ferrous Tt H-NOX with 1 equiv O2 and following the time course of A430 (Figure S4A). Using this method, the kon(O2) for Tt H-NOX was measured as 4.3 × 107 M−1 s−1 (Table 1). The koff(O2) from oxyferrous Tt H-NOX was measured by following the time course of A430, mixing oxyferrous Tt H-NOX with 25 mM Na2S2O4 using the latter to consume the dissociated O2 (Figure S4B). The kobs was 1.9 s−1 and showed no dependence on the concentration of Na2S2O4 (data not shown), indicating that the rate-limiting step was O2 dissociation and the kobs was the true koff(O2) of oxyferrous Tt H-NOX. The affinity of wt Tt H-NOX for O2 was calculated as KD(O2) = koff(O2)/kon(O2) = 4.4 × 10−8 M (Table 1). The measured kinetic parameters and affinity of wt Tt H-NOX to O2 in this study (Table 1) are all similar to the published values.20,22
When anaerobic wt Cb H-NOX was mixed with aerobic buffer, an oxyferrous complex formed immediately (Scheme 1, A ⇔ F) and the reaction finished in the dead time of the stopped-flow apparatus (Figure 6A). The first spectrum captured at 1.28 ms after mixing showed a Soret peak at 422 nm, and the absorption band at 564 nm of ferrous wt Cb H-NOX split into a α band at 583 nm and a β band at 549 nm, consistent with formation of an oxyferrous complex (Figure 6A). The method which enabled successful measurement of kon(O2) for wt Tt H-NOX (see above) failed for wt Cb H-NOX, since most of the reaction finished within the dead time of the stopped-flow apparatus under similar experimental conditions, by mixing wt Cb H-NOX with even stoichiometric O2. This was likely due to a very large kobs = kon(O2) + koff(O2), ≫ 600 s−1, of wt Cb H-NOX. Large koff(O2) also prevented fitting accurately to the time course of reaction III based on eq 3.
| (III) |
Since reaction III reached equilibrium quickly, preventing measuring kon(O2) and koff(O2) accurately, the KD(O2) of oxyferrous wt Cb H-NOX was measured in the stopped-flow by titrating wt Cb H-NOX with varying levels of O2, and the decrease of A432, due to the formation of oxyferrous complex (reaction III), versus [O2] was monitored (Figure 6B). The KD(O2) of wt Cb H-NOX was obtained by fitting to the plot of A432 versus [O2] to the following equation:
| (6) |
The affinity of wt Cb H-NOX for O2 obtained by this method was KD(O2) = 5.3 × 10−5 M (Table 1). Compared to wt Tt H-NOX, wt Cb H-NOX binds O2 about 1200 times more weakly. Nonetheless, an oxyferrous complex does form in Cb H-NOX under atmospheric O2 pressure. If the kon(O2) of wt Cb H-NOX is assumed similar to that of wt Tt H-NOX, koff(O2) of wt Cb H-NOX can be estimated: koff(O2) = KD(O2) × kon(O2) = ~2500 s−1, providing an estimation for how fast reaction III reaches equilibrium (Scheme 1, A ⇔ F).
Figure 6.
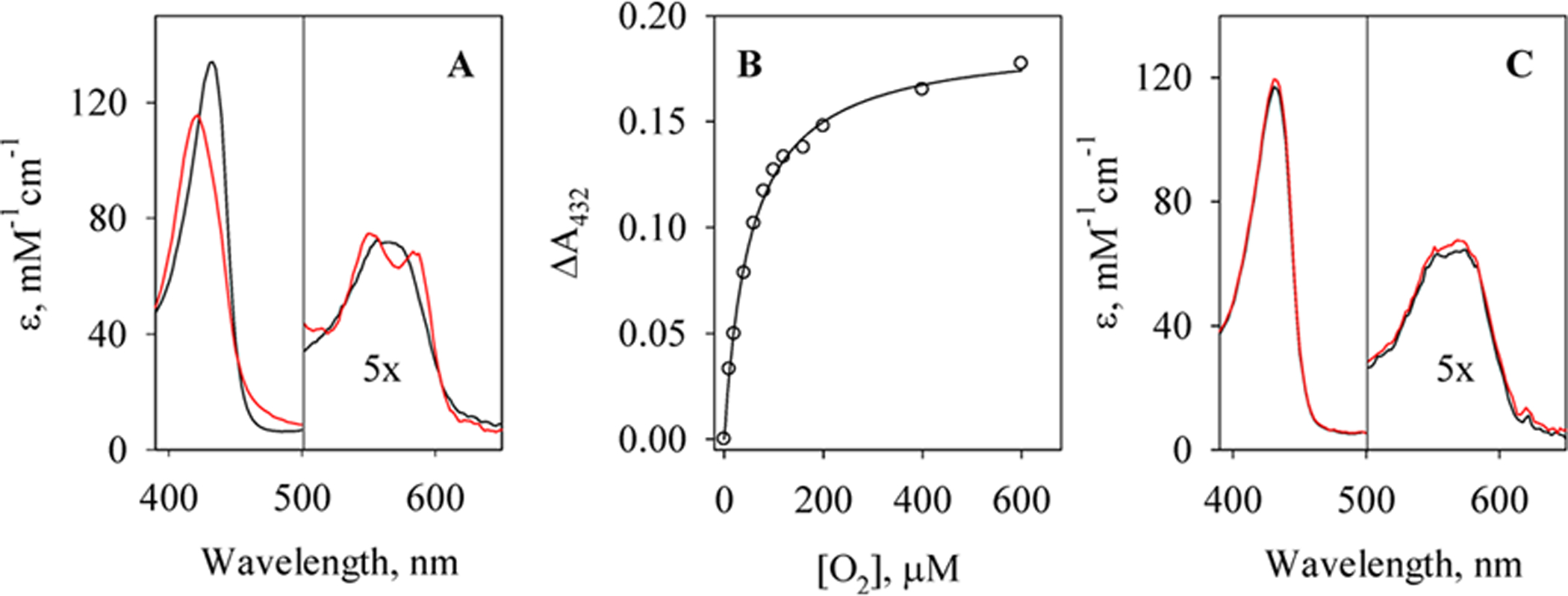
Interaction of O2 with wt and Y139F Cb H-NOXs. (A) Spectra of 3.6 μM anaerobic ferrous Cb H-NOX (black) and the first spectrum of rapid-scan reaction with aerobic buffers (red), [O2]final ≈ 120 μM. The y-axis represents the extinction coefficients. (B) The change of A432, the Soret of Cb H-NOX, in the reaction of 3.3 μM Cb H-NOX with various concentrations of O2 (circle) and its fit using eq 6 (solid line). (C) Spectra of 2.2 μM anaerobic Y139F Cb H-NOX before (black) and after (red) mixing with O2-saturated buffer, [O2]final ≈ 600 μM. The y-axis represents the extinction coefficients.
Mutating Y139 to phenylalanine excludes Y139F Cb H-NOX from binding O2, as no formation of any oxyferrous complex was observed when anaerobic Y139F Cb H-NOX was reacted with O2-saturated buffer, with a [O2] = 600 μM (Figure 6C). The same mutation in Tt H-NOX, Y140F, also abolishes the formation of any oxyferrous complex in Y140F Tt H-NOX.23 Both ferrous wt and Y139F Cb H-NOXs auto-oxidized to ferric state after prolonged incubation with O2, presumably through transient association with O2 (data not shown).
DISCUSSION
When the logarithms of true KD(NO), KD(CO), and KD(O2) of Cb and Tt H-NOXs are plotted in “sliding scale plot”, their log KD(NO)-log KD(CO) lines parallel with those of sGC, Ns, Vc H-NOXs and heme model compound (Figure 7). Therefore, KD(NO) and KD(CO) of both Cb and Tt HOXs obey the “sliding scale rule”. On the other hand, compared to that of Ns H-NOX, the log KD(CO)-log KD(O2) lines in both Cb and Tt H-NOXs deviate downward from the extensions of their log KD(NO)-log KD(CO) lines (Figure 7), reflecting their enhanced O2 affinity. The measured KD(O2)’s of wt Cb and Tt H-NOXs, 5.3 × 10−5 M and 4.4 × 10−8 M, respectively, much lower than the values estimated by linear extrapolation from log KD(NO) via log KD(CO), ~5.3 × 10−3 M for Cb H-NOX and ~1.0 × 10−3 M for Tt H-NOX, respectively. The deviation of its KD(O2) in Tt H-NOX from the “sliding scale rule” has been attributed to the stabilization of its oxyferrous complexes by its distal hydrogen bonding networks.3,11 Such a distal hydrogen bonding network likely exists in Cb H-NOX due to the conservation of the several key amino acid residues,6 resulting in stabilized oxyferrous complex and enhanced affinity for O2. Compared to the previous study on the gaseous ligand binding of Cb H-NOX,6 two major differences are noticeable: (1) a much lower KD(NO) was measured previously (Figure 7, dark yellow circle and dashed line) and (2) oxyferrous Cb H-NOX was observable and characterized in this study but not reported previously.6 The possible reasons for these differences are discussed below.
Figure 7.
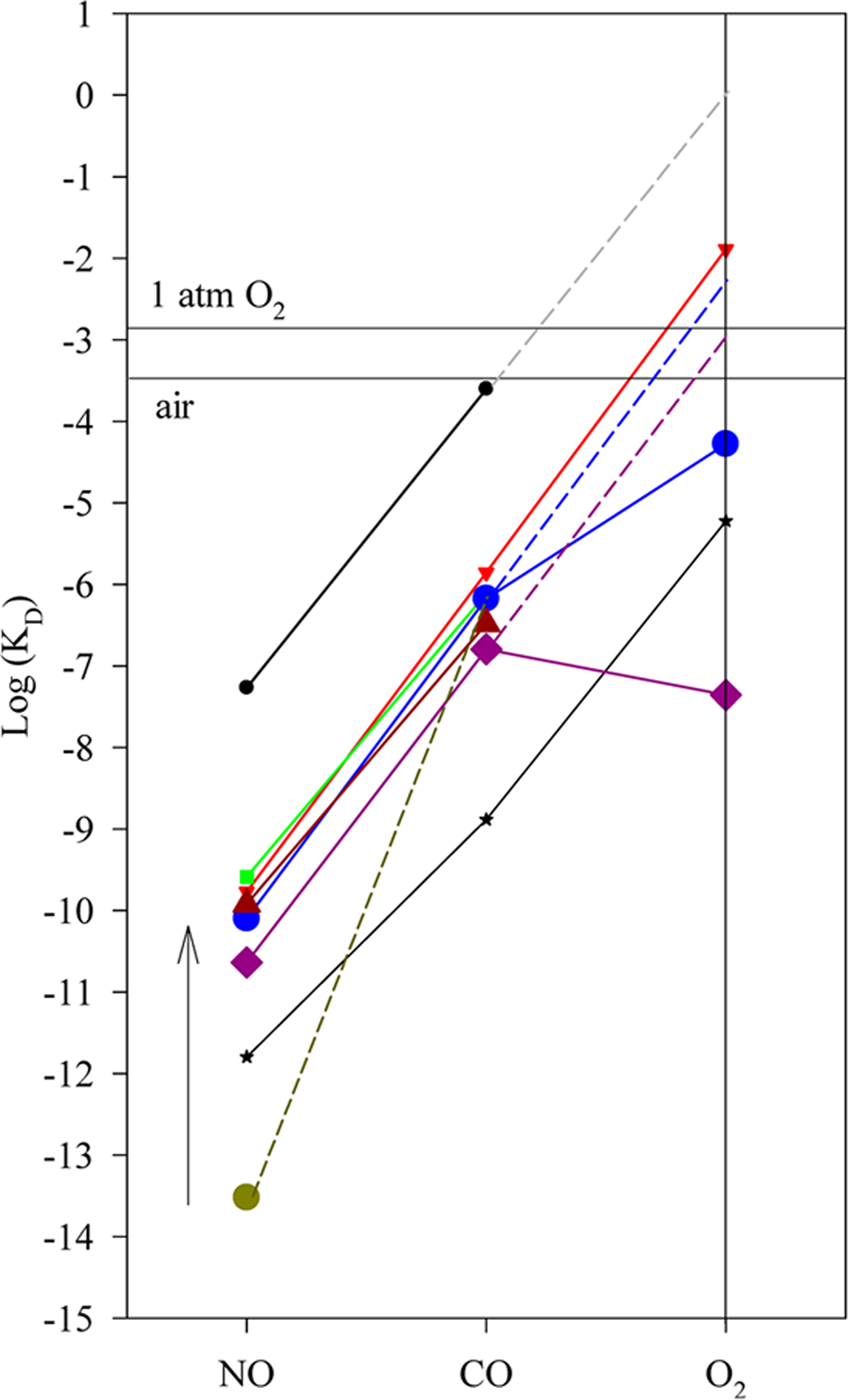
Relationship of log KD’s for H-NOXs versus ligand type. The measured logarithm values of KD(NO), KD(CO), and KD(O2) of H-NOXs are plotted versus ligand type. KD’s measured for sGC and heme model ferrous PPIX(1-MeIm) are also plotted for comparisons. Wt Cb H-NOX (blue circle); Y139F Cb H-NOX (brown triangle); Tt H-NOX (purple diamond); Vc H-NOX (green square); Ns H-NOX (red triangle); sGC (black circle) (an extrapolation from sGC log KD(NO)-log KD(CO) line, gray dashed line, predicts its KD(O2) ≈ 1.1 M) and ferrous PPIX (1-MeIm) (black star). The KD(NO) (dark yellow circle) of Cb H-NOX previously measured6 is connected to its KD(CO) by the broken line; the change between the KD(NO) measured in this study, and the previously measured value is represented by the arrow. The blue and purple dashed lines represent the prediction of KD(O2)’s of Cb and Tt H-NOXs, respectively, based on the extrapolation from their log KD(NO)-log KD(CO) lines.
NO Binding to Cb H-NOX.
To calculate the true KD(NO) in Cb H-NOX, both kon(NO) and koff(NO) should be of the same chemical reaction, namely, those for 6c NO-heme-His complex formation (Scheme 1, A ⇔ C):
| (IV) |
In the previous study, the kon(NO) of ~108 M−1 s−1 was not determined experimentally but an estimated value based on the assumption of diffusion limitation.6 This is a good assumption and confirmed by the kon(NO) measured experimentally in this study (Table 1), and the kon(NO) of Cb H-NOX is comparable to those of many other bacterial H-NOXs and hemoproteins.5,6,11 The femtomolar binding of NO to Cb H-NOX measured previously,6 3.0 × 10−14 M which is several orders tighter than the KD(NO)’s of other H-NOXs,5,12 was majorly due to the very slow koff(NO), measured by following the decay of the EPR signal of a 5c NO-heme complex observed in the purified Cb H-NOX.6 The reported KD(NO) was thus an apparent value, KD,apparent(NO) = koff(5c NO-heme)/kon(6c NO-heme-His). Since koff(NO) of a 5c NO-heme complex is usually significantly smaller than that of a 6c NO-heme-His complex (Table 1), the apparent KD,apparent(NO) previously calculated is much smaller than the true KD(NO) (reaction IV). Therefore, the previously reported value of KD(NO) was greatly underestimated for Cb H-NOX. A similar case of misrepresenting the KD(NO)(6c) of a hemoprotein with an apparent KD,apparent(NO) was sGC.3 In that case, koff = 6 × 10−4 s−1, for 5c NO-heme, was used to calculate a KD,apparent(NO) = 4.2 × 10−12 M, which was misinterpreted as the KD(NO) of sGC.24,25 The KD(NO)(6c) of sGC, 5.4 × 10−8 M, was later obtained by measuring the koff(NO) of its 6c NO-sGC complex, 27 s−1.11 Combining the results from this and previous studies, it is now clear that Cb H-NOX binds NO with a KD(NO) of 8.0 × 10−11 M (Table 1), which is comparable to the KD(NO)’s of other bacterial H-NOXs. On the other hand, dissociation of NO from Cb H-NOX may significantly slow down due to essentially irreversible conversion of its initial 6c NO-heme-His complex to a 5c NO-heme complex.
The koff(NO) of 5c NO-heme complex measured previously6 is about 3 orders of magnitude slower than that measured in this study (Table 1). One possible reason for the extremely slow koff(NO) measured previously is the inclusion of reducing agent, 15 mM β-mercaptoethanol (βME) in the buffer. βME effectively lowered the concentration of O2, which served as the only scavenging reagent for dissociated NO, resulting in significant rebinding of NO and therefore very slow decay of EPR signal of NO-heme complex. The presence of 85–90% unliganded Cb H-NOX in the previous EPR sample6 also slowed down the observed decay of the EPR signal. This portion of Cb H-NOX was present in the ferrous state when the protein was purified in the presence βME.6 In the EPR measurement of koff(NO), the reassociation of NO, which was present due to the lack of scavenger, with the unliganded ferrous Cb H-NOX may be prominent and led to further underestimation of the true koff(5c NO-heme). Slow koff(NO) of 5c NO-heme complex, lack of NO scavenger, and presence of prominent unliganded ferrous heme with open distal binding pocket collectively enabled the 5c NO-heme formed in vivo survive the purification in the previous study.6 No EPR signal of NO-heme complex(es) was observed in the purified Cb H-NOX in this study. Since no reducing agent was used during purification, Cb H-NOX purified in this study was in its ferric state, confirmed by no further reaction after adding ferricyanide (data not shown). NO completely dissociated from ferric Cb H-NOX due to the much weaker affinity of NO for ferric heme than for ferrous heme. Another minor factor which further reduces koff(NO) of the 5c NO-heme complex measured in the previous study was that the dissociation was monitored at relatively low temperature, 277 K,6 about 20 degrees lower than the experimental temperature in this study.
O2 Binding to Cb H-NOX.
In this study, it is clearly demonstrated that Cb H-NOX binds O2 under normal atmospheric pressure, but no oxyferrous Cb H-NOX complex was previously observed.6 One possible reason is that the auto-oxidation of oxyferrous Cb H-NOX is faster than that of Tt H-NOX. In fact, resting wt Cb H-NOX was purified in its ferric state, and resting wt Tt H-NOX was purified as an oxyferrous complex (Figure S5). However, since reducing reagent βME was included in the buffers for purifying wt Cb H-NOX previously,6 a more plausible reason for not detecting any oxyferrous Cb H-NOX previously was that βME decreased [O2] in the buffers to a level much lower than the KD(O2) of Cb H-NOX.
In the crystal structure of Tt H-NOX complexed with O2, the oxyferrous heme is stabilized by a network of hydrogen bonding including the primary interaction from the hydroxyl group of Y140 to O2 and the secondary ones involving W9 and N74 to Y140, fixing the orientation of Y140. Such a hydrogen bonding network enhances the affinity of Tt H-NOX for O2, confirmed because Y140F mutation eliminates O2 binding in Y140F Tt H-NOX.7,23 The distal heme pocket in Cb H-NOX is very similar to that in Tt H-NOX, revealed by structural comparison and sequence alignment between these two H-NOXs.6 The similarities include (1) conservation of the key residues involved in hydrogen bond network in Cb H-NOX, namely, Y139, W9, and N73; (2) conservation of residues which define the polarity of distal heme pocket, L144, F78, and I5 in Tt H-NOX and L143, F77, and V5 in Cb H-NOX. The similarity of the binding pockets in Cb and Tt H-NOXs suggests that they have similar kon(O2)’s, and the oxyferrous complexes in both H-NOXs are stabilized by hydrogen bonding interactions through their distal tyrosine residues.26 The importance of Y139 in stabilizing the oxyferrous complex in Cb H-NOX, just as Y140 in Tt H-NOX, is underscored by the undetection of oxyferrous complex in Y139F Cb and Y140F Tt H-NOXs.23 On the other hand, despite the similarity of the distal heme pockets in Cb and Tt H-NOXs, their KD(O2) constants exhibit a 3-order difference. The much larger KD(O2) of Cb H-NOX is likely due to its very large koff(O2), suggesting that the hydrogen bonding interactions for stabilizing the oxyferrous complex is weaker in Cb H-NOX compared to those in Tt H-NOX. Although Cb H-NOX is predicted to have similar distal hydrogen bonding constellation as Tt H-NOX, the unconserved residues around its heme pocket may introduce conformational flexibility which weakens distal hydrogen bonding leading to a less stable oxyferrous complex. A molecular dynamic simulation suggests that the dynamics of the protein conformation around the O2 binding site is different in Cb and Tt H-NOXs, providing some insights into the difference between the KD(O2)’s of these two H-NOXs’.27 On the other hand, such conformational flexibility is not expected to affect the bindings of CO and NO significantly based on the study showing that electrostatic hydrogen bonding donor at the distal heme pocket provides discriminative stabilization of 100:10:1 for O2:NO:CO in Mb.28 Cb H-NOX does exhibit KD(CO) and KD(NO) which are only slightly higher than those of Tt H-NOX. Although Cb H-NOX is capable of forming an oxyferrous complex under atmospheric pressure, its KD(O2) is more than 600-fold higher than [O2] of the strict anaerobic conditions under which C. botulinum germinates, [O2] < 2 ppm (or 8.2 × 10−8 M at 25 °C).29 Therefore, obligate anaerobe C. botulinum unlikely depends on Cb H-NOX to sense the level of oxygen.
Contrary to its stabilizing effect for oxyferrous Cb H-NOX, distal Y139, seems to destabilize the Fe–His bond in the 6c NO-heme-His complex in Cb H-NOX, as less 6c NO-heme-His converts to 5c NO-heme complex in Y139F Cb H-NOX, indicated by both optical and EPR data (Figures 1 and 4). A more stable 6c NO-heme-His complex in Y139F Cb H-NOX was previously reported based on optical data6 and substantiated by our EPR study (Figures 4 and 5). On the other hand, mutations of the corresponding distal tyrosine in Tt H-NOX, Y140L, and Y140F, do not affect the coordination number of NO complex as demonstrated in previous UV–vis optical study20 and by our EPR data. Interestingly, F142Y mutation, which introduces a distal tyrosine into one of the H-NOXs from Legionella pneumophila (L2 H-NOX), stabilizes its 6c NO-heme-His complex.20 It is still not clear why distal tyrosine in different H-NOXs have different effects on the stability of the Fe–His bond in their 6c NO-heme-His complexes, as DFT calculations suggest that hydrogen bond energy of a phenol group to NO is similar in both 5c and 6c heme-NO complexes.30 Detailed resonance Raman spectroscopy studies on the NO complexes of Cb and Tt H-NOXs and their distal tyrosine to phenylalanine mutants are underway and may provide new insights into this issue. EPR evidence in this study further reveals that distal tyrosine may tune the heme geometry of the 6c NO-heme-His complex in an H-NOX. This is demonstrated by the decreased rhombicity of the 6c NO-heme-His complex in Y140F Tt H-NOX (Figure 5).
Supplementary Material
Funding
This work was supported by NIH Grant RO1 HL 095820 to A.-L.T.
ABBREVIATIONS
- H-NOX
heme nitric oxide and oxygen binding protein
- Cb H-NOX
H-NOX from Clostridium botulinum
- L2 H-NOX
H-NOX from Legionella pneumophila
- Ns H-NOX
H-NOX from Nostoc punctiforme
- Tt H-NOX
H-NOX from Thermoanaerobacter tengcongensis
- Vc H-NOX
H-NOX from Vibrio cholera
- sGC
soluble guanylyl cyclase
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.5b00994.
(1) CO and NO binding parameters of Y140F Tt H-NOX (Table S1); (2) binding of CO to wt Cb H-NOX (Figure S1); (3) binding of CO to Y139F Cb H-NOX (Figure S2); (4) computer fitting of the NO binding to the 6c NO-heme-His complex in Cb H-NOX (Figure S3); (5) kinetics of the formation/dissociation of O2 complex of Tt H-NOX (Figure S4); and (6) optical spectra of resting wt Cb and Tt H-NOXs (Figure S5) (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Boon EM, and Marletta MA (2005) Ligand specificity of H-NOX domains: from sGC to bacterial NO sensors. J. Inorg. Biochem 99, 892–902. [DOI] [PubMed] [Google Scholar]
- (2).Shimizu T, Huang D, Yan F, Stranava M, Bartosova M, Fojtikova V, and Martinkova M (2015) Gaseous O2, NO, and CO in signal transduction: structure and function relationships of heme-based gas sensors and heme-redox sensors. Chem. Rev 115, 6491–6533. [DOI] [PubMed] [Google Scholar]
- (3).Tsai A-L, Martin E, Berka V, and Olson JS (2012) How do heme-protein sensors exclude oxygen? Lessons learned from cytochrome c’, Nostoc puntiforme heme nitric oxide/oxygen-binding domain, and soluble guanylyl cyclase. Antioxid. Redox Signaling 17, 1246–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Derbyshire ER, and Marletta MA (2009) Biochemistry of soluble guanylate cyclase. Handb Exp Pharmacol 191, 17–31. [DOI] [PubMed] [Google Scholar]
- (5).Wu G, Liu W, Berka V, and Tsai A-L (2013) The Selectivity of Vibrio cholerae H-NOX for Gaseous Ligands Follows the ″Sliding Scale Rule″ Hypothesis. Ligand Interactions with both Ferrous and Ferric Vc H-NOX. Biochemistry 52, 9432–9446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Nioche P, Berka V, Vipond J, Minton N, Tsai A-L, and Raman CS (2004) Femtomolar sensitivity of a NO sensor from Clostridium botulinum. Science 306, 1550–1553. [DOI] [PubMed] [Google Scholar]
- (7).Pellicena P, Karow DS, Boon EM, Marletta MA, and Kuriyan J (2004) Crystal structure of an oxygen-binding heme domain related to soluble guanylate cyclases. Proc. Natl. Acad. Sci. U. S. A 101, 12854–12859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Ma X, Sayed N, Beuve A, and van den Akker F (2007) NO and CO differentially activate soluble guanylyl cyclase via a heme pivot-bend mechanism. EMBO J. 26, 578–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Herzik MA Jr., Jonnalagadda R, Kuriyan J, and Marletta MA (2014) Structural insights into the role of iron-histidine bond cleavage in nitric oxide-induced activation of H-NOX gas sensor proteins. Proc. Natl. Acad. Sci. U. S. A 111, E4156–4164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Erbil WK, Price MS, Wemmer DE, and Marletta MA (2009) A structural basis for H-NOX signaling in Shewanella oneidensis by trapping a histidine kinase inhibitory conformation. Proc. Natl. Acad. Sci. U. S. A 106, 19753–19760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Tsai A-L, Berka V, Martin E, and Olson JS (2012) A ″sliding scale rule″ for selectivity among NO, CO, and O(2) by heme protein sensors. Biochemistry 51, 172–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Tsai A-L, Berka V, Martin F, Ma X, van den Akker F, Fabian M, and Olson JS (2010) Is Nostoc H-NOX a NO sensor or redox switch? Biochemistry 49, 6587–6599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Brent RP (1973) Algorithms for Minimization without Derivatives, Prentice-Hall, Englewood Cliffs, NJ. [Google Scholar]
- (14).Olson JS (1981) Stopped-flow, rapid mixing measurements of ligand binding to hemoglobin and red cells. Methods Enzymol. 76, 631–651. [DOI] [PubMed] [Google Scholar]
- (15).Winter MB, Herzik MA Jr., Kuriyan J, and Marletta MA (2011) Tunnels modulate ligand flux in a heme nitric oxide/oxygen binding (H-NOX) domain. Proc. Natl. Acad. Sci. U. S. A 108, E881–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Martin E, Berka V, Tsai A-L, and Murad F (2005) Soluble guanylyl cyclase: the nitric oxide receptor. Methods Enzymol. 396, 478–492. [DOI] [PubMed] [Google Scholar]
- (17).Andrew CR, George SJ, Lawson DM, and Eady RR (2002) Six- to five-coordinate heme-nitrosyl conversion in cytochrome c’ and its relevance to guanylate cyclase. Biochemistry 41, 2353–2360. [DOI] [PubMed] [Google Scholar]
- (18).Tsai A-L, Berka V, Sharina I, and Martin E (2011) Dynamic ligand exchange in soluble guanylyl cyclase (sGC): implications for sGC regulation and desensitization. J. Biol. Chem 286, 43182–43192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Boon EM, Davis JH, Tran R, Karow DS, Huang SH, Pan D, Miazgowicz MM, Mathies RA, and Marletta MA (2006) Nitric oxide binding to prokaryotic homologs of the soluble guanylate cyclase beta1 H-NOX domain. J. Biol. Chem 281, 21892–21902. [DOI] [PubMed] [Google Scholar]
- (20).Boon EM, Huang SH, and Marletta MA (2005) A molecular basis for NO selectivity in soluble guanylate cyclase. Nat. Chem. Biol 1, 53–59. [DOI] [PubMed] [Google Scholar]
- (21).Karow DS, Pan D, Tran R, Pellicena P, Presley A, Mathies RA, and Marletta MA (2004) Spectroscopic characterization of the soluble guanylate cyclase-like heme domains from Vibrio cholerae and Thermoanaerobacter tengcongensis. Biochemistry 43, 10203–10211. [DOI] [PubMed] [Google Scholar]
- (22).Weinert EE, Phillips-Piro CM, Tran R, Mathies RA, and Marletta MA (2011) Controlling conformational flexibility of an O2-binding H-NOX domain. Biochemistry 50, 6832–6840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Boon EM, and Marletta MA (2006) Sensitive and selective detection of nitric oxide using an H-NOX domain. J. Am. Chem. Soc 128, 10022–10023. [DOI] [PubMed] [Google Scholar]
- (24).Zhao Y, Brandish PE, Ballou DP, and Marletta MA (1999) A molecular basis for nitric oxide sensing by soluble guanylate cyclase. Proc. Natl. Acad. Sci. U. S. A 96, 14753–14758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Kharitonov VG, Russwurm M, Magde D, Sharma VS, and Koesling D (1997) Dissociation of nitric oxide from soluble guanylate cyclase. Biochem. Biophys. Res. Commun 239, 284–286. [DOI] [PubMed] [Google Scholar]
- (26).Springer BA, Sligar SG, Olson JS, and Phillips GN Jr. (1994) Mechanisms of Ligand Recognition in Myoglobin. Chem. Rev 94, 699–714. [Google Scholar]
- (27).Menyhard DK (2011) Conformational selection mechanism governs oxygen ligation to H-NOX proteins. Bioorg. Med. Chem. Lett 21, 3523–3526. [DOI] [PubMed] [Google Scholar]
- (28).Olson JS, and Phillips GN Jr. (1997) Myoglobin discriminates between O2, NO, and CO by electrostatic interactions with the bound ligand. J. Biol. Inorg. Chem 2, 544–552. [Google Scholar]
- (29).Young-Perkins KE, and Merson RL (1987) Clostridium botulinum spore germination, outgrowth, and toxin production below 4.6; interactions between pH, total acidity, and buffering capacity. J. Food Sci 52, 1084–1088. [Google Scholar]
- (30).Tangen E, Svadberg A, and Ghosh A (2005) Toward modeling H-NOX domains: a DFT study of heme-NO complexes as hydrogen bond acceptors. Inorg. Chem 44, 7802–7805. [DOI] [PubMed] [Google Scholar]
- (31).Yoo BK, Lamarre I, Rappaport F, Nioche P, Raman CS, Martin JL, and Negrerie M (2012) Picosecond to second dynamics reveals a structural transition in Clostridium botulinum NO-sensor triggered by the activator BAY-41–2272. ACS Chem. Biol 7, 2046–2054. [DOI] [PubMed] [Google Scholar]
- (32).Martin E, Berka V, Sharina I, and Tsai A-L (2012) Mechanism of binding of NO to soluble guanylyl cyclase: implication for the second NO binding to the heme proximal site. Biochemistry 51, 2737–2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.